

ABSTRACT
Argonaute (Ago) proteins are of key importance in many cellular processes. In eukaryotes, Ago can induce translational repression followed by deadenylation and degradation of mRNA molecules through base pairing of microRNAs (miRNAs) with a complementary target on a mRNA sequence. In bacteria, Ago eliminates foreign DNA through base pairing of siDNA (small interfering DNA) with a target on a DNA sequence. Effective targeting activities of Ago require fast recognition of the cognate target sequence among numerous off-target sites. Other target search proteins such as transcription factors (TFs) are known to rely on facilitated diffusion for this goal, but it is undetermined to what extent these small nucleic acid-guided proteins utilize this mechanism. Here, we review recent single-molecule studies on Ago target search. We discuss the consequences of the recent findings on the search mechanism. Furthermore, we discuss the open standing research questions that need to be addressed for a complete picture of facilitated target search by small nucleic acids.
Introduction
Sequence-specific recognition of nucleic acids by proteins is of great importance in cellular development and gene regulation [1]. Since the discovery of regulatory proteins that target specific DNA sequences, it was questioned how these proteins are able to recognize DNA targets among numerous other sequences in a fast yet specific manner. The most intensively studied target search protein is the E. coli lac repressor. For this protein, an extraordinary high binding rate has been observed of 1010 M−1 s−1. This binding rate is a factor of 100 faster than what theory would predict for collisions driven by three-dimensional (3D) diffusion (Einstein-Smoluchowski limit) [2,3]. While this result puzzled many for years, Berg et al. devised a theoretical framework that introduced the facilitated diffusion mechanism: the protein diffuses in 3D before binding non-specifically to a DNA strand, after which the protein diffuses laterally in one dimension (1D) along the strand to find its target [4]. As the dimensionality of the problem has been partially reduced from three dimensions to one dimension, a higher association rate is expected through this mechanism.
Since their seminal work, a new field surrounding target search has been developed. Interesting theoretical predictions have been obtained, such as the speed-stability paradox and the optimal partitioning of the different diffusional modes [5,6]. Experimentally, restriction enzymes such as EcoRV have been studied through a biochemical assay, where the binding rate of the protein was measured as the function of the overall length of the DNA [7]. Non-specific sequences around a target site act here as an antenna, by providing an initial binding site for the protein from solution. After binding to the non-specific sequence, the protein would move laterally to the target site. Thereby, observation of an increased binding rate for a longer DNA construct corresponds well with the facilitated diffusion model.
Biochemical studies rely heavily on theoretical assumptions as they can only measure average binding kinetics. Single-molecule methods provide an elegant solution as they visualize the different kinetic steps directly [8]. In recent years, single-molecule experimental groups have been able to observe with improving spatiotemporal resolution the target search mechanism of transcription factors [9], DNA repair proteins [10,11], zinc finger nucleases [12], TALEN [13] and the homologous recombination protein RecA [14,15]. Interestingly, some proteins have been found to make use of facilitated diffusion, while others seem to make use of 3D diffusion only. What determines whether a protein moves laterally is not known at this point. Certain considerations such as the type of substrate, biological function and necessity of timely regulation are likely factors that have to be taken into account.
More recently, target recognition for certain proteins was found to be mediated by small nucleic acid molecules. Small RNA molecules, which are loaded into proteins such as Argonaute (Ago) [16–18] and Clustered Regularly Interspaced Palindromic Repeats (CRISPR)-associated proteins [19,20], are utilized as a guide for the recognition of complementary sequences. These nucleic acid-guided proteins are expected to use a different search mechanism than transcription factors or restriction enzymes, the latters of which rely on interrogation of existing features of the DNA grooves. This is because there are no constraints on the sequence identity of the guide and therefore any sequence can be targeted. As a result of that, the amount of possible targets is greatly expanded, and the task of finding the right target in a timely manner becomes more complex.
Here we summarize the findings of recent single-molecule studies on Ago-mediated target search in the context of the theoretical framework. We provide a perspective of facilitated diffusion in target search with respect to its biological function.
Mechanisms of facilitated diffusion
In the context of facilitated diffusion, Berg et al. have proposed several mechanisms which are conducive for target search (Figure 1) [4]. Fundamentally, all modes of target search are driven by thermal energy, and therefore random motion is key in the searching process. The interactions between protein and non-specific nucleic acid sequences are governed through electrostatics, and the ions that screen the negatively charged nucleic acids (counterions) play an important role. For clarity’s sake, here are the definitions that we employ in our review (Table 1):
Figure 1.
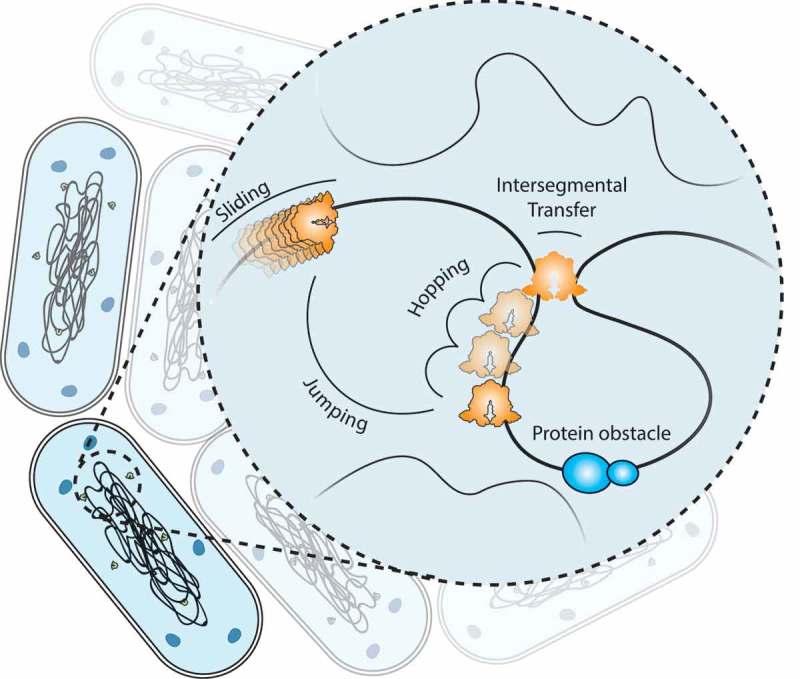
Target search mechanisms inside a cell.
Facilitated diffusion consists of cycles of non-specific binding to the DNA strand after three-dimensional (3D) search. This is followed by lateral diffusive motions along the DNA strand: (1) sliding, characterized by tight interactions with the strand. (2) Hopping consists of short dissociations away from the strand, however the movement is correlated along the strand. (3) Intersegmental transfer allows a protein with multiple binding sites to momentarily bind first to one than the other strand in a hand-over process. Lastly (4) jumping or 3D-diffusion allows the protein to diffuse in an uncorrelated manner to new DNA sites.
Table 1.
Description of different mechanisms of facilitated diffusion.
| Name | Description |
|---|---|
| Jumps/3D search | The protein explores the space in the cytosol or solution through 3D Brownian motion. Non-specific binding is followed by the protein dissociating. Subsequent binding to other sites occurs in an uncorrelated manner. |
| Sliding | The protein binds non-specifically to DNA/RNA and undergoes a movement which is characterized by a tight interaction with the nucleic acid molecule. The protein stays associated to the substrate at all times. In the words of Berg, no net displacement of counterions takes place here and therefore the time spent on non-specific substrate is not affected by a change in the ionic concentration in the surroundings. |
| Hopping | This mode is characterized by micro-dissociations from the DNA/RNA strand. While the method of diffusion is similar to 3D search, the difference is that the movements are correlated along the contour of the strand. Contrary to sliding, not every base is scanned in the effectively covered distance. Since the protein dissociates momentarily from the strand, condensation of counterions is allowed to occur. Hence the time spent on non-specific DNA is expected to decrease by increased ionic strength. |
| Intersegmental transfer | The protein with multiple binding sites is bound to one strand. In a hand-over process, the protein can be momentarily bound to two strands through interaction with its binding site, after which it moves to the other strand. |
Single-molecule techniques
How are we able to distinguish between these processes that happen on a nanometre length scale at millisecond timescales? In single-molecule target search studies, DNA–protein interactions are visualized through DNA curtains [21], flow stretch assays, Förster Resonance Energy Transfer (FRET) [22], single cell imaging [23] and force spectroscopy methods such as magnetic tweezers, optical tweezers, and atomic force spectroscopy [24,25].
These techniques provide great information on the kinetics of individual molecules. However, technical limitations could withhold one from observing the full dynamics. For example, many camera-based single-molecule fluorescence studies rely on the transmission of a fluorescence signal through the imaging system onto the pixels of the camera. The point-like fluorescent signals are mapped on the pixels of the camera as a spread-out image called the point spread function (PSF) of ~200 nm width [26], giving rise to an uncertainty in position. Since the shape of the PSF is known, one can still estimate the true position of the particle. But the finite amount of photons emitted from the fluorescent object – which is limited by photobleaching – determines the accuracy in position: the more photons are collected from a static source, the more accurate the estimation of its true position will be. As the protein of interest will not be static during target search but will undergo many fast movements, the camera needs to be fast enough to capture these dynamics. Most camera-based approaches collect fluorescent light at an acquisition frequency of 10–100 ms time resolution. Therefore, this frame rate and the number of photons collected during each time bin define the time resolution at which one can probe protein DNA interactions. For a more detailed introduction on this, we refer the reader to the many reviews available in the field [22,26].
The method of flow stretch assays, namely DNA curtains, provides the most intuitive visualization of target search. By labelling the protein of interest with a fluorescent probe, one is able to track the position of the protein on the DNA strand in presence of roadblocks and co-factors [10,21,27] (Figure 2(a)). From the position of the protein at different timepoints, one can directly observe whether lateral diffusion takes place (Figure 2(b)), and if so, derive the effective diffusion coefficient. The distinction between sliding and hopping is made by changing the strength of the ionic solution. As stated in Table 1, during sliding no net displacement of counterions takes place, and therefore a change in diffusion coefficient is not expected. In contrast, during hopping, proteins are expected to diffuse faster along the strand at a higher salt concentration, since less time is spent on each non-specific binding site on a strand.
Figure 2.
