

SUMMARY
Pathogenic and other cytoplasmic DNAs activate the cGAS-STING pathway to induce inflammation via transcriptional activation by IRF3 and NFκB, but the functional consequences of exposing cGAS to chromosomes upon mitotic nuclear envelope breakdown are unknown. Here we show that nucleosomes competitively inhibit DNA-dependent cGAS activation, and that the cGAS-STING pathway is not effectively activated during normal mitosis. However, during mitotic arrest, low level cGAS-dependent IRF3 phosphorylation slowly accumulates without triggering inflammation. Phosphorylated IRF3, independently of its DNA-binding domain, stimulates apoptosis through alleviating Bcl-xL-dependent suppression of mitochondrial outer membrane permeabilization. We propose that slow accumulation of phosphorylated IRF3, normally not sufficient for inducing inflammation, can trigger transcription-independent induction of apoptosis upon mitotic aberrations. Accordingly, expression of cGAS and IRF3 in cancer cells makes mouse xenograft tumors responsive to the anti-mitotic agent taxol. TCGA data for non-small cell lung cancer patients also suggest an effect of cGAS expression on taxane response.
Graphical Abstract
In Brief
Activation of the cytoplasmic DNA sensor cGAS is inhibited by nucleosomes during normal mitosis but during prolonged mitotic arrest, cGAS functions to promote cell death via slowly-accumulating phosphorylation of IRF3.
INTRODUCTION
Chromosomal DNA is sequestered from the cytoplasm by the nuclear envelope during interphase, but in mitosis, the nuclear envelope breaks down, and chromosomes promote spindle assembly (Zierhut and Funabiki, 2015). As spindle assembly and functional nuclear formation depend on nucleosomes, we speculated that this nucleosome-dependency ensured that assembly of these structures is limited to self-chromosomes, discriminating them from foreign pathogenic DNA (Zierhut and Funabiki, 2015; Zierhut et al., 2014). Unlike fertilization, when nucleosome-free sperm DNA is rapidly chromatinized in egg cytoplasm, in somatic cells invading pathogenic DNA is sensed by pattern-recognition receptors such as cGAS (Chen et al., 2016; Sun et al., 2013) (Figure 1). Upon binding to DNA, cGAS is activated to synthesize a cyclic GMP-AMP dinucleotide, cGAMP (Ablasser et al., 2013; Chen et al., 2016; Gao et al., 2013a; Sun et al., 2013; Wu et al., 2013). cGAMP recognition by STING (Chen et al., 2016; Ishikawa and Barber, 2008; Wu et al., 2013) triggers activation of the transcription factors IRF3 and NFκB (Chen et al., 2016; Sun et al., 2013; Wu et al., 2013), which induce inflammation through genes such as the type I interferon IFNβ (Chen et al., 2016). The cGAS pathway can also promote apoptosis by transcriptional activation of apoptotic regulators, as well as by a transcription-independent role of IRF3 (Chattopadhyay et al., 2010; Gaidt et al., 2017; Gulen et al., 2017; Heylbroeck et al., 2000; Nakhaei et al., 2012; Petrasek et al., 2013; Tang et al., 2016).
Figure 1. Nucleosomes bind cGAS with higher affinity than does naked DNA but inhibit cGAS catalytic activity.

(A) Schematics of cGAS signaling.
(B) Beads with or without naked DNA/nucleosomes were incubated with 35S-labelled cGAS. Proteins and DNA associated with beads were detected by fluorometry and Coomassie brilliant blue (CBB) (top), and SYBR-Safe (bottom).
(C) Gel mobility shift binding analysis with purified cGAS and naked DNA or mononucleosomes. Means and SEM (n ≥ 3). See Figure S1C for an example gel.
(D and E) Binding analysis of purified wild type cGAS or cGAS mutated in the DNA binding domain (KRKK mutant) for naked DNA or mononucleosomes. Means and SEM (n ≥ 3).
(F) His-tagged H2A–B or H3–H4 were incubated with cGAS and Talon beads. Proteins associated with beads were detected by CBB. H2A-B_ap*A, acidic patch to alanine mutant; H2A-B_ap*KR, acidic patch to lysine/arginine mutant.
(G–I) Thin-layer chromatography analysis of cGAS activity (see also Figure S1D). (G) Typical example using either naked DNA or nucleosomes as cGAS stimulator. (H) Apparent Kcat of cGAS. Mean values and SEM (n=3). (I) Apparent Kcat of cGAS with naked DNA and increasing concentrations of mononucleosomes. Mean values and SEM (n=3).
See also Figure S1.
How does chromosomal DNA normally insulate itself from cGAS? While the nuclear envelope may limit access of cGAS to chromosomes in interphase, cGAS can be recruited to chromosomes during mitosis (Gentili et al., 2019; Harding et al., 2017; Yang et al., 2017). Given the stability of cGAMP (Eaglesham et al., 2019), unless cGAS is silenced during mitosis, it could trigger inflammatory transcription upon mitotic exit. Similarly, in interphase, cases have been reported, where cGAS can reside in the nucleus, but cGAS activation is attenuated (Gentili et al., 2019). The mechanism underlying this attenuation is not understood.
Taxane drugs, such as taxol (paclitaxel), which stabilize microtubules and interfere with mitosis, are commonly used in cancer chemotherapy. However, how microtubule stabilization is transferred into anti-cancer effects has remained elusive (Mitchison et al., 2017; Mitchison, 2012; Weaver, 2014). When proliferating tissue culture cells are treated with taxol, they are arrested in mitosis by the spindle assembly checkpoint (London and Biggins, 2014). These cells eventually become committed to one of the two fates; exiting mitosis with failed chromosome segregation (“slippage”), or apoptotic cell death while arrested in mitosis (Gascoigne and Taylor, 2008; Topham and Taylor, 2013; Yang et al., 2009). This fate decision is made stochastically, and the probability of these choices differs widely amongst cell lines (Gascoigne and Taylor, 2008). Two competing mechanisms determine the timing of slippage and apoptosis. Slippage timing is affected by checkpoint strength, sustaining cyclin B above threshold levels (Collin et al., 2013). Apoptosis timing is determined by the rate at which “death signals” accumulate until threshold levels (Gascoigne and Taylor, 2008; Topham and Taylor, 2013). Apoptosis during mitotic arrest is executed by mitochondrial outer membrane permeabilization (MOMP) and subsequent caspase activation in a p53 independent manner (Blagosklonny, 2007; Topham and Taylor, 2013; Topham et al., 2015). Whichever mechanism reaches its threshold earlier manifests itself. However, the mechanisms that define the kinetics of death signal accumulation and threshold are poorly understood.
Here, we reveal that nucleosomes have a higher affinity for cGAS than naked DNA, but suppress DNA-dependent stimulation of cGAS activity. Accordingly, chromosomes are refractory to the innate immune system during normal mitotic progression. However, when cells arrest in mitosis, cGAS-dependent IRF3 phosphorylation slowly accumulates, and triggers intrinsic apoptosis through releasing Bcl-xL-dependent suppression of MOMP by a transcription-independent mechanism. Mouse xenograft experiments, as well as data from The Cancer Genome Atlas (TCGA), indicate a role for cGAS in the tumor response to taxane treatment.
RESULTS
Nucleosomes competitively suppress DNA-induced cGAS activation
Although it has been reported that chromatin can interact with cGAS (Mackenzie et al., 2017), the functional implication was unclear. We found that cGAS binds nucleosome arrays much better than naked DNA in vitro (Figure. 1B), and that even in physiological M-phase Xenopus egg extracts, did nucleosomes bind cGAS (Figure S1A). Quantitative gel-shift analysis using purified components further demonstrated that recombinant cGAS (Figure S1B) has ~2-fold higher affinity for mononucleosomes than for naked DNA (Figures 1C and S1C). Mutating DNA binding residues (Civril et al., 2013) greatly reduced affinity for naked DNA (KRKK mutant, Figure 1D), but only modestly for nucleosomes (Figure 1E), suggesting that mechanisms of cGAS binding to naked DNA and to nucleosomes are distinct. Indeed, cGAS bound the purified H2A–H2B heterodimer (Figure 1F), but mutating the acidic patch of H2A–H2B, which is frequently targeted by nucleosome binding proteins, to either alanine or lysine/arginine abrogated the interaction (ap*A and ap*KR Figure 1F).
In agreement with a recent report (Mackenzie et al., 2017), nucleosomes were able to stimulate cGAMP synthesis by cGAS, but when compared to DNA-bound cGAS, nucleosome-bound cGAS showed a ~3-fold reduction in the apparent catalysis rate (Kcat, Figures 1G, 1H, S1D). When increasing amounts of nucleosomes were added to a constant amount of naked DNA and cGAS, nucleosomes competitively suppressed the capacity of naked DNA to activate cGAS (Figure 1I). These results suggest that, on chromosomes, nucleosomes could interfere with cGAS activation by nucleosome-free regions.
cGAS-dependent IRF3 phosphorylation slowly accumulates during mitotic arrest
Consistent with the interaction between cGAS and nucleosomes, cGAS is recruited to mitotic chromosomes (Gentili et al., 2019; Harding et al., 2017; Yang et al., 2017). Live imaging of GFP-tagged cGAS (GFP-cGAS) in HeLa cells showed that cGAS associates with chromosomes immediately after nuclear envelope breakdown (Figures S1E and S1F, Movie S1), and this localization was not affected by mutations in the cGAS DNA binding domain (Fig. S1G). Immunofluorescence of endogenous cGAS confirmed its localization to mitotic chromosomes in HeLa cells, and in telomerase-immortalized human BJ hTERT fibroblasts (Figures S1H-J).
To determine if chromosome-bound cGAS could activate downstream signaling during mitosis, we monitored IRF3 phosphorylation at Ser396, a widely used indicator of cGAS activation (Chen et al., 2016). We initially found little evidence for phosphorylation on the endogenous protein in HeLa cells, where STING function is compromised by papillomavirus E7 protein (Lau et al., 2015), but when detection sensitivity was increased by overexpressing GFP-tagged IRF3, a slow cGAS-dependent increase in phospho-Ser396 was revealed during mitotic arrest induced with taxol and nocodazole (Figures 2A, B and S2A), in a manner dependent on the IRF3 kinase TBK1(Chen et al., 2016) (Figure 2B). Mitotic, cGAS-dependent, phosphorylation was also observed on endogenous IRF3 at Ser396 after enrichment by immunoprecipitation, as well as Ser386 (Schoggins et al., 2014) (Figure 2C, D and S2B). Association of cGAS with mitotic chromosomes, and mitotic IRF3 phosphorylation was also observed in untransformed human embryonic stem cells (Figures S2C and S2D).
Figure 2. The cGAS pathway is inactive during normal mitosis but is activated late in mitotic arrest.

(A) Western blot analysis of IRF3 phosphorylation of the indicated HeLa cells, harvested either in G2 or after the indicated times during mitotic arrest in 500 nM taxol, 10 μM proTAME.
(B) Western blot analysis of GFP-IRF3 S396 phosphorylation in mitotic arrest using the indicated cell lines (top). Bottom, verification of CRISPR-Cas9 disruptions.
(C) Western blot analysis of S396 phosphorylation of endogenous IRF3 immunoprecipitated from G2 arrested HeLa cells, or from cells at the indicated time points during arrest in 500 nM taxol, 10 μM proTAME.
(D) Western blot analysis of IRF3 S386 phosphorylation in HeLa cells harvested either in G2 or after the indicated times during arrest in 500 nM taxol, 10 μM proTAME.
(E) Fraction of cells (determined by live microscopy) of the indicated cell line and treatment that die in an unperturbed mitosis (n=30-50 for each sample).
(F) Length of mitosis (nuclear envelope breakdown - anaphase) determined by 15 min-interval time-lapse microscopy (n=30-50 for each sample). Red line: median.
siCNTRL, non-targeting control siRNA; sicGAS, siRNA targeting cGAS.
See also Figure S2.
In BJ hTERT, which maintains an intact cGAS-STING pathway (Ablasser et al., 2013; Konno et al., 2013), levels of mitotic IRF3 phosphorylation, albeit detectable, appear to be highly suppressed compared to the level achieved by DNA transfection of asynchronous cells (Figures S2E and S2F). Other known consequences of cGAS signaling, such as IκBα degradation and phosphorylation of c-Jun kinase (JNK) can also be stimulated during mitotic arrest, but only slowly (Figures S2G-S2J) (Abe and Barber, 2014).
Recently, mitotic defects were indirectly linked to cGAS activation in the following interphase (Dou et al., 2017; Glück et al., 2017; Harding et al., 2017; Mackenzie et al., 2017; Yang et al., 2017). Micronuclei generated from aberrant mitosis can activate cGAS due to their compromised nuclear envelopes. However, in our experiments, cells were treated with high taxol concentrations (500 nM), and additionally with proTAME, an inhibitor of the anaphase promoting complex (Zeng et al., 2010), resulting in tight cell cycle arrest that prevented slippage and micronuclei formation (see below, Figure 4D). Therefore, the observed cGAS-dependent IRF3 phosphorylation did not arise from micronuclei, but was specific to mitotic cells. Overall, we conclude that the cGAS pathway, despite chromosomal enrichment of cGAS during mitosis, requires long periods of time for activation.
Figure 4. cGAS promotes cell death in mitosis.

(A) Western blot analysis of cGAS levels. Dilution series from extracts made from siCNTRL cells were used to assess cGAS depletion. The vertical line indicates removed irrelevant lanes. (B–J) Time-lapse microscopy analysis of mitotic cell death. Cells were released from G2 arrest into M phase with the indicated drugs, and duration of mitosis until death or slippage was monitored. Red lines, median. siCNTRL, non-targeting control siRNA; sicGAS, siRNA targeting cGAS.
(B and C) Fraction of mitotic cell death of HeLa cells in 10 nM taxol. (B) Means and range (error bars) from three experiments (n=60 each). (C) Timing of mitotic cell death and slippage of individual cells (n=60 each).
(D and E) Timing of cell death in HeLa cells released into 500 nM taxol 10 μM proTAME. (D) Timing of death of individual cells (n=60). (E) Means and SD from six experiments (dots are medians from each experiment).
(F and G) Timing of cell death in BJ hTERT released into 500 nM taxol 10 μM proTAME. (F) Timing of death in individual cells (n=80). (G) Means and SD from six experiments (dots are medians from each experiment).
(H) Timing of mitotic cell death and slippage of ARPE-19 hTERT cells (n=40 each) released into 500 nM taxol 10 μM proTAME. One of two replicates shown.
(I) Complementation analysis in HeLa cells. Timing of mitotic cell death of individual HeLa cells (n ~50 each) was analyzed. cGAScat, catalytic mutant. cGASdna, DNA binding-induced catalytic activity mutant. See Figure S4J for Western blot of cGAS. One of two replicates shown.
(J) Analysis of mitotic lifespan of individual cells of cGAS disruption mutants (n=60) released into 500 nM taxol 10 μM proTAME. Cell lines generated with two different short guide RNAs (sg #1 and sg #2), as well as clones from these that express wild type GFP-cGAS were used. Untr., no siRNA treatment. See Figure S4L for Western blot of cGAS levels.
See also Figure S4.
cGAS affects cell fate during mitotic arrest
cGAS binding to chromosomes may not have any consequences during normal mitosis, which usually completes within 30 min, whereas cGAS signaling occurs over multiple hours (Sun et al., 2013) (Figures 2A and 2C). Indeed, cGAS depletion affected neither viability nor duration of unperturbed mitosis (Figures 2E and 2F). However, when cells are arrested in mitosis, even low cGAS activity may have functional consequences. Because cGAS protein levels vary drastically between cell lines (Figure 3A), and because cGAS and its downstream effectors STING and IRF3 had been suggested to promote cell death (Chattopadhyay et al., 2010; Gaidt et al., 2017; Nakhaei et al., 2012; Petrasek et al., 2013), we tested whether cGAS affects the decision between slippage and death. By tracking individual mitotic cells by live imaging (Gascoigne and Taylor, 2008; Wheelock et al., 2017), we monitored death rates as well as mitotic lifespans of individual cells.
Figure 3. cGAS expression correlates with taxol sensitivity in a panel of breast cancer cells.

(A) Western blot analysis of cGAS and STING in the indicated cell lines. The vertical line indicates removed irrelevant lanes.
(B-F) Time-lapse microscopy analysis of mitotic cell death in the indicated asynchronous populations following the indicated treatments.
(B) Fraction mitotic cell death in 10 nM taxol (n=50 for each sample). Averages and range are shown from two experiments.
(C) Fraction mitotic cell death in 500 nM taxol (n=45 for each sample). Data from two (HCC1143, MDA-MB-157) or three experiments (all others, averages and range plotted).
(D) Mitotic cell death frequency of unperturbed mitosis (n=40 for each sample).
(E) Effect of cGAS knockdown on mitotic cell death (10 nM taxol) in HCC1143, MDA-MB-157 and BT549 treated with the indicated siRNA (n=60 for each sample). Data from two (HCC1143, MDA-MB-157, averages and range plotted) or one experiment (BT549).
(F) Effect of cGAS knockdown on mitotic cell death (500 nM taxol, n=60 for each sample) in MDA-MB-231 treated with the indicated siRNA.
siCNTRL, non-targeting control siRNA; sicGAS, siRNA targeting cGAS.
See also Figure S3.
Since breast cancer is frequently treated with taxanes (Jordan and Wilson, 2004), we first analyzed seven breast cancer cell lines (Figure 3A), four of which expressed cGAS, and three of which did not. Three of the four cGAS-expressing cell lines (HCC1143, MDA-MB-231, BT549) showed stimulation of IRF3 phosphorylation during mitotic arrest (Figures S3A-D), while the fourth cGAS-expressing cell line, MDA-MB-157, had comparable basal IRF3 phosphorylation in an asynchronous population. When exposed to 10 nM taxol, similar to the estimated concentration within tumors during clinical treatment (Mitchison, 2012; Weaver, 2014), the three cGAS-negative cell lines did not die during mitotic arrest, rather slipping into interphase eventually. In contrast, three out of four cGAS-positive cell lines were prone to die in mitosis (Figures 3B and S3E). In the fourth cGAS-positive cell line, MDA-MB-231, mitotic cell death appears to be prevented by faster slippage (Figure S3E). When taxol was increased to 500 nM, arrest times were generally extended in cGAS negative cell lines and in MDA-MB-231, increasing the chance to undergo mitotic cell death. However, cells of the cGAS-negative cell lines CAL51 and T47D were still more likely to undergo slippage, which generally occurred later than death (Figures 3C and S3F). Therefore, the reduced tendency of mitotic cell death in cGAS-negative cell lines is not caused by faster slippage. No difference in mitotic viability was observed in the absence of taxol (Figure 3D). siRNA-mediated depletion of cGAS reduced mitotic mortality by ~50% in the three most sensitive cell lines (Figures 3E and S3G), with cGAS knockdown generally resulting in an extension of mitotic lifespan (Figure S3H). cGAS knockdown in MDA-MB-231 reduced mitotic cell death following treatment with high taxol concentrations (Figures 3F, S3I and S3J). Another cGAS expressing cell line, HBL-100, also showed high mitotic mortality in taxol in a manner partially dependent on cGAS (Figures S3K-S3N). Altogether, the expression of cGAS correlates with susceptibility to die during taxol-induced mitotic arrest in a number of cell lines.
cGAS activity promotes cell death during mitotic arrest
To characterize the mechanism by which cGAS promotes mitotic cell death in detail, we focused on two cell lines, HeLa Flp-In T-Rex (referred to as “HeLa” hereafter) (Gassmann et al., 2010; Tighe et al., 2008), which were used to inducibly express siRNA-resistant, wild type or mutant GFP-cGAS, and BJ-hTERT fibroblasts. Similar to our observations in breast cancer cells, cGAS knockdown reduced mitotic cell death in HeLa cells treated with 10 nM taxol (Figures 4A and 4B). This effect was not due to accelerated slippage, since cGAS depletion extended mitotic lifespan (Figure 4C).
