

Abstract
PALB2 (Partner and Localizer of BRCA2) was first identified as a BRCA2-interacting protein. Subsequently, PALB2 has been recognized as a cog in the cellular machinery for DNA repair by homologous recombination (HR). PALB2 also mediates S and G2 DNA damage checkpoints, and has an apparent function in protecting transcriptionally active genes from genotoxic stress. PALB2 also interacts with, is localized by, and functions downstream of BRCA1. Further, PALB2 interacts with other essential effectors of HR, including RAD51 and RAD51C, as well as BRCA2. Consistent with its function in HR and its interaction with key HR proteins, PALB2-deficient cells are hypersensitive to ionizing radiation and DNA interstrand crosslinking agents such as mitomycin C and cisplatin. Mechanistically, PALB2 is required for HR by mediating the recruitment of BRCA2 and the RAD51 recombinase to sites of DNA damage. Similar to bi-allelic loss-of-function mutations of BRCA1, BRCA2, RAD51 and RAD51C, bi-allelic mutations in PALB2 cause Fanconi anemia (FA), a rare childhood disorder which is associated with progressive bone marrow failure, congenital anomalies, and a predisposition to leukemia and solid tumors. Due to their close functional relationship, bi-allelic mutations of PALB2 and BRCA2 cause particularly severe forms of FA, called FANCN and FANCD1, both characterized by severe congenital abnormalities and very early onset of various cancers. This includes acute leukemias, Wilms tumor, medulloblastoma and neuroblastomas. Also, heterozygous germ-line mutations of PALB2, like mutations in several other essential HR genes listed above, yield an increased susceptibility to breast and pancreatic cancer.
Keywords: Fanconi anemia, Breast Cancer Susceptibility, Tumor Suppressor, Homologous Recombination, DNA Repair
Identity
Other names
FANCN, FLJ21816, LOC79728, PNCA3
HGNC (Hugo)
PALB2
Location
16p12.2
Local order
As outlined by NCBI (Gene), coding genes most proximal to PALB2 on 16p12.2, in the centromeric to telomeric direction, are ERN2 (endoplasmic reticulum to nucleus signalling 2), PLK1 (polo-like kinase 1), DCTN5 (dynactin subunit 5), PALB2, NDUFAB1 (NADH:ubiquinone oxidoreductase subunit AB1), UBFD1 (ubiquitin family domain containing 1), and EARS2 (glutamyl-tRNA synthetase 2).
DNA/RNA
The PALB2 protein was identified as a BRCA2-interacting protein using mass spectrometry. This was found to correspond to gene locus 79728 (LOC79728), which encodes putative protein FLJ21816 (Xia et al., 2006). In the same study, the first cDNA clone for human PALB2 was generated using RT-PCR. The human transcript includes 13 exons arranged as diagrammed below:
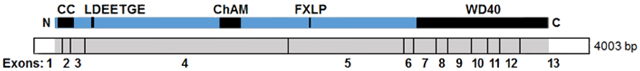
Exon structure of the human PALB2 gene. Exons are delineated by a vertical black line. The coding sequences are shown in grey, while non-coding sequences in exons 1 and 13 are shown in white. (Above) The encoded protein is shown in blue with key domains and/or motifs that mediate interactions shown in black. CC, coiled-coil; ChAM, chromatin association motif; LDEETGE, extended EDGE motif
Description
The human PALB2 gene (13 exons) spans 38.14 kb.
Transcription
Absent confirmed splice variants, the full-length transcript of PALB2 is 4,003 bp.
Protein
PALB2 has a large number of interactions with other DNA damage response proteins that function in DNA repair by homologous recombination, as illustrated below and reviewed elsewhere (Park et al., 2014b). This includes interactions with BRCA1, BRCA2, RAD51, RAD51C and XRCC3. In this way, PALB2 functions in a large network of HR proteins and seems to have a key role in coordinating their function (Park et al., 2014b). In particular, direct binding to BRCA1 mediates PALB2 recruitment to DNA damage foci (Zhang et al., 2009a; Zhang et al., 2009b). Notably, KEAP1-dependent ubiquitination of the PALB2 coiled-coil domain suppresses HR in G1 by inhibiting the interaction of PALB2 with BRCA1 (Orthwein et al., 2015). Importantly, direct interactions of the N- and C-termini of PALB2 with BRCA1 and BRCA2, respectively, physically links these tumor suppressor proteins (Sy et al., 2009b; Zhang et al., 2009a; Zhang et al., 2009b).
PALB2 also directly interacts with MORF4L1 (MRG15); this interaction appears to be independent of the interaction of MRG15 with the TIP60 histone methyltransferase complex (Hayakawa et al., 2010; Sy et al., 2009a). Additionally, PALB2 interacts with KEAP1, a sensor of oxidative stress (Ma et al., 2012).
