

Summary
The NRF2 transcription factor controls a cell stress program that is implicated in cancer and there is great interest in targeting NRF2 for therapy. We show that NRF2 activity depends on Fructosamine-3-kinase (FN3K) - a kinase that triggers protein de-glycation. In its absence, NRF2 is extensively glycated, unstable, and defective at binding to small MAF proteins and transcriptional activation. Moreover, the development of hepatocellular carcinoma triggered by MYC and Keap1 inactivation depends on FN3K in vivo. N-acetyl cysteine treatment partially rescues the effects of FN3K loss on NRF2 driven tumor phenotypes indicating a key role for NRF2-mediated redox balance. Mass spectrometry reveals that other proteins undergo FN3K-sensitive glycation, including translation factors, heat shock proteins, and histones. How glycation affects their functions remains to be defined. In summary, our study reveals a surprising role for the glycation of cellular proteins and implicates FN3K as targetable modulator of NRF2 activity in cancer.
In Brief
We identify a new biological role for glucose dependent glycation and enzymatic de-glycation in controlling the oncogenic transcription factor NRF2 and potentially other proteins.
Graphical Abstract
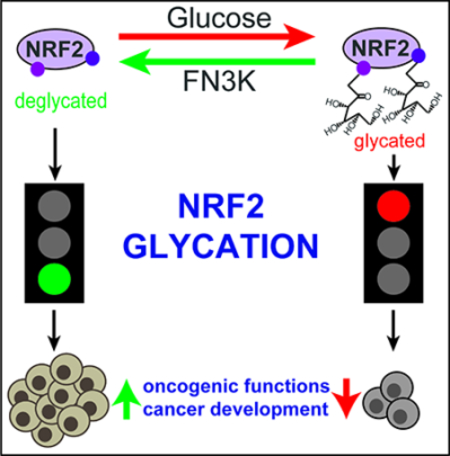
Introduction
The transcription factor NRF2 (encoded by Nuclear factor erythroid-derived 2-like 2 gene; NFE2L2) controls an antioxidant and cell stress program implicated in cancer and drug resistance (Bai et al., 2016; Jaramillo and Zhang, 2013; Kang and Hyun, 2017; Lin et al., 2016; Ma, 2013; Menegon et al., 2016; Rojo de la Vega et al., 2018; Wang et al., 2008). KEAP1 is a key regulator of NRF2 stability and is mutated in many cancers. NRF2 activates the transcription of antioxidant response element (ARE)-bearing genes involved in glutathione (GSH) production, redox balance, xenobiotic detoxification, and cellular anabolic metabolism (Lin et al., 2016; Rojo de la Vega et al., 2018). GSH production protects against reactive oxygen species (ROS) produced during oncogene driven cell proliferation or upon exposure to radiation and alkylating agents (DeNicola et al., 2011; Harris et al., 2015; Zanotto-Filho et al., 2016; Zhou et al., 2013). In principle, enhanced ROS neutralization can protect healthy cells from tumor development, or alternatively an improved redox balance may facilitate the proliferation of cancerous cells (Takahashi et al., 2018). Indeed, while loss of Keap1 promotes KRAS driven lung cancer in vivo, earlier studies found that NRF2 protected animals from carcinogen-induced lung cancer (Bauer et al., 2011; Menegon et al., 2016; Romero et al., 2017; Satoh et al., 2016; Satoh et al., 2013; Sporn and Liby, 2012).
The Cancer Genome Atlas (TCGA) reports mutually exclusive mutations of NRF2 and its E3 ubiquitin ligase complex and associated factors (KEAP1, CUL3, and CAND1) in hepatocellular carcinoma (HCC)(Cerami et al., 2012) (Sanchez-Vega et al., 2018). NRF2 activation has also been linked to chemotherapy resistance and this likely reflects NRF2’s anti-oxidant action (Wang et al., 2008). More recently NRF2 activating KEAP1 mutations have also been reported upon relapse from EGFR inhibitor (EGFRi) therapy in non-small cell lung cancer (NSCLC) patients (Yu et al., 2018). The prevalence of NRF2 activating mutations across many solid tumors and their impact on therapy indicate that strategies to target NRF2 are urgently needed.
Protein glycation refers to the non-enzymatic attachment of reducing monosaccharides like ribose, glucose, and glucose 6-phosphate to basic amino acids (lysine, arginine, histidine) to form fructosamines in a Maillard reaction (Fortpied et al., 2006; Takahashi, 2015; Zhang et al., 2009). De-glycation, the removal of attached sugars, is triggered by Fructosamine-3-kinase (FN3K), a rather unique kinase that directly phosphorylates the attached sugar and destabilizes the link (Van Schaftingen et al., 2012). The basic amino acids (lysine, arginine, histidine) affected by glycation often reside in accessible and functionally relevant domains such that glycation-induced changes in structure and charge may affect protein functions (Takahashi, 2015; Van Schaftingen et al., 2012). Glycation is distinct from enzymatic glycosylation or the much slower formation of advanced glycation end products (AGE) implicated in inflammation and late complications of diabetes (Moremen et al., 2012; Takahashi, 2015; Veiga da-Cunha et al., 2006; Zhang et al., 2009). The best-known example of a glycated protein is hemoglobin HbA1c that tracks with blood glucose levels and is used in diabetes management (Wareham and Pfister, 2010); other examples are glycated insulin and serum albumin (Abdel-Wahab et al., 1997; Anguizola et al., 2013; Hunter et al., 2003). Little is known about the effect of glycation on cellular proteins or its potential role in cancer.
Results
Genetics and oncogenic function of NRF2 activation in liver cancer
TCGA data indicate mutational NRF2 activation in ~15% of human HCCs (Figures S1A and S1B). We tested the role of these mutations in a murine model of MYC-driven HCC. Briefly, we observed that CRISPR/Cas9 induced activating mutations in Nrf2 (n = 12), and loss of the NRF2 regulators Keap1 (2 sgRNAs; n = 11), Cul3 (n = 6), and Cand1 (n = 12) all caused rapid development of aggressive HCCs in vivo (p ≤ 0.05 for Keap1, Cul3, and Cand1) (Figures 1A and 1B). Notably, sequencing and T7 endonuclease assays show that the CRISPR-induced oncogenic lesions in the murine model closely resemble those seen in human HCCs (Figures S1C and S1D). H&E staining and immunohistochemistry show MYC/sgKeap1 induced tumors express HNF4a and Ki67, markers of highly proliferative HCC, with some glandular features (Figure S1E). Gene and protein expression studies further confirm activation of NRF2 target genes (Figures S1F–S1H; Table S1). Moreover, MYC/sgKeap1 driven tumors depend on continuous NRF2 expression (n = 10, p < 0.05) (Figures 1C, S1I, and S1J). L-butathionine sulfoximine (BSO) inhibits the NRF2 target gene γ-glutamylcysteine synthetase (GCS) and increases the ratio of oxidized to reduced glutathione in vivo (n ≥ 7, p = 0.003) (Figure S1K). BSO treatment hampers the development of MYC/sgKeap1 driven HCCs in vivo indicating a key role for NRF2’s redox function in HCC development (n = 9, p < 0.002) (Figure 1D). Hence, NRF2 activation promotes the development of MYC-driven liver cancers in large part through a redox sensitive mechanism in vivo.
Figure 1: NRF2 has an oncogenic function in hepatocellular carcinoma.

A) Kaplan-Meier survival analysis of mice hydrodynamically injected with the MYC transposon system and the indicated NRF2 activating guide RNAs (sgRNA) and Cas9; B) Representative diseased livers from mice injected with MYC and sgKeap1 or sgNrf2 (targeting the Keap1 interacting ETGE domain); C) In vivo growth of murine MYC/sgKeap1 tumors transduced with Nrf2-targeted or control shRNAs; Data from HCC lines 1 & 2 were combined and plotted (n=5 each); D) Kaplan-Meier survival analysis of mice that were hydrodynamically injected with the MYC transposon system and an sgRNA/Cas9 targeting Keap1 and subsequently treated with glutathione synthesis inhibitor BSO. (* indicates p-value < 0.05 by two-tailed student t test). See also Figure S1 and Table S1.
Next, we examined the genomic context of NRF2 activating lesions. Briefly, we queried a pan-cancer genome-wide SELECT analysis for genetic lesions linked to NRF2 (Mina et al., 2017)(see Methods). We found a highly significant mutual exclusive relationship between NRF2 activation and EGFR mutations across all cancers and in treatment-naïve patients (Figures 2A, 2B, and S2A; Table S2). Prior analyses on smaller cohorts had yielded only ambiguous results (Arbour et al., 2018; Frank et al., 2018; Sasaki et al., 2013). Consistent with an overlapping function, a comparison of gene expression data from human EGFR and NRF2 activated NSCLC and HCCs reveals a similar NRF2 signature (Figures S2B, S2C). Moreover, EGFR ligands (EGF or TGFα) stimulate NRF2 expression in a MEK-dependent (trametinib sensitive) manner, while EGFR knockdown as well as erlotinib and trametinib treatments decrease NRF2 expression and activity in EGFR mutant human NSCLC cells (H3255: EGFRL858R and PC9: EGFRΔ E746-A750) (Figures 2C, 2D, and S2D – S2J) (DeNicola et al., 2011; Wang et al., 2008). This effect is relevant to therapy as in vitro KEAP1 loss impairs the signaling and cell death effects of erlotinib in EGFR mutant H3255 and PC9 cells (Figures 2E, 2F, and S2K–S2M). Consistently, a constitutively active NRF2 (NRF2E79V) activates ERK in EGFR wild type HepG2 cells (Figure S2N)(Takahashi et al., 2018). Notably, a recent study observed new KEAP1 mutations upon relapse form EGFR inhibitor treatment In NSCLC patients (Frank et al., 2018). Hence, NRF2 is an oncogene in solid tumors with implications in resistance to cancer therapy.
Figure 2: Mutual exclusive activation of NRF2 and EGFR pathways in human cancers.

A) Unbiased, pan-cancer analysis of mutations that are significantly related (co-occurring or mutual exclusive) with KEAP1 and NRF2 using the SELECT algorithm (see text and methods for details); B) Oncoprint showing mutual exclusive relation between NRF2/ KEAP1 and EGFR mutations in human cancers; C) Nuclear extracts from HepG2 cells treated with NRF2 inducer DLS (1 μM, 24 hours), EGF (10 ng/mL, 24 hours), TGFα (1 nM, 24 hours) or DMSO and probed with antibodies against NRF2 and lamin B; D) Nuclear (upper panel) and cytoplasmic extracts (lower panel) from H3255 (EGFRL858R) cells transduced with EGFR-specific shRNA or control and immunoblotted with indicated antibodies; E) Viability of isogenic H3255 cells transduced with sgRNAs targeting KEAP1 or LacZ (control) and treated with erlotinib; error bars represent SD from 3 replicates; F) Lysates from paired PC9 cells transduced with indicated sgRNA-Cas9 constructs and treated with DMSO or erlotinib (10 nM, 6 hours) probed with the indicated antibodies. (* indicates p-value < 0.05 by two-tailed student t test). See also Figure S2 and Table S2.
A CRISPR/Cas9 screen to identify requirements for NRF2 activation
In order to identify potentially targetable requirements for NRF2 activation by ROS stress, we developed a screenable assay where NRF2 drives the expression of the HSV-TK suicide gene. Briefly, our lentiviral construct encodes HSV-TK and a luciferase reporter under transcriptional control of four tandem, NRF2 responsive AREs, such that NRF2 activation will render cells sensitive to ganciclovir treatment (Figures 3A and 3B). We confirmed assay performance with chemical NRF2 activators tert-butylhydroquinone (tBHQ) and DL sulforaphane (DLS) that activate luciferase and induce ganciclovir sensitivity over the baseline levels (DMSO controls) in FL5.12 cells (Figures S3A–S3C). We performed a pooled, cell-based screen with a genome-scale CRISPR library searching for cells that escaped NRF2 induced ganciclovir sensitivity. We identified genomically integrated sgRNAs in the surviving cell population by deep-sequencing before and after NRF2 activation with tBHQ (see methods) (Figure 3C; Table S3). To identify the most significant hits we established stringent criteria of at least two sgRNAs enriched and showing an average 3-fold change across all sgRNAs and replicates (see Methods). As expected, loss of the negative NRF2 regulator Keap1 enhanced NRF2 activity and caused ganciclovir sensitivity (Figure 3C). We also identified sgRNAs against seven genes whose inactivation was protective (Figure 3C). These include Fn3k, fructosamine-3-kinase, involved in protein de-glycation, the uncoupling protein 2 (Ucp2) involved in mitochondrial metabolism and a regulator of a known ganciclovir exporter (Hu and Liu, 2010; Wang et al., 2017), Rasgef1c, a guanine nucleotide exchange factor for RAS-like GTPases, and the E2f8 transcription factor that interacts directly with Nrf2 and Cul3 promoters(Kent et al., 2016) (Figure 3C). We confirmed these effects using two sgRNAs against E2f8, Rasgef1c, and Fn3k in murine FL5.12 cells and for FN3K we tested two additional shRNAs in human HepG2 cells (Figures S3D–S3G). Hence, we identify and validate important regulators of NRF2 activation that warrant further characterization.
Figure 3: Genome-wide CRISPR screen identifies the de-glycating kinase FN3K as a requirement for NRF2 function.

A) Diagram of our strategy for a genome-wide screen against NRF2 driven expression of the HSV-TK suicide gene; B) Map of the lentiviral vector directing ARE-controlled HSV-TK and luciferase expression; C) Change in sgRNA library representation comparing untreated cells and cells treated with the NRF2 inducer tBHQ and ganciclovir; D) Predicted sites of NRF2 protein glycation using indicated algorithms; TAD: Transactivation domain; E) Phenyl borate affinity purification and immunoblotting reveals NRF2 glycation upon FN3K knockdown in KEAP1 mutant Huh1 cells; values on top refers to % of glycated NRF2 represented by the ratio of NRF2 signal intensity in PB-bound (PB) to the sum PB-bound and flow through (FT); F) Immunoblot for nuclear (upper panel) and cytoplasmic (lower panel) levels of the indicated proteins in KEAP1 wild type HepG2 cells transduced and treated as indicated; G) Chromatin immunoprecipitation (ChIP) on indicated HepG2 nuclear lysates with anti-NRF2 antibody followed by amplification of indicated promoters; shown as % of input DNA and error bar is SD of 4 replicates; H) Viability of HepG2 cells untreated or treated with H2O2 (400 μM, 24 hours) with and with without pre-incubation with NAC (10 mM, 3 hours); mean of 9 replicates ± SD. (* indicates p-value < 0.05 by two-tailed student t test). See also Figure S3 and Table S3.
