

ABSTRACT
To investigate the dynamics of circRNA expression in pig testes, we designed specific strategies to individually study circRNA production from intron lariats and circRNAs originating from back-splicing of two exons. By applying these methods on seven Total-RNA-seq datasets sampled during the testicular puberty, we detected 126 introns in 114 genes able to produce circRNAs and 5,236 exonic circRNAs produced by 2,516 genes. Comparing our RNA-seq datasets to datasets from the literature (embryonic cortex and postnatal muscle stages) revealed highly abundant intronic and exonic circRNAs in one sample each in pubertal testis and embryonic cortex, respectively. This abundance was due to higher production of circRNA by the same genes in comparison to other testis samples, rather than to the recruitment of new genes. No global relationship between circRNA and mRNA production was found. We propose ExoCirc-9244 (SMARCA5) as a marker of a particular stage in testis, which is characterized by a very low plasma estradiol level and a high abundance of circRNA in testis. We hypothesize that the abundance of testicular circRNA is associated with an abrupt switch of the cellular process to overcome a particular challenge that may have arisen in the early stages of steroid production. We also hypothesize that, in certain circumstances, isoforms and circular transcripts from different genes share functions and that a global regulation of circRNA production is established. Our data indicate that this massive production of circRNAs is much more related to the structure of the genes generating circRNAs than to their function.
Abbreviations: PE: Paired Ends; CR: chimeric Read; SR: Split Read; circRNA: circular RNA; NC: non conventional; ExoCirc-RNA: exonic circular RNA; IntroLCirc-: name of a porcine intronic lariat circRNA; ExoCirc-: name of a porcine exonic circRNA; IntronCircle-: name of a porcine intron circle; sisRNA: stable intronic sequence RNA; P: porcine breed Pietrain; LW: porcine breed Large White; RT: reverse transcription/reverse transcriptase; Total-RNA-seq: RNA-seq obtained from total RNA after ribosomal depletion; mRNA-seq: RNA-seq of poly(A) transcripts; TPM: transcripts per million; CR-PM: chimeric reads per million; RBP: RNA binding protein; miRNA: micro RNA; E2: estradiol; DHT: dihydrotestesterone
KEYWORDS: Circular RNAs, intronic circRNA, pubertal testis, regulation circRNA production, lariat, sisRNA, abundance in circRNA, steroids production, multi-exon circRNA, SMARCA5
Introduction
The discovery of RNA molecules with a circular configuration dates back four decades [1]. In the past few years, advances in next generation sequencing has enabled genome-wide detection and quantification of the expression of circular RNAs (circRNAs). This is a novel type of predominantly noncoding RNA that forms a covalently closed continuous loop and is highly represented in the eukaryotic transcriptome [2]. CircRNAs are very stable but not polyadenylated [2]. Contrary to what happens during canonical splicing, a circRNA with covalently linked ends is generated when a pre-mRNA undergoes ‘back-splicing’ to join a splice donor to an upstream splice acceptor (the end of an exon is joined to the beginning of upstream exon) [2]. Most circRNAs include only exonic sequences and are called exonic circRNA, but circRNAs containing only intronic sequences also exist [3,4].
When Zhang et al. (2013) studied RNA-seq data obtained after depletion of both ribosomal sequences and polyAdenylated transcripts with a focus on intronic transcripts, they observed that many were circular [5]. These intronic circRNAs, derived from lariat (intronic sequence spliced from pre-mRNA), are included in the vast family of stable intronic sequences RNAs (sisRNAs) [6,7]. The circular junction is the 2ʹ-5ʹ covalent bond generated after the branching step during splicing [8,9], and sequences correspond to a lariat without a tail [5,8–10]. Another type of circRNA containing entire intronic sequences (named intron circles) and with a classical 3ʹ-5ʹ covalent bond has recently been characterized [8,9].
With regard to their potential functions, several mechanisms of circRNA action have been identified, but mostly only for a few specific elements [11]. Consequently conflicts and controversies in these different views have now emerged [12]. The major functions commonly attributed to (exonic) circRNAs are miRNA sponge activity [13], competitive RBP-binding/sequestration, and micropeptide/microprotein expression [14,15]. CircRNAs may also affect splicing (direct regulation and/or competitive mRNA processing) [2,11,14]. Even though the impact of circRNAs containing intronic sequences on the transcription of their host gene (nuclear function) has been reported in numerous reviews [2,11,15,16], it is probably limited to a few intronic circRNAs [5,17], potentially because many intronic circRNAs have a function in the cytoplasm where they migrate, as recently demonstrated [8]. Two studies conducted using the same datasets demonstrated the remarkable richness in circRNA in the brain compared to other tissues but without agreeing on their number [18,19]. In addition, a multitude of circRNAs have been discovered in different stages of embryonic development [20–23]. In Xenopus tropicalis, intronic circRNAs (mainly circular sisRNA) persisted after egg fertilization and during early embryogenesis [8,10,22]. CircRNAs were originally found to increase in brain during aging [16,20,24–27] but not in heart [26] or muscle [28]. It is thus clear that these important molecules can be highly expressed and dynamically modulated even though the reason is not fully understood [2,24,25,29].
In the perspective to find alternatives to surgical castration of male piglets because of animal welfare concerns, it is important to understand the links between meat odor (boar taint) and sexual maturation. Raising non-castrated male pigs and slaughtering them before meat odor is expressed in the course of sexual maturity is an alternative currently under consideration. However, it should be noted that European pig breeds generally reach full sexual maturity at about eight months and are in puberty when they are usually slaughtered at about six months. Due to the probable variation in sexual maturity during puberty, it is crucial to study the testis during this period to understand variations in steroid-genesis and meat odor in male slaughter pigs [30]. The examination of seven transcriptome datasets of testicular RNA depleted in ribosomal sequences (Total-RNA-seq) to study the non-coding transcripts, we identified some intronic circRNAs. Knowing the possible variation in circRNA patterns during development and their possible abundance in this particular tissue [19,31,32], we decided to perform a genome-wide analysis of circRNAs in the testes of pigs during puberty. A number of bioinformatic approaches have been proposed to detect circRNAs in RNA-seq data [33] but to the best of our knowledge, none of them are suitable for the identification of intronic circRNAs. In contrast to several intronic circRNAs reported in different studies without specific limitation to the intronic fragments involved [19,31,32], we wanted to focus specifically on intronic circRNAs that may originate from the lariat mechanism described above (circular sisRNAs) or from the circularization of the full-length intron (intron circles). After defining our strategy, we chose to implement it on a Galaxy platform [34]. We first developed a tool focused on the detection of lariat-derived intronic circRNAs and circular sisRNAs, followed by a second tool dedicated to the identification of exonic circRNAs. Our study was organized as follows: (1) In the first analysis, we applied both our tools to Total-RNA-seq datasets from the testes of seven animals sampled during the pubertal period, and examined each identified circRNA independently. We characterized testicular circRNAs of intronic (circular sisRNA and intron circles) or exonic (exonic circRNAs) origin. (2) In the second analysis, we focused on the global production of circRNAs by tissue and on genes able to produce circRNA in order to gain insight into the regulation of circRNA production. We then extended the study to publicly available RNA-seq datasets to perform quantitative and comparative analyses. From these two analyses, we conclude that collective functions across different circRNAs may exist, as opposed to – or in addition to – functions of individual circRNAs.
Results
Strategies to characterize circRNAs
In designing our strategy, we were inspired by the DCC circRNA prediction tool [35] which is focuses on the identification of exonic circRNAs. Our approach consists in applying a series of filters to the output of the STAR read mapper [36]. As the mapping of the two segments of a chimeric read (CR) on the genome allows for the direct identification of the genomic boundaries of the circularized segment, the main objective is to select chimeric reads, where both segments map to the same strand, in inverted order (Figure 1(a)) and in the intronic/exonic region (Figures 1(b & c). We further only retain circRNAs supported by at least five CRs in at least one sample (animal). All the steps of this strategy are described in Suppl. Doc. S1.
Figure 1.
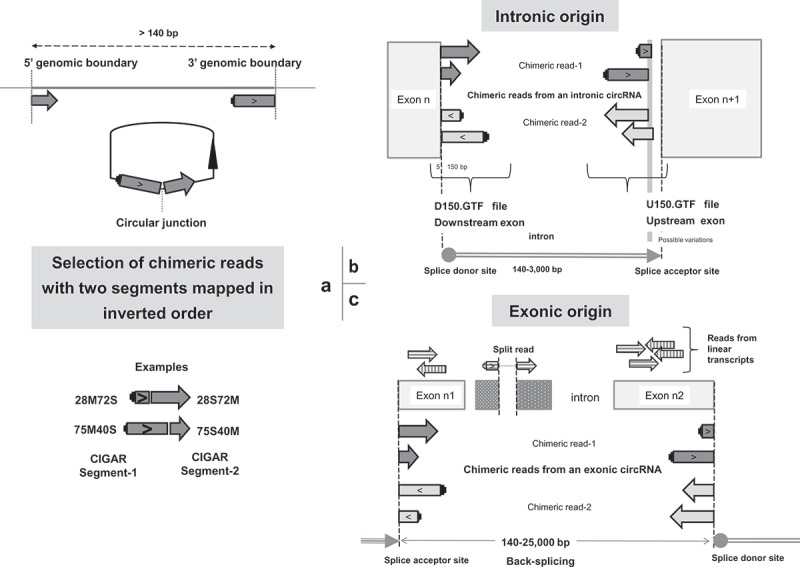
Strategy for the identification of circular RNAs. (a) Selection of chimeric reads (CRs): identifying circular junctions. Mapping of the two segments of a CR allows the direct identification of the circRNA junctions. As segment-1 maps downstream from segment-2, we qualify this read as mapping in ‘inverted order’. This selection is based on the CIGAR string of both fragments (see examples at the bottom of the figure). (b) Characterization of intronic circular RNAs. Each segment was mapped relative to the annotation using two GTF files describing respectively regions starting 5 bp upstream and ending 150 bp downstream from the end of an exon (D150.GTF) and regions starting 150 bp upstream and ending 5 bp downstream from the start of the following exon (U150.GTF). (c) Characterization of exonic circular RNA.
