

ABSTRACT
Evidence from yeast and mammals argues the existence of cross-talk between transcription and mRNA decay. Stabilization of transcripts upon depletion of mRNA decay factors generally leads to no changes in mRNA abundance, attributing this to decreased transcription rates. We show that knockdown of human XRN1, CNOT6 and ETF1 genes in HepG2 cells led to significant alteration in stability of specific mRNAs, alterations in half-life were inversely associated with transcription rates, mostly not resulting in changes in abundance. We demonstrate the existence of the gene expression buffering mechanism in human cells that responds to both transcript stabilization and destabilization to maintain mRNA abundance via altered transcription rates and may involve translation. We propose that this buffering may hold novel cancer therapeutic targets.
KEYWORDS: Gene expression, human cells, cancer, coupling, mRNA decay, transcription, translation, regulation
Introduction
Maintenance of mRNA abundance is vital for regulation of eukaryotic gene expression. It has long been viewed that transcription in the nucleus and mRNA decay in the cytoplasm determine the steady-state mRNA level as independent processes by merely supplying and degrading transcripts in their respective compartments. Over the last decade, pioneering work in yeast and mammals has revealed that these two processes, which are separated spatially and temporally are tightly coupled and can communicate with each other [1,2]. More specifically, the steady-state level of mRNA is highly resistant to perturbations in decay rate, as the rate of transcription can be adjusted to maintain the mRNA steady-state level. The mRNA decay machinery has been shown to play a direct role in buffering the abundance of transcripts by regulating its levels via coordinating both degradation of mRNA and the rate of transcription [1].
In S. cerevisiae, the fate of mRNA in the cytoplasm has been shown to be pre-determined in the nucleus [1]. Growing lines of evidence strongly suggest that the fate of the mRNA in the cytoplasm can be determined in a promoter-dependent manner [3–5]. Promoter sequences influence the loading of nascent mRNA with markers [3]. These markers could be proteins that contribute to the mRNP formation (e.g. a protein kinase Dbf2p) or potentially non-coding RNAs. It has been demonstrated that switching the promoter sequence of the highly turned over RPL30 transcript with the ACT1 promoter stabilizes RPL30 mRNA [6]. Similarly, switching the promoter sequences of CWL2 and SWI5 genes with the ACT1 promoter prevented cell-cycle progression-mediated decay, stabilizing the transcript, and this was attributed to changes in mRNP formation [4]. Dbf2p (functionally associated with the CCR4/Not complex, the conserved eukaryotic deadenylase complex) was shown to be imprinted onto CLW2 and SWI5 transcripts in a promoter-dependent manner to promote rapid mRNA decay upon cell cycle progression, suggesting a promoter-dependent formation of the nascent mRNP [4]. Furthermore, RNA Pol II subunits have been shown to influence events downstream of transcription. The dimeric RNA Pol II Rpb4/7 sub-complex is imprinted onto a specific range of yeast mRNAs in an RNA Poll II-dependent manner [5]. Rpb4/7 co-transcriptionally binds to the nascent transcript and remains bound through nuclear export [7] and subsequently regulates translation [8], localization [9] and decay in the cytoplasm [10]. It was shown that cytoplasmic functions of Rpb4 were dependent on Not5, a subunit of the Ccr4-Not complex [11]. Collectively these data argue the existence of tight coupling between transcription, translation and decay of mRNA in yeast.
Stabilization of mRNAs in the cytoplasm can in turn influence transcription events in the nucleus. The 5ʹ-3ʹ mRNA decay pathway appears to degrade the majority of yeast mRNAs, with deletion or inactivation of exonucleolytic activity of XRN1 (eXoRiboNuclease 1) resulted in globally reduced decay rates [1]. Stabilization of mRNA did not affect its total abundance as the rate of transcription was reduced which resulted in maintenance of steady-state mRNA levels [1,12,13]. It was found that cytoplasmic 5ʹ-3ʹ decay factors can be localized to the nucleus in an XRN1-dependent manner and XRN1 can bind to transcription start sites promoting initiation and elongation of transcription. Similarly, knockdown of PARN (the mammalian PolyA specific RiboNuclease) in mouse myoblasts, a subset of transcripts was stabilized while their rate of transcription was downregulated, resulting in either no changes or a small decrease in transcript steady-state levels [14]. Consistently, deletion of components of the Ccr4-Not complex in yeast did not lead to significant changes in abundance of transcripts, as stabilization of mRNAs was met with inverse changes to the rate of transcription [15]. Together these data suggest that both 5ʹ-3ʹ and 3ʹ-5ʹ mRNA decay factors may signal changes in transcript stability in the cytoplasm back to the nucleus leading to alterations in transcription rates. This is consistent with transcription and mRNA decay in yeast and mammals coordinating activities of the opposing process across the nuclear membrane to maintain steady-state mRNA levels in the cytoplasm. However, whether this buffering mechanism maintains the abundance of mRNA in human cells and involves translational factors is still unknown.
In this study, we tested the existence of an mRNA steady-state buffering mechanism in human liver carcinoma HepG2 cells in response to shRNA-mediated knockdown (KD) of the 5ʹ-3ʹ exonuclease XRN1, the general deadenylase and human Ccr4 homologue CNOT6 and the translation release factor 1, ETF1. We found that XRN1-mediated buffering maintains the steady-state abundance of specific mRNAs via coordination of degradation and transcription in human cells and propose that this is a conserved eukaryotic feature. Upon CNOT6 knockdown-mediated destabilization of mRNA, steady-state levels can be maintained, and we attribute this to an increased rate of transcription. The translational machinery can also play a role in preserving abundance of specific mRNAs, or a subset of transcripts, albeit with a lesser level of accuracy. The presented results reveal the existence of an inverse relationship between stability and transcription, a compensatory mechanism in human cells which responds to defects in mRNA decay or translation machineries.
Results
shRNA knockdowns
shRNA constructs with the pLKO.1 backbone were packaged into lentiviral particles and used to knockdown XRN1, CNOT6 and ETF1 genes in the human hepatocellular carcinoma HepG2 cell line. A control cell line was generated by transduction with a scrambled target sequence. Knockdown of XRN1, CNOT6 and ETF1 gene expression in the human hepatocellular-carcinoma HepG2 cell line was confirmed at the level of both mRNA and protein (Fig. 1A and 1B for XRN1; Fig. 2A and 2B for CNOT6 and Fig. 3A and 3B for ETF1).
Figure 1.
