

ABSTRACT
The innate immune system is the frontline host protection against pathogens. Effective antiviral immunity is elicited upon recognition of viral RNAs by the host pattern recognition receptors. One of the major viral RNA sensors is retinoic acid inducible gene-1, which triggers the production of interferons (IFNs). In turn, this protective response requires another viral sensor and immunity factor interferon-inducible protein kinase RNA activator (PACT/PRKRA). Here, we report the identification and characterization of a novel antisense PACT gene that expresses a non-coding RNA in a convergent and interferon-inducible manner. Publicly available gene structure and expression data revealed that this gene, that we termed ASPACT, overlaps with the 3′ -end of the PACT locus and is highly expressed during viral infection. Our results confirm the IFN-β-inducibility of ASPACT, which is dependent on STAT-1/2. We further discovered that downregulation of ASPACT impacts both the expression and localization of the PACT transcript. At the transcription level, ChIP and ChIRP assays demonstrated that the ASPACT non-coding RNA occupies distinct chromatin regions of PACT gene and is important for promoter recruitment of the epigenetic silencer HDAC1. In parallel, ASPACT was also found to mediate nuclear retention of the PACT mRNA via direct RNA–RNA interaction, as revealed by RNA antisense purification assay. In summary, our results support the model that the non-coding RNA ASPACT acts as a negative regulator of PACT at multiple levels, and reveal a novel regulator of the viral counteractive response.
KEYWORDS: Antisense non-coding RNA, PACT, interferon, epigenetic regulation, posttranscriptional regulation
Introduction
The innate immune system is the cornerstone of host protection against pathogens, a counteractive mechanism that ensures organism survival against foreign invasion. In response to the unsolicited entry of virus, host cells trigger signalling cascades that are transduced via cytokines and type-I interferon (IFN) proteins [1] upregulating IFN-stimulated genes (ISGs) [2]. Effective antiviral immunity requires several viral RNA sensors expressed by the host cells, such as retinoic acid inducible gene-1 (RIG-I), melanoma differentiation-associated protein 5 (MDA5), and laboratory of genetics and physiology 2 (LGP2), that trigger the production of type-I IFNs and activate innate immune responses. These proteins similarly require direct interaction with another viral sensor and immunity factor, interferon-inducible protein kinase RNA activator (PACT/PRKRA), for full activation [3–6]. Interestingly, in addition to the two double-stranded RNA binding domains (dsRBDs), PACT has a C-terminal domain that has been implicated in binding to and modulating interferon-induced protein kinase RNA (PKR) activity [7,8]. In this capacity, PACT enhances PKR self-phosphorylation, phosphorylation of eIF-2α and consequently, inhibition of translation in the event of viral infection [9]. However, despite its pleiotropic role and biological significance, the mechanisms involved the regulation of PACT’s activity are not well understood.
The Encyclopedia of DNA Elements (ENCODE) project concluded that more than 80% of DNA is functional, and interestingly, a considerable proportion (62%) of functional DNA is transcribed into non-coding RNAs [10]. Cumulative evidence shows that encoded non-coding transcripts, particularly in the form of long non-coding RNAs (lncRNAs), are important regulators of gene expression in various physiological and pathological processes. In this regard, widespread changes in the expression of lncRNAs during the innate immunity response and T cell development and activation have been reported [11]. For instance, long intergenic non-coding RNA (lincRNA-)-Cox2 reportedly represses basal expression of ISGs by associating with heterogeneous nuclear ribonucleoproteins (HNRNPs) [12]. HNRNPL, a member of the HNRNPs family, is known to impart gene regulation by associating with HNRNPL-related immunoregulatory lincRNA (THRIL), which is implicated in the expression of tumor necrosis factor in human monocytes [13]. These studies thus highlight the hierarchy and complexity of gene regulation and emphasize the emergence of lncRNAs in the modulation of immunity-related gene expression and host response.
One important and prevalent class of lncRNAs is the antisense transcripts, whose expression is in the opposite direction and share complementarity with part of the sense gene. Despite the lack of coding potential, antisense lncRNAs have been shown to impact distinct steps of the sense gene expression and, consequently, their associated biological functions [14]. In this functional regard, RNA–RNA and/or RNA–DNA complementarity and interactions are a key determinant underlying the regulatory effect of antisense RNAs. For example, duplex formation between sense and antisense RNAs in the cytoplasm could modulate sense mRNA stability and translation efficiency [15], whereas similar interactions in the nucleus could influence sense mRNA alternative splicing, editing, and transportation [16]. Moreover, lncRNA–DNA formation could create a scaffold for the recruitment of chromatin modifiers, such as PRC2 and HDAC1, thereby altering the epigenetic status of the sense gene [17]. The diverse mechanisms of lncRNA-mediated regulation of protein-coding gene expression support their functional relevance in shaping the transcriptome.
The present study reports the identification and characterization of a novel antisense PACT gene that expresses a non-coding RNA in a convergent and interferon-inducible manner. This antisense gene, that we have termed ASPACT, overlaps with the 3′ -end of the PACT locus and is highly expressed in the context of virus infection. We further reveal discovered that ASPACT negatively impacts both the expression and localization of the PACT transcript. At the transcription level, chromatin-bound ASPACT non-coding RNA is important for the recruitment of the epigenetic silencer HDAC1 to the PACT promoter. In parallel, ASPACT was also found to sequester PACT mRNA in the nucleus via direct RNA–RNA interaction. Taken together, our results demonstrate that non-coding RNA ASPACT acts as a negative regulator of PACT at multiple levels, and provide substantial evidence for the implication of the interplay between antisense lncRNA and sense mRNA gene pair in the host immune system.
Materials and methods
Cell culture, transfection, antisense oligodeoxynucleotides (ODNs), and plasmid
Human HeLa and HEK293 cells were cultured in DMEM supplemented with 10% heat-inactivated fetal bovine serum. For HEK293 cells, 1× NEAA and 1 mM sodium pyruvate were further added to the medium. All media and reagents were purchased from Thermo Fisher Scientific. The cells were incubated at 37°C with 5% CO2 in a humidified incubator. For transient knockdown of ASPACT (48 h), all ODNs (sequences shown in Supplementary Table) were transfected with 50 nM (final concentration) using Lipofetamine 2000 (Thermo Fisher Scientific). For overexpression of ASPACT, its transcript sequence was first amplified by RT-PCR, ligated into cloning vector using the HE Swift Cloning Kit (TOOLS Life Science, Taiwan), and subsequently sub-cloned into the expression vector pcDNA3.1(-). Expression construct was then delivered to cells using Lipofetamine 2000.
Northern blot analysis
Total RNA was isolated using TRIzol reagent (Ambion) from cultured cells. RNA was boiled in Glyoxal Sample Load Dye (Ambion), separated by 2% agarose gel electrophoresis, and transferred onto Nylon membrane in 20× SSC. Membrane was crosslinked by ultraviolet irradiation and hybridized overnight at 68°C with DIG-UTP-labelled RNA probes in hybridization buffer (50% formamide, 5× SSC, 0.02% SDS, and 4× Denhardt’s solution). The membrane was washed twice with low stringency buffer (2× SSC and 0.1% SDS) for 10 min at room temperature and then twice with high stringency buffer (0.1× SSC and 0.1% SDS) for 10 min at 68°C. Signals were visualized by chemiluminescent assay (CDP-Star; Roche). Primers used for generating probes are shown in Supplementary Table.
Western blot analysis
Whole-cell lysate, prepared in RIPA buffer [50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 0.5% deoxycholate, 0.1% SDS, 5 mM EDTA, and 1% NP-40], were separated by SDS-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred onto PVDF membrane. Primary antibodies against ACTIN (sc-47,778), GAPDH (sc-32,233), IRF1 (sc-497), RNA Pol II (sc-47,701), and PKR (sc-708) were purchased from Santa Cruz Biotechnology. Antibodies from Abcam (PACT, ab31967; p-PKR, ab32036), Cell Signaling Technology (EZH2, 3147S; p-STAT1, 9177; RIG-I, 3743), Millipore (HDAC1, 05–100), GeneDirex (Lamin A/C, GTX101127), and Enzo Life Sciences (MDA5, AT113) were also used for recognizing the respective protein factors. HRP-conjugated secondary antibodies for immunoblotting were obtained from Vector Laboratories.