Characterization of circRNAs in porcine testes
For our first analysis, seven Total-RNA-seq datasets originating from seven animals sampled when steroid production had just started (Suppl. Doc. T2-A, see Materials and Methods) were obtained. Each dataset consisted of between 70 million (Animal-65) and 88 million (Animal-05) stranded read pairs (Table 1). The dataset of Animal-31 differed from the others by a very low proportion of reads mapping to regions of protein coding transcripts (Table 1).
Table 1.
Intronic and exonic circular RNA detected on the 7 testicular datasets.
| Selected CR as coming from intronic circRNA | Validated Intronic CircRNAs | Name of animal | Dataset from testis | N. of reads available | N. of reads uniq. mapped on coding-protein transcripts | Validated exonic circRNAs | Selected CR as coming from exonic circRNAs |
|---|---|---|---|---|---|---|---|
| 1.566 | 16 + 2 NC (1) | Animal-05 | Total-RNA-seq | 178.4 106 (2) | 109.7 106 | 1.663 | 40.281 |
| 6.159 | 118 + 3 NC (1) | Animal-31 | Total-RNA-seq | 146.1 106 (2) | 39.7 106 | 4.862 | 115.235 |
| 1.096 | 21 | Animal-54 | Total-RNA-seq | 173.2 106 (2) | 112.6 106 | 1.18 | 30,280 |
| 966 | 20 + 1 NC (1) | Animal-73 | Total-RNA-seq | 156.2 106 (2) | 94.5 106 | 1.075 | 28.166 |
| 898 | 19 | Animal-16 | Total-RNA-seq | 152.2 106 (2) | 101.0 106 | 1.01 | 26.663 |
| 645 | 16 | Animal-65 | Total-RNA-seq | 140.5 106 (2) | 82.9 106 | 671 | 18.251 |
| 1.185 | 15 | Animal-93 | Total-RNA-seq | 151.5 106 (2) | 100.5 106 | 1.084 | 27.121 |
| <5 | Animal-05 | mRNA-seq | 23.86 106 (3) | 19.14 106 | 25 | ||
| <5 | Animal-31 | mRNA-seq | 25.77 106 (3) | 20.73 106 | 36 |
(1) NC: non-conventional intronic circRNA or intron circle
(2) reads of 100 & 125 bp
(3) Reads of 150 bp
Intronic circRNAs
In order to identify circRNA originating from intronic sequences, the pipeline described in Suppl. Doc. S1 (Fig. S1-A, left part) was applied and allowed us to detect between 645 (Animal-65) and 6,159 (Animal-31) CRs originating from intronic circRNAs (Table 1). After automatic grouping based on their genomic coordinates, we often observed several sub-groups of CRs, which seem to point to a single intronic origin. The example described in Suppl. Doc. F3-A showed a variation in the CR of a particular circRNA with respect to the 5ʹ boundary of the circular junction due to inclusion of different parts of the intron 3ʹ end (see also Figure 1(b)). We added a manual step to check and group the CRs (Suppl. Doc. S1), and after the examination of the seven datasets, 153 regions able to produce intronic circRNA were retained.
For each intronic circRNA bearing region we examined the consistency of all available data and information. With a single exception, all circRNAs originally indicated by the intronic circRNA pipeline were indeed intronic. As expected, the 5ʹ boundary of the map of one segment of a particular CR corresponded to the 5ʹend of the intron. As a circRNA is a very stable transcript, we expected substantial full-length coverage across the entire intronic sequence. To exclude false positives, we visually inspected intronic regions where very few CR had been detected. After examining whole intronic regions supported by less than eight CRs across all seven datasets on the IGV (Integrative Genome Viewer), we excluded 27 intronic sequences with very few reads mapped inside the intron, although the respective circular junction met the criterion of at least five spanning CRs (Suppl. Doc. S4-A). Three regions appeared to produce full-length circRNAs (Suppl. Doc. S4-B, S4-C & S4-D). In the 123 others, the 3ʹ boundary of the second segment map was located approximately 5 bases away from the 3ʹ end of the intron (Suppl. Doc. T2-B, example Suppl. Doc. S4-E). These features are compatible with an intronic lariat origin.
We retained 123 regions able to produce intronic lariat circRNAs (circular sisRNAs) and three non-conventional (NC) circRNAs including full length intronic sequences (Suppl. Doc. T2-B). Among these 123 intronic lariat circRNAs, 118 were found in Animal-31 whereas only 15 to 21 were detected in the six other animals (Table 1). Only six circRNAs were identified in all the animals and 86 circRNAs were present in only one animal (81 in Animal-31). For the intronic lariat circRNA named IntroLCirc-70 and originating from PEX10, the highest number of CRs was obtained across all seven datasets.
Exonic circRNAs
By applying an approach similar to the one developed for intronic circRNAs, we identified 5,241 exonic circRNAs (ExoCirc-RNA) in our dataset (Suppl. Doc. S1, Fig. S1-A, right part). During the process, we chose to only retain the coordinates of each ExoCirc-RNA. Before validating an ExoCirc-RNA and by only using the coordinates, we identified genes and transcripts involved in the back-splicing events that generated them. When an ExoCirc-RNA was associated with two different genes (very rare situations), we chose between them by examining human orthologous genes. We considered that an ExoCirc-RNA could only originate from a back-splicing event between two exons from the same gene, but tolerated ExoCirc-RNAs that originated from two different transcripts (95 ExoCirc-RNAs (2%) were found to originate from two different transcripts of the same gene). Finally, we preferred to exclude five ExoCirc-RNAs for which annotation problems were suspected (data not shown). We validated 5,236 ExoCirc-RNAs originating from 2,516 genes (2,624 transcripts) (Suppl. Table 1). We identified 8,469 single exons involved in the circular junctions of multi-exon circRNAs, termed ‘extreme exons’ in the sequel (Figure 2(a)). Among the 5,236 ExoCirc-RNAs, we noted the presence of 213 single-exon circRNAs (Figure 2(b)). Exons capable of constituting these single-exon circRNAs are significantly longer than those that can only be included in multi-exon circRNAs (646.8 ± 543.5 bp and 146.8 ± 156.5 bp; Wilcoxon test: p-value < 0.0001; Figure 2(c)).
Figure 2.
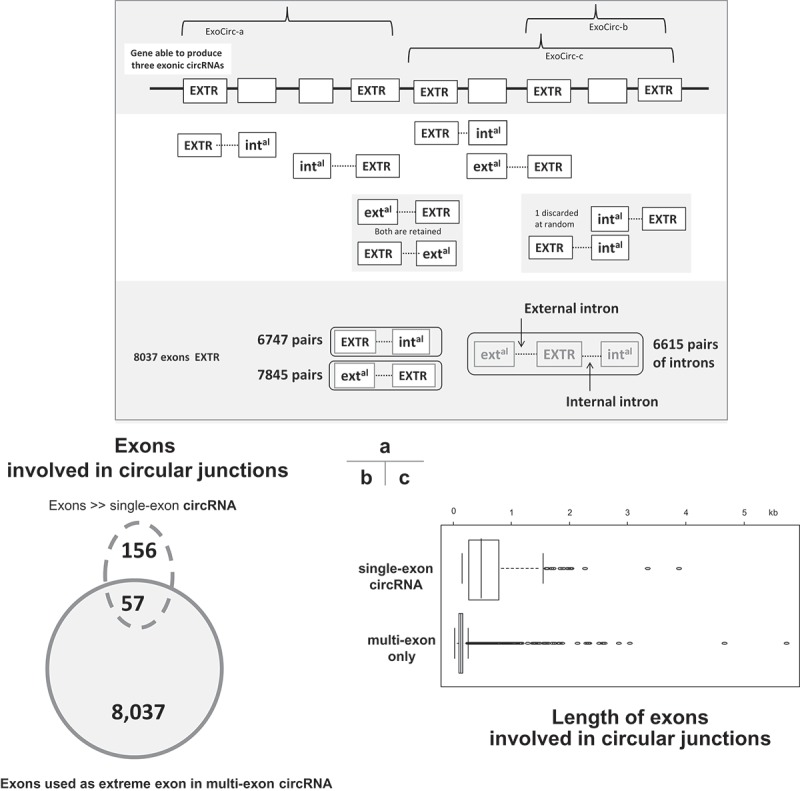
Illustrations of some features of circRNAs characterized in porcine pubertal testis. (a) Schematic representation of the identification of proximal exons and introns. At the top of the figure, we show a gene made of nine exons (boxes) able to produce three exonic circRNAs. Each exon involved in the back-splicing (named extreme, [EXTR]) at the origin of these ExoCirc-RNAs was identified. For each extreme exon, the two proximal exons (internal [intal] and external [extal]) and the two proximal introns were identified. The number of cases we found for each configuration is given at the bottom of the figure. (b) Exons involved in circular junctions. (c) Comparison of the length of exons involved in circular junctions.
It is interesting to note that 4,862 different exonic circRNA were identified in Animal-31, whereas only 671 exonic circRNAs were identified in Animal-65 (Table 1). ExoCirc-7094 (from CCT3) was the exonic circRNA with the highest number of specific CRs (1,739) across all seven datasets (Suppl. Doc. T2-C). ExoCirc-9244 (SMARCA5) and ExoCirc-4239 (WDR27) showed the highest animal-specificity with respect to the number of CRs (see Animal-31, Suppl. Doc. T2-C). In Total-RNA-seq datasets from Animal-31 and Animal-05, respectively, 2902 and 367 CR were identified per million reads considered (CR-PM, Suppl. Doc. T2-D). As reads 100 bp and 125 bp in length were mixed, we can only suggest a particularly high abundance of circRNAs in the dataset from Animal-31 compared to the six other datasets (Suppl. Doc. T2-D).
Evaluation of background noise
In order to evaluate the background noise of our experiments, we applied our two pipelines to polyA-selected mRNA-seq datasets (RNA-seq of poly(A) transcripts) from the testis of Animal-31 and 05. A very low number of CR were identified as originating from exonic circRNA and none from intronic circRNA (Table 1). In the mRNA-seq datasets, 1.20 and 1.88 CR-PM were identified, in Animal-31 and −05, respectively (Suppl. Doc. T2-D). This indicates that the background noise was about 0.1% (1.20 and 1.88 vs/2,902 and 367 CR-PM). The application of both pipelines on mRNA-seq datasets confirmed that the identified CRs were not the result of a linear concatenation of the transcripts.