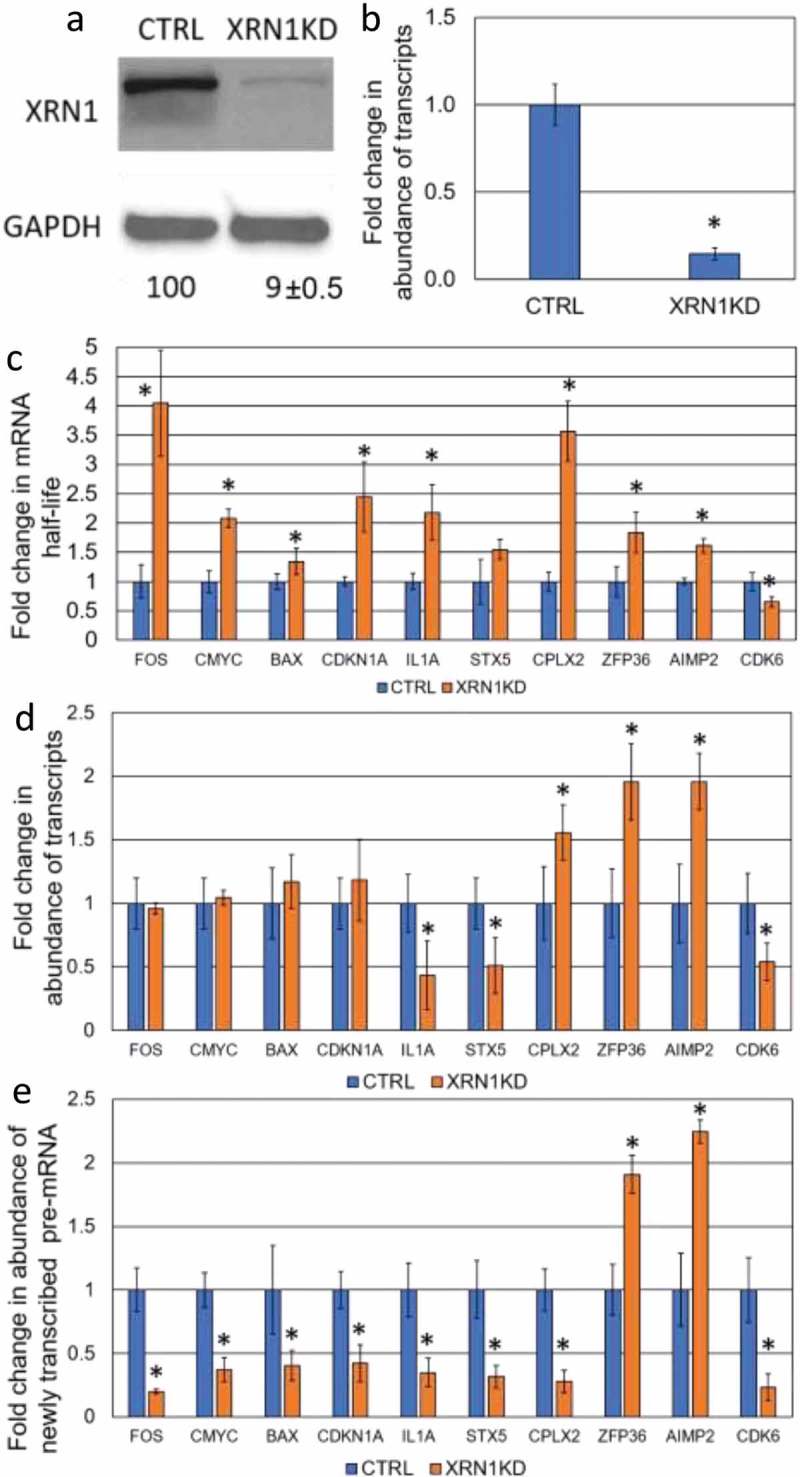
Determination the effect of XRN1-KD on the steady-state level of specific transcripts. (a) The level of XRN1 protein expression upon shRNA-mediated knockdown in CTRL (control) and XRN1-KD cell lines. The protein level was determined by western blotting and normalized to that of GAPDH and the average percentage of relative expression is shown below each lane with the standard deviation derived from three independent experiments. (b) qRT-PCR showing the fold change of the Xrn1 transcript in CTRL and XRN1-KD cell lines for comparison, error bars indicate deviation from three independent experiments and asterisks indicate significant differences (p < 0.05). (c) The fold change in mRNA stability for each transcript was assessed by normalization of the half-life of mRNA in XRN1-KD to that in CTRL cell lines. The half-life of each mRNA was determined by qRT-PCR. (d) The fold change in abundance of transcripts following XRN1 depletion was measured by qRT-PCR in XRN1-KD and normalized to that in CTRL cell lines. (e) The abundance of newly transcribed pre-mRNAs was assessed by qRT-PCR and normalized to the abundance of 7SL RNA. The fold change in the pre-mRNA was measured by normalization of the abundance of each pre-mRNA in XRN1-KD to that in CTRL cell lines. The error bars in (c), (d) and (e) represent the standard error of the mean measured from three independent experiments and three technical repeats and asterisks indicate statistically significant differences (p < 0.05).
Figure 2.