To verify if cGAS depletion delayed cell death, we blocked slippage using 500 nM taxol and proTAME (Figure S4A). Depleting cGAS in HeLa cells (Figures 4A, 4D, 4E, S4B, Movie S1), or BJ-hTERT cells (Figures 4A, 4F and 4G) extended mitotic lifespan. cGAS depletion also extended lifespan in another HeLa line, which had an overall longer lifespan than HeLa Flp-In T-REx (Figure S4B). cGAS depletion also delayed death during mitotic arrest by nocodazole (Figures S4C-S4E), and by proTAME alone (Figures S4F-S4I). In contrast, ARPE-19 hTERT retinal pigment epithelium cells, which express very little cGAS (Figure 4A), tolerated mitotic arrest extremely well, and cGAS siRNA treatment did not have any impact on mitotic lifespan (Figure 4H).
Multiple lines of evidence indicate that cGAS siRNA extends mitotic lifespan due to reduced cGAS activity. First, the phenotype can be reversed by expressing siRNA resistant wild type cGAS (GFP-cGASwt), but not by two catalytic mutants, GFP-cGAScat (E225A D227A) mutated in the catalytic pocket, or GFP-cGASDNA (K407E K411A) in which DNA binding does not induce catalysis (Civril et al., 2013) (Figures 4I and S4J). Second, control siRNA by itself did not affect timing of death (Figure S4K). Third, CRISPR-Cas9 mediated disruption of cGAS with two different short guide RNAs caused phenotypes identical to cGAS knockdown (Figures 4J and S4L). cGAS does not appear to be a general death accelerator, as its depletion did not affect timing of death after UV irradiation (Figures S4M and S4N). Altogether, these results indicate that cGAS promotes death during mitotic arrest.
cGAS promotes apoptosis by suppressing Bcl-xL-dependent inhibition of MOMP
Mitotic cell death was always accompanied by caspase-3/7 activation (Figures S5A-S5C), indicating that cGAS acts by accelerating apoptosis. Consistently, the caspase inhibitor ZVAD delayed mitotic cell death (Figures S5D), and increased the surviving fraction of cells (Figure S5E), whereas the necroptosis inhibitor Nec-1s (Takahashi et al., 2012) had no such effect (Figure S5E). A crucial step in apoptosis is MOMP, mediated by the Bax protein family (Figure S5A). This releases SMAC and cytochrome C from mitochondria, and activates the effector caspases, caspase-3 and caspase-7 (Figure S5A). cGAS depletion delayed Bax activation (Figures 5A-C) and release of SMAC and cytochrome C (Figures 5D-5F and S5F-S5K), suggesting that cGAS promotes apoptosis by regulating MOMP.
Figure 5. cGAS promotes mitotic cell death by accelerating MOMP.

(A–C) Bax activation in the indicated cell lines released from G2 into 500 nM taxol 10 μM proTAME. Cells were stained with the 6A7 antibody recognizing activated Bax. (A and B) Quantifications of HeLa cells (A, mean and range from two experiments) and BJ hTERT cells (B). (C) Representative images of HeLa cells at 14 h. Arrowheads, cells positive for activated Bax.
(D–F) SMAC release from mitochondria in the indicated cell lines released from G2 into 500 nM taxol 10 μM proTAME. Cells were stained for SMAC. (D and E) Quantifications of HeLa cells (D, mean and range from two experiments) and BJ hTERT cells (E). (F) Representative images of HeLa cells at 14 h. Arrowheads, cells with delocalized SMAC. Note that delocalization manifests itself as weak signal intensity. See Figure S5J for SMAC staining of untreated mitotic cells.
siCNTRL, non-targeting control siRNA; sicGAS, siRNA targeting cGAS.
See also Figure S5.
Two members of the anti-apoptotic Bcl2 family, Bcl-xL and Mcl1, are the major inhibitors of MOMP during mitosis (Figure S5A) (Shi et al., 2011; Topham et al., 2015), and Mcl1 was suggested to be the timer for mitotic death because it is degraded during mitotic arrest (Harley et al., 2010; Sloss et al., 2016; Topham and Taylor, 2013). However, Mcl1 started degrading hours before death, independently of cGAS (Figures S5L-S5N). In contrast, BcL-xL was more stable, and cGAS depletion mildly increased BcL-xL levels in mitotic HeLa cells (Figures S5L and S5N). In BJ hTERT, Bcl-xL gradually decreased during mitotic arrest in a manner dependent on cGAS (Figures S5M and S5N).
To determine if cGAS operates on the Mcl1 or Bcl-xL branch, we used inhibitors to these proteins (S63845 and ABT-263, respectively). Either inhibitor accelerated the timing of death in HeLa cells, and simultaneous treatment with both inhibitors immediately triggered apoptosis before mitotic entry (Figure S5O), confirming that Mcl1 and Bcl-xL represent the major mechanisms to suppress apoptosis in this cell line. In contrast, a specific inhibitor of Bcl-2, ABT-199 (Souers et al., 2013), had no effect on mitotic cell death (Figure S5P). While depletion of cGAS delayed the timing of mitotic death in cells treated with the Mcl1 inhibitor, it did not do so for cells treated with the Bcl-xL inhibitor (Figure S5O), suggesting that cGAS operates upstream of Bcl-xL, but independently of Mcl1. Altogether, the simplest explanation is that the cGAS pathway directly or indirectly overcomes the Bcl-xL-dependent suppression of Bax activation and MOMP.
The cGAS-cGAMP-STING-IRF3 axis promotes mitotic cell death independently of transcription induction
As the cGAS-STING-IRF3 pathway could promote death through regulating apoptotic protein expression by p53-dependent and p53-independent mechanisms (Goubau et al., 2009; Gulen et al., 2017; Knowlton et al., 2012; Lallemand et al., 2007), steady-state cGAS signaling could promote mitotic death either by changing gene expression directly, or through IFNβ-stimulated JAK-STAT signaling (Wang et al., 2017). However, an involvement of p53 is unlikely since mitotic cell death is independent of p53 (Blagosklonny, 2007), and cGAS promotes mitotic cell death in cell lines with defective p53 (HeLa, HCC1143, MDA-MB-157, MDA-MB-231, BT549, Figures 3B-3F and 4B-4D). Furthermore, cGAS depletion did not cause substantial changes in levels of any apoptotic protein tested (Figure 6A) or of an interferon-stimulated gene, IRF1 (Figure 6B). Treating BJ hTERT cells with neutralizing antibodies to the IFNAR2 subunit of the IFNβ receptor (Wu et al., 2018) abrogated the capacity of IFNβ to induce IRF1 (Figure 6B) but did not affect mitotic lifespan (Figure 6C), and neither did treatment with a pan JAK inhibitor, pyridone 6 (Thompson et al., 2002) (Figure S6A). Thus, cGAS regulates mitotic cell death independently of IFNβ production.
Figure 6. The cGAS-cGAMP-STING-IRF3 axis promotes mitotic cell death independently of transcriptional induction.

(A) Western blot analysis (means and SEM) of the indicated proteins in the indicated cell lines arrested in G2.
(B and C) Treatment of cells with neutralizing anti-IFNAR 2 antibodies. (B) Western blot analysis of BJ hTERT cells following IFNβ addition with and without anti-IFNAR2 antibodies.
(C) Mitotic lifespan of the indicated BJ hTERT cells (n=34-40 each) grown in the presence of the indicated antibodies and released from G2 arrest into 500 nM taxol 10 μM proTAME.
(D and E) Mitotic lifespan of cells (n=50 each) arrested in G2 and released into 500 nM taxol 10 μM proTAME. After 1.5 h (HeLa Cells, D) or 4 h (BJ hTERT cells, E), cGAMP was added to 33 μM for the indicated cells. Only cells that had already entered mitosis were analyzed.
(F) IFNB1 expression by quantitative PCR in the indicated cells. +DNA, asynchronous cells transfected with naked DNA. Cells were released from G2 arrest into media containing the indicated drugs. Mean and range from two experiments.
(G and H) Analysis of transcription inhibition. (G) Mitotic lifespan of cells (n=60 each) arrested in G2 and released into 500 nM taxol 10 μM proTAME. After 1.5 h (HeLa) or 4 h (BJ hTERT), triptolide (TRP) was added to 10 μM for the indicated cells. Only cells that had already entered mitosis were included in the analyzed. (H) Verification of transcription inhibition by TRP. Asynchronous HeLa cells treated with 10 μM TRP were stained with antibodies against serine 2 phosphorylation of the C-terminal tail of RNA polymerase II (CTD S2ph).
(I) Mitotic lifespan of the indicated cells (n=60 each) arrested in G2 and released into 500 nM taxol 10 μM proTAME. See Figure S6G for Western blot analysis.
(J) IRF3 rescue constructs. DBD, DNA-binding domain (gold). RD, regulatory domain (light gray). Region around S396 (dark gray).
(K–M) Mitotic lifespan of an IRF3 disruption cell line containing rescue constructs expressing doxycycline-inducible wild type GFP-IRF3 (K), a mutant lacking the DNA binding domain (L) or a mutant expressing GFP-IRF3 carrying the S396A mutation (M). See Figures S6H-S6J for Western blot. Cells were arrested in G2 and released into 500 nM taxol 10 μM proTAME (n=60 each).
Red lines, median. siCNTRL, non-targeting control siRNA; sicGAS, siRNA targeting cGAS; Untr., no siRNA treatment.
See also Figure S6.
Our earlier data had indicated that cGAMP synthesis by cGAS is important for limiting mitotic lifespan (Figure 4I). If cGAS promotes death through cGAMP, cGAMP addition may substitute for cGAS and reduce mitotic lifespan. Although cGAMP membrane transversal is inefficient (Gao et al., 2013b; Konno et al., 2013; Wu et al., 2013), simple addition to the medium of cells having entered mitotic arrest partially rescued the timing of cell death in both HeLa and BJ-hTERT cells depleted for cGAS (Figures 6D and 6E), supporting a direct role of cGAS during mitotic arrest, not in the preceding interphase. cGAMP addition could not induce IRF3 phosphorylation in ARPE-19 hTERT cells (Figure S6B), and accordingly, cGAMP addition or cGAS overexpression had only modest, if any, effects on mitotic cell death of ARPE-19 hTERT cells (Figures S6C-6E), indicating that cGAMP treatment does not induce death by nonspecific cytotoxicity. Altogether, these observations suggest that cGAMP signaling promotes mitotic cell death directly in mitosis.
The major known consequence of cGAS activation and cGAMP production is stimulation of transcription factors, such as IRF3. Although transcription is generally shut off in mitosis (Prescott and Bender, 1962), low level mitotic transcription may occur (Palozola et al., 2017). However, expression of the IRF3 targets IFNB1, CXCL10 and IRF1 (Chen et al., 2016), was not induced during mitosis in BJ-hTERT, whilst DNA transfection in asynchronous cells stimulated their transcription in a manner dependent on cGAS (Figures 6F, S6F and S6G). Furthermore, the transcription inhibitor triptolide (Bensaude, 2011) did not extend mitotic lifespan (Figures 6G and 6H), as recently reported (Novais-Cruz et al., 2018). Altogether, these data indicate that cGAS activation in mitosis does not induce an IRF3-dependent transcription response.
IRF3 was reported to induce apoptosis independently of transcription during RNA-virus induced apoptosis (known as RIPA) by directly activating the proapoptotic protein Bax, and inhibiting the anti-apoptotic protein XIAP (Chattopadhyay et al., 2010; Nakhaei et al., 2012; Petrasek et al., 2013). Indeed, disruption of either STING or IRF3 extended mitotic lifespan similarly to cGAS depletion (Figures S6H and S6I), and these effects could be reversed by wild type STING or IRF3 (Figures 6I-6K; S6J and S6K). To further characterize the role of IRF3, we generated cells expressing mutant versions of IRF3 (Figure 6J). An IRF3 construct incapable of transcriptional induction due to lack of the DNA-binding domain (ΔDBD) (Chattopadhyay et al., 2010) rescued the extended lifespan of IRF3 disruption (Figures 6L and S6L), supporting the transcription-independent mechanism of apoptosis. In contrast to RIPA, which depends on key ubiquitylation sites on IRF3 whilst being independent of IRF3 phosphorylation (Chattopadhyay et al., 2016), mutation of the RIPA ubiquitylation sites only partially compromised mitotic apoptosis (Figures S6M and S6N), whereas S396 of IRF3, which is phosphorylated during mitotic arrest in a manner-dependent on cGAS (Figures 2A, 2C and S2A), was required for timely mitotic cell death (Figures 6M and S6O). Accordingly, disruption of the IRF3 S396 kinase TBK1 also extended mitotic lifespan (Figures S6P and S6Q). Altogether, we conclude that induction of cGAS-dependent phosphorylation of IRF3 during mitotic arrest promotes apoptosis independently of transcription induction by IRF3.
cGAS promotes the taxol response in a mouse xenograft cancer model
To assess if the cGAS pathway within tumors could affect taxol chemotherapy, we performed a mouse xenograft experiment (Figure S7A). Control or cGAS-disrupted HeLa cells (Figure 4J) were injected subcutaneously into immunocompromised NSG mice (Shultz et al., 2005). Once tumors had become palpable, taxol or DMSO was administered weekly. Immunofluorescence of surgically resected tumors confirmed that cGAS was exclusively present in the cytoplasm of interphase wild type cells, but enriched on chromosomes during mitosis (Figure S7B). Taxol delayed the growth of wild type tumors, but not that of cGAS-depleted tumors (Figures 7A, 7B, S7C). Furthermore, wild type tumors treated with taxol, but not control-treated tumors or cGAS-disrupted tumors, showed large areas of apoptosis (Figures 7C-7F and S7D).
Figure 7. The cGAS pathway promotes the response to taxol in a mouse xenograft cancer model, and correlates with increased survival in lung and ovarian cancer patients.

.(A-F) Xenograft tumors formed by wild type or cGAS-disrupted (sg_cGAS) HeLa cells injected into immunocompromised (NSG) mice were subjected to weekly treatments with taxol or DMSO (see also Figure S7A).
(A and B) Growth curves for the indicated tumors and treatments (mean and SEM; n=6 for wt + DMSO; n=8 for wt +Taxol; n=6 for sg_cGAS + DMSO; n=5 for sg_cGAS + taxol).
(C–E) Apoptosis in the indicated tumor samples as determined with antibodies recognizing the cleaved (active) form of caspase-3. (C and D) sample images for the indicated tumors and treatments (scale bar, 100 μm). (E) quantifications of cleaved caspase-3 signal in taxol treated samples divided by the signal in DMSO treated samples (means and SD).
(F) Western blot analysis of tumors recovered at the end of the experiment.
(G and H) Survival curves of non-small cell lung (G) and ovarian (H) cancer patients from the TCGA database stratified according to treatment (taxane vs. other) and expression levels of cGAS (cutoff: median). See Table S1 and Table S2 for patient numbers.
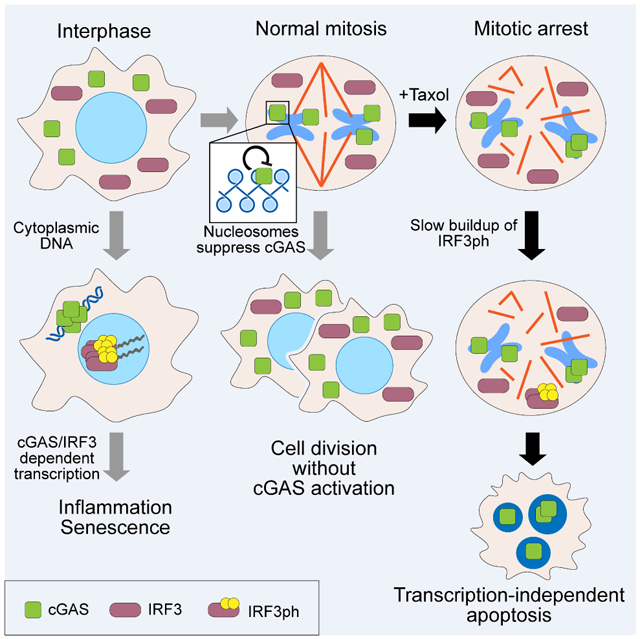
(I) Model for cGAS functions during infection and mitosis. Left: Due to a high transcriptional potential in interphase, activation of cGAS by cytoplasmic DNA predominantly results in inflammation. Middle: During normal mitosis, cGAS associates with mitotic chromosomes, but is inhibited by nucleosomes, and phosphorylated IRF3 is not generated. Right: During mitotic arrest, phosphorylated IRF3 slowly accumulates. Due to a high apoptotic potential but a low transcriptional potential, apoptosis rather than inflammation is induced.
To determine if cGAS worked through IRF3 in the xenograft model, we repeated these experiments with IRF3-disrupted cells and their derivatives expressing doxycycline-inducible wild type GFP-IRF3 or GFP-IRF3_ΔDBD (Figure 4K and 4L). Taxol treatment again delayed the growth of wild type tumors (Figure S7Ei), but not IRF3-disrupted tumors (Figure S7Eii). Tumors of the wild type GFP-IRF3 and ΔDBD rescue lines mimicked the IRF3 disrupted parental cell line in the absence of doxycycline (Figure S7Eiv and S7Evi), but doxycycline-induced-expression of wild type GFP-IRF3 rescued the taxol response (Figure S7Eiii). Similarly, re-expression of GFP-IRF3_ΔDBD allowed taxol to delay tumor growth (Figure S7Ev), suggesting that cGAS signaling to IRF3 can mediate the response to taxol independently of IRF3-dependent transcription, a finding that is also in agreement with the suppression of the cGAS pathway in interphase HeLa cells (Lau et al., 2015). However, the taxol response by GFP-IRF3_ΔDBD required a longer time to manifest itself (Figure S7Ev), suggesting that some transcription-dependent mechanisms may perhaps also be involved.