PALB2 is essential for embryonic development; homozygous knockout of PALB2 in mice disrupts the normal differentiation of mesoderm and results in embryonic lethality by E9.5 (Bowman-Colin et al., 2013; Rantakari et al., 2010).
Description
PALB2 contains a coiled-coil domain at its N-terminus from amino acids (a.a.) 9–44, which mediates interaction with BRCA1 (Sy et al., 2009b; Zhang et al., 2009a; Zhang et al., 2009b). A nearby sequence present at a.a. 88–94 in human PALB2 is responsible for interaction with KEAP1 (Ma et al., 2012). PALB2 contains a Chromatin Association Motif (ChAM) from a.a. 395–446 and a FXLP mofif from a.a. 612–615, which binds to MRG15, thereby promoting the interaction of PALB2 with chromatin (Bleuyard et al., 2012; Hayakawa et al., 2010; Xie et al., 2012). The C-terminal WD40 domain of PALB2, from a.a. 867–1186, directly binds BRCA2 (Oliver et al., 2009), RAD51 (Buisson et al., 2010), RAD51C and XRCC3 (Park et al., 2014a), POLE (pol η) (Buisson et al., 2014) and RNF168 (Luijsterburg et al., 2017). Within the WD40 domain, there is a hidden nuclear protein export signal from a.a. 928–945 (Pauty et al., 2017).

Key domains in the PALB2 protein and interactions they mediate. Functional domains (or motifs) are shown in black and are identified above the diagram; the amino acids that each domain spans is noted in parentheses. Known interactions which are mediated by the particular domain or motif are shown beneath the diagram.
Expression
According to The Human Protein Atlas (online), PALB2 is ubiquitously expressed to varying degrees across different tissues including the brain, bone marrow, spleen, lung, liver, pancreas, stomach, kidney, testis, ovary and skin.
Localisation
PALB2 localises to nuclei in both chromatin and the nucleoplasm during interphase (Xia et al., 2006). In untreated populations of human cancer cells, the majority of cells display a dispersed non-nucleolar signal while a subset of cells also display DNA damage foci. Treatment with agents that induce DNA damage and/or replication stress increases the assembly of nuclear DNA damage foci. The assembly of PALB2 nuclear foci requires interaction of the protein with BRCA1 (Zhang et al., 2009a; Zhang et al., 2009b). Additionally, the recruitment of PALB2 into foci is also promoted by MDC1, RNF8, UIMC1 (RAP80) and ABRAXAS1 (Abraxas), all of which are involved in the recruitment of BRCA1 (Zhang et al., 2012), and by RNF168 (Luijsterburg et al., 2017). MRG15, PALB2 phosphorylated at S59 and hypophosphorylated at S64, the APRIN cohesion factor and phosphorylated RPA2 also promote the recruitment of PALB2 to sites of DNA damage (Brough et al., 2012; Buisson et al., 2017; Hayakawa et al., 2010; Murphy et al., 2014).
Function
PALB2 acts as a typical cancer suppressor gene. Mono-allelic loss-of-function germline mutations are associated with an increased risk of developing breast cancer (Antoniou et al., 2014; Erkko et al., 2007; Rahman et al., 2007) and pancreatic cancer (Jones et al., 2009). Bi-allelic mutations in PALB2 (FANCN) cause a severe form of Fanconi anemia, subtype FA-N, with early onset of acute myeloid leukemia, medulloblastoma, neuroblastoma and often Wilms’ tumor, leading to early death in the first decade of life (Reid et al., 2007).
PALB2 is believed to act as a tumor suppressor protein by mediating DNA repair and thereby suppressing genome instability (Park et al., 2014b). Importantly, PALB2/FANCN-deficient cells have largely reduced levels of wild-type BRCA2 protein (Xia et al., 2007; Xia et al., 2006), reflective of a role for PALB2 in stabilizing the BRCA2 protein. Therefore, the phenotypes of these cells, as well as the clinical phenotypes of FA-N patients, are very similar to those of cells from patients with a BRCA2/FANCD1 deficiency.
As demonstrated by employing reporter constructs integrated into human cells, PALB2 has an important role in mediating the repair of DNA double-strand breaks (DSBs) by homologous recombination (HR) (Xia et al., 2006). While not specifically tested for PALB2, its partner BRCA2 has an additional role in mediating HR in response to DNA interstrand crosslinks (ICLs); ICLs are specifically repaired by FA proteins (Nakanishi et al., 2011). Consistent with a requirement for PALB2 in DNA repair by HR, and due to largely reduced BRCA2 protein levels, PALB2-deficient cells are hypersensitive to DNA interstrand crosslinking agents such as mitomycin C (MMC) and cisplatin (Xia et al., 2007; Xia et al., 2006), and to ionizing radiation (IR) (Park et al., 2014a). As further support for a role in DNA repair, PALB2-deficient cells are also hypersensitive to poly-ADP ribose polymerase (PARP) inhibitors (Buisson et al., 2010) and to aldehydes (Ghosh et al., 2014).
