

ABSTRACT
Liver-specific deletion of autophagy-related genes in mice leads to hepatomegaly, liver injury and spontaneous liver tumorigenesis. Accumulating evidence indicates that p62/SQSTM1-mediated NFE2L2/Nrf2/(nuclear factor, erythroid 2 like 2) activation plays a critical role in promoting liver injury and tumorigenesis in autophagy-defective livers. However, the mechanisms of how persistent NFE2L2 activation induces liver injury and tumorigenesis are unknown. In a recent study, it was found that deletion of Mtor (mechanistic target of rapamycin kinase) or Rptor/Raptor attenuates hepatomegaly and liver injury in young liver-specific atg5 knockout mice but accelerates liver tumorigenesis in old mice likely due to feedback AKT activation. Overall, these findings suggest that both hyper- and hypo-activation of MTOR are detrimental to the liver resulting in the development of liver tumors. A balanced MTOR activity is critical to maintain the normal physiological functions of the liver, and caution should be exercised when treating hepatocellular carcinomas using MTOR inhibitors.
Abbreviations: Atg5: autophgy related 5; DKO: double-knockout; HCC: hepatocellular carcinoma; INS: insulin; INSR: insulin receptor; KEAP1: kelch-like ECH-associated protein 1; KO: knockout; MTOR: mechanistic target of rapamycin kinase; NFE2L2: nuclear factor, erythroid 2 like 2; raptor: regulatory associated protein of MTOR, complex 1; SQSTM1: sequestosome 1: tsc1: TSC complex subunit 1.
KEYWORDS: AKT, Atg5, hepatocellular carcinoma, Nrf2, proteotoxicity
The incidence and mortality rate of hepatocellular carcinoma (HCC) is increasing worldwide as a result of hepatitis B or C virus infection, chronic alcohol consumption, and obesity related non-alcoholic fatty liver diseases. The current treatment options for HCC are very limited due to an incomplete understanding of the mechanisms of HCC pathogenesis. A chronic multi-hit process has been a proposed mechanism for HCC that includes hepatocyte death, inflammation, fibrosis, cirrhosis, cycles of cell death and compensatory hepatocyte proliferation, which lead to genetic changes and neoplastic transformation resulting in liver tumor formation. Autophagy is a lysosomal degradation pathway, which plays a complex role in the development of tumors, either suppressing or promoting tumors, depending on the state of tumor formation. Genetic ablation of Atg5 (autophagy related 5) or Atg7 specifically in mouse livers leads to spontaneous liver tumorigenesis, supporting the notion that autophagy acts as a liver tumor suppressor. Remarkably, liver-specific atg5 (L-atg5) or atg7 knockout (KO) mice have increased hepatocyte death, hepatomegaly, inflammation, fibrosis, cirrhosis, compensatory hepatocyte proliferation and liver tumors, which are typical features that aptly mimic the pathogenesis of human HCC development. Thus, the autophagy-deficient mouse model may serve as an ideal model to study the fundamental mechanisms of HCC development. Earlier studies reveal that SQSTM1/p62 (sequestosome 1)-mediated persistent NFE2L2/Nrf2 (nuclear factor, erythroid 2 like 2) activation contributes to the liver injury and tumorigenesis in autophagy-deficient mouse livers. This notion is supported by the convincing evidence that deletion