Effect of cGAS expression on survival of taxane-treated lung and ovarian cancer patients
Finally, we investigated a possible correlation between the cGAS pathway and the response to taxane chemotherapy in human non-small cell lung and ovarian cancer patients using data recorded within TCGA (Cancer Genome Atlas Network, 2012). Patients were stratified according to RNA levels of cGAS within their tumors, as well as according to whether or not they were treated with taxanes (Tables S1 and S2). Taxane-treated non-small cell lung cancer patients whose tumors expressed high levels of cGAS displayed prolonged survival when compared to patients with low-expressing tumors (Figure 7G). In contrast, for patients who received other types of therapy but were never treated with taxanes, expression levels of cGAS did not show any apparent correlation with survival (Figure 7G). For ovarian cancer patients, the scarcity of taxane-free patients made it difficult to assess the correlation between cGAS and taxane efficacy, but a similar trend of cGAS expression and prolonged survival was also observed among patients treated with taxanes (Figure 7H). These observations are consistent with the idea that cGAS is important for taxane therapy in lung, and perhaps ovarian cancer patients. We note, however, that this analysis is complicated by the fact that taxanes are not given as a monotherapy, but in combination with other chemotherapeutic agents (Tables S1 and S2). Furthermore, cGAS expression may in part reflect and/or cause immune cell infiltration into tumors, although the effects would still be specific for patients that were treated with taxanes. Despite these caveats, our data altogether suggest that the cGAS pathway contributes to the effect of taxane chemotherapy.
DISCUSSION
Our findings suggest that the organization of genomic DNA into nucleosomes prevents spikes in cGAS signaling during normal mitosis when cGAS is localized to chromosomes. This mechanism may also explain why cGAS activation upon enrichment on micronuclei is not as potent as activation with other stimuli such as viral DNA or transfected DNA (Harding et al., 2017; Mackenzie et al., 2017; Spektor et al., 2017). Since DNA length and bending are important for cGAS activation (Andreeva et al., 2017), it is possible that nucleosomes interfere with formation of such DNA structures. However, its higher affinity for nucleosomes than for naked DNA, and interaction with H2A–H2B suggest that cGAS interacts with nucleosomes in a configuration where it cannot readily be activated. Nucleosomes suppress cGAS lacking its disordered N-terminus, demonstrating that the suppression can act independently of its regulation via liquid phase condensation (Du and Chen, 2018), although nucleosome binding may also affect phase separation. Overall, our data are consistent with the idea that nucleosomes act as a hallmark of self-DNA distinguishing it from pathogenic DNA (Zierhut and Funabiki, 2015; Zierhut et al., 2014).
As phosphorylated IRF3 slowly accumulates during mitotic arrest, and all tested manipulations that block IRF3 phosphorylation delayed mitotic cell death, we suggest that phosphorylated IRF3 acts as a death signal during mitotic arrest. Mitotic IRF3 phosphorylation levels are much lower than what can be achieved through cytoplasmic DNA in interphase, but can still affect the fate decision between death and slippage. Although the detailed mechanism remains unclear, phosphorylated IRF3 appears to promote apoptosis through suppressing Bcl-xL in a manner independent of its transcriptional capacity. We propose that the cGAS pathway operates in two distinct modules depending on the transcriptional competence and apoptotic potential of the cell (Figure 7I): inflammatory signaling in transcriptionally competent interphase cells that have low apoptotic potential, and induction of apoptosis in transcriptionally attenuated mitotic cells that have high apoptotic potential.
Gradual accumulation of phosphorylated IRF3 may be due to the slow buildup of cGAMP synthesized by chromosomal cGAS, since nucleosomes do not completely inhibit cGAS. Additionally, although cGAS operates upstream of MOMP and caspase induced DNA damage, it is possible that pre-apoptotic mitotic stresses such as mitophagy and DNA damage contribute to cGAS activation (Doménech et al., 2015; Orth et al., 2012; Orthwein et al., 2014). As cGAS-depletion delayed, but not blocked, initiation of apoptosis, other mechanisms exist to influence the timing of mitotic cell death, such as degradation of Mcl-1 (Harley et al., 2010; Sloss et al., 2016), Cdk1-dependent caspase phosphorylation (Allan and Clarke, 2007; Andersen et al., 2009), mitophagy (Doménech et al., 2015), and transcriptional control of apoptotic regulators in the preceding interphase (Engel et al., 2016; Topham et al., 2015; Topham and Taylor, 2013). Mitotic duration before apoptosis can be affected by the cGAS pathway and any of these other partially redundant mechanisms.
Our xenograft experiments indicate that cGAS and IRF3 are important for suppression of tumor growth by taxol, although it remains unclear if this is directly mediated through mitotic cell death. The cGAS pathway is also expected to become activated in slipped cells due to the generation of micronuclei, and, within tumors, may mediate inflammation, senescence-like gene expression patterns, and metastasis (Bakhoum et al., 2018; Barber, 2015; Harding et al., 2017; Mackenzie et al., 2017; Mitchison et al., 2017). Both apoptotic cells and senescent cells can modulate the cancer microenvironment, in ways that may be detrimental or beneficial for the patient (Ichim and Tait, 2016; Sieben et al., 2018), and thus it will be important to understand how cGAS activation in tumors affects the immune system. Regardless of the exact mechanism, our analysis of TCGA data suggests that cGAS/STING/IRF3 expression within tumors contributes to the response to taxanes in lung and ovarian cancer patients. Since taxane treatment is regularly combined with other therapies, future studies specifically designed to assess the taxane response are needed to determine potential benefits of using the cGAS pathway as a predictive biomarker or as a combinatory therapeutic target for taxane-based treatment. As certain types of DNA damage may lead to mitotic arrest and mitotic cell death (Garner et al., 2013), cGAS may also influence the response to other types of chemotherapy. Further investigation of cGAS function in the response to mitotic defects will contribute to our understanding of tumor development and chemotherapy response.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Hironori Funabiki (funabih@rockefeller.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cells
The identity of all cell lines was validated using polymorphic short tandem repeat loci (Memorial Sloan Kettering Cancer Center). Note that the identity of cell line HBL-100, despite its high risk of contamination was correctly verified. We used it as a further example of a cGAS-expressing cell line (in addition to the seven others that we used), and any potential issues about its origin are irrelevant for this purpose. Mycoplasma contamination was monitored by fluorescence microscopy or PCR (Universal Mycoplasma Detection Kit, ATCC). HeLa Flp-In T-Rex cells stably expressing H2B-RFP (referred to as HeLa cells throughout this manuscript, a gift of A. Desai and R. Gassmann) (Gassmann et al., 2010), HeLa CCL-2 cells (a gift from I. Nakagawa), BT474, BT549, HBL-100, CAL51, MDA-MB-157, MDA-MB-231 and T47D cells (gifts of K. Birsoy) were grown in DMEM (Gibco 11995065) supplemented with 10% tet-tested FBS (Atlanta Biologicals) and penicillin-streptomycin (100 u/ml, Gibco). HCC1143 cells (a gift of K. Birsoy) were grown in RPMI 1640 medium (Life Technologies SH30027FS) supplemented with 10% tet-tested FBS (Atlanta Biologicals) and penicillin-streptomycin (100 u/ml, Gibco). BJ hTERT cells (a gift of T. de Lange) were grown in DMEM (Gibco 11995065) supplemented with 10% tet-tested FBS (Atlanta Biologicals), penicillin-streptomycin (100 u/ml, Gibco) and 10% medium 199 (Sigma M4530). ARPE-19 hTERT cells (a gift of T. de Lange) were grown in DMEM-F12 (Life Technologies 10565-018) supplemented with 10% tet-tested FBS (Atlanta Biologicals) and penicillin-streptomycin (100 u/ml, Gibco). RUES2 human embryonic stem cells XX female; US National Institutes of Health, human ESC registry no. 0013) were, as previously described (Yoney et al., 2018), grown in HUESM medium conditioned by mouse embryonic fibroblasts and containing 20 ng/ml bFGF. hES cell usage is covered by protocol #2019-03-004, approved by the Tri-Institutional Stem Cell Initiative Embryonic Stem Cell Research Oversight Committee.
HeLa Cell lines expressing GFP-tagged versions of cGAS, IRF3 and STING were generated using plasmid pcDNA5/FRT/TO as a backbone following instructions based on the Flp-In system (Invitrogen). ARPE-19 hTERT cell lines expressing GFP-tagged versions of cGAS were generated using the piggyBac transposon system. cGAScat represents the E225A D225A double mutant, cGASDNA represents the K407E K411A double mutant, and the cGAS KKKR mutant is the K407E K411A K173E R176E quadruple mutant. Plasmid construction details are available on request.
siRNA knockdowns were performed using the reverse transfection procedure. Briefly, siRNA was mixed with media without FBS (Opti-MEM, Life Technologies # 51985-034) and Lipofectamine RNAiMAX (Invitrogen # 13778075) at an siRNA concentration of 120 nM (10 nM final concentration once cells and media were added). Lipofectamine RNAiMAX was added at 1.2 pl/ml of final volume. The mixture was incubated for 15 min, and cells were then added in media without antibiotics. Media was changed the next day. cGAS siRNA (# L-015607-02, ON-TARGET plus SMARTpool) was obtained from Dharmacon. Non-targeting control siRNA (# D-001810–01, ON-TARGET plus) was obtained from Dharmacon.
For synchronous release into mitotic arrest, cells were pre-synchronized in S phase for 18 h with 2.5 mM thymidine. Cells were then washed twice in PBS and once in media, and cells were then released from the S phase arrest for 2 h in media without drugs. RO3306 (9 μM final) was then added to capture cells in G2. After 8–10 h, cells were washed twice with PBS, once with media containing 500 nM taxol before further incubation in media containing 500 nM taxol and 10 μM proTAME. Alternatively, 1.7 μM nocodazole was used instead of taxol. If BJ hTERT, or ARPE-19 hTERT cells were prepared for time-lapse microscopy, SiR-DNA (125 nM final concentration) was added to the media 2 h before release from G2 arrest, and to the final media containing taxol and proTAME to allow for visualization of DNA. Similarly, Sir-DNA was added to the media of breast cancer cells 2 h before addition of taxol and the start of time-lapse imaging. For induction of GFP-cGAS, GFP-IRF3 or GFP-STING, doxycycline was added to a final concentration of 100 ng/ml ~24 h before release from G2 arrest. To visualize Caspase 3/7 activity, IncuCyte Caspase-3/7 Reagent (Essen Bioscience # 4440) was added to the media at 1:2000 dilution.
If cells were prepared for time-lapse imaging, cells were subjected to siRNA knockdown at the same time as they were plated at ~30% confluence onto glass bottom culture dishes (MatTek P35GC-1.5-14-C). Cells were grown over night before addition of thymidine. To limit phototoxicity, the final imaging media did not contain phenol red.
Xenopus laevis frogs
To prepare egg extracts, eggs were collected from the female African frog Xenopus laevis. Mature female frogs larger than 9 cm were purchased from NASCO, and housed in the dedicated water tanks with temperature controlled at 16-20 °C at the Rockefeller University Comparative Bioscience Center (CBC). Prior to ovulation, small numbers of frogs were transferred at the certified satellite facility with the temperature control in the Funabiki lab. The procedure is covered by IACUC protocol # 17051.
Mice
We used 7 weeks old male NOD-scid IL2Rgnul1 (NSG) mice (Jackson Laboratory) to avoid variability caused by estrous cycles in females, and to eliminate immune responses that could affect tumor growth in a complex manner. Animals were monitored twice a day for 48 h, and daily for the first 5 days following tumor cell inoculation or primary tumor resection for signs of bleeding or distress (distress was defined as problems with any of the following functions: ambulation, eating, drinking, defecation, urination, or breathing). No mice displayed these symptoms. Similarly, mice were monitored for problems in ambulation, eating, drinking, defecation, urination, or breathing, but these symptoms did not appear. The procedure is covered by IACUC protocol #18038-H.
METHOD DETAILS
Antibodies
Bad was detected with #9239 from Cell Signaling Technology (1:250 dilution for Western blotting); Bak was detected with #12105 from Cell Signaling Technology (1:250 dilution for Western blotting); active Bax was detected with antibody 6A7 (Thermo Fisher; 5 μg/ml for immunofluorescence); Bax was detected with antibody 50599-2-lg (Proteintech, 1:1000 dilution for Western blotting); Bcl-xL was detected with #2764 from Cell Signaling Technology (1:250 dilution for Western blotting); Bim was detected with #2819 from Cell Signaling Technology (1:250 dilution for Western blotting); c-IAP1 was detected with #7065 (Cell Signaling Technology, 1:500 for Western blotting); Casp-2 was detected with ab179520 (abcam, 1:1000 for Western blotting); Casp-7 was detected with ab201959 (abcam, 1:500 for Western blotting); Casp-9 was detected with ab202068 (abcam, 1:1000 for Western blotting); cGAS was detected with #15102 from Cell Signaling Technology (1:500 dilution for Western blot, 1:400 dilution for immunofluorescence); cleaved casp-3 was detected with # 9661 from Cell Signaling Technology (1:500 for immunofluorescence); cytochrome C was detected with ab110325 (abcam; 1:250 dilution for immunofluorescence); anti-IFNAR2 antibody was obtained from EMD Millipore (MAB1155); IκBα was detected with #9242 (Cell Signaling Technology, 1:500 for Western blotting); IRF1 was detected with #8478 (Cell Signaling Technology, 1:500 for Western blotting); IRF3 (S396ph) was detected with #4947 from Cell Signaling Technology (1:500 dilution for Western blotting); IRF3 (S386ph) was detected with ab76493 (abcam; 1:1000 dilution for Western blotting); IRF3 (total) was detected with ab68481 (abcam; 1:1000 dilution for Western blotting) or 11312-1-AP (Proteintech; 1:1000 dilution for Western blotting. This antibody was used for the ΔDBD construct, as the abcam antibody does not recognize it); IRF3 was immunoprecipitated with 11312-1-AP (Proteintech); Mcl1 was detected with #4572 from Cell Signaling Technology (1:500 dilution for Western Blotting); mouse IgG2a isotype control antibody was obtained from Sigma-Aldrich (M5409); JNK2 was detected with #9258 (Cell Signaling Technology, 1:500 for Western blotting); phospho-SAPK/JNK (T183/Y185) was detected with #4668 (Cell Signaling Technology, 1:500 dilution for Western blotting); phospho-CDK substrate motif was detected with #9477 (Cell Signaling Technology, 1:500 dilution for Western blotting); PUMA was detected with #12450 from Cell Signaling Technology (1:250 for Western blotting); RNA pol II CTD S2ph was detected with ab5095 (abcam, 1:500 for immunofluorescence); SMAC/DIABLO was detected with #15108 (Cell Signaling Technology, 1:200 dilution for immunofluorescence); STING was detected with D2P2F (#13647) from Cell Signaling Technology (1:500 dilution for Western blotting); TBK1 was detected with ab40676 (abcam, 1:1000 for Western blotting); α tubulin was detected with T9026 from Sigma (1:20000 dilution for Western blotting); XIAP was detected with #2045 (Cell Signaling Technology, 1:500 for Western blotting). For Western blotting, IRDye 800CW and 680LT secondary antibodies were used at 50 ng/ml and detected on an Odyssey infrared imaging system (LI-COR). For immunofluorescence microscopy, Alexa-488, Alexa-555 and Cy5 coupled secondary antibodies were used (Jackson ImmunoResearch).
DNA transfections to induce cGAS signaling
Cells were grown close to confluence on 6 cm dishes. Media was removed from cells and replaced with 2.4 ml of media containing no antibiotics. To generate the transfection mix, 7.5 μg of pAS696 (Guse et al., 2011) were combined with 0.3 ml Opti-MEM (Life Technologies # 51985-034). Separately, 25 μl Lipofectamine 2000 (Life Technologies, # 11668) were mixed with 0.3 ml Opti-MEM. Both mixtures were incubated at room temperature for 5 min, combined and incubated at room temperature for 20 min before being added to the cell dish. Cells were harvested after 2 h.
CRISPR-Cas9 mediated gene disruptions.
Sequences encoding short guide RNAs (sgRNAs) were cloned into pX330 (Cong et al., 2013), and transfected into cells. Cells were grown for five days, and then plated for single colonies. Multiple candidate clones were picked and tested for gene disruption by western blot. SgRNA sequences were as follows: cGAS sg#1: CTGCCCCCAAGGCTTCCGCA; cGAS sg#2: GGCGCCCCTGGCATTCCGTG; IRF3 sg#1: GGTTGCGTTTAGCAGAGGAC; IRF3 sg#2: ACCTCTCCGGACACCAATGG; STING sg#1: GCTGGGACTGCTGTTAAACG; STING sg#2: GCAAGCATCCAAGTGAAGGG; TBK1 sg#1: CATAAGCTTCCTTCGTCCAG; TBK1 sg#2: ACAGTGTATAAACTCCCACA.
Western blots.
All cells harvested during mitotic arrest were collected using mitotic shake-off. Other cells were removed from dishes by trypsin treatment (Life Technologies # 25300-054), washed in PBS and resuspended in 2x sample buffer (125 mM Tris-Cl [pH 6.8 at 22 °C]; 2% SDS; 0.7 M 2-mercaptoethanol) and boiled for 5 min. Samples were then subjected to water bath sonication, spun down, and sample buffer containing glycerol and bromophenol blue were added to result in a final concentration of 6.25% glycerol and 25 μg/ml bromophenol blue. Samples were again boiled and subjected to gel electrophoresis and western blotting. Alternatively, samples were lysed in lysis buffer (150 nM NaCl; 3 mM MgCl2; 0.5 % Triton X-100; 1 mM EGTA; 1 mM DTT; 50 mM Na-Hepes [pH 7.5 at 4 °C]; 1 × phosphatase inhibitor cocktail 3 [Sigma Aldrich P0044]; 1x complete protease inhibitor cocktail [Roche 11873580001]; 10 μg/ml LPC [leupeptin, pepstatin, chymostatin) by rotation at 4 °C for 20 min. Samples were spun at 4 °C for 10 min (16.000 g) and diluted in 3 X sample buffer (see above).
For checking siRNA efficiency, cells treated identically to the cells that were used for microscopy were harvested just before release into taxol.
Recombinant proteins.