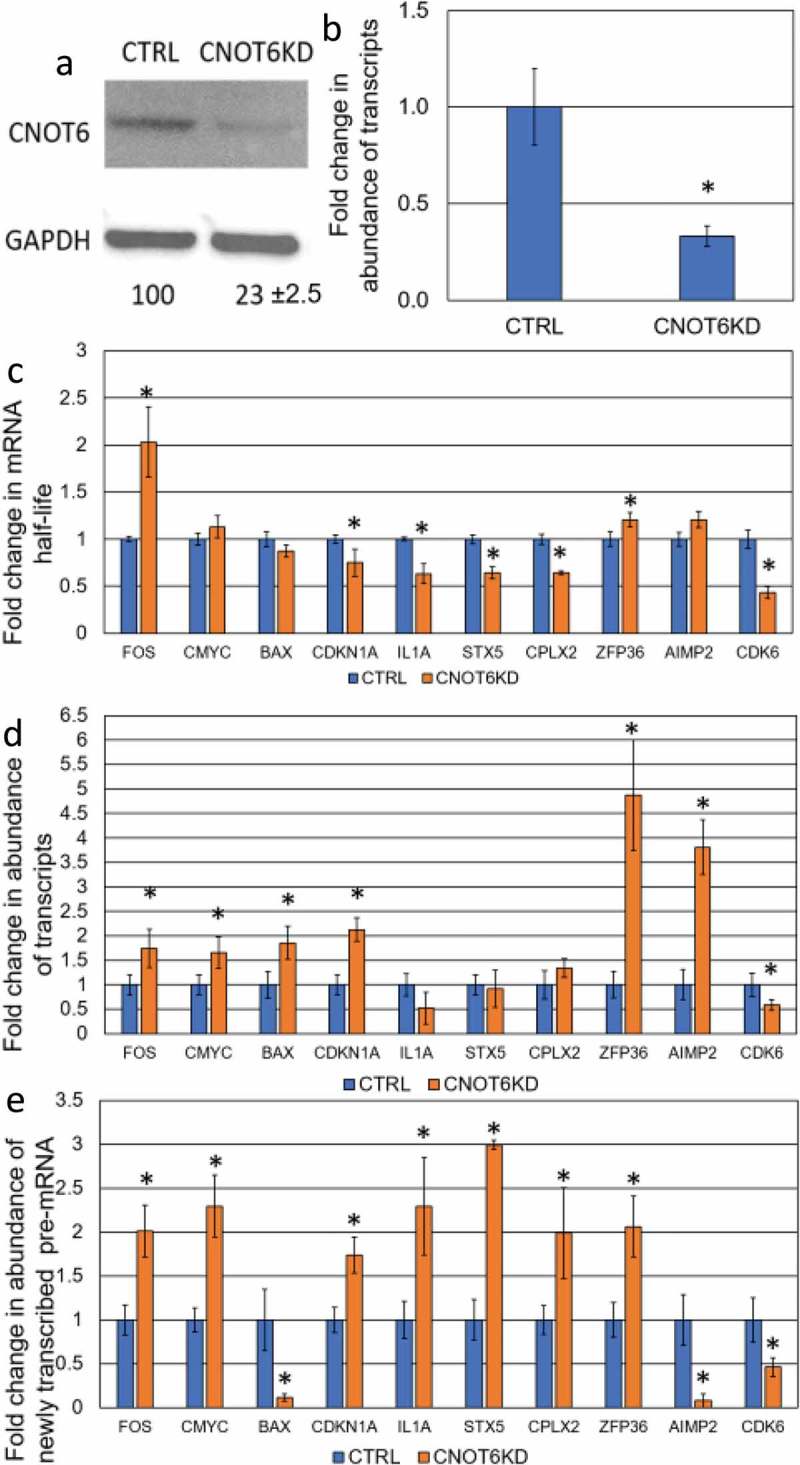
Determination the effect of CNOT6-KD on the steady-state level of specific mRNAs. (a) The level of CNOT6 protein and (b) the Cnot6 mRNA expression upon shRNA-mediated knockdown in CTRL and CNOT6-KD cell lines. Details described as in Fig. 1. (c) The fold change in transcript stability for each mRNA was measured as described in Fig. 1. (d) The abundance of transcripts following CNOT6 depletion was assessed as described in Fig. 1. (e) The abundance of newly transcribed pre-mRNAs was determined as described in Fig. 1.
Figure 3.
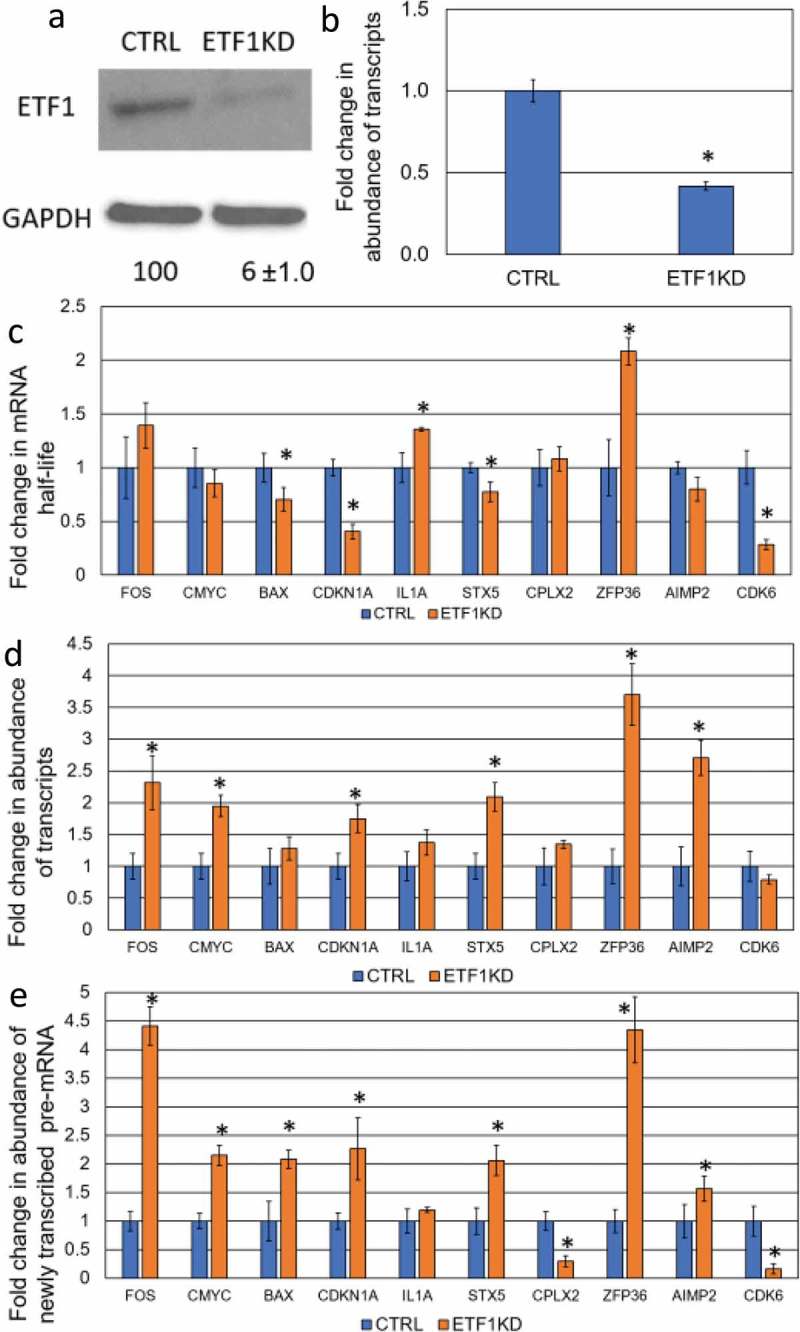
Determination the effect of ETF1-KD on the steady-state level of specific mRNAs. (a) The level of ETF1 protein and (b) the Etf1 transcript expression upon shRNA-mediated knockdown in CTRL and ETF1-KD cell lines. Details as described in Fig. 1. (c) The fold change in transcript stability for each mRNA was measured as described in Fig. 1. (d) The abundance of transcripts following ETF1 depletion was assessed as described in Fig. 1. (e) The abundance of newly transcribed pre-mRNAs was determined as described in Fig. 1.