RNA extraction, reverse transcription (RT)-PCR, and quantitative PCR (qPCR)
Total RNA was isolated by TRIzol reagent (Ambion), and reverse transcribed into complementary DNA (cDNA) by MML-V reverse transcriptase (Thermo Fisher Scientific) with random hexamer. Individual gene expression was detected by real-time quantitative PCR (Bio-Rad CFX Connect Real-Time SYBR-Green PCR system) with specific primers, and quantitatively analysed by CFX Manager Software (Bio-Rad). Relative gene expression was determined by normalization to the abundance of housekeeping gene expression, with the control group being represented as reference point. All results were obtained from at least three independent experiments and presented as mean and SEM (error bars), and statistical significance was measured by Student’s t test and presented as P-value. Sequences of primers used in all PCR-related experiments are listed in Supplementary Table.
Subcellular fractionation
Cells were washed and harvested with PBS, and centrifuged at 1,000 x g for 10 min. After supernatant removal, the nuclear fractionation buffer [10 mM Tris-HCl (pH 7.8), 140 mM NaCl, 1.5 mM MgCl2, 0.5% NP-40, and 3 Unit/ml RNaseOUT (Thermo Fisher Scientific)] was added to the pellet. The suspended pellet was incubated on ice for 4 min, and subsequently centrifuged at 500 x g for 4 min. The supernatant was collected as the cytoplasmic fraction. The pellet was further washed with fractionation buffer twice, centrifuged at 3,000 rpm for 5 min at 4°C, and used as the nuclear fraction. Total RNAs of the cytoplasmic and nuclear fractions were extracted by the TRIzol reagent, and subsequently reverse transcribed to cDNA. Target gene expression in the cDNA sample was determined by qPCR, with U48 and 7SL as nuclear and cytoplasmic markers, respectively.
Luciferase reporter assay
The chimeric reporter constructs were generated by fusing the ASPACT promoter sequences with the luciferase gene (pGL3-Basic Vector). For the mutant construct, interferon-stimulated response element (ISRE) within the ASPACT promoter was mutated by PCR-based nucleotide substitution using the following primers: forward, 5′–CATGTATCAGTAGCAGAGTTTCTT; reverse, 5′–AAGAAACTCTGCTACTGATACATG (altered nucleotides are underlined). For reporter assay, the indicated plasmids were co-transfected with β-gal expression vector (Renilla luciferase vector in the case of IFN-β-Luc reporter) into HeLa cells for 2 days. Transfected cells were washed with PBS, and collected by Reporter Lysis 5× Buffer (Promega). Luciferases and β-gal activities in the transfectants were measured by Dual-Luciferase Reporter Assay System (Promega), and relative luciferase activity was obtained by normalization to β-gal or Renilla luciferase intensity. All samples were analyzed in triplicates, and four independent experiments were performed.
RNA-protein complex immunoprecipitation assay (RNA-IP)
RNA-IP was performed essentially as described previously [18]. Briefly, cells were washed and harvested using ice-cold polysomal lysis buffer [100 mM KCl, 5 mM MgCl2, 10 mM HEPES (pH 7.0), 0.5% NP-40, 1 mM DTT, 50 Unit/ml RNaseOUT (Invitrogen), protease inhibitor cocktail (Roche)]. Lysates were passed through 27G needle ten times and centrifuged at 12,000 x g for 15 min at 4°C. Supernatants were collected and precleared by Dynabeads Protein G (Thermo Fisher Scientific) for 1 h at 4°C. The precleared lysates were then incubated with control IgG or specific antibody overnight at 4°C. Antibody-conjugated lysates were subsequently incubated with Dynabeads Protein G for 1 h at 4°C. Precipitated complexes were washed with polysomal lysis buffer, and treated with 10 Unit DNase I (Fermentas) at 37°C for 15 min. The immunoprecipitated RNAs were extracted by TRIzol, reverse transcribed to cDNA, and finally analysed by qPCR.
Chromatin immunoprecipitation assay (ChIP)
ChIP was performed as described before [19]. Transfected cells were subjected to formaldehyde crosslinking, chromatin fragmentation, and immunoprecipitation. After protein removal, the eluted DNA was quantified by qPCR assay using primers corresponding to the target genomic loci (Supplementary Table).
Detection of nascent RNA synthesis rate
Metabolic labelling was used in the analysis of nascent RNA synthesis, procedures of which were performed as described previously [20]. 500 μM 4-thiouridine (4-sU) was added to live cells for 30 min, and total RNA was isolated by TRIzol and dissolved in TE buffer [10 mM Tris-HCl, 1 mM EDTA (pH 7.5)]. Isolated RNA was incubated with thiol-specific biotinylated (biotin-HPDP) for 1.5 h, extracted by Phase Lock Gel (QuantaBio) with chloroform-isoamyl alcohol (24:1), and then resuspended with TE buffer. Biotinylated RNA was immobilized by streptavidin coupled magnetic beads (Thermo Fisher Scientific) in a 30-min incubation, and subsequently washed with binding and washing buffer [5 mM Tris-HCl (pH 7.5), 0.5 mM EDTA, and 1 M NaCl] and eluted with 100 mM DTT. Eluted RNA sample was purified with RNeasy Mini Kit (QIAGEN), and converted into cDNA by reverse transcription. Nascent RNA synthesis rate was determined by qPCR assay, with EF1A expression as the internal control.
Design and synthesis of RNA capturing probes
Target RNAs were captured using antisense single-stranded DNA (asDNA) biotinylated probes. For capturing long RNAs (>250 nucleotides), pools of asDNA oligos with 20 nt complementary to ASPACT RNA were designed using the online probe designer at http://www.singlemoleculefish.com [21]. Specificity was confirmed by alignment to the human genome using the BLAT tool. All probes (sequences listed in Supplementary Table) were biotinylated at the 3′-end and synthesized by MDBio, Inc. (Taiwan).
Chromatin isolation by RNA purification (ChIRP)
ChIRP was performed as previously described [22] with slight modifications. Cells were crosslinked with 1% glutaraldehyde in PBS for 10 min at room temperature, followed by 125 mM glycine quenching for 5 min. Crosslinked cells were resuspended in lysis buffer [50 mM Tris-HCl (pH7.0), 10 mM EDTA, 1% SDS, 1 mM PMSF, and 0.1 Unit/μl RNaseOUT] and sonicated using Bioruptor (Diagenode), yielding RNA fragments of 100 ~ 500 bp. Fragmented chromatin was diluted in two-time volume of hybridization buffer [750 mM NaCl, 1% SDS, 50 mM Tris-HCl (pH7.0), 1 mM EDTA, 15% formamide, 1 mM PMSF, and 0.1 Unit/μl RNaseOUT]. asDNA probes of 100 pmol were added to 3 ml of diluted chromatin and incubated for 4 h at 37°C with rotation. The probes-hybridized RNAs were subsequently immobilized with streptavidin coupled magnetic T1 beads (Thermo Fisher Scientific) for 30 min at 37°C. Beads:biotin-probe:RNA:chromatin complexes were captured magnetically and washed five times with wash buffer (2× SSC, 0.5% SDS, and 1 mM PMSF). After last wash buffer was removed, beads were subjected to different elution protocols. For RNA extraction, washed complexes were incubated with RNA PK buffer [100 mM NaCl, 10 mM Tris-HCl (pH7.0), 1 mM EDTA, 0.5% SDS, and 0.2 μg/μl proteinase K] for 45 min at 65°C, followed by boiling for 15 min, and trizol:chloroform extraction. Eluted RNA was quantified by qPCR assay. For DNA elution, washed complexes were resuspended in 3× volume of DNA elution buffer (50 mM NaHCO3, 1% SDS, 200 mM NaCl, 100 μg/ml RNase A, and 0.1 Unit/μl RNase H) and incubated for 30 min at 37°C with rotation. Eluted chromatin was then treated with 0.2 μg/μl proteinase K for 45 min at 65°C and further purified for qPCR analysis.