FN3K-sensitive glycation impairs NRF2 activity
We decided to focus on FN3K and explore the role of NRF2 protein glycation. First, we used two in silico analysis tools - Net glycated (Johansen et al., 2006) and Gly-PseAAC (Xu et al., 2017) - to ask if NRF2 might undergo glycation. Both algorithms predicted extensive glycation of multiple residues in NRF2’s N- and C-terminal domains. These correspond to the KEAP1 interaction and the small Musculoaponeurotic Fibrosarcoma protein (sMAF) binding regions, respectively (Figure 3D). Phenylborate (PB) binds to glycated (and with lesser affinity to glycosylated) proteins and PB affinity purification can be used to isolate such modified proteins. Upon FN3K knockdown using two shRNAs we readily detect glycated NRF2 protein by PB affinity purification on lysates from KEAP1N414Y mutant Huh1 HCC cells (Figures 3E, S3H, and S3I). Comparison of PB enriched NRF2 and NRF2 in the flow through (FT) allows quantification and shows that approximately 29% of NRF2 is glycated in FN3K deficient cells (Figure 3E). Moreover, NRF2 glycation increases with glucose levels (Figure S3J). Note that β-actin is a substrate for glycosylation or O-GlcNAcylation reactions (Terman and Kashina, 2013), and glycosylated β-actin can bind to PB columns, but these modifications are not affected by FN3K (Figure S3H).
Next, we tested how glycation affects NRF2 function in KEAP1 wild type cells. Upon NRF2 stimulation with DLS we find that loss of FN3K impairs induction and nuclear accumulation of NRF2 and this results in loss of target protein expression (NQO1, TXNRD1, GPX2) (Figures 3F and S3K). This corresponds to reduced NRF2 occupancy at endogenous AREs in the promoters of target genes (NQO1, TXNRD1) (Figure 3G), and results in an increased sensitivity to oxidative stress by H2O2 (400 μM, 24 hours) (Figure 3H). Pre-treatment with the ROS scavenger and GSH precursor N-acetyl cysteine (NAC) reverses H2O2 and DLS toxicity and restores glutathione balance in FN3K deficient HepG2 and H3255 cells, respectively (Figures 3H and S3L). Hence, FN3K deficiency increases NRF2 glycation and impairs its ability to counter ROS stress in liver and lung cancer cells.
Glycation decreases NRF2 stability in KEAP1 proficient cells
To accurately measure whether glycation affects NRF2 stability in KEAP1 competent cells we utilized a highly sensitive assay based on the detection of an NRF2-nanoluciferase fusion protein. Upon FN3K knockdown we find that NRF2-nanoluciferase is destabilized (>50%) in KEAP1 wild type HepG2 cells, by contrast NRF2 stability is unaltered in KEAP1 mutant Huh1 cells (Figure 4A). Flow cytometric detection of NRF2 using a PE-labeled antibody has higher sensitivity than immunoblots and permits measurement of baseline NRF2 levels in HepG2 cells. It readily detects DLS induction and knockdown of NRF2 confirming antibody specificity (Figure S4A). FACS-based detection also reveals reduction in baseline NRF2 upon knockdown of FN3K with two different shRNAs in HepG2 cells (Figures S4B–S4D). H3255 cells (KEAP1/NRF2 wild type and EGFR mutant) have higher levels of NRF2 and both FACS assay and immunoblot show reduction of NRF2 protein upon knockdown of either FN3K or NRF2 (Figures S4E and S4F). Moreover, FN3K deficiency leads to increased proteasomal and MG132-sensitive degradation of the glycated NRF2 protein (Figure S4G). While we have seen that EGFR/MEK pathway induce NRF2 transcription (Figures S2H and S2I) it does not alter FN3K expression and conversely, FN3K affects the NRF2 protein and not its mRNA levels (Figures S4H–S4J).
Figure 4: NRF2 glycation suppresses its oncogenic functions in KEAP1 wild type and mutant cell.

A) Measuring NRF2 stability using an NRF2-nanoluciferase fusion protein in lysates of KEAP1 proficient HepG2 and KEAP1 mutant Huh1 cells transduced with control or shRNA against FN3K; mean of n=6 for HepG2 and n=3 for Huh1 cells ± SD; B) Relative expression of ~80 antioxidative genes in KEAP1N414Y mutant Huh1 cells transduced with control or FN3K shRNAs; average of four replicates (n=2 for each FN3K-specific shRNAs) relative to control shRNA; C) Unsupervised clustering of total proteomics data from indicated Huh1 cell lysates; GSEA analysis of over- and underrepresented proteins in FN3K-deficient cells shows reduction of NRF2 target proteins (top) and proteins involved in xenobiotic metabolism (bottom); D) Luminescence-based quantification of oxidized and reduced glutathione in KEAP1N414Y Huh1 cells expressing control vector or shRNA against FN3K; error bar represents SD from n ≥ 5 replicates; E) Nuclear extracts from Huh1 cells with control vector or FN3K knockdown immunoprecipitated with NRF2 or IgG antibodies and probed for MAFG and β-actin; nuclear lysates (bottom) were loaded on a separate gel and probed with αNRF2 and αLamin B as indicated; F) Schematic of KEAP1-dependent and independent mechanisms of NRF2 inhibition by glycation. (*denotes two-tailed t test calculated p-value of <0.05). See also Figure S4 and Table S4.
Glycation impairs NRF2 function in KEAP1 deficient cells
We noticed that glycation also affects NRF2 function in KEAP1 mutant cells, for example gene and protein expression analyses of FN3K deficient and control Huh1 liver cancer cells (KEAP1N414Y) showed loss of NRF2 targets and resultant redox imbalance as indicated by increased glutathione oxidation (Figures 4B–4D; Table S4). Moreover, hyperglycemia-induced increase in NRF2 glycation (Figure S3J) corresponds to loss of its target genes in Huh1 cells, which is restored by enforced FN3K expression (Figures S4K–S4M). These effects are not related to NRF2 stability (Figure 4A). Instead, we find that FN3K loss impairs the interaction with small MAF proteins in KEAP1 deficient cells. Briefly, co-immunoprecipitation revealed that NRF2 binds to MAFG protein and that this interaction is largely abolished upon FN3K knockdown using two different shRNAs in two KEAP1 mutant lines (Huh1, H460) without affecting its nuclear localization (Figures 4E, S4N, and S4O). We confirmed that knockdown of FN3K affects NRF2 target gene and protein expression at physiological (1 g/L) glucose levels (Figures S4P and S4Q). We also noticed that NRF2 increases cellular glucose levels in a 2-deoxyglucose based reporter assay, which is expected to increase glycation and potentially switch on feedback regulation (Diez-Sampedro et al., 2003; Mitsuishi et al., 2012) (Figures S4R and S4S). Together, glycation of NRF2 has KEAP1 dependent and KEAP1 independent effects on NRF2 stability and transcriptional activity (Figure 4F).
Mass spectrometric mapping and quantification of NRF2 glycation
Quantification of protein glycation by PB affinity chromatography (Figures 3E and S3H) is limited by the capacity of the column, low sensitivity, and detection of only the early Amadori adducts. However, glycation of lysine and arginine residues impairs trypsin cleavage (Shapiro et al., 1980), and this is detectable by mass spectrometry (MS) with much higher sensitivity. Specifically, trypsin digest of a glycated protein is expected to result in longer peptides if glycation conceals the trypsin cleavage site or an increased peptide mass corresponding to the mass of the attached sugar if glycation occurs within the peptide. To design a targeted MS approach, we first compared trypsin digests of in vitro glycated (5 or 10 g/L glucose for 14 days) or unglycated recombinant NRF2 protein. Upon glycation, we could readily detect reduced trypsin cleavage at C-terminal NRF2 residues K462, K472, K487, R499, K543, K554, and R569 but not the R587 residue (Figure 5A). Chymotrypsin cleaves at carboxyl side of aromatic amino acids (tyrosine, phenylalanine, tryptophan), and AspN cleavage occurs N-terminal to aspartic acid, therefore these digests are not affected by glycation (Figures S5A and S5B). A notable exception is loss of NRF2 D570 cleavage by AspN that likely reflects steric hindrance from R569 glycation (Figure S5B). Hence, trypsin digest and shotgun MS reveal changes in peptide cleavage patterns consistent with lysine/arginine glycation.
Figure 5: Mapping and quantifying NRF2 glycation by mass spectrometry.

A) Mass spectrometric identification of tryptic peptides from unglycated (upper) and in vitro glycated recombinant NRF2 (lower); peptides that decreased in abundance upon in vitro glycation are shown in black while those that are unaffected in red; B) AUC analysis of indicated tryptic peptides generated from non-glycated and in vitro glycated NRF2; data are represented relative to R25-R34 peptide and error bar represents SD from 3 replicates; C) Spectral plot of K487-R499 peptide from unmodified (left) and in vitro glycated NRF2 (right, 5 g/L glucose for 14 days); D) Spectral intensity graphs of indicated peptides obtained from tryptic digest of immunoprecipitated nuclear NRF2 from control and FN3K deficient Huh1 cells; E) Normalized AUC analysis of indicated tryptic peptides generated by digesting immunoprecipitated NRF2 from parental (n=2) or FN3K-silenced Huh1 cells (n=3) as indicated; error bar represents SD. (* indicates p-value < 0.05). See also Figures S5, S6, and Table S5.
To directly confirm lysine modification by glucose we performed Higher-energy Collisional Dissociation (HCD) based fragmentation and MS on glycated and unglycated NRF2. First, we optimized enzyme digests and independent NRF2 digestion with trypsin, chymotrypsin, and Glu-C (cleaves at C-terminals of aspartate and glutamate) collectively covered >90% of the protein (Figure S5C). Next, in vitro glycation of NRF2 (10 g/L glucose for ~3 days) followed by HCD fragmentation/MS revealed changes in the mass of lysines K462, K472, K487, and K574 by +162 consistent with an attached glucose (Figure S6; Table S5). For example, the expected mass:charge of R460-K472 peptide is 675.4 when not glycated, which increases to 756.4 when K462 is glycated giving the difference of ~81 (Figures S6A, S6B, and S6H; Table S5). Since the charge of both peptides is +2 the absolute mass difference is ~+162. Further HCD fragmentation of that peptide shows the m/z of Y11+2H ion (corresponding to K462) to be 720.9, which is +81 higher than the expected ~640 when not glycated giving the absolute increase of mass by ~162 (Table S5). We observed similar changes in lysine mass by ~+162 for K472, K487 and K574 (Figures S6C–S6H; see Table S5 for corresponding fragmentation profiles). Moreover, we see a decline in terminal cleavage of residues K487 and R569 in glycated NRF2 consistent with their covalent modification by glucose adduct (Figure S5D).
To precisely quantify NRF2 glycation we next devised a targeted parallel reaction monitoring (PRM)-based MS approach. We focused on nine NRF2 peptides that showed a near-perfect linear detection across NRF2 quantities (K487-R499, K462-K472, R25-R34, R34-R42, K206-K219, K428-K438, K449-R456, R569-R587, R587-K596) (Figures S5E and S5F). Importantly, the K487-R499 peptide is robustly detected in the unglycated and completely lost in the in vitro glycated NRF2 protein digest (Figures 5B and 5C). Other peptides show similar changes in their trypsin digest pattern and the area under curve (AUC) analysis reveals a time and concentration dependent decrease in two additional NRF2 peptides (K462-K472 and R569-R587) upon in vitro glycation (Figure 5B). We cannot precisely quantitate K574 glycation by this method since we do not see any peptides generated by cleavage at this site (owing to proline at position 575). Moreover, we readily detect loss of tryptic peptides K487-R499 and R569-R587 and to a lesser degree that of K462-K472 after in vitro glycation of NRF2 with 1 g/L glucose for 3 hours (Figure S5G), this is in accordance with physiological levels of glucose and the reported half-life of NRF2 in cancer cells (Walker-Samuel et al., 2013; Zhang, 2006).
Next, we applied this quantitative method to NRF2 isolated form isogenic pairs of FN3K proficient and deficient Huh1 liver cancer cells. We measured three potentially glycated NRF2 peptides (K462-K472, K487-R499, and R569-R587) and three control peptides (R25-R34 and R34-R42, and R587-K596) (Table S5). The AUC analysis on three biological replicates reveals a 50% decline in cleavage efficiencies for the K462-K472 and K487-R499 peptides, and a 40% drop for the R569-R587 peptide (Figures 5D and 5E). The discrepancy between PB and MS in quantifying NRF2 glycation likely reflects binding capacity of the PB columns and the higher sensitivity of MS. Mutating all six C-terminal glycation sites (K462, K472, K487, R499, R569, R587) to alanine (NRF2E79V-A6) resulted in a non-functional NRF2 protein signifying their importance in NRF2 function and precluding functional studies (Figure S5H). Together, these data reveal extensive glucose dependent and FN3K sensitive NRF2 glycation in cancer cells.
The role of FN3K-sensitive NRF2 glycation in liver cancer in vivo
Next, we wanted to test the role for NRF2 glycation in relevant tumor settings in vivo. We probed the requirement for Fn3k in MYC/sgKeap1 driven HCC by co-delivery of sgRNAs against Fn3k or GFP as control (Figure 6A). As expected, animals receiving the Keap1/GFP sgRNA combination rapidly developed multifocal liver tumors. By contrast, mice injected with Keap1/Fn3k sgRNAs developed significantly fewer and smaller tumors (n = 11 tumors, avg. volumes: Keap1/GFP – 127 mm3, Keap1/Fn3k – 17 mm3, p < 0.05) (Figures 6B and 6C). Moreover, we noticed that most tumors arising in animals that received the Fn3k sgRNA had escaped complete FN3K inactivation and retained one or both copies of the gene (Figure S7A). This indicates a requirement for FN3K in MYC and NRF2 driven HCCs in vivo.
