

Summary
Class switch recombination (CSR) is a DNA recombination reaction that diversifies the effector functions of antibodies. CSR occurs via the formation and non-homologous end joining (NHEJ) repair of programmed DNA double-strand breaks (DSBs) at the immunoglobulin heavy chain locus. The DNA repair factors 53BP1 and Rif1 promote NHEJ and CSR by protecting DSBs against resection. However, to what extent repression of DNA end resection contributes to CSR is unknown. Here, we show that B lymphocytes devoid of 53BP1-Rif1-dependent DSB end protection activity undergo robust CSR. Inactivation of specific sets of phospho-sites within 53BP1 N-terminal SQ/TQ motifs abrogates Rif1 recruitment and inhibition of resection but only mildly reduces CSR. Furthermore, mutations within 53BP1 oligomerization domain abolish CSR without substantially affecting DNA end processing. Thus, inhibition of DNA end resection does not correlate with CSR efficiency, indicating that regulation of DSB processing is not a key determinant step in CSR.
Keywords: class switch recombination, NHEJ, DNA end resection, 53BP1, Rif1
Graphical Abstract
Highlights
-
•
53BP1 oligomerization is largely dispensable for inhibition of DSB resection
-
•
53BP1 higher order oligomerization is a pre-requisite for CSR
-
•
B lymphocytes devoid of 53BP1-Rif1 DSB end protection activity undergo robust CSR
-
•
53BP1-mediated DSB end mobility is dispensable for CSR
Class switch recombination (CSR) occurs via non-homologous end joining (NHEJ) of programmed DNA double-strand breaks (DSBs) at the immunoglobulin heavy chain locus. Sundaravinayagam et al. show that inhibition of DSB resection does not correlate with CSR efficiency and that regulation of DSB processing plays a limited role during CSR.
Introduction
The diversity of human immunoglobulin (Ig) gene repertoire is shaped by antibody diversification processes occurring at different stages of B lymphocytes development (Dudley et al., 2005, Methot and Di Noia, 2017). Class switch recombination (CSR) is a DNA rearrangement reaction occurring in mature B lymphocytes, which replaces the constant portion of the IgM heavy chain with one of the alternative isotypes (IgG, IgA, or IgE). As a consequence, this process alters the Ig effector function without affecting its antigen specificity (Chaudhuri and Alt, 2004, Methot and Di Noia, 2017). CSR is crucial to the establishment of a proper immune response because it enhances the capability of antigen-specific Igs to effectively dispose of the pathogens (Durandy et al., 2013).
The Igh locus of mature B lymphocytes spans more than 250 kb and comprises a rearranged VDJ region and a series of exon sets encoding the different constant (C) regions (Cμ, Cγ3, Cγ1, Cγ2b, Cγ2a, Cε, and Cα in mice), which define the Ig isotypes. CSR occurs through a deletional recombination event that replaces the μ constant gene of the Ig heavy chain with one of the several downstream constant regions. The reaction is initiated in G1 by the B cell-specific enzyme activation-induced deaminase (AID), which targets highly repetitive stretches of DNA preceding each constant region (switch [S] regions) in a transcription-dependent manner (Chaudhuri et al., 2003, Dickerson et al., 2003, Muramatsu et al., 2000, Ramiro et al., 2003, Revy et al., 2000). AID activity ultimately leads to the formation of DNA double-strand breaks (DSBs) (Boboila et al., 2012, Methot and Di Noia, 2017). During CSR, breaks at the donor Sμ region and one of the downstream acceptor switch regions are repaired by components of the non-homologous end joining (NHEJ) DSB repair pathway. NHEJ directly ligates CSR DSBs after minimal DNA end processing, resulting in deletion of the intervening sequence and expression of the newly switched heavy chain (Casellas et al., 1998, Chapman et al., 2013, Di Virgilio et al., 2013, Escribano-Díaz et al., 2013, Manis et al., 1998, Manis et al., 2004, Pan-Hammarström et al., 2005, Reina-San-Martin et al., 2003, Ward et al., 2004, Yan et al., 2007).
Productive joining of distal breaks in donor and acceptor switch regions represents only one of the possible outcomes of AID-induced DSB repair. Switch region breaks can also be internally re-joined, leading to unproductive intra-switch recombination (Alt et al., 1982, Bottaro et al., 1998, Dudley et al., 2002, Gu et al., 1993, Hummel et al., 1987, Reina-San-Martin et al., 2003, Reina-San-Martin et al., 2004, Winter et al., 1987). Given the repetitive nature of the switch regions, intra-S recombination events are favored by limited resection, which, combined with the intra-S break proxymity, skews repair toward the preferential use of the exposed microhomologies (microhomology-mediated end joining [MMEJ]) (Boboila et al., 2010, Bothmer et al., 2013, Dunnick et al., 1993, Yan et al., 2007, Yancopoulos et al., 1986).
The balance between NHEJ-mediated CSR and MMEJ-intra-S recombination is controlled by the DNA end resection inhibitory function of 53BP1 (Bothmer et al., 2010, Bothmer et al., 2011, Bothmer et al., 2013, Manis et al., 2004, Reina-San-Martin et al., 2007, Ward et al., 2004), a DSB repair protein that has emerged as a key regulator of DNA end processing (Panier and Boulton, 2014, Zimmermann and de Lange, 2014). 53BP1 binds damaged chromatin and recruits its phospho-dependent interactor Rif1, which protects DSB ends against processing by the DNA resection-promoting factor CtIP (Chapman et al., 2013, Di Virgilio et al., 2013, Escribano-Díaz et al., 2013, Feng et al., 2013, Zimmermann et al., 2013). This function is mediated by the downstream REV7-SHLD1-SHLD2-SHLD3 (shieldin) and CTC1-STN1-TEN1 (CST) complexes, which localize to sites of DNA damage and counteract DSB end resection (Dev et al., 2018, Findlay et al., 2018, Ghezraoui et al., 2018, Gupta et al., 2018, Mirman et al., 2018, Noordermeer et al., 2018, Tomida et al., 2018). In addition to CSR, the DNA end resection inhibitory function of 53BP1-RIF1-shieldin promotes fusion of telomeres that lack shelterin protection, and end joining of unrepaired chromatid breaks into radial chromosomes in Brca1-deficient cells (Bunting et al., 2010, Chapman et al., 2013, Dev et al., 2018, Dimitrova et al., 2008, Ghezraoui et al., 2018, Gupta et al., 2018, Mirman et al., 2018, Sfeir and de Lange, 2012, Zimmermann et al., 2013). These aberrant DNA repair reactions require 53BP1-dependent DSB end mobility in addition to its DNA end protection function (Dimitrova et al., 2008, Lottersberger et al., 2015).
Protection of AID-induced DSBs against resection is thought to play a central role during CSR. However, the actual extent to which 53BP1-Rif1-dependent repression of DNA resection is required for CSR is not known. Here, we dissected the contribution of the different functions of 53BP1 to CSR. We show that CSR is only marginally dependent on 53BP1-Rif1-mediated inhibition of DNA end processing, whereas the formation of higher order 53BP1 oligomers is crucial for the reaction. Furthermore, CSR does not require the DSB-induced chromatin mobility function of 53BP1. This sets CSR apart from the other DNA repair reactions mediated by 53BP1, fusion of deprotected telomeres and formation of radial chromosomes.
Results
Disruption of 53BP1 Higher Order Oligomerization Abrogates CSR
Deletion of amino acids 1231–1270 of human 53BP1 interferes with 53BP1 oligomerization and abrogates its ability to rescue CSR in 53BP1-deficient B lymphocytes (Bothmer et al., 2011, Ward et al., 2006, Zgheib et al., 2009). This amino acid stretch contains predicted secondary structure elements (Figures 1A and S1A), suggesting the presence of a folded oligomerization domain (OD) in this region. To dissect how 53BP1 multimerization supports CSR, we fused the peptide sequence encompassing amino acids 1233–1288 to the maltose-binding protein (MBP) (Figures S1B and S1C) and assessed the oligomerization status of the fusion protein by gel filtration analysis coupled to right-angle light scattering (RALS). In these experiments, MBP-OD eluted as a tetramer (Figure 1B; MBP-ODWT). Mutation of the evolutionarily conserved residue D1256 led to a dimeric species, whereas the deletion of the OD core (amino acids 1254–1258) rendered the protein monomeric (Figure 1B). In agreement with these data, co-immunoprecipitation analysis of differentially tagged 53BP1 constructs showed that deletion of the OD core destabilized 53BP1 oligomerization more severely than the D1256A mutation (Figure 1C). These findings indicate that the OD acts as an independent tetramerization module, and D1256 is part of an interface that mediates dimerization of 53BP1 dimers. Notably, this analysis provided us with a tool to dissect the role of 53BP1 dimerization and tetramerization for CSR and the regulation of DNA end resection.
Figure 1.

Disruption of 53BP1 Higher Order Oligomerization Abrogates CSR without Affecting DSB End Protection
(A) Schematic representation of 53BP1 protein domains and OD mutants. LC8, LC8 (8 kDa light chain dynein)-binding motif; OD, oligomerization domain; Tudor, Tudor domain; UDR, ubiquitylation-dependent recruitment motif; ΔBRCT, BRCA1 C terminus domains-truncated.
(B) Gel filtration coupled to right-angle light scattering (RALS) analysis of the OD constructs fused to N-terminal MBP. UV absorbance is shown in red-blue-green-brown and refers to the left y axis, whereas the molecular mass of the constructs as deduced by RALS is shown in the corresponding brighter colors and refers to the right y axis. Results are representative of two independent experiments. See also Figure S1.
(C) Western blot (WB) analysis of anti-HA immunoprecipitates from irradiated (20 Gy; 1 h recovery time) BOSC23 cells transfected with the indicated 53BP1-3xFlag/-3xHA constructs. Numbers underneath each lane indicate relative quantification of oligomutant versus WT 53BP1-3xFlag proteins in anti-HA immunoprecipitates (53BP1WT-3xFlag set to 1). Results are representative of two independent experiments. EV, empty vector.
(D) Top: representative flow cytometry plots measuring CSR to IgG1 in WT and 53bp1−/− splenocytes transduced with the indicated constructs. Numbers in the plots refer to the percentage of switched cells (IgG1+ cells). Bottom: summary dot plot for four independent experiments. CSR efficiencies within each experiment were normalized to the CSR value of 53BP1WT-reconstituted cells, which was set to 100%.
(E) Representative WB analysis of 53bp1−/− B lymphocytes reconstituted with the indicated 53BP1 viral constructs.
(F and G) Automated IRIF quantification of immunofluorescent staining for 53BP1 (F) and Rif1 (G) in irradiated (10 Gy; 2 h recovery time) 53bp1−/− iMEFs reconstituted with the indicated constructs. The top graph within each panel shows a representative experiment, and the bottom graph summarizes the mean values of three experiments performed on three independent sets of reconstituted cell lines. Each symbol in the top graph represents a single cell, and the number of scored cells was ≥200 per sample. The mean is indicated. Either 53BP1mTudor or 53BP1mUDR was included in parallel to EV as an additional control in the individual repeats. The representative experiments in (F) and (G), top graphs, used 53BP1mTudor. See also Figure S2B for representative immunofluorescent staining images.