Perturbations to mRNA decay machinery do not always lead to mRNA stabilization in human HepG2 cells
Based on analysis in both S. cerevisiae and mice, stabilization of transcripts upon knockdown/knockout of RNA decay factors does not necessarily lead to a corresponding increase in the level of the respective mRNA [1,14–16]. To test whether this is the case in human cells, ten genes related to different cellular functions, such as tumorigenesis (proto-oncogenes and apoptosis-related genes), cell cycle as well as inflammatory factors and non-cancerous genes were randomly selected for this study. The transcript half-life was determined by monitoring the change in transcript abundance, after inhibition of transcription with actinomycin D, over a time course of 2 hours. A comparison was made between the HepG2 control cells and two knockdown derivatives; XRN1-KD and CNOT6-KD (Table 1). As expected, enhanced stability of mRNA was observed for eight transcripts in XRN1-KD cell lines, whilst stability of the IL1A transcript was unchanged and stability of the CDK6 mRNA was decreased (Table 1; Fig. 1C). Surprisingly, knockdown of CNOT6 extended the half-life of only FOS and ZFP36 mRNAs, whilst the stability of the c-MYC, BAX, and AIMP2 transcripts was not altered but CDKN1A, IL1A, STX5, CPLX2 and CDK6 transcripts were all destabilized (Table 1; Fig. 2C). From these data, we concluded that XRN1 plays a major role in the degradation of most transcripts in human HepG2 cells whilst the loss of CNOT6 increased the production of specific pre-mRNA (Fig. 2E) in response to enhanced degradation of respective transcripts (Fig. 2C).
Table 1.
The half-lives (T1/2) of 10 mRNAs in CTRL cells and upon knockdown of XRN1, CNOT6 and ETF1.
| Gene ID | Gene | Description | CTRL T½ | XRN1KD T½ | p values | Fold change XRN1 | CNOT6KD T½ | p values | Fold change CNOT6 | ETF1KD T½ | p values | Fold change ETF1 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2353 | FOS | FBJ Murine Osteosarcoma Viral Oncogene | 12 ± 1 | 58 ± 14 | 0.0076 | 4.05 | 30 ± 5 | 0.0218 | 2.03 | 21 ± 4 | 0.6072 | 1.39 |
| 4609 | CMYC | V-Myc Avian Myelocytomatosis Viral Oncogene | 26 ± 6 | 53 ± 15 | 0.0043 | 2.08 | 26 ± 5 | 0.4515 | 1.13 | 21 ± 6 | 0.2127 | 0.85 |
| 3552 | IL1A | Interleukin 1 | 35 ± 8 | 49 ± 10 | 0.0789 | 1.34 | 24 ± 6 | 0.0396 | 0.63 | 46 ± 8 | 0.0328 | 1.36 |
| 581 | BAX | Bcl-2 related protein | 44 ± 8 | 82 ± 22 | 0.0330 | 2.44 | 39 ± 7 | 0.4860 | 0.87 | 31 ± 10 | 0.0368 | 0.71 |
| 1026 | CDKN1A | Inhibitor of CDK2, 4, 6, p21 | 49 ± 15 | 83 ± 25 | 0.0216 | 2.18 | 33 ± 2 | 0.0238 | 0.75 | 22 ± 2 | 0.0083 | 0.40 |
| 6811 | STX5 | Syntaxin 5 | 27 ± 6 | 41 ± 13 | 0.0372 | 1.55 | 21 ± 2 | 0.0304 | 0.64 | 22 ± 5 | 0.0320 | 0.77 |
| 10,814 | CPLX2 | Complexin 2 | 27 ± 5 | 94 ± 21 | 0.0003 | 3.56 | 22 ± 1 | 0.0031 | 0.65 | 36 ± 10 | 0.0846 | 1.08 |
| 7538 | ZFP36 | Tristetraproline | 35 ± 6 | 51 ± 2 | 0.0301 | 1.84 | 45 ± 5 | 0.0191 | 1.2 | 75 ± 13 | <0.001 | 2.08 |
| 7965 | AIMP2 | Aminoacyl tRNA synthetase int. protein | 50 ± 5 | 79 ± 9 | <0.001 | 1.61 | 63 ± 11 | 0.0681 | 1.21 | 39 ± 12 | 0.0905 | 0.80 |
| 1021 | CDK6 | Cyclin-Dependent Kinase 6 | 107 ± 9 | 78 ± 23 | 0.0252 | 0.65 | 51 ± 13 | <0.001 | 0.43 | 35 ± 16 | <0.001 | 0.28 |
XRN1-mediated buffering exists in HepG2 cells
It has been shown that Xrn1 plays a key role in buffering the abundance of transcripts in S. cerevisiae. We found that six transcripts with enhanced stability in the XRN1-KD cell line, FOS, c-MYC, BAX, CDKN1A, STX5 and CPLX2 (Fig. 1C) also showed reduced transcription rates, based on the accumulation of pulse-labelled unspliced transcript (pre-mRNA) (Fig. 1E). For these transcripts, mRNA abundance was not significantly altered with the exception of CPLX2, which had an enhanced steady-state level (Fig. 1D). Interestingly, the ZFP36 and AIMP2 transcripts showed both stabilization (Fig. 1C) and enhanced transcription (Fig. 1E) and, hence increased steady-state levels in the XRN1-KD cell line (Fig. 1D), this suggests that not all transcripts are under the regulation of XRN1-mediated buffering. For the CDK6 transcript, knockdown of XRN1 led to destabilization, a lower rate of transcription and decreased mRNA abundance (Fig. 1C, D and E). From these data, we conclude that abundance of most transcripts tested in this study is regulated in an XRN1-dependent manner.
mRNA buffering mechanism responds to CNOT6-dependent mRNA destabilization
As previously stated, we tested the effect of CNOT6 knockdown on transcript half-life. Surprisingly, we observed destabilization CDKN1A, IL1A, STX5, CPLX2 and CDK6 transcripts (Fig. 2C) and this was associated with enhanced transcription (Fig. 2E). The steady-state level of IL1A, STX5 and CPLX2 transcripts in CNOT6-KD was maintained and the abundance of CDKN1A transcript increased (Fig. 2D). This supports the existence of a buffering mechanism, which can be triggered in response to CNOT6-KD and resembles the previous observation in mice that steady-state levels are not always accurately maintained [14]. Like the XRN1 knockdown phenotype, the CDK6 transcript was destabilized and had a lower transcription rate, as well as lower abundance (Fig. 2C, D and E). Only FOS and ZFP36 mRNAs showed a significant increase in stability and mirrored the transcription and abundance phenotypes of ZFP36 and AIMP2 transcripts in XRN1-KD, with both enhanced transcription rates and steady-state levels being observed (Fig. 2C, D and E). We concluded that mRNA-buffering is active, and responsive, to both enhanced degradation and stabilization by inversely altering transcription rates, such that the level of specific mRNA can be maintained by multiple factors, or via a pathway-specific mechanism.