Figure 6: FN3K is required for proliferation of NRF2-driven tumors in vivo.

A) Diagram of dual gene-targeting strategy in murine HCCs; B) Ultrasound of murine livers 4 weeks after injection with indicated plasmids; tumors are marked in red; C) Ex vivo images of livers from animals injected with indicated sgRNA combinations; D) Subcutaneous Huh1 xenografts with and without FN3K knockdown and NAC treatment as indicated measured ~30 days after implantation; E) Decreased NRF2 target protein expression by immunoblot on the FN3K deficient Huh1 tumors from panel 5D; F) Phenyl borate enrichment and immunoblot shows NRF2 glycation in the FN3K deficient Huh1 xenografts. See also Figure S7 and Table S6.
FN3K knockdown similarly diminished engraftment of fully transformed, Keap1/KEAP1 mutant murine and human HCC cells in vivo (n ≥10, p < 0.05) (Figures 6D and S7B). We also tested if Fn3k could act as an oncogenic driver in the liver by enforcing its expression but found it was not sufficient to drive HCC development with MYC (Figure S7C). This suggests that FN3K is required to maintain NRF2 in the unglycated and active state and that it alone is not sufficient to activate NRF2. We observed the same FN3K requirement in lung cancer engraftment of KEAP1 mutant (KEAP1D236H) H460 and EGFR mutant (EGFRL8585R) H3225 NSCLC cells in vivo (Figures S7D and S7E). Moreover, knock down of FN3K was able to restore, at least in large part, the erlotinib sensitivity of sgKEAP1 expressing EGFR mutant human NSCLC lines (H3255 and PC9) (Figure S7F). Hence, FN3K deficiency blocks NRF2’s pro-oncogenic and drug resistance effects in models of lung and liver cancer in vivo.
Next, we wanted to determine the relevant contribution of NRF2 glycation to the effects of FN3K inactivation in NRF2 activated tumors. First, we observed that Fn3k knockdown leads to a striking reduction of NRF2 target proteins in murine MYC/sgKeap1 HCC liver tumor isografts and similarly in three pairs of FN3K-proficient and FN3K-deficient human xenografts (Huh1, H460, and H3255) (Figures 6E and S7G–S7J; Table S6). Moreover, we readily detect glycated NRF2 on PB columns from FN3K deficient Huh1 xenografts (Figure 6F). This defect is further reflected in depletion of reduced glutathione in the FN3K deficient tumors in vivo (Figures S7K–S7M). NAC treatment (40 mM) restores the redox balance as indicated by normalized glutathione levels in FN3K deficient xenografts (Figure S7M). NAC treatment also partially restores the engraftment of FN3K-deficient Huh1 tumor cells in NSG animals (n=15, p<0.05) (Figures 6D, S7K, and S7L). This partial rescue of tumor growth by NAC treatment likely reflects non-oxidative NRF2 functions and additional glycation targets. Further, we find that the stabilized form of NRF2, NRF2E79V is able to protect FN3K deficient Huh1 cells from the effects of H2O2 (25 μM) exposure indicating that NRF2 is an important mediator of the sensitivity to oxidative stress in FN3K deficient cells (Figure S7N). Together, FN3K emerges as a targetable vulnerability in NRF2-driven cancers.
Defining the glycated proteome in liver cancer cells
Glycation of intracellular proteins has not been studied in cancer and we expect that other proteins are also subject to FN3K-sensitive glycation. To more broadly identify FN3K-sensitive protein glycation we performed MS on PB purified and unpurified lysates from isogenic pairs of FN3K deficient and parental Huh1 human liver cancer cells (Figure 7A). Briefly, we identify 110 proteins that show highly significant (p-value < 0.05) differential FN3K-sensitive glycation (Figures 7B, S8A and S8B; Table S7). The MS technology is primarily able to detect highly abundant proteins and we see differential glycation of proteins that relate to diverse biological processes including mRNA translation, the unfolded protein response, and the COP9 signalosome (Figure 7C). The extent of glycation ranges from < 10% to ~40% with a median of ~6%, which is in agreement with glycation levels observed in Fn3k−/− mice (Veiga da-Cunha et al., 2006) (Figure S8A; Table S7). The reasons for this range are not clear and may relate to protein stability and accessibility. Examples of significantly glycated proteins include metabolic enzymes such as LDHA and LDHC, translational factors (eIF4A1), HSP90 whose glycation has been observed previously (Nokin et al., 2016), and to a lesser degree histone proteins H2 and H3 (Figures 7D and S8C). We confirmed FN3K-sensitve glycation of LDHA by immunoblot on PB affinity purified lysates (Figure S8D). Moreover, we readily detect the expected extensions in tryptic digests by MS for LDHA, histone H3, and the translation factor eIF4A1 where glycated lysines and arginines flag some of the cleavage sites (Figures 7D, S8C, S8E, and S8F). Hence, many proteins - in addition to NRF2 - undergo glycation and the biological effects of this mark are currently unknown.
Figure 7: Analyzing the glycated proteome in liver cancer.

A) Experimental strategy for MS identification of differentially glycated proteins in isogenic FN3K proficient and deficient Huh1 cells B) Heat map showing significantly glycated proteins in FN3K-deficient Huh1 cells; C) Gene ontology analysis identifies pathways significantly enriched in proteins susceptible to FN3K-sensitive glycation; D) Glycation patterns for LDHA; terminally glycated lysine residues that cause peptide elongations upon FN3K knockdown are marked in red while internal residues are shown in green and underlined. See also Figure S8 and Table S7.
Discussion
NRF2 function requires removal of sugar adducts
We report a surprising role for protein glycation in cancer. Glycation refers to the chemical modification of proteins with sugars (glucose, ribose, glucose-6-phosphate) and is countered by the kinase FN3K that triggers de-glycation by phosphorylating the attached sugar. Specifically, we observed that the oncogenic NRF2 transcription factor is sensitive to glycation and an informative genetic screen revealed that NRF2 activity requires the action of FN3K kinase. FN3K phosphorylates monosaccharides that are attached to proteins and triggers their spontaneous removal. FN3K is the only known mammalian kinase with this unusual function (Clements et al., 2006; Van Schaftingen et al., 2012). We find that glycation enhances KEAP1 mediated NRF2 degradation. In addition, NRF2 glycation impairs its interaction with the transcription co-factors, sMAF proteins. In this manner, glycation affects NRF2 function in both KEAP1 proficient and mutant cells.
Cellular metabolites can directly influence transcription factor activity
Mass spectrometry confirms the sugar adducts on NRF2 and enables precise mapping and quantification. In the absence of FN3K the NRF2 C-terminal sites show up to 50% glycation, well above the median ~6% found in our proteomic analysis and reported in Fn3k−/− mouse (Veiga da-Cunha et al., 2006), accounting for sensitivity of NRF2 mutant cells to FN3K inhibition. Glycation can occur independently at all sites and while we do not know the functional impact of single glycation events the fraction of glycated NRF2 may be even higher. Importantly, glycation kinetics of NRF2 are in line with intracellular glucose concentration and half-life of NRF2 in cancer cells (Walker-Samuel et al., 2013; Zhang, 2006). Other metabolic intermediates can also affect this pathway, for example methylglyoxal (MGO), a by-product of glycolysis, and itaconate, a by-product of the TCA cycle, modify KEAP1 and indirectly influence NRF2 activity (Bollong et al., 2018; Mills et al., 2018). The related NRF1 protein undergoes N-linked asparagine glycosylation and subsequent deglycosylation by PNG-1 converts asparagine to aspartate and thereby promoting its activity (Lehrbach et al., 2019). It will be intriguing to investigate to what extent fluctuations in cellular and organismal metabolism are reflected in these posttranslational modifications and how they ultimately adjust protein functions.
The glycated proteome in cancer cells
Glycation is a chemical, non-enzymatic reaction that can affect other cellular proteins depending on their ‘glycatibility’ (spatial and chemical context of basic amino acids) and susceptibility to FN3K mediated de-glycation (Johansen et al., 2006; Venkatraman et al., 2001). Mass spectrometry reveals that in liver cancer cells ~100 proteins show extensive and FN3K sensitive glycation. These include relatively abundant (and detectable) proteins such as translation factors (eIF4A1, eIF1, eIF3G), heat shock proteins (HSP90AA1, HSP90AA4), enzymes in glucose metabolism (LDHA, LDHC), several DNA and RNA binding proteins such as transcription factors (NRF2), replication and repair proteins (HELB, MCM3), splicing factors (SRSF7, PUF60), and also histone proteins. Histone glycation sites (H3K115, H2BK108) can also undergo other modifications and their interrelationship is not known (Chatterjee et al., 2015; Galligan et al., 2018). In this regard, recent studies implicate histone and DNA modification with MGO in gene expression and DNA repair (Galligan et al., 2018; Richarme et al., 2017; Zheng et al., 2019). We find that NRF2 is very sensitive to post-translational sugar modifications and this suggests that glycation can also affect the function of other cellular proteins.
FN3K is a candidate drug target in NRF2 driven cancers
NRF2 is activated by mutations in up to 30% of solid tumors including lung, liver, bladder, head and neck, esophageal, and pancreatic cancers and is considered an important driver of oncogenesis (Jaramillo and Zhang, 2013; Menegon et al., 2016; Ngo et al., 2017; Rojo de la Vega et al., 2018; Romero et al., 2017). Intriguingly, TCGA data also reveal less-frequent somatic mutations affecting NRF2 glycation sites (R499W, R569C, R569H) in endometrial, melanoma, and colon cancer although their function is not yet known. Our findings indicate that FN3K is required, although not sufficient for NRF2 activity. This suggests small molecule inhibitors of the kinase FN3K may be able to maintain NRF2 in the glycated and inactive state. Our genetic data support a requirement for FN3K in NRF2 activated lung and liver cancers, and NAC rescue experiments indicate that the redox balancing function of NRF2 is an important target of FN3K loss. Notably, FN3K knockout mice show high levels of glycated proteins and develop normally indicating that FN3K inhibition may be well-tolerated (Veiga da-Cunha et al., 2006). Together, FN3K emerges as a synthetic vulnerability in certain cell stress states including in NRF2 activated cancers.
STAR Methods
LEAD CONTACT AND MATERIALS AVAILIBILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact of this study, Hans-Guido Wendel (wendelh@mskcc.org). This study did not generate new unique reagents.
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Cell Lines
HepG2, Huh1, mouse HCCs, and 293T cells were maintained in DMEM (5 g/L glucose) while H3255, PC9, H460, and FL5.12 lines were maintained in RPMI-1640 (2 g/L glucose). All culture media were supplemented with heat inactivated fetal bovine serum (10%), L-glutamine (2 mM), penicillin/streptomycin (1%), and plasmocin (5 μg/mL). WEHI conditioned media (10%) and IL3 (1 ng/mL) were also added to FL5.12 culture medium. Primary mouse MYC/sgKeap1 HCC lines were generated by first digesting tumor suspension with collagenase IV (1 mg/mL)/dispase II (3 mg/mL) enzyme mix (1 hour at 37°C) and subsequently plating the digested suspension on collagen coated plates (100 μg/mL). After establishment, primary HCC lines were maintained on collagen coated plates in complete DMEM growth media. HEK-293T, H3255, and HepG2 cells were bought from ATCC and no cell lines used in this study were found in the commonly misidentified cell line database maintained by ICLAC and NCBI Biosample. Cell lines were tested for mycoplasma contamination and maintained in prophylactic dose of plasmocin. For lentivirus transductions, virus prepared in 293T cells transfected with psPAX2, pVSV.G, and the target construct (2:1:2) was either concentrated (Lenti-X concentrator, Clonetech) or not as needed and cells were transduced in the presence of polybrene (4–8 μg/mL).
Animal Studies
All mouse experiments were conducted in accordance with Memorial Sloan Kettering Cancer Center’s Institutional Animal Care and Use Committee (IACUC) approved protocols. All mice were housed at the MSKCC animal facility and Research Animal Resource Center (RARC) provided husbandry and clinical care. Hydrodynamic injections to model HCC were done in wild type C57/BL6 female mice as done previously (Tschaharganeh et al., 2014). Briefly, plasmid mix consisting of MYC transposon (10 μg), sleeping beauty transposase (2 μg), and sgRNA/Cas9 containing plasmid (10 μg) was injected in a single mouse in 2 mL saline. BSO treatments were done as described previously(Kramer et al., 1989). For mouse HCC isograft experiment, ~5 million cells in 50% matrigel were injected subcutaneously in nude mice. For Huh1, H460, and H3255 xenografts, ~5 million cells in 50% matrigel were subcutaneously transplanted into immunodeficient NOD/SCID/IL2Rγ−/− (NSG) mice. N-acetyl cysteine (NAC) was added in drinking water (40 mM) and replaced daily. Tumor measurements were done periodically and volumes were calculated using the formula 1/6*π*length*width2/1000. Animals were randomized prior to treatments and tumor measurements were done in a blinded manner i.e. person injecting/measuring tumors did not know the experimental conditions. Unless otherwise stated we used ≥ 5 age-matched (7–8 weeks) female mice for all in vivo experiments (Jackson laboratories).
METHODS DETAILS
Gene and Protein expression
Total RNA was isolated using mRNA easy kit (Qiagen) and reverse transcribed using M-MuLV reverse transcriptase (NEB) following priming by oligo-dT. Quantitative PCR was performed using TaqMan fast master mix (ABI) on QuantStudio Flex detection system (ABI) using gene specific primer/probe sets. PCR analysis was performed using 2−ΔΔCT method. For quantitative PCR array, DNA-free RNA was reverse transcribed and gene expression was quantitated using RT2 profiler oxidative stress array. The CT values were normalized to all house-keeping genes included in the array and analyzed using 2−ΔΔCT method.
RNA expression analysis was accomplished as previously described (Ortega-Molina et al., 2015). Briefly, DNA free RNA was subjected to library preparation using Illumina TrueSeq RNA kit V2 according to manufacturer’s protocol and sequenced on HiSeq2500 platform (50 bp single end reads). The trimmed reads were aligned to the mouse genome MM10 using rnaStar aligner and expression count matrix was generated from the aligned reads using HTSeq. Normalization and differential expression analysis was done on the raw count matrix using DESeq in R/Bioconductor package.
