

Abstract
Adenosine triphosphate phosphoribosyltransferase (ATP-PRT) catalyzes the first step in histidine biosynthesis, a pathway essential to microorganisms and a validated target for antimicrobial drug design. The ATP-PRT enzyme catalyzes the reversible substitution reaction between phosphoribosyl pyrophosphate and ATP. The enzyme exists in two structurally distinct forms, a short- and a long-form enzyme. These forms share a catalytic core dimer but bear completely different allosteric domains and thus distinct quaternary assemblies. Understanding enzymatic transition states can provide essential information on the reaction mechanisms and insight into how differences in domain structure influence the reaction chemistry, as well as providing a template for inhibitor design. In this study, the transition state structures for ATP-PRT enzymes from Campylobacter jejuni and Mycobacterium tuberculosis (long-form enzymes) and from Lactococcus lactis (short-form) were determined and compared. Intrinsic kinetic isotope effects (KIEs) were obtained at reaction sensitive positions for the reverse reaction using phosphonoacetic acid, an alternative substrate to the natural substrate pyrophosphate. The experimental KIEs demonstrated mechanistic similarities between the three enzymes and provided experimental boundaries for quantum chemical calculations to characterize the transition states. Predicted transition state structures support a dissociative reaction mechanism with a DN*AN‡ transition state. Weak interactions from the incoming nucleophile and a fully dissociated ATP adenine are predicted regardless of the difference in overall structure and quaternary assembly. These studies establish that despite significant differences in the quaternary assembly and regulatory machinery between ATP-PRT enzymes from different sources, the reaction chemistry and catalytic mechanism are conserved.
Graphical Abstract
The histidine biosynthetic pathway is essential in bacteria, fungi, and plants, whereas mammals lack this pathway and require histidine in their diet. This difference makes enzymes of the histidine biosynthesis pathway attractive antimicrobial targets. A histidine auxotroph, lacking a central 0.7-kbp fragment of the hisDC gene, of Mycobacterium tuberculosis (Mtu) did not survive single amino acid starvation experiments in an environment similar to that encountered in the intercellular compartment where the bacteria reside.1-3 In addition, inhibition of histidinol dehydrogenase (HisD), showed growth inhibition for Brucella suis in minimal media and in human macrophages.4 These results further validate the ATP-PRT enzyme as a suitable drug target.
Histidine biosynthesis comprises 10 enzymatic steps with the first step of the pathway catalyzed by adenosine triphosphate phosphoribosyltransferase (ATP-PRT).5 The enzyme catalyzes a substitution reaction between adenosine triphosphate (ATP) and phosphoribosyl pyrophosphate (PRPP) to give phosphoribosyl-ATP (PRATP; Figure 1A). Nine additional enzymatic steps yield the end product histidine. The ATP-PRT enzyme is allosterically inhibited by histidine, providing control for histidine biosynthesis in response to the cellular histidine concentration. ATP-PRT belongs to the phosphoribosyltransferase (PRT) enzyme family, which catalyzes the transfer of ribose 5-phosphate from PRPP to a nitrogenous, and commonly aromatic, base. The PRT family is divided into four protein types based on sequence and structure. Classification of type I PRT enzymes is based on a shared core domain responsible for PRPP binding. Additional loops, and in some cases an additional domain, are responsible for binding the nucleophile.6 The shared core domain contains a characteristic 13 amino acid sequence motif responsible for PRPP binding.6 Type II and III PRTs have novel folds compared to type I PRTs, and the classification of these enzymes as part of the PRT family is based on their reaction chemistry. ATP-PRT is a type IV PRT and exists in two structurally distinct forms, a long- and a short-form enzyme (Figure 1B and C). The two forms share a catalytic core, comprised of two mixed α/β domains. Two of these catalytic cores come together to deliver a catalytic dimer in both the long- and short-form enzymes. The two enzyme forms differ in their allosteric machinery and quaternary assembly.7 The long-form enzyme is a homohexamer with each chain comprised of the catalytic core, and a covalent C-terminal regulatory domain containing the allosteric binding site for histidine (Figure 1B).8 The short-form is a hetero-octamer containing two catalytic dimers that associate with a second discrete domain (denoted HisZ) for allosteric regulation by the binding of histidine (Figure 1C).7
Figure 1.

(A) The reaction catalyzed by the ATP-PRT enzyme between PRPP and ATP. (B) ATP-PRT long form with the catalytic core in blue and the covalently linked regulatory domain in yellow (PDB: 4YB6). (C) ATP-PRT short form with the catalytic core in blue and the regulatory unit in red (PDB: 1Z7N).
The active site is found in the cleft of the two domains (domain I and II) of the catalytic core, in both short- and long-form ATP-PRT proteins. Crystal structures of ATP-PRT long-form with substrate ATP and natural inhibitors His, AMP, and a His/AMP combination, as well as for the short-form enzyme with PRPP bound at the active site, have helped identify key residues in the binding sites.8-11 Domain II shares parts of the signature amino acid sequence also found for type I PRT enzymes responsible for PRPP binding. ATP-PRT, however, lacks the flexible hood domain observed to sit over the PRPP binding site in type I enzymes. Domain I is predominantly responsible for ATP binding with the binding site only partially conserved across both enzyme forms.