(H) Schematic representation of the BrdU assay for DNA end resection. BrdU treated cells were analyzed for BrdU content under native (not denatured) conditions to estimate resection levels following NCS treatment. Adapted from (Tkáč et al., 2016).
(I) Representative BrdU fluorescence-activated cell sorting (FACS) plots showing levels of resection in NCS-treated 53bp1−/− iMEF reconstituted with the indicated constructs.
(J) Summary graph showing quantification of the BrdU FACS data represented in (I) for four experiments performed on three independent sets of reconstituted cell lines. Numbers above each bar indicate the fold mean BrdU intensity over WT, which was set to 1.
Significance in (D), (F), (G), and (J) was calculated using the Mann-Whitney U test, and error bars represent SD. ∗p ≤ 0.05, ∗∗p ≤ 0.01, ∗∗∗p ≤ 0.001, and ∗∗∗∗p ≤ 0.0001; ns, not significant. See also Figure S3.
To assess how the differential assembly of 53BP1 influenced its ability to support CSR, we compared the capability of 53BP1ΔOligo, 53BP1ΔCore, and 53BP1D1256A to rescue CSR in 53bp1−/− B cells. We used primary cultures of splenocytes and monitored CSR ex vivo to IgG1 in response to stimulation with LPS and IL-4. As expected, 53bp1−/− B cells exhibited 10-fold lower levels of CSR compared with wild-type (WT) cells, and reconstitution with 53BP1WT efficiently rescued CSR (Figures 1D and 1E). On the contrary, none of the OD mutants significantly elevated CSR efficiency above the levels of the empty vector (EV) control or of a 53BP1 mutant that cannot associate with chromatin (53BP1mTudor) (Figure 1D). We concluded that the integrity of 53BP1 higher order oligomers is a pre-requisite for CSR.
Oligomerization-Impaired 53BP1 Is Able to Counteract DSB End Resection
In undamaged cells, 53BP1 associates with its direct binding partner Tudor interacting repair regulator (TIRR), which binds to 53BP1 tandem Tudor domain and masks the H4K20me2 binding surface (Botuyan et al., 2018, Dai et al., 2018, Drané et al., 2017, Wang et al., 2018, Zhang et al., 2017). Upon DNA damage, the 53BP1-TIRR complex dissociates (Drané et al., 2017, Zhang et al., 2017), and 53BP1 is recruited to the damaged chromatin via the direct bivalent interaction of its Tudor domain and ubiquitination-dependent recruitment (UDR) motif with H4K20me2 and H2AK15ub histone modifications, respectively (Botuyan et al., 2006, Fradet-Turcotte et al., 2013, Huyen et al., 2004, Sanders et al., 2004). 53BP1 engages the modified nucleosomes as a dimer in a pincer-like binding mode, and this interaction arrangement has been postulated to enable its DNA end resection inhibitory activity (Fradet-Turcotte et al., 2013, Wilson et al., 2016).
To assess to what extent impaired oligomerization of 53BP1 affects DNA end protection against resection, we first examined the ability of 53BP1 OD mutants to recruit Rif1 to sites of ionizing radiation (IR)-induced DNA damage. To this end, we reconstituted 53bp1−/− immortalized mouse embryonic fibroblasts (iMEFs) with 53BP1WT and OD mutant constructs (Figure S2A) and monitored 53BP1 and Rif1 irradiation-induced foci (IRIF) formation by high-throughput microscopy quantification. As expected, the constitutive association of 53BP1 with chromatin (53BP1mTudor) and its DNA damage-induced recruitment (53BP1mUDR) were necessary for efficient 53BP1 IRIF formation (Figures 1F and S2B). In contrast, mutations or deletions disrupting the OD assembly or deletion of the entire OD only partially affected 53BP1 localization to DSBs (Figures 1F and S2B). Furthermore, the OD mutants were either as proficient as 53BP1WT (53BP1D1256A) or only minimally affected (53BP1ΔOligo and 53BP1ΔCore) in recruiting Rif1 to damaged chromatin (Figures 1G and S2B). This indicates that the reduced capability of 53BP1 to localize to sites of DNA damage does not significantly interfere with its ability to recruit Rif1.
Next, we tested the capability of these mutants to repress resection of DNA breaks induced by the radiomimetic drug neocarzinostatin (NCS). To this end, we used the 5-bromo-2′-deoxyuridine (BrdU) DNA end resection assay, which measures the levels of BrdU exposed by DNA processing as a quantitative readout for DNA end resection (Figure 1H). In agreement with the well-established role of 53BP1 in the inhibition of DNA end resection, cells transduced with EVor the 53BP1mTudor mutant displayed a high BrdU signal (Figures 1I and 1J), which is indicative of extensive DNA processing. Reconstitution with 53BP1WT rescued the phenotype (Figures 1I and 1J). OD mutant-expressing iMEFs exhibited only slightly increased BrdU levels compared with 53BP1WT (Figures 1I and 1J), indicating that assembly via the OD does not have a major impact on 53BP1-mediated inhibition of DNA end processing.
To assess for DNA end resection at the single nucleotide level, we used CRISPR/Cas9 nuclease and AID as DNA break-inducing source in CH12 cells. CH12 is a murine B cell lymphoma line that expresses AID and undergoes efficient CSR to IgA following cytokine stimulation (Nakamura et al., 1996). We used CRISPR-Cas9 somatic gene targeting to generate a CH12 cell line that harbored a deletion in the 53bp1 locus encompassing the OD core (53bp1Δ6 CH12) (Figures S3A and S3B). This cell line recapitulated the severe CSR deficiency (Figure S3C) of oligomutant-reconstituted primary B cells (Figure 1D). We first monitored processing of Cas9-induced breaks at the Rosa26 locus using a well-established DSB end resection assay (Rosa26-ERA; Figure S3D). 53bp1Δ6 CH12 cells were as proficient as WT cells in preventing resection of CRISPR-Cas9-induced DSBs (Figure S3D). Next, we compared the frequency and length of microhomologies (MHs) at Sμ-Sα junctions in cells activated to undergo CSR. 53BP1 deficiency results in CSR junctions skewed toward the preferential use of MHs as a consequence of increased resection of AID-induced breaks (Panchakshari et al., 2018). In agreement with this observation, 53bp1−/− CH12 cells displayed the expected bias toward MH-dependent end joining, with twice as many junctions as in WT cells presenting ≥3 bp MH (Figure S3E; Table S1). In contrast, Sμ-Sα ligation events with ≥3 bp MH were only modestly increased in 53bp1Δ6 cells (Figure S3E; Table S1). In line with these results, 53bp1Δ6 CH12 cells accumulated reduced levels of the single-stranded binding protein complex RPA at donor and acceptor switch regions compared with 53bp1−/− CH12 (Figure S3F).
Altogether, these findings indicate that the assembly of 53BP1 higher order oligomers via the OD is largely dispensable for inhibition of DSB resection yet essential for CSR.
DSB End Protection-Deficient B Lymphocytes Undergo Robust CSR
Following the generation of AID-induced breaks, 53BP1 recruits Rif1 to protect switch region breaks against resection (Chapman et al., 2013, Di Virgilio et al., 2013, Escribano-Díaz et al., 2013). Given the severe defect of the OD core mutants to support CSR despite minimally affected DNA end protection capabilities, we next examined to what extent the regulation of DNA end resection contributed to 53BP1’s role in CSR.
53BP1’s ability to inhibit DNA end resection is dependent on phosphorylation of the N-terminal SQ/TQ sites, as a mutant bearing alanine substitutions of all 28 N-terminal motifs (53BP128A; Figure 2A) cannot interact with Rif1 and support CSR despite being normally recruited to sites of DNA damage (Bothmer et al., 2011, Chapman et al., 2013, Di Virgilio et al., 2013, Escribano-Díaz et al., 2013). However, 53BP128A is also impaired in DNA damage-induced chromatin mobility, a Rif1-independent function of 53BP1 that is required for both telomere fusion and radial formation (Dimitrova et al., 2008, Lottersberger et al., 2013, Lottersberger et al., 2015, Zimmermann et al., 2013). To assess the specific contribution of Rif1-mediated inhibition of DSB resection to CSR, we generated 53BP1 mutants that harbored alanine substitutions in the SQ/TQ sites within the N-terminal DNA end protection domain (Pro domain; Figure 2A) but retained all SQ/TQ sites in the mobility domain (Mob domain; Figure 2A). This yielded 53BP1ΔPro, which contained mutations in the first 15 N-terminal SQ/TQ sites, and 53BP1ΔRif1, which preserved the SQ sites in the PTIP-binding region (Figure 2A).
Figure 2.

Inactivation of 53BP1 SQ/TQ Sites T302 to S625 Is Sufficient to Abolish Rif1-Dependent Inhibition of DNA End Resection
(A) Top: schematic representation of 53BP1 protein with the position of the 28 N-terminal SQ/TQ motifs indicated as black circles. Bottom: schematic representation of the 53BP1 phospho mutants with S/TQ sites indicated as black circles and S/TQ-to-AQ substitutions marked in light gray. Pro, DNA end protection SQ/TQ set; Mob, DNA mobility SQ/TQ set.
(B and C) Automated IRIF quantification of immunofluorescent staining for 53BP1 (B) and Rif1 (C) in irradiated (10 Gy; 2 h recovery time) 53bp1−/− iMEFs reconstituted with the indicated constructs. The top graph within each panel shows a representative experiment, and the bottom graph summarizes the mean values of three experiments performed on three independent sets of reconstituted cell lines. Each symbol in the top graph represents a single cell, and the number of scored cells was ≥200 per sample. The mean is indicated. See also Figure S2D for representative immunofluorescent staining images.
(D) Representative BrdU FACS plots showing levels of resection in NCS-treated 53bp1−/− iMEFs reconstituted with the indicated constructs.
(E) Summary graph showing quantification of the BrdU FACS data represented in (D) for six experiments performed on three independent sets of reconstituted cell lines. Numbers above each bar indicate the fold mean BrdU intensity over WT, which was set to 1.
Significance in (B), (C), and (E) was calculated using the Mann-Whitney U test, and error bars represent SD. ∗∗p ≤ 0.01 and ∗∗∗∗p ≤ 0.0001; ns, not significant. See also Figure S4B.
To assess whether abrogation of SQ/TQ phosphorylation in the 53BP1ΔPro and 53BP1ΔRif1 mutants interfered with Rif1 interaction and recruitment to DSBs, we reconstituted 53bp1−/− iMEFs with 53BP1WT and 53BP1 phospho mutants (Figure S2C), and monitored 53BP1 and Rif1 foci formation after irradiation. In agreement with the observation that N-terminal phosphorylation is dispensable for recruitment of 53BP1 to sites of DNA damage, 53BP128A (Bothmer et al., 2011), 53BP1ΔPro and 53BP1ΔRif1 foci were readily detectable following irradiation (Figures 2B and S2D). However, Rif1 did not form IRIF in 53BP1ΔPro and 53BP1ΔRif1-reconstituted cells (Figures 2C and S2D), thus indicating that both phospho mutants cannot support Rif1 relocalization to DSBs.