As a mediator of HR, PALB2 recruits BRCA2 and the RAD51 recombinase to sites of DNA damage (Xia et al., 2006). Additionally, PALB2 stabilizes BRCA2 present in chromatin (Xia et al., 2006). Biochemical experiments demonstrate that PALB2 also directly binds DNA and promotes strand invasion necessary to initiate HR (Buisson et al., 2010; Dray et al., 2010). In this process, PALB2 decreases inhibition of D-loop formation mediated by RPA and enhances HR by stabilizing RAD51 filaments. Also, PALB2 interacts with pol η, thereby promoting DNA synthesis at D-loops (Buisson et al., 2014).
PALB2 has additional roles in other facets of the DNA damage response, beyond its role in mediating HR. Among these, PALB2 promotes maintenance of G2 checkpoint arrest in response to DNA damage (Menzel et al., 2011). PALB2 is also required for chromosome stability. PALB2-deficient cells display increased breaks and radials in response to DNA damage (Bowman-Colin et al., 2013). Further, PALB2 has a role in protecting the cell from replication stress. Carriers of PALB2 mutations display increased firing of dormant replication origins (Nikkila et al., 2013) and mice with a single amino acid knock-in of in Brca2, p.Gly25Arg, which is deficient for binding to PALB2, display decreased fork stability in response to hydroxyurea (Hartford et al., 2016). Homozygosity of these Brca2 knock-in mice, and also hemizygosity in combination with Palb2 and Trp53 heterozygosity, results in defects in body size, fertility, meiosis and genome stability, and also increases tumor susceptibility (Hartford et al., 2016). Further, via its chromodomain, MRG15 targets PALB2 to actively transcribed genes and protects them from DNA damage induced by camptothecin (Bleuyard et al., 2017).
Homology
Based on HomoloGene (NCBI), the following are homologs of the human PALB2 gene (NP_078951.2, 1186 a.a.): Chimpanzee (Pan troglodytes) XP_510877.2, 1186 a.a. Rhesus monkey (Macaca mulatta) XP_001095569.2, 1135 a.a. Dog (Canis lupus familiaris) XP_850671.2, 1195 a.a. Mouse (Mus musculus) NP_001074707.1, 1104 a.a. Rat (Rattus norvegicus) NP_001178532.1, 1110 a.a. Chicken (Gallus gallus) XP_004945321.1, 1341 a.a.
Mutations
Germ-line frameshift, splice site and nonsense mutations that result in truncation of at least part of the C-terminal WD40 domain of PALB2, which binds BRCA2, are linked to Fanconi anemia (Reid et al., 2007; Xia et al., 2007), as well as breast cancer and pancreatic cancer (Erkko et al., 2007; Jones et al., 2009; Rahman et al., 2007; Tischkowitz et al., 2007). Consistent with the role of PALB2 as an interaction partner in a large DNA repair network, these mutations disrupt the binding of PALB2 with BRCA2 and other partners, thereby diminishing DNA repair by HR.
Germ-line, as well as acquired missense mutations, have also been reported in breast cancer patients (Casadei et al., 2011). At the time of writing, 2023 distinct variations were listed in Clinvar (https://www.ncbi.nlm.nih.gov/clinvar/?term=palb2%5Bgene%5D) for PALB2, with 244 frameshift, 110 nonsense, 43 splice site, 14 near gene/UTR, and 966 missense alterations listed. Notably, the clinical significance, as well as the functional significance, of most of these amino acid exchanges is unknown. These are therefore termed variants of uncertain significance (VUS). However, we have demonstrated by functional tests in reconstituted PALB2-deficient FA cells that the p.L939W and p.L1143P variants in the WD40 domain of PALB2 decrease the efficiency of HR and confer partial resistance to IR, when compared to cells reconstituted with wild-type PALB2 (Park et al., 2014a). More recently, it has been demonstrated that the c.104T>C (p.L35P) missense mutant in the N-terminus of PALB2 segregates with malignancies in a family with a strong history of breast cancer (Foo et al., 2017). The p.L35P mutant protein completely abrogates the interaction of PALB2 with BRCA1 and therefore shows no protein activity in HR assays or in assays of cellular resistance to platinum and PARP inhibitors. The findings with p.L35P demonstrate that missense mutations in PALB2 can be pathogenic. Considering the large number of PALB2 VUS, and given the importance of PALB2 functionality for determining prognosis and treatment stratification in patients, significant efforts should be undertaken to systematically determine the functional consequences of such variants on defined cellular functions. Determination of the effects of PALB2 VUS on cellular sensitivity to PARP inhibitors, cisplatin and related drugs, and irradiation is particularly important, since PALB2 promotes cellular resistance to each of these therapeutic agents.