Nucleosomes were prepared from pre-formed H2A–H2B dimers and H3–H4 tetramers. Our previously published methods were used for histone purification (Zierhut et al., 2014). Briefly, histones H2A, H2B, H3 and H4 were individually expressed in E. coli under conditions in which they formed inclusion bodies. Histones were then individually purified under denaturing conditions (6 M Guanidine HCl; 500 mM NaCl; 50 mM Tris-Cl [pH 8 at 22 °C]; 5 mM 2-mercaptoethanol; 7.5 mM imidazole), refolded into H2A–H2B dimers or H3-H4 tetramers by dialysis into 20 mM MOPS; 500 mM NaCl; 2% glycerol; 1 mM EDTA; 5 mM 2-mercaptoethanol; pH 7 at 22°C, and purified using size-exclusion chromatography.
cGAS was purified using a modified version of published procedures (Gao et al., 2013a). Similar to previous work (Ablasser et al., 2013; Gao et al., 2013a; Kranzusch et al., 2013), we generated cGAS lacking its N terminal (amino acids 1-150) unstructured tail. This truncation had previously been shown to be catalytically active (Ablasser et al., 2013; Gao et al., 2013a; Kranzusch et al., 2013), and to fully substitute for full-length cGAS in interferon induction in response to cytoplasmic DNA (Sun et al., 2013). The DNA-binding KRKK mutant carried K173E R176E K407E K411A. We expressed these proteins in E. coli with an N-terminal MBP tag and a C-terminal 10xHis tag, with the tags separated from the rest of the protein with TEV protease recognition sites. E. coli Rosetta 2 (DE3 pLysS) cells containing cGAS expression plasmids were grown in 1.5 x TBG-M9 medium (15 g/l tryptone; 7.5 g/l yeast extract; 5 g/l NaCl; 0.15 g/l MgSO4; 1.5 g/l NH4Cl; 3 g/l KH2PO4; 6 g/l Na2HPO4; 0.4 % glucose) until an OD600 of ~0.9, and protein expression was induced at 18 °C with 0.6 mM IPTG for 20 h. Cells were then lysed by sonication in wash/lysis buffer (50 mM Tris-Cl [pH 7.5 at 4 °C]; 0.3 M NaCl; 10 mM 2-mercaptoethanol) supplemented with 1 mM PMSF; 0.25 mg/ml lysozyme; 10 μg/ml leupeptin; 10 μg/ml pepstatin; 10 μg/ml chymostatin. The fusion protein was enriched on amylose resin (NEB), and eluted by treatment with TEV protease. To remove TEV protease (which itself was His-tagged), the eluate was also run over a Ni-NTA column (Qiagen). Proteins were further purified on a HiTrap heparin column (GE Healthcare) on an ÄKTA FPLC system (GE Healthcare) using a salt gradient for elution that was established with wash/lysis buffer as low salt buffer and wash/lysis buffer containing 1 M NaCl as high salt buffer. Peak fractions were subjected to a final purification step by size-exclusion chromatography using a Superdex 200 10/300 GL column (GE Healthcare) on an ÄKTA FPLC system (GE Healthcare) in 20 mM Tris-Cl [pH 7.5 at 4 °C]; 0.3 M NaCl; 1 mM DTT. Peak fractions were combined, concentrated, and stored at −20 °C in the same buffer containing 48% glycerol.
Preparation of nucleosome arrays and mononucleosomes for binding assays
DNA substrates used for nucleosome arrays and for their naked DNA counterparts were generated as described previously (Guse et al., 2011; Zierhut et al., 2014). Briefly, plasmid pAS696 containing 19 tandem copies of the “601” nucleosome-positioning sequence (Lowary and Widom, 1998) was digested with HaeII, DraI, EcoRI and XbaI to remove the 601 array and to digest the vector backbone into smaller pieces. The array was then separated from the vector backbone fragments using PEG precipitation. To facilitate coupling to streptavidin beads, ends were filled in with Klenow fragment (NEB) in the presence of dCTP, biotin-14-dATP (Invitrogen), thio-dTTP and thio-dGTP (both ChemCyte). This resulted in biotinylation of both ends of the linear array. Arrays were separated from unincorporated nucleotides using Sephadex G-50 Nick columns (GE Healthcare).
To generate the DNA used for mononucleosomes or single copies of the naked 601 sequence, pAS696 was digested with AvaI (which cuts in between 601 monomers), and the fragment was separated from the vector backbone using PEG precipitation.
Nucleosomes were assembled on these substrates using salt dialysis following published procedures (Guse et al., 2011; Zierhut et al., 2014). 50 μl reactions were prepared containing 10 μg of DNA, and histones H2A–H2B and H3–H4 at empirically determined concentrations that varied slightly with each batch (but usually in slight excess over DNA), 2 M NaCl and 1x TE. Mixtures were moved to dialysis buttons (Hampton Research), submerged in 500 ml high salt buffer (10 mM Tris-Cl [pH 7 at 22 °C]; 1 mM EDTA; 2 M NaCl; 5 mM 2-mercaptoethanol; 0.01 % Triton X-100) and salt was reduced through dialysis over an exponential gradient established by pumping in 2 l of low salt buffer 1 (as high salt buffer but 50 mM NaCl) whilst removing excess buffer from the original 500 ml over 1-3 days at 4 °C. Nucleosomes were then dialyzed into low salt buffer 2 (10 mM Tris-Cl [pH 7 at 22 °C]; 0.25 mM EDTA; 100 mM NaCl; 1 mM TCEP). Mononucleosomes that were used for cGAMP synthesis assays were further dialyzed into 20 mM Tris (pH 7.5 at 22 °C), 150 mM NaCl, 1 mM DTT, 10 mM MgCl2. The quality of nucleosome formation on arrays was assessed by digestion with AvaI (which cuts in between 601 monomers on the array) followed by native gel electrophoresis in 5% polyacrylamide using 0.5x TBE as running buffer, as described previously (Guse et al., 2011). Quality of mononucleosome formation was determined by native gel electrophoresis without prior digestion.
Generation of beads coupled to naked DNA or nucleosome arrays
For both nucleosome beads and naked DNA beads, nucleosomes were initially coupled to Dynabeads M280 Streptavidin (Invitrogen). Nucleosomes containing a total amount of 330 ng of DNA were coupled to 2 μl beads in 75 μl of buffer containing 2.5 % polyvinylalcohol, 150 mM NaCl, 50 mM Tris-Cl (pH8 at 22 °C), 0.25 mM EDTA, 0.05 % Triton X-100. Coupling was carried out under agitation at room temperature for 2 h. Beads were then washed four times in low salt wash buffer 150 mM NaCl, 50 mM Tris-Cl (pH8 at 22 °C), 0.25 mM EDTA, 0.05 % Triton X-100. For naked DNA beads, nucleosomes were subsequently stripped by incubation under agitation at 20 °C in 2 M NaCl, 50 mM Tris-Cl (pH8 at 22 °C), 0.25 mM EDTA, 0.05 % Triton X-100 for 10 min. Beads were then washed four times in the same buffer, and once in low salt wash buffer.
Cell-free Xenopus egg extracts
Cytostatic factor (CSF) M phase arrested X. laevis egg extracts were prepared as previously described (Murray, 1991). Histones H3 and H4 were depleted using antibodies recognizing acetylated lysine 12 on histone H4 using our previously described method (Zierhut et al., 2014). The protocol for work with X. laevis was approved by The Rockefeller University Institutional Animal Care and Use Committee.
Nucleosome/naked DNA bead binding assays
For bead pull downs in the absence of egg extract, nucleosome beads, chromatin beads and uncoupled beads were washed twice in binding buffer (20 mM Hepes pH 7.7; 200 mM NaCl; 0.05 % Triton X-100; 0.5 mM TCEP pH 7.5). 35S labeled full-length cGAS was generated with the TnT Coupled Reticulocyte Lysate System (Promega) according to the manufacturer’s instructions. TnT reactions were diluted 1:10 in binding buffer supplemented with BSA to a final concentration of 0.25 μg/μl, and beads were incubated in 20 μl of this mixture at 20 °C for 45 min under agitation. Beads were then washed in binding buffer, bound proteins were eluted with SDS sample buffer, and analyzed by gel electrophoresis followed by staining with coomassie brilliant blue and fluorography. In parallel, DNA was extracted from another set of beads subjected to an identical procedure to determine whether nucleosome beads and DNA beads contained equal amounts of DNA. To this end, beads were incubated in 500 μl Stop solution 2 (20 mM Tris pH 8.0 at 22 °C; 20 mM EDTA; 0.5% SDS; 1 mg/ml Proteinase K [Roche]) at 37 °C for 45 min. DNA was then extracted using phenol/chloroform, precipitated with ethanol/NaOAc in the presence of glycogen, and run on an agarose gel. Bands were visualized using SYBR-safe (Invitrogen), and quantified using ImageJ (v. 2.0.0-rc-43/1.51k).
For bead pull downs from egg extract, nucleosome beads, chromatin beads and uncoupled beads were washed twice with CSF-XB (100 mM KCl; 1 mM MgCl2; 50 mM sucrose; 5 mM EGTA; 10 mM Hepes pH 8), and then mixed with 20 μl egg extract containing 35S labeled full-length cGAS generated as above (1:10 dilution of TnT reaction in egg extract). Cycloheximide (Sigma) was added to a final concentration of 100 μg/ml to prevent labeling of other proteins within the extract. The mixture was then incubated at 16 °C under rotation for 45 min, diluted 10 fold in CSF-XB, and beads were recovered. Beads were washed 3 times in CSF-XB containing 0.05% Triton X-100. Bound proteins were eluted with SDS sample buffer and analyzed by gel electrophoresis. The gel was stained with coomassie brilliant blue and cGAS was detected with fluorography. Control beads to determine loading amounts were prepared as above with the following exceptions: Mixtures were diluted 10 fold in CSF-XB, beads were recovered, resuspended in 250μl Stop buffer 1 (20 mM Tris pH 8.0 at 22 °C; 20 mM EDTA; 0.5% SDS; 50 μg/ml RNase A [Qiagen]) and incubated at 37 °C for 25 min. 250 μl of Stop buffer 2 was then added and beads were incubated at 37 °C for another 25 min. DNA was extracted with phenol/chloroform, precipitated with ethanol/NaOAc, dissolved in 1x TE containing 50 μg/ml RNase A, and incubated at 37 °C for 20 min before being analyzed as above.
In-solution nucleosome/naked DNA binding assays
Binding reactions contained mononucleosomes or 601 monomers at 20 nM and recombinant cGAS at the concentrations indicated in Fig. 2C in a total of 20 μl of low salt buffer 2 (10 mM Tris-Cl [pH 7 at 22 °C]; 0.25 mM EDTA; 100 mM NaCl; 1 mM TCEP). Reactions were allowed to proceed for 1 h at room temperature. Products were separated by native polyacrylamide gel electrophoresis (5% polyacrylamide in 0.5x TBE). Gels were stained with SYBR-Safe to visualize DNA, and disappearance of the band corresponding to naked DNA or nucleosomes was quantified using ImageJ. Binding affinity was determined with GraphPad Prism (v. 5.0a) using non-linear regression analysis assuming specific one site binding.
cGAMP synthesis assays
cGAMP synthesis assays were performed in 20 mM Tris (pH 7.5 at 22 °C), 150 mM NaCl, 1 mM DTT, 10 mM MgCl2. Reactions were performed under saturation conditions and contained 1 μM cGAS and 230 nM mononucleosomes or monomer of naked 601 sequence. cGAS was first pre-incubated with naked DNA or mononucleosomes at room temperature for 30 min. ATP and GTP were then added to a final concentration of 1 mM each to start the reaction. At the same time, GTP labeled at the alpha position with 33P was also added to a final concentration of 33 nM. Reactions (10 μl total volume) were incubated at 37 °C and 1.5 μl samples were taken at the indicated time points. Unreacted nucleotides were digested with 4 u alkaline phosphatase (calf intestinal phosphatase, NEB) at 37 °C for 30min, and products were separated via thin-layer chromatography using PEI cellulose F coated sheets (EMD Millipore # 1055790001) with 1.2 M KH2PO4 (pH 3.8) as running buffer. Plates were dried at 65 °C, analyzed by fluorometry, and images were quantified using ImageJ. Initial reaction velocities were determined using linear regression analysis of the 10 min, 20 min and 30 min time points. We confirmed that under our conditions, all cGAS molecules were saturated with nucleosomes or DNA by titration experiments. Thus, the values obtained for our initial reaction velocities represent maximum reaction velocities (Vmax). Vmax values were used to determine Kcat values.
Histone H2A–B and H3–H4 binding assays
Assays were performed similar to previously published methods (Fujita et al., 2015). His-tagged H2A–B dimers and H3–H4 tetramers (50 nM each) were mixed with 50 nM recombinant cGAS and 5 μl of Dynabeads Talon (Invitrogen) in 200 μl of binding buffer (20 mM Tris-Cl [pH 8 at 4 °C]; 0.2M NaCl; 60 mM imidazole; 0.3% Tween-20; 0.01 μg/μl BSA) and incubated under agitation at 4 °C. Beads were washed three times in wash buffer (as binding buffer, but containing only 30 mM imidazole and not containing BSA), and bound proteins were eluted with SDS sample buffer, and analyzed by gel electrophoresis followed by staining with coomassie brilliant blue. H2A-B_ap*A mutants were H2A E56A E61A E64A D90A E91A E92A; H2B E113A; H2A-B_ap*KR mutants were (H2A E56K E61K E64K D90R E91R E92K; H2B E113K.
IRF3 immunoprecipitation
For each time point of immunoprecipitations, cells were removed from a 10 cm dish, washed once in PBS and lysed with 400 μl of lysis buffer (150 mM NaCl; 0.5% NP-40; 50 mM Tris-Cl [pH 7.5 at 4 °C]; 3 mM EDTA; 3 mM EGTA; 1 x phosphatase inhibitor cocktail 3 [Sigma Aldrich P0044]; 1x complete protease inhibitor cocktail [Roche 11873580001]; 10 μg/ml LPC [leupeptin, pepstatin, chymostatin] by incubation with rotation at 4 °C for 10 min. The lysate was sonicated in a bioruptor water bath (10 min at 4 °C, with repeated cycles of 1 min on at medium setting, with 30 sec breaks). The lysate was then spun for 5 min at 16000 g, 4 °C, snap-frozen in liquid nitrogen, and stored at −80 °C until use.
Western blotting was first used to compare IRF3 concentrations between samples and to adjust input amounts. For each sample of the experiment from Figure 2C, 11 μg of anti-IRF3 antibody (55 μl of 11312-1-AP, Proteintech) was coupled to 55 μl of protein A Dynabeads (Invitrogen) in wash/coupling buffer (0.1M Na-Hepes [pH 8.0 at 22 °C], 150 mM NaCl) by incubation with rotation at 4 °C over night. Beads were washed three times in wash/coupling buffer, moved to a new tube and bound antibody was crosslinked to the beads by 30 min incubation with rotation at room temperature in 4 mM BS3 (PI21585, Fisher Scientific). The reaction was then quenched with two 15 min incubations in wash/coupling buffer supplemented with 0.1 M Tris-Cl [pH 8 at 22 °C]. Beads were washed three times in wash/coupling buffer, moved to a new tube and washed two more times in lysis buffer. Beads were then mixed with 200 μl lysate, and incubated for 1 h under rotation at 4 °C. Beads were washed once with lysis buffer, moved to a new tube with a second wash, and bound proteins were eluted by incubation for 5 min at room temperature in 20 μl sample buffer (250 mM Tris-Cl [pH 6.8 at 22 °C]; 4% SDS; 1.4 M 2-mercaptoethanol; 13% glycerol; 50 μg/ml bromophenol blue; 0.1 M NaCl). Samples were boiled and analyzed by gel electrophoresis/western blotting.
Immunofluorescence microscopy
For visualizing cGAS, cells were grown on coverslips coated in poly-D-lysine (Sigma P1024, coverslips were coated by incubation for ~30 min in a 1 mg/ml solution). Coverslips were washed once in PBS, and subsequently cells were fixed with paraformaldehyde (Electron Microscopy Services # RT 157-8; 2% in PBS) by incubation for 20 min. Coverslips were washed three times in PBS and cells were then permeabilized for 5 min by incubation with PBS containing 0.2% Triton X-100 (Bio-Rad). Following three washes in PBS, coverslips were blocked in PBS-T (PBS plus 0.1% Tween-20 (Bio-Rad)) containing 5% BSA (Sigma A7906). Antibody incubations (1 h each) were performed in the same solution. Coverslips were washed five times in PBS-T after each antibody incubation and subsequently mounted in Vectashield with DAPI (Vector Laboratories; H-1200).
To visualize activated Bax, cytochrome C and SMAC/DIABLO, cells were collected and washed once in PBS. Cells were then fixed for 10 min at room temperature in 1 x PBS; 3.7% formaldehyde, 0.2% Triton X-100. Fixed cells were washed once in PBS, resuspended in TBS (150 mM NaCl; 150 mM Tris-Cl [pH 7.5 at 22 °C]), and spun onto coverslips using a StatSpin CytoFuge 2 (1000 rpm, 4 min). Cover slips were stained as described above.
For tumor immunofluorescence, see description in detailed section “Tumor Immunofluorescence” below.
In all cases except tumor samples, microscopy was performed using a Delta Vision Spectris setup (Applied Precision) consisting of an Olympus IX71 wide-field inverted fluorescence microscope, and a Photometrics CoolSnap HQ camera (Roper Scientific). Images were captured at 0.2 μm Z steps, processed by iterative constrained deconvolution using SoftWoRx (Applied Precision), and analyzed in ImageJ (v. 2.0.0-rc-43/1.51k). Sum projections are shown throughout the figures, except where noted. For tumor samples, images were acquired using the same setup but single images were taken.
Time-lapse microscopy
Live cell imaging was performed in an LCV110U VivaView FL incubator microscope (Olympus) at 37 °C in 5% CO2. The setup also consisted of an X-Cite-exacte illumination source (Excelitas Technologies) and an Orca-R2 CCD camera (Hamatsu). Images were acquired with a 20 x objective every 15 min for 48-72 h, in some cases with multiple Z steps (~2 μM step sizes; ImageJ was used to generate projections when Z slices were taken). Individual cells were manually tracked using the CellCognition software(Held et al., 2010) (v 1.5.2). Timing of death or slippage into interphase were determined morphologically (slippage was defined by cell flattening, and initiation of death was defined by the characteristic morphological changes reminiscing of blebbing and cell explosion).
UV-viability assays
Cells were seeded onto 6-well plates at ~20% confluence and allowed to attach for ~10 h. Cells were then washed twice in PBS and irradiated using an XL-1000 Spectrolinker UV crosslinker (Spectronics Corporation). Media was added and cells were grown under standard conditions. At the indicated time points, the supernatant (enriched for dead cells) was transferred to a tube and the dish was washed once in PBS, with the wash also moved to the same tube. The remaining cells were trypsinized and also moved to the same tube. Cells were spun down, resuspended in 200 μl PBS and mixed with 50 μl 0.4% Trypan blue. The fraction of dead cells was then determined using cell counting.
cDNA preparation and quantitative PCR
cDNA was prepared as follows. ~ 3 × 106 cells were harvested and resuspended in 1 ml TRIzol (Life Technologies, # 15596018). This mixture was frozen at −80 °C until use. For RNA extraction, 0.2 ml chloroform were added after thawing, and samples were thoroughly mixed. Samples were allowed to stand at room temperature for 2 min before centrifugation for 18 min (10.000 g; 4 °C). The aqueous phase was removed and mixed with an equal volume of ethanol. After mixing, samples were loaded onto RNeasy columns (Qiagen), washed once with buffer RW1 and twice with buffer RPE. Samples were eluted using 50 μl of nuclease-free water. ~ 300 ng RNA of each sample were used to generate cDNA using the Transcriptor First Strand cDNA Synthesis kit (Roche, # 04 379 012 001) according to the manufacturer’s instructions (both Anchored-oligo (dT)18 as well as random hexamer primers were used).