ATP-PRT reversibly catalyzes the substitution reaction between PRPP and ATP.12 Like all known PRT enzymes, this reaction depends on magnesium. Overall, the ATP-PRT enzyme follows an ordered sequential reaction mechanism with ATP binding first followed by PRPP. Pyrophosphate (PPi) is the first product released, followed by PRATP.13,14 The reaction proceeds with overall inversion of the stereochemistry at the site of the substitution. Preliminary kinetic isotope effect (KIE) work was used by Goitein et al. in 1978 to identify the reaction mechanisms of a number of PRT enzymes, including the ATP-PRT (long-form) from yeast. The results for ATP-PRT gave inconclusive results.15 Since then, the transition state analysis has been accomplished for several type I PRTs (bacterial, protozoan, and human orotate PRTs) 16,17 and a type II PRT (nicotinamide phosphoribosyltransferase (NAMPT)),18 with transition states ranging from associative for NAMPT to early and late dissociative (type I PRTs). ATP-PRT differs from these examples in that purine N1 is a nucleophilic center. It shows two distinct allosteric regulatory mechanisms, and the effect of these on the catalytic mechanism has not been shown.
The use of experimental intrinsic KIEs at reaction sensitive positions in combination with computational chemistry to match the experimental and calculated data provides atomic level detail on bond angles, bond distances, and charge distribution of the transition state (TS) of the enzyme-catalyzed chemical reaction.19 The TS structure also provides a blueprint with vital information for the design of (stable) TS analogues shown to bind their target enzyme with high specificity and potency.20,21 Here, we present the first example of a full TS analysis on type IV PRT enzymes. Our studies probe the reaction mechanisms of ATP-PRT enzymes from Campylobacter jejuni (CjeATP-PRT) and Mycobacterium tuberculosis (MtuATP-PRT), representing the long form, and from Lactococcus lactis (Lla), providing a short-form enzyme. Our results reveal that these enzymes go through a dissociative DN*AN‡ TS with a fully dissociated ATP purine ring and little involvement of the incoming nucleophile.
RESULTS AND DISCUSSION
Intrinsic KIEs and Commitment to Catalysis (Cf and Cr).
When measuring isotope effects by internal competition, the experimental KIEs include contributions from commitment to catalysis, which can lead to suppression or complete masking of experimental KIEs in some cases. The work by Goitein et al. on the primary 14C and α-secondary 3H KIEs on both the forward and reverse reaction of ATP-PRT from yeast was inconclusive, as a result of unrecognized commitment to catalysis problems.15 Commitment to catalysis problems have been successfully reduced through different approaches, such as working away from physiological pH or by using substrates that exhibit a decreased reaction rate and an increased steady state Km value, allowing the expression of the KIEs on the reaction chemistry to be observed.18,22,23 Additionally, for substrate and product stability reasons, the reaction was studied in reverse. To this end, the ATP-PRT catalyzed reaction was studied using phosphonoacetic acid (PA) as an alternative to substrate pyrophosphate for the reverse reaction. PA has been successfully employed in similar studies on type I PRT enzymes by Tao et al.16 Transition state information has been used to design potent PRT inhibitors.24
The required isotopically labeled PRATP was generated using enzymatic transformations from D-ribose to give [1′-14C] and [1′-3H]. [5′-14C] and [5′-3H]PRATP, containing isotope labels distal to the reaction site, were prepared for use as remote labels. The [5′-14C] KIE is assumed to be at unity as it is three bonds away from the reaction center, within the ribosyl group, and does not directly participate in the reaction chemistry.18,25 [1-15N, 5′-14C] and [6-15N, 5′-14C]PRATP included a silent remote 5′-14C to enable tracking of 15N at the position of interest through scintillation counting.15,26 In a typical competitive KIE experiment, substrate labeled at a reaction sensitive position, for example [1′-14C]PRATP, was combined with an appropriate remote label to track the light isotope at the position of interest, in this case, [5′-3H]PRATP. The experimental KIE was determined from the change in 14C/3H ratio for d-β-5-phosphoribosyl-1-phosphonoacetic acid (PRPA), isolated from PRATP on a charcoal column, at both partial and full chemical conversions (eq S1). Where the 5′-3H remote label was used, the observed KIEs were corrected for a small [5′-3H] KIE (eq S2) to give the experimental values shown in Table 1. The remote 5′-3H position was not further included in the TS analysis.
Table 1.
KIE Results for ATP-PRT Enzymes a
| MtuATP-PRT | CjeATP-PRT | LlaATP-PRT | |
|---|---|---|---|
| 1′-3H | 1.250 ± 0.008 (10) | 1.212 ± 0.002 (12) | 1.147 ± 0.006 (12) |
| 1′-14C | 1.028 ± 0.001 (8) | 1.029 ± 0.002 (10) | 1.031 ± 0.001 (8) |
| 1-15N, 5′-14C | 1.024 ± 0.001 (8) | 0.995 ± 0.003 (8) | 1.028 ± 0.001 (8) |
| 6-15N, 5′-14C | 1.012 ± 0.001 (8) | 1.005 ± 0.001 (8) | 0.996 ± 0.003 (8) |
| 5′-3H | 1.017 ± 0.001 (8) | 1.009 ± 0.001 (8) | 1.003 ± 0.001 (8) |
The number of independent KIE measurements is in parentheses. Errors on each experiment are given as standard error of the mean.