Epigenetics
Hypermethylation of CpG islands in the PALB2 promoter has been observed in a subset of cases of inherited and sporadic breast cancer, and in ovarian cancer (Potapova et al., 2008).
Implicated in
Germ-line bi-allelic and heterozygous loss-of-mutations in PALB2 are associated with different clinical disorders and outcomes. Bi-allelic inactivating mutations of PALB2 result in Fanconi anemia subtype N (FA-N, gene: FANCN), while heterozygous inheritance of a deleterious PALB2 mutation increases the lifetime risk of developing breast and pancreatic cancer. Loss of heterozygosity has not been consistently detected in tumors that develop in carriers of heterozygous PALB2 mutations (Hartley et al., 2014). How much germ-line mutations of PALB2 increase the risk of developing other malignancies, such as ovarian or lung cancers, remains to be determined (Phuah et al., 2013).
Fanconi Anemia (FA)
Just seven months after the identification and characterization of PALB2 as a novel BRCA2 binding protein (Xia et al., 2006), two independent groups identified a total of eight FA patients with biallelic mutations in PALB2: [(Xia et al., 2007): n=1, (Reid et al., 2007): n=7]. These patients exhibited a severe FA phenotype with pronounced congenital abnormalities and a high incidence of malignancies before age seven that was similar to the phenotype described for BRCA2-deficient FANCD1 patients (Alter et al., 2007; Hirsch et al., 2004; Wagner et al., 2004). Notably, these eight children developed 12 distinct malignancies (5X medulloblastoma, 3X Wilms tumors, 2X acute myeloid leukemia, 1X neuroblastoma and 1X hemangioendothelioma) before five years of age. One German patient experienced three different malignancies at 12 months of age, and there were cases of breast and pancreatic cancers present in the families (Reid et al., 2007). PALB2 was the 12th identified FA gene, defining the FA-N complementation group.
At present, 22 FA or FA-like genes have been identified (Nepal et al., 2017). Except for the X-linked FANCB and the autosomal dominant RAD51 (FANCR), FA genes are autosomal recessive tumor suppressor genes. Based on the central activation step in the FA pathway, the monoubiquitination of the FANCD2/FANCI protein dimer, one can distinguish the so-called early (or upstream) FA genes, FANC -A, -B, -C, -E, -F, -G, -L, -M, UBE2T (FANC-T) with no ubiquitination of FANCD2/I when mutated (Mamrak et al., 2017; Nepal et al., 2017). In contrast, late/downstream FA genes are not required for monoubiquitination of FANCD2 and FANCI. These late genes include BRCA2 (FANCD1), BRIP1 (FANCJ), FANCN/PALB2,/RAD51C (FANCO), RAD51 (FANCR), BRCA1 (FANCS), XRCC2 (FANCU), MAD2L2 (FANCV/polTheta) and RFWD3 (FANCW). Most FA patients have bi-allelic mutations in the upstream FA genes, especially FANCA, FANCC and FANCG, and show the characteristic clinical features of FA. These clinical features include progressive bone marrow failure around 7.6 years of age, various congenital anomalies, and a predisposition to acute myeloid leukemia and an assortment of solid tumors that occur in the second and third decade of life (Kutler et al., 2003). Congenital anomalies observed in FA patients can include microcephaly, short stature, skin pigmentation defects, hypogonadism, and radial ray anomalies. A significant number of FA patients also experience endocrine abnormalities (Rose et al., 2012).
In contrast to other FA complementation groups, FA patients that harbour biallelic mutations in PALB2/FANCN or its partner BRCA2/FANCD1 show clinically indistinguishable phenotypes of severe FA characterized by a very early onset and high penetrance of cancers before age seven (Reid et al., 2007). More than 90% of patients succumb to their malignancies before ten years of age. Notably, the spectrum of cancers found in FA patients from the FANCN/PALB2 and FANCD1/BRCA2 complementation groups is different than for other FA complementation groups, including frequent occurrences of medulloblastoma, Wilms tumor, neuroblastoma and hepatoblastomas (Alter et al., 2007; Tischkowitz and Xia, 2010). Bone marrow failure is usually not observed in FA-N patients (Reid et al., 2007).