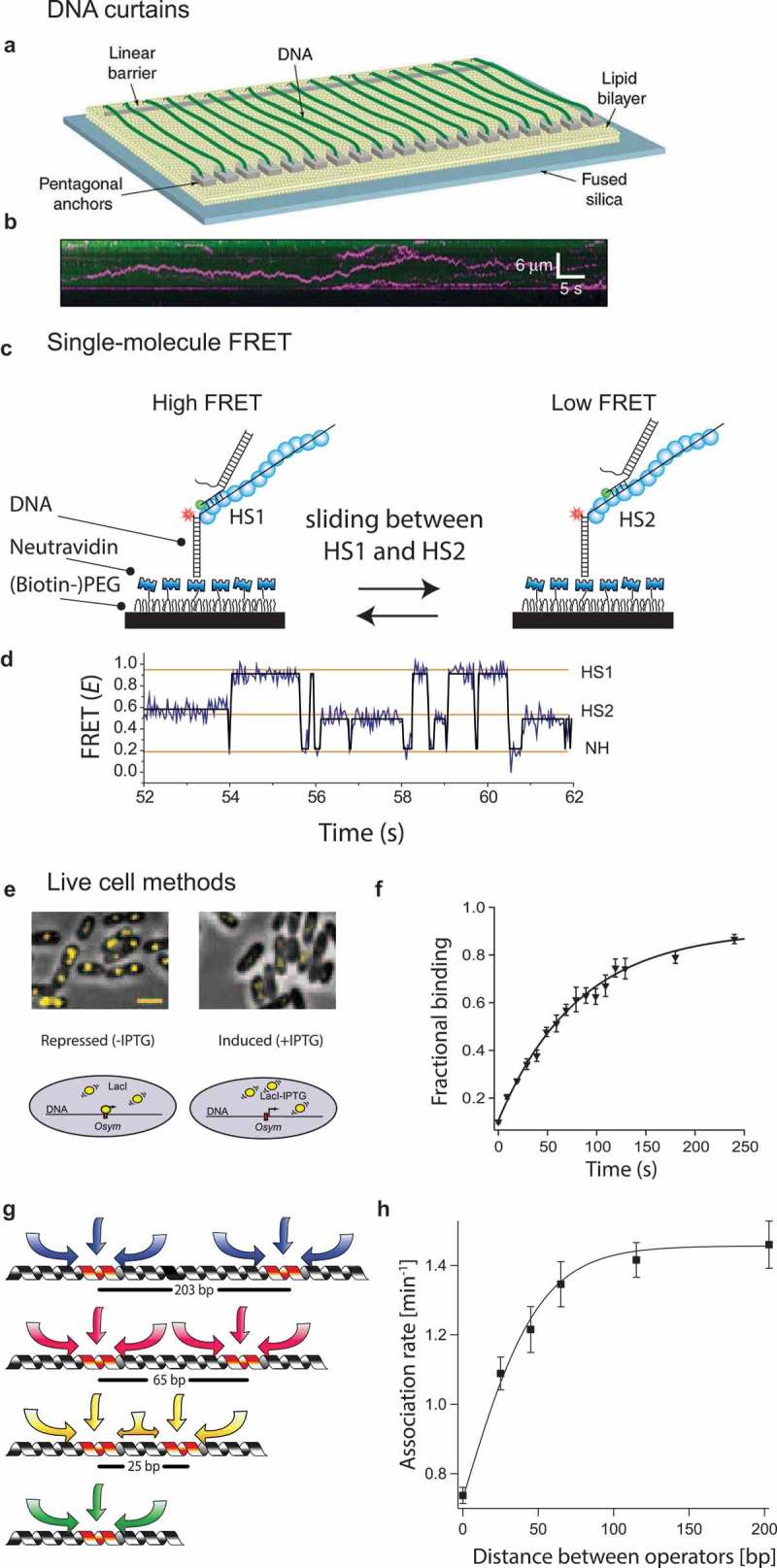
Fluorescence-based single-molecule techniques for studying target search.
(a) Diagram of the nanofabricated DNA curtain device that contains a barrier against lipid diffusion for stretching the DNA. Here, flow is used to stretch the DNA strand. On the other side, a pentagonal structure is used to anchor the other end of the strand [10]. As a result of this, hundreds of DNA strands are aligned in parallel and can be imaged simultaneously. (b) The DNA curtains are visualized through YOYO-1 dye staining. A fluorescent probe (pink) is attached to the protein of interest, and the position (vertical axis) of said protein is tracked in time (horizontal axis). By imaging both the DNA strand and the fluorescent probe, one can visualize how it travels along the DNA strand [10]. (c) Single-molecule FRET assay showing a RecA filament (blue) containing two homology sites. Recognition of homology site 1 (HS1) or homology site 2 (HS2) results in the appearance of a high FRET state and an intermediate FRET state, respectively, [14]. (d) Single-molecule time trace showing FRET for an immobilized ssDNA with two identical homology sequences HS1 and HS2. Docking of double-stranded DNA at a location outside a FRET sensitive regime results in a low FRET (NH) state [14]. (e) Single operator binding assay used by the Elf lab [9]. (Top) Overlays of E. coli cells in phase contrast and with fluorescently labelled LacI (yellow). In absence of IPTG (left), LacI is able to bind to the single operator site LacOsym, resulting in a diffraction-limited spot. In the presence of IPTG (300 μM) (right), LacI is unable to bind due to the competition with IPTG and diffuses too rapidly resulting in diffusional smears [9]. (f) Graph plotting the fraction of stable LacI binding vs the time after removal of the IPTG [9]. (g) The mean sliding length is determined by placing two identical targets in varying distances. If the mean sliding length of said targets overlaps, the LacI protein will effectively only sense one target, resulting in a decreased association rate [9]. (h) Rate of binding plotted against the inter-target distance [9]. Permission has been obtained for the above figures. Copyright 2010 Nature.
This technique has been able to uncover the nature of target search for a wide variety of proteins [10,15,28]. It provides for a great in-depth characterization of target search on a large length scale interrogation of sequences. However, due to the large size of the PSF and the thermal fluctuations affecting the position of the DNA strands, observation of proteins on DNA is generally limited to a resolution of ~250 bp. As it cannot be ruled out that lateral movements take place within their observations [15,29], other high-resolution techniques should be used complementarily.
Single-molecule Förster Resonance Energy Transfer (smFRET) provides high spatiotemporal resolution. FRET is an energy transfer process between two fluorophores, where due to dipole–dipole interactions, energy from a donor fluorophore is transferred to an acceptor fluorophore if they are within a few nanometres. The FRET efficiency E, (the ratio IA/(IA+ID), where IA is the intensity of acceptor signals and ID is that of donor signals) is given by E = 1/(1+(R/R0)6, where R0 is the characteristic distance of the dye pair (Förster radius) and is typically a few nanometres. A change of dye-pair distance results in a measurable change in the ratio of intensities of donor and acceptor fluorophore. A seminal smFRET study that investigated target search was on the RecA protein [14], where two identical homology sites were placed on a DNA construct (Figure 2(c)). The design was such that binding to one homology site resulted in a higher FRET efficiency compared to binding the other homology site. The rationale behind it was that while FRET provides high spatiotemporal resolution (~nm at 0.1 s timescale), the dynamics of both 3D and lateral diffusion were expected to occur on a much faster timescale (millisecond timescale). Through the use of local energy traps, one could momentarily trap the RecA nucleofilament at the sites and characterize the nature of their interactions (Figure 2(c) bottom).
While most in vitro techniques allow one to probe the mechanisms, it is important to know what the effect would be of the presence of cellular proteins and the crowding environment in physiological conditions [30]. Live cell imaging allows one to study single-molecule-facilitated diffusion inside a living cell. The first study of single-molecule live cell imaging was performed on a transcription factor, the lac repressor, LacI, which acts on the operator of lac genes [9]. Binding of LacI to the operator site prevents expression of the lac operon genes that metabolize lactose. However, by adding Isopropyl-β-D-1-thiogalactopyranoside (IPTG), a molecular reagent that binds to the lac repressor, one can prevent LacI from binding to the operator site. Removing IPTG from the solution allows LacI to bind once more to the operator site.
The authors of this study used fluorescently labelled LacI to study target search. In the presence of IPTG, the LacI repressor cannot bind to the target site (Figure 2(d)). By measuring the association rate after removing the IPTG inducer, they could measure the average time of a single LacI molecule to find its target. The unbound molecules diffuse too fast to be recorded while bound molecules a stable signal in time and space (Figure 2(e)). When two targets are placed close to each other, the two targets will appear as one target if the distance between two targets is smaller than the mean sliding distance (Figure 2(f)). At distances longer than ~50 bp, the targets were perceived as independent targets, but at distances shorter than ~50 bp, the association rate was comparable to single target association rate kinetics (Figure 2(g)). Additionally, to find out whether LacI was able to bypass protein roadblocks, a TetR protein was bound next to one of the targets. In the presence of TetR, the association rate was significantly affected indicating that LacI is not able to bypass roadblocks through sliding only.
As a whole, the aforementioned studies show that single-molecule methods provide understanding in the molecular processes that govern facilitated diffusion. Similar methods may provide key insights for Ago-mediated target search.
Architecture and mechanism of Ago
RNA silencing
Eukaryotic Agos are the key proteins in a process termed RNA silencing where small RNAs inhibit gene translation and induce deadenylation and degradation by targeting complementary mRNA molecules. Two types of small RNAs are central in gene silencing: microRNA (miRNA) and small interfering RNA (siRNA). MiRNAs only require partial base pairing as a first step in gene silencing whereas siRNA molecules have a perfect complementarity to their target RNA, after which Ago slices the mRNA target. MicroRNA originates from endogenous transcribed RNAs, and siRNAs are typically from exogenous double-stranded RNAs. The biogenesis of miRNA and siRNA is beyond the scope of this review. For a detailed description of these processes, we refer to [31,32]. Below we discuss briefly the silencing pathway involving miRNA.
The Ago and the guide RNA together form the core targeting element of the assembly, known as the RNA induced silencing complex (RISC). After binding to the target sequence, this complex initiates gene silencing through translational repression and mRNA decay. In the case of the latter, Ago recruits the scaffolding protein GW182 (TNCR6A/B/C in humans). In human Argonaute 2 (hAGO2), this occurs through the interaction of the tryptophan binding pocket on Ago with the scaffold protein [33–35]. The scaffold protein in turn bridges interactions with downstream effectors such as CCR4-NOT which recruit the translational repressor and decapping activator DDX6 [35–39]. While the effect of gene regulation is often shown to be subtle [40], gene silencing has been shown to be essential in gene regulation and cell development [41,42]. Dysregulation of miRNA is a cause of cancer in many cases [43,44], with miRNAs acting as tumour suppressors [45]. Besides the insight in gene regulatory processes that these findings provide, the potential of using these small RNAs for therapeutics applications has also been harvested, with the first miRNA therapeutics reaching phase I and II in clinical trials [46].