Biological validation of the circular character
In order to validate a selected number of circRNAs by RT-qPCR analysis, we designed divergent primers for circRNA detection (Figure 3(a)): PCR amplicons span the circular junction of circRNAs and cannot originate from linear transcripts (see list in Suppl. Doc. T2-E). Nevertheless, this amplicon could originate from a linear transcript resulting from trans-splicing (Figure 3(a)). Resistance to RNase R was used to rule out this possibility and to prove that RNA is indeed circular since this enzyme digests all linear RNAs but not circRNAs [37].
Figure 3.
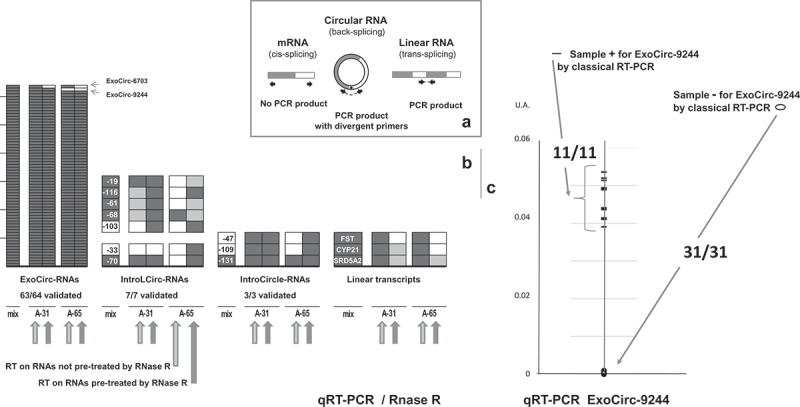
Biological experiments. (a) Schematic representation of divergent primer design and its interests. This primer pair enables amplification not only of circRNA but also of linear RNA when the latter originates from trans-splicing. Resistance to RNase R was used to rule out this possibility. (b) Results of RT-PCR. Each small box represents a PCR test and they were stacked. Five tests were performed for each type of transcript tested, on a mix of cDNAs and without/with pre-treatment of RNAs by RNAse R before the reverse transcription of RNAs from animals A-31 and A-65. When an amplicon of the expected size was observed, the box is colored black. Each negative result (white boxes) observed on cDNAs obtained after RNase R pre-treatment was confirmed by a second test (data not shown). When only a reduction in amplification was observed, the box is colored grey. In the second experiment (framed in dotted lines) concerning the RNase R resistance test of IntroLcirc-19, −116, −61,-68, −103, RT-PCRs were performed in parallel with two Taq polymerases. When positive amplification was observed with only one, the box is colored grey. (c) Evaluation of the level of ExoCirc-9244 by qRT-PCR. All 42 samples considered were previously tested for ExoCirc-9244 by RT-PCR.
PCR amplification with these divergent primer pairs was compared using cDNAs from Animal-31 and A-65 (named respectively RT_A-31 and RT_A-65) obtained after reverse transcription (RT) on RNAs treated with RNase R or untreated (Figure 3(b)). Three linear transcripts (FST, CYP21, SRD5A2) known to be expressed in testis (Suppl. Doc. T2-E) [30] were analyzed in parallel to control the digestion of linear transcripts by RNase R. As expected, we observed no amplification of FST when the RT was performed on RNA treated by RNAse R, we can thus conclude that RNase R digestion of FST transcripts was complete in RT-samples from both animals. For CYP21, the extinction of amplification due to digestion was observed only in RT_A-65, and for SRD5A2, we observed only a decrease in the intensity of the amplification, reflecting only partial digestion of these linear transcripts (Figure 3(b)). Thus, our data demonstrate that the digestion of linear RNAs by RNase R varies depending on the linear transcripts considered.
All 64 tested exonic circRNAs amplified in a single distinct product of the expected size. Among the 64 exonic circRNA tested for resistance to RNase R, only ExoCirc-6703 did not resist (Figure 3(b)). No attenuation of the amplification was observed, and on the contrary in several cases, an accentuation was observed when the RT-PCR was performed on RNA treated by RNase R. In conclusion, the pipeline developed to characterize exonic circRNAs might lead to the identification of 1.55% false positive elements, which is an excellent result compared to the proportion of false positives found using other tools for circRNA prediction [33].
In the first experiment to test circRNA resistance to RNase R, we also included three intronic lariat circRNAs and three NC intronic circRNA (Figure 3(b)). No result was observed with IntroLCirc-103 (data not shown in Figure 3(b)). We tested two other reverse transcriptase enzymes, but the best reverse transcription (linear and circRNA) was observed with our original choice. Except for one NC intronic circRNA and one intronic lariat circRNA, we observed odd results for the three others: only RNA pretreated by RNase R allowed the amplification of these circular elements in RT_A-65 (Figure 3(b)). For IntroLCirc-33, it was the unique positive result that we achieved to obtain. We suspect that the pretreatment of RNAs by RNase R, prior to cDNA synthesis by reverse transcription, led to a favorable ratio of primer to potential binding sites and subsequently to better reverse transcription and amplification. A second experiment relating to RNase R resistance confirmed the circular nature of five further intronic circRNAs (Figure 3(b)) and underlined the difficulty involved in obtaining an amplicon with intronic circRNA, because frequently they have only been obtained with one of the two polymerases tested and/or only after RNase R.
Identification of animals analogous to animal-31
To identify other animals with particular circRNA features analogous to Animal-31 (with an exceptionally high abundance of circRNAs), four exonic circRNAs were tested. We selected ExoCirc-9244, ExoCirc-4239, ExoCirc-1865 and ExoCirc-4237, because their CR patterns were previously found to differ significantly in Animal-31 and in the other animals (Suppl. Doc. T2-C). The four circRNAs were tested by PCR on RT-samples from 76 animals, including the seven animals in the first subset (analyzed by RNA-seq). Only one exonic circRNA harbored an interesting pattern: PCR products from ExoCirc-9244 were detected in RT-samples from 12 animals (Animal-31 + 11 additional animals), while 64 other individuals (including Animal-05, 16, −54, −65, 73 and −93) did not express ExoCirc-9244.
We noted a significant difference in the estradiol (E2) level between these two subsets of animals with or without ExoCirc-9244 testicular expression (means 14.91 and 28.26 pg/ml; Wilcoxon test: p-value = 0.0021) (Suppl. Doc. F3-B1). The characteristics of the additional 11 animals expressing ExoCirc-9244 in their testicular transcriptome are reported in Suppl. Doc. T2-A. A qRT-PCR confirmed our results concerning the presence/absence of ExoCirc-9244 in 42 animals chosen among the 76 animals originally tested using standard RT-PCR (Figure 3(c)). Samples expressing ExoCirc-9244 had low steroid synthesis potential (Suppl. Doc. F3-B2).
Total production of circrnas in testis, muscle and embryonic cortex tissues
In addition to the data analysis focusing on the expression of single specific circRNAs, we conducted a second analysis based on a comparative investigation of Total-RNA-seq datasets from three porcine tissues. Before performing quantitative analysis, we needed to make sure we were using datasets that are reliable and were obtained using analogous experimental pipelines to enable comparison of the datasets. Thus, we added publicly available RNA-seq datasets from stranded porcine Total RNAseq libraries from brain (five time-points during fetal porcine development of cortex [21]), from muscle (three postnatal time-points [31]) and from testis (240 days [31]) to our seven testis datasets. To ensure conditions were exactly the same, we limited our seven testis datasets to subsets corresponding to 2 × 100 bp paired-end reads (Table 2). In total, we considered 16 datasets, of which five (the three samples of muscle, testis 240 days and testis from Animal-31) were characterized by a high ratio of multi-mapped reads (Table 2, Suppl. Doc. F3-C) that can be attributed to ribosomal sequences. This result of the analysis could not be associated with a specific tissue, the circRNA content or the kit used for depletion of ribosomal sequences (designed for human sequences) (Suppl. Doc. F3-C).
Table 2.
Intronic and exonic circular RNA detected on all the 16 datasets, 7 from this study and 9 from the literature.