RNA antisense purification (RAP-RNA)
RAP-RNA was performed essentially based on a previous report [23], with some modifications. Approximately five million cells were crosslinked with 2% formaldehyde and solubilized using GuSCN hybridization buffer [20 mM Tris-HCl (pH7.5), 7 mM EDTA, 3 mM EGTA, 150 mM LiCl, 1% NP-40, 0.2% N-laurysarcosine, 0.1% sodium deoxycholate, 3 M guanidine thiocyanate, and 2.5 mM DTT]. Solubilized lysates were subsequently sonicated, yielding RNA fragments of ~150 nt, and further incubated with 100 pmol asDNA probes per experiments in GuSCN hybridization buffer at 37°C for 3 h with rotation. The probes-hybridized RNAs were subsequently immobilized with streptavidin coated magnetic C1 beads (Thermo Fisher Scientific) for 30 min at 37°C. RNA complexes were washed using GuSCN wash buffer [20 mM Tris-HCl (pH7.5), 7 mM EDTA, 1% NP-40, 0.2% N-laurysarcosine, 0.1% sodium deoxycholate, 3 M guanidine thiocyanate, and 2.5 mM DTT] six times at 45°C. To release the RNA, beads were heated for 30 min at 37°C with rotation. Beads were then magnetically separated using the DYNAL (Life Technologies) and supernatants containing eluted target RNA were digested by the 1 μg/μl proteinase K for 1 h at 65°C to remove all proteins. The remaining nucleic acids were then purified by SILANE beads (Thermo Fisher Scientific). DNA probes were removed by digestion with DNase I. Isolated RNA was quantitatively determined by qPCR assay.
5′- and 3′-rapid amplification cDNA ends (RACE)
RACE was performed using a 5′/3′ RACE kit, 2nd generation (Roche) according to the manufacturer’s instructions. Briefly, 2 μg of total RNA isolated from HeLa cells was used for each RT-PCR, followed by nested PCR reactions. Primer sequences are listed in Supplementary Table.
Isolation of ribosome nascent chain-mRNA (RNC-mRNA) complex
Experimental procedures of RNC-mRNA complex extraction were based on a previous report, with some modifications [24]. Briefly, cells were pre-treated with 0.1 μg/μl cycloheximide for 15 min, and subsequently separated into nuclear and cytosolic fractions. The cytosolic RNC-mRNA complex was further isolated by centrifuging at 425,000 × g for 5 h at 4°C with sucrose solution. Sucrose solution contained 33% sucrose, ribosome buffer [20 mM HEPES/KOH (pH 7.4), 15 mM MgCl2, and 200 mM KCl], 2 mM dithiothreitol, and 0.1 μg/μl cycloheximide. The sediment fraction was washed twice with ribosome buffer, from which TRIzol reagent was then used to isolate RNA. Collected RNA was reverse transcribed into cDNA sample, and the abundance of target gene was quantified by qPCR experiment. EF1A expression was analysed as the control.
Results
ASPACT is highly expressed during viral infection
PACT acts as a critical component in the signalling cascade involved in the RIG-I-dependent antiviral response and is thus intricately controlled. Intriguingly, genomic annotations available in the UCSC Genome Browser revealed an unknown antisense gene (AC009948.5) in the vicinity of the PACT locus (Figure 1(a)). The transcript is located on the plus-strand of chromosome 2 and overlaps with the 3′-end of the PACT transcript encoded on the minus strand. The exon–intron organization of the encoded gene suggested that three possible transcript variants [V1 (1.3 kbp), V2 (1.4 kbp), and V3 (2.3 kbp)] could arise due to alternative splicing. To further elucidate its expression and possible correlation with innate immunity, we examined the expression profiles of AC009948.5 using microarray datasets archived in the Gene Expression Omnibus (GEO). We found that AC009948.5 exhibits altered expression patterns in response to viral infection: elevated levels were noted in the blood from patients with Dengue virus (DENV) infection (Figure 1(b)) [25] and in pandemic influenza virus-infected normal human bronchial epithelial cells (Figure 1(c)) [26]. Intriguingly, the PACT transcript, which is encoded on the opposite strand, showed distinct expression patterns (Figure 1(d,e)). These findings raised the possibility that this antisense transcript naturally exists in the cells and is highly expressed in the context of viral infection. Next, open reading frame (ORF) prediction using the Coding Potential Calculator (CPC) algorithm [27] failed to reveal a reliable ORF in the transcript unit (with a negative coding potential score of −1.06711) (data not shown), thus categorizing ASPACT as a non-coding RNA. We hereafter termed this novel antisense transcript antisense long non-coding RNA of PACT (or ASPACT).
Figure 1.
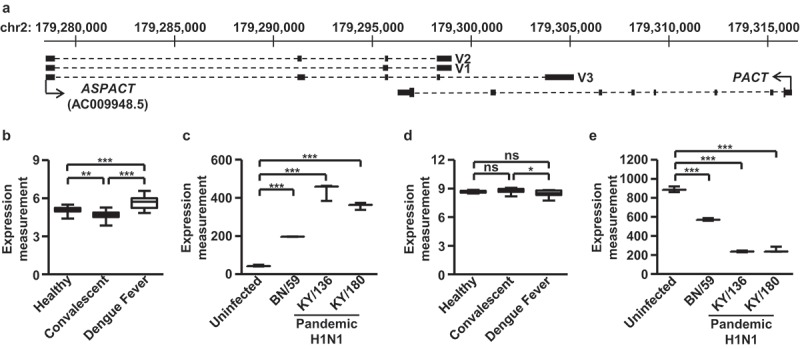
ASPACT is highly expressed during viral infection. a Gene structure and orientation of ASPACT and PACT genes. The ASPACT gene encodes three splice variants: V1, V2, and V3 (identified exons shown as black rectangles). The arrows were represented as the orientation of transcription for ASPACT and PACT. b–e Expression levels of ASPACT and PACT were profiled on the basis of various microarray datasets archived in the GEO. Alterations in the indicated biological contexts are shown by box plots: b, d blood samples from patients with acute DENV infection (n = 18), during convalescence (n = 19), and healthy controls (n = 9); c, e well-differentiated primary lung bronchial epithelial cells 36 h after infection with seasonal H1N1 BN/59, pandemic H1N1 KY/136 and KY/180 (n = 3). In all panels, data represent the mean and SEM (error bars). Significant differences were verified by Student’s t test (ns: P > 0.05, *: P < 0.05, **: P < 0.01, ***: P < 0.001).
Characterization of the transcript unit and regulation of ASPACT expression
To experimentally verify the expression and spatial localization of ASPACT transcript in cells, we first performed Northern blot analysis using total RNA isolated from different cellular compartments of HeLa cells. Using an ASPACT-specific (exons 2–3) probe, strong hybridizing signals were detected in the nuclei (Figure 2(a)), indicating a distinct subcellular distribution of the antisense transcript. To further refine the transcript structure of this novel gene, we carried out 5′- and 3′-rapid amplification cDNA ends (RACE) combined with polymerase chain reaction (PCR) and Sanger sequencing analyses using region-specific primers (Supplementary Fig. S1A, B). Our results revealed that the 1.4 kbp transcript (V2) is the major form encoded by ASPACT, while the long (V3; 2.3 kbp) and short (V1; 1.3 kbp) variant form were undetectable by the end-point PCR and Northern blot methods (Figure 2(a) and Supplementary Fig. S1C). Analogous to most lncRNAs, ASPACT contains a 5′-cap and 3′-poly(A)-tail (data not shown) and predominantly exists in the nucleus.
Figure 2.
