

Abstract
The canonical model of RB-mediated tumour suppression developed over the past 30 years is based on the regulation of E2F transcription factors to restrict cell cycle progression. Several additional functions have been proposed for RB, on the basis of which a non-canonical RB pathway can be described. Mechanistically, the non-canonical RB pathway promotes histone modification and regulates chromosome structure in a manner distinct from cell cycle regulation. These functions have implications for chemotherapy response and resistance to targeted anticancer agents. This Opinion offers a framework to guide future studies of RB in basic and clinical research.
The mechanism by which RB interacts with E2F family members E2F1–E2F5 to regulate cell cycle entry has been studied in great detail1–3. In its simplest form, RB represses transcription of E2F family member target genes in the G1 phase of the cell cycle1–3. Transcriptional repression in this paradigm occurs through RB binding to the transactivation domain of these E2Fs. Heterodimerization of the respective E2F family member with a dimeric partner (DP) subunit localizes this complex to E2F target gene promoters that contain the consensus DNA binding element TTTCGCGC1–3 (FIG. 1a). Growth factor signals stimulate the assembly and activation of cyclin D and its associated cyclin-dependent kinases (CDK4 or CDK6) that initially phosphorylate RB in the G1 phase of the cell cycle1–3. Cyclin E-CDK2 complexes phosphorylate RB in late G1, which releases the respective E2F family member from RB, leading to the activation of transcriptional targets to advance the cell cycle1–3 (FIG. 1a). Frequent genome alterations affecting the function of RB, cyclins, CDKs and CDK inhibitors have underscored the central importance of this regulatory mechanism in cancer, a circuit that has become known as the RB pathway4,5 (BOX 1). The abundance of mutations affecting members of this pathway, and the resulting lack of proliferative control, has led to the idea that RB pathway disruption is a hallmark of cancer6. In this article, we refer to RB-E2F transcriptional control that is regulated by cyclin-CDK complexes as the canonical function of RB (FIG. 1a).
Fig. 1 |. Canonical RB-E2F regulation and mechanisms of CDK resistance.

a | An illustration of our definition of canonical RB function is presented. In this model, growth factors signal the expression and activation of D-type cyclins. Cyclin D-cyclin-dependent kinase 4 (CDK4) or CDK6 and cyclin E-CDK2 act to hyperphosphorylate RB. Members of the E2F family of transcription factors and their dimeric partners (DPs) are released to activate the expression of genes that advance the cell cycle. This signalling pathway can be blocked by CDK inhibitor 2A (CDKN2A)-mediated inhibition of cyclin D, which is associated with CDK activity, b | RB can escape CDK hyperphosphorylation when its RxL motif is bound by protein phosphatase 1 (PP1) or when the RxL motif is acetylated or methylated, collectively depicted as RxL motif inhibition. This prevents substrate recognition and ensures retention of RB function. c | RB and transcription factor E2F1 can interact through molecular contacts that are distinct from RB and other E2F family members. In this interaction, CDK hyperphosphorylation of RB is unable to disrupt E2F1 binding. Similarly, ultraviolet light or osmotic shock can activate p38 MAPK to phosphorylate RB. Once phosphorylated, this stimulates E2F1 binding, even when RB is hyperphosphorylated by CDKs. d | RB-E2F complexes can recruit chromatin-modifying enzymes such as enhancer of zeste homologue 2 (EZH2) to mediate patterns of methylation of histone H3 lysine 27 (H3K27me3) at promoters. RB is removed from these locations by CDK hyperphosphorylation at the onset of S phase, but histone tail modification patterns are preserved during DNA replication, enabling stable transmission of the epigenetic state to the next cell generation. In the ensuing G1 phase, RB is dephosphorylated by PP1A and RB-E2F recruitment of histone methyltransferase activity reinforces patterns of modification in the ensuing cell cycle.
Box 1 |. RB pathway and genetic alterations.