Knockdown of translation termination factor 1 triggers a buffering response
The current data suggest the existence of crosstalk between transcription, stability and translation [8]. However, whether translation or its factors play a role in the maintenance of mRNA steady-state levels is not yet known. Hence, we decided to test the involvement of a member of the translational apparatus in this buffering response and investigated mRNA stability, transcription and abundance in response to ETF1-KD (eRF1 in yeast), that is responsible for recognition of the stop codon (Table 1; Fig. 3). Stability of four transcripts, BAX, CDKN1A, STX5 and CDK6 decreased while no changes in half-life were observed for FOS, c-MYC, CPLX2 and AIMP2 transcripts (Fig. 3C). Transcript stability of IL1A and ZFP36 increased in response to knockdown of ETF1 (Fig. 3C). These data suggest that translation may be interconnected with mRNA stability in human cells as previously shown in yeast [13,17].
ETF1 knockdown-mediated destabilization also triggers a buffering response as the rate of transcription of specific genes increased in ETF-1 KD cell lines (Fig. 3E) resembling the CNOT6-KD phenotype. Like XRN1-KD and CNOT6-KD, stability and transcription of CDK6 was decreased, suggesting that some transcripts are either exempt from buffering, or regulated in a pathway-specific manner. While the level of the BAX transcript was not changed, abundance of both CDKN1A and STX5 mRNAs was increased, suggesting the enhanced rate of transcription was not precisely coordinated in response to ETF1-KD (Fig. 3E). The stabilized ZFP36 transcript also had an enhanced rated of transcription as well as overall level of abundance (Fig. 3E, D), whilst stabilization of IL1A correlated with an increase in transcription (Fig. 3C, E). We conclude that translation factors may be a part of the buffering mechanism, which adjusts the abundance of specific transcripts in response to changes in stability, via increased transcription rates.
Discussion
We investigated the existence of a gene expression regulatory mechanism in human cells, namely the buffering mechanism which maintains the steady-state levels of mRNAs, in response to perturbation of the mRNA decay. We found in HepG2 cells, upon KD of XRN1, CNOT6 or ETF1, changes in stability of specific transcripts do not always lead to changes in the level of their abundance, this is attributed to inverse-changes in the rate of transcription. XRN1-KD mediated stabilization was coupled with a diminished rate of transcription and hence, resulted in minimal changes in abundance of several mRNAs consistent with previous findings in yeast and mammals [1,13,15,16]. However, observed destabilization of specific transcripts in CNOT6-KD and ETF1-KD cells is coupled with enhanced rates of mRNA synthesis that can maintain the pre-KD steady-state level of mRNAs. Importantly, it appears that the expression of some genes is regulated by multiple decay or translation factors, whilst expression of other genes seems to be regulated in a decay-factor or pathway-specific manner.
The level of mRNA is determined by the balance between transcription and mRNA decay [1]. Traditionally, it has been the view that the main role of mRNA decay machinery in the cytoplasm was to act at the end of the mRNA life. It is now established that mRNA stability is integral to regulation of gene expression and is critical to the quick adaptive response upon changes in environmental signals [18]. Furthermore, it was recently discovered that knockout or disruption of decay factors, such as Xrn1, Ccr4, Caf1 in yeast, and PARN in mice myoblasts, leads to transcript stabilization but does not result in changes in the total level of expression for many mRNAs as the rate of transcription was diminished [1,14,15]. These data have revealed the existence of a cross-talk between mRNA decay and transcription which is responsible for buffering mRNA abundances. We have shown that equivalent mechanisms exist in human cells, may involve translation and are likely to be conserved in eukaryotes.
The presented data shows that most transcripts tested (8 out of 10) in HepG2 cells are degraded in an XRN1-dependent manner which is consistent with findings in yeast [1]. Also consistent with previous findings, in human cells upon XRN1-KD-mediated transcript stabilization, a robust feedback mechanism, which prevents substantial changes in abundances of specific mRNAs, was observed suggesting a conserved nature of buffering of mRNA abundance in eukaryotes [1]. In yeast, Xrn1 has been shown to be localized in the nucleus and directly enhances initiation and elongation of transcription by preferentially binding to chromatin of transcriptionally active genes [1]. It was found that the nuclear localization of XRN1 is linked to its exonucleolytic activity; shuttling of other decay factors, such as Pat1 and Lsm1 (which were also shown to affect transcription rates), is dependent on XRN1 ability to fully degrade mRNA [1]. This suggests that XRN1 is a major transcript decay-checkpoint factor that balances mRNA decay with transcription rates and hence, appears to be the master coordinator of this gene expression buffering mechanism. However, it was also observed that upon XRN1 depletion, other major decay factors can shuttle in an XRN1-independent manner and impact transcription rates, suggesting some redundancies in the buffering mechanism.