For immunoblotting total lysates were prepared in RIPA buffer supplemented with protease and phosphatase inhibitors, treated with benzonase, separated on 4–12% Tris-acrylamide gels (Invitrogen), and transferred on to a nitrocellulose membrane. Whenever required nuclear and cytoplasmic fractionations were performed using NE-PER extraction kit (Thermo Scientific, PI78833). Antibodies used in this study are described in Resource Table. Proteins were visualized using LI-COR or enhanced chemiluminescence detection after incubation with applicable secondary antibodies. All antibody incubations were done in PBS blocking buffer (LI-COR).
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| NQO1 | CST | Cat#62262 |
| TXNRD1 | CST | Cat#15140 |
| GPX2 | R&D | Cat#MAB5470 |
| NRF2 | R&D | Cat#MAB3925 |
| FN3K | Invitrogen | Cat#PA5–28603 |
| EGFR | CST | Cat#4267 |
| phosphoERK | CST | Cat#4370 |
| tERK | CST | Cat#9102 |
| KEAP1 | CST | Cat#8047 |
| MAFG | R&D | Cat#MAB3924 |
| LDHA | CST | Cat#3582 |
| Histone H3 | Abcam | Cat#ab1791 |
| Lamin B | Santa Cruz | Cat#sc-374015 |
| β-actin | Sigma-Aldrich | Cat#A5441 |
| Tubulin | Sigma-Aldrich | Cat#T5168 |
| GAPDH | CST | Cat#2118 |
| Goat anti-rabbit-IR800 | LI-COR | Cat#926–32211 |
| Goat anti-mouse-IR680 | LI-COR | Cat#926–68070 |
| Donkey anti-goat-IR800 | LI-COR | Cat#925–32214 |
| Quick western IR680 | LI-COR | Cat#926–68100 |
| goat anti-mouse-HRP | Santa Cruz | Cat#sc-2005 |
| PE-conjugated NRF2 | CST | Cat#14409 |
| ChIP/IP NRF2 antibody | CST | Cat#14596 |
| IgG | CST | Cat#2729 |
| IHC: Ki67 | Abcam | Cat#ab16667 |
| IHC: HNF4a | Santa Cruz | Cat#sc-6556 |
| IHC: MYC | Abcam | Cat#ab32072 |
| HUMAN qPCR Probes | ||
| NQO1 | Life Technologies | Hs01045993_g1 |
| TXNRD1 | Life Technologies | Hs00917067_m1 |
| GPX2 | Life Technologies | Hs01591589_m1 |
| GCLC | Life Technologies | Hs00155249_m1 |
| FN3K | Life Technologies | Hs00223368_m1 |
| NRF2 | Life Technologies | Hs00975961_g1 |
| GUSB | Life Technologies | 4333767 |
| ACTB | Life Technologies | 4331182 |
| GAPDH | Life Technologies | 4332649 |
| MOUSE qPCR Probes | ||
| Nrf2 | Life Technologies | Mm00477784_m1 |
| Actb | Life Technologies | Mm00607939_s1 |
| Bacterial and Virus Strains | ||
| Stable Competent cells | NEB | Cat#C3040H |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant human NRF2 | Origene | Cat#TP760529 |
| Erlotinib | Selleck Chemicals | Cat#S1023 |
| MG-132 | Sigma-Aldrich | Cat#M7449 |
| Trametinib | Selleck Chemicals | Cat#S2673 |
| NAC | Sigma-Aldrich | Cat#A7250 |
| BSO | Sigma-Aldrich | Cat#B2640 |
| Ganciclovir | Sigma-Aldrich | Cat#G2536 |
| Critical Commercial Assays | ||
| CellTiter-Glo reagent | Promega | Cat#G9242 |
| Nano-Glo assay | Promega | Cat#N1110 |
| GSH/GSSG-Glo assay | Promega | Cat#V6611 |
| Glucose uptake-Glo assay | Promega | Cat#J1342 |
| T7 assay | NEB | Cat#M0302 |
| Genomic DNA | Promega | Cat#A1120 |
| Mini-Column Cellufine PB Affinity Chromatography | AMS biotechnology | Cat#202–51 |
| Oxidative stress Array | Qiagen | Cat#330231 |
| FAST Taqman Mastermix | Applied Biosystems | Cat#4444557 |
| FAST Sybr Green Mastermix | Applied Biosystems | Cat#4385612 |
| Fix/Perm Buffer | Invitrogen | Cat#00–5523-00 |
| NE-PER Kit | Thermo Scientific | Cat#78833 |
| H & E and IHC slides | Histowiz.com | N/A |
| Experimental Models: Cell Lines | ||
| H3255 | ATCC | Cat#CRL-2882; RRID: CVCL_0262 |
| H460 | Lowe lab | RRID: CVCL_0459 |
| HepG2 | ATCC | Cat#HB-8065; RRID: CVCL_0027 |
| 293T | ATCC | Cat#CRL-3216; RRID: CVCL_0063 |
| FL5–12 | (McKearn et al., 1985) | RRID: CVCL_0262 |
| Huh1 | Tschaharganeh lab | RRID: CVCL_02956 |
| PC9 | Lowe lab | RRID: CVCL_B260 |
| MYC/sgKeap1 mHCC line | This study | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J | Jackson Laboratory | Stock # 000664; RRID: IMSR_JAX000664 |
| Nude | Jackson Laboratory | Stock # 007850; RRID: IMSR_JAX007850 |
| NOD/SCID/IL2Rγ−/− (NSG) | Jackson Laboratory | Stock # 005557; RRID: IMSR_JAX005557 |
| Oligonucleotides | ||
| List of oligos | See Table S7 | N/A |
| Recombinant DNA | ||
| pNLF1-NRF2 (NRF2-nanoluc fusion) | Promega | N1391 |
| psPAX2 | Trono lab | Addgene # 12260 |
| psVSV.G | Wendel lab | N/A |
| LentiCRISPR V2 | (Sanjana et al., 2014) | Addgene # 52961 |
| pKLV Lentiviral backbone | (Koike-Yusa et al., 2014) | Addgene # 62348 |
| sgRNA library | (Koike-Yusa et al., 2014) | Addgene # 50947 |
| Software and Algorithms | ||
| Prism V7 | GraphPad software | https://www.graphpad.com |
| DAVID | N/A | https://david.ncifcrf.gov |
| cBioPortal | N/A | www.cBioPortal.org |
| Image studio Lite | Li-COR | https://www.licor.com/bio/image-studio-lite/ |
| Scaffold 4.8.4 | N/A | www.proteomesoftware.com/products/scaffold/ |
| ExPASy | N/A | www.expasy.org |
| R Package DESeq | N/A | https://www.huber.embl.de/users/anders/DESeq/ |
| Deposited Data | ||
| RNA sequencing data | This study | GEO accession GSE133160 |
| Proteomic data | This study | ProteomeXchange ID PXD014334 |
| Other Reagents | ||
| Magnetic protein A/G beads | Life Technologies | Cat#88803 |
| Purecol Collagen | Fisher Scientific | Cat#50360230 |
| MuMLV-RT | NEB | Cat#M0253 |
| RPMI 1650 | MSKCC Media Core | N/A |
| DMEM-HG | MSKCC Media Core | N/A |
| DMEM w/o glucose | Life Technologies | Cat#11966025 |
| Glucose | Life Technologies | Cat#A249001 |
| FBS | Sigma-Aldrich | Cat#F8317 |
| IL3 | PeproTech, Inc | Cat#213–13 |
| Puromycin | Life Technologies | Cat#A1113803 |
| Polybrene | Sigma Aldrich | Cat#TR-1003 |
| EGF | Sigma Aldrich | Cat#E9644 |
| TGFα | PeproTech, Inc | Cat#100–16A |
For flow cytometry, cells were fixed in fixation/permeabilization buffer as per manufacturer’s instruction and stained with either isotype control (1:100) or PE-conjugated NRF2 antibody (1:100). Samples were analyzed on Guava bench-top flow cytometer. IHC was performed as described previously (Tschaharganeh et al., 2014).
In vitro glycation and PB affinity chromatography
Recombinant human NRF2 (Origene) was glycated in vitro in PBS supplemented with 1, 5 or 10 g/L glucose at 37°C for 3 hours, 3 days, or 14 days. Subsequently, NRF2 was acetone precipitated, separated on 4–12% Tris-acrylamide gel (Invitrogen), and analyzed by LC/MS. For direct detection of glycation, in vitro glycated NRF2 was directly processed for proteolytic cleavage without precipitation/PAGE.
Phenyl borate (PB) has been used to isolate glycated proteins (Fluckiger and Gallop, 1984). Briefly, 1.5–2 mg of nucleic acid free whole cell lysates were processed for PB affinity purification as per manufacturer’s instruction. Protein lysates were run through the 1 mL PB columns, after which columns were washed at least three times with wash buffer (~10 mL each) and proteins trapped in the columns were competitively eluted using 1M Sorbitol. The eluted proteins were precipitated using ice-cold acetone, resuspended in protein loading buffer, and subjected to immunoblotting or mass spectrometry. 2.5–20% of lysates prior to PB enrichment were used as input for immunoblotting. For Figure 4, 1.5 mg protein was used for PB/MS and 75 μg for total MS. We corrected for this difference during analysis.
Chromatin and protein immunoprecipitations
Quantitative ChIP PCR was done on ~15–20 million cells as described previously(Banito et al., 2018). Briefly, following cross-linking nuclei were isolated and gDNA was sheared by sonication (Covaris machine, 25 mins at 4°C). ChIP was performed by incubating fragmented DNA with either NRF2 antibody or IgG at 4°C ON. The complexes were pur ified using magnetic protein A/G beads and following elution and reverse cross-linking DNA was isolated using PCR purification columns (Qiagen). qPCR was performed on ChIP DNA using SYBR green chemistry (Fast MM, ABI). The primers used are shown in Resource Table (Reichard et al., 2007; Torrente et al., 2017). For immunoprecipitation and co-immunoprecipitation, we used the nuclear Co-IP kit (Active Motif) and followed stringent IP and wash conditions. Immunoprecipitations were carried out using ~2 mg of pre-cleared H460 or Huh1 nuclear lysates that were incubated with NRF2 antibody (1:50) at 4°C ON and pulled down with magnetic protein A/G beads. After extensive washes as per manufacturer’s instructions antibody bound proteins were eluted in loading buffer, separated on 4–12% Tris-acrylamide gel, and either immunoblotted or processed for mass spectrometry (90–125 KDa gel sections) as required. For immunoblotting, 3% or 10% lysates were used as input.
Viability, luciferase and T7 endonuclease assays
For all relevant proliferation assays, we used CellTiter-Glo reagent (Promega) as per manufacturer’s protocol. Luciferase assays were performed using Firefly luciferase assay kit as per manufacturer’s protocol (Promega). Luciferase assay values were either normalized to total protein content measured by Bradford assay or viable cells measured by CellTiter-Glo. Reduced and oxidized glutathione levels were measured using luminescence-based assay (Promega, V6611) as per manufacturer’s instructions. pNLF1-NRF2 (NRF2-nanoluc fusion, Promega) was used to assess NRF2 stability. For each biological replicate relevant reporter assays were done in duplicate.
To detect CRISPR-induced mutations at target loci we first harvested genomic DNA (Promega), amplified the target region, and performed T7 assay (NEB) as per manufacturer’s protocol.
Genome-wide CRISPR screening
FL5.12 cells (maintained in RPMI-1640 containing 2 g/L glucose) were first transduced with lentivirus to express Cas9 in a doxycycline-inducible manner then with genome-wide CRISPR library(Koike-Yusa et al., 2014) and ARE-reporter construct. The library transduction efficiency was kept to ~25% and screening was done at ~100X library coverage. Cas9 was induced with doxycycline (1 μg/mL) for 5 days following which cells were sequentially treated with tBHQ (2 μM) for 24 hours and ganciclovir (1.25 μM) for 48 hours. After three cycles of tBHQ and ganciclovir treatment, gDNA was isolated from surviving cells and sgRNA region was amplified using the following primer pair F: GACTATCATATGCTTACCGT & R: AAGTGCCCAGCGGGGCTGCT. The gel-purified amplicon was barcoded and subjected to deep sequencing using MiSeq platform using Illumina sequencing protocol (150 bp single end reads). Cells harvested after doxycycline but prior to tBHQ/ganciclovir treatment were used as control for initial sgRNA representation. Screen was done in triplicate with each replicate transduced independently with the sgRNA library.
All reads were trimmed using fastx_trimmer from fastx_toolkit (version 0.0.13) to extract the region of interest, then collapsed with fastx_collapser to obtain a tab delimited table of raw counts. This table of counts was then joined with the sgRNA library sequences, using a home-brewed python script, which reads through both files (read count and library sequences) and looks for a match in the sequence to assign the count to the corresponding sgRNA. The resulting table, containing the read count, sgRNA sequence, sgRNA name and associated gene, serves as an input for the Differential Analysis, performed using the R package DESeq.
Parameters used for Deseq were the local fit in estimating dispersion:
cdsA = estimateDispersions( cdsA,fitType=“local” )
The nbinomTest was used for differential analysis:
resSample1 = nbinomTest( cdsA, “untreated”, “treated” )
Gene was identified as significantly enriched if it fulfilled following criteria:
total normalized reads > 4X the overall coverage (top ~2% of sequencing reads)
Gene enriched in all three replicates with p-value ≤ 0.05 and average FC > 3
≥ 2 sgRNAs were enriched in all the replicates with a subset showing >10X FC and high sequence coverage (top 10%, ≥ 5X sequencing coverage) (with the exception of Rasgef1c and Rps6ka5 that had only 1 sgRNA enriched in one of the three replicate but had 4 and 2 sgRNAs enrich in the other replicates and fulfilled all the other conditions)(Koike-Yusa et al., 2014). Genes that showed enrichment due to their tumor suppressive function were ignored during analysis.