The forward commitments to catalysis were measured for the three enzymes using the isotope trapping method developed by Rose.27 Isotopically labeled PRATP was incubated with enzyme followed by the addition of a large excess of unlabeled PRATP and PA, the second substrate. The amount of labeled PRPA formed was then used to calculate the commitment factor (eqs S3-S5).27 CjeATP-PRT gave a forward commitment factor of 0.029, and a forward commitment factor of 0.027 was found for the LlaATP-PRT enzyme. After correction of the experimental KIEs using eq S6 (SI), the values are within the experimental errors, confirming that the experimental KIE values are near intrinsic (Table 1). Commitment to catalysis experiments was not accomplished with MtuATP-PRT, as insufficient catalytic conversion was observed under the conditions used to measure the commitment factors. Given the large 1′-3H KIE of 1.25 and a 1-15N KIE near the theoretical maximum, as well as the observation of low forward commitments for the other two enzymes, the experimental KIEs for MtuATP-PRT were assumed to be near or equal to intrinsic values. The large KIE values for all three enzymes also indicate that reverse commitments are negligible under our experimental conditions.
For all three enzymes, large 1′-3H KIEs (15–25%) and small to moderate 1′-14C KIEs (~3%) indicate a dissociative reaction mechanism with low bond order to the nucleophile (PA) and leaving group (ATP) at the TS. This is further supported by the large 1-15N KIE, which arises from the extent of C1′–N1 bond dissociation at the TS. MtuATP-PRT and LlaATP-PRT gave 1-15N KIEs within the range expected for a fully dissociated purine base.28
Computation of Transition States.
A TS model for ATP-PRT consistent with the experimental KIE data was developed by systematically matching the experimental KIEs to those calculated from DFT calculations. Calculations were carried out in vacuo according to well-established methods which have been tested rigorously for small molecule KIEs.17,29-31 TS structures determined using this approach have led to useful representations of enzymatic TSs and are supported by tight binding of TS analogues designed based on these structures.17,24
The calculated KIEs are obtained from vibrational differences between a reactant state (ground state (GS)) model and the TS model using the ISOEFF98 program.32 The crystal structure of the CjeATP-PRT core mutant (PDB: 5UBG) with PRATP bound was used for the initial geometry of the GS model. The PRATP structure was truncated at the C1”—N9 bond to reduce computational cost, which is a recognized approach to evaluate enzyme transition states.31 The optimized reactant structure that gave the lowest overall energy and that contained no imaginary frequencies was selected as the reactant geometry for the KIE calculations. To find the theoretical maximum magnitude of the 1-15N and 6-15N KIE, the adenine base was modeled as fully dissociated, at which the KIE is equivalent to the equilibrium isotope effects (EIE) and the effects of charge and tautomer state on the 1-15N and 6-15N EIE were investigated (Table 2 and Figure 2).
Table 2.
Calculated EIEs for a Fully Dissociated Adenine
| base structure | 1-15N | 6-15N |
|---|---|---|
| H9–amino | 1.016 | 0.989 |
| H3–imino (E) | 1.019 | 1.000 |
| H3–imino (Z) | 1.022 | 0.999 |
| H3–amino | 1.014 | 0.988 |
| H7–amino | 1.017 | 0.996 |
Figure 2.

Investigated adenine base structures.
With the nitrogen isotope effects suggesting a dissociated nitrogenous base (discussed in more detail below), the TS is expected to resemble the PRPA species. Crystal structures can provide good starting coordinates for TS model generation. However, no high resolution crystal structure with PRPA, or a related molecule, bound to the active site of an ATP-PRT enzyme was available. Given the highly conserved binding mode of PRPP for type I PRTs and the high levels of structural conservation around the PRPP binding site for type IV (ATP-PRT) and type I PRTs, a similar binding mode for PRPP was assumed for the ATP-PRT enzyme as indicated by an overlay of this conserved region (Figure 4C). A crystal structure of a type I PRT was considered a reasonable starting point for the TS model generation. Thus, the crystal structure of adenine PRT from Giardia lamblia (PDB: 1L1R) with PRPP bound was used as a starting model. The model included the catalytically essential Mg2+ ion and two coordinated water molecules to satisfy the octahedral coordination of magnesium. The pyrophosphate was replaced in silico by phosphonoacetic acid. Additionally, the 5′-phosphate group orientation was frozen in the TS models by constraining the O4′–C4′–C5′–H5′ and the C4′–C5′–O5′–P′ dihedral angles to 172.7° and 108.5°, respectively, as observed in the input crystal structure (PDB: 1L1R). A series of TS models was generated by systematically altering the 1′C–OPA bond distance along the reaction coordinate from 1.60 to 3.00 Å in point calculations and the corresponding KIEs calculated for each model.
Figure 4.

(A) ATP bound to the active site of CjeATP-PRT (PDB: 4YB7) with the main chain in yellow and H-bonds as yellow dashed lines. (B) ATP bound to the active site of MtuATP-PRT (PDB: 5U99) with the main chain in green and second chain in purple, H-bonds as yellow dashed lines. (C) Overlay of the conserved phosphate binding loop between PRPP bound Giardia lamblia adenine PRT (PDB: 1L1R), ATP bound CjeATP-PRT (PDB: 4YB7), and ATP bound MtuATP-PRT (PDB: 5U99). (D) A partial sequence alignment with pink highlighted regions showing low levels of conservation between the three enzymes leading to changes in the binding mode of ATP. Cyan highlighted residues show high levels of conservation.