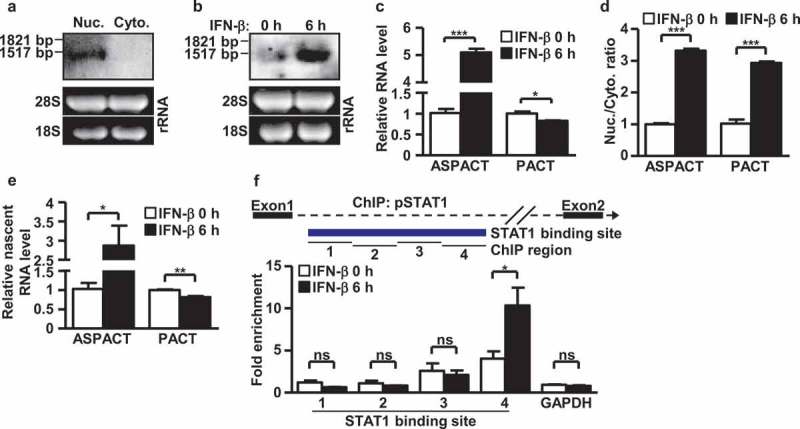
Structure and expression of the ASPACT transcript unit. a Expression of the ASPACT RNA transcript (1.4 kbp) was detected by Northern blot assay using total RNA isolated from nuclei and cytoplasm compartments of HeLa cells. The loading control is shown in the lower panel; 28S: 4.8 kbp, 18S: 1.9 kbp. mRNA sizes are indicated in kbp. b Interferon inducibility of ASPACT RNA expression was detected by Northern blot assay using IFN-treated HeLa cells. The loading control is shown in the lower panel: 28S: 4.8 kbp, 18S: 1.9 kbp. c After IFN-β treatment for 6 h, total RNA was extracted from HeLa cells for RT-qPCR analysis of ASPACT and PACT expression. Expression levels were normalized to an internal control, EF1A, and are presented relative to the mock sample. d Changes in subcellular distribution of ASPACT and PACT transcripts. After IFN-β treatment for 6 h, HeLa cells were subjected to subcellular fractionation, and the expression of transcripts in each fraction was evaluated by qPCR. e Nuclear run-on assay was performed to measure levels of newly synthesized PACT and ASPACT RNAs in IFN-treated HeLa cells, followed by qPCR analysis. Prior to fold-change calculations, target gene expression levels were normalized to that of EF1A. The mock treatment is represented as the reference. f ChIP assay was utilized to demonstrate pSTAT1 occupancy of ASPACT chromatin in IFN-treated HeLa cells. Precipitated DNA fragments were analysed by qPCR using primers corresponding to different regions of ASPACT chromatin, and normalized to the value of IgG. A schematic of the qPCR amplicon in the pSTAT1 binding region is depicted in the upper panel. In all panels, data represent the mean and SEM (error bars) from four independent experiments. Significant differences were verified by Student’s t test (ns: P > 0.05, *: P < 0.05, **: P < 0.01, ***: P < 0.001).
The immune role of PACT as well as the finding from our in silico analysis of infection-related upregulation of ASPACT promoted us to investigate whether ASPACT is an interferon-inducible gene. Interestingly, the promoter region of ASPACT harbors an IFN-stimulated response element (ISRE) and has been annotated in the ENCODE data (https://www.encodeproject.org/) to be associated with key transcription factors in IFN signalling, STAT1 and STAT2 (Supplementary Fig. S1C). To further resolve the regulation of ASPACT gene, we next examined its expression in response to interferon signaling. To this end, Northern blot and quantitative real-time RT-PCR (RT-qPCR) analyses of IFN-β-treated HeLa and HEK293 cells quantitatively showed an elevation in ASPACT expression level in the presence of this cytokine (Figure 2(b-d) and Supplementary Fig. S1D). Moreover, by analysing newly transcribed RNAs using nuclear run-on assay, we found an IFN-β-induced increase in the transcription rate of ASPACT gene. Conversely, nascent expression of the PACT transcript, as well as the abundance of its protein product, was slightly but consistently reduced (Figure 2(e) and Supplementary Fig. S1E, F). In line with the transcriptional upregulation, our ASPACT promoter reporter assay also showed that the upstream region was positively responsive to IFN-β treatment in an ISRE-dependent manner (Supplementary Fig. S1G). In addition, IFN-β treatment led to enhanced binding of RNA Pol II (Supplementary Fig. S1H) and pSTAT1 (Figure 2(f)) at the ASPACT promoter, as indicated by chromatin immunoprecipitation (ChIP) assay. Taken together, these data illustrate the IFN-β inducibility of ASPACT, which is driven by STAT1/2.
Negative role of ASPACT in the regulation of PACT expression
In light of the overlapping nature and converse expression of the ASPACT–PACT gene pair, as well as the IFN-β inducibility of ASPACT, we hypothesized reciprocal regulation between these two genes. To test this, we next characterized whether ASPACT plays a role in PACT expression. Oligodeoxynucleotides (ODNs) were designed and used to silence ASPACT expression in HeLa and HEK293 cells (Supplementary Fig. S2A, B). Knockdown of ASPACT subsequently resulted in a significant induction in PACT mRNA (Figure 3(a) and Supplementary Fig. S2C) as well as protein (Figure 3(b) and Supplementary Fig. S2D) expression. In contrast, ectopic overexpression of ASPACT-V2 (1.4 kbp) in HeLa and HEK293 cells (Supplementary Fig. S2E, F) diminished the levels of PACT mRNA and protein (Figure 3(c,d) and Supplementary Fig. S2G, H). These results provide evidence for negative regulation of PACT by the ASPACT transcript.
Figure 3.
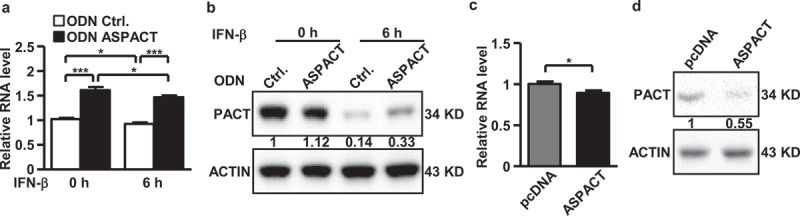
Regulatory role of ASPACT in PACT expression. a Expression of ASPACT was abolished by oligodeoxynucleotide-based knockdown in HeLa cells. Cells were then treated with IFN-β for 6 h to mimic viral infection, and subjected to gene expression analysis. The RNA abundance of PACT was analysed by qPCR and normalized to EF1A level. b Protein levels of PACT in ASPACT knockdown and IFN-treated HeLa cells were measured by Western blot assay using ACTIN as an internal control. c, d ASPACT expression and control vectors were transfected into HeLa cells, and subsequently collected for RNA (c) and protein (d) expression analysis. PACT RNA abundance was analysed by qPCR and normalized to EF1A levels. PACT protein expression was measured by Western blot assay, using ACTIN as the internal control. In all panels, data represent the means and SEM (error bars) from four independent experiments. Significant differences were verified by Student’s t test (ns: P > 0.05, *: P < 0.05, **: P < 0.01, ***: P < 0.001).
ASPACT impacts the transcriptional and epigenetic status of PACT
Antisense lncRNAs located in the nucleus may act as regulators of complementary mRNA expression via two known mechanisms – transcriptional interference to hinder transcription, or complementary binding to sense mRNA to manipulate transcript localization [28]. We sought to delineate possible modes of ASPACT-mediated antagonism of PACT. We first focused on the transcriptional aspect of gene regulation. For that purpose, we conducted nuclear run-on assays and characterized the effects of ASPACT misexpression on the transcriptional rate of PACT. Our results were consistent with the notion that ASPACT inhibits transcription of the PACT gene. Knockdown of ASPACT in HeLa and HEK293 cells led to upregulation of nascent PACT RNA (Figure 4(a) and Supplementary Fig. S3A), whereas expression of exogenous ASPACT abrogated new synthesis of PACT transcripts (Figure 4(b) and Supplementary Fig. S3B). Further ChIP analysis demonstrated a declined occupancy by RNA Pol II of the PACT promoter in ASPACT overexpressing cells (Figure 4(c) and Supplementary Fig. S3C), providing additional support for ASPACT-induced transcriptional suppression of PACT.
Figure 4.