CNOT6-KD led to destabilization of five out of ten tested transcripts. Consistent with yeast data, depletion of CNOT6 did not result in changes in abundance of specific mRNAs which is attributed to adjustment of transcription rates to balance changes in decay rates [15]. Abundance of three mRNAs were regulated by a CNOT6-KD-mediated buffering response, suggesting that transcription of different genes can be regulated by different decay factors; this specificity could be dependent on primary routes of mRNA decay. These data raise an important question: why does depletion of CNOT6, a decay factor which generally initiates mRNA decay, result in destabilization of transcripts? CNOT6 is a conserved general eukaryotic deadenylase [19] which plays a key role in removal of poly(A) binding protein (PABP) exposing the poly(A) tail to Caf1 which degrades PABP-free poly(A) tail [20,21]. Depletion of CNOT6 may prevent or delay the removal of PABP and subsequent shortening of the poly(A) tail by Caf1 and hence, delay poly(A) tail length dependent decapping. Prevention of poly(A) tail length dependent decapping could lead to activation of alternative decay pathways, such as poly(A) tail length-independent decapping, which could behave more aggressively, including activation of specific 5ʹ-3ʹ mRNA decay factors; this is consistent with mRNA decay pathway redundancy [22]. Thus, it is imperative to investigate the state of the 5ʹ and 3ʹ ends of mRNA in response to CNOT6 KD, as transcript destabilization could be a result of an indirect effect of stabilization of other decay factor mRNA. It cannot be ruled out that CNOT6 could be functioning primarily in the nucleus [19] as a potential repressor of transcription, we observed a dramatic increase in the level of pre-mRNA, enhanced transcription in turn could signal to cytoplasmic destabilization to maintain the steady-state mRNA level. Future work should aim to investigate CNOT6 targets on a genome-wide scale in order to elucidate its effect on stability of different mRNAs in cancerous and non-cancerous cells. Consistent with our findings, in mice 24 transcripts were found to be destabilized upon depletion of PARN deadenylase [14]. It was suggested that this could be due to knock-on effects of some PARN-KD mediated stabilized transcripts (e.g. EDC3, the decapping enhancer protein 3) or off-target effects of shRNA constructs used to knockdown PARN in mice [14], whether or not buffering was active was not investigated for these destabilized transcripts. It was not observed whether destabilization of specific transcripts is a result of overactive alternative pathways (e.g. overexpression of XRN1); if this is the case, this buffering response could solely be attributed to robust XRN1-mediated signalling, as opposed to CNOT6 mediated buffering. In yeast, depletion of Ccr4 led to stabilization of transcripts and maintenance of steady-state mRNA levels due to adjustment to the rate of transcription [15], however it is also not known whether deletion of this deadenylase led to overexpression of other mRNA decay factors. Such discrepancies, despite the conserved nature of Ccr4 throughout all eukaryotes, may in part be due to the cancerous nature of HepG2 cells and a genome-wide investigation of the transcriptome of CNOT6-KD should help to clarify this phenomenon.
Translation is an integral part of gene expression and it was previously proposed that gene expression is circular [1], which implies that all stages of this pathway in the cytoplasm can signal back to transcription in order to adjust the pattern of expression. Therefore, it was important in this study to test the involvement of the translational apparatus in this buffering mechanism. We chose the translation release factor ETF1, eukaryotic release factor 1 and three out of the ten tested transcripts were destabilized upon ETF1-KD. Surprisingly, the destabilized transcripts also displayed an enhanced level of transcription, resembling the CNOT6-KD phenotype, such that the level of abundance of these mRNAs was not significantly affected. Whilst the translational efficiency of the tested mRNAs in this cell line was not tested, the presented data is consistent with the notion that translation is intimately linked to mRNA stability [23]. At this stage it is difficult to explain the observed destabilization of certain transcripts, although it is feasible that depletion of ETF1, the protein which is involved in the recognition of the stop codon, may result in a prolonged ‘pausing’ of the ribosome on the stop codon; this pausing, in turn, may trigger a no-go decay pathway [24] or activation of a nonsense-mediated decay like response which is known to be a global regulator of translational fidelity [25–27]. Enhanced activity of some specific decay factors, as a potential off-target effect of the ETF1-KD, could also play a part. Nevertheless, ETF1-KD-dependent alteration of transcript stability activates a buffering response via inverse adjustment of transcription rates resembling the CNOT6-KD like phenotype. This argues that ETF1 is involved in steady-state buffering and that translation may be intrinsically linked to transcription.
Abundance of CPLX2 and CDKN1A mRNAs was seen to be buffered in both XRN1-KD and CNOT6-KD cell lines, whilst some other transcripts only had a buffering response to knockdown of specific factors. Despite the opposite alteration in their stability (stabilization in XRN1-KD vs destabilization in CNOT6-KD) the steady-state level of both mRNAs in these KDs was maintained. This suggests that mRNA decay factors may play additional roles in degradation, such as coordinating the ‘optimal’ mRNA decay rate, or decay polarity as previously shown in yeast and plants [28,29].
The observed down-regulation of the stability and transcription of CDK6, a key regulator of the G1/S cell cycle transition [30], in all three KD derivatives shows that the abundance of some transcripts may not be regulated by this buffering mechanism. Instead, expression of these genes may be controlled by their effectors such as a cyclin-dependent kinase inhibitor [31] the abundance of which is subject to the buffering mechanism, i.e. we found the level of the CDKN1A transcript is buffered. This suggests a potential hierarchy in regulation of gene expression, which is yet to be fully understood.
The observed buffering of transcript abundance does not always result in precise maintenance of steady-state mRNA levels, such that the abundance of some transcripts is changed upon decay/translation factor KD despite inverse-changes in transcription rate. This phenomenon has been noted previously in mammals upon PARN-KD [14] and in yeast, in response to depletion of Xrn1 [1]. Whilst such inaccuracy could be merely due to overactive or repressed transcription, it is still not clear why for some transcripts the level of expression is maintained more accurately than for others, implying the possible existence of different mechanisms of regulation of transcription in response to alteration of stability. It also cannot be ruled out that such inaccuracy may be due to signalling from changes in the rate of translation. Either of these possibilities could hold true, but the finding of the present study, that translation is also involved in the buffering mechanism, adds another dimension to the evidence for gene expression cross talk. This suggests that translation may also contribute to determination of the rate of transcription and degradation, providing the cell with an additional level of protection against perturbation in gene expression steps.
HepG2 is a cancer cell line and does not represent wild-type cell homeostasis, such that its gene expression signature differs from non-cancerous epithelial liver cells [32]. Whilst it is highly likely that steady-state levels of transcripts would also be buffered in physiological cells by the same buffering mechanism, the patterns of abundance are likely to be different. The half-lives of c-FOS and c-MYC mRNAs in HepG2 cells were found to be consistent with those previously found in other cancerous cell lines, including HeLa (cervix carcinoma), MCF7 (breast carcinoma) and Daudi (Burkitt lymphoma) [33,34], and their transcript abundance is subject to regulation by XRN1-dependent buffering mechanism. This suggests that expression of specific oncogenes during cancers is tightly regulated in order to maintain the cancerous phenotype and can involve the buffering mechanism. Hence, elements of the buffering mechanism could represent targets for novel therapies for cancer.
Material and methods
Cell culture
The human epithelial hepatocellular carcinoma cell line HepG2 (HB-8065) was obtained from the American Type Culture Collection (ATCC). HepG2 was cultured in high-glucose Dulbecco’s Modified Eagle’s Media DMEM, supplemented with 10% heat-inactivated Fetal Bovine Serum (FBS) and 1% penicillin/streptomycin grown in 5% CO2 at 37⁰C as per ATCC’s recommendation. The human epithelial embryonic kidney cell line HEK293t (CRL32-16) was obtained from ATCC. HEK293t was cultured in high-glucose DMEM, supplemented with 10% FBS, 2mM L-glutamine, and grown in 5% CO2 at 37⁰C.