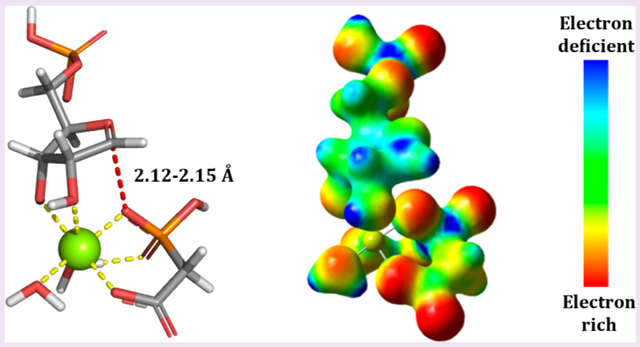
Bond distances with minimal bond order to the incoming PA nucleophile matched the intrinsic KIE values best (Figure 3). Low bond order is reflected by small imaginary frequencies which have no significant effect on the 1′-14C KIEs. This structure predicts that the calculated KIEs will be similar to EIEs and the TS structure is closely related to a fully dissociated stabilized ribooxocarbenium ion intermediate. The subsequent TS structures were modeled accordingly, with the models optimized as TS intermediates. Structures with a bond distance of 2.12, 2.14, and 2.15 Å for LlaATP-PRT, CjeATP-PRT, and MtuATP-PRT, respectively, gave the best match between calculated and experimental KIEs for 1′-14C (Table 3). The match between the model and experimental KIEs is further supported by predicted large 1′-3H KIEs, which were in close agreement for the CjeATP-PRT and MtuATP-PRT enzymes. The predicted 1′-3H KIE was slightly larger for LlaATP-PRT. This difference can arise from van der Waals interactions and/or base orientation with neighboring catalytic site groups, both of which are difficult to capture in the TS model. For example, steric crowding at the 1′-3H position at the transition state is known to decrease out-of-plane modes and thereby decrease the size of the KIE, as observed for LlaATP-PRT.33
Figure 3.

TS structures for LlaATP-PRT, CjeATP-PRT, and MtuATP-PRT in stick and electrostatic potential surface (ESPS) representation. TSs were determined in vacuo using the B3LYP functional and the 6-31*G(d,p) basis set implemented in Gaussian 09. The ESPSs were generated using the Cube program part of the Gaussian program package. Coloring indicates electron rich (red) to electron deficient (blue) through a color gradient.
Table 3.
Bond Distances That Gave the Closest Match between Calculated and Experimental KIEs
| 1′C–OPA bond distance | 1′-14C KIE calculated | 1′-14C KIE experimental | 1′-3H KIE calculated | 1′-3H KIE experimental | |
|---|---|---|---|---|---|
| LlaATP-PRT | 2.12 Å | 1.031 | 1.031 ± 0.001 | 1.227 | 1.147 ± 0.008 |
| CjeATP-PRT | 2.14 Å | 1.029 | 1.029 ± 0.001 | 1.236 | 1.212 ± 0.002 |
| MtuATP-PRT | 2.15 Å | 1.028 | 1.028 ± 0.002 | 1.240 | 1.250 ± 0.008 |
The 1-15N and 6-15N KIEs and Involvement of the Leaving Group at the Transition State.
Large 1-15N intrinsic KIEs were measured for MtuATP-PRT and LlaATP-PRT at 1.024 and 1.028, respectively. These values suggest that there is a substantial increase in vibrational freedom following dissociation of the N-glycosidic bond. The 6-15N isotope effect near unity at 0.996 ± 0.003 for LlaATP-PRT suggests no significant change in vibrational freedom upon dissociation at this position, whereas the MtuATP-PRT enzyme gave a significant change with a 6-15N isotope effect of 1.012 ± 0.001.
The formation of an anionic adenine base was excluded based on pKa considerations and the lack of residues at the active site able to stabilize this negatively charged species (Figure 4). A positively charged leaving group was ruled out based on the calculated 1-15N KIEs of 1.014 and 1.017 (Table 2, H3-amino and H7-amino, respectively), being nearly 1% lower than the experimental value. A neutral leaving group as the H3 tautomer is in good agreement with the experimental values, with the best match to the 1-15N and 6-15N KIEs being the H3–imino (Z) adenine ring for the MtuATP-PRT and LlaATP-PRT enzymes. The generation of a neutral adenine would require a proton donor. To validate the computation data, substrate bound crystal data were explored.
X-ray crystal data for the CjeATP-PRT enzyme with ATP bound at the active site has been reported previously.8 Furthermore, as part of this study, a structure with ATP bound to MtuATP-PRT (PDB: 5U99) was obtained, providing insight into the environment of the purine base. While the ATP binding sites for all three enzymes share overall shape and architecture, there are differences in the residues that contribute to binding (Figure 4). These differences give rise to changes in binding mode, particularly for the triphosphate moiety of ATP. There are also differences in residues that interact directly with the adenine ring. The crystal structure of MtuATP-PRT revealed a water molecule near to the N3 of ATP held in place by Mg2+, and no further polar interactions from active site residues or resolved waters were observed in other parts of the adenine ring, supporting the H3–imino (Z) at the TS.
No crystal structure of LlaATP-PRT with ATP bound is available, so a comparison was made to the CjeATP-PRT and MtuATP-PRT, as these enzymes both share sequence similarity to the LlaATP-PRT enzyme. Like the CjeATP-PRT (and unlike MtuATP-PRT), an arginine (Arg10) sits over the adenine ring system to stabilize binding (Figure 4C). How the active site arginine affects the nitrogenous base during the TS is difficult to say, given the observation of a large 1-15N isotope effect for the LlaATP-PRT enzyme and a KIE at unity for CjeATP-PRT. Although it is noted that this additional interaction does not appear to alter the binding mode of ATP between CjeATP-PRT and MtuATP-PRT. The binding site for the triphosphate moiety (Lys50, Pro51) more closely aligns with features of MtuATP-PRT than CjeATP-PRT. This change places the Mg2+ and coordinated water molecule close to the adenine ring described above. These results also support a fully dissociated adenine at the TS for LlaATP-PRT.