The architecture of argonaute
Ago is a nucleoprotein that consists of a bilobed structure where the guide strand is partially located inside a cleft (Figure 3(a)) [47–49]. Structural, biochemical and computational data suggest that the guide miRNA is divided into five functional domains: the 5ʹ anchor, the seed, the mid-region, the 3ʹ supplementary region and the 3ʹ end region (Figure 3(b)) [50]. Functionally speaking, the seed is arranged such that nucleotides 2–8 (from the 5ʹ end) are arranged in an A-form-like geometry, and this region is responsible for stable target binding [40]. The pre-arranged helix allows Ago to bind without entropic costs to the target strand, providing stable binding once the complementary sequence is recognized. Computational analysis has shown that many functional/canonical miRNA targets only require seed pairing. However, matches in the supplementary region appear to be of importance as well for RNA silencing [51,52]. Additionally, once binding to the seed has occurred, the guide-target base pairing propagates to the 3ʹ end of the guide if complementarity exists between guide and target [53].
Figure 3.
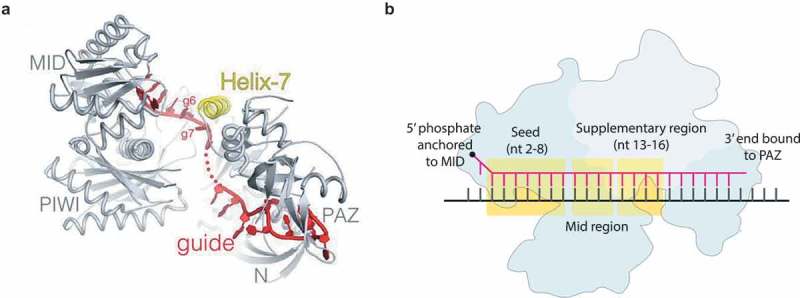
Argonaute undergoes a conformational change as required by the speed-stability paradox.
(a) Cartoon representation of hAgo2-miRNA complex based on the wild-type hAgo2 structure (PDB ID: 4OLA). The different domains of hAgo2 (grey) are indicated. The miRNA guide is indicated in red, while the helix-7 fragment is indicated in yellow [66]. (B) Schematic drawing of miRNA, which is divided into five regions: the 5ʹ end anchored in a MID binding pocket, the Seed region (nt 2–8), the Mid-region where cleavage occurs on the opposing strand between nucleotide 10 and 11, the Supplementary region which supplements the binding of the seed domain. Lastly, the PAZ domain anchors the 3ʹ end of the guide [66]. Permission has been obtained for the above figures. Copyright 2017 John Wiley and Sons.
Prokaryotic Agos
While the role of Ago in eukaryotes is well characterized, the role of prokaryotic Agos (pAgos) remained elusive for years. In recent years, it has been proposed that pAgos are involved in host defence rather than gene regulation, as some pAgos utilize DNA or RNA guide to target single-stranded DNA (ssDNA) instead of RNA [54,55]. For instance, reduced plasmid transformation efficiency and intracellular plasmid concentrations have been observed for Thermus thermophilus (TtAgo), Pyrococcus furiosus (PfAgo) and Methanocaldococcus jannaschii (MjAgo) [56–59]. pAgos were found to target ssDNA in vitro, not dsDNA and currently it is not known how this process takes place in vivo as ssDNA is rarely present there. In the case of thermophilic prokaryotes (such as TtAgo), it is thought that local melting of AT-rich regions would contribute to accessibility for effective cleavage of the target. At the same time, ssDNA may very well be the target of pAgos that cleave ssDNA. Recently, ssDNA viruses have been found to be abundant in certain environments such as seawater, fresh water, soil [60–62]. Here, a defence system that exclusively targets ssDNA would be highly beneficial for the survival of the host.
Two-mode search of Ago
In order to find a target in a timely manner, a protein needs to bind non-specifically to nucleic acids, search rapidly for the associated target, and bind strongly to a target site. The search of targets needs to happen in both a fast and specific manner, yet this cannot happen at the same time, since specificity imposes as a rule that the energy barriers become too high for lateral diffusion [5]. The paradox is solved by assuming that the protein has two states of binding: a search mode, in which the energy barriers it encounters are minimal, enabling smooth lateral diffusion, and a recognition mode, which is characterized by high affinity and slow diffusion (Figure 4(a)). The encountered energy landscape in the recognition mode (Erecognition in Figure 4(b)) is on average higher than the mean energy level landscape in the search mode (Esearch), so that the protein spends more time in the search mode than in the recognition mode. A key idea here is that the energy landscape the protein encounters during the search mode is well-correlated with the energy landscape in the recognition mode and that an energy gap exists between the two modes. So the deep minimum in the recognition mode would correspond to a more shallow minimum in the search mode. When the protein is trapped inside one of these energy levels during the search mode, it will likely transition into a recognition mode. Effectively, this results in a pre-selection at the minima of the binding landscape, since here it’s more likely that a transition will happen from the search mode to the recognition mode. Even if the conformational transition rate from the search mode to the recognition mode is low [63], a gain in search speed is still predicted [6].
Figure 4.
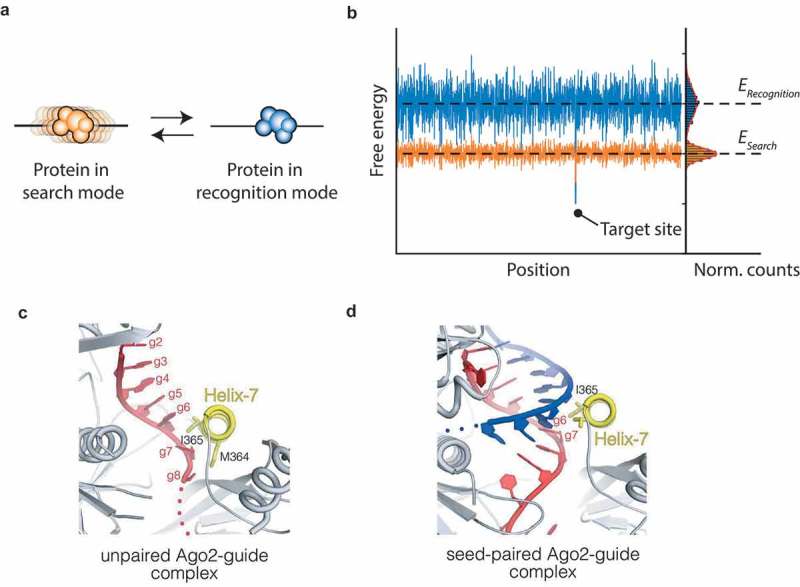
Argonaute undergoes a conformational change, as required by the speed-stability paradox.
(a) The speed stability paradox posited by Slutsky et al. In the search mode (orange), the protein is able to diffuse laterally without encountering significant energy barriers. Once it encounters a potential target site (indicated by the deeper energy level in the binding energy landscape right), it may switch to a recognition mode (blue). In this mode, the specificity of the protein is increased and the protein will not diffuse. (b) The energy landscape as encountered by a protein in the search mode (orange) and a protein in the recognition mode (blue). In the search mode, the landscape that the protein encounters is shallow and the variance in energy levels is small. The deeper energy levels in the recognition mode prevent the protein from diffusing laterally. At a potential target site, it’s more likely for the protein to switch from a search mode to a recognition mode, since the energy level of the former is higher than the latter. (c) Close-up view of the seed region shows the pre-formed helix of nt 2–6. Helix-7 disrupts base stacking by intercalating itself between g6 and g7 [66]. (PDB ID: 4OLA) (d) Close-up view of the seed region in the event of fully base-paired seed. Helix-7 undergoes a conformational change here, docking into the minor groove of the seed-paired complex [66]. (PDB ID: 4W5O). Permission has been obtained for the above figures. Copyright 2017 John Wiley and Sons.
Experimental evidence of two-state search
Theoretically, it was posited that Ago target search may be mediated by such binding modes [64] and there is structural evidence to support this. Since the seed of the guide strand is pre-arranged in a helical manner (Figure 3(a)), this suggests that initial target recognition and perhaps also initial target search commences at this region. From the crystal structure of hAgo2, it has been posited that guide nucleotide 2–5 of hAgo2 is used for initial recognition of target sites [65]. A kink introduced between g6-g7 breaks the A-form structure of the helix, and this is caused by insertion of the residues Ile-365 and Met-364 of α-helix-7 between the bases of 6 and 7 (Figure 4(c)) [65]. Base pairing beyond nucleotide 7 requires a conformational change of helix-7 to accommodate. At the same time, this conformational change stabilizes the base pairing of nucleotides 6 and 7 of the guide (Figure 4(d)) after which base pairing of additional nucleotides can also take place [66].
Fluorescence single-molecule in vitro studies have given further proof of this two-state binding mode. In all the single molecule studies mentioned here, the target strand with fluorophore is immobilized on the surface. A second fluorophore is then attached to the miRNA guide which is loaded into the core-RISC. Through single-molecule FRET or colocalization of both dyes, the binding and unbinding rate can be studied for various base pairing sequences (Figure 5). In the case of smFRET, either a donor fluorophore or an acceptor fluorophore can be immobilized on the surface [67,68] (Figures 5(a, d)). The guide strand that contains the complementary dye required for FRET is loaded into Ago. High FRET indicates binding of the Ago-guide complex to the target site, since the dyes must be in close proximity for energy transfer to occur. From the length of the high FRET signal, one can obtain the dissociation rate (Figure 5(b)). Likewise, by measuring the time between introducing Ago-guide complex to the chamber and first binding to a target, one can obtain the binding rate (Figure 5(c)). Additionally, in the case of Salomon’s assay (Figure 5(g)), an RNA target was tagged with 17 dyes attached to the 3ʹ end, so that cleavage events can be readily distinguished from photobleaching [69].
Figure 5.