Lentiviral packaging of shRNA plasmids and generation of knockdown strains
Transcript knockdown was undertaken using shRNA constructs specific to XRN1, CNOT6 and ETF1 transcripts packaged into lentivirus particles [14]. Validated shRNA constructs were chosen from the Broad Institute’s RNAi consortium (Table S2). Constructs had specific target sequences inserted between the AgeI and EcoRI restriction sites of the pLKO.1 plasmid (Addgene #10,878, a kind gift from David Root) resulting in recombinant plasmids of around 7.5kb in length. The plasmid constructs were packaged into lentivirus particles using the HEK293t cell line as a host, as previously described [35]. The pLJM1-EGFP plasmid (Addgene #19,319, a kind gift from David Sabatini) was used as a positive control for transfection and transduction. Transduced cells were selected for using puromycin (3µg/ml). Details of shRNA knockdown are provided in Supplementary Materials and Methods.
Extraction and manipulation of RNA and qRT-pcr
Total RNA was isolated using Trisure (Bioline) as per the manufacturer instructions. Briefly, the cell monolayer was washed with ice-cold PBS and 1 ml Trisure reagent was added per 10cm2 surface area. After 5 minutes of gentle agitation, the cell lysate was collected and mixed with 0.2 ml of chloroform per 1 ml Trisure. The samples were centrifuged at 12000xg at 4⁰C for 10 minutes. The upper aqueous phase was mixed with an equal volume of chloroform, vortexed and centrifuged as previously. RNA in the upper phase was precipitated with equal volume of cold isopropanol. The pellet was air-dried, washed twice with 70% EtOH and resuspended in 10mM Tris, pH7.4. Total RNA was treated with DNase Turbo DNase (Invitrogen) as per manufacturer recommendation.
To determine half-lives of transcripts, an mRNA stability time-course was undertaken. Proliferating cultures were treated with actinomycin D (8µg/ml) for 30 minutes prior to the start of time course [14] with time points of T0, 30 minutes, 60 minutes and 120 minutes. Total RNA was extracted as described previously.
RNA samples were reverse transcribed using the Tetro cDNA synthesis kit using N6 random hexamer primers (Bioline) as per manufacturer recommendations. The relative expression of transcripts was measured by quantitative (q)PCR using Biorad iTaq 2x Supermix and the CFX-Connect RealTime PCR detection system. Three housekeeping genes were used for normalization: Succinate Dehydrogenase subunit A (SDHA), Glyceraldehyde 3-Phosphate Dehydrodenase (GAPDH) and Tyrosine 3-Monooxygenase/Tryptophan 5-Monooxygenase Activation protein-Zeta (YWHAZ). Housekeeping genes were chosen by use of the geNorm kit by Primer Design, the variance in expression between strains was analysed using qbase+ software as per manufactory recommendation. Expression of the three chosen RNA Pol II housekeeping genes was normalized to 18S rRNA (RNA Pol I transcript) and no differences in the abundance between each KDs and control cell lines was observed. Primer sequences are listed in Table S1A.
Metabolic labelling of RNA for measurement of transcription rates. To measure the rate of transcription, RNA was metabolically labelled using 4sU and extracted, followed by isolation of pre-mRNA using streptavidin magnetic beads as described previously [14]. The level of pre-mRNA abundance was measured by qRT-PCR using a primer pair with the amplicon spanning an intron-exon junction of the target gene. The level of pre-mRNA abundance was normalized to that of 7SL rRNA as described previously [14]. Primer sequences are listed in Table S1B. Details of pre-mRNA isolation are described in Supplementary Materials and Methods.
Protein extraction and western blot
Protein extracts were prepared using the detergent-based cell lysis RIPA buffer (10mM Tris-Cl pH 8.0, 1mM EDTA, 1% Triton X-100, 1% Sodium deoxycholate, 0.1% SDS, 140mM NaCl and 1mM protease inhibitor cocktail (Sigma)). Western blot was undertaken as previously described [14]. Primary antibodies are listed in Table S3. Detailed protocol is described in Supplementary Materials and Methods.
Data analysis
The fold changes in abundance, half-life and pre-mRNA between KD cell lines and CTRL cells were determined by qRT-PCR [36]. A paired T-test was conducted for each ΔCT value for each gene per strain (n ≥ 3) for total and pre-mRNA samples. The P-values for half-life analysis were calculated by paired T-tests which were conducted to compare the half-lives between the CTRL and strain cell lines (n ≥ 3). For determination of half-life, time points were fit to a nonlinear least squares model with confidence intervals (R2) >75% and half-lives were extrapolated from the decay curve [37]. Average half-life curves are presented in Supplementary Fig. S1. The truncated product method [38] was applied on all P-values in this study to determine whether there was bias from testing of multiple hypotheses. The truncated product method P-value was <0.0001 which is an indicator that statistically significant results were not biased by multiple comparison. The relative level of protein abundance was determined by densitometry analysis using the ImageJ software, protein abundance was normalized to that of GAPDH.
During the revision of the manuscript, a new publication on the involvement of translation in buffering in yeast by Blasco-Moreno et al. 2019 [39] has emerged which is consistent with the findings of this study.
Funding Statement
This work was supported by the Coventry University [N/A]; Centre for Sports, Exercise and Life Sciences, Coventry University [N/A].