CjeATP-PRT ase gave a 1-15N isotope effect near unity of 0.995 ± 0.003 (Table 1). An isotope effect near unity indicates no change in the sum of bond vibrational interactions between the GS and the TS through bond breaking and different forms of equilibrated adenine bases (Figure 2). Loss of the N1-ribosyl group at the TS is therefore replaced by adenine ring conjugation or polarization changes that compensate for N1 bond changes.18,34 Given the similarities of the active sites of LlaATP-PRT and CjeATP-PRT around the adenine binding pocket in the crystal structures, the moderate 1′-14C KIE, and large α-secondary 3H isotope effect suggesting a dissociated transition state, this result must be explained through compensation by several bonding changes distinct to CjeATP-PRT at the TS.
Transition State Structures and 1′-14C and 1′-3H KIEs.
Transition state models with calculated KIEs that closely matched the primary 1′-14C intrinsic KIE were selected, as this position directly reports on the reaction mechanism. Additionally, this position is insensitive to geometry changes resulting from binding and is not expected to be affected by hydrogen bonding interactions at the active site.35 In the case of N-ribosyltransferase enzymes, moderate 14C isotope effects in the range of 1.01–1.06 suggest a dissociative reaction mechanism with small but significant interactions from either the nucleophile or leaving group at the TS. The 1′-14C KIEs are supported by α-secondary or 1′-3H KIEs, with larger isotope effects consistent with a dissociative reaction mechanism. All ATP-PRT enzymes gave a primary isotope effect in the range of 1.028–1.031, suggesting closely related TSs that are consistent with a dissociative reaction mechanism, which are again supported by large 1′-3H KIEs of 1.15–1.25 as well as large 1-15N KIEs.
A transition state with minimal bond order to the attacking phosphonoacetic acid with a C1′-OPA bond distance of 2.12 Å for LlaATP-PRT, 2.14 Å for CjeATP-PRT, and 2.15 Å for MtuATP-PRT gave the best match between experimental and calculated KIEs. This TS corresponds to a dissociative reaction mechanism and a Dn*AN‡ TS for the three enzymes, following IUPAC conventions and based on the length of the C1′-OPA bond being less than ≤2.2 Å and no participation by ATP.36,37 Going from the ground state to the TS, there is a positive charge build-up at the C1′ position as indicated by the change in Mulliken charge of −0.560 at the ground state and 0.253–0.290 at the TSs. The ribose ring oxygen stabilizes the positive charge, resulting in the generation of a double bond characteristic, as this center undergoes a hybridization change from sp3 toward sp2, characteristic for a carbocation species. The Mulliken charge again highlights this change with reduced negativity at the oxygen atom from −0.302 in the GS to −0.013 to 0.019 at the TS. The double bond characteristic is also reflected in the C1′-O4′ bond length, which is significantly shortened in all three TSs. The catalytically essential divalent magnesium cation plays a crucial role in stabilizing charge build up at the nucleophilic oxygen as well as anchoring the ribooxocarbenium ion through the 2,3-hydroxyls. Furthermore, the magnesium serves to anchor the correct position of the incoming nucleophile by coordinating the nucleophilic oxygen of the phosphonate and carboxylic acid functionality.
Analysis of the reaction mechanism and the prediction of the TS for ATP-PRT enables the design of inhibitors. Successful inhibitor design should at least capture two key features. First, the inhibitor should mimic the ribooxocarbenium ion charge distribution. Second, an inhibitor should include features of a neutral ATP base, with appropriate bond distances (≥3 Å) to the ribooxocarbenium analogue. Including these features will help provide for selectivity for the ATP-PRT enzyme over other enzymes involving PRPP as a substrate.
Conclusions.
The results described here present the first detailed description of the reaction catalyzed by ATP-PRT and provide the first accurate representation of the transition state structure for type IV PRT enzymes. Experimental KIE analysis revealed that all three enzymes studied displayed highly dissociative reaction mechanisms with low bond order to the phophonoacetic acid nucleophile and no significant bond order to the ATP adenine leaving group, and the ribosyl moiety was found to have significant carbocation characteristics.
Variation among the TSs for enzymes catalyzing the same reaction is not uncommon. For example, significant differences between the TSs have been reported for human and bovine purine nucleoside phosphorylase (PNP).29,30,34 Although, significantly different in reaction mechanism, the enzymes share 87% sequence identity. Comparatively, P. falciparum and human OPRT share only 26% identity in amino acid sequence but display very similar TSs.17 The ATP-PRT long and short form are different from the given examples in that they share a common catalytic core but have distinct regulatory mechanisms and quaternary assemblies. Overall, the three ATP-PRT enzymes share ~30% sequence identity, regardless of the difference in structure. On the basis of the experimental KIE results, the TSs for Mtu, Cje, and LlaATP-PRT were predicted to be very similar to C1′–OPA bond distances within 0.03 Å across the three enzymes and all three enzymes going through a DN*AN‡‡ TS. The similarities of results and the close resemblance of the TS for the ATP-PRT long and short forms suggest that the reaction chemistry is independent of the regulatory domains, mechanism, and overall quaternary structure. These findings support the proposal that both forms of ATP-PRT share a common ancestor consisting of a catalytic core and that the challenge of regulation for the control of this metabolically demanding pathway was overcome through divergent evolution.