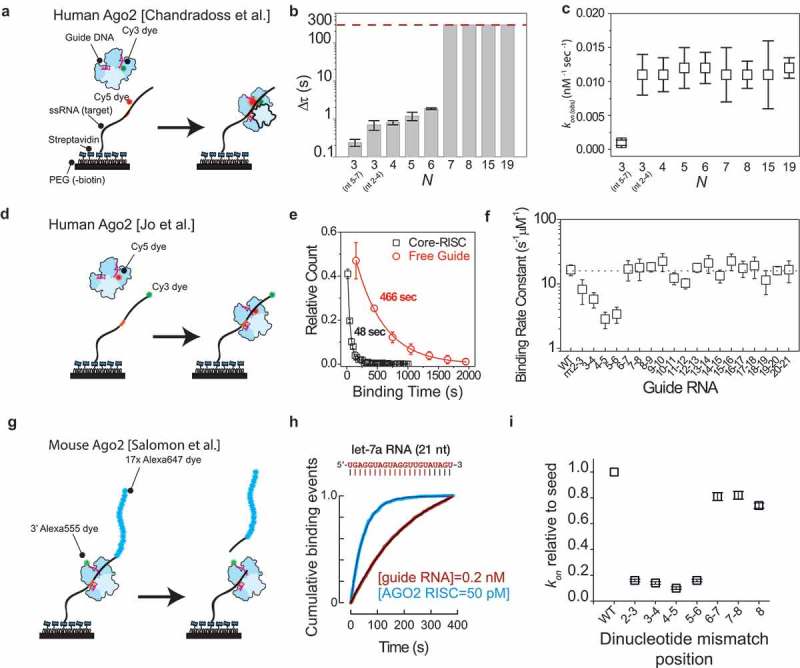
Studies of the initial search and recognition of AGO2 through single-molecule fluorescence techniques.
(a) Schematic drawing of the experimental assay used by Chandradoss et al. [67]. Target RNA is immobilized on the surface through biotin-streptavidin conjugation. The target RNA is labelled with an acceptor-dye (Cy5) while the donor dye (Cy3) is located on the miRNA guide. In absence of hAgo2-RISC binding to the strand, no signal is observed. Once it binds to the minimal target motif, the proximity of donor and acceptor induces Forster resonance energy transfer (FRET), resulting in a high acceptor signal. The duration of the high acceptor signal can be used to estimate the dwell time Δt. (b) The dwelltime Δt plotted versus the number N of seed-paired bases. The dashed line indicates the upper limit of dwell time estimation [67]. (c) The binding rate plotted for various values of N of base pairs [67]. (d) Schematic drawing of the single-molecule assay by Jo et al. where through FRET of the Cy5 and the Cy3 dye the binding can be ascertained [68]. (e) Dwell time distribution of core-RISC (black squares) and free let7 miRNA (red circles) fitted with a single-exponential decay [68]. (f) Binding rate plotted versus dinucleotide-mismatched guide RNAs [68]. (g) Schematic assay for Salomon et al. [69]. Seventeen Alexa647 dyes are attached to the 5ʹ end of the target RNA to distinguish cleavage from photobleaching. (h) Comparison of target binding rates (kon) for 21 nt sequences for let-7a RNA and miR-21 RNA. Inset shows a representative intensity trace [69]. (i) Comparison of target binding rate for let-7a sequences with complete seed-matched pairing or seed-matched pairing bearing dinucleotide mismatches [69]. Permission has been obtained for the above figures. Copyright 2015 Elsevier.
From single molecule fluorescence assays, it was found that Ago accelerated the binding rate greatly, compared to binding with guide RNA only, for both hAgo2 and mouse Ago2 (Figure 5(e, h)). The rationale is that pre-arranging the seed would result in a higher probability of successful binding to the target strand, hence effectively increasing the binding rate of the complex. Dinucleotide mismatches in the seed were found to be detrimental to the binding rate for both mouse Ago2 and hAgo2 (Figure 5(f, i)) [68–70]. Disruption of base pairing in the seed would also often result in quick dissociation of mouse RISC [69], showing that seed-pairing is essential for target recognition. Furthermore, it was shown that hAgo2 utilizes a part of the seed (nt 2–4) for initial target search, since shrinking the seed pairing from 2–8 to 2–4 did not change the binding rate (Figure 5(c)) [67]. Varying the seed pairing from 2–4 to 5–7 did reduce the binding rate significantly [67,69], indicating that the 2–4 seed motif is essential for initial recognition.
Transition to recognition mode
Beyond initial recognition, the transition from an initial search mode to a recognition mode of hAgo2 was hinted at through single-molecule FRET. As stated before, the crystal structure of hAgo2 suggests that a conformational change of helix-7 is required for stable base pairing beyond N = 6. In single-molecule FRET, through extending complementarity in base pairing from nucleotide 2–4 to 2–19, a significant increase in binding time was observed between N = 6 and N = 7 (Figure 5(b)). This suggests that a conformational change took place that the strengthened seed-target interactions, as is required for the speed-stability paradox for fast and specific targeting [67]. Furthermore, mutants, in which either the helix-7 is lacking or where two helix-7 residues are mutated, have shown to, respectively, decrease both the on-rate and the off-rate, indicating that it fulfils additional functionality by rapidly dismissing off-targets while the search itself can be accelerated by prepaying the entropic costs of arranging the guide in a helical manner [66].
In short, the first single-molecule fluorescence studies provided key insights in recognition through visualization of transient kinetics that bulk and static methods could not provide.
Target search of Ago
Hidden rapid dynamics in target search
How does one envision target search of Ago to take place? The minimal RISC complex would be expected to bind non-specifically to a random position on the target strand, before moving to the target site. This would result in a gradual change in the FRET value. However, the single molecule data of the aforementioned in vitro assays contained only stable traces. The absence of such signature indicates that either the complex binds directly to target from solution, or more likely, that the dynamics take place on a timescale that occur much faster than the acquisition time of 100 ms. In order to characterize the dynamics, one would need to resort to stronger energy traps. The first study that investigated the nature of Ago target search was inspired by the RecA target search assay [14], where two identical strong binding sites were placed on an RNA construct (Figure 6(a)) [67]. Similarly to the RecA assay, binding to one sub-seed target site was designed to result in a higher FRET efficiency compared to binding to the other site. If only one target site was present on the RNA strand, only one FRET state could be observed. If two targets are present on one strand, one did not only observe the addition of binding signatures with a lower FRET state, but also a shuttling signature: the transition from one binding site to the other without interruption (Figure 6(b)) [67]
Figure 6.
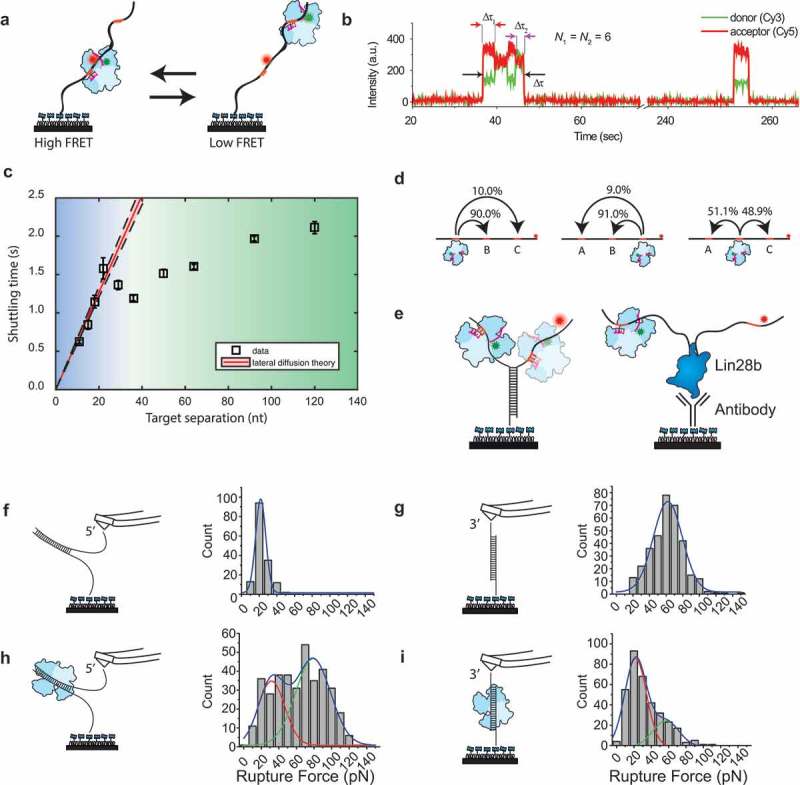
Single-molecule experiments on ago target search.
(a) Tandem target assay used by Chandradoss et al. [67]. The RNA target contains two identical targets, where the Cy5 dye is placed closer to the bottom target. During the experiments, hAgo2 shuttles between the two target sites. (b) A typical shuttling trace for N = 6 (nt 2–7). Binding to one target brings the donor dye of the guide closer to the acceptor dye, resulting in a higher FRET value. This way, binding to one target can be readily distinguished from binding to the distal target [67]. (c) Shuttling time of CbAgo plotted versus the distance between two identical targets. The red line indicates a lateral diffusion fit derived from a minimal kinetic model. The data diverge from the kinetic theory beyond the blue region and follows a different trend in the green region [72]. (d) Triple target assay used to visualize whether CbAgo skips over the middle target when translocating from site A to site C. Percentages indicate the relative amount of transitions from an initial state to a final state versus the total number of transitions [72]. (e) Left: a Y-fork construct that prevents CbAgo to slide from the target on one strand to the other. Right: Lin28 is immobilized as a protein blockade [72]. (f) DNA unzipping with AFM in absence of TtAgo. The AFM was retracted with a velocity of 200 nm/s. The rupture force is plotted with a Gaussian probability distribution fitted on top of it (blue) centred at 21.7 pN [75]. (g) DNA shearing with AFM in absence of TtAgo, with a fit centred at 62.0 pN [75]. (h) DNA unzipping with AFM in presence of TtAgo, fitted with a peak centred at 31.7 pN and 78.9 pN [75]. (i) DNA shearing with AFM in presence of TtAgo fitted with a peak centred at 21.9 pN and 56.8 pN [75]. Permission has been obtained for the above figures. Copyright 2018 American Chemical Society.
Since it appeared that the Ago complex did not dissociate from the strand during translocation from one target to the other target, it was suggested that lateral diffusion took place between the two targets [67]. This has also been observed for a pAgo from the mesophilic bacterium Clostridium butyricum (CbAgo) [71,72]. Kinetic modelling suggested that if lateral diffusion would place, a linear relation would exist between distance and shuttling time. This is what was observed, supporting the lateral diffusion model (Figure 6(c)).
A point worthy of note here is that the residence times for individual targets were found to be much shorter than the residence time of a single target [72]. This indicates that apparent stable binding traces contain rapid excursions away from and back to the target site. These events are typically too transient to be captured by camera-based fluorescence microscopy. In the presence of a second target site however, the probability of recapture is affected, since the protein now has a non-zero probability to end up in the second trap and hence the lifetime per individual state decreases. In general, this subtle behaviour could extend to other protein–DNA interactions as well, which in turn would necessitate a re-evaluation stable data traces of current and older single molecule literature. Additional proof for lateral diffusion of pAgo was shown by adding a third target between the two targets (Figure 6(d)). Over 90% of the translocations that occurred between the outermost targets happened through the middle site.