On a cellular level, FA including the FA-N/PALB2 complementation group is a chromosome instability syndrome. Notably, FA patients are hypersensitive to agents which induce DNA interstrand crosslinks (ICLs). Specifically, cells from FA patients display a characteristic spontaneous and ICL-induced chromosome instability; this phenotype is typically utilized to diagnose FA (Auerbach, 2009). Recently, without functional testing, next generation sequencing based strategies have also been employed to diagnose FA in patients and at the same time identify the defective gene (De Rocco et al., 2014). Additionally, cells from FA patients display accumulation in G2-M of the cell cycle in response to ICLs (Bogliolo and Surralles, 2015). Other cellular functions affected in cells with defects in the FA pathway include sensitivity to aldehydes and oxygen, excessive cytokine production, and defects in the spindle assembly checkpoint, autophagy, cellular reprograming, unwinding of quadruplex and triplex DNA and microsatellite instability (Bogliolo and Surralles, 2015). Correction of the cellular phenotypes by expression of the appropriate FA gene can be utilized to determine the FA complementation group (Chandra et al., 2005; Hanenberg et al., 2002; Virts et al., 2015). For patients with a deficiency for FANCA, stem cell gene therapy might become a new treatment option (Hanenberg et al., 2017).
Breast Cancer
Back-to-back with the identification of PALB2 as the 12th FA gene, Rahman et al. reported the identification of 10 out of 923 individuals (1.1%) from familial breast cancer pedigrees with monoallelic loss-of-function germ-line mutations in PALB2 (Rahman et al., 2007). A lower frequency of PALB2 germ-line mutation (0.5 to 1%) was found in patients with or without a positive family history (Erkko et al., 2007; Foulkes et al., 2007). Further, founder mutations were detected in Finland (Erkko et al., 2007) and Canada (Foulkes et al., 2007).
The largest study to date included 311 women and 51 men from 154 families with loss-of-function germ-line PALB2 mutations, of whom 229 women and 7 men developed breast cancer. Biostatistical analyses revealed that the risk of developing breast cancer for PALB2 mutation carriers was increased by a factor of 9.07 (95% CI, 5.72 to 14.39) when compared to the breast cancer incidence in the general population (Antoniou et al., 2014). The cumulative risk of female heterozygous PALB2 germ-line mutation carriers to develop breast cancer by the age of 70 was as high as 35%. Thus, along with BRCA1 and BRCA2, PALB2 is among the genes that confer the highest breast cancer risk when mutated. In a recent German study of 5589 breast cancer patients without mutations in BRCA1/2, loss-of-function mutations in PALB2 accounted for 1.15% of cases and were also significantly associated with bilateral breast cancer occurrence (Hauke et al., 2018). Only 8 out of 40 patents with PALB2 germ-line mutations belonged to the triple negative breast cancer subtype (Hauke et al., 2018).
Breast cancer causing mutations of PALB2 include established nonsense, frameshift and splice site mutations, which all are thought to compromise the role of PALB2 in cellular responses to DNA damage. However, it is noteworthy that almost 50% of the PALB2 sequence alterations listed in ClinVar are missense variations of unknown clinical and functional significance. Importantly, loss of function of PALB2 is synthetically lethal with radiation, PARP inhibitors, and cisplatin and related compounds. While tumors, which are driven by mutations in PALB2, typically have loss of function of PALB2, normal tissues retain a functional copy of this gene. As such, radiation and/or PARP inhibitors or platinum compounds may be particularly effective against tumor cells with bi-allelic PALB2 mutations.
Interestingly, the relative risk for ovarian cancer was only increased non-significantly to 2.31 for PALB2 mutation carriers (Antoniou et al., 2014). This is surprising, as the PALB2 protein physically interacts with the products of other ovarian cancer susceptibility genes, specifically BRCA1, BRCA2 and RAD51C (Park et al., 2014b).
Pancreatic Cancer
While BRCA2, which encodes a partner of the PALB2 protein, is the most frequently mutated gene in hereditary pancreatic cancer (Shindo et al., 2017; Zhen et al., 2015), mutation of PALB2 is also an important cause of this disease (Jones et al., 2009). To date, truncating loss-of-function germ-line mutations in PALB2 have been associated with the development of pancreatic cancer. In some pedigrees of families with inherited pancreatic cancer, breast and other cancers have also been observed (Blanco et al., 2013; Zhen et al., 2015).
Contributor Information
Helmut Hanenberg, Department of Pediatrics III, University Children’s Hospital Essen, University Duisburg-Essen, Essen Germany;.