We next tested whether the inability of 53BP1ΔPro and 53BP1ΔRif1 to recruit Rif1 to sites of DNA damage translated into impaired DNA end resection inhibition. iMEFs reconstituted with Rif1 interaction-deficient 53BP1 mutants displayed the same increased levels of resection as cells lacking 53BP1 (EV transduced) or cells expressing a 53BP1 mutant defective in DSB-induced chromatin recruitment (53BP1mUDR) (Figures 2D and 2E). These experiments confirm that Rif1 is the major effector in the regulation of DNA end resection downstream 53BP1. Altogether, these results indicate that abrogation of phosphorylation at SQ/TQ sites T302 to S625 is sufficient to abolish Rif1 recruitment to DSBs and 53BP1-dependent DNA end protection.
To assess to what extent 53BP1-Rif1-mediated regulation of DNA end processing contributes to CSR, we reconstituted 53bp1−/− splenocytes with 53BP1ΔPro and 53BP1ΔRif1 (Figures 3A–3C). Phosphomutant-expressing splenocyte cultures displayed the same proliferation capability as their WT counterpart (Figure S4A). We first assessed 53BP1ΔPro and 53BP1ΔRif1 proficiency to interact with Rif1 following DNA damage by co-immunoprecipitation analysis. In agreement with the inability to recruit Rif1 to IR-induced DSBs in MEFs (Figures 2C and S2D), 53BP1ΔPro and 53BP1ΔRif1 failed to co-immunoprecipitate Rif1 following IR in B cells (Figure S4B). Next, we assessed CSR efficiency ex vivo in response to stimulation conditions that induce class switching to different isotypes. We monitored switching to IgG1, IgG2b, and IgG3 because CSR to these isotypes is robust, and thus it ensures a large dynamic range and a more accurate detection of even subtle differences in CSR levels. Whereas CSR was abrogated to all tested isotypes in 53bp1−/− B cells transduced with EV or 53BP1mUDR, Rif1 interaction-deficient 53BP1 rescued CSR to a considerable extent under all stimulation conditions (Figures 3D–3F; mean values of 72%, 66%, and 65% of WT levels for IgG1, IgG2b, and IgG3, respectively, for 53BP1ΔRif1). Taken together, these results indicate that 53BP1-Rif1 DSB end protection plays a limited role in 53BP1’s ability to support CSR.
Figure 3.

DSB End Protection-Deficient B Lymphocytes Undergo Robust CSR
(A–C) Representative WB analysis of 53bp1−/− B lymphocytes reconstituted with the indicated 53BP1 viral constructs and stimulated to induce CSR to IgG1 (A), IgG2b (B), or IgG3 (C). Asterisk indicates an aspecific band used for internal normalization of protein levels. Numbers underneath each lane indicate mean values from relative quantification of 53BP1 WT and mutant protein levels in reconstituted 53bp1−/− splenocytes cultures compared with endogenous protein in the WT B cell culture. Quantification summarizes WB analyses of three independently reconstituted cultures per stimulation condition.
(D–F) Top: representative flow cytometry plots measuring CSR to IgG1 (D), IgG2b (E), and IgG3 (F) in primary cultures of 53BP1 construct-reconstituted 53bp1−/− B lymphocytes. Bottom: summary dot plot for three independent experiments. CSR efficiencies within each experiment were normalized to the CSR value of 53BP1WT-reconstituted cells, which was set to 100%.
Significance was calculated using the Mann-Whitney U test, and error bars represent SD. ∗p ≤ 0.05 and ∗∗p ≤ 0.01.
53BP1-Dependent DSB Mobility Is Dispensable for CSR
53BP1 mediates the mobility of deprotected telomeres and DSBs (Dimitrova et al., 2008, Lottersberger et al., 2015). This function contributes to telomere fusion following ablation of the shelterin component TRF2, and generation of radial chromosomes in BRCA1-deficient cells (Dimitrova et al., 2008, Lottersberger et al., 2015), the other two well-established DNA repair transactions that depend on 53BP1. Alanine substitutions of an internal set of SQ/TQ motifs (53BP1ΔMob; Figure 2A) abrogate 53BP1-dependent DNA end mobility without affecting its ability to repress hyper-resection of deprotected telomeres (Lottersberger et al., 2015). In agreement with these data, 53BP1ΔMob rescued Rif1 foci formation in irradiated 53bp1−/− iMEFs to WT levels (Figures 2C and S2D) and was proficient in protecting DSBs against resection (Figures 2D and 2E).
Given the limited dependency of CSR on 53BP1 regulation of DNA end processing, we tested whether 53BP1-mediated DSB mobility contributed to CSR. To do so, we measured the CSR efficiency of 53bp1−/− splenocytes reconstituted with 53BP1ΔMob mutant. 53BP1ΔMob rescued CSR to WT levels (Figures 4A and 4B), thus indicating that inactivation of DNA mobility SQ/TQ sites did not affect 53BP1’s ability to support CSR. We concluded that 53BP1-mediated DSB end mobility is dispensable for CSR. In this regard, CSR differs from the other DNA repair transactions that are dependent on 53BP1, fusion of deprotected telomeres and radial formation, which both require the DNA end mobility activity in addition to the inhibition of DNA end resection (Figure 4C).
Figure 4.

53BP1-Mediated DSB Mobility Is Dispensable for CSR
(A) Top: representative flow cytometry plots measuring CSR to IgG1 in WT and 53bp1−/− splenocytes transduced with the indicated constructs. Bottom: summary dot plot for four independent experiments. CSR efficiencies within each experiment were normalized to the CSR value of 53BP1WT-reconstituted cells, which was set to 100%. Significance was calculated using the Mann-Whitney U test, and error bars represent SD. ∗p ≤ 0.05; ns, not significant.
(B) Representative WB analysis of 53bp1−/− B lymphocytes reconstituted with the indicated 53BP1 viral constructs.
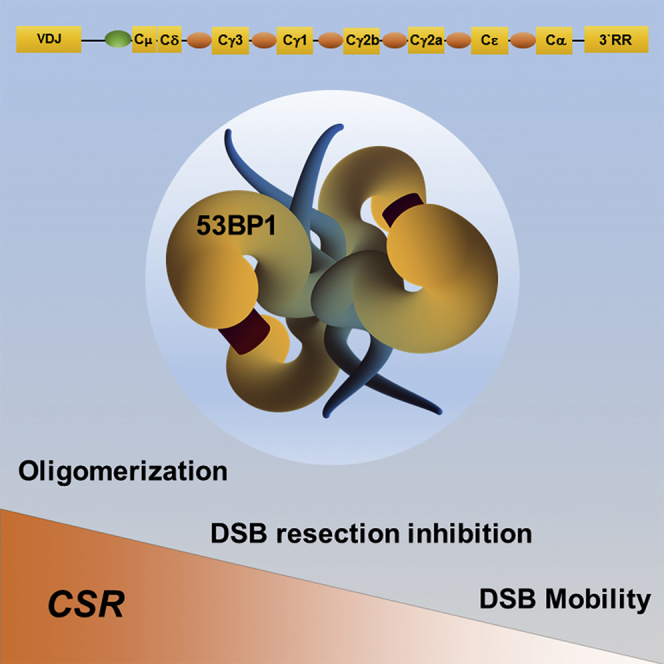
(C) Schematic representation of a 53BP1 tetramer. The blue appendages represent the N-terminal intrinsically disordered regions of each monomer and contain the Pro and Mob sets of SQ/TQ motifs. The UDR and Tudor domains of the 53BP1 monomers assemble into two nucleosome-binding dimers that are represented as yellow pincers. The relative contributions played by the regulation of DNA end processing, DSB mobility, and higher order oligomerization activities to CSR are indicated in red.
Discussion
Whereas CSR is predominantly dependent on NHEJ, the repetitive nature of the switch regions favors MMEJ-mediated intra-S recombination, and the ability of 53BP1-Rif1 to counteract DNA end processing is believed to contribute substantially to the severe CSR defects caused by deficiency in these DSB repair factors. Here, we showed that B lymphocytes devoid of 53BP1-Rif1-mediated DNA end protection can undergo robust class switching. Conversely, defects in higher order multimerization completely abolish 53BP1’s ability to support CSR without having a major impact on its DNA end resection inhibitory function. Considering the near complete abrogation of CSR in 53BP1- and Rif1-deficient B cells (Chapman et al., 2013, Di Virgilio et al., 2013, Escribano-Díaz et al., 2013, Manis et al., 2004, Ward et al., 2004), the major implication of these findings is that 53BP1 and Rif1 contribute multiple functions to CSR, with the regulation of DNA end resection activity only marginally influencing the process. This would explain the surprising observation that inhibition of CtIP-mediated end resection only negligibly rescues the CSR defect in 53BP1 and Rif1-deficient B lymphocytes (Bothmer et al., 2013, Di Virgilio et al., 2013).
Despite a partially defective association with damaged chromatin, 53BP1 OD mutants supported considerable Rif1 recruitment and inhibition of DNA end resection. These results are reminiscent of the behavior of OD-impaired 53BP1 at dysfunctional telomeres (Lottersberger et al., 2013). 53BP1ΔOligo exhibited only a mild localization defect to deprotected telomere-induced foci (TIFs) but was fully proficient with regard to counteracting CtIP-mediated telomere resection (Lottersberger et al., 2013). These observations raise the possibility that an independent oligomerization module may compensate for mutations within the OD and support sufficient levels of 53BP1 recruitment to damaged sites to ensure repression of DNA end processing (DSBs or dysfunctional telomeres). Indeed, during the preparation of this paper, another study reported that binding of the dimerization-inducing protein DYNLL1 to the LC8 motif in 53BP1 promoted OD-independent oligomerization and chromatin association (Becker et al., 2018). Given the milder chromatin recruitment defect of the 53BP1 LC8 mutant (Becker et al., 2018), the OD is likely to play a key role in the stabilization of 53BP1 oligomers.
We have found that the OD provides a tetramerization interface, and efficient higher order 53BP1 multimerization is essential for CSR. A single mutation within the OD core that still supports dimerization of the OD module abolishes CSR. Interestingly, deletion of the OD had a minor effect on the end joining of deprotected telomeres that specifically concerned the longest multichromosome fusion events, which might be limited by a less efficient interaction of distally located telomeric ends (Lottersberger et al., 2013). These findings are in agreement with a model in which 53BP1 dimers represent the minimum nucleosome-binding unit to allow DNA damage-dependent recruitment and inhibition of DNA end resection, whereas tetramerization (and higher order assembly) provides a tethering activity to bridge and stabilize distally located DNA ends (Figure 4C), including AID-induced DSBs in donor and acceptor switch regions or spatially separated telomeric ends. Consistent with this idea, 53BP1 facilitates long-range DNA end joining during V(D)J recombination (Difilippantonio et al., 2008). The combined contribution of DSB end protection and distal break tethering to CSR explains the increase in MMEJ-mediated intra-S region repair events in 53BP1-deficient B cells (Reina-San-Martin et al., 2007).
CSR is a complex process that relies on the coordinated interplay of chromatin reorganization, transcription, programmed DNA damage, and DSB repair (Chaudhuri and Alt, 2004, Methot and Di Noia, 2017, Yewdell and Chaudhuri, 2017). It is not at all surprising that specific factors could contribute multiple functions within the same or even different steps of the reaction. Indeed, additional Igh locus-specific functions have been described for 53BP1, including enforcement of orientation-specific joining of AID breaks and a structural role in the maintenance of the locus architecture (Dong et al., 2015, Feldman et al., 2017, Rocha et al., 2016). The extent to which these functions contribute to CSR is currently unknown. However, these and our findings suggest that, in addition to preventing resection of AID-induced breaks via Rif1 and the shieldin complex, 53BP1 plays the key role of maintaining the Igh locus in a configuration “primed” for productive CSR events.