METHODS
Protein Expression and Purification.
CjeATP-PRT and LlaATP-PRT were expressed and purified according to published procedures.7,8 MtuATP-PRT was expressed and purified following a modified procedure.9 A tobacco etch virus (TEV) cleavage site was included at the N-terminus, and the His purification tag was removed after the metal affinity chromatography step by incubating the enzyme at 37 °C for 2 h in the presence of TEV protease, followed by purification on a HiPrep 26/60 Sephacryl S-200 HR size exclusion chromatography column. Saccharomyces cerevisiae APRTase, EcoRK, and MtuPRPPase were expressed and purified according to previously reported procedures.38-40
Crystallization of MtuATP-PRT and Structure Determination.
Co-crystallization was achieved according to previously established conditions (1.6 M magnesium sulfate and 0.1 M MES, pH 6.5)9 with the addition of 10 mM ATP. Crystals were grown under vapor diffusion by the hanging drop method (drop size 2—4 μL and 1:1 condition and 20 mg mL−1 protein). Crystals formed within 12–24 h and grew as uniform triangular prisms of various sizes. Crystals were cryo-protected with 20% glycerol, flash frozen, and stored in pucks for data collection at the MX beamlines at the Australian Synchrotron.41 Data processing was carried out using XDS42 followed by scaling using the program AIMLESS. Molecular replacement using the apo structure (PDB: 1NH7) as the search model was used to solve the initial phasing implemented in the MR_Phaser program part of the CCP4 program suite43 followed by further model building and refinement in COOT and Refmac5, PDB code: 5U99 (see Supporting Information (SI) for full crystal parameters).43,44
Isotopic Labeling of PRATP and Purification.
The [1′-3H]-PRATP, [1′-14C]PRATP, [5′-3H]PRATP, and [5′-14C]PRATP were synthesized by coupled enzymatic synthesis from [1-3H]ribose, [1-14C]ribose, [6-3H]glucose, or[6-14C]glucose, respectively (American Radiolabeled Chemicals Inc.). [1-15N]Adenine was synthesized from 15N-formamide according to a modified procedure.45 [6-15N]-Adenine was synthesized in three steps from 6-chloropurine and 15NH4Cl according to a procedure by Milecki et al.46 (15N labels were purchased from Cambridge Isotope Laboratories Inc.). Labeled ATP was obtained from PRPP and the appropriate isotopically labeled adenine through coupled enzymatic synthesis.34 [1-14N, 5′-3H]PRATP and [6-14N, 5′-14C]PRATP were then obtained following the general procedure described for single isotopically labeled PRATP but replacing ATP for the appropriately labeled substrate (see Supporting Information for full details, S1–S3).
Measurement of Kinetic Isotope Effects.
The KIEs were measured under competitive conditions using dual channel liquid scintillation counting.18 A detailed description of the experimental procedure can be found in the Supporting Information, S4. In short, a typical experiment included four reactions that were quenched at partial conversion (20–40%), two 100% conversion reactions, and a negative control containing all components except for the enzyme to account for nonenzymatic degradation. KIEs for each position were determined from at least two independent experiments with a minimum of eight replicates in total. Reliability of the results is reported as standard error of the mean (SEM).
Forward Commitment to Catalysis by Isotope Trapping.
The forward commitment to catalysis, or the partitioning of the Michaelis complex to the product relative to substrate release (kcat/koff), was measured by isotope trapping.27 A detailed experimental section can be found in the Supporting Information (S5).
Computational Modeling of the Transition State Structure.
The crystal structure of PRPP bound to adenine phosphoribosyltransferase from Giardia lamblia (PDB: 1L1R) including Mg2+ and two water molecules, to complete the octahedral coordination of magnesium, was used to generate the starting point for the TS calculation. The pyrophosphate was replaced in silico with phosphonoacetic acid. All hybrid density functional theory calculations were carried out in vacuo using the B3LYP density functional with the 6-31*G(d,p) basis set.17,29-31 Bond frequencies were calculated at the same level of theory. A structure was considered optimized if only one imaginary frequency corresponding to the reaction coordinate was observed and the standard convergence criteria within Gaussian 09 were reached.47 The ISOEFF98 program was used to generate the calculated KIEs for these TS structures.32 The ESPSs were calculated using the Cube program part of the Gaussian 09 program suite and visualized using GaussView 3.0.47
Supplementary Material
ACKNOWLEDGMENTS
We thank G. Mittelstad for his help with the expression of the CjeATP-PRT enzyme and E. Livingston for providing the LlaATP-PRT enzyme.
Funding
This work was supported by the Maurice Wilkins Centre Fund, the UoC Doctoral Scholarship, the IRL Sir Roy McKenzie Trust, and National Institutes of Health research grant GM041916.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.7b00484.