SELECT and additional computational analysis
We ran the SELECT analysis(Mina et al., 2017) on The Cancer Genome Atlas PanCanAtlas cohort(Sanchez-Vega et al., 2018) including somatic point mutations and GISTIC gene-level copy number calls for a total of 9,125 samples from 32 different tumor types. Somatic mutations and copy number alterations were distilled into the genomic alteration matrix (GAM) required by SELECT, using the list of 505 selected functional events (SFEs) defined in (Mina et al., 2017). This collection of events includes both hotspots and copy number variants affecting both tumor suppressor and oncogenes. To identify co-occurrence and mutual exclusivity patterns between alterations affecting the NRF2 pathway and other functional genomic events we merged somatic point mutations affecting NRF2 and KEAP1 genes (NRF2 meta-SFE). SELECT was then run using the same parameters as in the original work and significant patterns involving the NRF2 meta-SFE were extracted. The significance threshold (t = 0.24) was determined as previously described(Mina et al., 2017).
GSEA and DAVID analysis: For human data, we compared publicly available (TCGA) RNA expression data between EGFR-mutant and all patients excluding those with KEAP1, CUL3, and NRF2 mutations and identified significant mRNA changes between the two groups (p-value < 0.1). We did the same analysis using the differential expression list obtained by comparing NRF2-stabilized patient samples (KEAP1, CUL3, and NRF2 mutation) and all samples (excluding EGFR-mutant cases). GSEA was conducted in pre-ranked mode using default parameters and enrichment of NRF2 targets was determined using gene list from Malhotra et al.(Malhotra et al., 2010). In addition, we performed enrichment analysis for KEGG and GO terms using MSigDB. For DAVID analysis, we used pre-ranked up- or down-regulated gene set according to the experimental conditions and identified GO and KEGG pathways enriched using medium stringency and default parameters. Unsupervised clustering of murine HCC tumors was performed using Morpheus online tool and Euclidean distance metric setting.
Mass Spectrometry
RP-nanoLC-MS/MS
Proteins were separated by SDS/PAGE, and stained with Brilliant Blue R Stain; and 15 gel sections excised with in situ trypsin digestion of polypeptides in each gel slice was performed as described previously(Shevchenko et al., 2006). The tryptic peptides were desalted by using stage tips as described by Rappsilber et al.(Rappsilber et al., 2003) using C18 Empore Solid Phase Extraction Disk (3M) placed at the end of a 200uL pipette tip. The purified peptides were diluted to 0.1% formic acid, and each gel section was analyzed separately by microcapillary LC with tandem MS by using the NanoAcquity system (Waters) with a 100-μm inner diameter × 10-cm length C18 column (1.7 μm BEH130; Waters) configured with a 180-μm × 2-cm trap column coupled to a Q-Exactive Plus mass spectrometer (Thermo Fisher). Trapping was performed at 15 μL/min buffer A for 1 min and elution with a 50% linear acetonitrile gradient over 120 minutes. Similarly, 5–10 μg recombinant human non-glycated and in vitro glycated NRF2 was purified by SDS-PAGE, visualized with coomassie staining, enzymatically digested with trypsin, chymotrypsin, or AspN enzymes, desalted, and analyzed by shotgun LC-MS/MS sequencing as described above. To directly detect glycation, three replicates of ~2.8 μg in vitro glycated NRF2 (10 g/L glucose, 3 days) were reduced with 5mM TCEP (56°C, 30min), alkylated with 11mM iodoacetamide (RT, 30min, dark), and quenched with 6mM DTT (RT, 15min, dark). Replicates were digested at a 1:10 enzyme to substrate ratio with trypsin (37°C), chymotrypsin (25°C), or Glu-C (37°C ), overnight with a urea concentration of 0.5M. Enzymatic activity was quenched with the addition of TFA to about 1%. Peptides were desalted by a StageTip method by Rappsilber using a C18 disk (3M Empore Solid Phase Extraction Disk). Prior to loading samples, StageTips were conditioned with 50μL washes of acetonitrile, 50% acetonitrile in 0.1% formic acid, then 0.1% formic acid. After washing, samples were loaded then washed with 0.1% formic acid. Elutions were collected with two 30μL washes with 50% acetonitrile in 0.1% formic acid, dried down by Speedvac, and reconstituted in 0.1% formic acid for LC-MS/MS analysis. MS data were collected in data dependent acquisition mode. Full scan MS1 spectra were acquired over 380–1600 ms at a resolution of 70,000 (m/z 400) with automatic gain control (AGC) at 3 × 106 ions, The top 15 most intense precursor ions were selected for HCD fragmentation performed at normalized collision energy (NCE) 25% with target ion accumulation value of 5 × 10(4). MS/MS spectra were collected with resolution of 17,500.
Targeted LC-MS/MS quantitation of NRF2 glycation
To quantitate NRF2 glycation by mass spectrometry, we first used Skyline (v 4.142) a freely available software (available at http://sky-line.maccosslab.org) to identify unique tryptic peptides present in NRF2 but absent in the rest of the human proteome. Peptides that contained cysteine or methionine were excluded. We then quantitated those peptides using targeted parallel reaction monitoring (PRM) on a high resolution and accurate mass quadrupole-orbitrap mass spectrometer. First, 1.4, 0.7, 0.35, and 0.18 μg NRF2 was used to generate standard curves for nine unique peptides. Coomassie stained gel bands were excised, reduced with DTT, alkylated with IAA, and digested with trypsin overnight at 37°C. Peptides were then desalted usi ng C18 zip tips, dried by vacuum centrifugation, and reconstituted in 10μL 0.1% formic acid. A 1μL injection was used for targeted PRM analysis of the NRF2 peptides for standard curve. The targeted PRM analysis of NRF2 was performed on the identical LC-MS system as described in detail above but adjusted for PRM mode. The QE Plus was operated in PRM mode with full MS scans performed with the following parameters: resolution: 70,000; AGC target: 3e6; maximum IT: 200 ms; scan range: 150 to 2,000 m/z. Targeted MS2 scans (PRM) were performed on m/z of 501.7644, 514.2458, 575.8347, 716.3832, 700.6559, and 494.7767 m/z with the following parameters: resolution: 17,500; AGC target: 2e5; maximum IT: 600ms; isolation window: 1.5 m/z; nce: 27. For each peptide, 4 to 6 singly charged y-type fragment ions near or above the precursor mass were used for the PRM quantitation. Thermo Xcalibur version 2.2 was used to analyze and integrate the LC-PRM data. Chromatographic peaks with 10 p.p.m. mass tolerance and retention time within 60 seconds of the determined elution time. ICIS integration algorithm was used to integrate the area under the curve for each product ion within 10 ppm and plotted as a summed intensity.
Database Search
The LC-MS/MS .raw files were processed using Mascot and searched for proteins against the SwissProt protein database for human (version 2.6.1.100 and downloaded July 5th, 2017) and mouse (version 2.3.02 and downloaded September 6th, 2016), respectively. Carbamidomethylation of C was set as a fixed modification and the following variable modifications were allowed: oxidation (M), N-terminal protein acetylation, deamidation (N and Q), and glycation (K and R). Search parameters specified an MS tolerance of 10 ppm, an MS/MS tolerance at 0.080 Da, and full trypsin digestion allowing ≤ two missed cleavages. False discovery rate was restricted to 1% at both protein and peptide level and normalized protein intensities were obtained using Scaffold (4.8.4). Glycated lysines were detected by measuring the increase in m/z ratios corresponding to the mass of +162.0528 for fructosamine (early product of glycation) or that of downstream advanced adducts such as +58.0054 for carboxymethyl lysines (CML) or +72.0211 for carboxyethyl lysines (CEL)(Marotta et al., 2003). The expected m/z ratios (Figure S6H) were calculated using ExPASy.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data are presented as mean and error bars represent standard deviation (SD) from ≥ 3 biological replicates, unless otherwise stated in the figure legends. All statistical analyses were done using GraphPad Prism (v.7.0). Kaplan-Meier Log-rank analysis was performed to compare mouse survival and unpaired two-tailed parametric t test with Welch’s correction was used for all other statistical analyses. Modified t test and negative binomial test with Benjamini-Hochberg correction were performed for genetic screen and RNAseq analysis, respectively using the DESeq package. All statistical details including number of biological replicates (n) can be found in results and/or figure legends.
DATA AND CODE AVAILABILITY
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD014334. RNA sequencing data are available via GEO accession GSE133160.
Supplementary Material
Figure S1: Oncogenic role of NRF2 in liver cancer (Related to Figure 1 and Table S1)
A) Lollipop plot showing mutation spectrum of NRF2, KEAP1, CUL3, and CAND1 in human HCC; NRF2 mutations localize to the N-terminal ETGE and DLG motifs (labeled in red) that are docking sites for KEAP1/CUL3-dependent E3 Ubiquitin ligase complex while those affecting KEAP1, CUL3, and CAND1 are spread across their ORF and are detrimental to their function; B) Oncoprint showing mutual exclusive gain-of function alterations in NRF2 and loss of its negative regulators KEAP1, CUL3, and CAND1 in HCC; C) Detection of site-specific in-frame stabilizing/activating Nrf2 mutations in the murine HCCs; sequencing showed CRISPR-induced in-frame indels disrupting the ETGE motif and includes a mutation that resembled the alternate splice pattern leading to exon 2 loss in human HCCs (Tumor-1)(Goldstein et al., 2016); D) T7 endonuclease assay confirms gene editing at the indicated loci; red arrows point to T7 cleavage products; # indicates a non-specific product; E) H & E staining and IHC of MYC/sgKeap1 murine HCCs with indicated antibodies; boundary defining normal (N) and tumor (T) cells is marked in red; F) Unsupervised clustering of RNA sequencing data from murine HCCs reveals activation of a common signature by Nrf2, Keap1, and Cul3 mutations; G) Gene set enrichment analysis (GSEA) reveals activation of canonical NRF2 target genes in murine MYC/sgNrf2, MYC/sgKeap1, and MYC/sgCul3 HCCs compared to MYC/sgGFP control HCCs; H) Immunoblot analysis of indicated NRF2 targets in murine HCCs driven by MYC/sgGFP, MYC/sgKeap1, MYC/sgNrf2, MYC/sgCand1 or MYC/sgCul3; I) Nrf2 knockdown by shRNA measured by qRT-PCR; J) Immunoblot of indicated NRF2 targets in murine HCC lines 1 & 2 transduced with control or Nrf2 shRNAs; K) Quantification of reduced and oxidized glutathione in control (n=7 tumors) or BSO treated (n=8 tumors) mice (n=4 mice for each condition) following hydrodynamic injection with MYC transposon and sgKeap1/Cas9 constructs. (* p-value <0.05 by two-tailed student t test).
Figure S4: FN3K regulates NRF2 stability and links its function to cell’ nutrient levels (Related to Figure 4 and Table S4)
A) Indicated HepG2 cells were probed with isotype control or PE-conjugated αNRF2 antibody and subjected to flow cytometry; B and C) Flow cytometry-based detection of NRF2 in control, shFN3K-1 (B), or shFN3K-2 (C) transduced HepG2 cells; D) Quantification of NRF2 expression in parental and FN3K-silenced HepG2 cells from B (shFN3K-1) and C (shFN3K-2) (n=6); E and F) Expression of NRF2 in Parental, FN3K-, or NRF2-silenced H3255 cells as detected by immunoblotting (E) or flow cytometry (F); G) Lysates from parental and FN3K-silenced H3255 cells treated with either DMSO or MG132 (10 μM, ~6 hours) were immunoblotted and probed for NRF2 expression; H) Quantitative PCR to measure FN3K and NRF2 expression in indicated HepG2 cells; mean and SD of n=3; I) Quantitative PCR to assess expression of FN3K, NRF2, and NRF2 targets NQO1 and GPX2 in H3255 cells transduced as indicated; mean and SD of n=3 replicates; J) Immunoblot to assess the expression of NRF2 and FN3K in DMSO or trametinib treated H3255 cells (25 nM, 24 hours); K) Quantitative PCR to assess expression of indicated NRF2 target genes in Huh1 cells cultured in 5 or 10 g/L glucose; error bar represent SD from 3 replicates; L) Lysates from control or FN3K overexpressing Huh1 cells were immunoblotted and probed with indicated antibodies; M) NQO1 expression in control and FN3K overexpressing Huh1 cells cultured in 5 or 10 g/L glucose as indicated; mean and SD of 5 replicates; N) Immunoblot analysis of NRF2-bound fraction and total nuclear lysates (3%) from FN3K proficient and deficient H460 cells (KEAP1D236H); O) FN3K and NQO1 expression in cells used in immunoprecipitation shown in panel S4N; P) Immunoblot of indicated Huh1 cells cultured in 5 or 1 g/L glucose and probed with αNQO1, αTXNRD1, and αactin; Q) Relative expression of indicated NRF2 target genes in parental and FN3K-silenced Huh1 cells growing in 1 g/L glucose; data represents mean and SD from 3 replicates; R) Glucose uptake in NRF2E79V/ΔNEH1 and NRF2E79V expressing KEAP1wt HepG2 cells; Y-axis represents mean of 4 replicates ± SD; S) Glucose uptake in parental and NRF2-deficeint KEAP1N414Y Huh1 HCC cells; Y-axis represents mean of 4 replicates ± SD. (* p-value < 0.05 by two-tailed student t test).
Figure S5: Quantifying NRF2 glycation (Related to Figures 5, S6, and Table S5)
A and B) Shotgun mass spec derived peptide profile of non-glycated (right) or glycated (left) NRF2 cleaved with chymotrypsin (A) or AspN (B); C) Mass spec coverage of recombinant NRF2 cleaved with trypsin, chymotrypsin, or Glu-C; colored area corresponds to regions that were detected; D) Number of tryptic cleavages originating or terminating at the indicated K/R in control or in vitro glycated NRF2 (related to Figure S6); E and F) Parallel reaction monitoring (PRM)-derived standard curve of area under the curve (AUC) generated for indicated tryptic peptides following digestion of increasing amounts of unglycated NRF2; G) AUC analysis of indicated tryptic peptides generated from non-glycated and in vitro glycated NRF2 (1 g/L glucose, 3 hours); data are represented relative to R34-R42 peptide and error bar represents SD from ≥ 4 replicates; H) Quantitative analysis of NRF2 target genes TXNRD1 and GCLC in control, glycation competent NRF2E79V, or glycation deficient NRF2E79V-A6 expressing HepG2 cells; mean and SD from n=3 replicates. (* p-value < 0.05 by two-tailed student t test).