Paul R. Andreassen, Division of Experimental Hematology & Cancer Biology, Cancer and Blood Diseases Institute, Cincinnati Children’s Hospital Medical Center, Cincinnati OH, USA;
References
- Bleuyard JY, Fournier M, Nakato R, Couturier AM, Katou Y, Ralf C, Hester SS, Dominguez D, Rhodes D, Humphrey TC, Shirahige K, Esashi F. MRG15-mediated tethering of PALB2 to unperturbed chromatin protects active genes from genotoxic stress. Proc Natl Acad Sci U S A. 2017. July 18;114(29):7671–7676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman-Colin C, Xia B, Bunting S, Klijn C, Drost R, Bouwman P, Fineman L, Chen X, Culhane AC, Cai H, Rodig SJ, Bronson RT, Jonkers J, Nussenzweig A, Kanellopoulou C, Livingston DM. Palb2 synergizes with Trp53 to suppress mammary tumor formation in a model of inherited breast cancer. Proc Natl Acad Sci U S A. 2013. May 21;110(21):8632–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brough R, Bajrami I, Vatcheva R, Natrajan R, Reis-Filho JS, Lord CJ, Ashworth A. APRIN is a cell cycle specific BRCA2-interacting protein required for genome integrity and a predictor of outcome after chemotherapy in breast cancer. EMBO J. 2012. March 7;31(5):1160–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisson R, Dion-Côté AM, Coulombe Y, Launay H, Cai H, Stasiak AZ, Stasiak A, Xia B, Masson JY. Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat Struct Mol Biol. 2010. October;17(10):1247–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisson R, Niraj J, Rodrigue A, Ho CK, Kreuzer J, Foo TK, Hardy EJ, Dellaire G, Haas W, Xia B, Masson JY, Zou L. Coupling of Homologous Recombination and the Checkpoint by ATR. Mol Cell. 2017. January 19;65(2):336–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadei S, Norquist BM, Walsh T, Stray S, Mandell JB, Lee MK, Stamatoyannopoulos JA, King MC. Contribution of inherited mutations in the BRCA2-interacting protein PALB2 to familial breast cancer. Cancer Res. 2011. March 15;71(6):2222–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Levran O, Jurickova I, Maas C, Kapur R, Schindler D, Henry R, Milton K, Batish SD, Cancelas JA, Hanenberg H, Auerbach AD, Williams DA. A rapid method for retrovirus-mediated identification of complementation groups in Fanconi anemia patients. Mol Ther. 2005. November;12(5):976–84 [DOI] [PubMed] [Google Scholar]
- Dray E, Etchin J, Wiese C, Saro D, Williams GJ, Hammel M, Yu X, Galkin VE, Liu D, Tsai MS, Sy SM, Schild D, Egelman E, Chen J, Sung P. Enhancement of RAD51 recombinase activity by the tumor suppressor PALB2. Nat Struct Mol Biol. 2010. October;17(10):1255–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erkko H, Xia B, Nikkilä J, Schleutker J, Syrjäkoski K, Mannermaa A, Kallioniemi A, Pylkäs K, Karppinen SM, Rapakko K, Miron A, Sheng Q, Li G, Mattila H, Bell DW, Haber DA, Grip M, Reiman M, Jukkola-Vuorinen A, Mustonen A, Kere J, Aaltonen LA, Kosma VM, Kataja V, Soini Y, Drapkin RI, Livingston DM, Winqvist R. A recurrent mutation in PALB2 in Finnish cancer families. Nature. 2007. March 15;446(7133):316–9 [DOI] [PubMed] [Google Scholar]
- Foo TK, Tischkowitz M, Simhadri S, Boshari T, Zayed N, Burke KA, Berman SH, Blecua P, Riaz N, Huo Y, Ding YC, Neuhausen SL, Weigelt B, Reis-Filho JS, Foulkes WD, Xia B. Compromised BRCA1-PALB2 interaction is associated with breast cancer risk. Oncogene. 2017. July 20;36(29):4161–4170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Sur S, Yerram SR, Rago C, Bhunia AK, Hossain MZ, Paun BC, Ren YR, Iacobuzio-Donahue CA, Azad NA, Kern SE. Hypersensitivities for acetaldehyde and other agents among cancer cells null for clinically relevant Fanconi anemia genes. Am J Pathol. 2014. January;184(1):260–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartford SA, Chittela R, Ding X, Vyas A, Martin B, Burkett S, Haines DC, Southon E, Tessarollo L, Sharan SK. Interaction with PALB2 Is Essential for Maintenance of Genomic Integrity by BRCA2. PLoS Genet. 2016. August;12(8):e1006236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley T, Cavallone L, Sabbaghian N, Silva-Smith R, Hamel N, Aleynikova O, Smith E, Hastings V, Pinto P, Tischkowitz M, Tomiak E, Foulkes WD. Mutation analysis of PALB2 in BRCA1 and BRCA2-negative breast and/or ovarian cancer families from Eastern Ontario, Canada. Hered Cancer Clin Pract. 