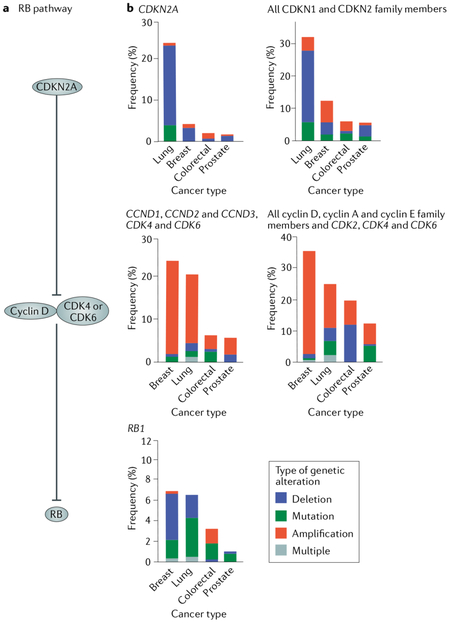
The RB pathway in cancer refers to a signalling pathway in which RB is the target of regulation by phosphorylation by cyclin D and cyclin-dependent kinase 4 (CDK4) or CDK6, which in turn is subject to negative regulation by the CDK inhibitor 2A (CDKN2A) and its family members4,120 (see the figure, part a). An extended view of this pathway includes the cyclin E or cyclin A-CDK2 axis and its inhibitors (p21 and its family). In all cases, alterations to RB regulators lead to its hyperphosphorylation and cell cycle progression. The basic concept of this pathway is that cell cycle control is disrupted through loss-of-function mutations in either RB1 or CDKN2A. Alternatively, gain-of-function mutations in genes encoding D-type cyclins (CCND1, CCND2 and CCND3) or CDK4 or CDK6 constitutively phosphorylate and inactivate RB. Tabulation of genetic alterations to this pathway have indicated that RB pathway disruption is widespread in cancer6. Cancer genomic studies indicate that near-ubiquitous RB pathway mutations can be found in some cancer types. The Cancer Genome Atlas data illustrating RB pathway mutations for lung adenocarcinoma88, prostate carcinoma98, colorectal adenocarcinoma121 and invasive breast carcinomas122 are shown (see the figure, part b). The left-most graphs depict mutation frequency in these cancer types for the best-known RB pathway components, whereas the right-most graphs show an expanded view of RB regulation in which genetic alterations of all CDK inhibitors and G1 CDKs are included. These data illustrate that the majority of lung and breast cancers possess recognizable loss-of-function changes in RB1 and CDKN2A or gain-of-function alterations in genes encoding D-type cyclins and associated CDKs88. Alternatively, colorectal and prostate carcinomas show relatively few RB pathway alterations. This finding illustrates the challenges in accounting for loss of proliferative control in some cancer types using RB pathway mutations.
Biochemical experiments and clinical studies alike have generated data that are not easily reconciled with the canonical RB pathway. For example, chromatin immunoprecipitation and sequencing (ChIP-seq) studies have revealed that RB occupies not only E2F-regulated gene promoters but also transcriptional enhancers and repetitive DNA sequences7–9. Several studies have also detected RB complexes on chromatin in S phase, even though hyperphosphorylation of RB by CDKs has been previously shown to decrease binding of RB to E2F target gene promoters10–13. In addition, multiple clinical studies have indicated that loss of RB protein expression, as opposed to alterations of other RB pathway components, is uniquely correlated with improved response to chemotherapeutics3,14–20. Further, different cancer types often show mutations of unique RB pathway components, suggesting that the effects of mutations in different RB pathway components are not functionally equivalent3. In some cancers, RB1 gene loss has also been observed to occur later in disease progression21–24 or is found more prevalently in metastasis25–27, when deregulated cell proliferation has already been established. Although RB-like protein 1 (RBL1, also known as p107) and RBL2 (also known as p130) also regulate E2F and the cell cycle, mutations inactivating the encoding genes are less common in tumours than mutations inactivating RB1 (REFS23,24,28). Similarly, engineered mutations in the murine Rbl1 and Rbl2 genes give weaker tumour phenotypes than mutations in Rbl (REF2). Each of these observations has several potential explanations, which are not fully explored in this article. Overall, however, they suggest that RB has functions that extend beyond the control of classic E2F targets or the regulation of cell proliferation. Importantly, these functions may be clinically relevant for patients with cancer.