Acknowledgments
PS was supported by the Health Life Sciences Faculty research Centre for Sport, Exercise and Life Sciences, Coventry University. We would like to thank S. Tompsett and H. Kikuchi for technical assistance.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental material
Supplementary data for this article can be accessed here
References
- [1].Haimovich G, Medina D, Causse S, et al. Gene expression is circular: factors for mRNA degradation also foster mRNA synthesis. Cell. 2013;153:1000–1011. [DOI] [PubMed] [Google Scholar]
- [2].Braun KA, Young ET.. Coupling mRNA synthesis and decay. Mol Cell Biol. 2014;34:4078–4087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bellofatto V, Wilusz J. Transcription and mRNA stability: parental guidance suggested. Cell. 2011;147:1438–1439. [DOI] [PubMed] [Google Scholar]
- [4].Trcek T, Larson DR, Moldón A, et al. Single-molecule mRNA decay measurements reveal promoter regulated mRNA stability in yeast. Cell. 2011;23:1484–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Goler-Baron V, Selitrennik M., Barkai O, et al. Transcription in the nucleus and mRNA decay in the cytoplasm are coupled processes. Genes Dev. 2008;22:2022–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bregman A, Avraham-Kelbert M, Barkai O, et al. Promoter elements regulate cytoplasmic mRNA decay. Cell. 2011;147:1473–1483. [DOI] [PubMed] [Google Scholar]
- [7].Farago M, Nahari T, Hammel C, et al. Rpb4p, a subunit of RNA polymerase II, mediates mRNA export during stress. Mol Biol Cell. 2003;14:2744–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Harel-Sharvit L, Eldad N, Haimovich G, et al. RNA polymerase II subunits link transcription and mRNA decay to translation. Cell. 2010;143:552–563. [DOI] [PubMed] [Google Scholar]
- [9].Lotan R, Bar-On V. G., Harel-Sharvit L, et al. The RNA polymerase II subunit Rpb4p mediates decay of a specific class of mRNAs. Gen & Dev. 2005;19:3004–3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lotan R, Goler-Baron V, Duek L, et al. The Rpb7p subunit of yeast RNA polymerase II plays roles in the two major cytoplasmic mRNA decay mechanisms. J Cell Biol. 2007;178:1133–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Villanyi Z, Ribaud V, Kassem S, et al. The Not5 subunit of the Ccr4-not complex connects transcription and translation. PLOS Genet. 2014;10:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Anderson JS, Parker R. The 30 to 50 degradation of yeast mRNAs is a general mechanism for mRNA turnover that requires the SKI2 DEVH box protein and 30 to 50 exonucleases of the exosome complex. Embo J. 1998;17:1497–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Parker R. RNA degradation in saccharomyces cerevisiae. Genet. 2012;191:671–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lee JE, Lee JY, Trembly J, et al. The PARN deadenylase targets a discrete set of mRNAs for decay and regulates cell motility in mouse myoblasts. PLoS Genet. 2012;8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Sun M, Schwalb B, Schulz D, et al. Comparative dynamic transcriptome analysis (cDTA) reveals mutual feedback between mRNA synthesis and degradation. Genome Res. 2012;22:1350–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sun M, Schwalb B, Pirkl N, et al. Global analysis of eukaryotic mRNA degradation reveals Xrn1-dependent buffering of transcript levels. Mol Cell. 2013;52:52–62. [DOI] [PubMed] [Google Scholar]
- [17].Decker CJ, Parker R. P-bodies and stress granules: possible roles in the control of translation and mRNA degradation. Cold Spring Harb Perspect Biol. 2012;4:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Houseley J, Tollervey D. The many pathways of RNA degradation. Cell. 2009;136:763–776. [DOI] [PubMed] [Google Scholar]
- [19].Collart MA, Panasenko OO. The Ccr4 – not complex. Gene. 2012;492:42–53. [DOI] [PubMed] [Google Scholar]
- [20].Webster MW, Chen Y-H, Stowell JAW, et al. mRNA deadenylation is coupled to translation rates by the differential activities of Ccr4-Not nucleases. Mol Cell. 2018;70:1089–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Yi H, Park J, Ha M, et al. PABP cooperates with the CCR4-NOT complex to promote mRNA deadenylation and block precocious decay. Mol Cell. 2018;70:1081–1088. [DOI] [PubMed] [Google Scholar]
- [22].Van-Hoof A, Lennertz P, Parker R. Three conserved members of the RNase D family have unique and overlapping functions in the processing of 5S, 5.8S, U4, U5, RNase MRP and RNase P RNAs in yeast. Embo J. 2000;19:1357–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Inada T, Makino S. Novel Roles of the multi-functional CCR4-NOT complex in post-transcriptional regulation. Front Genet. 2014;5:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Doma MK, Parker R. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature. 2006;440:61–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Celik A, He F, Jacobsen A. NMD monitors Translational Fidelity 24/7. Current Genet. 2017;63:1007–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Carnes J, Jacobsen M, Leinwand L, et al. Stop codon suppression via inhibition of eRF1 expression. Rna. 2003;9:648–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Deanna M, Janzen DM, Geballe AP. The effect of eukaryotic release factor depletion on translation termination in human cell lines. Nucleic Acids Res. 2004;32:4491–4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Pilkington GR, Parker R. Pat1 contains distinct functional domains that promote P-body assembly and activation of decapping. Mol Cell Biol. 2007;28:1298–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Sement F, Ferrier E, Zuber H, et al. Uridylation Prevents 3ʹ Trimming of Oligoadenylated mRNAs. Nucleic Acids Res. 2013;41:7115–7127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sherr CJ. G1 phase progression: cycling on cue. Cell. 1994;79:551–555. [DOI] [PubMed] [Google Scholar]
- [31].Morgan DO. Principles of CDK regulation. Nature. 1995;374:131–134. [DOI] [PubMed] [Google Scholar]
- [32].Tyakht AV, Song Q, Jia G, et al. RNA-Seq gene expression profiling of HepG2 cells: the influence of experimental factors and comparison with liver tissue. BMC Genomics. 2014;15:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Schiavi SC, Belasco JG, Greenberg ME. Regulation of proto-oncogene mRNA stability. Biochbnica et Biophysica Acta. 1992;1114:95–106. [DOI] [PubMed] [Google Scholar]
- [34].Dani C, Blanchard JM, Piechaczyk M, et al. Extreme instability of myc mRNA in normal and transformed human cells. Proc Natl Acad Sci USA. 1984;81:7046–7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].The Computational Biology and Functional Genomics Laboratory (2012) shRNA Lentivirus Production using HEK293T cells and FuGENE. [cited 2018 Jan 20].
- [36].Livak KJ, Schmittgen TD. Analysis of relative gene expression data using realtime quantitative PCR and the 2DDCT method. Methods. 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- [37].Wang Y, Liu CL, Storey JD, et al. Precision and functional specificity in mRNA decay. Proc Natl Acad Sci USA. 2002;99:5860–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Zaykin DV, Zhivotovsky LA, Westfall PH, et al. Truncated product method for combining P-values. Genet Epidem. 2002;22:170–185. [DOI] [PubMed] [Google Scholar]
- [39].Blasco-Moreno B, de Campos-Mata L, Böttcher R, et al. The exonuclease Xrn1 activates transcription and translation of mRNAs encoding membrane proteins. Nat Comm. 2019;10:1298–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- The Computational Biology and Functional Genomics Laboratory (2012) shRNA Lentivirus Production using HEK293T cells and FuGENE. [cited 2018 Jan 20].