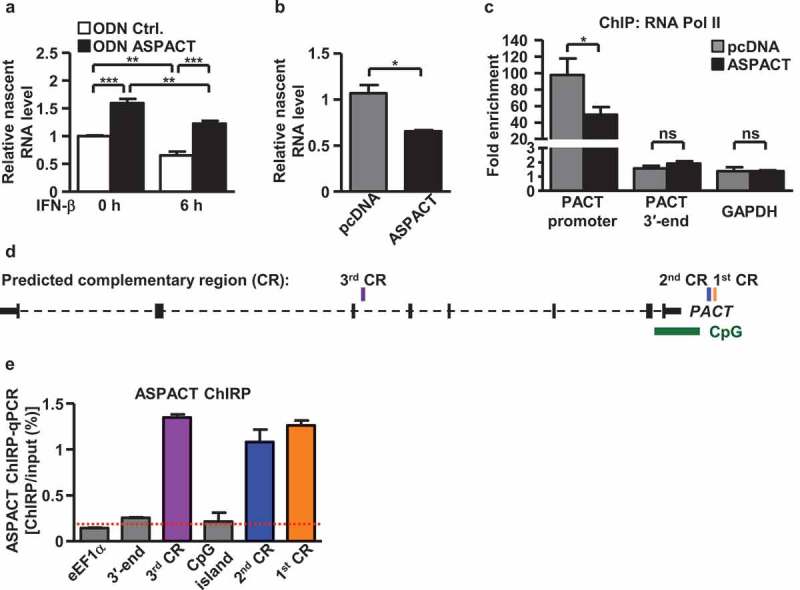
ASPACT negatively regulates PACT expression at the transcriptional level. a, b Nuclear run-on assay was performed to measure levels of nascent PACT RNA in ASPACT knockdown and IFN-treated (a) or ASPACT overexpressing (b) HeLa cells, followed by qPCR analysis. Prior to fold-change calculations, target gene expression levels were normalized to that of EF1A. The mock treatment represents as the reference. c ChIP assay was performed to demonstrate RNA Pol II occupancy of the PACT promoter chromatin. Precipitated DNA fragments were analysed by qPCR and normalized to the value of IgG. The extent of occupancy was compared between control and ASPACT-overexpressing HeLa cells. d Predicted complementary region (CR) in the PACT gene locus with potential correspondence to the ASPACT sequence. Three distinct CRs were uncovered based on high sequence alignment scores, and are represented as rectangles in relation to the CpG island and the gene body of PACT. e ChIRP analysis of ASPACT binding to the PACT chromatin. Retrieval of PACT chromatin DNA was validated by qPCR analysis of the indicated regions (using EF1A as the control). Results are expressed as retrieval rate of chromatin DNA relative to the input. For the bar graphs shown in this figure, data represent the means and SEM (error bars) from four independent experiments. Significant differences were verified by Student’s t test (ns: P > 0.05, *: P < 0.05, **: P < 0.01, ***: P < 0.001).
Given that antisense lncRNAs may modulate gene expression via complementary complexing with promoter sequence of target gene [29,30], we aimed to elucidate whether ASPACT alters PACT transcription via RNA–DNA interactions. Alignment of the ASPACT transcript sequence with the PACT gene unit uncovered three potential complementary regions (CRs) with high binding scores (Supplementary Fig. S3D–F): two CRs located on a CpG island upstream of PACT, and a third CR in the middle of PACT gene body (Figure 4(d)). Next, to demonstrate that this predicted sequence complementarity functionally translated into RNA–chromatin interaction, we employed the Chromatin Isolation of RNA Purification analysis (ChIRP) method [22]. We designed and synthesized antisense DNA (asDNA) probes complementary to the ASPACT RNA sequence and tiled across the entire length of ASPACT (1.4 kbp). As a control, we ensured that asDNA probes could efficiently pull down chromatin-bound ASPACT RNA by showing that ~65% of ASPACT RNA was specifically retrieved (Supplementary Fig. S3G, H). Further ChIRP-qPCR analysis of copurified chromatin DNA confirmed the specific presence of PACT gene fragments, affirming the physical association of ASPACT RNA transcript with the chromatin regions of the PACT CpG island and gene body (Figure 4(e) and Supplementary Fig. S3I).
lncRNAs could also act as modular scaffolds to recruit and assemble multiple protein complexes involved in gene regulation [31,32]. Having established the suppressive and putatively chromatin-based role of ASPACT in PACT modulation, we next aimed to address the possible epigenetic function of the ASPACT transcript. We used RNA immunoprecipitation assay (RNA-IP) to explore the binding of ASPACT RNA by the epigenetic silencers, HDAC1 and EZH2, two known RNA-binding chromatin regulators. Both anti-HDAC1 and anti-EZH2 immunoprecipitated RNAs were analysed using qPCR to demonstrate the specific presence of ASPACT. ASPACT was found in abundant association with these immunocomplexes (Figure 5(a,b)), indicating the unique assemblage of ASPACT with the epigenetic silencers. To further decipher the functional outcome of this interaction, ChIP assays were utilized to monitor the effect of ASPACT on chromatin association of these protein factors. Our results indicated that HDAC1 occupied the PACT promoter and intragenic regions (downstream of the third CR) and that ASPACT overexpression augmented this binding of HDAC1 (Figure 5(c) and Supplementary Fig. S4A). By contrast, despite the association with the ASPACT transcript, no significant association with PACT chromatin region was detectable for EZH2 (Figure 5(d) and Supplementary Fig. S4B). Taken together, these data strongly link ASPACT to PACT chromatin status as a possible transcription-inhibitory RNA scaffold.
Figure 5.
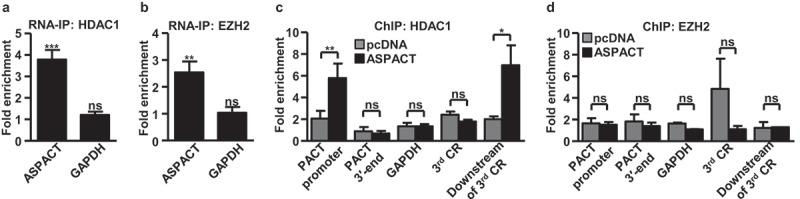
ASPACT associates with HDAC1 and mediates its promoter recruitment. a, b The protein–RNA interaction of HDAC1 (a) and EZH2 (b) with ASPACT was demonstrated by RNA-IP assay. HeLa cell lysates were collected and subsequently immunoprecipitated with specific antibodies, and the precipitated RNA was quantified by qPCR and normalized to the value of IgG. GAPDH served as negative control. c, d Extent of HDAC1 (c) and EZH2 (d) association with the PACT chromatin locus in ASPACT overexpressing cells was examined by ChIP assay. The precipitated DNA fragments corresponding to the promoter and 3′-end regions were analysed by qPCR and normalized to the value of IgG. GAPDH served as a negative control. In all panels, data represent the means and SEM (error bars) from four independent experiments. Significant differences were verified by Student’s t test (ns: P > 0.05, *: P < 0.05, **: P < 0.01, ***: P < 0.001).
ASPACT mediates nuclear retention of PACT mrna via direct RNA–RNA interaction
In addition to chromatin-based control, previous studies have also identified several nuclear antisense lncRNAs as transcript spatial regulators through RNA–RNA interactions [33]. Our above studies of ASPACT’s IFN-β inducibility showed simultaneous accumulation of both ASPACT and PACT RNAs in the nuclear compartment of interferon-treated cells (Figure 2(d)), suggesting a role of the nuclear ASPACT in PACT mRNA localization. To further elucidate this intermolecular mechanism of transcript spatial modulation and to assess whether ASPACT may be linked to subcellular localization of PACT mRNA, we performed subcellular fractionation combined with qPCR-based gene expression analyses. The extent of compartment separation was controlled by measuring the levels of nuclear and cytosolic marker genes, U48 and 7SL, respectively (Supplementary Fig. S5A–D). Interestingly, we found that abolition of ASPACT expression via ODNs in IFN-β-treated cells reduced the relative abundance of nuclear pools of PACT mRNA (Figure 6(a) and Supplementary Fig. S5E). Corresponding with the cytoplasm-biased distribution of PACT mRNA, expression levels of PACT protein were incidentally increased (Figure 6(b)). Furthermore, these changes in transcript distribution were reversed by overexpression of ASPACT (Figure 6(c,d) and Supplementary Fig. S5F). Collectively, these data hint at a role for ASPACT in interfering proper PACT mRNA distribution.
Figure 6.