| Number of Genes producing |
Number of Genes producing |
|||||||
|---|---|---|---|---|---|---|---|---|
| N. of Reads available (100 bp) |
Reads multi-mapped | Samples | Reads considered(1) | CR-PM(2) Intronic | CR-PM(2) Exonic | Classification | Intronic cirRNAs | Exonic circRNAs |
| 26.69 106 | 2.74 106 | Embryonic Cortex 42 days | 16.73 106 | 3 | 556 | Intermediate | 36 | 2,406 |
| 21.41 106 | 4.28 106 | Embryonic Cortex 60 days | 9.68 106 | 8 | 1,037 | Abundant | 64 | 2,494 |
| 26.63 106 | 2.52 106 | Embryonic Cortex 80 days | 15.88 106 | 4 | 233 | Basic | 43 | 1,593 |
| 48.52 106 | 4.48 106 | Embryonic Cortex 100 days | 24.26 106 | 2.5 | 188 | Basic | 55 | 1,879 |
| 53.99 106 | 4.36 106 | Embryonic Cortex 115 days | 29.50 106 | 1.3 | 137 | Basic | 29 | 1,627 |
| 64.82 106 | 9.66 106 | Testis Animal-05 | 40.50 106 | 13 | 340 | Basic | 365 | 3,416 |
| 37.14 106 | 17.86 106 | Testis Animal-31 | 10.21 106 | 140 | 2,704 | Abundant | 530 | 4,338 |
| 34.60 106 | 4.40 106 | Testis Animal-54 | 22.46 106 | 8 | 220 | Basic | 108 | 2,105 |
| 52.68 106 | 6.42 106 | Testis Animal-73 | 34.85 106 | 9 | 260 | Basic | 154 | 2,729 |
| 34.28 106 | 3.46 106 | Testis Animal-16 | 23.01 106 | 11 | 249 | Basic | 147 | 2,254 |
| 39.32 106 | 3.60 106 | Testis Animal-65 | 26.45 106 | 7 | 245 | Basic | 106 | 2,417 |
| 30.92 106 | 4.26 106 | Testis Animal-93 | 19.51 106 | 8 | 267 | Basic | 104 | 2,131 |
| 83.71 106 | 25.68 106 | Testis 240 days | 41.34 106 | 8 | 298 | Basic | 109 | 1,854 |
| 77;22 106 | 32.08 106 | Muscle 0 day | 27.96 106 | 6 | 121 | 18 | 498 | |
| 75.86 106 | 27.28 106 | Muscle 30 days | 14.50 106 | 2.4 | 93 | 12 | 402 | |
| 73.54 106 | 29.32 106 | Muscle 240 days | 23.58 106 | 7 | 102 | 25 | 501 |
(1) To compare results, we chose to perform the normalization with the number of reads uniquely mapped by STAR and in a region delimited by a protein-coding transcript
(2) Number of CR from circRNAs per million of considered reads
In order to estimate the global production of circRNAs, which was the objective of the second analysis, we focused on the joint analysis of the number of CRs identified in intronic or exonic circRNAs after using both the tools developed in our study (Suppl. Doc. S1). With the Testis_A-31 dataset (Testis from Animal-31), we were able to quantify a total production of 2,704 and 140 CR-PM of exonic and intronic circRNAs, respectively, while with the other datasets, we noted 220 to 340 and 7 to 13 of exonic and intronic cirRNAs, respectively (Table 2). Due to the very distinct circRNA pattern in Testis_A-31 compared to the other testis datasets, we assigned this sample the status ‘abundant’ referring to the quantity of circRNA. In contrast, the other samples were considered as ‘basic’. In embryonic cortex, the profile at 60 days (E60) was very distinct compared to that found at 80, 100 and 115 days of embryonic life, while an intermediate situation was observed at 42 days (Table 2). Thus, we also assigned the status ‘abundant’ to the embryonic cortex sampled at E60, which produced 1,037 and 8 CR-PM from exonic and intronic circRNAs, respectively. Embryonic cortex sampled at 80, 100 and 115 days had the lowest ability to produce intronic circRNA (< 4 CR-PM, Table 2). Intronic circRNAs were very rare, especially in embryonic cortex sampled at 100 and 115 days. These results suggest that the ratio of intronic to exonic circRNA may be lower in embryonic cortex than in testis or muscle. No variation in the number of circRNAs was observed between the three muscle datasets.
Genes producing circRNAs
Genes producing circRNAs in testis
In pubertal testis, only 2,516 genes were able to produce 5,236 exonic circRNAs (Suppl. Table 1), and 1,147 genes (45%) were able to produce 2 to 17 exonic circRNAs. In contrast, 92,272 and 32,914 exonic circRNAs produced from 11,801 and 7,790 genes have been reported in CircBase and CircRNAdb, respectively in humans (http://www.circbase.org/; http://202.195.183.4:8000/circrnadb/circRNADb.php). Of the 2,516 genes producing exonic circRNAs in our study on pigs, 2,445 were annotated (Sscrofa11.1). Thereby, among the genes identified here as being able to produce exonic circular transcripts, more than 80% (2,068/2,445 in CircBase and 2,002/2,445 in CircRNAdb) have been previously reported as being able to produce exonic circRNAs in humans.
When we examined the list of genes able to produced circRNA (2,245/2,516 were known in humans) using g:Profiler (https://biit.cs.ut.ee/gprofiler/gost) enrichment of genes involved in cell cycle and organelle biogenesis was observed. We also observed enrichment of genes associated with testicular gametogenesis (data not shown). Nevertheless, given the number of genes examined, these deviations are probably only a reflection of the particularities of the pubertal testicular transcriptome. Among genes known to be involved in the testicular metabolism of steroids, only STAR and HSD17B4 were identified as being able to produce circRNAs in pubertal testis (Suppl. Table 1).
The 123 intronic circRNAs and the three NC intronic circRNAs identified in testicular datasets were produced from 114 genes (Figure 4(a)). Eight genes were able to produce two intronic circRNAs (CCHCR1, FANCA, FER1L5, PITRM1, PPIL2, RANGAP1, RNF25, and SPAG5), and DNAH17 was able to produce five intronic circRNAs (Suppl. Doc. T2-B). The 23 genes identified as being capable of producing both intronic and exonic circRNAs in testes accounted for a fifth of the 114 genes capable of producing intronic circRNAs. In testis, 2,607 individual genes were able to produce intronic and/or exonic circRNAs (Figure 4(a)).
Figure 4.
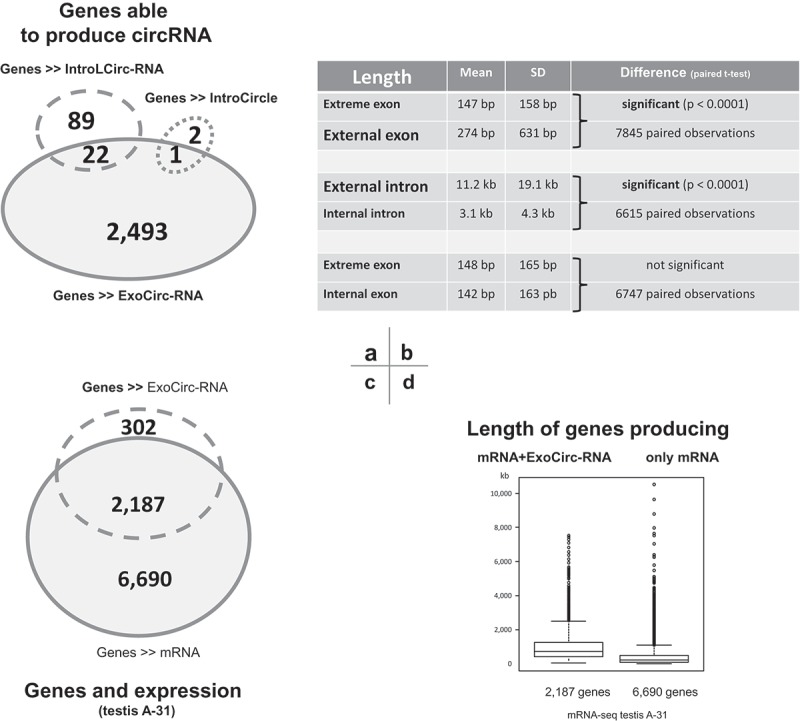
Illustrations of some features of genes able to produce circRNAs in pubertal testis. (a) Overlap of genes able to produce different types of circRNAs. (b) Comparison of the length of exons/introns from multi-exon circRNAs (c) Overlap of genes able to produce linear and/or circular transcripts in testis of animal-31. (d) Comparison of the lengths of genes able to produce linear and circular transcripts with genes that only produce linear transcripts.
We chose to only retain the 5,023 multi-exon circRNAs (Figure 2(b)) to study the proximal environment of each exon involved in a circular junction (= extreme exon). For each extreme exon, we identified the two proximal exons (internal and external) and the two proximal introns (Figure 2(a)). The comparison of 7,845 pairs [extreme exon – external exon] showed that extreme exons are significantly smaller than their external exon counterparts (Figure 4(b)). The comparison of 6,615 pairs [internal intron – external intron] showed that external introns are significantly longer than internal introns (Figure 4(b)).
By examining the mRNA-seq data available for Animal-31, we retained 8,877 genes with high expression of linear transcripts (RSEM>10). In this list, 84% of genes able to produce ExoCirc-RNAs were found (Figure 4(c)). These 2,187 genes were significantly longer than the 6,090 genes that only produced mRNAs (105.8 ± 97.3 kb and 44.1 ± 67.8 kb; Wilcoxon test: p-value < 0.001) (Figure 4(d)), which is consistent with [38].
Genes producing circRNAs in testis, muscle and embryonic cortex tissues
Next, we compared the list of genes able to produce circRNAs in the three tissues, testis, muscle and embryonic cortex. For each of the 16 RNA-seq datasets, the hosting (or parent) gene of each CR was identified. The results of several datasets from one tissue were merged if they were from the same tissue and embryonic or postnatal stage, and lists containing the name of hosting genes and the number of CR-PM were drawn up first (Suppl. Doc. F3-D). Even though only 49 genes were able to produce exonic circRNAs in all tissues (Suppl. Doc. F3-D1), it is interesting to note that the gene CSPP1 (Centrosome and spindle pole associated protein 1) was in top position on all three tissue-specific lists of genes with high circRNA production potential (1/89 muscle, 1/681 cortex, 3/678 testis, 2/4,338 Testis_A-31). This gene is functionally involved in cell division.
The Testis_240d dataset did not differ from the six pubertal datasets classified as ‘basic’ in terms of the number of CR-PM produced (Table 2), the total number of genes involved (Table 2), or the number of genes already observed in our first analysis (69% for Testis_240d while 67–71% for Testis_A-16, A-54, A-65, A-73, A-93). Thus, we assigned Testis_240d the status ‘basic’ based on the level of circRNA production. Next, we compared the production of circRNAs by specific genes in different developmental/ontogenetic stages in testis and in embryonic cortex. A positive correlation was found for testicular production of single exonic circRNAs between the ‘abundant’ and ‘basic’ status: exonic circRNAs most frequently in the ‘abundant’ status were at top of the list of expression levels in the ‘basic’ status.
When we considered 2,052 genes and only compared Animal-05 representing the ‘basic’ status and Animal-31 representing the ‘abundant’ status, the results were similar (Pearson r = 0.86 with p-value < 0.001, Figure 5(a)), and when we considered 678 genes and seven samples representing the ‘basic’ status and Animal −31, representing the ‘abundant’ status (Pearson r = 0.85 with p-value < 0.001, Figure 5(b)). Testicular production of exonic circRNA was at least six-fold higher in Animal-31 than in Animal-05. When genes producing exonic circRNAs were ranked in decreasing order of their production of exonic circRNAs for the ‘basic’ status, the genes ranked in basically the same order as for the ‘abundant’ status in testis (Figures 5(a & b) and in embryonic cortex (Figure 5(c)). In these analyses, the list of genes contained a number of false positives that we were able to narrow down when we used multiple datasets (Figure 5(b)). As we noted that these false positives are very rarely in the top on the testicular list of circRNAs, we decided to limit the comparison of genes identified as potentially responsible for producing circRNA in the embryonic cortex to the top 100 for analysis, as shown in Figure 5(c). Among the 2,494 genes capable of producing circRNAs in Cortex_E60, only 21% had not produced circRNAs in pubertal testis (Testis_A-31). When we examined the 4,338 genes producing circRNA in pubertal testis, we found that we had identified 45% as producers of exonic circRNA in the embryonic cortex (Cortex_E60) (Suppl. Doc. F3-D4). This suggests a higher proportion of tissue-specific exonic circRNA producing genes in testis than in embryonic cortex. This result is consistent with published results concerning circRNA in testis [19,31,32]. Nevertheless, this feature is probably only a reflection of the difference between the testicular transcriptome, which is a differentiated tissue considered here in a developmental period, and the transcriptome of the cortex which is here considered as an embryonic tissue.