Physiological relevance
Inside the cell, crowding affects both binding and dissociation rate [30]. Mimicking the cellular conditions through the use of PEG showed that the occurrence of shuttling increased with more crowding agent, up to 90% of the time for 10% PEG [67]. It would therefore be expected that in vivo crowding would increase the affinity of Ago with the target strand.
In cellular environments, proteins and secondary structures are likely to form road blockades, potentially blocking proteins that diffuse laterally along the strand [9]. Remarkably, Ago from the mesophile Clostridium butyricum was not found to be impeded in any way by secondary structures or protein barriers placed between the two target sites (Figure 6(i)) [72]. This suggests that sliding is not a dominant mechanism in the target search of Ago and also that the target search process itself is robust: the time to search for a target does not depend on the presence of secondary structures. Structural and functional similarity between eAgo and pAgo could suggest that this behaviour is not only conserved for pAgo, but for also for eAgos. If that is the case, then this process may allow the protein to search along RNA without being hindered by complex secondary structures of RNA.
Ago glides and makes intersegmental jumps while searching for its target
The characterization of target search through pAgo also uncovered an additional mechanism: intersegmental jumps. This mode was first characterized for EcoRV restriction enzyme [73] and is known to allow proteins to quickly travel to different segments of the same (or different) nucleic acid molecule to search for its complementary sequence without compromising for redundant lateral diffusive behaviour [5,6,74].
It was found that the shuttling time did not increase linearly for larger distances between targets (Figure 6(c)) suggesting that an additional mechanism was at work. Since ssDNA is quite flexible, it was thought that this long-range mechanism would be akin to intersegmental jumps. To test this out, the authors took a construct from the region where it behaved according to lateral diffusion model and a construct from the green region. The two shuttling times of the two constructs were compared at varying ionic strength [72]. If the lateral movements of Ago consisted of hopping, this would become apparent in the sensitivity to changing salt concentration. Likewise, during intersegmental jumps Ago also dissociates from the strand, hence it also is sensitive to a change in ionic strength. Surprisingly, it was found that the shuttling rate for the short construct did not exhibit the salt-dependent behaviour that one would expect for hopping, rather only the long construct did. Since Ago is able to bypass secondary structures and proteins with ease, this indicates that during the lateral diffusion process, few counterions are expunged while it is still able to glide over secondary structures.
Evidence of lateral diffusion through atomic force microscopy
Additional proof for lateral diffusion has been found through the use of atomic force microscopy (AFM) with TtAgo [75]. Two different kinds of constructs were designed: in the first design the dsDNA was unzipped (Figure 6(c)), whereas in the second design the dsDNA base-pairing was ruptured through a shearing force (Figure 6(d)). Intuitively one would expect unzipping to be more favourable than shearing, and this was indeed what was found. More interestingly, through comparison of the required force both in absence and presence of TtAgo, it was found that the Ago actively remodels the energy landscape such that the required shearing force was lowered in presence of Ago (Figure 6(e)) while the required unzipping force was increased (Figure 6(f)) [75]. In summary, Ago seems to prefer a lateral movement over an unzipping movement, as the energetic barrier for a lateral movement is lowered. These results provide support to the idea that one-dimensional diffusion along the strand is more efficient than three-dimensional diffusion.
Is Ago target search time optimal?
The first fluorescence studies have uncovered the mechanism of interactions of the Ago-guide complex with a target strand. However, quantitative understanding is still lacking at this point. One of the outstanding questions is how the gliding and intersegmental jumps are temporally divided for Ago to maximize its target search speed. While theoretical studies have predicted that the optimal search time for a target consists of equal time spent in 1D and 3D diffusion [5], this has proven to be not always the case. Some proteins have been found to have different distributions in vivo [76–78], where a tenfold or hundredfold more time is spent being bound to nucleic acid strands rather than being diffusing in solution. It will be of interest to see why this is the case for some proteins and whether Ago is one of them. We speculate that, when the tandem target assay is used, the mean first passage time between two targets could be used to infer to what extend Ago partitions its search process into lateral diffusion and intersegmental jumps.
Next, it is not always well understood how redundancy and efficiency in target recognition are coupled to each other. Intrinsically, lateral target search is redundant by its very diffusive nature. As an example, the human oxoguanine DNA glycosylase 1 was found to have an effective diffusion coefficient of 5 × 106 bp2/s [28]. The barrier in energy landscape along the DNA sequence was on the order of kBT, indicating that lateral diffusion is not limited by the roughness [5]. Persistent contact with DNA indicates that DNA segments are scanned multiple times resulting in an inefficient mechanism. However, the redundancy in target search may be to compensate for the inefficiency of target recognition (i.e. multiple attempts are necessary to recognize the target, but with sliding/hopping the target is bypassed multiple times due to 1D diffusion.). It has been observed that the loosely interacting mode of Ago-guide complex with the nucleic acid strand could potentially allow the protein to skip over bases, implying that multiple scanning attempts might be needed for the protein to recognize a cognate target site.
How physiologically relevant is the loosely associated searching mode of Ago? The ability of Ago proteins to bypass secondary structures without any impedance suggests that the search behaviour itself is robust. Secondary structures in mRNA occur frequently in vivo [79–82], providing many functional elements essential for regulation of various post-transcriptional mechanisms [83–86]. In the 3ʹ UTR, where many miRNA target sites are located [40], the RNA structure is more structured than in coding regions [80]. Unimpeded target search allows Ago to efficiently scan for target sites without it being trapped between dsRNA segments. Future measurements in vivo should point out to what extentthese weak interactions help Ago speed up its searching process.
CRISPR-associated proteins use a different strategy
To what extent would the target search mechanism of Ago be conserved among other small RNA-guided systems? The most widely known class of nucleic acid-guided endonucleases are derived from the CRISPR immunity system. As an immunity system against genetic elements from bacteriophages and plasmids, prokaryotes insert short fragments of the foreign DNA into their own genome, the so-called CRISPR array [87,88]. Spacers from this array are transcribed and processed into short RNA fragments, termed CRISPR RNAs (crRNAs). CRISPR-associated (Cas) proteins utilize the crRNAs to target foreign complementary DNA targets, called protospacers, after which cleavage occurs, either by recruitment of other Cas proteins or by direct slicing by the targeting proteins themselves [88]. The protospacer sequences targeted by Cas proteins are flanked by a short sequence motif, referred to as the protospacer adjacent motif (PAM) [88]. Cas proteins use these motifs to distinguish foreign DNA from endogenous DNA.
Differences between Cas proteins and Ago
The first single-molecule studies on Cas proteins, such as the E. coli Cascade complex [89] and Streptococcus pyogenes Cas9 (spCas9) [90], suggest that the contribution of lateral diffusion of Cas proteins is much smaller than what has been observed of restriction enzymes, repair enzymes and other proteins that interact with double-stranded DNA [89–91]. This difference is not unexpected since the price to be paid for the flexibility of programming the guide, is that the double-stranded region of DNA needs to be opened up by the Cas proteins for interrogation, which is energetically costly.
Eukaryotic Ago proteins do not suffer from this energetic cost as they target single-stranded RNA. In this sense, prokaryotic Agos seem to have more in common with CRISPR proteins, as they are thought to be involved as well in host defence and require high fidelity recognition of dsDNA [55]. However, the prokaryotic Agos studied so far seem to interact only with single-stranded DNA, and it is currently unknown how they access double-stranded binding sites in vivo. In some prokaryotes, the genes of pAgos seem to cluster with genes encoding for nucleases and helicases [92] and it has been posited that pAgo-associated helicases could potentially assist the Ago to unwind the double-stranded segments, thereby allowing the endonuclease to access the single-stranded DNA molecule for interrogation. At the same time, recent findings indicate that the presence of ssDNA viruses are abundant in certain environments. Here, targeting ssDNA through facilitated diffusion would be highly beneficial for pAgo.
Reduction in search complexity helps for 3D target search
In contrast to Ago target search, the target search of Cas protein such as Cas9 requires melting of DNA. It would be unfeasible for Cas proteins to randomly melt DNA sites all over the genome for interrogation. The aforementioned Cas-proteins have been found to interact longer with PAM sequences compared to non-specific sequences, which allows the Cas protein to have enough time to interrogate the sequence [89,90]. Thus, the PAM sequence recognition, which is critically important for the distinction between self and non-self targeting, also serves as an extra pre-selection step for target recognition. Dividing the recognition process in two or multiple step manner not only circumvents the speed-stability problem in facilitated diffusion but also reduces the time needed to find the target through 3D diffusion only.
Here the term search complexity, a concept, which was introduced for RecA homology search [15], may account for the observed dominance of 3D diffusion for some small RNA-guided proteins [89,90,93]. The rationale is that short sequences tend to occur more often inside the genome while longer sequences will have fewer exact matches. A protein relying on initial recognition of a short sequence will only have to search through a small part of the genome, and the rest would be ignored.
For Cas proteins, the PAM sequence acts as a pre-selection mechanism filtering out the other sequences that are of no interest. For example, for a three-nucleotide sequence, such as a PAM site, roughly 10% of the genome would need to be interrogated, spending virtually no time on the remaining 90% sequences. In contrast, in the extreme case of a one-step recognition process with the full target of 22 nt, the target search would be inefficient. The probability of 22-nucleotide target occurring randomly would be extremely small: only once every 1.7 * 1013 base pairs. This is obviously advantageous in terms of uniqueness. However, the protein has to reject almost every site after opening up the strand for interrogation, and the time it takes to unbind from intermediate base pairing would make this strategy unfeasible [94]. This is again reminiscent of the speed-stability paradox posited by Mirny. The more specific the search will be, the more time the protein will spend on an off-target site. So, the key assumption of reduction in search complexity is that search complexity is proportional to the time it takes for the target to be localized. This assumes that differences in search complexity are coupled to differences in kinetics between short and long sequence base pairing between guide and target. In order for this concept to work, dissociation rates of short pairing sequences have to be substantially higher than those of long pairing sequences, which has been observed for Ago and Cas9 proteins [67,94,95].
Structural considerations of different target search proteins
What are the common structural features that determine whether a protein will slide or hop during lateral diffusion? It is assumed that some proteins maintain tight contact with the DNA/RNA substrate relying on their structural features whereas other proteins use only transient interactions. For example, the MutS repair enzyme consists of a clamp-like structure [96]. Once it is in a closed clamp-like conformation, this allows the protein to slide with high processivity along the dsDNA strand. Similarly, the Lac repressor is able to interrogate the grooves of the dsDNA thoroughly, through its structural form that facilitates interaction with the dsDNA groove [97]. It may be therefore unsurprising that some nucleic acid-guided endonucleases like Ago or Cas do not use sliding, since their structure does not facilitate these interactions with the DNA substrate in the first place.