Materials and methods, including, the synthesis of the isotopically labeled substrates, the coordinates of calculated structures (S7–S10), the coupled enzymatic synthesis of PRATP from D-ribose (S1), the coupled enzymatic synthesis of PRATP from glucose (S2), and the synthesis of [1-15N]adenine (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Parish T (2003) Starvation survival response of Mycobacterium tuberculosis. J. Bacteriol. 185, 6702–6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Bange FC, Brown AM, and Jacobs WR (1996) Leucine auxotrophy restricts growth of Mycobacterium bovis BCG in macrophages. Infect. Immun. 64, 1794–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Hondalus MK, Bardarov S, Russell R, Chan J, Jacobs WR, and Bloom BR (2000) Attenuation of and protection induced by a leucine auxotroph of Mycobacterium tuberculosis. Infect. Immun. 68, 2888–2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Abdo M-R, Joseph P, Mortier J, Turtaut F, Montero J-L, Masereel B, Köhler S, and Winum J-Y (2011) Anti-virulence strategy against Brucella suis: synthesis, biological evaluation and molecular modeling of selective histidinol dehydrogenase inhibitors. Org. Biomol. Chem. 9, 3681–3690. [DOI] [PubMed] [Google Scholar]
- (5).Alifano P, Fani R, Liò P, Lazcano A, Bazzicalupo M, Carlomagno MS, and Bruni CB (1996) Histidine biosynthetic pathway and genes: structure, regulation, and evolution. Microbiol. Rev.60, 44–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Sinha SC, and Smith JL (2001) The PRT protein family. Curr. Opin. Struct. Biol. 11, 733–739. [DOI] [PubMed] [Google Scholar]
- (7).Livingstone EK, Mittelstädt G, Given FM, and Parker EJ (2016) Independent catalysis of the short form HisG from Lactococcus lactis. FEBS Lett. 590, 2603. [DOI] [PubMed] [Google Scholar]
- (8).Mittelstaädt G, Moggré GJ, Panjikar S, Nazmi AR, and Parker EJ (2016) Campylobacter jejuni adenosine triphosphate phosphoribosyltransferase is an active hexamer that is allosterically controlled by the twisting of a regulatory tail. Protein Sci. 25, 1492–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Cho Y, Sharma V, and Sacchettini JC (2003) Crystal structure of ATP phosphoribosyltransferase from Mycobacterium tuberculosis. J. Biol. Chem. 278, 8333–8339. [DOI] [PubMed] [Google Scholar]
- (10).Lohkamp B, McDermott G, Campbell S. a., Coggins JR, and Lapthorn AJ (2004) The Structure of Escherichia coli ATP-phosphoribosyltransferase: Identification of Substrate Binding Sites and Mode of AMP Inhibition. J. Mol. Biol. 336, 131–144. [DOI] [PubMed] [Google Scholar]
- (11).Champagne KS, Piscitelli E, and Francklyn CS (2006) Substrate recognition by the hetero-octameric ATP phosphoribosyltransferase from Lactococcus lactis. Biochemistry 45, 14691–14696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ames BN, Martin RG, and Garry BJ (1961) The First Step of Histidine Biosynthesis. J. Biol. Chem. 236, 2019–2026. [PubMed] [Google Scholar]
- (13).Morton DP, and Parsons SM (1976) Biosynthetic direction substrate kinetics and product inhibition studies on the first enzyme of histidine biosynthesis, adenosine triphosphate phosphoribosyltransferase. Arch. Biochem. Biophys. 175, 677–686. [DOI] [PubMed] [Google Scholar]
- (14).Kleeman JE, and Parsons SM (1976) Reverse direction substrate kinetics and inhibition studies on the first enzyme of histidine biosynthesis, adenosine triphosphate phosphoribosyltransferase. Arch. Biochem. Biophys. 175, 687–693. [DOI] [PubMed] [Google Scholar]
- (15).Parsons SMP, Chelsky D, and Goitein RK (1978) Primary 14C and α Secondary 3H Substrate Effects for Some Phosphoribosyltransferases. J. Biol. Chem. 253, 2963–2971. [PubMed] [Google Scholar]
- (16).Tao W, Grubmeyer C, and Blanchard JS (1996) Transition state structure of Salmonella typhimurium orotate phosphoribosyltransferase. Biochemistry 35, 14–21. [DOI] [PubMed] [Google Scholar]
- (17).Zhang Y, Luo M, and Schramm VL (2009) Transition states of Plasmodium falciparum and human orotate phosphoribosyltransferases. J. Am. Chem. Soc. 131, 4685–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Burgos ES, Vetticatt MJ, and Schramm VL (2013) Recycling nicotinamide. the transition-state structure of human nicotinamide phosphoribosyltransferase. J. Am. Chem. Soc. 135, 3485–3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Schramm VL (2007) Enzymatic transition state theory and transition state analogue design. J. Biol. Chem. 282, 28297–300. [DOI] [PubMed] [Google Scholar]
- (20).Evans GB, Schramm VL, and Tyler PC (2015) The immucillins: Design, synthesis and application of transition-state analogues. Curr. Med. Chem. 22, 3897–3909. [DOI] [PubMed] [Google Scholar]
- (21).Wang S, Cameron SA, Clinch K, Evans GB, Wu Z, Schramm VL, and Tyler PC (2015) New Antibiotic Candidates against Helicobacter pylori. J. Am. Chem. Soc. 137, 14275–14280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Porter DJT, and Spector T (1993) Alternative substrates for calf intestinal adenosine deaminase. A pre-steady-state kinetic analysis. J. Biol. Chem. 268, 2480–2485. [PubMed] [Google Scholar]
- (23).Cleland WW (2005) The use of isotope effects to determine enzyme mechanisms. Arch. Biochem. Biophys. 433, 2–12. [DOI] [PubMed] [Google Scholar]