No additional role during CSR has been described for Rif1 so far. However, we recently performed a proteomic-based screen to identify Rif1 interactors in primary B lymphocytes undergoing CSR and identified several transcriptional regulators in addition to genome stability factors (Delgado-Benito et al., 2018). This raises the intriguing possibility that Rif1 might contribute to the transcriptional control of CSR. Such a hypothesis is in line with several studies supporting Rif1 role as a transcriptional regulator in yeast and mammals (Dan et al., 2014, Daxinger et al., 2013, Hardy et al., 1992, Kim et al., 2008, Li et al., 2017, Loh et al., 2006, Smith et al., 1999, Tanaka et al., 2016, Wang et al., 2006) and represents an exciting new direction of investigation into the coordinated control of pre- and post-DSB phases of CSR.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rat anti-CD180 (RP/14) | BD Biosciences | Cat# 552128, RRID: AB_394343 |
| Goat anti-IgA-PE | Southern Biotech | Cat# 1040-09 |
| Mouse anti-CD40 Clone HM40-3 | BioLegend | Cat# 102902, RRID:AB_312945 |
| Rabbit anti-53BP1 | Novus Biologicals | Cat# NB100-304, RRID:AB_10003037 |
| Mouse anti-Flag M2 | Sigma-Aldrich | Cat# F3165, RRID: AB_259529 |
| Mouse anti-Flag M2 (HRP conjugated) | Sigma-Aldrich | Cat# A8592, RRID: AB_439702 |
| Rabbit anti-Rif1 | Di Virgilio et al., 2013 | N/A |
| Rabbit anti-Tubulin | Abcam | Cat# ab4074 RRID:AB_2288001 |
| Rat anti-IgG1-APC Clone X56 | BD Biosciences | Cat# 550874, RRID: AB_398470 |
| Rat anti-IgG3-BIOT Clone R40-82 | BD Biosciences | Cat# 553401, RRID: AB_394838 |
| Rat anti-IgG2b-PE Clone RMG2b-1 | BioLegend | Cat# 406707, RRID: AB_2563380 |
| Goat anti-rabbit Alexa546 | Invitrogen | Cat# A-11035, RRID: AB_2534093 |
| Goat anti-mouse Alexa488 | Invitrogen | Cat# A-11029, RRID: AB_2534088 |
| Mouse anti-BrdU APC antibody (Clone Bu20a) | BioLegend | Cat# 339808 RRID:AB_10895898 |
| Mouse anti-HA probe antibody (F-7) | Santa Cruz | Cat# sc-7392 RRID:AB_627809 |
| Rabbit anti-HA-tag (C29F4) | Cell Signaling | Cat# 3724S RRID:AB_1549585 |
| Mouse-anti-RPA2 (12F3.3) | Novus Biologicals | Cat# NB100-158 RRID:AB_2180491 |
| normal mouse IgG | Santa Cruz | Cat# sc-2025 RRID:AB_737182 |
| Bacterial and Virus Strains | ||
| E.coli BL21 Rosetta 2 | Novagen | Cat# 71400 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| RPMI 1640 | Life Technologies | Cat# 21875091 |
| HEPES | Life Technologies | Cat# 15630056 |
| Sodium Pyruvate | Life Technologies | Cat# 11360039 |
| Antibiotic Antimycotic | Life Technologies | Cat# 15240062 |
| L-Glutamine | Life Technologies | Cat# 25030024 |
| 2-Mercaptoethanol | Life Technologies | Cat# 21985023 |
| DMEM | Life Technologies | Cat# 41965062 |
| Penicillin-Streptomycin | Life Technologies | Cat# 15140122 |
| ExpressTM medium | Novagen | Cat# 71491 |
| AEBSF (4-(2-Aminoethyl)benzensulfonylfluorid) | Carl Roth | Cat# A154 |
| cOmpleteTM protease inhibitor | Roche | Cat# 11697498001 |
| Benzonase | Sigma | Cat# E1014-25KU |
| Sinapinic acid | Sigma-Aldrich | Cat# #D7927 |
| Cyano-4-hydroxycinnamic acid | Sigma-Aldrich | Cat# #C2020 |
| Rat anti-CD43 (Ly-48) Microbeads | Miltenyi Biotec | Cat# 130-049-801 |
| LPS | Sigma-Aldrich | Cat# L2630 |
| IL-4 (mouse recombinant) | Sigma-Aldrich | Cat# I1020 |
| BAFF (human recombinant) | PeproTech | Cat# 310-13 |
| TGFβ-1 (mouse recombinant) | R&D Systems | Cat# 7666-MB-005/CF |
| Puromycin dihydrochloride | Sigma-Aldrich | Cat# P8833 |
| RIPA buffer | Sigma | Cat# R0278 |
| Complete EDTA-free protease inhibitor cocktail | Roche | Cat# 11873580001 |
| CellTrace Violet | Thermofisher | Cat# C34557 |
| Paraformaldehyde | Sigma-Aldrich | Cat# P6148 |
| Hoechst 33258 | Thermofisher | Cat# H3569 |
| Neocarzinostatin (NCS) | Sigma | Cat# N9162 |
| 16% Formaldehyde Methanol-free | Thermo scientific | Cat# 28908 |
| Dynabeads Protein G | ThermoFisher | Cat# 10004D |
| FuGENE® HD Transfection Reagent | Promega | Cat# E2312 |
| Igepal CA-630 | Sigma | Cat# I8896 |
| Pierce Phosphatase Inhibitor Mini Tablets | ThermoFisher | Cat# A32957 |
| Phenylmethylsulfonyl fluoride | Sigma-Aldrich | Cat# 10837091001 |
| Pepstatin A | Merck | Cat# P5318 |
| Critical Commercial Assays | ||
| APC BrdU Flow Kit | BD Biosciences | Cat# 552598 |
| Neon Transfection System, 100 μl Kit | Invitrogen | Cat# MPK10025 |
| Expand Long Template PCR System | Sigma-Aldrich | Cat# 11681834001 |
| NucleoSpin DNA Purification Kit | Macherey-Nagel | Cat# 740499 |
| TOPO TA Cloning Kit | Invitrogen | Cat# 450641 |
| Luna® Universal One-Step RT-qPCR Kit | NEB | Cat# E3005S |
| Deposited Data | ||
| Original scans and IF images | This paper; Mendeley Data | https://data.mendeley.com/datasets/n7jxbpddnf/draft?a=d8838dbd-c9e9-40d5-b467-0150330356a1 |
| Experimental Models: Cell Lines | ||
| Murine: CH12 (CH12F3) - WT, parental | Nakamura et al., 1996 | N/A |
| Murine: 53bp1−/− CH12 | Delgado-Benito et al., 2018 | N/A |
| Murine: 53bp1Δ6 CH12 | This paper | N/A |
| Murine: 53bp1−/− iMEFs | Bothmer et al., 2011 | N/A |
| Murine: WT iMEFs | Bothmer et al., 2011 | N/A |
| Human: BOSC23 | Pear et al., 1993 | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: 53bp1−/− (and derived splenocytes cultures) | Ward et al., 2003 | N/A |
| Oligonucleotides | ||
| 53BP1ΔBRCT-3xHA construct cloning | See Table S2 | N/A |
| 53BP1ΔPRO- & 53BP1ΔMOB-3xFlag construct cloning | See Table S2 | N/A |
| CRISPR-Cas9 gene targeting | See Table S2 | N/A |
| RPA ChIP-qPCR | See Table S2 | N/A |
| End resection assay | See Table S2 | N/A |
| Switch junctional analysis | See Table S2 | N/A |
| Recombinant DNA | ||
| Plasmid: pET21b+ | Novagen | Cat# 69741 |
| Plasmid: pMAL-C2X | Addgene | Cat# #75286 |
| Plasmid: pMX-53BP1ΔBRCT-3xFlag | Bothmer et al., 2011 | N/A |
| Plasmid: pMX-53BP1ΔOligo-3xFlag | Ward et al., 2006 | N/A |
| Plasmid: pMX-53BP1ΔCore-3xFlag | This paper | N/A |
| Plasmid: pMX-53BP1D1256A-3xFlag | Zgheib et al., 2009 | N/A |
| Plasmid: pMX-53BP1mTudor-3xFlag | Botuyan et al., 2006 | N/A |
| Plasmid: pMX-53BP128A-3xFlag | Bothmer et al., 2011 | N/A |
| Plasmid: pMX-53BP1ΔPro-3xFlag | This paper | N/A |
| Plasmid: pMX-53BP1ΔRif1-3xFlag | This paper | N/A |
| Plasmid: pMX-53BP1ΔMob-3xFlag | Lottersberger et al., 2015 | N/A |
| Plasmid: pMX-53BP1mUDR-3xFlag | Fradet-Turcotte et al., 2013 | N/A |
| Plasmid: pMX-53BP1ΔBRCT-3xHA | This paper | N/A |
| Plasmid: pX458 | Addgene | Cat# #48138 |
| Plasmid: pX458-Cas9D10A | This paper | N/A |
| Software and Algorithms | ||
| BZ-H4A/Advanced Analysis Software | Keyence | N/A |
| BZ-H4C/Hybrid Cell Count | Keyence | N/A |
| BZ-H4CM/Macro Cell Count | Keyence | N/A |
| ImageJ | NIH | N/A |
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michela Di Virgilio (michela.divirgilio@mdc-berlin.de).
Experimental Model and Subject Details
Sources of mouse models and cell lines used in the study are reported below and in the Key Resources Table.
Mice strains
53bp1−/− mice (B6;129-Trp53bp1tm1Jc/J) were previously described (Ward et al., 2003). 6 to 20 weeks old animals were used for the study, and age-matched groups were employed within each experiment. Both male and female mice were used for the experiments. Mice were maintained in a specific pathogen-free (SPF) barrier facility under standardized conditions (20+/−2°C temperature; 55% ± 15% humidity) on a 12 h light/12 h dark cycle. All experiments were performed in compliance with EU Directive 2010/63/EU, and in agreement with protocols approved by Landesamt für Gesundheit und Soziales (LAGeSo, Berlin).
Primary cell cultures
Splenocytes cultures from 53bp1−/− mice were grown in RPMI 1640 medium (Life Technologies) supplemented with 10% fetal bovine serum (FBS), 10 mM HEPES (Life Technologies), 1 mM Sodium Pyruvate (Life Technologies), 1X Antibiotic Antimycotic (Life Technologies), 2 mM L-Glutamine (Life Technologies), and 1X 2-Mercaptoethanol (Life Technologies) at 37°C and 5% CO2 levels.
Cell lines
The cell lines employed for this study are: CH12 (CH12F3, murine, (Nakamura et al., 1996)), 53bp1−/− (Trp53bp1−/−) CH12 (murine, (Delgado-Benito et al., 2018)), 53bp1Δ6 CH12 (murine, this paper), WT and 53bp1−/− iMEFs (murine, (Bothmer et al., 2011)), and BOSC23 (human, (Pear et al., 1993)). CH12 cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS), 10 mM HEPES, 1 mM Sodium Pyruvate, 1X Antibiotic Antimycotic, 2 mM L-Glutamine, and 1X 2-Mercaptoethanol at 37°C and 5% CO2 levels. iMEFs and BOSC23 cells were cultured in DMEM medium (Life Technologies) supplemented with 10% FBS, 2 mM L-Glutamine, and Penicillin-Streptomycin (Life Technologies) at 37°C and 5% CO2 levels.