Interestingly, the degree of lateral diffusion seems to vary from organism to organism even though they are orthologs. Cascade from T. fusca shows lateral diffusive behaviour [98] whereas E. Coli Cascade does not [89] since the latter complex lacks a positive patch in the Cse1 subunit. The question remains why this would be conserved in T. fusca but not in E. coli if they are functionally similar and require the same rapid response in case of a foreign genomic invasion. Is lateral diffusion always required for a protein to find its target in a timely manner? Remarkably, Cas12a shows lateral diffusion over microns at a remarkable coefficient of a micron per second, while it does not enclose the double-stranded DNA [99]. Crystal structures may give us hints towards understanding the nature of lateral diffusion. Single molecule kinetics in conjunction with molecular dynamics simulations may give us an idea on which common features are required.
Future perspectives on Ago target search
While much has been uncovered in the last decades about small RNA/DNA-mediated target search, there are still many long-standing questions of interest. The molecular nature of targeting through small RNAs has been uncovered to some extent, but the mechanistic picture is still incomplete.
For example, while it is now known that Ago and CRISPR proteins use lateral diffusion, it is not known what the effect of sequence on target search is. That is, to what degree is the facilitated diffusion reliant on the interaction between guide–target base pairing as opposed to an interaction between only the protein and target substrate? For some Cas proteins, which movement is driven by 3D diffusion, the prime determinant in target search would be the PAM recognition step. For Ago proteins, which rely on seed recognition, potential sub-seed-matching sequences could slowdown the search process while the absence of such sequences could speed it up. It is known that circular RNA (circRNA) can act as a sponge for miRNA, strongly suppressing miR-7 activity [100]. Cognate seed target sites were found to be responsible for this, but it is not known if and how shorter sub-seed sequences would influence the search dynamics. Perhaps other sequences have evolved over time to ensure temporal control of post-transcriptional gene-regulation by subtly tuning the search time. A high spatiotemporal single molecule technique is required in order to visualize the effect of sequence on diffusion at the length scale of dozens of base pairs.
So far, single-molecule Ago target search studies have not focused on the physiological environment of the cells such as the difference between eukaryotic cells and prokaryotic cells. For example, if a prokaryotic Ago is indeed involved in host defence against foreign genetic elements, is the protein then at all times present for surveillance? That seems unlikely: the probability for pAgo to encounter and target its own genome for chopping or slicing would be much higher than necessary and this would have fatal consequences. Besides the biological function, there is also the size difference between eukaryotic cells (~10–100 μm) and prokaryotic cells (~ μm). The cytoplasmic volume that Ago has to search through is orders of magnitude different, and this may affect the temporal distribution of 1D target search and 3D target search. Live cell studies, similar to the LacI studies of target search, are needed to validate and verify the questions here.
For eukaryotic Agos, the effect of secondary and tertiary structures of mRNA on target search remains largely unexplored. These structures often have physiological regulatory functions [86,101,102] and reliable prediction of RNA structures remains a challenge as in vivo and in silico analyses often differ [81,82]. It has been shown previously that RNA structures that base pair with the target completely abolish silencing [103], suggesting that RISC cannot overcome target inaccessibility by itself. However, miRNA binding sites have been uncovered in some viral sequences [104,105]. In addition, miR-159 target sites have also been shown to be reliant on the presence of stem loops, as disruption of adjacent stem loops seemed to attenuate miR159 targeting efficacy [106]. The functional role of secondary structures in the context of miRNA silencing is therefore far from understood.
Lastly, most studies have focussed on a minimal RISC, consisting of Ago and guide. However in vivo, mRNA silencing happens through translational repression and mRNA decay [107]. Several components in this silencing machinery have been uncovered [108,109], of which the scaffold protein GW182 (TNRC6 in mammals) family is the key component. This protein has been elusive in structural studies due to its intrinsically unstructured nature and therefore insight through biophysical studies has been limited. Recently, a link between the novel field of biological phase separations and RNAi has been uncovered through the molecular interactions of AGO and TNCR6. Promoted by multivalent interactions between glycine/tryptophan domain and the tryptophan binding pockets of hAGO2, condensation of miRISC was found to form in vitro and in vivo [110]. The finding suggests that phase separation of miRISC could contribute to repression of mRNA targets through sequestration of the targets inside the droplets. Currently, the biophysical properties of RISC condensates remain unexplored, and the nature of how these individual components of the condensate interact with each other is still an elusive question. Furthermore, until now Ago target search has been studied in an in vitro environment. How well would the results from the published studies translate to a phase-separated environment, where RISC exists in a high local concentration?
In conclusion, we have noted the recent leaps in knowledge of the kinetics of target search and target recognition of individual regulatory complexes. However, the biophysical mechanisms that govern the interplay of these slicing/silencing proteins and their degradation machinery have been largely unexplored. Future studies will allow us to paint a complete picture of targeting by small RNAs in a cellular context.
Funding Statement
C.J. was supported by Vidi (864.014.002) of the Netherlands Organization for Scientific research Nederlandse Organisatie voor Wetenschappelijk Onderzoek [864.14.002].
Disclosure of potential conflicts of interest
No potential conflict of interest was reported by the authors.
References
- [1].Pauli A, Rinn JL, Schier AF.. Non-coding RNAs as regulators of embryogenesis. Nat Rev Genet. 2011;12(2):136–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Riggs AD, Bourgeois S, Cohn M, The lac represser-operator interaction: III. Kinetic studies. J Mol Biol [Internet] 1970. November 14 [cited 2018 September27];53(3):401–417. Available from: https://www.sciencedirect.com/science/article/pii/0022283670900744?via%3Dihub [DOI] [PubMed] [Google Scholar]
- [3].Riggs AD, Suzuki H, Bourgeois S, Lac repressor-operator interaction: I. Equilibrium studies. J Mol Biol [Internet] 1970. February 28 [cited 2018 September27];48(1):67–83. Available from: https://www.sciencedirect.com/science/article/pii/0022283670902196?via%3Dihub [DOI] [PubMed] [Google Scholar]
- [4].Berg OG, Winter RB, von Hippel PH. Diffusion-driven mechanisms of protein translocation on nucleic acids. 1. Models and theory. Biochemistry. 1981;20(24):6929–6948. [DOI] [PubMed] [Google Scholar]
- [5].Slutsky M, Mirny LA. Kinetics of protein-DNA interaction: facilitated target location in sequence-dependent potential. Biophys J. 2004;87(6):4021–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Mirny L, Slutsky M, Wunderlich Z, et al. How a protein searches for its site on DNA: the mechanism of facilitated diffusion. J Phys A Math Theor [Internet] 2009;42(43):434013 Available from: http://stacks.iop.org/1751-8121/42/i=43/a=434013?key=crossref.c3b19cfa3ebb32a80370e612c091028f [Google Scholar]
- [7].Stanford NP, Szczelkun MD, Marko JF, et al. One- and three-dimensional pathways for proteins to reach specific DNA sites. Embo J. 2000;19(23):6546–6557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Shimamoto N. One-dimensional diffusion of proteins along DNA. J Biol Chem [Internet] 1999. May 28;274(22):15293–15296. Available from: http://www.jbc.org/lookup/doi/10.1074/jbc.274.22.15293 [DOI] [PubMed] [Google Scholar]
- [9].Hammar P, Leroy P, Mahmutovic A, et al. The lac repressor displays facilitated diffusion in living cells. Science [Internet] 2012. June 22;336(6088):1595–1598. Available from: http://science.sciencemag.org/content/sci/336/6088/1595.full.pdf [DOI] [PubMed] [Google Scholar]
- [10].Gorman J, Chowdhury A, Surtees JA, et al. Dynamic basis for one-dimensional DNA scanning by the mismatch repair complex Msh2-Msh6. Mol Cell. 2007;28(3):359–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gorman J, Plys AJ, Visnapuu M-L, et al. Visualizing one-dimensional diffusion of eukaryotic DNA repair factors along a chromatin lattice. Nat Struct Mol Biol [Internet] 2010;17(8):932–938. Available from: http://www.nature.com/doifinder/10.1038/nsmb.1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zandarashvili L, Esadze A, Vuzman D, et al. Balancing between affinity and speed in target DNA search by zinc-finger proteins via modulation of dynamic conformational ensemble. Proc Natl Acad Sci [Internet] 2015;112(37):E5142–9. Available from: http://www.pnas.org/lookup/doi/10.1073/pnas.1507726112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cuculis L, Abil Z, Zhao H, et al. Direct observation of TALE protein dynamics reveals a two-state search mechanism. Nat Commun [Internet] 2015;6:7277 Available from: http://www.nature.com/doifinder/10.1038/ncomms8277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ragunathan K, Liu C, Ha T. RecA filament sliding on DNA facilitates homology search. Elife. 2012;2012(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Qi Z, Redding S, Lee JY, et al. DNA sequence alignment by microhomology sampling during homologous recombination. Cell [Internet] 2015;160(5):856–869. DOI: 10.1016/j.cell.2015.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lau NC, Lim LP, Weinstein EG, et al. An abundant class of tiny RNAs with probable regulatory roles in Caenorhabditis elegans. Science [Internet] 2001. October 26;294(5543):858LP–862. Available from: http://science.sciencemag.org/content/294/5543/858.abstract [DOI] [PubMed] [Google Scholar]
- [17].Lagos-Quintana M, Rauhut R, Lendeckel W, et al. Identification of novel genes coding for small expressed RNAs. Science [Internet] 2001. October 26;294(5543):853LP–858. Available from: http://science.sciencemag.org/content/294/5543/853.abstract [DOI] [PubMed] [Google Scholar]
- [18].Lee RC, Ambros V. An extensive class of small RNAs in Caenorhabditis elegans. Science [Internet] 2001. October 26;294(5543):862LP–864. Available from: http://science.sciencemag.org/content/294/5543/862.abstract [DOI] [PubMed] [Google Scholar]