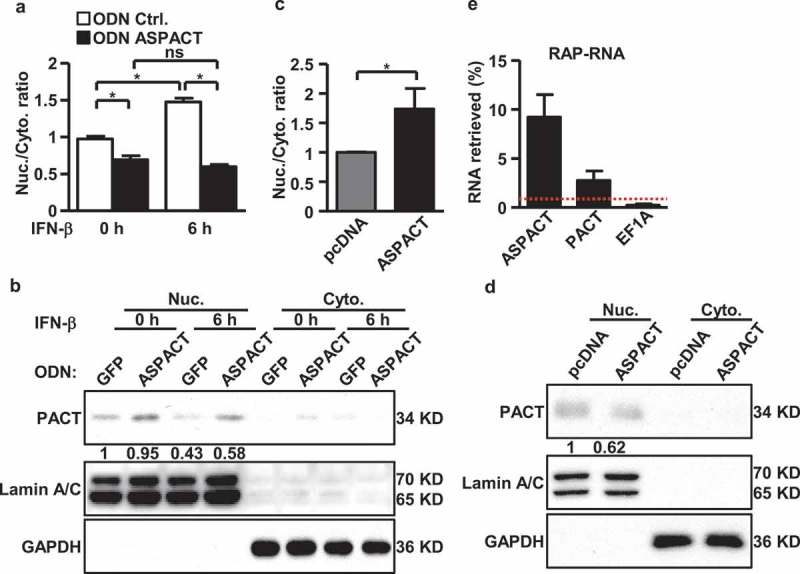
ASPACT mediates nuclear retention of PACT mRNA via direct RNA–RNA interaction. a–c ASPACT knockdown HeLa cells were treated with IFN-β for 6 h then subjected to subcellular fractionation. The abundance of PACT transcript in each fraction was assessed by qPCR, and normalized to U48 and 7SL, individually (a). Western blot analysis was also performed to profile the expression of PACT protein (Lamin A/C and GAPDH serving as nuclear and cytosolic markers, respectively) (b). C, D Expression alterations of PACT RNA (c) and protein (d) in response to ectopic overexpression of ASPACT. Transfected HeLa cells were fractionated into nuclear and cytosol compartments and assessed for RNA and protein expression levels via qPCR and immunoblotting, respectively. E RT-qPCR was used to measure the percentage of the total cellular ASPACT, PACT, or negative control EF1A recovered after RAP-RNA of ASPACT from HeLa cells (see Methods section). Values correspond to the amount of RNA in the elution divided by that in the input lysate. In all panels, data represent the means and SEM (error bars) from four independent experiments. Significant differences were verified by Student’s t test (ns: P > 0.05, *: P < 0.05, **: P < 0.01, ***: P < 0.001).
To further clarify whether intermolecular interactions were involved in this spatial regulation, we next carried out RNA antisense purification (RAP-RNA) to characterize RNA–RNA complex formation via in vivo crosslinking [23]. Similar to the ChIRP assay, ASPACT-complementary asDNA probes were used to isolate ASPACT and crosslinked RNAs from HeLa cells. RT-qPCR analysis was then performed to quantitatively detect the association of PACT mRNA. Our results then showed that ASPACT RAP-RNA was enriched for PACT mRNA (Figure 6(e)), indicating an in vivo interaction between these two RNA molecules. This interaction was specific as the pull-down reaction did not uncover the abundant EF1A mRNA (Figure 6(e)). Furthermore, based on the extent of pull-down enrichment, the RAP assay also identified PACT mRNA (exons 3–4) as a possible strong interaction region of ASPACT. These data are consistent with the scenario that ASPACT could also target PACT expression by altering the subcellular distribution of the PACT transcript. Finally, to resolve the relative contribution of transcriptional vs. post-transcriptional aspects of ASPACT’s action to PACT expression, we examined PACT mRNA and protein levels in the context of HDAC inhibition, which reverses epigenetic suppression (Supplementary Fig. S5G). We subsequently found that TSA treatment on ASPACT-overexpressing cells relieved the expression suppression on the PACT mRNA but not protein (Supplementary Fig. S5H, I). While these observations reinforced the coordination between HDAC and ASPACT in the epigenetic regulation of PACT expression, they also implied that regulation at the post-transcriptional level seems to have a greater impact on the overall abundance of PACT protein.
ASPACT is functionally linked to innate immunity
PACT has been implicated in the cellular response to viral infection [34]. Due to the IFN inducibility of ASPACT and its established role as a negative regulator of PACT expression, we speculated that ASPACT may also represent a critical component of the host innate immunity. To test this, we assessed the expression of several innate immunity genes that encode the cytoplasmic viral RNA sensors, the interferon signaling-associated, interferon stimulated genes (ISGs), or type-I IFNs in the ASPACT mis-expressing cells. ASPACT knockdown cells were treated with IFN-β and subsequently harvested for gene expression analysis by qPCR. The levels of these genes – cytoplasmic sensors (MDA5 and LGP2), interferon regulatory factors (IRF1, IRF7, and IRF9), STAT1/2, PKR, IFITM3, and type-I IFNs (IFNA1, IFNA4, and IFNB1) – were increased in the absence of ASPACT (Figure 7(a-d)). By contrast, expression of exogenous ASPACT in the presence of poly(I:C), which mimics viral infection, downregulated their transcript levels (Supplementary Fig. S6A–D). Similarly to ISGs alteration, protein levels of RIG-I, ISG15, PKR, pPKR, and MDA5 were elevated in ASPACT-knockdown, IFN-β-treated HeLa cells, indicative of heightened interferon signalling (Figure 7(e)). Conversely, overexpression of ASPACT abrogated the upregulation of these molecules (Figure 7(f) and Supplementary Fig. S6E).
Figure 7.
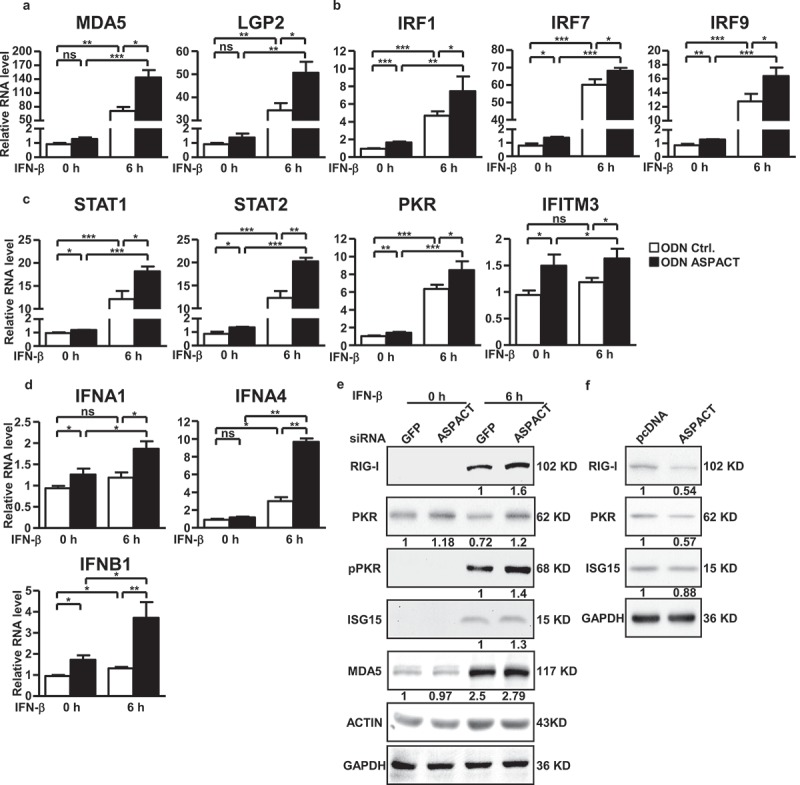
Functional link of ASPACT to the innate immunity. a–e ASPACT knockdown HeLa cells were treated with or without IFN-β for 6 h and subsequently harvested for RT-qPCR (a–d) or Western blot (e) analyses of the indicated ISGs. mRNA expression was normalized to the level of EF1A, while ACTIN and GAPDH were used as the loading control in immunoblotting. F HeLa cells were transfected with ASPACT expression and control vectors and subsequently collected for RIG-I, PKR, and ISG15 and expression analysis via Western blot assay (using GAPDH as the internal control). For the bar graphs shown in this figure, data represent the means and SEM (error bars) from four independent experiments. Significant differences were verified by Student’s t test (ns: P > 0.05, *: P < 0.05, **: P < 0.01, ***: P < 0.001).