Method Details
Protein expression
The nucleotide-sequence encoding residues 1233-1288 of human 53BP1 (Uniprot Q12888) was amplified by PCR and cloned into a modified pET21b+ vector (Novagen #69741) encoding N-terminal MBP, followed by a 24 amino acid long linker. Site-directed mutagenesis was used to introduce the indicated mutations. The plasmids were transformed into E.coli BL21 Rosetta 2 (Novagen) and cells were grown at 37°C in ExpressTM medium (Novagen) until an optical density at a wavelength of 600 nm (OD600) of 0.9, when the temperature was switched to 18°C for overnight expression. Alternatively, protein was produced in LB medium by inducing protein expression at an OD600 of 0.4 with 1 mM IPTG and 4 h incubation at 37°C. Cells were harvested by centrifugation and cell pellets stored at −20°C until purification. Thawed cells were resuspended in 20 mM HEPES (or 20 mM Tris-HCl) pH 7.5, 150 mM NaCl, 1 mM DTT with AEBSF (4-(2-Aminoethyl)benzensulfonylfluorid) and cOmpleteTM protease inhibitor (Roche) and lysed with a fluidizer (Microfluidics). Cell lysates were centrifuged at 35,000 rpm for 45 min at 4°C. The filtered supernatant was then incubated with 25 U/ml Benzonase for 25 min at 4°C to reduce nucleic acid contaminations and subsequently applied to an equilibrated MBP affinity column (GE). The column was washed with 20 column volumes (CV) of resuspension buffer at 4°C. Protein was eluted with 10 CVs of 20 mM HEPES (or 20 mM Tris-HCl) pH 7.5, 150 mM NaCl, 1 mM DTT, 40 mM maltose. The eluted protein was concentrated to 20 mg/ml using 10 kDa cutoff Amicon spin filters, followed by size exclusion chromatography on a Superdex S200 in running buffer containing 20 mM HEPES (or 20 mM Tris-HCl) pH 7.5, 150 mM NaCl, 1 mM DTT. Pooled fractions were concentrated in 10 kDa cutoff Amicon spin filters and frozen in small aliquots. Protein size was verified using MALDI-TOF with a 4-point calibration standard and sinapinic acid or α-cyano-4-hydroxycinnamic acid as matrix.
RALS analysis
RALS analysis was performed on an analytical Superdex S200 10/300 column in running buffer containing 20 mM HEPES pH 7.5, 150 mM NaCl, 1 mM DTT, using an ÄKTApurifier and an online Malvern Viscotek RALS system (VE 3580 RI detector and 270 Dual detector). All protein samples were diluted to 2 mg/ml concentration in running buffer. 50 μl of each sample was applied and run at a flowrate of 0.5 ml/min. Data was recorded with UNICORN and OmniSEC 5.00 software and analyzed in OmniSEC 5.00 software. MBP expressed from plasmid pMAL-C2X was used as a control. Molecular weight (MW) of MBP-OD: 49.5 kDa. The clear shift in the elution volume of MBP (MW: 42.5 kDa) and MBP-ODΔCore (MW: 48.6 kDa) in Figure 1B is likely caused by the more extended shape of MBP-ODΔCore.
Cell cultures and retroviral infection
B lymphocytes were isolated from mouse spleens using anti-CD43 MicroBeads (Miltenyi Biotec) and stimulated to undergo class switching with 25 μg/ml LPS (Sigma-Aldrich) and 5 ng/ml of mouse recombinant IL-4 (Sigma-Aldrich) for CSR to IgG1; 25 μg/ml LPS, 10 ng/ml BAFF (PeproTech) and 2 ng/ml TGFβ (R&D Systems) for CSR to IgG2b; or 25 μg/ml LPS for CSR to IgG3. Cultures were supplemented with 0.5 μg/ml RP105 (BD) under all stimulation conditions. CH12 cells (Nakamura et al., 1996) were stimulated to undergo CSR to IgA by treatment with 0.5 μg/ml αCD40 (BioLegend), 5 ng/ml TGFβ and 5 ng/ml of mouse recombinant IL-4 for 48 h.
pMX-53BP1 retroviral vectors were generated by cloning the cDNA for human 53BP1ΔBRCT (aa 1-1710) into a pMX-based vector with a C-terminal 3XFlag tag. 53BP1ΔBRCT backbone was employed as WT and for mutagenesis purposes since its reduced size allows efficient retroviral packaging and infection of B cells, while functionally behaving as the full-length protein in CSR, radials formation, and fusion of deprotected telomeres (Bothmer et al., 2011, Lottersberger et al., 2013). The 53BP1 mutants employed in this study are: 53BP1ΔOligo (Δ1231-1270, (Ward et al., 2006)), 53BP1ΔCore (Δ1254-1258, this study), 53BP1D1256A (Zgheib et al., 2009), 53BP1mTudor (D1521R, (Botuyan et al., 2006)), 53BP128A (S6A, S13A, S25A, S29A, S105A, S166A, S176A, S178A, T302A, S437A, S452A, S523A, T543A, S580A, S625A, S674A, T696A, S698A, S784A, S831A, T855A, S892A, S1068A, S1086A, S1104A, S1148A, T1171A, S1219A, (Bothmer et al., 2011)), 53BP1ΔPro (S6A, S13A, S25A, S29A, S105A, S166A, S176A, S178A, T302A, S437A, S452A, S523A, T543A, S580A, S625A, this study), 53BP1ΔRif1 (T302A, S437A, S452A, S523A, T543A, S580A, S625A, this study), 53BP1ΔMob (S674A, T696A, S698A, S784A, S831A, T855A, S892A, S1068A, S1086A, S1104A, S1148A, T1171A, S1219A, (Lottersberger et al., 2015)), and 53BP1mUDR (L1619A, (Fradet-Turcotte et al., 2013)). 53BP1ΔPro was generated by combining the sequence containing the first 15 mutated SQ/TQ to SA/TA sites from 53BP128A and the sequence containing the following 13 SQ/TQ sites from 53BP1WT into pMX vector using a new SgrAI site introduced via silent mutations at nucleotides 1893 and1896 of human 53BP1 coding sequence. 53BP1ΔMob was generated by combining the sequence containing the first 15 SQ/TQ sites from 53BP1WT and the sequence containing the following 13 mutated SQ/TQ to SA/TA sites from 53BP128A into pMX vector using the same strategy employed for 53BP1ΔPro. Primers used for 53BP1ΔPro/53BP1ΔMob cloning are listed in Table S2. 53BP1ΔRif1 was generated by subcloning the sequence encompassing the first 8 SQ sites from pMX-53BP1WT into pMX-53BP1ΔPro using BsrGI cloning site. 3x haemagglutinin (3xHA) tag was cloned at the C terminus of 53BP1ΔBRCT by two sequential PCR rounds using the primers listed in Table S2.
Splenocytes infections were performed as it follows. The 293T derivative cell line BOSC23 (Pear et al., 1993) was transfected with pCL-Eco and pMX-53BP1 retroviral vectors using FuGENE® HD Transfection Reagent (Promega) to generate viral particle-containing supernatants. B cells were transduced twice with the viral supernatant, one and two days after isolation, and CSR was assessed 96 h post-activation. To control for any difference in the expression of the transduced constructs within each reconstitution experiment, 53BP1 protein levels were routinely monitored by western blot analysis, as it has been previously described (Bothmer et al., 2011, Callen et al., 2013, Di Virgilio et al., 2013, Fradet-Turcotte et al., 2013). iMEFs were transduced as described for splenocytes above, with the exception that only one round of infection was employed one day after seeding, and 2 μg/ml Puromycin (Sigma-Aldrich) was added to the cultures 24 h after infection.
Generation of 53BP1 oligo mutant expressing CH12 cell lines
CH12 clonal derivatives were generated via electroporation with gRNA pairs and nickase Cas9-based plasmid. Tandem U6 cassettes were cloned into a mutated version of pX458 expressing Cas9D10A. Single GFP-positive cells were sorted in 96-well plates and clones were allowed to grow for 12 days before expansion. Clones were validated at the level of genomic scar and protein expression. The sequences of the gRNAs employed in this study are listed in Table S2. Controls for 53bp1−/− and 53BP1 oligo mutants-expressing clonal derivatives included both WT cultures and clones derived from targeting CH12 with random sequences not present in mouse genome. CH12 cell lines were activated for 48 h with αCD40, IL-4, and TGFβ to induce CSR to IgA.
Western blot analysis and Co-IP assays
Western blot analysis of protein levels was performed on whole cell lysates prepared by lysis in RIPA buffer (Sigma) supplemented with Complete EDTA free proteinase inhibitors (Roche).
For co-immunoprecipitation of differentially tagged 53BP1 proteins, BOSC23 cells were co-transfected with equal amounts of pMX-53BP1WT-3xHA and pMX-53BP1-3xFlag (WT, ΔOligo, ΔCore, or D1256A) using FuGENE® HD Transfection Reagent. 36 hours post-transfection, cells were irradiated (20 Gy) and left to recover for 1 hour. Cells were lysed in lysis buffer (20 mM Tris HCl, 150 mM NaCl, 0.5% Igepal CA-630 (Sigma), 1.5 mM MgCl2) supplemented with Complete EDTA-free protease inhibitor cocktail, Pepstatin A (Merck), phenylmethylsulfonyl fluoride (PMSF) (Sigma), Pierce Phosphatase Inhibitors (ThermoFisher), and Benzonase (Sigma). Lysates were clarified by 15 min centrifugation at 13,000 rpm at 4°C, and incubated with magnetic beads (DynaBeads Protein G, Thermo Fisher) conjugated with anti-HA antibody (Santa Cruz) for 1 h at 4 C. Proteins were eluted by incubation in NuPAGE LDS sample buffer (Invitrogen) supplemented with 45 mM DTT for 10 min at 72°C.
For 53BP1-Rif1 co-immunoprecipitation analysis, 53bp1−/− splenocytes were activated with 25 μg/ml LPS, 5 ng/ml of mouse recombinant IL-4, and 0.5 μg/ml RP105, and infected with pMX-53BP1-3xFlag retroviral constructs as described above. At 96 h post-activation, cells were irradiated (20Gy) and left to recover for 1 hour. Cells were harvested, and washed two times with 1x PBS and one time with polyvinylpyrrolidone (PVP) buffer (20 mM HEPES, 1.2% PVP 40, Complete EDTA-free protease inhibitor cocktail, Pierce Phosphatase Inhibitors, 0.5 mM DTT, Pepstatin A, and PMSF). Cells were lysed in lysis buffer (10 mM Tris HCl, 100 mM NaCl, 0.05% Igepal CA-630, 1 mM MgCl2) supplemented with Complete EDTA-free protease inhibitor cocktail, Pierce Phosphatase Inhibitors, Pepstatin A, PMSF, and Benzonase. Lysates were clarified by 15 min centrifugation at 13,000 rpm at 4°C, and incubated with magnetic beads (DynaBeads Protein G, Thermo Fisher) conjugated with anti-Flag M2 antibody (Sigma-Aldrich) for 1 h at 4°C. Proteins were eluted by incubation in NuPAGE LDS sample buffer supplemented with 45 mM DTT for 10 min at 72°C.