Mechanistic studies have also revealed that RB has a multitude of molecular functions. These functions involve the regulation of inflammation29, mitochondria and metabolism30,31, autophagy32, apoptosis33 and stemness34,35. Importantly, these functions have been linked to CDK-dependent E2F target gene transcription and cell cycle regulation31,36,37 and thus are related to the canonical RB pathway. Still, one simple fact about RB is obscured by the complexity of available studies, which is that RB is primarily located in the nucleus and associated with chromatin38. The pool of RB that controls cell cycle progression through targeting of E2F-regulated promoters (the canonical RB function) (FIG. 1a) is undoubtedly one important facet of RB action. In this article, we propose that there is a second pool of chromatin-associated RB that performs non-canonical functions. This non-canonical activity of RB impacts the organization of heterochromatin, promotes chromosome integrity and may be independent of CDK-mediated RB phosphorylation. We propose that this second pool of RB may help to explain many of the features of RB activity that do not fit with canonical RB pathway regulation. Importantly, this non-canonical activity of RB may explain recent data on the effects of RB loss and their impact on therapeutic response and acquired resistance14,16–27. We appreciate that the molecular details of the non-canonical functions of RB are still poorly understood and that, therefore, categorizing RB-dependent phenotypes as the result of canonical or non-canonical RB activities is an imperfect approach. Nevertheless, we hope that this interpretation of the RB literature will help to stimulate the appreciation and investigation into a largely unexplored area of cancer biology and complement our current knowledge of the role of RB in cell cycle control.
RB function in proliferating cells
The current understanding of RB has long been shaped by the concept that phosphorylation on multiple sites by cyclin D-associated and cyclin E-associated CDKs, so-called hyperphosphorylation, leads to dissociation of RB from chromatin at the beginning of S phase and supposedly renders it functionless for the remainder of the cell cycle28. Results showing that RB loss affects the biology of proliferating cells suggest that this model is incomplete and that RB retains activity even in cells that have high CDK activity and in which RB is phosphorylated. There are several mechanisms that may explain how RB retains activity in these conditions, as described below.
RB can be protected from phosphorylation-dependent effects.
Most cyclin-CDK complexes require a basic amino acid patch near the carboxyl terminus of RB to dock and recognize it as a substrate39. A number of studies have described acetylation or methylation of lysine residues in this patch that may inhibit CDK recognition and RB phosphorylation40–42. In addition, serine/threonine-protein phosphatase 2 (PP2) has been observed to protect RB from phosphorylation-dependent dissociation from chromatin in S phase10. Protein phosphatase 1 (PP1) has also been shown to recognize a peptide motif that overlaps with the cyclin docking site in the carboxyl terminus of RB43. This not only facilitates dephosphorylation but also sterically inhibits cyclin-CDK recognition and therefore phosphorylation of RB (FIG. 1b). Collectively, these studies illustrate that mechanisms exist to protect RB from the inactivating effects of CDKs, even when CDK activity is high at the end of G1 and during S phase.
Hyperphosphorylated RB can bind E2F1.
It has also been demonstrated that RB retains some function when hyperphosphorylated by CDKs. RB interacts with transcription factor E2F1 through molecular contacts, in particular, via the marked box domain of E2F1 (REFS13,44,45). This interaction is resistant to disruption by CDK phosphorylation, allowing some sites on RB to be phosphorylated without disrupting this interaction13,46. This interaction between RB and E2F1 results in histone H3 lysine 27 trimethylation (H3K27me3) at repetitive genomic sequences7. By contrast, the interaction of RB with other E2F family members is disrupted by CDK-mediated phosphorylation, thereby promoting E2F gene transcription28. Crystal structures of RBL1 interacting with the marked box domain of E2F5 reveal that the respective interaction site contains a CDK phosphorylation site, whereas the RB-E2F1 marked box interaction site does not contain a CDK phosphorylation site47, suggesting a reason why this complex is not disrupted by CDK activity. Also, the RB-E2F1 complex has low binding affinity for a consensus E2F promoter DNA element compared with the high binding affinity of E2F1 in uncomplexed form48,49, suggesting a reason why RB-E2F1 complexes in S phase do not bind to and regulate E2F target genes. These data argue for a qualitatively unique interaction between RB and E2F1 that is not shared with other E2F family proteins. Importantly, a mutant mouse line bearing an F832A substitution in its RB protein is unable to form this unique RB-E2F1 interaction7. Cells from these mice possess normal regulation of E2F target genes, enter into S phase normally but misexpress repetitive sequences and are cancer prone7. This result highlights the surprising impact of the CDK-resistant interaction of RB with E2F1.
It has also recently been demonstrated that phosphorylation of RB by p38 MAPK on a site distinct from the E2F1 interaction site can actually stimulate E2F1 binding to RB at yet another location in the RB polypeptide and lead to complex formation at promoter regions of E2F target genes50. This p38 MAPK phosphorylation event bypasses CDK control of RB and thus offers another mechanistic explanation for RB function that occurs in the presence of high CDK activity and hyperphosphorylation (FIG. 1c).