To provide a more sensitive readout for PACT activity as a means to complement our gene expression analyses, we further performed the IFN-β-Luc reporter assay. To this end, reporter activity was measured in the presence of MDA5 and/or ASPACT knockdown in the HeLa cells (Supplementary Fig. S6F). Our results in this part then revealed that MDA5-induced type I IFN response could be further enhanced when ASPACT was down-regulated, thus strengthening the negative impact of ASPACT on PACT signalling. These results collectively provide strong evidence for ASPACT-mediated regulation of PACT is functionally coordinated with host immune response. Together, our results uncover a novel regulator of the viral counteractive response that mitigates PACT and immune-associated activities.
Discussion
The intracellular antivirus signalling pathway serves as a protective mechanism involved in the host defence against exogenous stress. In this elaborate, dsRNA-dependent signalling network, PACT serves as a critical switch. The present study provides evidence to further our understanding of this pathway by uncovering a new component that closely regulates the expression of this RNA-binding factor. ASPACT acts at distinct steps during the expression of the PACT gene and presumably serves to fine-tune PACT activity in an interferon-responsive feedback manner. While numerous lncRNAs have been implicated in important respects of immunity, such as production of cytokines, cell activation, and differentiation [35], it remains unclear whether an antisense RNA and its interplay with the sense mRNA gene could contribute to immune-related signalling. By acting at the early stages of the PACT expression, this antisense RNA-mediated control may maintain a robust balance between antiviral signalling versus inflammatory stress. Intriguingly, lncRNAs have rarely been recognized to possess dual regulatory roles at both the transcriptional and posttranscriptional levels. In this regard, one notable example is the tumorigenesis-related lncRNA, HOTTIP, which reportedly exerts twofold regulatory effects on the HOXA13 expression [36]. A novel aspect of our findings is thus the identification of the multifunctionality of ASPACT in terms of its control of PACT expression.
The dynamic, context-dependent protein interactomes of PACT constitute an important determinant underlying the multifaceted nature of PACT’s role in dsRNA-dependent signalling pathway. In addition to RIG-I and PKR, PACT is also known to interact with the components of the RNA-induced silencing complex. In this capacity, PACT, in coordination with transactivation response RNA binding protein (TRBP), binds to Dicer and is required for the RNA interference pathway [37,38]. TRBP is paralogous and most similar to PACT among members of dsRNA-binding protein (dsRBP) family. Interestingly, TRBP reportedly forms heterodimers with PACT and colocalizes at the perinuclear region through their dsRBDs [39]. In addition, their simultaneous association with PKR contributes to a collaborative yet antagonistic action in innate immunity; while TRBP exerts an inhibitory effect on PKR, PACT is required for activation of PKR in the absence of dsRNA [40]. This PACT activity towards PKR activation is responsive to stress stimulation (dsRNA or reactive oxygen species) that triggers dissociation of the TRBP–PACT heterodimers, indicating that TRBP acts as a stress-associated modulator of PACT. While the engagement of different protein partners is seemingly critical for the PACT-based functional switch, the exact mechanism underlying the regulation of PACT, particularly at the transcription level, is not fully understood. Our study thus provides evidence for another regulatory pathway of PACT activity, namely, the multistep action of the antisense RNA, ASPACT.
An interesting finding from our study is the discovery that, despite the potential association with both HDAC1 and EZH2 (Figure 5(a,b)), ASPACT is important for targeting only HDAC1 to the PACT gene. This observation, similar to the PRC2-independent transcription repressive role of HOTAIR RNA, implies that ASPACT could exert distinct effects via differential complexes with epigenetic silencers and further suggests there could be additional targets under trans-regulation by ASPACT. In this functional regard, ASPACT may act as a ‘selector element’ [41] that confers discriminating binding of chromatin modifiers and/or transcriptional regulators to target regions. This selective nature of its mode of action is also supported by our observation of the unique targeting of ASPACT RNA to distinct chromatin regions of the PACT locus, in particular the promoter and gene body regions. Intriguingly, this functional attribute is shared among lncRNAs, which have been shown to exhibit widespread yet specific RNA–chromatin interaction patterns across functional elements of the genome [42]. Further investigation of the gene regulatory function of ASPACT on a genome-wide scale could be achieved using global approaches such as ChIRP-seq.
Functional implications of lncRNA transcripts in posttranscriptional gene regulation have also been extensively documented [43]. Analogous to the classical miRNA–mRNA and snRNA–pre-mRNA complementarity, direct intermolecular RNA–RNA interactions contribute considerably to this RNA biology. Adding to the complexity of lncRNA-directed gene regulation, a protein scaffold functionality that is primarily mediated via indirect association with protein intermediates has also been ascribed to lncRNAs. Well-known examples include hNEAT1 in the nuclear paraspeckles [44] and Malat1 that recruits SRSF1 to active gene loci for alternative splicing of pre-mRNAs [45]. The results of our RAP–RNA assay lends support to the notion of a direct interaction between ASPACT and PACT RNAs (Figure 6(e)). Interestingly, unlike the splicing factor, Malat1, ASPACT presumably hybridizes with the mature form of PACT RNA, as PACT-encoded introns were not detectable in the ASPACT pull-down (data not shown). Based on our observations, one potential functional outcome of this inter-transcript interaction could be the spatial preservation of PACT mRNA in the nucleus. However, whether and how ASPACT mediates this spatial control by recruiting or sterically hindering proteins related to nucleocytoplasmic trafficking or nuclear retention remains unknown, and additional systematic interactomic and/or functional screens are required. Interestingly, despite this change in subcellular distribution, we found via ribosome nascent chain-mRNA analysis that ASPACT exerts no effect on the polysomal association of PACT mRNA, thus excluding translational regulation as a possible alternative mechanism underlying ASPACT’s action (Supplementary Fig. S7). Finally, given that ASPACT was found to also affect the expression of MDA5 and LGP2 mRNAs (Figure 7(a) and Supplementary Fig. S6A), it remains formally possible that ASPACT could also target additional dsRNA sensors in a capacity that is not related to PACT.
Funding Statement
This work was supported by the Chang Gung Memorial Hospital [BMRP960]; Chang Gung Memorial Hospital [BMRPF45]; Chang Gung Memorial Hospital, Linkou [CMRPD1F0443]; Chang Gung Memorial Hospital, Linkou [CMRPD1H0371]; Chang Gung Memorial Hospital, Linkou [CMRPD1F0573]; Chang Gung Memorial Hospital, Linkou [CMRPD1H0261]; Chang Gung Memorial Hospital, Linkou [CMRPD1H0022]; Ministry of Science and Technology, Taiwan [107-2320-B-182-042-MY 3]; Ministry of Science and Technology, Taiwan [105-2314-B-182-061-MY4]; Ministry of Science and Technology, Taiwan [108-3017-F-182-001]; Ministry of Science and Technology, Taiwan [106-2320-B-182-035-MY3].
Acknowledgments
We are grateful to members of the BC-MT laboratory for critical reading of the article and important discussions. This work was supported by grants from the Ministry of Science and Technology of Taiwan (MOST106–2320-B-182–035-MY3 to HL; MOST105-2314-B-182-061-MY4 and MOST107-2320-B-182-042-MY3 to BC-MT), Chang Gung Memorial Hospital (CMRPD1F0573, CMRPD1H0371, and BMRPF45 to HL; CMRPD1F0443, CMRPD1H0022, CMRPD1H0261, and BMRP960 to BC-MT), and the Ministry of Education of Taiwan. This work was also financially supported by the Research Center for Emerging Viral Infections from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) in Taiwan and the Ministry of Science and Technology (MOST), Taiwan (MOST108-3017-F-182-001).