Figure 5.
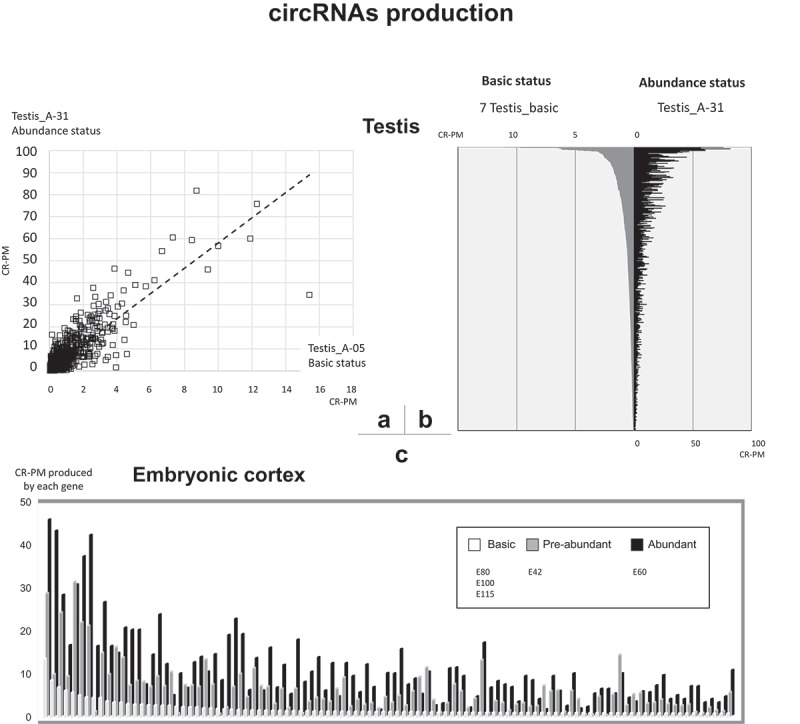
Illustrations of abundance in circular RNAs. (a) Relationships of circRNA production in testis (A-05 and A31). 2052 genes able to produce ExoCirc-RNA were considered (all validated) (b) Relationships of circRNA production between A31 and the 7 basic samples (Testis_A-05, A-16, A-54, A-65, A-73, A-93 and 240d). 678 genes able to produce ExoCirc-RNA were considered (650 were validated) (c) Relationships of circRNA production between embryonic cortex samples. Only the 100 genes identified as the best producers of exonic circRNAs were used for this histogram. The basic situation (E80, E100, and E115) is represented in the front histogram (in white). The background histogram (in black) corresponds to E60 and the middle histogram (in grey) to E42.
Relationships between mRNA and circRNA productions
We began by visually inspecting read data of the Total-RNA-seq on the IGV. If a gene is able to produce a single exonic circRNA with a small number of exons, we observed an accumulation of reads only on the relevant exons (Figure 6(a), Suppl. Doc. S4-F & S4-G). In contrast, we never observed an alteration to the respective mRNA-seq profile. We were unable to confirm that the production of an exonic circRNA leads to a skipping of the respective exons in the mRNA, as suggested by Görlach and Holdenrieder [29]. Concerning the genes that contribute to the production of intronic circRNA, the transcription of exons located downstream from the intronic circRNA did not appear to be affected (Suppl. Doc. S4-H & S4-J).
Figure 6.
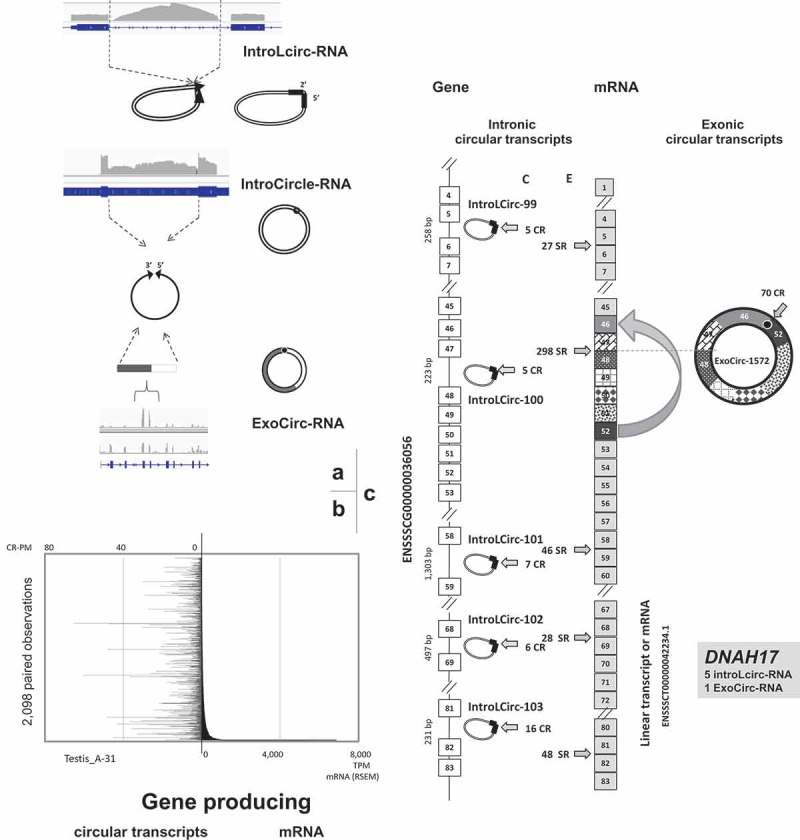
Production of circRNAs. (a) IGV views and schematic representation of the genesis of each type of circRNA. From top to bottom: intronic circRNA. intron circle, Exonic circRNA. (b) Absence of a relationship between circRNA and mRNA production (c) The DNAH17 gene produces six circRNAs: five intronic lariat circRNAs (IntroLcirc-99, −100,-101, −102, and −103) and one exonic circRNA (ExoCirc-1572). E: number of split reads (SR) that characterized the (exonic) linear transcript.
We examined the circRNA production in the embryonic cortex by 13 genes chosen among the highest producers of exonic circRNAs in testis samples with the status ‘abundant’ (Suppl. Doc. T2-F). We found five genes capable of producing a large quantity of exonic circRNAs in testis and in embryonic cortex (Suppl. Doc. T2-F) (PTP3A1, PICALM, MATR3, CSPP1, and PRPF40A) and four genes producing circRNA, which appeared to be relatively specific to the testis (KDM4B, HTR4, CCT3, and UPF2). CCT3 is the gene producing ExoCirc-7094, the most frequent exonic circRNA identified in pubertal testis (Suppl. Doc. T2-C). We examined available knowledge on these nine genes in humans regarding the expression of their exonic linear transcripts, (by analyzing 53 tissues in the Genotype-Tissue Expression (GTEx) Project [39]). The CYP11A1 gene involved in steroid metabolism was added to this comparison. The five genes common to testis and embryonic cortex appear to be ubiquitously and constitutively expressed at low (PRPF40A) or very low expression levels (MATR3, CSPP1). The comparison also indicated that these four genes, which were identified as the biggest producers of circRNAs, have few links with steroid metabolism. In every tissue explored in the GTEx project, ubiquitous low (UPF2 and CCT3) or very low (HTR4) expression was observed (Suppl. Doc. T2-G). Even though the expression of the KDM4B gene is higher in testes than in other human tissues (Suppl. Doc. T2-G), this gene is not really involved in steroid metabolism: it is known to interact with transcription factors and in association with nuclear receptors such as steroid receptors to suppress or promote activation of target genes [40]. Only CCT3 appeared to be able to produce a significant quantity of canonical linear transcripts (at a threshold of the 10% of the best producers of linear transcripts). All these observations support the absence of an association between the production of mRNAs and circRNAs and highlight the fact that genes that appear to be the largest contributors of circRNAs are not the largest sources of linear transcripts.
Finally, we investigated the relationship between circRNA production in testis and the production of linear transcripts by analyzing mRNA-seq datasets from Animal-31. More than 16,000 genes were identified as producers of linear transcripts in the testis of Animal-31 and we retained 8,877 genes with significant expression of linear transcripts (RSEM>10). Among them, 2,098 genes were characterized by their ability to produce at least one validated ExoCircRNA (first analysis), and only 23 were in the top 10% of the producers of linear transcripts in Animal-31. Simultaneous co-production of circular and linear transcripts involved 24% of active genes in the testicular transcriptome (Figure 4(a)). No association was found between production profiles of exonic circRNAs and mRNAs (Figure 6(b)).
It was not possible to directly compare the quantity of mRNA evaluated in mRNA-seq data and the quantity of circRNA measured as CR-PM in Total-RNA-seq. In order to quantitatively compare the production of intronic circRNA and mRNA, we analyzed the dataset from Animal-31 (Total-RNA-seq, described in Table 1), because it contains a substantial number of intronic circRNAs. We considered the number of linear reads spanning the exon-exon junction from the exonic transcript (split reads: SR) in Total-RNA-seq as representative of the production of mRNA (Figure 1(c)). The evaluation was performed on exons adjacent to the intron from which the circRNA originated. We used the number of reads characterizing the exonic (linear) transcript (E) and the number of CR characterizing the circRNA (C) to evaluate the ratio between the two types of transcripts (Suppl. Doc. T2-B). Among the 116 intronic circRNAs examined (and without production of exonic circRNA in the same region), the E/C ratio was very high (>4) for 57 regions (Suppl. Doc. T2-B). For example, all the data obtained concerning DNAH17 (Figure 6(c)) were consistent and compatible with the production of five intronic and one exonic circRNAs (and Suppl. Doc. S4-K & S4-L). After visually inspecting many regions responsible for the production of intronic circRNAs (see Suppl. Doc. S4-K) on IGV, we expected a larger number of genes with a very low calculated E/C ratio (only 8 with E/C < 0.25). Furthermore, when we analyzed regions generating intronic circRNA, we noted that the accumulation of reads in intronic regions is not always correlated with the number of CR detected (Suppl. Doc. S4-K).