2 μl of each cDNA reaction were used for quantitative PCR with the LightCycler 480 SYBR Green I Master mix (Roche, # 04 707 516 001), carried out on an iCycler (Bio-Rad) equipped with the IQ 5 detection system. CXCL10 primer sequences were AGGAACCTCCAGTCTCAGCA and ATTTTGCTCCCCTCTGGTTT; interferon beta (IFNB1) primer sequences were AGGACAGGATGAACTTTGAC and TGATAGACATTAGCCAGGAG; IRF1 primer sequences were ATGCCCATCACTCGGATGC and CCCTGCTTTGTATCGGCCTG. Readings were normalized to GAPDH using primers AATCCCATCACCATCTTCCA and TGGACTCCACGACGTACTCA. The following temperature program was used: 5 min 95 °C; 50 x (10 sec 95 °C; 20 sec 52 °C; 20 sec 72 °C).
IFNAR-neutralization and JAK inhibition experiments
For the analysis of the addition of IFNβ receptor (IFNAR) neutralizing antibodies on mitotic cell death, anti IFNAR2 antibody or mouse IgG2a control antibody was added to a final concentration of 10 μg/ml at the same time as cells were shifted to medium containing thymidine. All subsequently used media contained the same concentrations of these respective antibodies. As control for the effective inhibition of the interferon receptor, cells were pre-treated with 10 μg/ml anti-IFNAR2 antibody or IgG2a control antibodies for 24 h and subsequently, IFNβ was added to 0.1 ng/ml. Cells were harvested after 4 h and processed for Western blotting.
For the JAK inhibition experiment (Figure S6A), 100 nM pyridone 6 (Thompson et al., 2002) was added to the media at the same time as thymidine was added. All subsequent media changes included pyridone 6.
Primary tumor assays in mouse xenografts
Cells (wild type HeLa Flp-In T-Rex, sg_cGAS #1 (Figure 4J), sg_IRF3 #2 (Figure S6I) or the rescue lines expressing GFP-IRF3 (clone 1, Figure 6K) or GFP-IRF3-ΔDBD (clone 2, Figure 6L), respectively) were subcutaneously injected (1×10~6 cells in 0.1ml) into each of the flanks of mice anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg) prior to the procedure. For the experiment from Figure 7, four mice were used for each cohort (wild type + DMSO; wild type + taxol; sg_cGAS + DMSO; sg_cGAS + taxol), resulting in a total of ~8 tumors for each cohort. For the experiment in Figure S7E, four mice were used for each cohort. All mice were fed with Purina Rodent Chow #5001 (Research Diet, Inc). The doxycycline treated cohort were fed with the same but containing 625 mg doxycycline per kg of chow starting 48 hours prior to the tumor cell injection (65 mg to 85 mg/kg mouse body weight/day). Littermates were randomly assigned to each cohort. Tumor growth was monitored with a digital caliper every week. The last measurement was performed one week after administration of the last drug dose. Treatment was started once tumors reached the threshold size of 50 mm3. Tumors were removed 48 h after administration of the last dose of taxol/DMSO for Western blotting, and 48 h after administration of the second dose of taxol/DMSO for immunofluorescence.
Tumor preparations for Western blotting and immunofluorescence
For Western blotting, tumors were surgically removed, separated from dead and non-cancerous tissue, and cut into a cell slurry. The slurry was treated with collagenase IV (Worthington, LS004188) and subsequently, cells were pelleted by centrifugation. The cell pellet was resuspended in ACK buffer (Lonza, 10-548E), and cells were separated on a 40%/22% stepwise OptiPrep gradient (Sigma, D1556). Viable tumor cells from the top of the gradient were diluted in 80 ml of D10F media, and pelleted. The cell suspension was then passed twice through a 100 μm cell strainer (Falcon, 352350), and once through a 70 μm cell strainer (Falcon, 352360). Cells were pelleted again, and tumor cells were separated from mouse cells using the Mouse Cell Depletion Kit from Miltenyi Biotec (#130-104-694) according to the manufacturer’s instructions (cells were separated with LS columns in the final step, Miltenyi Biotec #130-042-401).
For immunofluorescence, tumors were surgically removed and fixed in 4% paraformaldehyde over night. Tumors were washed twice in PBS and incubated in 70% ethanol over night. Samples were subsequently embedded in paraffin and 10 μm slices were cut onto glass slides with a microtome (Histoserv, Inc.).
Tumor immunofluorescence
Paraffin was removed from samples by 1 min incubation in xylenes. Samples were stepwise rehydrated by 1 min incubations in 1:1 mixture of xylenes/ethanol, followed by 100% ethanol, 90% ethanol, 70% ethanol, 50 % ethanol in water. Slides were then incubated in PBS, moved to 10 mM Na-citrate (pH 6) and antigens were recovered by boiling in a microwave for 3 min followed by a wash step in PBS.
Samples were blocked in naive serum (Vector Laboratories) over night at 4 °C, and incubated over night in primary antibody. Samples were washed three times in PBS and incubated for one hour in secondary antibody. Samples were washed three more times in PBS and mounted in ProLong gold (Invitrogen) with DAPI. Microscopy was performed using a Delta Vision Spectris setup (Applied Precision) consisting of an Olympus IX71 wide-field inverted fluorescence microscope, and a Photometrics CoolSnap HQ camera (Roper Scientific). Images were captured with a 10 x objective using SoftWoRx (Applied Precision), and processed and analyzed in ImageJ (v. 2.0.0-rc-43/1.51k). Whole tumor images were tiled using a 4 x objective and stitched using arivis Vision 4D (v. 3.0). To determine how variable the apoptotic response was throughout a tumor, whole tumor images were separated into individual regions of equal size and analyzed.
TCGA data analysis
Clinical data (gdac.broadinstitute.org/runs/stddata__2016_01_28/data/LUSC/20160128/gdac.broadinstitute.org_LUSC.Merge_Clinical.Level_1.2016012800.0.0.tar.gz and gdac.broadinstitute.org/runs/stddata__2016_01_28/data/LUAD/20160128/gdac.broadinstitute.org_LUAD.Merge_Clinical.Level_1.2016012800.0.0.tar.gz and gdac.broadinstitute.org/runs/stddata__2016_01_28/data/OV/20160128/gdac.broadinstitute.org_OV.Merge_Clinical.Level_1.2016012800.0.0.tar.gz) and RNA expression data with normalized RSEM values (gdac.broadinstitute.org/runs/stddata__2016_01_28/data/LUAD/20160128/gdac.broadinstitute.org_LUAD.Merge_rnaseqv2__illuminahiseq_rnaseqv2__unc_edu__Level_3__RSEM_genes_normalized__data.Level_3.2016012800.0.0.tar.gz and gdac.broadinstitute.org/runs/stddata__2016_01_28/data/LUSC/20160128/gdac.broadinstitute.org_LUSC.Merge_rnaseqv2__illuminahiseq_rnaseqv2__unc_edu__Level_3__RSEM_genes_normalized__data.Level_3.2016012800.0.0.tar.gz and gdac.broadinstitute.org/runs/stddata__2016_01_28/data/OV/20160128/gdac.broadinstitute.org_OV.Merge_rnaseqv2__illuminahiseq_rnaseqv2__unc_edu__Level_3__RSEM_genes__data.Level_3.2016012800.0.0.tar.gz) of the non-small cell lung cancer datasets (TCGA-LUAD and TCGA-LUSC) and the ovarian cancer dataset (TCGA-OV) were retrieved from FireBrowse (http://firebrowse.org) (Berger et al., 2018; Cancer Genome Atlas Network, 2012; Ciriello et al., 2015). 288 lung cancer patients and 343 ovarian cancer patients had associated comprehensive follow-up records, RNA expression data and records of clinical treatment. Of these, 107 lung cancer patients and 315 ovarian cancer patients were treated with taxane-based therapy, including paclitaxel, docetaxel and taxotere. The other 181 lung cancer and 28 ovarian cancer patients did not receive taxane-based treatment, but received other types of therapies. The curated clinical information is recorded in Table S1 and Table S2.
CGAS expression levels were extracted and underwent z-score transformation by using the following equation: Z-score = (normalized RSEM value – average of normalized RSEM value in normal tissue) / standard deviation of normalized RSEM value in normal tissue. Patients were stratified along the median. Kaplan-Meier survival curves were drawn with the surminer R package (https://CRAN.R-project.org/package=survminer). Significance was evaluated by using the log rank test.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance was determined with GraphPad Prism (v. 5.0a) using a built-in unpaired t-test function (Figures 1H, 1I, S2H, S2J, 4B, 4E, 4G; S6C; S7E; as determined by F-test, no statistically significant differences in variances existed in any of these cases), a built-in Mann-Whitney U test function (Figures S3H, S3J, S3N, 4C, 4D, 4F, 4H-4J, S4A-S4F, S4H, S5D, S5O, S5P, 6C-6E, 6G, 6I, 6K-6M, S6A, S6I, S6N, S6P, 7A, 7E), or the Wilcoxon signed rank test (Figure 6A, comparison to a hypothetical distribution with a median of 1). For patient data analysis (Figures 7G-7J), the significance was also evaluated by using the Cox proportional hazards model (Cox, 1972) and adjusted for multiple testing using the Bonferroni method. Other statistical parameters, such as error bar information, n values and number of experiment repeats can be found in the figure captions. Note that for the mouse experiments, n values are indicated in the methods section.
Since mitotic cell death is a stochastic event, data variations are inherit to each cell line and unpredictable, and we therefore empirically determined the sample size that is sufficient to provide statistically significant differences. No statistical tests were used to predetermine sample sizes. No samples were excluded from any analyses. Investigators were not blinded during experiments and analysis. No randomization methods were used to determine how samples were allocated.
Patient data from TCGA
Please refer to Table S1 and Table S2 for detailed information.
Supplementary Material
Figure S1. cGAS interacts with nucleosomes and mitotic chromosomes. Related to Figure 1.
(A) cGAS associates with nucleosomes in M-phase Xenopus egg extracts. Beads coated with nucleosome arrays (Nucleosomes), naked DNA (Naked DNA) or just streptavidin (Beads) were incubated in egg extract containing 35S-labelled cGAS. Proteins associated with each set of beads were detected by fluorometry (upper panel). Note that the extract was depleted of endogenous histones H3–H4 to prevent nucleosome assembly on naked DNA. CBB, Coomassie brilliant blue. Streptavidin serves as an indicator for the amount of beads loaded. The strong CBB staining in the input lane is due to extract proteins migrating at this size. The lower panel shows amounts of DNA immobilized on the indicated beads.
(B) Coomassie stained gel of purified wild type and KKKR mutant cGAS.
(C) Example of the gel shift analysis with recombinant cGAS and naked DNA or mononucleosomes.
(D) Quantification of cGAMP generation by cGAS in conjunction with naked DNA or mononucleosomes.
(E–G) cGAS associates with mitotic chromosomes in living cells. Time-lapse analysis of HeLa cells stably expressing the indicated constructs released from G2 arrest into 500 nM taxol. NEBD, nuclear envelope breakdown. GFP-cGASDNA, K407E K411A DNA-binding mutant.
(H–J) cGAS associates with mitotic chromosomes. Images of HeLa cells (H) or BJ hTERT fibroblasts (I) treated with either siRNA to cGAS (sicGAS) or with control siRNA (siCNTRL), fixed and stained with anti-cGAS antibodies. Quantification of the fraction of mitotic figures that are positive for cGAS localization on chromosomes in either untreated cells (Untr.) or cells treated with 500 nM taxol for 4 h is shown in (J).
All graphs represent mean values and SEM from at least three independent experiments.
Figure S2. cGAS-signaling in mitosis. Related to Figure 2.
(A) Western blot analysis of IRF3 S396 phosphorylation in HeLa cells of the indicated type (with or without GFP-IRF3 overexpression) and siRNA treatment harvested either in G2 or after the indicated times during arrest in 1.7 μM nocodazole.
(B) Western blot analysis of IRF3 S386 phosphorylation in HeLa cells harvested either in G2 or after the indicated times during arrest in 1.7 μM nocodazole.
(C) Immunofluorescence analysis of cGAS staining on mitotic chromosomes in RUES2 hES cells.
(D) Mitotic phosphorylation of IRF3 at Ser386 in RUES hES cells. Cells were treated with 500 nM taxol for 6 h and mitotic cells were collected by shake-off. Non mitotic cells were subsequently removed by scraping. As control for cell cycle arrest, a phospho-CDK site Western blot is shown.
(E) Analysis of IRF3 phosphorylation in BJ hTERT fibroblasts. Samples were taken following DNA transfection (+DNA) at the indicated times after release into 500 nM taxol, 10 μM proTAME from G2. siCNTRL, control siRNA
(F) Western blot analysis of phosphorylation of IRF3 immunoprecipitated from G2 arrested cells, or from cells at the indicated time points during arrest in 500 nM taxol, 10 μM proTAME. siCNTRL, control siRNA.
(G and H) Western blot analysis of IκBα levels in BJ hTERT cells harvested either in G2 or after the indicated times during arrest in 0.5 μM taxol 10 μM proTAME. (G) Example gel. The vertical line indicates removed irrelevant lanes. (H) Quantification. Shown are mean values and SEM from four experiments. P values are indicated at the 16 h and 20 h time points. siCNTRL, control siRNA. sicGAS, cGAS siRNA.
(I and J) Western blot analysis of JNK2 phosphorylation (T183/Y185) in BJ hTERT cells harvested either in G2 or after the indicated times during arrest in 0.5 μM taxol 10 μM proTAME. (I) Example gel. (J) Quantification. Shown are mean values and SEM from six experiments. P values are indicated at the 16 h and 20 h time points. siCNTRL, control siRNA. sicGAS, cGAS siRNA.
Figure S3. Mi totic cell death in breast cancer cell lines and HBL-100. Related to Figure 3.
(A–D) Western blot analysis of IRF3 S386 phosphorylation in cells of the indicated cell lines harvested either asynchronously or after the indicated times following treatment with 500 nM taxol. Taxol treated cells were harvested by shake off. Note that HCC1143 grows too slowly to allow quantitative collection of mitotic cells at 12 h. As control for cell cycle arrest, phospho-CDK site Western blots are shown.
(E) Timing of mitotic cell death and slippage for individual cells (gray circles) of the indicated cell lines (n = 50 for each cell line) treated with 10 nM taxol. Analysis carried out by time-lapse microscopy. Red line, median.
(F) Timing of mitotic cell death and slippage for individual cells (gray circles) of the indicated cell lines (n = 45 for each cell line) treated with 500 nM taxol. Analysis carried out by time-lapse microscopy. Red line, median.
(G and H) Effect of cGAS knockdown on mitotic cell death. Cells were subjected to siRNA targeting to cGAS (sicGAS) or control siRNA (siCNTRL). Cells were treated with 10 nM taxol, and individual cells were tracked using time-lapse microscopy (n = 60 for each sample). (G), Western blot analysis. Percentages indicate dilution series of samples prepared from control siRNA treated cells. (H), Timing of mitotic cell death and slippage for individual cells of the indicated cell lines (n = 60 for each cell line). Red line, median.
(I and J) Effect of cGAS knockdown on mitotic cell death of MDA-MB-231 cells. Cells were subjected to siRNA targeting to cGAS (sicGAS) or control siRNA (siCNTRL). Cells were treated with 500 nM taxol, and individual cells were tracked using time-lapse microscopy (n = 60 for each sample). (I), Western blot analysis. Percentages indicate dilution series of samples prepared from control siRNA treated cells. (J), Timing of mitotic cell death and slippage for individual cells of the indicated cell lines (n = 60 for each cell line). Red line, median.
(K–N) The cGAS-expressing cell line HBL-100 is prone to death in mitosis. siCNTRL, control siRNA. (K) cGAS expression and knockdown efficiency in the HBL-100 cell line. Percentages indicate dilution series of samples prepared from control siRNA treated cells. (L), Fraction of mitotic cell death upon treatment with the indicated concentrations of taxol. Untr., untreated. (M) cGAS knockdown reduces cell death in 10 nM taxol. (N) cGAS knockdown extends mitotic lifespan in HBL-100 cells. Individual cells (grey dots, n = 60) were tracked using time-lapse microscopy. Red line, median.
Figure S4. cGAS-induced mitotic cell death in HeLa cells and BJ hTERT fibroblasts. Related to Figure 4.
(A) Asynchronous HeLa cells (left) or BJ hTERT fibroblasts (right) were treated with 500 nM taxol and mitotic cells were tracked using time-lapse microscopy. Each dot represents an individual cell (n = 19-49 for each condition). Red lines represent the median. siCNTRL, control siRNA; sicGAS, cGAS siRNA.
(B) HeLa CCL-2 cells have a longer mitotic lifespan that is further extended by cGAS depletion. Duration of mitosis until mitotic cell death was monitored by time-lapse microscopy. Each dot represents an individual cell (n = 40 for each condition). Red lines represent the median. siCNTRL, control siRNA.
(C–E) Time-lapse microscopy-based analysis of mitotic cell death in nocodazole-arrested cells. Cells of the indicated cell lines were released from G2 arrest into media containing the indicated drugs, and duration of mitosis until mitotic cell death was monitored. All red lines indicate the median. Each dot represents an individual cell (n = 35-60 for each condition). siCNTRL, control siRNA; sicGAS, cGAS siRNA.
(F and G) Time-lapse microscopy-based analysis of mitotic cell death in HeLa cells arrested with proTAME in the absence of taxol. Cells of the indicated cell lines were released from G2 arrest into media containing 10 μM proTAME, and duration of mitosis until mitotic cell death was monitored. (F) Timing of slippage and mitotic cell death. All red lines indicate the median. Each dot represents an individual cell (n = 60 for each condition). Note that for unknown reasons, proTAME arrests a large fraction of cells for only limited periods of time. (G) Fraction of the cells from (F) dying during mitotic arrest. siCNTRL, control siRNA; sicGAS, cGAS siRNA.
(H and I) Time-lapse microscopy-based analysis of mitotic cell death in BJ hTERT fibroblasts arrested with proTAME in the absence of taxol. Cells of the indicated cell lines were released from G2 arrest into media containing 10 μM proTAME, and duration of mitosis until mitotic cell death was monitored. (H) Timing of slippage and mitotic cell death. All red lines indicate the median. Each dot represents an individual cell (n = 60 for each condition). Note that for unknown reasons, proTAME arrests a large fraction of cells for only limited periods of time. (I) Fraction of the cells from (H) dying during mitotic arrest. siCNTRL, control siRNA; sicGAS, cGAS siRNA.
(J) cGAS levels in the experiment shown in Fig. 4I, as determined by Western blot analysis. siCNTRL, control siRNA. Percentages indicate dilution series of samples prepared from control siRNA treated cells. cGAScat, catalytic mutant. cGASDNA, DNA binding-induced catalytic activity mutant.
(K) Control siRNA (siCNTRL) treatment does not affect mitotic cell death. Cells were released from G2 arrest into M phase with the indicated drugs, and duration of mitosis until mitotic cell death was monitored. All red lines indicate the median. Each dot represents an individual cell (n = 60 for each condition).
(L) cGAS levels in the experiment shown in Fig. 4J, as determined by Western blot. The vertical line indicates removed irrelevant lanes.
(M and N) cGAS does not affect survival after UV irradiation. Hela cells (M) or BJ h-TERT fibroblasts (N) were irradiated with the indicated doses of UV, and cell viability was determined at the indicated time points. Mean values and range (error bars) from two experiments are shown. Note that for many of the data points, error bars are too small to be seen.
siCNTRL, control siRNA; sicGAS, cGAS siRNA.