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here
References
- [1].Ivashkiv LB, Donlin LT.. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14:36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Diamond MS, Farzan M. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat Rev Immunol. 2013;13:46–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Kok KH, Lui PY, Ng MH, et al. The double-stranded RNA-binding protein PACT functions as a cellular activator of RIG-I to facilitate innate antiviral response. Cell Host Microbe. 2011;9:299–309. [DOI] [PubMed] [Google Scholar]
- [4].Kowalinski E, Lunardi T, McCarthy AA, et al. Structural basis for the activation of innate immune pattern-recognition receptor RIG-I by viral RNA. Cell. 2011;147:423–435. [DOI] [PubMed] [Google Scholar]
- [5].Lui PY, Wong LR, Ho TH, et al. PACT facilitates RNA-induced activation of MDA5 by promoting MDA5 oligomerization. J Immunol. 2017;199:1846–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Miyamoto M, Komuro A. PACT is required for MDA5-mediated immunoresponses triggered by cardiovirus infection via interaction with LGP2. Biochem Biophys Res Commun. 2017;494:227–233. [DOI] [PubMed] [Google Scholar]
- [7].Li S, Peters GA, Ding K, et al. Molecular basis for PKR activation by PACT or dsRNA. Proc Natl Acad Sci U S A. 2006;103:10005–10010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Patel CV, Handy I, Goldsmith T, et al. PACT, a stress-modulated cellular activator of interferon-induced double-stranded RNA-activated protein kinase, PKR. J Biol Chem. 2000;275:37993–37998. [DOI] [PubMed] [Google Scholar]
- [9].Vaughn LS, Bragg DC, Sharma N, et al. Altered activation of protein kinase PKR and enhanced apoptosis in dystonia cells carrying a mutation in PKR activator protein PACT. J Biol Chem. 2015;290:22543–22557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Heward JA, Lindsay MA. Long non-coding RNAs in the regulation of the immune response. Trends Immunol. 2014;35:408–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Geisler S, Coller J. RNA in unexpected places: long non-coding RNA functions in diverse cellular contexts. Nat Rev Mol Cell Biol. 2013;14:699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Carpenter S, Aiello D, Atianand MK, et al. A long noncoding RNA mediates both activation and repression of immune response genes. Science. 2013;341:789–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Li Z, Chao TC, Chang KY, et al. The long noncoding RNA THRIL regulates TNFalpha expression through its interaction with hnRNPL. Proc Natl Acad Sci U S A. 2014;111:1002–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Villegas VE, Zaphiropoulos PG. Neighboring gene regulation by antisense long non-coding RNAs. Int J Mol Sci. 2015;16:3251–3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Rashid F, Shah A, Shan G. Long non-coding RNAs in the cytoplasm. Genomics Proteomics Bioinformatics. 2016;14:73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sun Q, Hao Q, Prasanth KV. Nuclear long noncoding RNAs: key regulators of gene expression. Trends Genet. 2017;34:142–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dykes IM, Emanueli C. Transcriptional and post-transcriptional gene regulation by long non-coding RNA. Genomics Proteomics Bioinformatics. 2017;15:177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hsieh CL, Liu H, Huang Y, et al. ADAR1 deaminase contributes to scheduled skeletal myogenesis progression via stage-specific functions. Cell Death Differ. 2014;21:707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yang CC, Liu H, Chen SL, et al. Epigenetic silencing of myogenic gene program by Myb-binding protein 1a suppresses myogenesis. Embo J. 2012;31:1739–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Dolken L. High resolution gene expression profiling of RNA synthesis, processing, and decay by metabolic labeling of newly transcribed RNA using 4-thiouridine. Methods Mol Biol. 2013;1064:91–100. [DOI] [PubMed] [Google Scholar]
- [21].Raj A, van den Bogaard P, Rifkin SA, et al. Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods. 2008;5:877–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Chu C, Qu K, Zhong FL, et al. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell. 2011;44:667–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Engreitz JM, Sirokman K, McDonel P, et al. RNA-RNA interactions enable specific targeting of noncoding RNAs to nascent Pre-mRNAs and chromatin sites. Cell. 2014;159:188–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wang T, Cui Y, Jin J, et al. Translating mRNAs strongly correlate to proteins in a multivariate manner and their translation ratios are phenotype specific. Nucleic Acids Res. 2013;41:4743–4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kwissa M, Nakaya HI, Onlamoon N, et al. Dengue virus infection induces expansion of a CD14(+)CD16(+) monocyte population that stimulates plasmablast differentiation. Cell Host Microbe. 2014;16:115–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gerlach RL, Camp JV, Chu YK, et al. Early host responses of seasonal and pandemic influenza A viruses in primary well-differentiated human lung epithelial cells. PLoS One. 2013;8:e78912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kang YJ, Yang DC, Kong L, et al. CPC2: a fast and accurate coding potential calculator based on sequence intrinsic features. Nucleic Acids Res. 2017;45:W12–W16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nat Rev Genet. 2016;17:47–62. [DOI] [PubMed] [Google Scholar]
- [29].Yap KL, Li S, Munoz-Cabello AM, et al. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol Cell. 2010;38:662–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Vance KW, Ponting CP. Transcriptional regulatory functions of nuclear long noncoding RNAs. Trends Genet. 2014;30:348–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kopp F, Mendell JT, Classification F. Experimental dissection of long noncoding RNAs. Cell. 2018;172:393–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Xu TP, Wang WY, Ma P, et al. Upregulation of the long noncoding RNA FOXD2-AS1 promotes carcinogenesis by epigenetically silencing EphB3 through EZH2 and LSD1, and predicts poor prognosis in gastric cancer. Oncogene. 2018. [DOI] [PubMed] [Google Scholar]
- [33].Tollervey JR, Curk T, Rogelj B, et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci. 2011;14:452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Luthra P, Ramanan P, Mire CE, et al. Mutual antagonism between the Ebola virus VP35 protein and the RIG-I activator PACT determines infection outcome. Cell Host Microbe. 2013;14:74–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Jiang R, Tang J, Chen Y, et al. The long noncoding RNA lnc-EGFR stimulates T-regulatory cells differentiation thus promoting hepatocellular carcinoma immune evasion. Nat Commun. 2017;8:15129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lin C, Wang Y, Wang Y, et al. Transcriptional and posttranscriptional regulation of HOXA13 by lncRNA HOTTIP facilitates tumorigenesis and metastasis in esophageal squamous carcinoma cells. Oncogene. 2017;36:5392–5406. [DOI] [PubMed] [Google Scholar]
- [37].Lee HY, Zhou K, Smith AM, et al. Differential roles of human Dicer-binding proteins TRBP and PACT in small RNA processing. Nucleic Acids Res. 2013;41:6568–6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Heyam A, Coupland CE, Degut C, et al. Conserved asymmetry underpins homodimerization of Dicer-associated double-stranded RNA-binding proteins. Nucleic Acids Res. 2017;45:12577–12584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Daniels SM, Gatignol A. The multiple functions of TRBP, at the hub of cell responses to viruses, stress, and cancer. Microbiol Mol Biol Rev. 2012;76:652–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Daher A, Laraki G, Singh M, et al. TRBP control of PACT-induced phosphorylation of protein kinase R is reversed by stress. Mol Cell Biol. 2009;29:254–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yoon JH, Abdelmohsen K, Kim J, et al. Scaffold function of long non-coding RNA HOTAIR in protein ubiquitination. Nat Commun. 2013;4:2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Marchese FP, Raimondi I, Huarte M. The multidimensional mechanisms of long noncoding RNA function. Genome Biol. 2017;18:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Yoon JH, Abdelmohsen K, Gorospe M. Posttranscriptional gene regulation by long noncoding RNA. J Mol Biol. 2013;425:3723–3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Yamazaki T, Souquere S, Chujo T, et al. Functional domains of NEAT1 architectural lncRNA induce paraspeckle assembly through phase separation. Mol Cell. 2018;70:1038–1053 e1037. [DOI] [PubMed] [Google Scholar]
- [45].Malakar P, Shilo A, Mogilevsky A, et al. Long noncoding RNA MALAT1 promotes hepatocellular carcinoma development by SRSF1 upregulation and mTOR activation. Cancer Res. 2017;77:1155–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.