Figure S6: Mapping NRF2 glycation (Related to Figures 5, S5, and Table S5)
A-G) MS2 spectral profiles of K463-K472 (non-glycated) (A), R460-K472 (K462 glycated) (B), K463-K487 (K472 glycated) (C), K487-R499 (non-glycated) (D), K472-R499 (K487 glycated) (E), R569-R587 (non-glycated) (F), and R569-R587 (K574 glycated) (G); the amino acid position denotes the tryptic cleavage sites (see Table S5 for fragmentation profile); H) Table showing increase in mass of R460-K472, K462-K487, K487-R499, and R569-R587 tryptic peptides due to glycation.
Figure S7: FN3K loss impairs NRF2 function in vivo (Related to Figure 6 and Table S6)
A) Targeted deep sequencing to assess frequency and nature of indels at indicated loci from four control (MYC with sgKeap1/sgGFP) and eleven experimental tumors (MYC with sgKeap1/sgFn3k); B) Tumor volumes of isografted murine MYC/sgKeap1-derived HCC line 1 transduced with either control or Fn3k-specific shRNA; C) Kaplan-Meier survival analysis of mice hydrodynamically injected with transposon combination of MYC with either GFP (control) or Fn3k-IRES-GFP (FN3K); D) Tumor volumes measured 30 days after subcutaneous transplantation of wild type or FN3K silenced KEAP1D236H H460 NSCLC cells; E) Tumor volume of EGFRL858R mutant H3255 lung cancer xenografts with and without FN3K knockdown at ~day 30 after implantation; F) FN3K proficient and deficient H3255 (black) and PC9 (blue) cells with and without KEAP1 editing as indicated were treated with DMSO or erlotinib (50 nM) and assessed for viability; data are represented as the ratio of erlotinib to DMSO and error bars indicate SD of 5 replicates; G) Unsupervised clustering of mass spectrometric proteome analyses of Fn3k-proficient (n=2) and deficient (n=4) murine MYC/sgKeap1 HCCs (tumors correspond to Extended Data Fig. 6B); H) GSEA identifies NRF2 targets as most significantly down-regulated proteins in Fn3k-deficient MYC/sgKeap1 HCCs in vivo; I) qRT-PCR analysis of NRF2 target genes NQO1, TXNRD1, GPX2, and GCLC in indicated H460 xenografts; normalized to the GUSB housekeeping gene and shown as average fold change over control shRNA (n=3 ± SD); J) Relative expression of NRF2 target genes NQO1, TXNRD1, GPX2, and GCLC in indicated H3255 xenografts by qRT-PCR normalized to GUSB and shown as average fold change over control shRNA (n=5 ± SD); K) Representative xenografts 20 days after transplantation with FN3K proficient and deficient Huh1 cells left untreated or treated with NAC (40 mM) as indicated; L) Tumor volumes of xenografts shown in panel S5K; M) Quantitation of reduced glutathione in Huh1 xenografts shown in panels S5K and S5L; error bars represent SD from 8 tumor sections harvested from 4 independent mice; N) Parental or FN3K-silenced Huh1 cells were transduced with transcriptionally active variant NRF2E79V-P2A-mCherry as indicated and grown in normal or H2O2 (25 μM) containing media; fraction of NRF2E79V expressing cells were analyzed at days 4 and 8 of H2O2 treatment by flow cytometric detection of linked mCherry reporter; data are represented as fold change in % of mCherry+ cells relative to untreated cells and SD is from n=4 replicates. (* p-value < 0.05 by two-tailed student t test).
Figure S8: The “glycated” proteome of Huh1 cells (Related to Figure 7 and Table S7)
A) Waterfall plot of glycated proteins ordered by increase in % of glycation; B) Immunoblot of total Huh1 extracts used in mass spec confirms that FN3K deficiency does not alter the overall expression of LDHA or histone H3 while it decreases that of NQO1 as expected; C) Mapped lysines within histones H3 (upper), H2B (middle), and eIF4A1 (lower) that are glycated in the absence of FN3K (indicated in red); peptide extensions for shFN3K samples are underlined; D) Immunoblot analysis of PB-enriched (PB-fraction), PB-depleted (FT fraction), and input lysates prepared from indicated Huh1 cells; E and F) MS/MS spectra of K22-K42 (E) and extended K22-K47 peptides (F) derived from LDHA from control or FN3K-silenced Huh1 cells, respectively; red lysine represents site of glycation and mass shift of +58 indicates carboxymethyl lysine modification, downstream product of glucose catalyzed glycation.
Supplemental Table 1 (Related to Figures 1 and S1): Gene expression analysis of NRF2 activated murine HCCs
Supplemental Table (Related to Figures 4 and S4): Gene and protein expression analysis from FN3K-proficient and deficient Huh1 cells.
Supplemental Table 5 (Related to Figures 5, S5, and S6): Fragmentation profiles of peptides shown in Figures S6 and characteristics of peptides shown in Figures 5D and 5E.
Supplemental Table 6 (Related to Figures 6 and S7): Mass spectrometric analysis of Fn3k-proficient and deficient murine MYC/sgKeap1 HCC isografts.
Supplemental Table 7 (Related to Figures 7 and S8): Mass spectrometric analysis of PB-enriched proteome from FN3K-proficient and deficient Huh1 cells and primer sequences used in this study.
Figure S2: Relationship between EGFR and NRF2 in cancer (Related to Figure 2 and Table S2)
A) Mutation spectrum of NRF2, KEAP1, and EGFR in TCGA datasets and used in our SELECT analysis of mutual exclusivity; B) Gene ontology analysis of EGFR-mutant (EGFR) and NRF2 or KEAP1 mutant (NRF2) human lung and liver cancers shows activation of the NRF2-controlled gene expression programs; C) GSEA shows activation of canonical NRF2-driven genes in human EGFR-activated (left) as well as NRF2 and KEAP1 mutated (right) NSCLC and HCCs; D and E) qRT-PCR for NRF2 target genes TXNRD1 (D) and NQO1 (E) on cDNA from HepG2 cells treated as indicated (10 ng/mL EGF or 1 nM TGFα for 48 hours); mean of at least 4 replicates ± SD; F and G) Effect of erlotinib treatment on indicated NRF2 target gene expression in EGFRL858R H3255 (10nM, 48 hours) (F) EGFRΔE746-A750 PC9 cells (25nM, 48 hours) (G) by qRT-PCR; average of 3 replicates ± SD; H) Relative NRF2 mRNA expression in control or EGF treated HepG2 cells (10 ng/mL, 48 hours) (n=3); I) Relative NRF2 mRNA expression in control or erlotinib treated H3255 cells (25 nM, 48 hours) (n=3); J) Immunoblot to assess NRF2 expression in erlotinib or trametinib treated H3255 cells (25 nM, 24 hours); K) Immunoblot confirming loss of KEAP1 in H3255 cells transduced with sgKeap1/Cas9 lentiviral cassette; L) Immunoblot analysis of H3255 cells transduced with indicated sgRNA/Cas9 construct and treated with DMSO or erlotinib (10 nM, ~6 hours); M) Quantification of decrease in ERK phosphorylation in sgGFP or sgKEAP1 transduced H3255 cells following treatment with erlotinib (10 nM, ~6 hours) (n=4); N) Parental and NRF2E79V expressing HepG2 cells probed for total and phosphorylated ERK. (* p-value <0.05 by two-tailed student t test).
Figure S3: FN3K is required for NRF2-driven transcription (Related to Figure 3 and Table S3)
A) Relative luciferase activity in FL5-12-ARE reporter cells treated with DMSO, DLS (1 μM), or tBHQ (2 μM) as indicated (n=2); B) Cell viability of FL5-12-ARE cells treated as in (A) and grown in the presence or absence of ganciclovir (1.25 μM, 48 hours) (n=2); C) Relative luciferase activity in FL5-12-ARE reporter cells with or without Nrf2 silencing and treated with tBHQ as indicated; D) Relative luciferase activity in FL5-12-ARE cells transduced with indicated sgRNA-Cas9 and stressed with tBHQ (2 μM, n=6); E) FL5-12-ARE reporter cells with and without Fn3k loss were treated with tBHQ, and luciferase activity was measured (2 μM, n=10); F) T7 endonuclease assay to confirm editing by sgFn3k-1 (top) and sgFn3k-2 (bottom); red arrows indicate T7 cleavage products; G) HepG2-ARE reporter cells with and without FN3K silencing were stressed with DLS or tBHQ, as indicated and luciferase activity was assayed (n=5); H) Immunoblot of PB-enriched lysates from FN3K proficient and deficient Huh1 probed with indicated antibodies; I) Quantitative PCR to confirm FN3K knockdown (KD) in Huh1 cells used in panel S2H; J) Immunoblot of PB-enriched lysates from Huh1 growing in 5 or 10 g/L glucose probed with indicated antibodies; K) NQO1 expression in KEAP1 wild type HepG2 cells treated with DMSO or DLS in presence or absence of FN3K knockdown; Average of 5 replicates ± SD; L) Quantification of reduced and oxidized glutathione from control, FN3K- or NRF2 deficient H3255 cells treated with DLS (2 μM, ~6 hours) with and without NAC pre-treatment (5 mM, ~3 hours) as indicated; error bars represent SD from ≥ 4 replicates. (* p-value <0.05 by two-tailed student t test).
Highlights.
Glucose modification (glycation) is an important mark on intracellular proteins.
Glycation and de-glycation regulate the oncogenic transcription factor NRF2.
NRF2-driven cancers depend on Fructosamine-3-kinase to de-glycate NRF2 in vivo.
Proteomics reveals glycation of several cellular proteins and metabolic enzymes.
Acknowledgments
We thank members of Integrated genomics core (Juan Li), the Antitumor assessment core (Elizabeth Peguero, Hongyan Li, Xiaoping Chen, Amber Bahr, and Craig McCarthy) and Microchemistry and Proteomics core (Amelia Gabler) at MSKCC. We also thank D. Tschaharganeh and A. Banito for reagents and assistance and N. Socci for data expression analysis. JL is a recipient of the Shulamit Katzman fellowship. EDS is supported by MSKCC core grant P30CA008748 and NIH U54 OD020355-01. SWL is an investigator of the Howard Hughes Medical Institute. GC is supported by the Giorgi-Cavaglieri Foundation. HGW is supported by the Lymphoma Research Foundation; Mr. William H. Goodwin and Mrs. Alice Goodwin and the Commonwealth Foundation for Cancer Research; the Center for Experimental Therapeutics at MSKCC; NIH grants RO1CA183876-03, R01CA207217-01, R01CA19038-03, PA50CA217694, and P50CA192937-01A1; the Starr Cancer Consortium; the Geoffrey Beene Cancer Research Center; a Leukemia and Lymphoma Society (LLS) SPORE grant; and the MSKCC Core Grant (P30 CA008748). HGW is a scholar of the Leukemia Lymphoma Society. Mouse illustration in Figure 5 was made using Biorender (app.biorender.com)
Footnotes
Declaration of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdel-Wahab YH, O’Harte FP, Barnett CR, and Flatt PR (1997). Characterization of insulin glycation in insulin-secreting cells maintained in tissue culture. The Journal of endocrinology 152, 59–67. [DOI] [PubMed] [Google Scholar]
- Anguizola J, Matsuda R, Barnaby OS, Hoy KS, Wa C, DeBolt E, Koke M, and Hage DS (2013). Review: Glycation of human serum albumin. Clinica chimica acta; international journal of clinical chemistry 425, 64–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbour KC, Jordan E, Kim HR, Dienstag J, Yu HA, Sanchez-Vega F, Lito P, Berger M, Solit DB, Hellmann M, et al. (2018). Effects of Co-occurring Genomic Alterations on Outcomes in Patients with KRAS-Mutant Non-Small Cell Lung Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 24, 334–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X, Chen Y, Hou X, Huang M, and Jin J (2016). Emerging role of NRF2 in chemoresistance by regulating drug-metabolizing enzymes and efflux transporters. Drug metabolism reviews 48, 541–567. [DOI] [PubMed] [Google Scholar]
- Banito A, Li X, Laporte AN, Roe JS, Sanchez-Vega F, Huang CH, Dancsok AR, Hatzi K, Chen CC, Tschaharganeh DF, et al. (2018). The SS18-SSX Oncoprotein Hijacks KDM2B-PRC1.1 to Drive Synovial Sarcoma. Cancer cell 33, 527–541 e528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer AK, Cho HY, Miller-Degraff L, Walker C, Helms K, Fostel J, Yamamoto M, and Kleeberger SR (2011). Targeted deletion of Nrf2 reduces urethane-induced lung tumor development in mice. PloS one 6, e26590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollong MJ, Lee G, Coukos JS, Yun H, Zambaldo C, Chang JW, Chin EN, Ahmad I, Chatterjee AK, Lairson LL, et al. (2018). A metabolite-derived protein modification integrates glycolysis with KEAP1-NRF2 signalling. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee N, North JA, Dechassa ML, Manohar M, Prasad R, Luger K, Ottesen JJ, Poirier MG, and Bartholomew B (2015). Histone Acetylation near the Nucleosome Dyad Axis Enhances Nucleosome Disassembly by RSC and SWI/SNF. Molecular and cellular biology 35, 4083–4092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements CM, McNally RS, Conti BJ, Mak TW, and Ting JP (2006). DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proceedings of the National Academy of Sciences of the United States of America 103, 15091–15096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, et al. (2011). Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluckiger R, and Gallop PM (1984). Measurement of nonenzymatic protein glycosylation. Methods in enzymology 106, 77–87. [DOI] [PubMed] [Google Scholar]
- Fortpied J, Maliekal P, Vertommen D, and Van Schaftingen E (2006). Magnesium-dependent phosphatase-1 is a protein-fructosamine-6-phosphatase potentially involved in glycation repair. The Journal of biological chemistry 281, 18378–18385. [DOI] [PubMed] [Google Scholar]
- Frank R, Scheffler M, Merkelbach-Bruse S, Ihle MA, Kron A, Rauer M, Ueckeroth F, Konig K, Michels S, Fischer R, et al. (2018). Clinical and Pathological Characteristics of KEAP1- and NFE2L2-Mutated Non-Small Cell Lung Carcinoma (NSCLC). Clinical cancer research : an official journal of the American Association for Cancer Research. [DOI] [PubMed] [Google Scholar]
- Galligan JJ, Wepy JA, Streeter MD, Kingsley PJ, Mitchener MM, Wauchope OR, Beavers WN, Rose KL, Wang T, Spiegel DA, et al. (2018). Methylglyoxal-derived posttranslational arginine modifications are abundant histone marks. Proceedings of the National Academy of Sciences of the United States of America 115, 9228–9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein LD, Lee J, Gnad F, Klijn C, Schaub A, Reeder J, Daemen A, Bakalarski CE, Holcomb T, Shames DS, et al. (2016). Recurrent Loss of NFE2L2 Exon 2 Is a Mechanism for Nrf2 Pathway Activation in Human Cancers. Cell reports 16, 2605–2617. [DOI] [PubMed] [Google Scholar]
- Harris IS, Treloar AE, Inoue S, Sasaki M, Gorrini C, Lee KC, Yung KY, Brenner D, Knobbe-Thomsen CB, Cox MA, et al. (2015). Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer cell 27, 211–222. [DOI] [PubMed] [Google Scholar]
- Hu W, and Liu W (2010). Side populations of glioblastoma cells are less sensitive to HSV-TK/GCV suicide gene therapy system than the non-side population. In vitro cellular & developmental biology Animal 46, 497–501. [DOI] [PubMed] [Google Scholar]
- Hunter SJ, Boyd AC, O’Harte FP, McKillop AM, Wiggam MI, Mooney MH, McCluskey JT, Lindsay JR, Ennis CN, Gamble R, et al. (2003). Demonstration of glycated insulin in human diabetic plasma and decreased biological activity assessed by euglycemic-hyperinsulinemic clamp technique in humans. Diabetes 52, 492–498. [DOI] [PubMed] [Google Scholar]
- Jaramillo MC, and Zhang DD (2013). The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes & development 27, 2179–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen MB, Kiemer L, and Brunak S (2006). Analysis and prediction of mammalian protein glycation. Glycobiology 16, 844–853. [DOI] [PubMed] [Google Scholar]
- Kang KA, and Hyun JW (2017). Oxidative Stress, Nrf2, and Epigenetic Modification Contribute to Anticancer Drug Resistance. Toxicological research 33, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent LN, Rakijas JB, Pandit SK, Westendorp B, Chen HZ, Huntington JT, Tang X, Bae S, Srivastava A, Senapati S, et al. (2016). E2f8 mediates tumor suppression in postnatal liver development. The Journal of clinical investigation 126, 2955–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike-Yusa H, Li Y, Tan EP, Velasco-Herrera Mdel C, and Yusa K (2014). Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nature biotechnology 32, 267–273. [DOI] [PubMed] [Google Scholar]
- Kramer RA, Soble M, Howes AE, and Montoya VP (1989). The effect of glutathione (GSH) depletion in vivo by buthionine sulfoximine (BSO) on the radiosensitization of SR 2508. International journal of radiation oncology, biology, physics 16, 1325–1329. [DOI] [PubMed] [Google Scholar]
- Lehrbach NJ, Breen PC, and Ruvkun G (2019). Protein Sequence Editing of SKN-1A/Nrf1 by Peptide:N-Glycanase Controls Proteasome Gene Expression. Cell 177, 737–750 e715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T-Y, Cantley LC, and DeNicola GM (2016). NRF2 Rewires Cellular Metabolism to Support the Antioxidant Response.