- (24).Zhang Y, Evans GB, Clinch K, Crump DR, Harris LD, Fröhlich RFG, Tyler PC, Hazleton KZ, Cassera MB, and Schramm VL (2013) Transition state analogues of plasmodium falciparum and human orotate phosphoribosyltransferases. J. Biol. Chem. 288, 34746–34754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Huang W, Li C, Li B, Umekawa M, Yamamoto K, Zhang X, and Wang L (2009) Glycosynthases Enable a Highly Efficient Chemoenzymatic Synthesis of N-Glycoproteins Carrying Intact Natural N-Glycans. J. Am. Chem. Soc. 131, 2214–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Rising KA, and Schramm VL (1994) Enzymatic Synthesis of NAD+ with the Specific Incorporation of Atomic Labels. J. Am. Chem. Soc. 116, 6531–6536. [Google Scholar]
- (27).Rose IA (1980) The isotope trapping method: Desorption rates of productive E.S complexes. Methods Enzymol 64, 47–59. [DOI] [PubMed] [Google Scholar]
- (28).Berti PJ, and McCann JAB (2006) Toward a Detailed Understanding of Base Excision Repair Enzymes: Transition State and Mechanistic Analyses of N -Glycoside Hydrolysis and N- Glycoside Transfer. Chem. Rev. 106, 506–555. [DOI] [PubMed] [Google Scholar]
- (29).Taylor Ringia EA, Tyler PC, Evans GB, Furneaux RH, Murkin AS, and Schramm VL (2006) Transition state analogue discrimination by related purine nucleoside phosphorylases. J. Am. Chem. Soc. 128, 7126–7127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kline PC, and Schramm VL (1993) Purine Nucleoside Phosphorylase. Catalytic Mechanism and Transition-State Analysis of the Arsenolysis Reaction. Biochemistry 32, 13212–13219. [DOI] [PubMed] [Google Scholar]
- (31).Hirschi JS, Takeya T, Hang C, and Singleton DA (2009) Transition State Geometry Measurements from 13C Isotope Effects. The Experimental Transition State for the Epoxidation of Alkenes with Oxaziridines. J. Am. Chem. Soc. 131, 2397–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Anisimov V, and Paneth P (1999) ISOEFF98. A program for studies of isotope effects using Hessian modifications. J. Math. Chem. 26, 75–86. [Google Scholar]
- (33).Lewis BE, and Schramm VL (2003) Binding equilibrium isotope effects for glucose at the catalytic domain of human brain hexokinase. J. Am. Chem. Soc. 125, 4785–4798. [DOI] [PubMed] [Google Scholar]
- (34).Lewandowicz A, and Schramm VL (2004) Transition state analysis for human and Plasmodium falciparum purine nucleoside phosphorylases. Biochemistry 43, 1458–1468. [DOI] [PubMed] [Google Scholar]
- (35).Lewis BE, and Schramm VL (2001) Conformational Equilibrium Isotope Effects in Glucose by 13 C NMR Spectroscopy and Computational Studies. J. Am. Chem. Soc. 123, 1327–1336. [DOI] [PubMed] [Google Scholar]
- (36).Guthrie RD, and Jencks WP (1989) IUPAC Recommendations for the Representation of Reaction Mechanisms. Acc. Chem. Res. 22, 343–349. [Google Scholar]
- (37).Schramm VL, and Shi W (2001) Atomic motion in enzymatic reaction coordinates. Curr. Opin. Struct. Biol. 11, 657–65. [DOI] [PubMed] [Google Scholar]
- (38).Chuvikovsky DV, Esipov RS, Skoblov YS, Chupova LA, Muravyova TI, Miroshnikov AI, Lapinjoki S, and Mikhailopulo IA (2006) Ribokinase from E. coli: expression, purification, and substrate specificity. Bioorg. Med. Chem. 14, 6327–32. [DOI] [PubMed] [Google Scholar]
- (39).Shi W, Tanaka KSE, Crother TR, Taylor MW, Almo SC, and Schramm VL (2001) Structural Analysis of Adenine Phosphoribosyltransferase from Saccharomyces cerevisiae. Biochemistry 40, 10800–10809. [DOI] [PubMed] [Google Scholar]
- (40).Singh V, Lee JE, Nunez S, Howell PL, and Schramm VL (2005) Transition State Structure of 5′-Methylthioadenosine/S-Adenosylhomocysteine Nucleosidase from Escherichia coli and Its Similarity to Transition State Analogues. Biochemistry 44, 11647–11659. [DOI] [PubMed] [Google Scholar]
- (41).Cowieson NP, Aragao D, Clift M, Ericsson DJ, Gee C, Harrop SJ, Mudie N, Panjikar S, Price JR, Riboldi-Tunnicliffe A, Williamson R, and Caradoc-Davies T (2015) MX1: A bending-magnet crystallography beamline serving both chemical and macro-molecular crystallography communities at the Australian Synchrotron.J.Synchrotron Radiat. 22, 187–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Kabsch W (2010) XDS. Acta Crystallogr Sect. D: Biol. Crystallogr. 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, and Wilson KS (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010) Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Apsel B, Blair JA, Gonzalez B, Nazif TM, Feldman ME, Aizenstein B, Hoffman R, Williams RL, Shokat KM, and Knight ZA (2008) Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat. Chem. Biol. 4, 691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Milecki J, Foldesi A, Fischer A, Adamiak RW, and Chattopadhyaya J (2001) Synthesis of multiply labelled ribonucleo-sides for sequence-specific labelling of oligo-RNA. J. Labelled Compd. Radiopharm. 44, 763–783. [Google Scholar]
- (47).Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr., Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, and Fox DJ (2009) Gaussian 09, Revision D.01, Gaussian Inc., Wallingford, CT. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.