of either Sqstm1 or Nfe2l2 in L-atg5 KO mice attenuates the liver injury, hepatomegaly and tumorigenesis that are found in L-atg5 KO mice. However, the protective effects of deletion of Nfe2l2 are more evident than the deletion of Sqstm1 in L-atg5 KO mice as the tumorigenesis is completely abolished in nfe2l2 atg5 double-knockout (DKO) mice, whereas only decreased tumor numbers are observed in sqstm1 atg5 DKO mice. Because SQSTM1 activates NFE2L2 via the non-canonical NFE2L2 pathway by the sequestration of KEAP1 (kelch-like ECH-associated protein 1), the improvement of the liver pathology in sqstm1 atg5 DKO mice is likely a secondary effect due to the impaired NFE2L2 activation resulting from the ablation of SQSTM1 in L-atg5 KO mice. These observations indicate that persistent NFE2L2 activation due to the accumulation of SQSTM1 plays a predominant role in the liver injury and tumorigenesis in autophagy-deficient livers. However, persistent NFE2L2 activation alone does not trigger liver pathogenesis because L-keap1 KO mice have almost healthy livers. Therefore, it is the combination of NFE2L2 activation coupled with autophagy deficiency that results in the hepatomegaly, fibrosis and tumorigenesis. As a transcription factor, persistent NFE2L2 activation increases expression of a large set of genes resulting in a robust increase of newly synthesized proteins. It is conceivable that L-atg5 KO mice may behave like a water sink with a clogged drain. Under normal conditions, the flow-in water (newly synthesized proteins) runs out of the sink through the drain (autophagy) efficiently without causing any damage. However, a clogged drain (deficiency of autophagy) leads to the overflow of water, which will likely worsen due to excessive water flow with a fully-opened faucet (persistent NFE2L2 activation) (Figure 1A). Like an over-flowing sink, autophagy-deficient livers with over-accumulated proteins may lead to increased proteotoxicity and hepatocyte cell death. If the ‘sink hypothesis’ is correct, blocking the protein input should be beneficial for the improvement of the pathogenesis of autophagy-defective livers.
Figure 1.
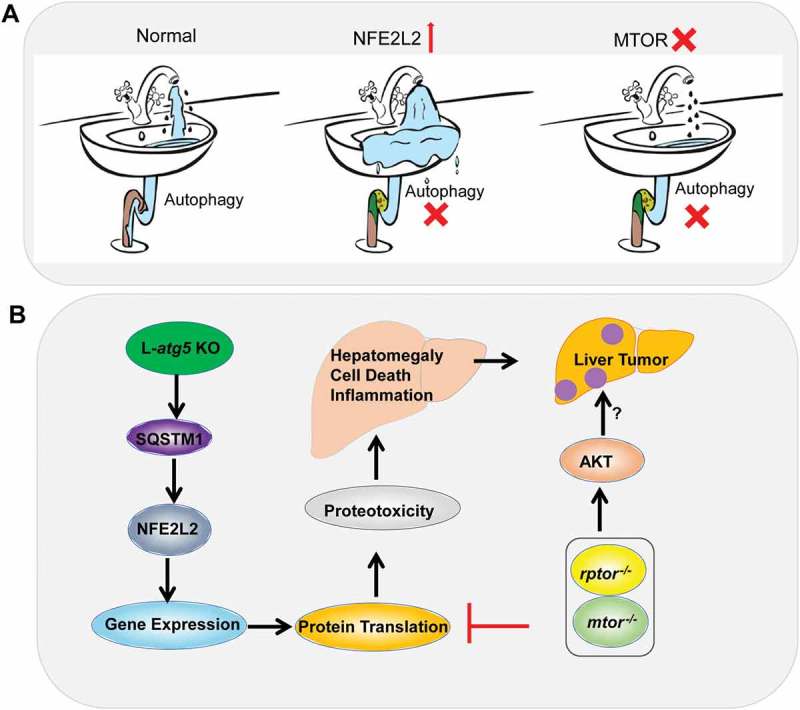
Hypothesis and molecular events of defective hepatic autophagy for liver pathogenesis. (A) A sink hypothesis. Under normal conditions, the incoming water (newly synthesized proteins) runs down the sink through the drain (autophagy) efficiently without causing any damage. A clogged drain (deficiency of autophagy) with excessive full water running (NFE2L2 activation) results in water overflow. A slow water stream (mtor deletion) slows down the water overflow (improves liver injury). (B) L-atg5 KO mice have impaired hepatic autophagy resulting in the accumulation of SQSTM1 and persistent NFE2L2 activation. Persistent NFE2L2 activation leads to increased expression of NFE2L2 target genes, protein translation and proteotoxicity, which results in cell death, hepatomegaly, chronic inflammation, fibrosis and hepatocellular adenoma. Deletion of either Mtor or Rptor decreases the general protein input resulting in decreased liver injury in young L-atg5 KO mice. However, deletion of either Mtor or Rptor also leads to the feedback activation of AKT and accelerates the development of liver tumors.