- Ma Q (2013). Role of nrf2 in oxidative stress and toxicity. Annual review of pharmacology and toxicology 53, 401–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra D, Portales-Casamar E, Singh A, Srivastava S, Arenillas D, Happel C, Shyr C, Wakabayashi N, Kensler TW, Wasserman WW, et al. (2010). Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic acids research 38, 5718–5734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marotta E, Lapolla A, Fedele D, Senesi A, Reitano R, Witt M, Seraglia R, and Traldi P (2003). Accurate mass measurements by Fourier transform mass spectrometry in the study of advanced glycation end products/peptides. Journal of mass spectrometry : JMS 38, 196–205. [DOI] [PubMed] [Google Scholar]
- McKearn JP, McCubrey J, and Fagg B (1985). Enrichment of hematopoietic precursor cells and cloning of multipotential B-lymphocyte precursors. Proceedings of the National Academy of Sciences of the United States of America 82, 7414–7418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menegon S, Columbano A, and Giordano S (2016). The Dual Roles of NRF2 in Cancer. Trends in molecular medicine 22, 578–593. [DOI] [PubMed] [Google Scholar]
- Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, Jedrychowski MP, Costa ASH, Higgins M, Hams E, et al. (2018). Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 556, 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mina M, Raynaud F, Tavernari D, Battistello E, Sungalee S, Saghafinia S, Laessle T, Sanchez-Vega F, Schultz N, Oricchio E, et al. (2017). Conditional Selection of Genomic Alterations Dictates Cancer Evolution and Oncogenic Dependencies. Cancer cell 32, 155–168 e156. [DOI] [PubMed] [Google Scholar]
- Moremen KW, Tiemeyer M, and Nairn AV (2012). Vertebrate protein glycosylation: diversity, synthesis and function. Nature reviews Molecular cell biology 13, 448–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo HKC, Kim DH, Cha YN, Na HK, and Surh YJ (2017). Nrf2 Mutagenic Activation Drives Hepatocarcinogenesis. Cancer research 77, 4797–4808. [DOI] [PubMed] [Google Scholar]
- Nokin MJ, Durieux F, Peixoto P, Chiavarina B, Peulen O, Blomme A, Turtoi A, Costanza B, Smargiasso N, Baiwir D, et al. (2016). Methylglyoxal, a glycolysis side-product, induces Hsp90 glycation and YAP-mediated tumor growth and metastasis. eLife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang Y, Zhao C, Jiang M, Hu D, Agirre X, Niesvizky I, et al. (2015). The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nature medicine 21, 1199–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Ishihama Y, and Mann M (2003). Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Analytical chemistry 75, 663–670. [DOI] [PubMed] [Google Scholar]
- Reichard JF, Motz GT, and Puga A (2007). Heme oxygenase-1 induction by NRF2 requires inactivation of the transcriptional repressor BACH1. Nucleic acids research 35, 7074–7086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richarme G, Liu C, Mihoub M, Abdallah J, Leger T, Joly N, Liebart JC, Jurkunas UV, Nadal M, Bouloc P, et al. (2017). Guanine glycation repair by DJ-1/Park7 and its bacterial homologs. Science 357, 208–211. [DOI] [PubMed] [Google Scholar]
- Rojo de la Vega M, Chapman E, and Zhang DD (2018). NRF2 and the Hallmarks of Cancer. Cancer cell 34, 21–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeBoeuf SE, Karakousi TR, Ellis DC, Bhutkar A, Sanchez-Rivera FJ, et al. (2017). Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nature medicine 23, 1362–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, Dimitriadoy S, Liu DL, Kantheti HS, Saghafinia S, et al. (2018). Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 173, 321–337 e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki H, Suzuki A, Shitara M, Okuda K, Hikosaka Y, Moriyama S, Yano M, and Fujii Y (2013). Keap1 mutations in lung cancer patients. Oncology letters 6, 719–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh H, Moriguchi T, Saigusa D, Baird L, Yu L, Rokutan H, Igarashi K, Ebina M, Shibata T, and Yamamoto M (2016). NRF2 Intensifies Host Defense Systems to Prevent Lung Carcinogenesis, but After Tumor Initiation Accelerates Malignant Cell Growth. Cancer research 76, 3088–3096. [DOI] [PubMed] [Google Scholar]
- Satoh H, Moriguchi T, Takai J, Ebina M, and Yamamoto M (2013). Nrf2 prevents initiation but accelerates progression through the Kras signaling pathway during lung carcinogenesis. Cancer research 73, 4158–4168. [DOI] [PubMed] [Google Scholar]
- Shapiro R, McManus MJ, Zalut C, and Bunn HF (1980). Sites of nonenzymatic glycosylation of human hemoglobin A. The Journal of biological chemistry 255, 3120–3127. [PubMed] [Google Scholar]
- Shevchenko A, Tomas H, Havlis J, Olsen JV, and Mann M (2006). In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nature protocols 1, 2856–2860. [DOI] [PubMed] [Google Scholar]
- Sporn MB, and Liby KT (2012). NRF2 and cancer: the good, the bad and the importance of context. Nature reviews Cancer 12, 564–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M (2015). Glycation of Proteins. In Glycoscience: Biology and Medicine, Taniguchi N, Endo T, Hart GW, Seeberger PH, Wong C-H, ed. (Springer; ). [Google Scholar]
- Takahashi N, Chen HY, Harris IS, Stover DG, Selfors LM, Bronson RT, Deraedt T, Cichowski K, Welm AL, Mori Y, et al. (2018). Cancer Cells Co-opt the Neuronal Redox-Sensing Channel TRPA1 to Promote Oxidative-Stress Tolerance. Cancer cell 33, 985–1003 e1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terman JR, and Kashina A (2013). Post-translational modification and regulation of actin. Current opinion in cell biology 25, 30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrente L, Sanchez C, Moreno R, Chowdhry S, Cabello P, Isono K, Koseki H, Honda T, Hayes JD, Dinkova-Kostova AT, et al. (2017). Crosstalk between NRF2 and HIPK2 shapes cytoprotective responses. Oncogene 36, 6204–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschaharganeh DF, Xue W, Calvisi DF, Evert M, Michurina TV, Dow LE, Banito A, Katz SF, Kastenhuber ER, Weissmueller S, et al. (2014). p53-dependent Nestin regulation links tumor suppression to cellular plasticity in liver cancer. Cell 158, 579–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Schaftingen E, Collard F, Wiame E, and Veiga-da-Cunha M (2012). Enzymatic repair of Amadori products. Amino acids 42, 1143–1150. [DOI] [PubMed] [Google Scholar]
- Veiga da-Cunha M, Jacquemin P, Delpierre G, Godfraind C, Theate I, Vertommen D, Clotman F, Lemaigre F, Devuyst O, and Van Schaftingen E (2006). Increased protein glycation in fructosamine 3-kinase-deficient mice. The Biochemical journal 399, 257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatraman J, Aggarwal K, and Balaram P (2001). Helical peptide models for protein glycation: proximity effects in catalysis of the Amadori rearrangement. Chemistry & biology 8, 611–625. [DOI] [PubMed] [Google Scholar]
- Walker-Samuel S, Ramasawmy R, Torrealdea F, Rega M, Rajkumar V, Johnson SP, Richardson S, Goncalves M, Parkes HG, Arstad E, et al. (2013). In vivo imaging of glucose uptake and metabolism in tumors. Nature medicine 19, 1067–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Li G, Yang Z, Wang L, Zhang L, Wang T, Zhang Y, Zhang S, Han Y, and Jia L (2017). Uncoupling protein 2 downregulation by hypoxia through repression of peroxisome proliferator-activated receptor gamma promotes chemoresistance of non-small cell lung cancer. Oncotarget 8, 8083–8094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XJ, Sun Z, Villeneuve NF, Zhang S, Zhao F, Li Y, Chen W, Yi X, Zheng W, Wondrak GT, et al. (2008). Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 29, 1235–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wareham NJ, and Pfister R (2010). Diabetes: glycated hemoglobin is a marker of diabetes and CVD risk. Nature reviews Cardiology 7, 367–368. [DOI] [PubMed] [Google Scholar]
- Xu Y, Li L, Ding J, Wu LY, Mai G, and Zhou F (2017). Gly-PseAAC: Identifying protein lysine glycation through sequences. Gene 602, 1–7. [DOI] [PubMed] [Google Scholar]
- Yu HA, Suzawa K, Jordan E, Zehir A, Ni A, Kim R, Kris MG, Hellmann MD, Li BT, Somwar R, et al. (2018). Concurrent Alterations in EGFR-Mutant Lung Cancers Associated with Resistance to EGFR Kinase Inhibitors and Characterization of MTOR as a Mediator of Resistance. Clinical cancer research : an official journal of the American Association for Cancer Research 24, 3108–3118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotto-Filho A, Masamsetti VP, Loranc E, Tonapi SS, Gorthi A, Bernard X, Goncalves RM, Moreira JC, Chen Y, and Bishop AJ (2016). Alkylating Agent-Induced NRF2 Blocks Endoplasmic Reticulum Stress-Mediated Apoptosis via Control of Glutathione Pools and Protein Thiol Homeostasis. Molecular cancer therapeutics 15, 3000–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DD (2006). Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug metabolism reviews 38, 769–789. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Ames JM, Smith RD, Baynes JW, and Metz TO (2009). A perspective on the Maillard reaction and the analysis of protein glycation by mass spectrometry: probing the pathogenesis of chronic disease. Journal of proteome research 8, 754–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q, Omans ND, Leicher R, Osunsade A, Agustinus AS, Finkin-Groner E, D’Ambrosio H, Liu B, Chandarlapaty S, Liu S, et al. (2019). Reversible histone glycation is associated with disease-related changes in chromatin architecture. Nat Commun 10, 1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Ye W, Shao Q, Zhang M, and Liang J (2013). Nrf2 is a potential therapeutic target in radioresistance in human cancer. Critical reviews in oncology/hematology 88, 706–715. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Oncogenic role of NRF2 in liver cancer (Related to Figure 1 and Table S1)
A) Lollipop plot showing mutation spectrum of NRF2, KEAP1, CUL3, and CAND1 in human HCC; NRF2 mutations localize to the N-terminal ETGE and DLG motifs (labeled in red) that are docking sites for KEAP1/CUL3-dependent E3 Ubiquitin ligase complex while those affecting KEAP1, CUL3, and CAND1 are spread across their ORF and are detrimental to their function; B) Oncoprint showing mutual exclusive gain-of function alterations in NRF2 and loss of its negative regulators KEAP1, CUL3, and CAND1 in HCC; C) Detection of site-specific in-frame stabilizing/activating Nrf2 mutations in the murine HCCs; sequencing showed CRISPR-induced in-frame indels disrupting the ETGE motif and includes a mutation that resembled the alternate splice pattern leading to exon 2 loss in human HCCs (Tumor-1)(Goldstein et al., 2016); D) T7 endonuclease assay confirms gene editing at the indicated loci; red arrows point to T7 cleavage products; # indicates a non-specific product; E) H & E staining and IHC of MYC/sgKeap1 murine HCCs with indicated antibodies; boundary defining normal (N) and tumor (T) cells is marked in red; F) Unsupervised clustering of RNA sequencing data from murine HCCs reveals activation of a common signature by Nrf2, Keap1, and Cul3 mutations; G) Gene set enrichment analysis (GSEA) reveals activation of canonical NRF2 target genes in murine MYC/sgNrf2, MYC/sgKeap1, and MYC/sgCul3 HCCs compared to MYC/sgGFP control HCCs; H) Immunoblot analysis of indicated NRF2 targets in murine HCCs driven by MYC/sgGFP, MYC/sgKeap1, MYC/sgNrf2, MYC/sgCand1 or MYC/sgCul3; I) Nrf2 knockdown by shRNA measured by qRT-PCR; J) Immunoblot of indicated NRF2 targets in murine HCC lines 1 & 2 transduced with control or Nrf2 shRNAs; K) Quantification of reduced and oxidized glutathione in control (n=7 tumors) or BSO treated (n=8 tumors) mice (n=4 mice for each condition) following hydrodynamic injection with MYC transposon and sgKeap1/Cas9 constructs. (* p-value <0.05 by two-tailed student t test).