The antibodies used for WB analysis are: anti-53BP1 (Novus Biological), anti-Flag M2 (HRP conjugated) (Sigma-Aldrich), anti-Rif1 (Di Virgilio et al., 2013), anti-Tubulin (Abcam), and anti-HA (Cell Signaling).
The indicated MWs of 214 and 190 kDa in the anti-53BP1 blots refer to the endogenous full-length 53BP1 and the retrovirally expressed ΔBRCT proteins, respectively.
Flow cytometry
For class switching assays, cell suspensions were stained with fluorochrome-conjugated anti-IgG1, anti-IgG3 (BD-Biosciences), anti-IgG2b (BioLegend), or anti-IgA (Southern Biotech). Samples were acquired on an LSRFortessa cell analyzer (BD-Biosciences). For cell proliferation analysis by cell tracking dye dilution, primary B cells were pulsed with 5 μM CellTrace Violet (Thermofisher) for 20 min at 37C.
Immunofluorescence
iMEFs were grown on coverslips overnight. Cells were irradiated (10 Gy IR) and allowed to recover for 2 h. Upon fixation with 4% paraformaldehyde (Sigma-Aldrich) and permeabilization with 0.5% Triton X-100, cells were stained with mouse anti-Flag M2 (Sigma-Aldrich), and rabbit anti-Rif1 serum (Di Virgilio et al., 2013) antibodies as primary antibodies, and with goat anti-rabbit Alexa546 and goat anti-mouse Alexa488 (Invitrogen) as secondary antibodies, alongside permeable nuclear dye (Hoechst dye 33258, ThermoFisher). Images were acquired using inverted LSM700 laser scanning confocal microscope (Zeiss) for the representative images in Figures S2B and S2D, and with inverted fluorescence phase contrast microscope Keyence BZ-X800E (Keyence) for the automated foci quantification in Figures 1F, 1G, 2B, and 2C.
BrdU assay for DNA end resection
The BrdU assay for DNA end resection was performed as previously described (Tkáč et al., 2016). Briefly, iMEFs were incubated with 30 μM BrdU for 24 h, followed by treatment with 200 ng/mL Neocarzinostatin (NCS) for 3 h (+NCS samples) or mock-treated (-NCS samples). Cells were trypsinized, washed with PBS, and fixed with 75% ethanol in PBS for 16 h at −20°C. Cells were washed with 0.1% Tween 20 in PBS following fixation and each subsequent incubation. Each sample was split into 2 aliquots, one of which was denatured using 2 N HCl for 45 min at 22°C in order to measure total BrdU incorporation. Blocking was performed in 5% fetal bovine serum (FBS) in PBS for 1 h at 22°C. BrdU exposed on single-stranded DNA (ssDNA) stretches as a consequence of DNA end resection was detected under native conditions by incubation with APC-conjugated anti-BrdU antibody (BioLegend) in PBS supplemented with 5% FBS for 1 h at 22°C. BrdU fluorescence intensity was acquired using the BD LSRFortessa flow cytometer (BD Biosciences). Resection levels in Figures 1J and 2E were expressed as the mean BrdU intensity of native NCS-treated cells normalized to the mean BrdU intensity value of the same cells under denatured conditions.
Rosa26 DNA end resection assay (Rosa26-ERA)
The end resection assay was performed as previously described (Delgado-Benito et al., 2018). Single guide RNAs targeting two sequences 2276 bp apart within the ROSA26 locus (gDSB-1 and gDSB-2) were cloned independently into pX458 plasmid (gDSB-1/2). CH12 cells were electroporated with a 1:1 mix of gDSB-1 and gDSB-2 constructs using the Neon Transfection System (Thermo Fisher Scientific). 24h later, GFP-positive cells were sorted and pellets were frozen. Genomic DNA was extracted according to standard protocols and individual repair junctions were amplified using nested PCR reactions. PCR products were extracted from agarose gel and sequenced. Single guide RNAs and primers used for the end resection assay are listed in Table S2.
RPA-ChIP
ChIP-qPCR for RPA was performed as it follows. 20 million cells were counted and stimulated for 30 h with 0.5 μg/ml αCD40, 5 ng/ml TGFβ, and 5 ng/ml of mouse recombinant IL-4. Cells were fixed with 1% Formaldehyde (Thermo Scientific) at 37°C for 10 min followed by addition of 1/20 volume of 2.5 M glycine (dissolved in PBS pH 7.4). Fixed cells were washed two times with ice cold PBS, centrifuged and aliquoted. Cells were resuspended in 800 μL of RIPA buffer supplemented with Complete EDTA free proteinase inhibitor and sonication was performed with a Covaris S220 Focused Ultrasonicator (peak value 105, duty factor 5, cycle/burst 200 for 20 minutes). Each ChIP was performed using 50-60 μg of chromatin. Chromatin fragments were pre-cleaned by incubation with Dynabeads Protein G (Thermofisher) with rotation at 4°C for 1 h, and immunoprecipitated with antibody-bound Dynabeads. 10 μg antibodies specific for RPA2 (Novus Biologicals) or normal mouse IgG (Santa Cruz) were used for each sample. DNA was amplified using Luna® Universal One-Step RT-qPCR Kit (NEB) on a StepOnePlus qPCR machine (Thermofisher). Primers used for RPA-ChIP analysis (Wiedemann et al., 2016) are listed in Table S2.
Switch Junction Analysis
For Sμ-Sα junctional analysis in CH12, cells were stimulated for 48 h with 0.5 μg/ml αCD40, 5 ng/ml TGFβ and 5 ng/ml of mouse recombinant IL-4, and sorted for IgA+ cells. Genomic DNA was extracted from the sorted population, and Sμ-Sα junctions were amplified by Expand long template PCR system (Sigma Aldrich). PCR conditions were: 38 cycles of denaturation 94°C for 15″, annealing 60°C for 30′, and elongation 68°C for 30′ (plus 30′ at each following cycle). PCR products were run on agarose gels, and fragments corresponding to 350-1000 bp were extracted with NucleoSpin DNA Purification Kit (Macherey-Nagel), cloned using TOPO-TA cloning kit (Invitrogen), and sequenced using M13 forward and reverse universal primers. Primers used for switch junction analysis (Lee-Theilen et al., 2011) are listed in Table S2.
Quantification and Statistical Analysis
The statistical significance of differences between groups/datasets was determined by the Mann–Whitney U test for all data presented in this study. Error bars in Figures 1D, 1J, 2E, 3D–3F, 4A, and S3C represent SD. Statistical details of experiments can also be found in the figure legends. Western blots were quantified using ImageJ image processing program (NIH). For quantification of oligomutant versus WT 53BP1-3xFlag proteins in anti-HA immunoprecipitates in Figure 1C, the signal associated with 53BP1-3xFlag proteins was normalized against the levels of 53BP1WT-3xHA in each corresponding pull-down. For quantification of 53BP1 protein levels in the western blot analyses in Figures 3A–3C, and repeats, the signal associated to 53BP1 was normalized with that of a aspecific reference band present in all lanes. IR-induced foci quantification in Figures 1F, 1G, and 2B, and 2C was performed using the BZ-H4A/Advanced Analysis Software, BZ-H4C/Hybrid Cell Count and BZ-H4CM/Macro Cell Count analysis applications (Keyence).
Data and Code Availability
Original imaging and western blot data used to generate any of the figure panels have been deposited at Mendeley: https://data.mendeley.com/datasets/n7jxbpddnf/draft?a=d8838dbd-c9e9-40d5-b467-0150330356a1
Acknowledgments
We thank all members of the Di Virgilio lab for their feedback and discussion; L. Keller and M. Driesner (Max Delbrück Center [MDC]) for support with cloning, end resection assay, and genotyping; R. Zinzen (MDC) for kindly granting access to the lab’s automated foci counting Keyence microscope; the MDC Advanced Light Microscopy (ALM) technology platform for its technical support with the immunofluorescence (IF) assays; the MDC FACS Core Facility and H.P. Rahn for assistance with cell sorting; the MDC Protein Production & Characterization Facility and A. Schütz for providing the MBP control; K. Faelber and J. Schlegel (MDC) for generating and analyzing the RALS data for the MBP control; and R. Pavri (Research Institute of Molecular Pathology [IMP], Vienna) and Y. Doksani (FIRC Institute for Molecular Oncology [IFOM], Milan) for critical reading of the manuscript. This work was supported by European Research Council (ERC) grant 638897 (M.D.V.) and by Helmholtz-Gemeinschaft Zukunftsthema “Immunology and Inflammation” (grant ZT-0027 to M.D.V. and O.D.). M.D.V. is a Helmholtz Young Investigators Group leader (Helmholtz Association).
Author Contributions
Conceptualization, M.D.V.; Investigation, D.S., A.R., M.A., D.T., S.B., T.S., V.D.-B., and V.C.; Formal Analysis, D.S. and M.D.V.; Writing – Original Draft, M.D.V.; Writing – Review & Editing, O.D. and M.D.V.; Visualization, M.D.V.; Supervision, O.D. and M.D.V.; Project Administration, M.D.V.; Funding Acquisition, O.D. and M.D.V.
Declaration of Interests
The authors declare no competing interests.
Published: August 6, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.06.035.