2014;12(1):19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa T, Zhang F, Hayakawa N, Ohtani Y, Shinmyozu K, Nakayama J, Andreassen PR. MRG15 binds directly to PALB2 and stimulates homology-directed repair of chromosomal breaks. J Cell Sci. 2010. April 1;123(Pt 7):1124–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S, Hruban RH, Kamiyama M, Borges M, Zhang X, Parsons DW, Lin JC, Palmisano E, Brune K, Jaffee EM, Iacobuzio-Donahue CA, Maitra A, Parmigiani G, Kern SE, Velculescu VE, Kinzler KW, Vogelstein B, Eshleman JR, Goggins M, Klein AP. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science. 2009. April 10;324(5924):217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luijsterburg MS, Typas D, Caron MC, Wiegant WW, van den Heuvel D, Boonen RA, Couturier AM, Mullenders LH, Masson JY, van Attikum H. A PALB2-interacting domain in RNF168 couples homologous recombination to DNA break-induced chromatin ubiquitylation. Elife. 2017. February 27;6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Cai H, Wu T, Sobhian B, Huo Y, Alcivar A, Mehta M, Cheung KL, Ganesan S, Kong AN, Zhang DD, Xia B. PALB2 interacts with KEAP1 to promote NRF2 nuclear accumulation and function. Mol Cell Biol. 2012. April;32(8):1506–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzel T, Nähse-Kumpf V, Kousholt AN, Klein DK, Lund-Andersen C, Lees M, Johansen JV, Syljuåsen RG, Sørensen CS. A genetic screen identifies BRCA2 and PALB2 as key regulators of G2 checkpoint maintenance. EMBO Rep. 2011. July 1;12(7):705–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy AK, Fitzgerald M, Ro T, Kim JH, Rabinowitsch AI, Chowdhury D, Schildkraut CL, Borowiec JA. Phosphorylated RPA recruits PALB2 to stalled DNA replication forks to facilitate fork recovery. J Cell Biol. 2014. August 18;206(4):493–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakanishi K, Cavallo F, Perrouault L, Giovannangeli C, Moynahan ME, Barchi M, Brunet E, Jasin M. Homology-directed Fanconi anemia pathway cross-link repair is dependent on DNA replication. Nat Struct Mol Biol. 2011. April;18(4):500–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nepal M, Che R, Zhang J, Ma C, Fei P. Fanconi Anemia Signaling and Cancer. Trends Cancer. 2017. December;3(12):840–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikkilä J, Parplys AC, Pylkäs K, Bose M, Huo Y, Borgmann K, Rapakko K, Nieminen P, Xia B, Pospiech H, Winqvist R. Heterozygous mutations in PALB2 cause DNA replication and damage response defects. Nat Commun. 2013;4:2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver AW, Swift S, Lord CJ, Ashworth A, Pearl LH. Structural basis for recruitment of BRCA2 by PALB2. EMBO Rep. 2009. September;10(9):990–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orthwein A, Noordermeer SM, Wilson MD, Landry S, Enchev RI, Sherker A, Munro M, Pinder J, Salsman J, Dellaire G, Xia B, Peter M, Durocher D. A mechanism for the suppression of homologous recombination in G1 cells. Nature. 2015. December 17;528(7582):422–6 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Park JY, Singh TR, Nassar N, Zhang F, Freund M, Hanenberg H, Meetei AR, Andreassen PR. Breast cancer-associated missense mutants of the PALB2 WD40 domain, which directly binds RAD51C, RAD51 and BRCA2, disrupt DNA repair. Oncogene. 2014. October 2;33(40):4803–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JY, Zhang F, Andreassen PR. PALB2: the hub of a network of tumor suppressors involved in DNA damage responses. Biochim Biophys Acta. 2014. August;1846(1):263–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauty J, Couturier AM, Rodrigue A, Caron MC, Coulombe Y, Dellaire G, Masson JY. Cancer-causing mutations in the tumor suppressor PALB2 reveal a novel cancer mechanism using a hidden nuclear export signal in the WD40 repeat motif. Nucleic Acids Res. 2017. March 17;45(5):2644–2657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phuah SY, Lee SY, Kang P, Kang IN, Yoon SY, Thong MK, Hartman M, Sng JH, Yip CH, Taib NA, Teo SH. Prevalence of PALB2 mutations in breast cancer patients in multiethnic Asian population in Malaysia and Singapore. PLoS One. 2013;8(8):e73638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potapova A, Hoffman AM, Godwin AK, Al-Saleem T, Cairns P. Promoter hypermethylation of the PALB2 susceptibility gene in inherited and sporadic breast and ovarian cancer. Cancer Res. 2008. February 15;68(4):998–1002 [DOI] [PubMed] [Google Scholar]