- [19].Jinek M, Chylinski K, Fonfara I, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science [Internet] 2012. August 17;337(6096):816LP–821. Available from: http://science.sciencemag.org/content/337/6096/816.abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Brouns SJJ, Jore MM, Lundgren M, et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science [Internet] 2008. August 15;321(5891):960LP–964. Available from: http://science.sciencemag.org/content/321/5891/960.abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Greene EC, Wind S, Fazio T, et al. DNA curtains for high-throughput single-molecule optical imaging. Methods Enzymol [Internet] 2010. January 1 [cited 2018 September27];472:293–315. Available from: https://www.sciencedirect.com/science/article/pii/S0076687910720061?via%3Dihub [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Joo C, Balci H, Ishitsuka Y, et al. Advances in single-molecule fluorescence methods for molecular biology. Annu Rev Biochem [Internet] 2008. June 1;77(1):51–76. DOI: 10.1146/annurev.biochem.77.070606.101543. [DOI] [PubMed] [Google Scholar]
- [23].Yu J. Single-molecule studies in live cells. Annu Rev Phys Chem [Internet] 2016. May 23;67(1):565–585. DOI: 10.1146/annurev-physchem-040215-112451. [DOI] [PubMed] [Google Scholar]
- [24].Neuman KC, Nagy A. Single-molecule force spectroscopy: optical tweezers, magnetic tweezers and atomic force microscopy. Nat Methods [Internet] 2008. May 29;5:491 DOI: 10.1038/nmeth.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dulin D, Lipfert J, Moolman MC, et al. Studying genomic processes at the single-molecule level: introducing the tools and applications. Nat Rev Genet [Internet] 2012. October 30;14:9 DOI: 10.1038/nrg3316. [DOI] [PubMed] [Google Scholar]
- [26].Fernández-Suárez M, AY Ting. Fluorescent probes for super-resolution imaging in living cells. Nat Rev Mol Cell Biol. 2008;9(12):929–943. [DOI] [PubMed] [Google Scholar]
- [27].Gorman J, Greene EC. Visualizing one-dimensional diffusion of proteins along DNA. Nat Struct Mol Biol. 2008;15(8):768–774. [DOI] [PubMed] [Google Scholar]
- [28].Blainey PC, van Oijen AM, Banerjee A, et al. A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. Proc Natl Acad Sci [Internet] 2006;103(15):5752–5757. Available from: http://www.pnas.org/cgi/doi/10.1073/pnas.0509723103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sternberg SH, LaFrance B, Kaplan M, et al. Conformational control of DNA target cleavage by CRISPR–cas9. Nature [Internet] 2015. October 28;527:110 DOI: 10.1038/nature15544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Li G-W, Berg OG, Elf J. Effects of macromolecular crowding and DNA looping on gene regulation kinetics. Nat Phys [Internet] 2009;5(4):294–297. DOI: 10.1038/nphys1222. [DOI] [Google Scholar]
- [31].Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol Internet]. 2014;15(8):509–524. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25027649. [DOI] [PubMed] [Google Scholar]
- [32].Carthew RW, Sontheimer EJ. Origins and mechanisms of miRNAs and siRNAs. Cell. 2009;136(4):642–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ding L, Spencer A, Morita K, et al. The developmental timing regulator AIN-1 Interacts with miRISCs and may target the Argonaute protein ALG-1 to cytoplasmic P bodies in C. elegans. Mol Cell [Internet] 2005. August 19 [cited 2018 September27];19(4):437–447. Available from: https://www.sciencedirect.com/science/article/pii/S1097276505014772?via%3Dihub [DOI] [PubMed] [Google Scholar]
- [34].Liu J, Rivas FV, Wohlschlegel J, et al. A role for the P-body component GW182 in microRNA function. Nat Cell Biol [Internet] 2005. November 13;7:1261 DOI: 10.1038/ncb1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Meister G, Landthaler M, Peters L, et al. Identification of novel Argonaute-associated proteins. Curr Biol [Internet] 2005. December 6 [cited 2018 September27];15(23):2149–2155. Available from: https://www.sciencedirect.com/science/article/pii/S0960982205013011?via%3Dihub [DOI] [PubMed] [Google Scholar]
- [36].Chekulaeva M, Mathys H, Zipprich JT, et al. miRNA repression involves GW182-mediated recruitment of CCR4–NOT through conserved W-containing motifs. Nat Struct Mol Biol [Internet] 2011. October 7;18:1218 DOI: 10.1038/nsmb.2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Braun JE, Huntzinger E, Fauser M, et al. GW182 proteins directly recruit cytoplasmic deadenylase complexes to miRNA targets. Mol Cell [Internet] 2011. October 7 [cited 2018 September28];44(1):120–133. Available from: https://www.sciencedirect.com/science/article/pii/S1097276511007246?via%3Dihub [DOI] [PubMed] [Google Scholar]
- [38].Behm-Ansmant I, Rehwinkel J, Doerks T, et al. mRNA degradation by miRNAs and GW182 requires both CCR4: NOT deadenylase and DCP1: DCP2 decapping complexes. Genes Dev. 2006;20:1885–1898 ST–MRNA degradation by miRNAs and GW1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Chen C-YA, Zheng D, Xia Z, et al. Ago–TNRC6 triggers microRNA-mediated decay by promoting two deadenylation steps. Nat Struct Mol Biol [Internet] 2009. October 18;16:1160 DOI: 10.1038/nsmb.1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Bernstein E, Kim SY, Carmell MA, et al. Dicer is essential for mouse development. Nat Genet. 2003;35(3):215–217. [DOI] [PubMed] [Google Scholar]
- [42].Chong MMW, Zhang G, Cheloufi S, et al. Canonical and alternate functions of the microRNA biogenesis machinery. Genes Dev. 2010;24(17):1951–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lin S, Gregory RI. MicroRNA biogenesis pathways in cancer. Nat Rev Cancer [Internet] 2015. May 22;15(6):321 DOI: 10.1038/nrc3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bracken CP, Scott HS, Goodall GJ. A network-biology perspective of microRNA function and dysfunction in cancer. Nat Rev Genet [Internet] 2016. October 31;17:719 DOI: 10.1038/nrg.2016.134. [DOI] [PubMed] [Google Scholar]
- [45].Takamizawa J, Konishi H, Yanagisawa K, et al. Reduced expression of the let-7 MicroRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res [Internet] 2004. June 1;64(11):3753LP–3756. Available from: http://cancerres.aacrjournals.org/content/64/11/3753.abstract [DOI] [PubMed] [Google Scholar]
- [46].Chakraborty C, Sharma AR, Sharma G, et al. Therapeutic miRNA and siRNA: moving from bench to clinic as next generation medicine. Mol Ther Nucleic Acids [Internet] 2017. September 15 [[cited 2018 September28]];8:132–143. Available from: https://www.sciencedirect.com/science/article/pii/S2162253117301907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wang Y, Sheng G, Juranek S, et al. Structure of the guide-strand-containing argonaute silencing complex. Nature [Internet] 2008;456(7219):209–213. DOI: 10.1038/nature07315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Nakanishi K, Weinberg DE, Bartel DP, et al. Structure of yeast Argonaute with guide RNA. Nature [Internet] 2012. May 18;486:368 DOI: 10.1038/nature11211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Schirle NT, MacRae IJ. The crystal structure of human Argonaute2. Science [Internet] 2012. May 25;336(6084):1037–1040. Available from: http://onlinelibrary.wiley.com/doi/10.1002/macp.1965.020830117/abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Wee LM, Flores-Jasso CF, Salomon WE, et al. Argonaute divides Its RNA guide into domains with distinct functions and RNA-binding properties. Cell. 2012;151(5):1055–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Agarwal V, Bell GW, Nam JW, et al. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4(AUGUST2015):1–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Grimson A, Farh KKH, Johnston WK, et al. MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol Cell. 2007;27(1):91–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Wang Y, Juranek S, Li H, et al. Nucleation, propagation and cleavage of target RNAs in ago silencing complexes. Nature [Internet] 2009;461(7265):754–761. Available from: http://www.ncbi.nlm.nih.gov/pubmed/19812667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Olovnikov I, Chan K, Sachidanandam R, et al. Bacterial Argonaute samples the transcriptome to identify foreign DNA. Mol Cell [Internet] 2013. September 12 [cited 2018 September28]; 51(5):594–605. DOI: 10.1016/j.molcel.2013.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Swarts DC, Jore MM, Westra ER, et al. DNA-guided DNA interference by a prokaryotic Argonaute. Nature [Internet] 2014;507(7491):258–261. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24531762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Swarts DC, Hegge JW, Hinojo I, et al. Argonaute of the archaeon pyrococcus furiosus is a DNA-guided nuclease that targets cognate DNA. Nucleic Acids Res. 2015;43(10):5120–5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Willkomm S, Oellig CA, Zander A, et al. Structural and mechanistic insights into an archaeal DNA-guided Argonaute protein. Nat Microbiol [Internet] 2017;2(March):17035 Available from: http://www.nature.com/articles/nmicrobiol201735 [DOI] [PubMed] [Google Scholar]
- [58].Zander A, Holzmeister P, Klose D, et al. Single-molecule FRET supports the two-state model of Argonaute action. RNA Biol [Internet] 2014;11(1):45–56. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3929424&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Zander A, Willkomm S, Ofer S, et al. Guide-independent DNA cleavage by archaeal Argonaute from methanocaldococcus jannaschii. Nat Microbiol [Internet] 2017;2(March):17034 Available from: http://www.nature.com/articles/nmicrobiol201734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Reavy B, Swanson MM, Cock PJA, et al. Distinct circular single-stranded DNA viruses exist in different soil types. Appl Environ Microbiol. 2015;81(12):3934–3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Labonté JM, Suttle CA. Previously unknown and highly divergent ssDNA viruses populate the oceans. Isme J. 2013;7(11):2169–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Yoshida M, Mochizuki T, Urayama SI, et al. Quantitative viral community DNA analysis reveals the dominance of single-stranded DNA viruses in offshore upper bathyal sediment from Tohoku, Japan. Front Microbiol. 2018;9(FEB):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kalodimos CG, Boelens R, Kaptein R. A residue-specific view of the association and dissociation pathway in protein-DNA recognition. Nat Struct Biol. 2002;9(3):193–197. [DOI] [PubMed] [Google Scholar]
- [64].Klein M, Chandradoss SD, Depken M, et al. Why Argonaute is needed to make microRNA target search fast and reliable. Semin Cell Dev Biol [Internet] 2017. May;65:20–28. Available from: https://linkinghub.elsevier.com/retrieve/pii/S1084952116301434 [DOI] [PubMed] [Google Scholar]