Figure S4: FN3K regulates NRF2 stability and links its function to cell’ nutrient levels (Related to Figure 4 and Table S4)
A) Indicated HepG2 cells were probed with isotype control or PE-conjugated αNRF2 antibody and subjected to flow cytometry; B and C) Flow cytometry-based detection of NRF2 in control, shFN3K-1 (B), or shFN3K-2 (C) transduced HepG2 cells; D) Quantification of NRF2 expression in parental and FN3K-silenced HepG2 cells from B (shFN3K-1) and C (shFN3K-2) (n=6); E and F) Expression of NRF2 in Parental, FN3K-, or NRF2-silenced H3255 cells as detected by immunoblotting (E) or flow cytometry (F); G) Lysates from parental and FN3K-silenced H3255 cells treated with either DMSO or MG132 (10 μM, ~6 hours) were immunoblotted and probed for NRF2 expression; H) Quantitative PCR to measure FN3K and NRF2 expression in indicated HepG2 cells; mean and SD of n=3; I) Quantitative PCR to assess expression of FN3K, NRF2, and NRF2 targets NQO1 and GPX2 in H3255 cells transduced as indicated; mean and SD of n=3 replicates; J) Immunoblot to assess the expression of NRF2 and FN3K in DMSO or trametinib treated H3255 cells (25 nM, 24 hours); K) Quantitative PCR to assess expression of indicated NRF2 target genes in Huh1 cells cultured in 5 or 10 g/L glucose; error bar represent SD from 3 replicates; L) Lysates from control or FN3K overexpressing Huh1 cells were immunoblotted and probed with indicated antibodies; M) NQO1 expression in control and FN3K overexpressing Huh1 cells cultured in 5 or 10 g/L glucose as indicated; mean and SD of 5 replicates; N) Immunoblot analysis of NRF2-bound fraction and total nuclear lysates (3%) from FN3K proficient and deficient H460 cells (KEAP1D236H); O) FN3K and NQO1 expression in cells used in immunoprecipitation shown in panel S4N; P) Immunoblot of indicated Huh1 cells cultured in 5 or 1 g/L glucose and probed with αNQO1, αTXNRD1, and αactin; Q) Relative expression of indicated NRF2 target genes in parental and FN3K-silenced Huh1 cells growing in 1 g/L glucose; data represents mean and SD from 3 replicates; R) Glucose uptake in NRF2E79V/ΔNEH1 and NRF2E79V expressing KEAP1wt HepG2 cells; Y-axis represents mean of 4 replicates ± SD; S) Glucose uptake in parental and NRF2-deficeint KEAP1N414Y Huh1 HCC cells; Y-axis represents mean of 4 replicates ± SD. (* p-value < 0.05 by two-tailed student t test).
Figure S5: Quantifying NRF2 glycation (Related to Figures 5, S6, and Table S5)
A and B) Shotgun mass spec derived peptide profile of non-glycated (right) or glycated (left) NRF2 cleaved with chymotrypsin (A) or AspN (B); C) Mass spec coverage of recombinant NRF2 cleaved with trypsin, chymotrypsin, or Glu-C; colored area corresponds to regions that were detected; D) Number of tryptic cleavages originating or terminating at the indicated K/R in control or in vitro glycated NRF2 (related to Figure S6); E and F) Parallel reaction monitoring (PRM)-derived standard curve of area under the curve (AUC) generated for indicated tryptic peptides following digestion of increasing amounts of unglycated NRF2; G) AUC analysis of indicated tryptic peptides generated from non-glycated and in vitro glycated NRF2 (1 g/L glucose, 3 hours); data are represented relative to R34-R42 peptide and error bar represents SD from ≥ 4 replicates; H) Quantitative analysis of NRF2 target genes TXNRD1 and GCLC in control, glycation competent NRF2E79V, or glycation deficient NRF2E79V-A6 expressing HepG2 cells; mean and SD from n=3 replicates. (* p-value < 0.05 by two-tailed student t test).
Figure S6: Mapping NRF2 glycation (Related to Figures 5, S5, and Table S5)
A-G) MS2 spectral profiles of K463-K472 (non-glycated) (A), R460-K472 (K462 glycated) (B), K463-K487 (K472 glycated) (C), K487-R499 (non-glycated) (D), K472-R499 (K487 glycated) (E), R569-R587 (non-glycated) (F), and R569-R587 (K574 glycated) (G); the amino acid position denotes the tryptic cleavage sites (see Table S5 for fragmentation profile); H) Table showing increase in mass of R460-K472, K462-K487, K487-R499, and R569-R587 tryptic peptides due to glycation.
Figure S7: FN3K loss impairs NRF2 function in vivo (Related to Figure 6 and Table S6)
A) Targeted deep sequencing to assess frequency and nature of indels at indicated loci from four control (MYC with sgKeap1/sgGFP) and eleven experimental tumors (MYC with sgKeap1/sgFn3k); B) Tumor volumes of isografted murine MYC/sgKeap1-derived HCC line 1 transduced with either control or Fn3k-specific shRNA; C) Kaplan-Meier survival analysis of mice hydrodynamically injected with transposon combination of MYC with either GFP (control) or Fn3k-IRES-GFP (FN3K); D) Tumor volumes measured 30 days after subcutaneous transplantation of wild type or FN3K silenced KEAP1D236H H460 NSCLC cells; E) Tumor volume of EGFRL858R mutant H3255 lung cancer xenografts with and without FN3K knockdown at ~day 30 after implantation; F) FN3K proficient and deficient H3255 (black) and PC9 (blue) cells with and without KEAP1 editing as indicated were treated with DMSO or erlotinib (50 nM) and assessed for viability; data are represented as the ratio of erlotinib to DMSO and error bars indicate SD of 5 replicates; G) Unsupervised clustering of mass spectrometric proteome analyses of Fn3k-proficient (n=2) and deficient (n=4) murine MYC/sgKeap1 HCCs (tumors correspond to Extended Data Fig. 6B); H) GSEA identifies NRF2 targets as most significantly down-regulated proteins in Fn3k-deficient MYC/sgKeap1 HCCs in vivo; I) qRT-PCR analysis of NRF2 target genes NQO1, TXNRD1, GPX2, and GCLC in indicated H460 xenografts; normalized to the GUSB housekeeping gene and shown as average fold change over control shRNA (n=3 ± SD); J) Relative expression of NRF2 target genes NQO1, TXNRD1, GPX2, and GCLC in indicated H3255 xenografts by qRT-PCR normalized to GUSB and shown as average fold change over control shRNA (n=5 ± SD); K) Representative xenografts 20 days after transplantation with FN3K proficient and deficient Huh1 cells left untreated or treated with NAC (40 mM) as indicated; L) Tumor volumes of xenografts shown in panel S5K; M) Quantitation of reduced glutathione in Huh1 xenografts shown in panels S5K and S5L; error bars represent SD from 8 tumor sections harvested from 4 independent mice; N) Parental or FN3K-silenced Huh1 cells were transduced with transcriptionally active variant NRF2E79V-P2A-mCherry as indicated and grown in normal or H2O2 (25 μM) containing media; fraction of NRF2E79V expressing cells were analyzed at days 4 and 8 of H2O2 treatment by flow cytometric detection of linked mCherry reporter; data are represented as fold change in % of mCherry+ cells relative to untreated cells and SD is from n=4 replicates. (* p-value < 0.05 by two-tailed student t test).
Figure S8: The “glycated” proteome of Huh1 cells (Related to Figure 7 and Table S7)
A) Waterfall plot of glycated proteins ordered by increase in % of glycation; B) Immunoblot of total Huh1 extracts used in mass spec confirms that FN3K deficiency does not alter the overall expression of LDHA or histone H3 while it decreases that of NQO1 as expected; C) Mapped lysines within histones H3 (upper), H2B (middle), and eIF4A1 (lower) that are glycated in the absence of FN3K (indicated in red); peptide extensions for shFN3K samples are underlined; D) Immunoblot analysis of PB-enriched (PB-fraction), PB-depleted (FT fraction), and input lysates prepared from indicated Huh1 cells; E and F) MS/MS spectra of K22-K42 (E) and extended K22-K47 peptides (F) derived from LDHA from control or FN3K-silenced Huh1 cells, respectively; red lysine represents site of glycation and mass shift of +58 indicates carboxymethyl lysine modification, downstream product of glucose catalyzed glycation.
Supplemental Table 1 (Related to Figures 1 and S1): Gene expression analysis of NRF2 activated murine HCCs
Supplemental Table (Related to Figures 4 and S4): Gene and protein expression analysis from FN3K-proficient and deficient Huh1 cells.
Supplemental Table 5 (Related to Figures 5, S5, and S6): Fragmentation profiles of peptides shown in Figures S6 and characteristics of peptides shown in Figures 5D and 5E.
Supplemental Table 6 (Related to Figures 6 and S7): Mass spectrometric analysis of Fn3k-proficient and deficient murine MYC/sgKeap1 HCC isografts.
Supplemental Table 7 (Related to Figures 7 and S8): Mass spectrometric analysis of PB-enriched proteome from FN3K-proficient and deficient Huh1 cells and primer sequences used in this study.
Figure S2: Relationship between EGFR and NRF2 in cancer (Related to Figure 2 and Table S2)
A) Mutation spectrum of NRF2, KEAP1, and EGFR in TCGA datasets and used in our SELECT analysis of mutual exclusivity; B) Gene ontology analysis of EGFR-mutant (EGFR) and NRF2 or KEAP1 mutant (NRF2) human lung and liver cancers shows activation of the NRF2-controlled gene expression programs; C) GSEA shows activation of canonical NRF2-driven genes in human EGFR-activated (left) as well as NRF2 and KEAP1 mutated (right) NSCLC and HCCs; D and E) qRT-PCR for NRF2 target genes TXNRD1 (D) and NQO1 (E) on cDNA from HepG2 cells treated as indicated (10 ng/mL EGF or 1 nM TGFα for 48 hours); mean of at least 4 replicates ± SD; F and G) Effect of erlotinib treatment on indicated NRF2 target gene expression in EGFRL858R H3255 (10nM, 48 hours) (F) EGFRΔE746-A750 PC9 cells (25nM, 48 hours) (G) by qRT-PCR; average of 3 replicates ± SD; H) Relative NRF2 mRNA expression in control or EGF treated HepG2 cells (10 ng/mL, 48 hours) (n=3); I) Relative NRF2 mRNA expression in control or erlotinib treated H3255 cells (25 nM, 48 hours) (n=3); J) Immunoblot to assess NRF2 expression in erlotinib or trametinib treated H3255 cells (25 nM, 24 hours); K) Immunoblot confirming loss of KEAP1 in H3255 cells transduced with sgKeap1/Cas9 lentiviral cassette; L) Immunoblot analysis of H3255 cells transduced with indicated sgRNA/Cas9 construct and treated with DMSO or erlotinib (10 nM, ~6 hours); M) Quantification of decrease in ERK phosphorylation in sgGFP or sgKEAP1 transduced H3255 cells following treatment with erlotinib (10 nM, ~6 hours) (n=4); N) Parental and NRF2E79V expressing HepG2 cells probed for total and phosphorylated ERK. (* p-value <0.05 by two-tailed student t test).
Figure S3: FN3K is required for NRF2-driven transcription (Related to Figure 3 and Table S3)
A) Relative luciferase activity in FL5-12-ARE reporter cells treated with DMSO, DLS (1 μM), or tBHQ (2 μM) as indicated (n=2); B) Cell viability of FL5-12-ARE cells treated as in (A) and grown in the presence or absence of ganciclovir (1.25 μM, 48 hours) (n=2); C) Relative luciferase activity in FL5-12-ARE reporter cells with or without Nrf2 silencing and treated with tBHQ as indicated; D) Relative luciferase activity in FL5-12-ARE cells transduced with indicated sgRNA-Cas9 and stressed with tBHQ (2 μM, n=6); E) FL5-12-ARE reporter cells with and without Fn3k loss were treated with tBHQ, and luciferase activity was measured (2 μM, n=10); F) T7 endonuclease assay to confirm editing by sgFn3k-1 (top) and sgFn3k-2 (bottom); red arrows indicate T7 cleavage products; G) HepG2-ARE reporter cells with and without FN3K silencing were stressed with DLS or tBHQ, as indicated and luciferase activity was assayed (n=5); H) Immunoblot of PB-enriched lysates from FN3K proficient and deficient Huh1 probed with indicated antibodies; I) Quantitative PCR to confirm FN3K knockdown (KD) in Huh1 cells used in panel S2H; J) Immunoblot of PB-enriched lysates from Huh1 growing in 5 or 10 g/L glucose probed with indicated antibodies; K) NQO1 expression in KEAP1 wild type HepG2 cells treated with DMSO or DLS in presence or absence of FN3K knockdown; Average of 5 replicates ± SD; L) Quantification of reduced and oxidized glutathione from control, FN3K- or NRF2 deficient H3255 cells treated with DLS (2 μM, ~6 hours) with and without NAC pre-treatment (5 mM, ~3 hours) as indicated; error bars represent SD from ≥ 4 replicates. (* p-value <0.05 by two-tailed student t test).
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD014334. RNA sequencing data are available via GEO accession GSE133160.