Discussion
The development of our circRNA detection workflow was primarily motivated by the lack of an available computational tool dedicated to the identification of intronic circRNAs. In contrast to most available multipurpose circRNA identification tools [33], we decided to integrate constraints concerning the localization of mapping, to restrict our study to circRNAs of known origin (intronic or exonic) and to focus on circRNAs produced by genes coding for proteins. Our approach may appear simple and rather rough but the circular junctions detected were rarely invalidated by visual IGV examinations. The important point was to identify only circRNAs with a high level of specificity, at the expense of sensitivity (this study is not intended to be exhaustive).
Although computational prediction of circRNAs has matured over the years, their detection requires complex computational workflows that, depending on the tissue and the conditions, typically yield candidate sets of hundreds or thousands of circRNA candidates [41]. The best illustration is two articles that proposing to analyze the same dataset but with two different pipelines [18,19]: 1,262 and more than 5,500 circRNAs were identified in the same rat testis dataset. Here, we found it easier to adapt the strategy developed for intronic circRNAs to identify exonic cirRNAs than to use several published pipelines as proposed by Hansen (2018) [33].
Only our pipeline dedicated to discovering exonic circRNA includes management of the strand orientation of the gene in the output, but none of the intronic circRNAs characterized in our study (Suppl. Doc. T2-B) resulted from antisense transcription. The 123 intronic lariat circRNAs could be classified as circular sisRNAs. The respective 123 introns were small (mean: 354 bp, median: 245 bp) as previously reported [5,10]. We would like to emphasize that the pipeline for intronic circRNA identification does not fully account for the position of the 5ʹ boundary of the circular junction. Nevertheless, the selected CR enabled the definition of standard porcine intronic lariat circRNAs with the 5ʹ boundary of the circular junction (Figure 1(a), Figure 6(a)), which is compatible with a circularization event limited by the branch-point as it is used in pre-mRNA splicing in humans [5]. We confirmed the unusual feature of intronic circRNAs characterized by Talhouarne and Gall (2018) [8]: a cytosine at the 5ʹ boundary of the circular junction (branch-point) was often found rather than the more frequent adenine in intronic sequences of sisRNAs.
Three NC circRNAs containing the entire sequence of the intron (Figure 1(b), Figure 6(a), Suppl. Doc. S4-K, S4-B, S4-C & S4-D) and Suppl. Doc. T2-B) were confirmed by PCR on cDNA from pubertal testes (Figure 3(b)). These observations are compatible with the consideration of these 3 NC intronic circRNA as intron circles as recently described [8,9]. Contrary to the intronic lariat circRNA, their circularization involves a classical 3ʹ-5ʹ link [9], which is of great advantage for their successful experimental confirmation by PCR on RT products (Figures 3b & 6a).
Very frequently, CR supporting a given intronic circRNA differed in their coordinates. This resulted from two phenomena: (1) Mapping by STAR includes uncertainty close to intronic sequence boundaries, and (2) the circRNA size variation due to variability at the 5ʹ boundary of circular junction observed on CR (Suppl. Doc. F3-A). These variations could be due to the limited fidelity of the reverse transcriptase noted upstream from a 2ʹ-5ʹ link [9].
The presence of a 2ʹ-5ʹ phosphodiester bond (and not a 3ʹ-5ʹ [42],) at the circular junction of intronic lariat circRNA (Figure 6(a)) may have consequences for the 3D structure of the circular molecule and may explain the poor accessibility for the reverse transcriptase, which would lead to under-representation of the intronic circular junction region among the reads [8,9]. Greater difficulties in amplifying the circular junction of circular sisRNA than of exonic circRNA on RT products could be linked to this feature. We tested three reverse transcriptase enzymes to circumvent this problem but which did not improve the amplification results (data not shown). Obviously, difficulties to amplify the circular junction of a circular sisRNA are common: other authors did not use a standard RT to work on sisRNA using PCR [8,10].
Until now, the brain was considered as the tissue with the largest quantity and diversity of circRNAs. The production of circRNAs could involve 20% of protein-coding genes [2,25]. In this study, we noted that 24% of protein-coding genes effectively produced linear and circular transcripts in pubertal testis (Figure 6(b)). Compared to other tissues, the conditions in the brain favor higher biogenesis (less editing, higher level of splicing factors, higher level of RBPs) and less degradation (fewer cellular divisions) [25]. It is always difficult to compare one’s results with those of other studies [19,31], but the present study confirms the high potential of testis to produce large amounts of circRNAs. Our study also confirms that it is not possible to dissociate quantity and diversity of exonic circRNAs [43] and underlines the fact that the same is also true for intronic circRNAs.
Here we show that ExoCirc-RNAs are typically produced by large genes that are also able to produce mRNAs (84%) (Figures 4(c & d)). Extreme exons are typically flanked by long introns (Figure 4(b)). We show that proximal external exons (not included in the ExoCirc-RNA, Figure 2(a)) are longer than extreme exons (involved in the circular junction of multi-exon circRNAs). Ragan et al. [38] showed that these features probably create preconditions for circRNA production by promoting looping interactions between flanking introns. We showed that exon exhibiting back-splicing between their two ends are longer than exons involved in multi-exon circRNAs. All our results are consistent with data published in humans [38] and in pigs [21] even though the comparisons were made differently [38]. The ability to produce circRNA seems to be mainly linked to the genomic structure of genes.
The results presented in Figure 5 (see also Suppl. Doc. T2-F) also show that genes that are the major contributors to the production of circRNAs are not major contributors of linear transcripts. In the literature, data are contradictory regarding quantitative relationships between circular and linear transcripts. Some previous reports assumed that circRNAs compete with pre-mRNA splicing [44,45], whereas Zhou et al [19] found a positive correlation between mRNA and circRNA production, even though this had been rejected in other studies [32,43]. It is important to underline that in the current study we avoided the problem of quantifying linear transcripts in Total-RNA-seq datasets [46] by using mRNA-seq data analyzed using RSEM [47]. Our study features an overall level of organization with its own logic for circRNA production that is not a simple by-product of transcription.
It is important to underline that the ‘abundant’ status includes a higher presence of both exonic and intronic circRNAs. The observation that five genes of the top ranked exonic circRNA with the status ‘abundant’ also ranked first in the embryonic cortex and in the pubertal testis (Suppl. Doc. T2-F) once again suggests that accumulation corresponds to a highly organized overall mechanism (already suggested by Kristensen et al. (2018) [43]), but is probably not completely specific to a particular tissue. However, even though the testis has two major specific functions (endocrine and reproductive) genes directly involved in steroid production did not contribute more than average to the production of circRNAs.
Even though more genes in the group with ‘abundant’ status produce circRNAs than in the group with ‘basic’ status (Suppl. Doc. F3-D3), the main reason for the high quantity of circRNAs is the increase in transcription and subsequent transcript circularization of the respective genes (Figure 5). The recruitment of new genes to increase circular transcript production appears to be only a marginal phenomenon. In addition to functions of individual circRNAs, it is conceivable that circRNAs have collective functions. For a number of functions, different isoforms of circRNAs from individual genes and even circular transcripts from different genes could be interchangeable.
CircRNAs were originally found in elevated concentrations during aging, resulting in their accumulation especially in the brain [16,24,25]. Until now, the global age-related accumulation of circRNAs and dynamic overall changes in their expression levels during development appeared to be specific to neural tissues [16,24,25]. Very recently Zhou et al. (2018) [19] reported a dynamic pattern of circRNA expression in testis during the postnatal life of rats and the pattern of circRNA expression appears to mirror the ontogenetic maturation of reproductive abilities. In pigs, six out of seven pubertal samples (5–6 months) covering the beginning of the period of increasing reproduction abilities in pigs were assigned a ‘basic’ status, but so was the sample from the animal aged 8 months. The ‘abundant’ status of one animal, defined by a burst (in quantity and in diversity) in the circRNA population compared to that in a ‘basic’ status, does not appear to be linked with the ontology of reproductive abilities. We also postulate that ExoCirc-9244, resulting from circularization between exon-16 and −12 of the porcine SMARCA5 gene, could be an indicator of the ‘abundant’ status in the pubertal testis. Among the 76 testicular samples analyzed by PCR, we identified 12 transcriptomes containing a large quantity of ExoCirc-9244, which were assigned the ‘abundant’ circRNA status. Furthermore, plasma estradiol levels were significantly lower in these 12 animals than in animals with no ExoCirc-9244 expression. We further confirmed the capacity of the 12 individuals to produce sexual steroids by monitoring the expression level of genes involved in steroidogenesis (Suppl. Doc. F3-B2). We think that these high ExoCirc-9244 producing animals are temporally and physiologically closer to the onset of testicular steroid production than the others. However, not all animals with a low E2 level were concerned (Suppl. Doc. F3-B1). We are not able to affirm that the burst of circRNA is limited to a particular period and/or that it occurs in only some but not all animals. In this context, it is interesting to note that in humans, a circRNA derived from SMARCA5 (circSMARCA5, hsa_circ_0001445, circularization of exon-16 and exon-15) was shown to be up-regulated in prostate cancer and was significantly induced after a DHT treatment [48]. Porcine ExoCirc-9244 is not exactly the porcine version of hsa_circ_0001445 because it originates from the circularization between exons 16 and 12. We found no CRs originating from ExoCirc-9244 in the five datasets from embryonic cortex, in the three muscle datasets or in the dataset from Testis_240d. The characteristics of ExoCirc-9244 originating from SMARCA5 provide additional arguments in favor of the existence of differences in steroid levels between the ‘basic’ and ‘abundant’ status in pubertal testis and we propose it as a marker of circRNA abundance in pubertal testis.