- [65].Schirle NT, Sheu-Gruttadauria J, MacRae IJ. Structural basis for microRNA targeting. Science [Internet] 2014;346(6209):608–613. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4313529&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Klum SM, Chandradoss SD, Schirle NT, et al. Helix‐7 in Argonaute2 shapes the microRNA seed region for rapid target recognition. Embo J [[Internet]]. 2017;e201796474 Available from: http://emboj.embopress.org/lookup/doi/10.15252/embj.201796474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Chandradoss SD, Schirle NT, Szczepaniak M, et al. A dynamic search process underlies MicroRNA targeting. Cell [Internet] 2015;162(1):96–107. DOI: 10.1016/j.cell.2015.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Jo MH, Shin S, Jung SR, et al. Human Argonaute 2 has diverse reaction pathways on target RNAs. Mol Cell [Internet] 2015;59(1):117–124. DOI: 10.1016/j.molcel.2015.04.027. [DOI] [PubMed] [Google Scholar]
- [69].Salomon WE, Jolly SM, Moore MJ, et al. Single-molecule imaging reveals that Argonaute reshapes the binding properties of its nucleic acid guides. Cell [Internet] 2015;162(1):84–95. DOI: 10.1016/j.cell.2015.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Yao C, Sasaki HM, Ueda T, et al. Single-molecule analysis of the target cleavage reaction by the drosophila RNAi enzyme complex. Mol Cell [Internet] 2015;59(1):125–132. DOI: 10.1016/j.molcel.2015.05.015. [DOI] [PubMed] [Google Scholar]
- [71].Hegge JW, Swarts DC, Chandradoss SD, et al. DNA-guided DNA cleavage at moderate temperatures by clostridium butyricum Argonaute. 2019. DOI: 10.1101/534206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Cui TJ, Klein M, Hegge JW, et al. Argonaute bypasses cellular obstacles without hindrance during target search. 2019. DOI: 10.1101/535575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].van Den Broek B, Lomholt MA, Kalisch S-MJ, et al. How DNA coiling enhances target localization by proteins. Proc Natl Acad Sci U S A [Internet] 2008;105(41):15738–15742. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2572962&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Kolomeisky AB. Physics of protein-DNA interactions: mechanisms of facilitated target search. Phys Chem Chem Phys. 2011;13(6):2088–2095. [DOI] [PubMed] [Google Scholar]
- [75].Song G, Chen H, Sheng G, et al. Argonaute facilitates the lateral diffusion of the guide along its target and prevents the guide from being pushed away by the ribosome. Biochemistry [Internet] 2018. April 17;57(15):2179–2183. DOI: 10.1021/acs.biochem.8b00213. [DOI] [PubMed] [Google Scholar]
- [76].Wang YM, Austin RH, Cox EC. Single molecule measurements of repressor protein 1D diffusion on DNA. Phys Rev Lett [Internet] 2006. July 27;97(4):48302 DOI: 10.1103/PhysRevLett.97.048302. [DOI] [PubMed] [Google Scholar]
- [77].Blainey PC, Luo G, Kou SC, et al. Nonspecifically bound proteins spin while diffusing along DNA. Nat Struct Mol Biol [Internet] 2009;16(12):1224–1229. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2889498&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Elf J, Li G-W, Xie XS. Probing transcription factor dynamics at the single-molecule level in a living cell. Science [Internet] 2007. May 25;316(5828):1191LP–1194. Available from: http://science.sciencemag.org/content/316/5828/1191.abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Ding Y, Tang Y, Kwok CK, et al. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature [Internet] 2014;505(7485):696–700. DOI: 10.1038/nature12756. [DOI] [PubMed] [Google Scholar]
- [80].Beaudoin J-D, Novoa EM, Vejnar CE, et al. Analyses of mRNA structure dynamics identify embryonic gene regulatory programs. Nat Struct Mol Biol. [Internet] 2018;1 Available from: http://www.nature.com/articles/s41594-018-0091-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Bevilacqua PC, Ritchey LE, Su Z, et al. Genome-wide analysis of RNA secondary structure. Annu Rev Genet [Internet] 2016;50(1):235–266. DOI: 10.1146/annurev-genet-120215-035034. [DOI] [PubMed] [Google Scholar]
- [82].Vandivier LE, Anderson SJ, Foley SW, et al. The conservation and function of RNA secondary structure in plants. Annu Rev Plant Biol [Internet] 2016;67(1):463–488. DOI: 10.1146/annurev-arplant-043015-111754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Warf MB, Berglund JA, Role of RNA structure in regulating pre-mRNA splicing. Trends Biochem Sci [Internet] 2010. March 1 [cited 2018 September28];35(3):169–178. Available from: https://www.sciencedirect.com/science/article/pii/S0968000409001960?via%3Dihub [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Martin KC, Ephrussi A. mRNA localization: gene expression in the spatial dimension. Cell [Internet] 2009. February 20 [cited 2018 September28];136(4):719–730. Available from: https://www.sciencedirect.com/science/article/pii/S0092867409001263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Ray PS, Jia J, Yao P, et al. A stress-responsive RNA switch regulates VEGFA expression. Nature [Internet] 2008. December 21;457:915 DOI: 10.1038/nature07598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Weingarten-Gabbay S, Elias-Kirma S, Nir R, et al. Systematic discovery of cap-independent translation sequences in human and viral genomes. Science [Internet] 2016. January 15;351(6270). Available from: http://science.sciencemag.org/content/351/6270/aad4939.abstract [DOI] [PubMed] [Google Scholar]
- [87].Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327(5962):167–170. [DOI] [PubMed] [Google Scholar]
- [88].Sorek R, Lawrence CM, Wiedenheft B. CRISPR-mediated adaptive immune systems in bacteria and archaea. Annu Rev Biochem [Internet] 2013;82(May):237–266. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23495939 [DOI] [PubMed] [Google Scholar]
- [89].Redding S, Sternberg SH, Marshall M, et al. Surveillance and processing of foreign DNA by the escherichia coli CRISPR-cas system. Cell [Internet] 2015;163(4):854–865. DOI: 10.1016/j.cell.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Sternberg SH, Redding S, Jinek M, et al. DNA interrogation by the CRISPR RNA-guided endonuclease cas9. Nature [Internet] 2014;507(7490):62–67. DOI: 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Globyte V, Lee SH, Bae T, et al. CRISPR/cas9 searches for a protospacer adjacent motif by lateral diffusion. Embo J [Internet] 2018. December 20;e99466 Available from: https://www.biorxiv.org/content/early/2018/02/13/264879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Swarts DC, Makarova K, Wang Y, et al. The evolutionary journey of Argonaute proteins. Nat Struct Mol Biol [Internet] 2014;21(9):743–753. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4691850&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Jones DL, Leroy P, Unoson C, et al. Kinetics of dCas9 target search in Escherichia coli. Science. 2017;357(6358):1420–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Singh D, Sternberg SH, Fei J, et al. Real-time observation of DNA recognition and rejection by the RNA-guided endonuclease cas9. Nat Commun. 2016;7:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Jung SR, Kim E, Shin S, et al. Mechanisms of the binding/dissociation Acceleration of the target–guide interaction by thermus thermophilus Argonaute. Bull Korean Chem Soc. 2018;39(2):167–173. [Google Scholar]
- [96].Obmolova G, Ban C, Hsieh P, et al. Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature [Internet] 2000. October 12;407:703 DOI: 10.1038/35037509. [DOI] [PubMed] [Google Scholar]
- [97].Schumacher MA, Choi KY, Zalkin H, et al. Crystal structure of LacI member, PurR, bound to DNA: minor groove binding by alpha helices. Science [Internet] 1994. November 4;266(5186):763LP–770. Available from: http://science.sciencemag.org/content/266/5186/763.abstract [DOI] [PubMed] [Google Scholar]
- [98].Dillard KE, Brown MW, Johnson NV, et al. Assembly and translocation of a CRISPR-cas primed acquisition complex. Cell [Internet] 2018;175(4):934–946. DOI: 10.1016/j.cell.2018.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Jeon Y, Choi YH, Jang Y, et al. Direct observation of DNA target searching and cleavage by CRISPR-cas12a. Nat Commun [Internet] 2018;9(1). DOI: 10.1038/s41467-018-05245-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Hansen TB, Jensen TI, Clausen BH, et al. Natural RNA circles function as efficient microRNA sponges. Nature [Internet] 2013;495(7441):384–388. Available from: http://www.nature.com/doifinder/10.1038/nature11993 [DOI] [PubMed] [Google Scholar]
- [101].Mandal M, Breaker RR. Gene regulation by riboswitches. Nat Rev Mol Cell Biol. 2004;5(6):451–463. [DOI] [PubMed] [Google Scholar]
- [102].Kedde M, Van Kouwenhove M, Zwart W, et al. A pumilio-induced RNA structure switch in p27-3′2 UTR controls miR-221 and miR-222 accessibility. Nat Cell Biol [Internet] 2010;12(10):1014–1020. DOI: 10.1038/ncb2105. [DOI] [PubMed] [Google Scholar]
- [103].Ameres SL, Martinez J, Schroeder R. Molecular basis for target RNA recognition and cleavage by human RISC. Cell. 2007;130(1):101–112. [DOI] [PubMed] [Google Scholar]
- [104].Scheel TKH, Luna JM, Liniger M, et al. A broad RNA virus survey reveals both miRNA dependence and functional sequestration. Cell Host Microbe [Internet] 2016. March 9 [cited 2018 September28];19(3):409–423. Available from: https://www.sciencedirect.com/science/article/pii/S1931312816300488?via%3Dihub [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Jopling CL, Schütz S, Sarnow P, Position-dependent function for a tandem MicroRNA miR-122-binding site located in the hepatitis C virus RNA genome. Cell Host Microbe [Internet] 2008. July 17 [cited 2018 September28];4(1):77–85. Available from: https://www.sciencedirect.com/science/article/pii/S193131280800173X?via%3Dihub [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Zheng Z, Reichel M, Deveson I, et al. Target RNA secondary structure is a major determinant of miR159 efficacy. Plant Physiol [Internet] 2017. July 1;174(3):1764LP–1778. Available from: http://www.plantphysiol.org/content/174/3/1764.abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Gebert LFR, MacRae IJ. Regulation of microRNA function in animals. Nat Rev Mol Cell Biol [Internet]. 2018;(Mid) Available from: http://www.nature.com/articles/s41580-018-0045-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Nuñez JK, Kranzusch PJ, Noeske J, et al. Cas1-cas2 complex formation mediates spacer acquisition during CRISPR-cas adaptive immunity. Nat Struct Mol Biol [Internet] 2014;21(6):528–534. Available from: http://www.nature.com/nsmb/journal/v21/n6/pdf/nsmb.2820.pdf [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet [Internet] 2015;16(7):421–433. DOI: 10.1038/nrg3965. [DOI] [PubMed] [Google Scholar]
- [110].Sheu-Gruttadauria J, MacRae IJ. Phase transitions in the assembly and function of human miRISC. Cell [Internet] 2018;173(4):946–957.e16. DOI: 10.1016/j.cell.2018.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]