Supplemental Information
References
- Alt F.W., Rosenberg N., Casanova R.J., Thomas E., Baltimore D. Immunoglobulin heavy-chain expression and class switching in a murine leukaemia cell line. Nature. 1982;296:325–331. doi: 10.1038/296325a0. [DOI] [PubMed] [Google Scholar]
- Becker J.R., Cuella-Martin R., Barazas M., Liu R., Oliveira C., Oliver A.W., Bilham K., Holt A.B., Blackford A.N., Heierhorst J. The ASCIZ-DYNLL1 axis promotes 53BP1-dependent non-homologous end joining and PARP inhibitor sensitivity. Nat. Commun. 2018;9:5406. doi: 10.1038/s41467-018-07855-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boboila C., Jankovic M., Yan C.T., Wang J.H., Wesemann D.R., Zhang T., Fazeli A., Feldman L., Nussenzweig A., Nussenzweig M., Alt F.W. Alternative end-joining catalyzes robust IgH locus deletions and translocations in the combined absence of ligase 4 and Ku70. Proc. Natl. Acad. Sci. U S A. 2010;107:3034–3039. doi: 10.1073/pnas.0915067107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boboila C., Alt F.W., Schwer B. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv. Immunol. 2012;116:1–49. doi: 10.1016/B978-0-12-394300-2.00001-6. [DOI] [PubMed] [Google Scholar]
- Bothmer A., Robbiani D.F., Feldhahn N., Gazumyan A., Nussenzweig A., Nussenzweig M.C. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J. Exp. Med. 2010;207:855–865. doi: 10.1084/jem.20100244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothmer A., Robbiani D.F., Di Virgilio M., Bunting S.F., Klein I.A., Feldhahn N., Barlow J., Chen H.T., Bosque D., Callen E. Regulation of DNA end joining, resection, and immunoglobulin class switch recombination by 53BP1. Mol. Cell. 2011;42:319–329. doi: 10.1016/j.molcel.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothmer A., Rommel P.C., Gazumyan A., Polato F., Reczek C.R., Muellenbeck M.F., Schaetzlein S., Edelmann W., Chen P.L., Brosh R.M., Jr. Mechanism of DNA resection during intrachromosomal recombination and immunoglobulin class switching. J. Exp. Med. 2013;210:115–123. doi: 10.1084/jem.20121975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottaro A., Young F., Chen J., Serwe M., Sablitzky F., Alt F.W. Deletion of the IgH intronic enhancer and associated matrix-attachment regions decreases, but does not abolish, class switching at the mu locus. Int. Immunol. 1998;10:799–806. doi: 10.1093/intimm/10.6.799. [DOI] [PubMed] [Google Scholar]
- Botuyan M.V., Lee J., Ward I.M., Kim J.E., Thompson J.R., Chen J., Mer G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127:1361–1373. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botuyan M.V., Cui G., Drané P., Oliveira C., Detappe A., Brault M.E., Parnandi N., Chaubey S., Thompson J.R., Bragantini B. Mechanism of 53BP1 activity regulation by RNA-binding TIRR and a designer protein. Nat. Struct. Mol. Biol. 2018;25:591–600. doi: 10.1038/s41594-018-0083-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting S.F., Callén E., Wong N., Chen H.T., Polato F., Gunn A., Bothmer A., Feldhahn N., Fernandez-Capetillo O., Cao L. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callen E., Di Virgilio M., Kruhlak M.J., Nieto-Soler M., Wong N., Chen H.T., Faryabi R.B., Polato F., Santos M., Starnes L.M. 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell. 2013;153:1266–1280. doi: 10.1016/j.cell.2013.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casellas R., Nussenzweig A., Wuerffel R., Pelanda R., Reichlin A., Suh H., Qin X.F., Besmer E., Kenter A., Rajewsky K., Nussenzweig M.C. Ku80 is required for immunoglobulin isotype switching. EMBO J. 1998;17:2404–2411. doi: 10.1093/emboj/17.8.2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman J.R., Barral P., Vannier J.B., Borel V., Steger M., Tomas-Loba A., Sartori A.A., Adams I.R., Batista F.D., Boulton S.J. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol. Cell. 2013;49:858–871. doi: 10.1016/j.molcel.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri J., Alt F.W. Class-switch recombination: interplay of transcription, DNA deamination and DNA repair. Nat. Rev. Immunol. 2004;4:541–552. doi: 10.1038/nri1395. [DOI] [PubMed] [Google Scholar]
- Chaudhuri J., Tian M., Khuong C., Chua K., Pinaud E., Alt F.W. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature. 2003;422:726–730. doi: 10.1038/nature01574. [DOI] [PubMed] [Google Scholar]
- Dai Y., Zhang A., Shan S., Gong Z., Zhou Z. Structural basis for recognition of 53BP1 tandem Tudor domain by TIRR. Nat. Commun. 2018;9:2123. doi: 10.1038/s41467-018-04557-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan J., Liu Y., Liu N., Chiourea M., Okuka M., Wu T., Ye X., Mou C., Wang L., Wang L. Rif1 maintains telomere length homeostasis of ESCs by mediating heterochromatin silencing. Dev. Cell. 2014;29:7–19. doi: 10.1016/j.devcel.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daxinger L., Harten S.K., Oey H., Epp T., Isbel L., Huang E., Whitelaw N., Apedaile A., Sorolla A., Yong J. An ENU mutagenesis screen identifies novel and known genes involved in epigenetic processes in the mouse. Genome Biol. 2013;14:R96. doi: 10.1186/gb-2013-14-9-r96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado-Benito V., Rosen D.B., Wang Q., Gazumyan A., Pai J.A., Oliveira T.Y., Sundaravinayagam D., Zhang W., Andreani M., Keller L. The chromatin reader ZMYND8 regulates Igh enhancers to promote immunoglobulin class switch recombination. Mol. Cell. 2018;72:636–649.e8. doi: 10.1016/j.molcel.2018.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dev H., Chiang T.W., Lescale C., de Krijger I., Martin A.G., Pilger D., Coates J., Sczaniecka-Clift M., Wei W., Ostermaier M. Shieldin complex promotes DNA end-joining and counters homologous recombination in BRCA1-null cells. Nat. Cell Biol. 2018;20:954–965. doi: 10.1038/s41556-018-0140-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Virgilio M., Callen E., Yamane A., Zhang W., Jankovic M., Gitlin A.D., Feldhahn N., Resch W., Oliveira T.Y., Chait B.T. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science. 2013;339:711–715. doi: 10.1126/science.1230624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickerson S.K., Market E., Besmer E., Papavasiliou F.N. AID mediates hypermutation by deaminating single stranded DNA. J. Exp. Med. 2003;197:1291–1296. doi: 10.1084/jem.20030481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Difilippantonio S., Gapud E., Wong N., Huang C.Y., Mahowald G., Chen H.T., Kruhlak M.J., Callen E., Livak F., Nussenzweig M.C. 53BP1 facilitates long-range DNA end-joining during V(D)J recombination. Nature. 2008;456:529–533. doi: 10.1038/nature07476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova N., Chen Y.C., Spector D.L., de Lange T. 53BP1 promotes non-homologous end joining of telomeres by increasing chromatin mobility. Nature. 2008;456:524–528. doi: 10.1038/nature07433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J., Panchakshari R.A., Zhang T., Zhang Y., Hu J., Volpi S.A., Meyers R.M., Ho Y.J., Du Z., Robbiani D.F. Orientation-specific joining of AID-initiated DNA breaks promotes antibody class switching. Nature. 2015;525:134–139. doi: 10.1038/nature14970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drané P., Brault M.E., Cui G., Meghani K., Chaubey S., Detappe A., Parnandi N., He Y., Zheng X.F., Botuyan M.V. TIRR regulates 53BP1 by masking its histone methyl-lysine binding function. Nature. 2017;543:211–216. doi: 10.1038/nature21358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley D.D., Manis J.P., Zarrin A.A., Kaylor L., Tian M., Alt F.W. Internal IgH class switch region deletions are position-independent and enhanced by AID expression. Proc. Natl. Acad. Sci. U S A. 2002;99:9984–9989. doi: 10.1073/pnas.152333499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley D.D., Chaudhuri J., Bassing C.H., Alt F.W. Mechanism and control of V(D)J recombination versus class switch recombination: similarities and differences. Adv. Immunol. 2005;86:43–112. doi: 10.1016/S0065-2776(04)86002-4. [DOI] [PubMed] [Google Scholar]
- Dunnick W., Hertz G.Z., Scappino L., Gritzmacher C. DNA sequences at immunoglobulin switch region recombination sites. Nucleic Acids Res. 1993;21:365–372. doi: 10.1093/nar/21.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durandy A., Kracker S., Fischer A. Primary antibody deficiencies. Nat. Rev. Immunol. 2013;13:519–533. doi: 10.1038/nri3466. [DOI] [PubMed] [Google Scholar]
- Escribano-Díaz C., Orthwein A., Fradet-Turcotte A., Xing M., Young J.T., Tkáč J., Cook M.A., Rosebrock A.P., Munro M., Canny M.D. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell. 2013;49:872–883. doi: 10.1016/j.molcel.2013.01.001. [DOI] [PubMed] [Google Scholar]
- Feldman S., Wuerffel R., Achour I., Wang L., Carpenter P.B., Kenter A.L. 53BP1 Contributes to Igh Locus Chromatin Topology during Class Switch Recombination. J. Immunol. 2017;198:2434–2444. doi: 10.4049/jimmunol.1601947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L., Fong K.W., Wang J., Wang W., Chen J. RIF1 counteracts BRCA1-mediated end resection during DNA repair. J. Biol. Chem. 2013;288:11135–11143. doi: 10.1074/jbc.M113.457440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findlay S., Heath J., Luo V.M., Malina A., Morin T., Coulombe Y., Djerir B., Li Z., Samiei A., Simo-Cheyou E. SHLD2/FAM35A co-operates with REV7 to coordinate DNA double-strand break repair pathway choice. EMBO J. 2018;37:e100158. doi: 10.15252/embj.2018100158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fradet-Turcotte A., Canny M.D., Escribano-Díaz C., Orthwein A., Leung C.C., Huang H., Landry M.C., Kitevski-LeBlanc J., Noordermeer S.M., Sicheri F., Durocher D. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature. 2013;499:50–54. doi: 10.1038/nature12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezraoui H., Oliveira C., Becker J.R., Bilham K., Moralli D., Anzilotti C., Fischer R., Deobagkar-Lele M., Sanchiz-Calvo M., Fueyo-Marcos E. 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature. 2018;560:122–127. doi: 10.1038/s41586-018-0362-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H., Zou Y.R., Rajewsky K. Independent control of immunoglobulin switch recombination at individual switch regions evidenced through Cre-loxP-mediated gene targeting. Cell. 1993;73:1155–1164. doi: 10.1016/0092-8674(93)90644-6. [DOI] [PubMed] [Google Scholar]
- Gupta R., Somyajit K., Narita T., Maskey E., Stanlie A., Kremer M., Typas D., Lammers M., Mailand N., Nussenzweig A. DNA repair network analysis reveals shieldin as a key regulator of NHEJ and PARP inhibitor sensitivity. Cell. 2018;173:972–988.e23. doi: 10.1016/j.cell.2018.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy C.F., Sussel L., Shore D. A RAP1-interacting protein involved in transcriptional silencing and telomere length regulation. Genes Dev. 1992;6:801–814. doi: 10.1101/gad.6.5.801. [DOI] [PubMed] [Google Scholar]
- Hummel M., Berry J.K., Dunnick W. Switch region content of hybridomas: the two spleen cell Igh loci tend to rearrange to the same isotype. J. Immunol. 1987;138:3539–3548. [PubMed] [Google Scholar]
- Huyen Y., Zgheib O., Ditullio R.A., Jr., Gorgoulis V.G., Zacharatos P., Petty T.J., Sheston E.A., Mellert H.S., Stavridi E.S., Halazonetis T.D. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature. 2004;432:406–411. doi: 10.1038/nature03114. [DOI] [PubMed] [Google Scholar]