Finally, we observed that circRNAs can also be abundantly expressed in pubertal testis and in embryonic cortex. These observations confirm that in brain, the expression of circRNAs changes temporally and spatially during neural development [22] with no apparent correlation with their linear host gene expression [27]. On the other hand, we observed no variation in the total production of circRNAs in muscle (similar observations have been reported in Rhesus Macaque [28]), yet another tissue in permanent renewal during postnatal life. We noted that the circRNA ‘abundant’ status includes a higher presence of both exonic and intronic circRNAs. The presence of a large number of intronic circRNAs has been reported in the cytoplasm during the oogenesis of Xenopus tropicalis [10]. Other situations or developmental periods/stages with abundance of exonic circRNA have been reported, in particular in platelets [49] and during human epidermal stem cell differentiation [43]. The data on the quantity of ExoCirc-9244 in pubertal testis in the present study suggests a link between the onset of steroid production and ‘abundant’ status of quantity of circRNAs. A large quantity of circRNAs may be produced at a particular stage during the development of embryonic cortex or pubertal testis, and ExoCirc-9244 seems to be an informative indicator of this process in pubertal testis. We suggest that the abundance of circRNAs could be linked to an abrupt switch of the cellular metabolism to cope with a particular challenge or with a sudden change in the cellular context [50]. The small quantity of most individual circRNAs indicates that in this situation with overall intronic and exonic circRNAs abundance, the tissues benefit from the collective functions of these transcripts rather than from specific action of distinct circRNAs. This hypothesis is also supported by our data indicating that the massive production of circRNAs is much more related to the structure of the gene than to its function. In agreement with the results of study by Liang et al [50] and underlined by Li et al. [12], we suggest that it is even possible that the circRNA products may not be important in themselves and that the most important point is the activation of the circRNA production pathway. Moreover, it is possible that in some circumstances the number of circRNAs would be more important than their individual characteristics.
Material and methods
Animals
The testes of 220 animals from a large framework experiment on genetic and genomic breeding values of entire male pigs were sampled at slaughtering. Animal husbandry and sample collection have already been described elsewhere [30]. Animals were usually slaughtered near 160 days (± 3 weeks) of age, namely during puberty. Pigs from European breeds (Pietrain, Large White) are sexually mature (testes able to produce sperm and hormones) at eight months. To choose animals among the 220 sampled, we used the plasmatic E2 level that enables the increasing endocrine abilities of the testes to be monitored [51,52]. This measurement was performed on all animals eight days before slaughter.
For RNA-seq experiments, we avoided choosing animals with an estradiol level below the limit of detection (85% of animals of the total population). A subset of four purebred Pietrain (P) and three crossbred Pietrain × Large White (PxLW) boars with a plasmatic testosterone level close to the mean of the entire population (1 to 2 ng/ml, data not shown) was selected. These values clearly showed that puberty had started (the production of steroids was just starting), but no animal could be considered as mature (testosterone > 15 ng/ml).
A second subset of 76 animals (including the seven animals in the first subset) originating from the same population was used for validation of circRNAs by PCR experiments. As a new method recently became available for E2 evaluation, we decided to measure the E2 of these 76 animals on plasma samples kept at −20°C. In this subset, E2 ranged between the limit of detection (7 pg/ml) and 80 pg/ml, and values between 110 and 400 pg/ml are usually found for animals aged 9 months (data not shown). No significant difference was observed between Pietrain (38 animals, mean E2 = 25.30 pg/ml) and PxLW (38 animals, mean E2 = 27.01 pg/ml) subpopulations. This subset of 76 animals was less homogeneous in terms of age of animals, even though the difference between breeds was not statistically significant (P: −0.14; PxLW: 0.30). Only the animals of interest for this study are described in Suppl. Doc. T2-A.
External datasets
We downloaded four Total-RNA-seq datasets obtained by Liang et al. (2017) [31] from Guizhou mini-pigs: testis postnatal 240 days (GSM1902350); skeletal muscle neonatal, at 0 day (GSM1902353), postnatal at 30 days (GSM1902354) and at 240 days (GSM1902354). Age at 30 days corresponds to slaughter just after weaning. Male 240 day-old pigs are considered to have completed puberty (production of sperm and hormones).
We downloaded five datasets obtained by Veno et al. (2015) [21] from the porcine embryonic cortex: 42 days (GSM1846497), 60 days (GSM1846498), 80 days (GSM1846499), 100 days (GSM1846500) and 115 days (GSM1846501). Gestation in the sow lasts 115 days.
From samples to reads
Samples for RNA preparation were disrupted, homogenized, and ground to a fine powder by rapid agitation for 1 min in a liquid nitrogen–cooled grinder with stainless steel beads before RNA extraction. Total RNA was extracted with TRIzol reagent (Invitrogen), purified using the NucleoSpin RNA kit (Macherey-Nagel), and treated with DNase to remove contaminating DNA.
For Illumina total RNA sequencing, rRNA depletion was done with the RiboMinus eukaryote kit [30](ThermoFisher scientific) according to the manufacturer’s recommendations. Measurements made with an Agilent Bioanalyser confirmed successful rRNA depletion. The Illumina stranded TruSeq sample preparation kit was used to generate libraries for stranded paired-end sequencing. After sequencing on HiSeq2500 or HiSeq2500-1T, two types of reads were available (100 and 125 bp from the same library) for each animal (Table 1, Suppl. Doc. T2-D). Data were processed to remove adapter sequences and reads with low sequence information. The reads Total-RNA-seq from seven pubertal testes are deposited in the NCBI under accession number PRJNA506525 (SRA BioProject).
A set of reads (12 million of PE 2X150bp) of mRNAseq was available for two samples (testis from Animal-31 and Animal-05) (Table 1). After a polyA+ selection, the Illumina TruSeq mRNA kit was used to generate libraries for stranded paired-end sequencing, which were sequenced on HiSeq3000.
Alignments
After Total-RNA-seq (RNA-seq obtained from total RNA after ribosomal depletion) or mRNA-seq (RNA-seq of poly(A) transcripts), we obtained a dataset of stranded PE reads consisting of two subsets (reads-1 and reads-2). All alignments were obtained using the fast and splice-aware read mapper Spliced Transcripts Alignment to a Reference (STAR) [36]. For STAR-SE (mates of each pair were mapped independently) and for STAR-PE, parameters proposed by Cheng et al. (2016) [35] were used. The genome assembly (Sscrofa11.1) used here corresponds to GenBank Assembly ID GCA_000003025.6. We used the gene annotation (Sscrofa.11.1.90.GTF) proposed by Ensembl.
Reads from mRNA-seq were mapped with STAR-PE and counted with the RNA-Seq by Expectation Maximization (RSEM) software v. 1.3 [47].
Total-RNA-seq analysis and identification of circRNAs
Mates of each RNA-seq read pair were mapped independently with STAR, and searched for chimeric reads (CR). All useful information concerning CR (Figure 1(a)) files and reads spanning each exon-exon junction (split reads, SR) can be found in auxiliary files generated by STAR.
Strategies were implemented in several workflows to create two pipelines running on the Galaxy platform in a local Sigenae/Genotoul in Toulouse (http://bioinfo.genotoul.fr/), but none of the tools incorporated in these pipelines are specific to this platform. A complete description of the strategies is provided in Suppl. Doc. S1.
We considered a circular transcript as characterized when it was supported by at least five CRs in one animal (Table 1). This threshold was not applied for other evaluations reported in Table 2. Following the study of Zhang et al. (2013) [5], we decided to limit the selection to intronic circRNAs < 3,000 bp. As the assembling of the porcine genome is not perfect, we decided to limit the selection to exonic circRNA < 25,000 bp. As we observed a lot of PE in which the two ends overlapped in almost all their sequences and where each read was chimeric (see Suppl. Doc. S4-M), we decided to require a minimum size between the mapping positions of the two CR segments in order to validate them as originating from a circRNA (Figures 1(b & c)).
PCR experiments with circRNAs
A set of 84 RT were produced from RNA from 76 individual animals using the High capacity cDNA Reverse Transcription kit (Applied Biosystems #4368814, random primers dN6) from 2 µg of RNA (total volume = 20 µl). This set included the 24 RNA already used in qPCR experiments [30] and another eight samples (including Animal-16, −31, −54, −65, and −73) with two independent RNA extractions. With the aim of obtaining a better reverse transcription of intronic circRNA, new RTs were performed with three reverse transcriptases: Applied Biosystems #4368814 kit/superscript II (Invitrogen #18064014)/superscript III (Invitrogen #18080093).
To evaluate resistance to RNase-R digestion, cDNAs were prepared in parallel after pretreatment with and without RNase R of two samples (Animal-31, −65). Total RNA (2 µg) was denatured at 65°C for 5 min and cooled on ice for 1 min, then 1X buffer RNase R, and either 20 units of RNase R (Epicentre) or nuclease-free water was added. Linear RNA was digested for 90 min at 37°C followed by inactivation for 3 min at 95°C (mixture of protocols proposed by [10,49]). RT (Applied Biosystems #4368814) was directly carried out after the inactivation step.
Primer pairs were designed for 7 intronic and 64 exonic circRNA and 3 intron circles (Suppl. Doc. T2-E). We designed divergent primers that face away from each other on the linear RNA, so that they can only amplify the circRNAs, and not linear RNAs with the same sequence (Figure 3(a)). The cycling protocol began with one cycle at 95°C for 4 minutes, followed by 42 PCR cycles at 95°C for 30 seconds, at 60°C for 30 seconds and at 72°C for 10 seconds. PCRs were performed with 1/50 µl of RT products.
PCR were usually performed with Taq polymerase from Promega (GoTaq flexi DNA polymerase, #M8829) in a volume of 15 µl. In the second experiment shown in Figure 3(b), the PrimeStar GXL DNA polymerase (Takara #R050A) was used in parallel in a volume of 25 µl. Quantitative PCRs were performed to evaluate the relative quantity of ExoCirc-9244 in cDNAs [30].