Overall, protection from CDK phosphorylation and retention of function after CDK phosphorylation are emerging concepts in RB research. It has also been demonstrated that hyperphosphorylated RB takes on new functions in regulating AKT-dependent signal transduction that monophosphorylated or unphosphorylated RB does not have51; although this study focused on RB interactions that occur in the cytoplasm, it is possible that additional interactions of nuclear RB are also stimulated by phosphorylation.
Chromatin replication preserves RB-dependent epigenetic marks.
An important but indirect mechanism that explains retention of RB function in S phase is inherent to chromatin replication mechanisms. Even though RB-E2F complexes are extensively disassembled at S phase entry, it is known that RB recruits epigenetic proteins such as the histone methyltransferase enhancer of zeste homologue 2 (EZH2) to direct H3K27me3 deposition at transcriptional enhancers and promoters of genes encoding the stem cell factors octamer-binding protein 4 (OCT4; also known as POU5F1) and SOX2 using a CDK-sensitive interaction with E2Fs7,8,52. This heterochromatin pattern is then preserved in S phase during DNA replication53,54. In turn, H3K27me3 locations are reinforced in daughter cells through another round of EZH2 recruitment by RB in the ensuing G1 (FIG. 1d). In this way, mechanisms that perpetuate epigenetic memory during DNA replication may sustain RB-dependent features without requiring the continuous physical presence of RB protein.
In sum, these studies indicate that RB can be protected from high levels of CDK activity (thereby maintaining a pool of hypophosphorylated RB), that hyperphosphorylated RB can retain function and that RB-dependent histone modification patterns are preserved throughout the cell cycle. These characteristics allow RB to exert activities in proliferating cells, for example, to shape cell-lineage commitment55. The ability of RB to function in a manner that is resistant to G1-S cell cycle regulation is fundamental to the epigenetic and chromosome structure functions that we discuss in this article.
RB maintains genome stability
RB occupies diverse genome locations.
E2F binding sites are often located in proximal promoters, and the canonical function of RB involves cell cycle-dependent chromatin changes in this local environment as part of the transcriptional control mechanism56. RB is able to localize to the genome in a sequence-independent manner, and this has come into focus with the discovery that non-homologous end joining57 (NHEJ) and homologous recombination58 (HR) repair mechanisms require RB for maximal function (FIG. 2a). These studies established that RB is physically recruited to the sites of DNA breaks during the repair process57,58. In a complementary manner, studies using ChIP-seq demonstrated that RB is targeted to many different genomic locations such as gene bodies during transcription9, transcriptional enhancers8 and broad categories of repetitive DNA elements including long interspersed nuclear elements (LINEs) and satellite repeats7,59,60.
Fig. 2 |.

2F family members and extensively throughout the genome at repetitive sequences and replication origins with transcription factor E2F1 (and likely other recruiting factors (RFs)) in a sequence-independent manner. b | The distribution of RB throughout the genome using RFs that are not sequence-specific leaves it poised to participate at sites of DNA breaks for non-homologous end joining (NHEJ) and homologous recombination (HR) repair in complex with X-ray repair cross-complementing protein 5 (XRCC5), XRCC6 or transcription activator BRG1. RB also recruits cohesin and condensin II complexes to replicating DNA and mitotic chromosomes for condensation and segregation. Lastly, RB directs histone deacetylation through the nucleosome-remodelling and deacetylase (NuRD) complex and histone H3 lysine 27 trimethylation (H3K27me3) using enhancer of zeste homologue 2 (EZH2) at repeats and enhancers. Ac, acetyl.
E2F1 participates in the genome-wide roles of RB.