- Rahman N, Seal S, Thompson D, Kelly P, Renwick A, Elliott A, Reid S, Spanova K, Barfoot R, Chagtai T, Jayatilake H, McGuffog L, Hanks S, Evans DG, Eccles D, Easton DF, Stratton MR. PALB2, which encodes a BRCA2-interacting protein, is a breast cancer susceptibility gene. Nat Genet. 2007. February;39(2):165–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rantakari P, Nikkilä J, Jokela H, Ola R, Pylkäs K, Lagerbohm H, Sainio K, Poutanen M, Winqvist R. Inactivation of Palb2 gene leads to mesoderm differentiation defect and early embryonic lethality in mice. Hum Mol Genet. 2010. August 1;19(15):3021–9 [DOI] [PubMed] [Google Scholar]
- Reid S, Schindler D, Hanenberg H, Barker K, Hanks S, Kalb R, Neveling K, Kelly P, Seal S, Freund M, Wurm M, Batish SD, Lach FP, Yetgin S, Neitzel H, Ariffin H, Tischkowitz M, Mathew CG, Auerbach AD, Rahman N. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet. 2007. February;39(2):162–4 [DOI] [PubMed] [Google Scholar]
- Sy SM, Huen MS, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair Proc Natl Acad Sci U S A 2009. April 28;106(17):7155–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischkowitz M, Xia B. PALB2/FANCN: recombining cancer and Fanconi anemia Cancer Res 2010. October 1;70(19):7353–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virts EL, Jankowska A, Mackay C, Glaas MF, Wiek C, Kelich SL, Lottmann N, Kennedy FM, Marchal C, Lehnert E, Scharf RE, Dufour C, Lanciotti M, Farruggia P, Santoro A, Savasan S, Scheckenbach K, Schipper J, Wagenmann M, Lewis T, Leffak M, Farlow JL, Foroud TM, Honisch E, Niederacher D, Chakraborty SC, Vance GH, Pruss D, Timms KM, Lanchbury JS, Alpi AF, Hanenberg H. AluY-mediated germline deletion, duplication and somatic stem cell reversion in UBE2T defines a new subtype of Fanconi anemia Hum Mol Genet 2015. September 15;24(18):5093–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner JE, Tolar J, Levran O, Scholl T, Deffenbaugh A, Satagopan J, Ben-Porat L, Mah K, Batish SD, Kutler DI, MacMillan ML, Hanenberg H, Auerbach AD. Germline mutations in BRCA2: shared genetic susceptibility to breast cancer, early onset leukemia, and Fanconi anemia Blood 2004. April 15;103(8):3226–9 [DOI] [PubMed] [Google Scholar]
- Xia B, Dorsman JC, Ameziane N, de Vries Y, Rooimans MA, Sheng Q, Pals G, Errami A, Gluckman E, Llera J, Wang W, Livingston DM, Joenje H, de Winter JP. Fanconi anemia is associated with a defect in the BRCA2 partner PALB2 Nat Genet 2007. February;39(2):159–61 [DOI] [PubMed] [Google Scholar]
- Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, Liu X, Jasin M, Couch FJ, Livingston DM. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2 Mol Cell 2006. June 23;22(6):719–29 [DOI] [PubMed] [Google Scholar]
- Xie T, Graveline R, Kumar GS, Zhang Y, Krishnan A, David G, Radhakrishnan I. Structural basis for molecular interactions involving MRG domains: implications in chromatin biology Structure 2012. January 11;20(1):151–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Bick G, Park JY, Andreassen PR. MDC1 and RNF8 function in a pathway that directs BRCA1-dependent localization of PALB2 required for homologous recombination J Cell Sci 2012. December 15;125(Pt 24):6049–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Fan Q, Ren K, Andreassen PR. PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2 Mol Cancer Res 2009. July;7(7):1110–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Ma J, Wu J, Ye L, Cai H, Xia B, Yu X. PALB2 links BRCA1 and BRCA2 in the DNA-damage response Curr Biol 2009. March 24;19(6):524–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen DB, Rabe KG, Gallinger S, Syngal S, Schwartz AG, Goggins MG, Hruban RH, Cote ML, McWilliams RR, Roberts NJ, Cannon-Albright LA, Li D, Moyes K, Wenstrup RJ, Hartman AR, Seminara D, Klein AP, Petersen GM. BRCA1, BRCA2, PALB2, and CDKN2A mutations in familial pancreatic cancer: a PACGENE study Genet Med 2015. July;17(7):569–77 [DOI] [PMC free article] [PubMed] [Google Scholar]