Figure S5. cGAS induces MOMP during mitotic arrest. Related to Figure 5.
(A) Schematic overview of the pathways leading to apoptotic cell death. Key to apoptosis is the activation of the effector caspases, caspase-3 and caspase-7. Caspase-3/7 activation is achieved by release of cytochrome C (CytC) and SMAC from mitochondria during mitochondrial outer membrane permeabilization (MOMP). MOMP itself is mediated by Bax activation through degradation of the Bax inhibitors Bcl-xL and Mcl1.
(B and C) Mitotic cell death is associated with caspase-3/7 activity. (B) Fraction of cells positive for IncuCyte caspase-3/7 assay signal during mitotic cell death. Note that timing of cell death is delayed by sicGAS treatment, though cells eventually still die with active caspase 3/7 when slippage is inhibited by proTAME. (C) Representative images of cells from (B) at times of mitotic cell death. Scale bar, 10 μm.
(D) The effect of the caspase inhibitor Z-VAD-FMK (ZVAD, 80 μM) was compared with sicGAS. Cells of the indicated cell lines were released from G2 arrest into media containing the indicated drugs. Timing of mitotic cell death of individual HeLa cells (grey dots, n = 50 each) was monitored. Red lines represent the median.
(E) Necroptosis does not contribute to cGAS-induced mitotic cell death. Cells were released from G2 arrest into media containing the indicated drugs. The fraction of cells dying during incubation with the indicated drugs is shown. The caspase inhibitor ZVAD was used at 80 μM; the necroptosis inhibitor Nec-1s was used at 10 μM.
(F–H) Analysis of release of cytochrome C (CytC) from mitochondria in the indicated cell lines released from G2 into 500 nM taxol 10 μM proTAME. Cells were fixed at the indicated time points and stained for cytochrome C. (F and G), Quantifications of HeLa cells (F) and BJ hTERT cells (G). (H), Representative images of HeLa cells at 14 h. Arrowheads indicate cells showing delocalized cytochrome C staining.
(I–K) Analysis of release of cytochrome C (CytC) and SMAC from mitochondria in the indicated cell lines released from G2 into 500 nM taxol in the absence of proTAME. For comparison, images of untreated cells harvested from asynchronous cultures by mitotic shake off are shown. (I) and (J), representative images from cells that were fixed at the indicated time points and stained for cytochrome C or SMAC, respectively. Arrowheads indicate cells showing delocalized cytochrome C or SMAC staining. (K), Quantifications.
(L–N) Analysis of Bcl-xL and Mcl1 degradation in the indicated cell lines. Cells were released from G2 arrest into 500 nM taxol 10 μM proTAME. (L and M) sample gels. Vertical lines indicate removed irrelevant lanes. (N) Quantifications. Error bars, SEM (n = 2 for Mcl1 and n = 4 for Bcl-xL).
(O) Bcl-xL, not Mcl1, is a major determinant of mitotic lifespan. Cells were released from G2 arrest into media containing the indicated drugs. Individual cells were tracked by time-lapse microscopy (n = ~50 cells for each condition). Mcl1 inh., 3 μM S63845. Bcl-xL inh., 1 μM ABT-263. Note that simultaneous inhibition of Bcl-xL and Mcl1 results in immediate cell death before cells can enter mitosis (indicated as N.A., not applicable).
(P) Bcl2 inhibition does not affect mitotic cell death. Cells were released from G2 arrest into media containing the indicated drugs. Individual cells were tracked by time-lapse microscopy (n = 60 cells for each condition). ABT-199 used at 100 nM; ABT-263 used at 1 μM.
siCNTRL, control siRNA.
Figure S6. Analysis of cGAS, STING, TBK1 and IRF3 functions in mitotic cell death. Related to Figure 6.
(A) Mitotic lifespan of BJ hTERT cells grown in the presence of 100 nM pyridone 6, and released from G2 arrest into 500 nM taxol, 10 μM proTAME. Individual cells (gray circles) were tracked by time-lapse microscopy (n = 60 for each condition).
(B–E) cGAS expression and cGAMP have only minor effect on ARPE-19 hTERT cells. (B), Western blot analysis of IRF3 phosphorylation following treatment of asynchronous cells with 33 μM cGAMP. (C), ARPE-19 hTERT cells were released from G2 arrest in the presence of taxol and proTAME. After three hours, cGAMP was added to 33 μM for the indicated cells. Untr., untreated. Mean and SEM from three experiments (n = 30 each) are shown. (D and E), Analysis of the effect of exogenous cGAS expression on viability of ARPE-19 hTERT cells in 500 nM taxol 10 μM proTAME. (D), Fraction of mitotic cell death. Individual cells (n = 30-60 each) were tracked using time-lapse microscopy. (E), Western blot analysis of protein expression. The same nomenclature for cGAS mutants is used as in Fig. 4I.
(F and G) cDNA prepared from the indicated cells was used to determine CXCL10 (F) or IRF1 (G) expression by quantitative PCR. +DNA, asynchronous cells transfected with naked DNA. Cells were released from G2 arrest into media containing the indicated drugs. Mean and range from two experiments are shown.
(H and I), STING and IRF3 promote cell death in mitosis. Analysis by Western blot (H) and time-lapse microscopy (I). Two clones for each guide RNA were analyzed. For (F), cells were released from G2 arrest into media containing the indicated drugs, and the time until mitotic cell death was monitored. All red lines indicate the median. Each dot represents an individual cell (n = 60 for each condition).
(J) Western blot analysis of STING expression in the experiment from Fig. 6I. Please note that background expression in the absence of doxycycline was too high and this condition was therefore not analyzed by microscopy.
(K and L), Western blot analysis of IRF3 levels in the experiments shown in Fig. 6K and 6L, respectively. The vertical line indicates removed irrelevant lanes.
(M and N) Analysis of rescue of IRF3 disruption with the K193R K313R K315R mutant that is defective in a form of transcription-independent cell death induction by IRF3. (M), Western blot analysis of IRF levels. (N) Time-lapse microscopy-based analysis of mitotic cell death for the indicated cell lines and treatments. Cells were released from G2 arrest into media containing the indicated drugs, and duration of mitosis until mitotic cell death was monitored. All red lines indicate the median. Each dot represents an individual cell (n = 35-60 for each condition).
(O) Western blot of IRF3 levels for the experiment shown in Fig. 6M.
(P and Q) Analysis of mitotic lifespan in CRISPR-mediated TBK1 disruptions generated with two different guide RNAs. Note that the cell line used to generate these disruptions is the same as is used in Fig. 2A, and also expresses GFP-IRF3. (P), Time-lapse microscopy-based analysis of mitotic cell death for the indicated cell lines. Cells were released from G2 arrest into media containing the indicated drugs, and duration of mitosis until mitotic cell death was monitored. All red lines indicate the median. Each dot represents an individual cell (n = 60 for each condition). (Q), Western blot analysis of TBK1 levels.
Figure S7. cGAS pathway roles in taxol response of mouse xenograft tumors. Related to Figure 7.
(A) Overview of the xenograft experiment from Figure 7A-7F and Figure S7A-S7E. NSG mice were subcutaneously injected bilaterally with either wild type or cGAS-disrupted (sg_cGAS) HeLa cells. After two weeks, mice were either treated with weekly taxol doses (10 mg/kg), or control treated with DMSO. Two days after the second taxol/DMSO administration, tumors were prepared for histological analysis. Two days after the third taxol/DMSO administration, tumors were prepared for Western blot analysis.
(B) Presence of cGAS on mitotic chromosomes and within the interphase cytoplasm in the indicated xenografts tumors. Arrowheads indicate cells undergoing mitosis (magnified views of the mitotic cells on the right). Scale bar, 10 μm.
(C) Representative tumors from the indicated genotypes and treatments. Scale bar, 1 cm.
(D) IgG isotype control staining of a slice of the cGAS wild type xenograft tumor. Scale bar, 100 μm.
(E) Immunocompromised (NSG) mice were injected with either wild type or IRF3-disrupted (sg_IRF3) HeLa cells, or with sg_IRF3 cells containing doxycycline-inducible rescue constructs expressing either GFP-IRF3 or GFP-IRF3_ΔDBD. After two weeks, mice were subjected to weekly treatments with taxol or DMSO as a control. After three weeks, tumors were recovered and analyzed (see also Figure S7A). Mice were fed either with or without doxycycline-containing food. Growth curves for the indicated tumors and treatments are shown (mean and SEM; n = 8 for wt + DMSO; n = 8 for wt + Taxol; n = 7 for sg_IRF3 + DMSO; n = 7 for sg_IRF3 + taxol; n = 8 for sg_IRF3 + IRF3 +DMSO −doxycycline; n = 8 for sg_IRF3 + IRF3 +taxol −doxycycline; n = 8 for sg_IRF3 + IRF3 +DMSO +doxycycline; n = 8 for sg_IRF3 + IRF3 +taxol +doxycycline; n = 8 for sg_IRF3 + IRF3_ΔDBD +DMSO −doxycycline; n = 8 for sg_IRF3 + IRF3_ΔDBD +taxol −doxycycline;) n = 7 for sg_IRF3 + IRF3_ΔDBD +DMSO +doxycycline; n = 8 for sg_IRF3 + TRF3_ΔDBD +taxol +doxycycline). ⁋: Experiment termination at the planned schedule. ‡: All but one mouse in this experimental group were found dead at day 25.
Movie S1: Time-lapse imaging of cGAS knockdown cells rescued with GFP or GFP-cGAS during mitotic arrest. Related to Figures 1 and 4.
cGAS-depleted HeLa cells expressing GFP (left) or siRNA resistant GFP-cGAS (right), and H2B-RFP were released from G2 into 500 nM taxol 10 μM proTAME. Images were taken every 15 min. Top: Overlays of H2B-RFP and GFP (left) or GFP-cGAS (right). Bottom: DIC channel images corresponding to the fluorescence images on top.
Table S1: Analysis of non-small cell lung cancer patient data from The Cancer Genome Atlas (TCGA). Related to Figure 7.
The table is broken into three tabs.
(Overview) Overview of the patients whose data was processed for Figure 7G. The table on the left divides patients by whether or not they received taxane-based treatment, the table on the right divides patients by whether or not they received DNA-damaging agents. LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma. Stage, number of patients for each cancer stage.
(Raw) Detailed data for each patient that was used for Figure 7G. Column “vitalDay” contains the day of death or day of last follow-up. CGAS column, z score for CGAS expression.
(Risk) Number of patients at risk for each cohort at each year.
Table S2: Analysis of ovarian cancer patient data from The Cancer Genome Atlas (TCGA). Related to Figure 7.
The table is broken into three tabs.
(Overview) Overview of the patients whose data was processed for Figure 7H. The table on the left divides patients by whether or not they received taxane-based treatment, the table on the right divides patients by whether or not they received DNA-damaging agents. Stage, number of patients for each cancer stage.
(Raw) Detailed data for each patient that was used for Figure 7H. Column “vitalDay” contains the day of death or day of last follow-up. CGAS column, z score for CGAS expression.
(Risk) Number of patients at risk for each cohort at each year.
Table S3: Oligonucleotides used in this study.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Bad | Cell Signaling Technology | Cat# 9239; RRID: AB_2062127 |
| Bak | Cell Signaling Technology | Cat# 12105; RRID: AB_2716685 |
| Active Bax 6A7 | Thermo Fisher | Cat# 14-6997-81; RRID: AB_468388 |
| Bax | Proteintech Group | Cat# 50599-2-lg |
| Bcl-xL | Cell Signaling Technology | Cat# 2764; RRID: AB_2228008 |
| Bim | Cell Signaling Technology | Cat# 2819; RRID: AB_10692515 |
| c-IAP1 | Cell Signaling Technology | Cat# 7065; RRID: AB_10890862 |
| Casp-2 | Abcam | Cat# ab179520 |
| Casp-7 | Abcam | Cat# ab201959 |
| Casp-9 | Abcam | Cat# ab202068 |
| cGAS | Cell Signaling Technology | Cat# 15102; RRID: AB_2732795 |
| Cleaved casp-3 | Cell Signaling Technology | Cat# 9661; RRID: AB_2341188 |
| Cytochrome C | Abcam | Cat# ab110325; RRID: AB_10864775 |
| IFNAR2 | Millipore | Cat# MAB1155; RRID: AB_2122758 |
| IκBα | Cell Signaling Technology | Cat# 9242; RRID: AB_331623 |
| IRF1 | Cell Signaling Technology | Cat# 8478; RRID: AB_10949108 |
| IRF3 (phospho Ser386) | Abcam | Cat# ab76493; RRID: AB_1523836 |
| IRF3 (phospho Ser396) | Cell Signaling Technology | Cat# 4947; RRID: AB_823547 |
| IRF3 | Abcam | Cat# ab68481; RRID: AB_11155653 |
| IRF3 | Proteintech Group | Cat# 11312-1-AP; RRID: AB_2127004 |
| JNK2 | Cell Signaling Technology | Cat# 9258; RRID: AB_2141027 |
| Phospho-JNK | Cell Signaling Technology | Cat# 4668; RRID: AB_823588 |
| Murine IgG2a Isotype Control | Sigma-Aldrich | Cat# M5409; RRID: AB_1163691 |
| Mcl1 | Cell Signaling Technology | Cat# 4572; RRID: AB_2281980 |
| Phospho-CDK substrate motif | Cell Signaling Technology | Cat# 9477; RRID: AB_2714143 |
| PUMA | Cell Signaling Technology | Cat# 12450 |
| RNA pol II CTD S2ph | Abcam | Cat# ab5095; RRID: AB_304749 |
| SMAC/Diablo | Cell Signaling Technology | Cat# 15108 |
| STING | Cell Signaling Technology | Cat# 13647; RRID: AB_2732796 |
| TBK1 | Abcam | Cat# ab40676; RRID: AB_776632 |
| α-Tubulin | Sigma-Aldrich | Cat# T9026; RRID: AB_477593 |
| XIAP | Cell Signaling Technology | Cat# 2045; RRID: AB_2214866 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| ABT-199 | Selleck Chemicals | Cat# S8048; CAS No. 1257044-40-8 |
| ABT-263 | Selleck Chemicals | Cat# S1001; CAS No. 923564-51-6 |
| 2'3'-cGAMP | InvivoGen | Cat# tlrl-nacga23; CAS No. 1441190-66-4 |
| Doxycycline hyclate | Sigma Aldrich | Cat# D9891; CAS No. 24390-14-5 |
| IFNβ | R & D Systems | Cat# 8499-IF-010/CF |
| Nec-1s | BioVision | Cat# 2263; CAS No. 852391-15-2 |
| Nocodazole | Sigma-Aldrich | Cat# M1404; CAS No. 31430-18-9 |
| Pyridone 6 | MedChemExpress | Cat# HY-14435; CAS No. 457081-03-7 |
| Paclitaxel (Taxol) | Calbiochem | Cat# 580555; CAS No. 33069-62-4 |
| proTAME | Boston Biochemicals | Cat# I-440 |
| RO-3306 | Sigma Aldrich | Cat# SML0569; CAS No. 872573-93-8 |
| S63845 | Selleck Chemicals | Cat# S8383; CAS No. 1799633-27-4 |
| SiR-DNA | Cyotskeleton | Cat# CY-SC007 |
| Thymidine | Sigma-Aldrich | Cat# T1895; CAS No. 50-89-5 |
| Triptolide | Invivogen | Cat# ant-tpl; CAS No. 38748-32-2 |
| Z-VAD-FMK (ZVAD) | EMD-Millipore | Cat# 627610; CAS No. 187389-52-2 |
| Critical Commercial Assays | ||
| RNeasy Mini Kit | QIAGEN | Cat# 74014 |
| Transcriptor First Strand cDNA Synthesis kit | Roche | Cat# 04379012001 |
| LiqhtCycler 480 SYBR Green I Master mix | Roche | Cat# 04707516001 |
| Incucyte Caspase-3/7 Reagent | Essen Bioscience | Cat# 4440 |
| Experimental Models: Cell Lines | ||
| ARPE-19 hTERT | T. de Lange | N/A |
| BJ hTERT | T. de Lange | N/A |
| BT474 | K. Birsoy | RRID: CVCL_0179 |
| BT549 | K. Birsoy | RRID: CVCL_1092 |
| CAL51 | K. Birsoy | RRID: CVCL_1110 |
| HBL-100 | K. Birsoy | RRID: CVCL_4362 |
| HCC1143 | K. Birsoy | RRID: CVCL_1245 |
| HeLa CCL-2 | I Nakagawa | RRID: CVCL_0030 |
| HeLa Flp-In T-Rex | A. Desai/R. Gassmann | N/A |
| MDA-MB-157 | K. Birsoy | RRID: CVCL_0618 |
| MDA-MB-231 | K. Birsoy | RRID: CVCL_0062 |
| RUES2 | A. Brivanlou | human ESC registry no. 0013; RRID: CVCL_VM29 |
| T47D | K. Birsoy | RRID: CVCL_0553 |
| Experimental Models: Organisms/Strains | ||
| Immunodeficient mice, NOD-scid IL2Rgnull (NSG), male | Jackson Laboratory | Cat# 005557 |
| Xenopus laevis frogs, female | NASCO | Cat# LM00535M |
| Oligonucleotides | ||
| cGAS siRNA ON-TARGET plus SMARTpool | Dharmacon | Cat# L-015607-02 |
| Non-targeting control siRNA | Dharmacon | Cat# D-001810-01 |
| sgRNAs, see Table S2 | This paper | N/A |
| QPCR primers, see Table S2 | (Wu et al., 2013; Wu et al., 2018; Yoney et al., 2018) | N/A |
| Recombinant DNA | ||
| pcDNA5/FRT/TO | ThermoFisher | Cat# V652020 |
| pX330-U6-Chimeric_BB-CBh-hSpCas9 | (Cong et al., 2013) | Addgene Cat# 42230 |
| cGAS bacterial expression plasmids | This paper | N/A |
| Software and Algorithms | ||
| CellCognition (v. 1.5.2) | (Held et al., 2010) | http://www.cellcognition-project.org/ |
| Graphpad Prism (v.5.0a) | GraphPad Software | https://www.graphpad.com/ |
| ImageJ (v. 2.0.0-rc-43/1.51k) | NIH | https://imagej.nih.gov/ij/index.html |
| SoftWoRx | Applied Precision | N/A |
| Vision4D | Arivis | https://www.arivis.com/en/imaging-science/arivis-vision4d |
Highlights for CELL-D-18-03062.