Although the targeting of RB to repetitive sequences and sites of DNA damage does not seem to require the consensus E2F element (TTTCGCGC), mechanistic studies show that RB often associates with these genome locations in a manner that depends on E2F1 (REFS7,58). Indeed, E2F1 is also recruited to sites of double-strand breaks58,61 and is essential for NHEJ61, HR58, fidelity of DNA replication, chromosome condensation59 and RB-dependent H3K27me3 deposition at repetitive sequences even without the presence of a consensus DNA binding site7. A mutation in RB that cripples its marked box domain interaction with E2F1 has been shown to disrupt binding of both RB and E2F1 to diverse categories of repetitive sequences7,59. The cooperative relationship between RB and E2F1 in localizing to repetitive sequences59 or to DNA breaks58, in which neither can localize without the other, might be mechanistically enabled by formation of the CDK phosphorylation-resistant RB-E2F1 complex59 (FIG. 1c). RB acetylation and methylation are induced by DNA damage40–42,62, and these modifications reduce CDK phosphorylation of RB42, further indicating that RB-E2F1 complexes may be sheltered from CDK activity when engaged in NHEJ and HR functions. Hyperphosphorylated RB also interacts with E2F1 in response to DNA breaks12,63. These observations further implicate RB-E2F1 interactions that are insensitive to CDK activity (FIG. 1). Although the E2F1 protein is capable of stimulating proliferation through activation of transcription of E2F target genes in a manner similar to other E2Fs, the effect of E2F1 loss is unique among E2Fs, as spontaneous DNA damage arises in response to E2F1 loss64, and E2f1-knockout mice are susceptible to cancer65; these effects are not observed when other E2F family members are inactivated66. For this reason, we consider these RB-E2F1-dependent epigenetic and genome stability roles as CDK-independent and as part of a non-canonical category of functions.
RB functions directly in DNA repair and chromatin regulation.
Once recruited to genomic repeats or DNA breaks, RB recruits regulators of chromatin structure that facilitate remodelling and higher-order packaging. At double-stranded DNA breaks, RB interacts with DNA end-binding proteins X-ray repair cross-complementing protein 5 (XRCC5) and XRCC6 to facilitate repair of DNA breaks through end joining57. In HR repair, RB recruits the DNA helicase transcription activator BRG1 to relax chromatin structure and promote end resection as part of the repair process58. Striking similarities have also been reported for mutants in the Arabidopsis spp. gene RBR1 (retinoblastoma-related 1), where RBR1 is required for recruitment of HR factors to sites of DNA breaks67,68. RB facilitates heterochromatin formation through recruitment of histone deacetylase complexes such as the nucleosome-remodelling and deacetylase (NuRD) complex and histone methyltransferases such as EZH2 to deacetylate and trimethylate H3K27 at repetitive sequences such as LINEs and satellites, leading to the silencing of their expression7,69.
RB-dependent deposition of H4K20me3 at centromeric and telomeric regions contributes to heterochromatin formation, recruitment of the structural maintenance of chromosome complex known as cohesin and genome stability at these regions70–72. On a whole-chromosome scale, RB can serve to recruit protein complexes involved in the structural maintenance of chromatin: condensin II and cohesin70,73. This is critical to the compaction and segregation of mitotic chromosomes in untransformed cells70,74. It has also been shown that failure to recruit these factors negatively affects DNA replication patterns earlier in the cell cycle during S phase and that insufficiently condensed chromosomes and mitotic errors may in part be due to DNA lesions that originate in defective replication59,70,75,76.
These studies show that, in addition to regulating proximal E2F-responsive cell cycle gene promoters, RB is distributed throughout the genome in a sequence-independent manner, often in complex with E2F1 (FIG. 2a). This leaves the question of how localization to DNA breaks or repeats is coordinated. A recent report of RB binding to histone H1 may offer some clues for future research77, but our understanding of this is very much just beginning. From these locations, RB is poised to participate in many fundamental processes of genome maintenance including DNA-break repair, DNA replication, chromosome condensation and heterochromatin formation (FIG. 2b). The diverse functions and molecular partners that RB uses suggest that additional studies are needed to understand how these partners are selected and the many functions are coordinated.
Importantly, chromosome instability phenotypes have been observed in RB-mutant embryonic stem cells, where there is no functional G1-S regulation by RB78–80. Because these repair and epigenetic functions adopt CDK-resistance mechanisms and involve somewhat direct RB participation (FIG. 1), we infer that the role of RB in these processes fits best with the non-canonical function of RB. However, it is important to consider that the well known RB-E2F cell cycle paradigm contributes to the expression of repair factors and spindle assembly checkpoint components81. We speculate that upon reduced activity of both canonical and non-canonical RB pathways, cells experience DNA damage and reorganization of heterochromatin distribution patterns on a genome-wide scale. We suggest that this state of genetic instability is key to the effects of RB loss on drug sensitivity and explains the cell-lineage alterations described in the ensuing sections.
RB loss sensitizes to chemotherapy
Defective RB function can influence cellular responses to a variety of cancer treatment approaches3. Among these, it is noteworthy that agents that serve to block proliferation, such as hormone receptor antagonists, depend on RB for action15,82,83, while RB-deficient cells are more sensitive to DNA-damaging agents84–87. We suggest that loss of both canonical cell cycle control and non-canonical regulation of chromatin structure make cells lacking RB uniquely sensitive to genotoxic agents3.