- Kim J., Chu J., Shen X., Wang J., Orkin S.H. An extended transcriptional network for pluripotency of embryonic stem cells. Cell. 2008;132:1049–1061. doi: 10.1016/j.cell.2008.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Theilen M., Matthews A.J., Kelly D., Zheng S., Chaudhuri J. CtIP promotes microhomology-mediated alternative end joining during class-switch recombination. Nat. Struct. Mol. Biol. 2011;18:75–79. doi: 10.1038/nsmb.1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P., Wang L., Bennett B.D., Wang J., Li J., Qin Y., Takaku M., Wade P.A., Wong J., Hu G. Rif1 promotes a repressive chromatin state to safeguard against endogenous retrovirus activation. Nucleic Acids Res. 2017;45:12723–12738. doi: 10.1093/nar/gkx884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh Y.H., Wu Q., Chew J.L., Vega V.B., Zhang W., Chen X., Bourque G., George J., Leong B., Liu J. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat. Genet. 2006;38:431–440. doi: 10.1038/ng1760. [DOI] [PubMed] [Google Scholar]
- Lottersberger F., Bothmer A., Robbiani D.F., Nussenzweig M.C., de Lange T. Role of 53BP1 oligomerization in regulating double-strand break repair. Proc. Natl. Acad. Sci. U S A. 2013;110:2146–2151. doi: 10.1073/pnas.1222617110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lottersberger F., Karssemeijer R.A., Dimitrova N., de Lange T. 53BP1 and the LINC complex promote microtubule-dependent DSB mobility and DNA repair. Cell. 2015;163:880–893. doi: 10.1016/j.cell.2015.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manis J.P., Gu Y., Lansford R., Sonoda E., Ferrini R., Davidson L., Rajewsky K., Alt F.W. Ku70 is required for late B cell development and immunoglobulin heavy chain class switching. J. Exp. Med. 1998;187:2081–2089. doi: 10.1084/jem.187.12.2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manis J.P., Morales J.C., Xia Z., Kutok J.L., Alt F.W., Carpenter P.B. 53BP1 links DNA damage-response pathways to immunoglobulin heavy chain class-switch recombination. Nat. Immunol. 2004;5:481–487. doi: 10.1038/ni1067. [DOI] [PubMed] [Google Scholar]
- Methot S.P., Di Noia J.M. Molecular mechanisms of somatic hypermutation and class switch recombination. Adv. Immunol. 2017;133:37–87. doi: 10.1016/bs.ai.2016.11.002. [DOI] [PubMed] [Google Scholar]
- Mirman Z., Lottersberger F., Takai H., Kibe T., Gong Y., Takai K., Bianchi A., Zimmermann M., Durocher D., de Lange T. 53BP1-RIF1-shieldin counteracts DSB resection through CST- and Polα-dependent fill-in. Nature. 2018;560:112–116. doi: 10.1038/s41586-018-0324-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu M., Kinoshita K., Fagarasan S., Yamada S., Shinkai Y., Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- Nakamura M., Kondo S., Sugai M., Nazarea M., Imamura S., Honjo T. High frequency class switching of an IgM+ B lymphoma clone CH12F3 to IgA+ cells. Int. Immunol. 1996;8:193–201. doi: 10.1093/intimm/8.2.193. [DOI] [PubMed] [Google Scholar]
- Noordermeer S.M., Adam S., Setiaputra D., Barazas M., Pettitt S.J., Ling A.K., Olivieri M., Álvarez-Quilón A., Moatti N., Zimmermann M. The shieldin complex mediates 53BP1-dependent DNA repair. Nature. 2018;560:117–121. doi: 10.1038/s41586-018-0340-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan-Hammarström Q., Jones A.M., Lähdesmäki A., Zhou W., Gatti R.A., Hammarström L., Gennery A.R., Ehrenstein M.R. Impact of DNA ligase IV on nonhomologous end joining pathways during class switch recombination in human cells. J. Exp. Med. 2005;201:189–194. doi: 10.1084/jem.20040772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchakshari R.A., Zhang X., Kumar V., Du Z., Wei P.C., Kao J., Dong J., Alt F.W. DNA double-strand break response factors influence end-joining features of IgH class switch and general translocation junctions. Proc. Natl. Acad. Sci. U S A. 2018;115:762–767. doi: 10.1073/pnas.1719988115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panier S., Boulton S.J. Double-strand break repair: 53BP1 comes into focus. Nat. Rev. Mol. Cell Biol. 2014;15:7–18. doi: 10.1038/nrm3719. [DOI] [PubMed] [Google Scholar]
- Pear W.S., Nolan G.P., Scott M.L., Baltimore D. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. U S A. 1993;90:8392–8396. doi: 10.1073/pnas.90.18.8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramiro A.R., Stavropoulos P., Jankovic M., Nussenzweig M.C. Transcription enhances AID-mediated cytidine deamination by exposing single-stranded DNA on the nontemplate strand. Nat. Immunol. 2003;4:452–456. doi: 10.1038/ni920. [DOI] [PubMed] [Google Scholar]
- Reina-San-Martin B., Difilippantonio S., Hanitsch L., Masilamani R.F., Nussenzweig A., Nussenzweig M.C. H2AX is required for recombination between immunoglobulin switch regions but not for intra-switch region recombination or somatic hypermutation. J. Exp. Med. 2003;197:1767–1778. doi: 10.1084/jem.20030569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reina-San-Martin B., Chen H.T., Nussenzweig A., Nussenzweig M.C. ATM is required for efficient recombination between immunoglobulin switch regions. J. Exp. Med. 2004;200:1103–1110. doi: 10.1084/jem.20041162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reina-San-Martin B., Chen J., Nussenzweig A., Nussenzweig M.C. Enhanced intra-switch region recombination during immunoglobulin class switch recombination in 53BP1-/- B cells. Eur. J. Immunol. 2007;37:235–239. doi: 10.1002/eji.200636789. [DOI] [PubMed] [Google Scholar]
- Revy P., Muto T., Levy Y., Geissmann F., Plebani A., Sanal O., Catalan N., Forveille M., Dufourcq-Labelouse R., Gennery A. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the hyper-IgM syndrome (HIGM2) Cell. 2000;102:565–575. doi: 10.1016/s0092-8674(00)00079-9. [DOI] [PubMed] [Google Scholar]
- Rocha P.P., Raviram R., Fu Y., Kim J., Luo V.M., Aljoufi A., Swanzey E., Pasquarella A., Balestrini A., Miraldi E.R. A damage-independent role for 53BP1 that impacts break order and Igh architecture during class switch recombination. Cell Rep. 2016;16:48–55. doi: 10.1016/j.celrep.2016.05.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders S.L., Portoso M., Mata J., Bähler J., Allshire R.C., Kouzarides T. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell. 2004;119:603–614. doi: 10.1016/j.cell.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Sfeir A., de Lange T. Removal of shelterin reveals the telomere end-protection problem. Science. 2012;336:593–597. doi: 10.1126/science.1218498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J.S., Caputo E., Boeke J.D. A genetic screen for ribosomal DNA silencing defects identifies multiple DNA replication and chromatin-modulating factors. Mol. Cell. Biol. 1999;19:3184–3197. doi: 10.1128/mcb.19.4.3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka H., Muto A., Shima H., Katoh Y., Sax N., Tajima S., Brydun A., Ikura T., Yoshizawa N., Masai H. Epigenetic regulation of the Blimp-1 gene (Prdm1) in B cells involves Bach2 and histone deacetylase 3. J. Biol. Chem. 2016;291:6316–6330. doi: 10.1074/jbc.M116.713842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkáč J., Xu G., Adhikary H., Young J.T.F., Gallo D., Escribano-Díaz C., Krietsch J., Orthwein A., Munro M., Sol W. HELB is a feedback inhibitor of DNA end resection. Mol. Cell. 2016;61:405–418. doi: 10.1016/j.molcel.2015.12.013. [DOI] [PubMed] [Google Scholar]
- Tomida J., Takata K.I., Bhetawal S., Person M.D., Chao H.P., Tang D.G., Wood R.D. FAM35A associates with REV7 and modulates DNA damage responses of normal and BRCA1-defective cells. EMBO J. 2018;37:e99543. doi: 10.15252/embj.201899543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Rao S., Chu J., Shen X., Levasseur D.N., Theunissen T.W., Orkin S.H. A protein interaction network for pluripotency of embryonic stem cells. Nature. 2006;444:364–368. doi: 10.1038/nature05284. [DOI] [PubMed] [Google Scholar]
- Wang J., Yuan Z., Cui Y., Xie R., Yang G., Kassab M.A., Wang M., Ma Y., Wu C., Yu X., Liu X. Molecular basis for the inhibition of the methyl-lysine binding function of 53BP1 by TIRR. Nat. Commun. 2018;9:2689. doi: 10.1038/s41467-018-05174-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward I.M., Minn K., van Deursen J., Chen J. p53 binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol. Cell. Biol. 2003;23:2556–2563. doi: 10.1128/MCB.23.7.2556-2563.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward I.M., Reina-San-Martin B., Olaru A., Minn K., Tamada K., Lau J.S., Cascalho M., Chen L., Nussenzweig A., Livak F. 53BP1 is required for class switch recombination. J. Cell Biol. 2004;165:459–464. doi: 10.1083/jcb.200403021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward I., Kim J.E., Minn K., Chini C.C., Mer G., Chen J. The tandem BRCT domain of 53BP1 is not required for its repair function. J. Biol. Chem. 2006;281:38472–38477. doi: 10.1074/jbc.M607577200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiedemann E.M., Peycheva M., Pavri R. DNA replication origins in immunoglobulin switch regions regulate class switch recombination in an R-loop-dependent manner. Cell Rep. 2016;17:2927–2942. doi: 10.1016/j.celrep.2016.11.041. [DOI] [PubMed] [Google Scholar]
- Wilson M.D., Benlekbir S., Fradet-Turcotte A., Sherker A., Julien J.P., McEwan A., Noordermeer S.M., Sicheri F., Rubinstein J.L., Durocher D. The structural basis of modified nucleosome recognition by 53BP1. Nature. 2016;536:100–103. doi: 10.1038/nature18951. [DOI] [PubMed] [Google Scholar]
- Winter E., Krawinkel U., Radbruch A. Directed Ig class switch recombination in activated murine B cells. EMBO J. 1987;6:1663–1671. doi: 10.1002/j.1460-2075.1987.tb02415.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan C.T., Boboila C., Souza E.K., Franco S., Hickernell T.R., Murphy M., Gumaste S., Geyer M., Zarrin A.A., Manis J.P. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature. 2007;449:478–482. doi: 10.1038/nature06020. [DOI] [PubMed] [Google Scholar]
- Yancopoulos G.D., DePinho R.A., Zimmerman K.A., Lutzker S.G., Rosenberg N., Alt F.W. Secondary genomic rearrangement events in pre-B cells: VHDJH replacement by a LINE-1 sequence and directed class switching. EMBO J. 1986;5:3259–3266. doi: 10.1002/j.1460-2075.1986.tb04637.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yewdell W.T., Chaudhuri J. A transcriptional serenAID: the role of noncoding RNAs in class switch recombination. Int. Immunol. 2017;29:183–196. doi: 10.1093/intimm/dxx027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zgheib O., Pataky K., Brugger J., Halazonetis T.D. An oligomerized 53BP1 tudor domain suffices for recognition of DNA double-strand breaks. Mol. Cell. Biol. 2009;29:1050–1058. doi: 10.1128/MCB.01011-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang A., Peng B., Huang P., Chen J., Gong Z. The p53-binding protein 1-Tudor-interacting repair regulator complex participates in the DNA damage response. J. Biol. Chem. 2017;292:6461–6467. doi: 10.1074/jbc.M117.777474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M., de Lange T. 53BP1: pro choice in DNA repair. Trends Cell Biol. 2014;24:108–117. doi: 10.1016/j.tcb.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M., Lottersberger F., Buonomo S.B., Sfeir A., de Lange T. 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science. 2013;339:700–704. doi: 10.1126/science.1231573. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original imaging and western blot data used to generate any of the figure panels have been deposited at Mendeley: https://data.mendeley.com/datasets/n7jxbpddnf/draft?a=d8838dbd-c9e9-40d5-b467-0150330356a1