Nucleosomes suppress DNA-induced cGAMP synthesis by cGAS
During mitotic arrest, cGAS promotes a slow buildup of IRF3 phosphorylation
Phospho-IRF3 promotes mitotic apoptosis independently of transcription induction
Xenograft experiments and patient data indicate a role for cGAS in taxol chemotherapy
ACKNOWLEDGMENTS
We thank K. Birsoy, A. Brivanlou, A. Desai, T. de Lange, R. Gassmann and I. Nakagawa for cell lines, G. Alushin, T. de Lange, C. Jenness, B. Sleckman, S. Tavazoie, D. Wynne and A. Yoney for reading of the manuscript and helpful suggestions; R. Benezra, for discussions and comments on the manuscript, C. Adura Alcaino, K. Birsoy, F. Glickman, L. Lama, S. Larisch, S. Nagata, B. Ostendorf, U. Schaefer, B. Sleckman, H. Steller, S. Tavazoie, J. Xue and members of the Funabiki lab for helpful discussions; A. Yoney for help with qPCR and hESCs; A. North and C. Pyrgaki for support with imaging. H.F (R01GM075249; R01GM125302; R35GM132111), the Bio-Imaging Resource Center (S10RR031855); and N.Y. (UL1TR001866) are supported by grants from the National Institutes of Health. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. N.Y. is supported by the Vincent Meyer Foundation and the Helmsley Charitable Trust. Patient analysis was based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/.
Footnotes
DECLARATION OF INTERESTS
The authors declare that no competing financial interests exist.
H.F. is affiliated with Graduate School of Medical Sciences, Weill Cornell Medicine, and Cell Biology Program, the Sloan Kettering Institute.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abe T, and Barber GN (2014). Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J Virol 88, 5328–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, RÖhl I, Hopfner K-P, Ludwig J, and Hornung V (2013). cGAS produces a 2“-5-”linked cyclic dinucleotide second messenger that activates STING. Nature 498, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan LA, and Clarke PR (2007). Phosphorylation of caspase-9 by CDK1/cyclin B1 protects mitotic cells against apoptosis. Mol Cell 26, 301–310. [DOI] [PubMed] [Google Scholar]
- Andersen JL, Johnson CE, Freel CD, Parrish AB, Day JL, Buchakjian MR, Nutt LK, Thompson JW, Moseley MA, and Kornbluth S (2009). Restraint of apoptosis during mitosis through interdomain phosphorylation of caspase-2. Embo J 28, 3216–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreeva L, Hiller B, Kostrewa D, Lässig C, de Oliveira Mann CC, Jan Drexler D, Maiser A, Gaidt M, Leonhardt H, Hornung V, et al. (2017). cGAS senses long and HMGB/TFAM-bound U-turn DNA by forming protein-DNA ladders. Nature 549, 394–398. [DOI] [PubMed] [Google Scholar]
- Bakhoum SF, Ngo B, Laughney AM, Cavallo J-A, Murphy CJ, Ly P, Shah P, Sriram RK, Watkins TBK, Taunk NK, et al. (2018). Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber GN (2015). STING: infection, inflammation and cancer. Nat. Rev. Immunol. 15, 760–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensaude O (2011). Inhibiting eukaryotic transcription: Which compound to choose? How to evaluate its activity? Transcription 2, 103–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger AC, Korkut A, Kanchi RS, Hegde AM, Lenoir W, Liu W, Liu Y, Fan H, Shen H, Ravikumar V, et al. (2018). A Comprehensive Pan-Cancer Molecular Study of Gynecologic and Breast Cancers. Cancer Cell 33, 690–705.e699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagosklonny MV (2007). Mitotic arrest and cell fate: why and how mitotic inhibition of transcription drives mutually exclusive events. Cell Cycle 6, 70–74. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Network (2012). Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay S, Kuzmanovic T, Zhang Y, Wetzel JL, and Sen GC (2016). Ubiquitination of the Transcription Factor IRF-3 Activates RIPA, the Apoptotic Pathway that Protects Mice from Viral Pathogenesis. Immunity 44, 1151–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chattopadhyay S, Marques JT, Yamashita M, Peters KL, Smith K, Desai A, Williams BRG, and Sen GC (2010). Viral apoptosis is induced by IRF-3-mediated activation of Bax. Embo J 29, 1762–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Sun L, and Chen ZJ (2016). Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 17, 1142–1149. [DOI] [PubMed] [Google Scholar]
- Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, Zhang H, McLellan M, Yau C, Kandoth C, et al. (2015). Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 163, 506–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civril F, Deimling T, de Oliveira Mann CC, Ablasser A, Moldt M, Witte G, Hornung V, and Hopfner K-P (2013). Structural mechanism of cytosolic DNA sensing by cGAS. Nature 498, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin P, Nashchekina O, Walker R, and Pines J (2013). The spindle assembly checkpoint works like a rheostat rather than a toggle switch. Nat Cell Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox DR (1972). Regression Models and Life-Tables. Journal of the Royal Statistical Society: Series B (Methodological) 34, 187–202. [Google Scholar]
- Doménech E, Maestre C, Esteban-Martínez L, Partida D, Pascual R, Fernández-Miranda G, Seco E, Campos-Olivas R, Pérez M, Megias D, et al. (2015). AMPK and PFKFB3 mediate glycolysis and survival in response to mitophagy during mitotic arrest. Nat Cell Biol 17, 1304–1316. [DOI] [PubMed] [Google Scholar]
- Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Z, et al. (2017). Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550, 402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du M, and Chen ZJ (2018). DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 361, 704–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaglesham JB, Pan Y, Kupper TS, and Kranzusch PJ (2019). Viral and metazoan poxins are cGAMP-specific nucleases that restrict cGAS-STING signalling. Nature 566, 259–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel K, Rudelius M, Slawska J, Jacobs L, Ahangarian Abhari B, Altmann B, Kurutz J, Rathakrishnan A, Fernández-Sáiz V, Brunner A, et al. (2016). USP9X stabilizes XIAP to regulate mitotic cell death and chemoresistance in aggressive B-cell lymphoma. EMBO Mol Med 8, 851–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita R, Otake K, Arimura Y, Horikoshi N, Miya Y, Shiga T, Osakabe A, Tachiwana H, Ohzeki J-I, Larionov V, et al. (2015). Stable complex formation of CENP-B with the CENP-A nucleosome. Nucleic Acids Res 43, 4909–4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidt MM, Ebert TS, Chauhan D, Ramshorn K, Pinci F, Zuber S, O'Duill F, Schmid-Burgk JL, Hoss F, Buhmann R, et al. (2017). The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell 171, 1110–1124.e1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, Serganov AA, Liu Y, Jones RA, Hartmann G, et al. (2013a). Cyclic [G(2',5')pA(3“,5”)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 153, 1094–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Ascano M, Zillinger T, Wang W, Dai P, Serganov AA, Gaffney BL, Shuman S, Jones RA, Deng L, et al. (2013b). Structure-function analysis of STING activation by c[G(2',5')pA(3“,5”)p] and targeting by antiviral DMXAA. Cell 154, 748–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner E, Kim Y, Lach FP, Kottemann MC, and Smogorzewska A (2013). Human GEN1 and the SLX4-associated nucleases MUS81 and SLX1 are essential for the resolution of replication-induced Holliday junctions. Cell Reports 5, 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gascoigne KE, and Taylor SS (2008). Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 14, 111–122. [DOI] [PubMed] [Google Scholar]
- Gassmann R, Holland AJ, Varma D, Wan X, Civril F, Cleveland DW, Oegema K, Salmon ED, and Desai A (2010). Removal of Spindly from microtubule-attached kinetochores controls spindle checkpoint silencing in human cells. Genes Dev 24, 957–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentili M, Lahaye X, Nadalin F, Nader GPF, Puig Lombardi E, Herve S, De Silva NS, Rookhuizen DC, Zueva E, Goudot C, et al. (2019). The N-Terminal Domain of cGAS Determines Preferential Association with Centromeric DNA and Innate Immune Activation in the Nucleus. Cell Reports 26, 2377–2393.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glück S, Guey B, Gulen MF, Wolter K, Kang T-W, Schmacke NA, Bridgeman A, Rehwinkel J, Zender L, and Ablasser A (2017). Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat Cell Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goubau D, Romieu-Mourez R, Solis M, Hernandez E, Mesplède T, Lin R, Leaman D, and Hiscott J (2009). Transcriptional re-programming of primary macrophages reveals distinct apoptotic and anti-tumoral functions of IRF-3 and IRF-7. Eur. J. Immunol. 39, 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulen MF, Koch U, Haag SM, Schuler F, Apetoh L, Villunger A, Radtke F, and Ablasser A (2017). Signalling strength determines proapoptotic functions of STING. Nat Commun 8, 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guse A, Carroll CW, Moree B, Fuller CJ, and Straight AF (2011). In vitro centromere and kinetochore assembly on defined chromatin templates. Nature 477, 354–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, and Greenberg RA (2017). Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley ME, Allan LA, Sanderson HS, and Clarke PR (2010). Phosphorylation of Mcl-1 by CDK1-cyclin B1 initiates its Cdc20-dependent destruction during mitotic arrest. Embo J 29, 2407–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Held M, Schmitz MHA, Fischer B, Walter T, Neumann B, Olma MH, Peter M, Ellenberg J, and Gerlich DW (2010). CellCognition: time-resolved phenotype annotation in high-throughput live cell imaging. Nat. Methods 7, 747–754. [DOI] [PubMed] [Google Scholar]
- Heylbroeck C, Balachandran S, Servant MJ, DeLuca C, Barber GN, Lin R, and Hiscott J (2000). The IRF-3 transcription factor mediates Sendai virus-induced apoptosis. J Virol 74, 3781–3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichim G, and Tait SWG (2016). A fate worse than death: apoptosis as an oncogenic process. Nat Rev Cancer 16, 539–548. [DOI] [PubMed] [Google Scholar]
- Ishikawa H, and Barber GN (2008). STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan MA, and Wilson L (2004). Microtubules as a target for anticancer drugs. Nat Rev Cancer 4, 253–265. [DOI] [PubMed] [Google Scholar]
- Knowlton JJ, Dermody TS, and Holm GH (2012). Apoptosis induced by mammalian reovirus is beta interferon (IFN) independent and enhanced by IFN regulatory factor 3- and NF-κB-dependent expression of Noxa. J Virol 86, 1650–1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno H, Konno K, and Barber GN (2013). Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell 155, 688–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranzusch PJ, Lee AS-Y, Berger JM, and Doudna JA (2013). Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Reports 3, 1362–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lallemand C, Blanchard B, Palmieri M, Lebon P, May E, and Tovey MG (2007). Single-stranded RNA viruses inactivate the transcriptional activity of p53 but induce NOXA-dependent apoptosis via post-translational modifications of IRF-1, IRF-3 and CREB. Oncogene 26, 328–338. [DOI] [PubMed] [Google Scholar]
- Lau L, Gray EE, Brunette RL, and Stetson DB (2015). DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science 350, 568–571. [DOI] [PubMed] [Google Scholar]
- London N, and Biggins S (2014). Signalling dynamics in the spindle checkpoint response. Nat Rev Mol Cell Biol 15, 736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowary PT, and Widom J (1998). New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J Mol Biol 276, 19–42. [DOI] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Martin C-A, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, et al. (2017). cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison TJ, Pineda J, Shi J, and Florian S (2017). Is inflammatory micronucleation the key to a successful anti-mitotic cancer drug? Open Biol 7, 170182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison TJ (2012). The proliferation rate paradox in antimitotic chemotherapy. Mol Biol Cell 23, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray AW (1991). Cell cycle extracts. Methods Cell Biol 36, 581–605. [PubMed] [Google Scholar]
- Nakhaei P, Sun Q, Solis M, Mesplède T, Bonneil É, Paz S, Lin R, and Hiscott J (2012). IκB kinase ε-dependent phosphorylation and degradation of X-linked inhibitor of apoptosis sensitizes cells to virus-induced apoptosis. J Virol 86, 726–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novais-Cruz M, Alba Abad M, van IJcken WF, Galjart N, Jeyaprakash AA, Maiato H, and Ferrás C (2018). Mitotic progression, arrest, exit or death relies on centromere structural integrity, rather than de novo transcription. Elife 7, 1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orth JD, Loewer A, Lahav G, and Mitchison TJ (2012). Prolonged mitotic arrest triggers partial activation of apoptosis, resulting in DNA damage and p53 induction. Mol Biol Cell 23, 567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orthwein A, Fradet-Turcotte A, Noordermeer SM, Canny MD, Brun CM, Strecker J, Escribano-Diaz C, and Durocher D (2014). Mitosis inhibits DNA double-strand break repair to guard against telomere fusions. Science 344, 189–193. [DOI] [PubMed] [Google Scholar]
- Palozola KC, Donahue G, Liu H, Grant GR, Becker JS, Cote A, Yu H, Raj A, and Zaret KS (2017). Mitotic transcription and waves of gene reactivation during mitotic exit. Science 358, 119–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrasek J, Iracheta-Vellve A, Csak T, Satishchandran A, Kodys K, Kurt-Jones EA, Fitzgerald KA, and Szabo G (2013). STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proceedings of the National Academy of Sciences 110, 16544–16549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott DM, and Bender MA (1962). Synthesis of RNA and protein during mitosis in mammalian tissue culture cells. Exp Cell Res 26, 260–268. [DOI] [PubMed] [Google Scholar]
- Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B, Eitson JL, Mar KB, Richardson RB, Ratushny AV, Litvak V, et al. (2014). Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505, 691–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Zhou Y, Huang H-C, and Mitchison TJ (2011). Navitoclax (ABT-263) accelerates apoptosis during drug-induced mitotic arrest by antagonizing Bcl-xL. Cancer Res 71, 4518–4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, Kotb M, Gillies SD, King M, Mangada J, et al. (2005). Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol 174, 6477–6489. [DOI] [PubMed] [Google Scholar]
- Sieben CJ, Sturmlechner I, van de Sluis B, and van Deursen JM (2018). Two-Step Senescence-Focused Cancer Therapies. Trends Cell Biol 28, 723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloss O, Topham C, Diez M, and Taylor S (2016). Mcl-1 dynamics influence mitotic slippage and death in mitosis. Oncotarget 7, 5176–5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, et al. (2013). ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med 19, 202–208. [DOI] [PubMed] [Google Scholar]
- Spektor A, Umbreit NT, and Pellman D (2017). Cell Biology: When Your Own Chromosomes Act like Foreign DNA. Curr Biol 27, R1228–R1231. [DOI] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, and Chen ZJ (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Duprez L, Grootjans S, Cauwels A, Nerinckx W, DuHadaway JB, Goossens V, Roelandt R, Van Hauwermeiren F, Libert C, et al. (2012). Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis 3, e437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang C-HA, Zundell JA, Ranatunga S, Lin C, Nefedova Y, Del Valle JR, and Hu C-CA (2016). Agonist-Mediated Activation of STING Induces Apoptosis in Malignant B Cells. Cancer Res 76, 2137–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JE, Cubbon RM, Cummings RT, Wicker LS, Frankshun R, Cunningham BR, Cameron PM, Meinke PT, Liverton N, Weng Y, et al. (2002). Photochemical preparation of a pyridone containing tetracycle: a Jak protein kinase inhibitor. Bioorg Med Chem Lett 12, 1219–1223. [DOI] [PubMed] [Google Scholar]
- Tighe A, Staples O, and Taylor S (2008). Mps1 kinase activity restrains anaphase during an unperturbed mitosis and targets Mad2 to kinetochores. J Cell Biol 181, 893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topham CH, and Taylor SS (2013). Mitosis and apoptosis: how is the balance set? Curr Opin Cell Biol 25, 780–785. [DOI] [PubMed] [Google Scholar]
- Topham C, Tighe A, Ly P, Bennett A, Sloss O, Nelson L, Ridgway RA, Huels D, Littler S, Schandl C, et al. (2015). MYC Is a Major Determinant of Mitotic Cell Fate. Cancer Cell 28, 129–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Xu L, Su J, Peppelenbosch MP, and Pan Q (2017). Transcriptional Regulation of Antiviral Interferon-Stimulated Genes. Trends Microbiol 25, 573–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver BA (2014). How Taxol/paclitaxel kills cancer cells. Mol Biol Cell 25, 2677–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheelock MS, Wynne DJ, Tseng BS, and Funabiki H (2017). Dual recognition of chromatin and microtubules by INCENP is important for mitotic progression. J Cell Biol 216, 925–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, and Chen ZJ (2013). Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Dao Thi VL, Huang Y, Billerbeck E, Saha D, Hoffmann H-H, Wang Y, Silva AV, Sarbanes S, Sun T, et al. (2018). Intrinsic Immunity Shapes Viral Resistance of Stem Cells. Cell 172, 423–438.e425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Ren J, Chen Q, and Chen ZJ (2017). cGAS is essential for cellular senescence. Proceedings of the National Academy of Sciences 9, 201705499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Kenny AE, Brito DA, and Rieder CL (2009). Cells satisfy the mitotic checkpoint in Taxol, and do so faster in concentrations that stabilize syntelic attachments. J Cell Biol 186, 675–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoney A, Etoc F, Ruzo A, Carroll T, Metzger JJ, Martyn I, Li S, Kirst C, Siggia ED, and Brivanlou AH (2018). WNT signaling memory is required for ACTIVIN to function as a morphogen in human gastruloids. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X, Sigoillot F, Gaur S, Choi S, Pfaff KL, Oh D-C, Hathaway N, Dimova N, Cuny GD, and King RW (2010). Pharmacologic inhibition of the anaphase-promoting complex induces a spindle checkpoint-dependent mitotic arrest in the absence of spindle damage. Cancer Cell 18, 382–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zierhut C, and Funabiki H (2015). Nucleosome functions in spindle assembly and nuclear envelope formation. Bioessays 37, 1074–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zierhut C, Jenness C, Kimura H, and Funabiki H (2014). Nucleosomal regulation of chromatin composition and nuclear assembly revealed by histone depletion. Nat Struct Mol Biol 21, 617–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. cGAS interacts with nucleosomes and mitotic chromosomes. Related to Figure 1.
(A) cGAS associates with nucleosomes in M-phase Xenopus egg extracts. Beads coated with nucleosome arrays (Nucleosomes), naked DNA (Naked DNA) or just streptavidin (Beads) were incubated in egg extract containing 35S-labelled cGAS. Proteins associated with each set of beads were detected by fluorometry (upper panel). Note that the extract was depleted of endogenous histones H3–H4 to prevent nucleosome assembly on naked DNA. CBB, Coomassie brilliant blue. Streptavidin serves as an indicator for the amount of beads loaded. The strong CBB staining in the input lane is due to extract proteins migrating at this size. The lower panel shows amounts of DNA immobilized on the indicated beads.
(B) Coomassie stained gel of purified wild type and KKKR mutant cGAS.
(C) Example of the gel shift analysis with recombinant cGAS and naked DNA or mononucleosomes.
(D) Quantification of cGAMP generation by cGAS in conjunction with naked DNA or mononucleosomes.
(E–G) cGAS associates with mitotic chromosomes in living cells. Time-lapse analysis of HeLa cells stably expressing the indicated constructs released from G2 arrest into 500 nM taxol. NEBD, nuclear envelope breakdown. GFP-cGASDNA, K407E K411A DNA-binding mutant.
(H–J) cGAS associates with mitotic chromosomes. Images of HeLa cells (H) or BJ hTERT fibroblasts (I) treated with either siRNA to cGAS (sicGAS) or with control siRNA (siCNTRL), fixed and stained with anti-cGAS antibodies. Quantification of the fraction of mitotic figures that are positive for cGAS localization on chromosomes in either untreated cells (Untr.) or cells treated with 500 nM taxol for 4 h is shown in (J).