Tools to analyze data
g:Profiler: https://biit.cs.ut.ee/gprofiler/gost
Databases
CircRNAdb: http://202.195.183.4:8000/circrnadb/circRNADb.php
CircBase http://www.circbase.org/
http://www.ensembl.org/Sus_scrofa/Info/Index
Expression atlas: https://www.ebi.ac.uk/gxa/home
We used knowledge relative to the Genotype-Tissue Expression (GTEx) Project [39]: RNA-seq from 53 human tissue samples (experiment accession: E-MTAB-5214).
Funding Statement
The investigations reported in this publication were supported by the Animal Genetics Division of INRA as part of the PigTRome project. Annie Robic acknowledges DAAD (Deutscher Akademischer Austauschdienst), which supported her research stay in FBN in 2017.
Acknowledgments
The authors are particularly grateful to the management and staff of Cooperl Arcatlantique for allowing them access to the Montfort Sur Meu slaughterhouse. We thank everyone from INRA PEGASE (F‐35590 Saint‐Gilles) who collected the samples. The authors greatly appreciate the excellent technical assistance provided by Raphaël Comte (INRA PEGASE). We thank the team who run the genomic platform of the Génopole Toulouse Midi-Pyrénées for their contribution to results. We are grateful to the Genotoul/bioinformatics platform Toulouse Midi-Pyrenees (Bioinfo Genotoul) for computing and storage resources.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental material
Supplemental data for this article can be accessed here.
References
- [1].Hsu MT, Coca-Prados M.. Electron microscopic evidence for the circular form of RNA in the cytoplasm of eukaryotic cells. Nature. 1979;280(5720):339–340. [DOI] [PubMed] [Google Scholar]
- [2].Holdt LM. Kohlmaier A and Teupser D. Molecular roles and function of circular RNAs in eukaryotic cells. Cell Mol Life Sci. 2018;75(6):1071–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Jeck WR, Sorrentino JA, Wang K, et al. Circular RNAs are abundant, conserved, and associated with ALU repeats. Rna. 2013;19(2):141–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Memczak S, Jens M, Elefsinioti A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495(7441):333–338. [DOI] [PubMed] [Google Scholar]
- [5].Zhang Y, Zhang XO, Chen T, et al. Circular intronic long noncoding RNAs. Mol Cell. 2013;51(6):792–806. [DOI] [PubMed] [Google Scholar]
- [6].Osman I. Tay ML and Pek JW. Stable intronic sequence RNAs (sisRNAs): a new layer of gene regulation. Cell Mol Life Sci. 2016;73(18):3507–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gardner EJ, Nizami ZF, Talbot CC Jr., et al Stable intronic sequence RNA (sisRNA), a new class of noncoding RNA from the oocyte nucleus of Xenopus tropicalis. Genes Dev. 2012;26(22):2550–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Talhouarne GJS, Gall JG. Lariat intronic RNAs in the cytoplasm of vertebrate cells. Proc Natl Acad Sci U S A. 2018;115(34):E7970–E77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Taggart AJ, Lin CL, Shrestha B, et al Large-scale analysis of branchpoint usage across species and cell lines. Genome Res. 2017;27(4):639–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Talhouarne GJ, Gall JG. Lariat intronic RNAs in the cytoplasm of Xenopus tropicalis oocytes. Rna. 2014;20(9):1476–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Huang S, Yang B, Chen BJ, et al The emerging role of circular RNAs in transcriptome regulation. Genomics. 2017;109(5–6):401–407. [DOI] [PubMed] [Google Scholar]
- [12].Li HM, Ma XL, Li HG. Intriguing circles: conflicts and controversies in circular RNA research. Wiley Interdiscip Rev RNA. 2019;e1538. [DOI] [PubMed] [Google Scholar]
- [13].Hansen TB, Jensen TI, Clausen BH, et al Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495(7441):384–388. [DOI] [PubMed] [Google Scholar]
- [14].Dhamija S, Menon MB Non-coding transcript variants of protein-coding genes – what are they good for? RNA Biol. 2018;1025–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wilusz JE. Circular RNAs: unexpected outputs of many protein-coding genes. RNA Biol. 2017;14(8):1007–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chen W, Schuman E. Circular RNAs in brain and other tissues: a functional enigma. Trends Neurosci. 2016;39(9):597–604. [DOI] [PubMed] [Google Scholar]
- [17].Li Z, Huang C, Bao C, et al Exon-intron circular RNAs regulate transcription in the nucleus. Nat Struct Mol Biol. 2015;22(3):256–264. [DOI] [PubMed] [Google Scholar]
- [18].Mahmoudi E, Cairns MJ. Circular RNAs are temporospatially regulated throughout development and ageing in the rat. Sci Rep. 2019;9(1):2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Zhou T, Xie X, Li M, et al Rat BodyMap transcriptomes reveal unique circular RNA features across tissue types and developmental stages. Rna. 2018;24:1443–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Westholm JO, Miura P, Olson S, et al Genome-wide analysis of drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep. 2014;9(5):1966–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Veno MT, Hansen TB, Veno ST, et al Spatio-temporal regulation of circular RNA expression during porcine embryonic brain development. Genome Biol. 2015;16:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Szabo L, Morey R, Palpant NJ, et al Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development. Genome Biol. 2015;16:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Dang Y, Yan L, Hu B, et al Tracing the expression of circular RNAs in human pre-implantation embryos. Genome Biol. 2016;17(1):130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Knupp D, Miura P. CircRNA accumulation: A new hallmark of aging? Mech Ageing Dev. 2018;173:71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hanan M. Soreq H and Kadener S. CircRNAs in the brain. RNA Biol. 2017;14(8):1028–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gruner H, Cortes-Lopez M, Cooper DA, et al CircRNA accumulation in the aging mouse brain. Sci Rep. 2016;6:38907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Rybak-Wolf A, Stottmeister C, Glazar P, et al Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol Cell. 2015;58(5):870–885. [DOI] [PubMed] [Google Scholar]
- [28].Abdelmohsen K, Panda AC, De S, et al Circular RNAs in monkey muscle: age-dependent changes. Aging (Albany NY). 2015;7(11):903–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gorlach A, Holdenrieder S. Circular RNA maps paving the road to biomarker development? J Mol Med. 2017;95(11):1137–1141. [DOI] [PubMed] [Google Scholar]
- [30].Robic A, Feve K, Riquet J, et al. Transcript levels of genes implicated in steroidogenesis in the testes and fat tissue in relation to androstenone accumulation in fat of pubertal pigs. Domest Anim Endocrinol. 2016;57:1–9. [DOI] [PubMed] [Google Scholar]
- [31].Liang G, Yang Y, Niu G, et al. Genome-wide profiling of Sus scrofa circular RNAs across nine organs and three developmental stages. DNA Res. 2017;24(5):523–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Dong WW, Li HM, Qing XR, et al. Identification and characterization of human testis derived circular RNAs and their existence in seminal plasma. Sci Rep. 2016;6:39080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hansen TB. Improved circRNA identification by combining prediction algorithms. Front Cell Dev Biol. 2018;6:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Afgan E, Baker D, Batut B, et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018;46(W1):W537–W44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Cheng J, Metge F, Dieterich C. Specific identification and quantification of circular RNAs from sequencing data. Bioinformatics. 2016;32(7):1094–1096. [DOI] [PubMed] [Google Scholar]
- [36].Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Suzuki H, Zuo Y, Wang J, et al. Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res. 2006;34(8):e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Ragan C, Goodall GJ, Shirokikh NE, et al. Insights into the biogenesis and potential functions of exonic circular RNA. Sci Rep. 2019;9(1):2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Project e Enhancing GTEx by bridging the gaps between genotype, gene expression, and disease. Nat Genet. 2017;49(12):1664–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Klein BJ, Piao L, Xi Y, et al. The histone-H3K4-specific demethylase KDM5B binds to its substrate and product through distinct PHD fingers. Cell Rep. 2014;6(2):325–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Jakobi T, Dieterich C. Computational approaches for circular RNA analysis. Wiley Interdiscip Rev RNA. 2019;10(3):e1528. [DOI] [PubMed] [Google Scholar]
- [42].Konarska MM, Grabowski PJ, Padgett RA, et al. Characterization of the branch site in lariat RNAs produced by splicing of mRNA precursors. Nature. 1985;313(6003):552–557. [DOI] [PubMed] [Google Scholar]
- [43].Kristensen LS, Okholm TLH, Veno MT, et al. Circular RNAs are abundantly expressed and upregulated during human epidermal stem cell differentiation. RNA Biol. 2018;15(2):280–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Salzman J, Gawad C, Wang PL, et al. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS One. 2012;7(2):e30733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ashwal-Fluss R, Meyer M, Pamudurti NR, et al circRNA biogenesis competes with pre-mRNA splicing. Mol Cell. 2014;56(1):55–66. [DOI] [PubMed] [Google Scholar]
- [46].Li M, Xie X, Zhou J, et al. Quantifying circular RNA expression from RNA-seq data using model-based framework. Bioinformatics. 2017;33(14):2131–2139. [DOI] [PubMed] [Google Scholar]
- [47].Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kong Z, Wan X, Zhang Y, et al. Androgen-responsive circular RNA circSMARCA5 is up-regulated and promotes cell proliferation in prostate cancer. Biochem Biophys Res Commun. 2017;493(3):1217–1223. [DOI] [PubMed] [Google Scholar]
- [49].Maass PG, Glazar P, Memczak S, et al. A map of human circular RNAs in clinically relevant tissues. J Mol Med. 2017;95(11):1179–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Liang D, Tatomer DC, Luo Z, et al. The output of protein-coding genes shifts to circular RNAs when the pre-mRNA processing machinery is limiting. Mol Cell. 2017;68(5):940–54 e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Zamaratskaia G, Babol J, Madej A et al. Age-related variation of plasma concentrations of skatole, androstenone, testosterone, oestradiol-17 beta, oestrone sulphate, dehydroepiandrosterone sulphate, triiodothyronine and IGF-1 in six entire male pigs. Reprod Domest Anim. 2004;39(3):168–172. doi: 10.1111/j.1439-0531.2004.00496.x [DOI] [PubMed] [Google Scholar]
- [52].Kanematsu N, Jin W, Watanabe G, et al Age-related changes of reproductive hormones in young Meishan boars. J Reprod Dev. 2006;52(5):651–656. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.