MTOR (mechanistic target of rapamycin kinase) is a key cellular nutrient sensor that regulates the cellular protein and lipid synthesis. In a recent study, we explored whether blunting of protein synthesis by deletion of Mtor would improve liver injury and tumorigenesis of the L-atg5 KO mice. Deletion of Mtor or Rptor/Raptor (regulatory associated protein of MTOR, complex 1) decreases hepatic levels of ubiquitinated proteins and attenuates NFE2L2 activation in L-atg5 KO mouse livers, which leads to decreased hepatomegaly, cell death and inflammation but not fibrosis in L-atg5 KO mice for the young mice at 2 months of age. These data generally support the ‘sink hypothesis’ that decreased production of hepatic proteins (decreased water flow) may indeed improve the liver pathogenesis of L-atg5 KO mice. Intriguingly, the protection against hepatomegaly, cell death and inflammation in the young L-atg5 KO mice by the deletion of Mtor or Rptor is gradually lost in old mice (age of 6–12 months), which is likely due to the chronic buildup of proteins over the longer time. Surprisingly, more than 50% of L-atg5 Mtor DKO and L-atg5 rptor DKO mice already develop spontaneous tumors but none of the L-atg5 KO mice have a tumor at 6 months of age. Our results are also in line with an early report that L-rptor KO mice have increased liver tumorigenesis when mice are challenged with the hepatic carcinogen diethylnitrosamine plus a high-fat diet. These data indicate that loss of MTOR, most likely MTORC1, promotes the liver tumorigenesis in autophagy-deficient mouse livers. These findings seem to be paradoxical to the common paradigm that at least 50% of HCCs are associated with increased MTOR activation. Indeed, L-tsc1 (TSC complex subunit 1) KO mice that have hyperactivation of MTOR also develop spontaneous liver tumors. How would decreased MTORC1 accelerate the tumorigenesis in autophagy-deficient livers? It is well known that the phosphoinositide 3-kinase-AKT pathway is frequently activated in human cancer including HCC. Growth factors such as INS (insulin) activate AKT and subsequently lead to the activation of MTORC1 via AKT-mediated phosphorylation and inhibition of the TSC1-TSC2 complex. However, MTORC1 activation can also inhibit AKT activation via the feed-back Inhibition of INSR (insulin receptor) substrates. Indeed, L-atg5 mtor DKO and L-atg5 rptor DKO mice have increased AKT activation compared with L-atg5 KO and matched wild-type mice. Future studies to blunt AKT pathways in L-atg5 mtor DKO and L-atg5 rptor DKO mice are needed to further confirm the causal role of AKT in promoting tumorigenesis of these mice.
Taken together, it seems that both hyper- and hypo-activation of MTOR may yield unwanted harmful effects to the liver resulting in the development of liver tumors, suggesting the balance of the MTOR activity is critical to maintain the normal physiological functions of the liver. Currently multiple clinical trials are using MTOR inhibitors in the treatment of cancers, including HCC; however, our results raise concern for these ongoing trials due to the potentially harmful side effects of chronic usage of MTOR inhibitors. The molecular events and signaling pathways that may contribute to the pathogenesis of autophagy-defective livers are summarized in Figure 1B.
Funding Statement
The research was supported in part by the NIAAA R01 AA020518, U01 AA024733, R21 AA027250 (W.X.D) and R21 AA026904 (H.M.N).