All graphs represent mean values and SEM from at least three independent experiments.
Figure S2. cGAS-signaling in mitosis. Related to Figure 2.
(A) Western blot analysis of IRF3 S396 phosphorylation in HeLa cells of the indicated type (with or without GFP-IRF3 overexpression) and siRNA treatment harvested either in G2 or after the indicated times during arrest in 1.7 μM nocodazole.
(B) Western blot analysis of IRF3 S386 phosphorylation in HeLa cells harvested either in G2 or after the indicated times during arrest in 1.7 μM nocodazole.
(C) Immunofluorescence analysis of cGAS staining on mitotic chromosomes in RUES2 hES cells.
(D) Mitotic phosphorylation of IRF3 at Ser386 in RUES hES cells. Cells were treated with 500 nM taxol for 6 h and mitotic cells were collected by shake-off. Non mitotic cells were subsequently removed by scraping. As control for cell cycle arrest, a phospho-CDK site Western blot is shown.
(E) Analysis of IRF3 phosphorylation in BJ hTERT fibroblasts. Samples were taken following DNA transfection (+DNA) at the indicated times after release into 500 nM taxol, 10 μM proTAME from G2. siCNTRL, control siRNA
(F) Western blot analysis of phosphorylation of IRF3 immunoprecipitated from G2 arrested cells, or from cells at the indicated time points during arrest in 500 nM taxol, 10 μM proTAME. siCNTRL, control siRNA.
(G and H) Western blot analysis of IκBα levels in BJ hTERT cells harvested either in G2 or after the indicated times during arrest in 0.5 μM taxol 10 μM proTAME. (G) Example gel. The vertical line indicates removed irrelevant lanes. (H) Quantification. Shown are mean values and SEM from four experiments. P values are indicated at the 16 h and 20 h time points. siCNTRL, control siRNA. sicGAS, cGAS siRNA.
(I and J) Western blot analysis of JNK2 phosphorylation (T183/Y185) in BJ hTERT cells harvested either in G2 or after the indicated times during arrest in 0.5 μM taxol 10 μM proTAME. (I) Example gel. (J) Quantification. Shown are mean values and SEM from six experiments. P values are indicated at the 16 h and 20 h time points. siCNTRL, control siRNA. sicGAS, cGAS siRNA.
Figure S3. Mi totic cell death in breast cancer cell lines and HBL-100. Related to Figure 3.
(A–D) Western blot analysis of IRF3 S386 phosphorylation in cells of the indicated cell lines harvested either asynchronously or after the indicated times following treatment with 500 nM taxol. Taxol treated cells were harvested by shake off. Note that HCC1143 grows too slowly to allow quantitative collection of mitotic cells at 12 h. As control for cell cycle arrest, phospho-CDK site Western blots are shown.
(E) Timing of mitotic cell death and slippage for individual cells (gray circles) of the indicated cell lines (n = 50 for each cell line) treated with 10 nM taxol. Analysis carried out by time-lapse microscopy. Red line, median.
(F) Timing of mitotic cell death and slippage for individual cells (gray circles) of the indicated cell lines (n = 45 for each cell line) treated with 500 nM taxol. Analysis carried out by time-lapse microscopy. Red line, median.
(G and H) Effect of cGAS knockdown on mitotic cell death. Cells were subjected to siRNA targeting to cGAS (sicGAS) or control siRNA (siCNTRL). Cells were treated with 10 nM taxol, and individual cells were tracked using time-lapse microscopy (n = 60 for each sample). (G), Western blot analysis. Percentages indicate dilution series of samples prepared from control siRNA treated cells. (H), Timing of mitotic cell death and slippage for individual cells of the indicated cell lines (n = 60 for each cell line). Red line, median.
(I and J) Effect of cGAS knockdown on mitotic cell death of MDA-MB-231 cells. Cells were subjected to siRNA targeting to cGAS (sicGAS) or control siRNA (siCNTRL). Cells were treated with 500 nM taxol, and individual cells were tracked using time-lapse microscopy (n = 60 for each sample). (I), Western blot analysis. Percentages indicate dilution series of samples prepared from control siRNA treated cells. (J), Timing of mitotic cell death and slippage for individual cells of the indicated cell lines (n = 60 for each cell line). Red line, median.
(K–N) The cGAS-expressing cell line HBL-100 is prone to death in mitosis. siCNTRL, control siRNA. (K) cGAS expression and knockdown efficiency in the HBL-100 cell line. Percentages indicate dilution series of samples prepared from control siRNA treated cells. (L), Fraction of mitotic cell death upon treatment with the indicated concentrations of taxol. Untr., untreated. (M) cGAS knockdown reduces cell death in 10 nM taxol. (N) cGAS knockdown extends mitotic lifespan in HBL-100 cells. Individual cells (grey dots, n = 60) were tracked using time-lapse microscopy. Red line, median.
Figure S4. cGAS-induced mitotic cell death in HeLa cells and BJ hTERT fibroblasts. Related to Figure 4.
(A) Asynchronous HeLa cells (left) or BJ hTERT fibroblasts (right) were treated with 500 nM taxol and mitotic cells were tracked using time-lapse microscopy. Each dot represents an individual cell (n = 19-49 for each condition). Red lines represent the median. siCNTRL, control siRNA; sicGAS, cGAS siRNA.
(B) HeLa CCL-2 cells have a longer mitotic lifespan that is further extended by cGAS depletion. Duration of mitosis until mitotic cell death was monitored by time-lapse microscopy. Each dot represents an individual cell (n = 40 for each condition). Red lines represent the median. siCNTRL, control siRNA.
(C–E) Time-lapse microscopy-based analysis of mitotic cell death in nocodazole-arrested cells. Cells of the indicated cell lines were released from G2 arrest into media containing the indicated drugs, and duration of mitosis until mitotic cell death was monitored. All red lines indicate the median. Each dot represents an individual cell (n = 35-60 for each condition). siCNTRL, control siRNA; sicGAS, cGAS siRNA.
(F and G) Time-lapse microscopy-based analysis of mitotic cell death in HeLa cells arrested with proTAME in the absence of taxol. Cells of the indicated cell lines were released from G2 arrest into media containing 10 μM proTAME, and duration of mitosis until mitotic cell death was monitored. (F) Timing of slippage and mitotic cell death. All red lines indicate the median. Each dot represents an individual cell (n = 60 for each condition). Note that for unknown reasons, proTAME arrests a large fraction of cells for only limited periods of time. (G) Fraction of the cells from (F) dying during mitotic arrest. siCNTRL, control siRNA; sicGAS, cGAS siRNA.
(H and I) Time-lapse microscopy-based analysis of mitotic cell death in BJ hTERT fibroblasts arrested with proTAME in the absence of taxol. Cells of the indicated cell lines were released from G2 arrest into media containing 10 μM proTAME, and duration of mitosis until mitotic cell death was monitored. (H) Timing of slippage and mitotic cell death. All red lines indicate the median. Each dot represents an individual cell (n = 60 for each condition). Note that for unknown reasons, proTAME arrests a large fraction of cells for only limited periods of time. (I) Fraction of the cells from (H) dying during mitotic arrest. siCNTRL, control siRNA; sicGAS, cGAS siRNA.
(J) cGAS levels in the experiment shown in Fig. 4I, as determined by Western blot analysis. siCNTRL, control siRNA. Percentages indicate dilution series of samples prepared from control siRNA treated cells. cGAScat, catalytic mutant. cGASDNA, DNA binding-induced catalytic activity mutant.
(K) Control siRNA (siCNTRL) treatment does not affect mitotic cell death. Cells were released from G2 arrest into M phase with the indicated drugs, and duration of mitosis until mitotic cell death was monitored. All red lines indicate the median. Each dot represents an individual cell (n = 60 for each condition).
(L) cGAS levels in the experiment shown in Fig. 4J, as determined by Western blot. The vertical line indicates removed irrelevant lanes.
(M and N) cGAS does not affect survival after UV irradiation. Hela cells (M) or BJ h-TERT fibroblasts (N) were irradiated with the indicated doses of UV, and cell viability was determined at the indicated time points. Mean values and range (error bars) from two experiments are shown. Note that for many of the data points, error bars are too small to be seen.
siCNTRL, control siRNA; sicGAS, cGAS siRNA.
Figure S5. cGAS induces MOMP during mitotic arrest. Related to Figure 5.
(A) Schematic overview of the pathways leading to apoptotic cell death. Key to apoptosis is the activation of the effector caspases, caspase-3 and caspase-7. Caspase-3/7 activation is achieved by release of cytochrome C (CytC) and SMAC from mitochondria during mitochondrial outer membrane permeabilization (MOMP). MOMP itself is mediated by Bax activation through degradation of the Bax inhibitors Bcl-xL and Mcl1.
(B and C) Mitotic cell death is associated with caspase-3/7 activity. (B) Fraction of cells positive for IncuCyte caspase-3/7 assay signal during mitotic cell death. Note that timing of cell death is delayed by sicGAS treatment, though cells eventually still die with active caspase 3/7 when slippage is inhibited by proTAME. (C) Representative images of cells from (B) at times of mitotic cell death. Scale bar, 10 μm.
(D) The effect of the caspase inhibitor Z-VAD-FMK (ZVAD, 80 μM) was compared with sicGAS. Cells of the indicated cell lines were released from G2 arrest into media containing the indicated drugs. Timing of mitotic cell death of individual HeLa cells (grey dots, n = 50 each) was monitored. Red lines represent the median.
(E) Necroptosis does not contribute to cGAS-induced mitotic cell death. Cells were released from G2 arrest into media containing the indicated drugs. The fraction of cells dying during incubation with the indicated drugs is shown. The caspase inhibitor ZVAD was used at 80 μM; the necroptosis inhibitor Nec-1s was used at 10 μM.
(F–H) Analysis of release of cytochrome C (CytC) from mitochondria in the indicated cell lines released from G2 into 500 nM taxol 10 μM proTAME. Cells were fixed at the indicated time points and stained for cytochrome C. (F and G), Quantifications of HeLa cells (F) and BJ hTERT cells (G). (H), Representative images of HeLa cells at 14 h. Arrowheads indicate cells showing delocalized cytochrome C staining.
(I–K) Analysis of release of cytochrome C (CytC) and SMAC from mitochondria in the indicated cell lines released from G2 into 500 nM taxol in the absence of proTAME. For comparison, images of untreated cells harvested from asynchronous cultures by mitotic shake off are shown. (I) and (J), representative images from cells that were fixed at the indicated time points and stained for cytochrome C or SMAC, respectively. Arrowheads indicate cells showing delocalized cytochrome C or SMAC staining. (K), Quantifications.
(L–N) Analysis of Bcl-xL and Mcl1 degradation in the indicated cell lines. Cells were released from G2 arrest into 500 nM taxol 10 μM proTAME. (L and M) sample gels. Vertical lines indicate removed irrelevant lanes. (N) Quantifications. Error bars, SEM (n = 2 for Mcl1 and n = 4 for Bcl-xL).
(O) Bcl-xL, not Mcl1, is a major determinant of mitotic lifespan. Cells were released from G2 arrest into media containing the indicated drugs. Individual cells were tracked by time-lapse microscopy (n = ~50 cells for each condition). Mcl1 inh., 3 μM S63845. Bcl-xL inh., 1 μM ABT-263. Note that simultaneous inhibition of Bcl-xL and Mcl1 results in immediate cell death before cells can enter mitosis (indicated as N.A., not applicable).
(P) Bcl2 inhibition does not affect mitotic cell death. Cells were released from G2 arrest into media containing the indicated drugs. Individual cells were tracked by time-lapse microscopy (n = 60 cells for each condition). ABT-199 used at 100 nM; ABT-263 used at 1 μM.
siCNTRL, control siRNA.
Figure S6. Analysis of cGAS, STING, TBK1 and IRF3 functions in mitotic cell death. Related to Figure 6.
(A) Mitotic lifespan of BJ hTERT cells grown in the presence of 100 nM pyridone 6, and released from G2 arrest into 500 nM taxol, 10 μM proTAME. Individual cells (gray circles) were tracked by time-lapse microscopy (n = 60 for each condition).
(B–E) cGAS expression and cGAMP have only minor effect on ARPE-19 hTERT cells. (B), Western blot analysis of IRF3 phosphorylation following treatment of asynchronous cells with 33 μM cGAMP. (C), ARPE-19 hTERT cells were released from G2 arrest in the presence of taxol and proTAME. After three hours, cGAMP was added to 33 μM for the indicated cells. Untr., untreated. Mean and SEM from three experiments (n = 30 each) are shown. (D and E), Analysis of the effect of exogenous cGAS expression on viability of ARPE-19 hTERT cells in 500 nM taxol 10 μM proTAME. (D), Fraction of mitotic cell death. Individual cells (n = 30-60 each) were tracked using time-lapse microscopy. (E), Western blot analysis of protein expression. The same nomenclature for cGAS mutants is used as in Fig. 4I.
(F and G) cDNA prepared from the indicated cells was used to determine CXCL10 (F) or IRF1 (G) expression by quantitative PCR. +DNA, asynchronous cells transfected with naked DNA. Cells were released from G2 arrest into media containing the indicated drugs. Mean and range from two experiments are shown.
(H and I), STING and IRF3 promote cell death in mitosis. Analysis by Western blot (H) and time-lapse microscopy (I). Two clones for each guide RNA were analyzed. For (F), cells were released from G2 arrest into media containing the indicated drugs, and the time until mitotic cell death was monitored. All red lines indicate the median. Each dot represents an individual cell (n = 60 for each condition).
(J) Western blot analysis of STING expression in the experiment from Fig. 6I. Please note that background expression in the absence of doxycycline was too high and this condition was therefore not analyzed by microscopy.
(K and L), Western blot analysis of IRF3 levels in the experiments shown in Fig. 6K and 6L, respectively. The vertical line indicates removed irrelevant lanes.
(M and N) Analysis of rescue of IRF3 disruption with the K193R K313R K315R mutant that is defective in a form of transcription-independent cell death induction by IRF3. (M), Western blot analysis of IRF levels. (N) Time-lapse microscopy-based analysis of mitotic cell death for the indicated cell lines and treatments. Cells were released from G2 arrest into media containing the indicated drugs, and duration of mitosis until mitotic cell death was monitored. All red lines indicate the median. Each dot represents an individual cell (n = 35-60 for each condition).
(O) Western blot of IRF3 levels for the experiment shown in Fig. 6M.
(P and Q) Analysis of mitotic lifespan in CRISPR-mediated TBK1 disruptions generated with two different guide RNAs. Note that the cell line used to generate these disruptions is the same as is used in Fig. 2A, and also expresses GFP-IRF3. (P), Time-lapse microscopy-based analysis of mitotic cell death for the indicated cell lines. Cells were released from G2 arrest into media containing the indicated drugs, and duration of mitosis until mitotic cell death was monitored. All red lines indicate the median. Each dot represents an individual cell (n = 60 for each condition). (Q), Western blot analysis of TBK1 levels.
Figure S7. cGAS pathway roles in taxol response of mouse xenograft tumors. Related to Figure 7.
(A) Overview of the xenograft experiment from Figure 7A-7F and Figure S7A-S7E. NSG mice were subcutaneously injected bilaterally with either wild type or cGAS-disrupted (sg_cGAS) HeLa cells. After two weeks, mice were either treated with weekly taxol doses (10 mg/kg), or control treated with DMSO. Two days after the second taxol/DMSO administration, tumors were prepared for histological analysis. Two days after the third taxol/DMSO administration, tumors were prepared for Western blot analysis.
(B) Presence of cGAS on mitotic chromosomes and within the interphase cytoplasm in the indicated xenografts tumors. Arrowheads indicate cells undergoing mitosis (magnified views of the mitotic cells on the right). Scale bar, 10 μm.
(C) Representative tumors from the indicated genotypes and treatments. Scale bar, 1 cm.
(D) IgG isotype control staining of a slice of the cGAS wild type xenograft tumor. Scale bar, 100 μm.
(E) Immunocompromised (NSG) mice were injected with either wild type or IRF3-disrupted (sg_IRF3) HeLa cells, or with sg_IRF3 cells containing doxycycline-inducible rescue constructs expressing either GFP-IRF3 or GFP-IRF3_ΔDBD. After two weeks, mice were subjected to weekly treatments with taxol or DMSO as a control. After three weeks, tumors were recovered and analyzed (see also Figure S7A). Mice were fed either with or without doxycycline-containing food. Growth curves for the indicated tumors and treatments are shown (mean and SEM; n = 8 for wt + DMSO; n = 8 for wt + Taxol; n = 7 for sg_IRF3 + DMSO; n = 7 for sg_IRF3 + taxol; n = 8 for sg_IRF3 + IRF3 +DMSO −doxycycline; n = 8 for sg_IRF3 + IRF3 +taxol −doxycycline; n = 8 for sg_IRF3 + IRF3 +DMSO +doxycycline; n = 8 for sg_IRF3 + IRF3 +taxol +doxycycline; n = 8 for sg_IRF3 + IRF3_ΔDBD +DMSO −doxycycline; n = 8 for sg_IRF3 + IRF3_ΔDBD +taxol −doxycycline;) n = 7 for sg_IRF3 + IRF3_ΔDBD +DMSO +doxycycline; n = 8 for sg_IRF3 + TRF3_ΔDBD +taxol +doxycycline). ⁋: Experiment termination at the planned schedule. ‡: All but one mouse in this experimental group were found dead at day 25.
Movie S1: Time-lapse imaging of cGAS knockdown cells rescued with GFP or GFP-cGAS during mitotic arrest. Related to Figures 1 and 4.
cGAS-depleted HeLa cells expressing GFP (left) or siRNA resistant GFP-cGAS (right), and H2B-RFP were released from G2 into 500 nM taxol 10 μM proTAME. Images were taken every 15 min. Top: Overlays of H2B-RFP and GFP (left) or GFP-cGAS (right). Bottom: DIC channel images corresponding to the fluorescence images on top.
Table S1: Analysis of non-small cell lung cancer patient data from The Cancer Genome Atlas (TCGA). Related to Figure 7.
The table is broken into three tabs.
(Overview) Overview of the patients whose data was processed for Figure 7G. The table on the left divides patients by whether or not they received taxane-based treatment, the table on the right divides patients by whether or not they received DNA-damaging agents. LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma. Stage, number of patients for each cancer stage.
(Raw) Detailed data for each patient that was used for Figure 7G. Column “vitalDay” contains the day of death or day of last follow-up. CGAS column, z score for CGAS expression.
(Risk) Number of patients at risk for each cohort at each year.
Table S2: Analysis of ovarian cancer patient data from The Cancer Genome Atlas (TCGA). Related to Figure 7.
The table is broken into three tabs.
(Overview) Overview of the patients whose data was processed for Figure 7H. The table on the left divides patients by whether or not they received taxane-based treatment, the table on the right divides patients by whether or not they received DNA-damaging agents. Stage, number of patients for each cancer stage.
(Raw) Detailed data for each patient that was used for Figure 7H. Column “vitalDay” contains the day of death or day of last follow-up. CGAS column, z score for CGAS expression.
(Risk) Number of patients at risk for each cohort at each year.
Table S3: Oligonucleotides used in this study.