RB loss improves chemotherapy and radiation response in lung cancer.
Analysis of lung adenocarcinomas, the most common form of non-small-cell lung cancer (NSCLC), offers insight into non-canonical RB roles in treatment response. Deregulated proliferation is likely influenced by mutations or copy number changes in genes encoding CDK inhibitor 2A (CDKN2A, also known as p16-INK4A), cyclin D, CDK4, CDK6 and RB that are commonplace in this cancer88 (BOX 1), and rapid proliferation creates sensitivity to DNA damage89. Loss of RB, but not other canonical pathway components, however, correlates with improved response to DNA damage agents85 and longer patient survival14,18,19. This indicates that RB loss is different from CDKN2A or cyclin-CDK alterations. Importantly, the proliferation indices of RB-positive and RB-negative, treatment naive lung adenocarcinomas are similar14, suggesting that differences in response cannot be explained by proliferation alone. A remarkably similar clinical scenario in which RB loss is not correlated with elevated levels of proliferation but is associated with unusually long survival following chemotherapy has been reported in serous ovarian cancer20. Another way to consider the role of RB in chemotherapy is that DNA damage agents and radiation induce an RB-dependent G1 cell cycle arrest in RB-positive but not RB-negative cancers3, thus explaining their sensitivity5,85. However, p53 is upstream of RB in the same DNA damage response cell cycle arrest checkpoint5, suggesting that failure of the p53-dependent DNA damage response should similarly increase treatment sensitivity to levels similar to those observed with RB loss. Because p53 status does not predict chemotherapy responsiveness in NSCLC85, it seems unlikely that the inability to arrest the cell cycle in response to DNA damage predicts a strong response to treatment85. Therefore, an alternative possibility for explaining the observed correlations of RB status and responsiveness to chemotherapy and DNA-damaging agents draws on the idea that RB has activities that extend beyond its role in the regulation of cell cycle progression. In particular, RB loss would compromise genome stability more directly through the roles of RB in promoting DNA repair and chromosome stability. Thereby, RB loss would create an inherent sensitivity to DNA damage-dependent therapies.
For several years the relationship between RB loss and DNA damage sensitivity has suggested a vulnerability that could be exploited if RB status was used as a guide in treatment choice3. These treatment response data have been difficult to explain when one views RB solely as a regulator of cell proliferation. By contrast, the non-canonical functions of RB provide an explanation that is simpler and more direct. When viewed from the perspective that RB is distributed throughout the genome and has roles that promote genome stability, the link between RB status and sensitivity to DNA-damaging agents seems logical. The idea that RB deficiency can be used to help predict sensitivity to DNA damage-based cancer treatment is therefore easily aligned to this mechanism of action.
RB deficiency in targeted therapy
Molecularly targeted cancer therapies promise to improve patient outcomes with reduced side effects by exploiting dependencies or vulnerabilities unique to cancer cells, and RB loss has been shown to confer resistance to targeted therapies. This is best illustrated by the reliance of CDK4 or CDK6 inhibitors such as palbociclib on RB function90. Blockade of D-type cyclin-CDK4 or CDK6 kinase activity is known to require RB for growth arrest91–93, and these agents were designed to re-activate the canonical RB pathway. However, the transdifferentiation phenotype of cancers relapsing from a range of molecularly targeted therapies suggests that RB loss is associated with acquired therapeutic resistance through means beyond altered cell cycle control. Understanding how RB loss contributes to resistance will be critical for realizing the potential of these targeted agents.
Resistance to anti-androgens in prostate adenocarcinoma.
Prostate cancers are uniquely dependent on the androgen receptor (AR) signalling axis for growth and survival. A common mechanism of acquired anti-androgen resistance is genetic alteration of AR itself, leading to persistent and sometimes ligand-independent AR signalling94. Newer anti-androgens like enzalutamide and abiraterone have been developed to overcome acquired resistance by improving AR signalling blockade95. However, unique forms of acquired resistance, characterized by transformation of the original AR-expressing adenocarcinoma into histological variants lacking AR, are increasingly observed in patients relapsing from these new anti-androgens96. One type of the AR-deficient variant undergoes histological transdifferentiation from the luminal epithelial phenotype of the original adenocarcinoma to a neuroendocrine phenotype97 (FIG. 3a). These treatment-acquired neuroendocrine prostate cancer variants share clonal origin with the pre-existing adenocarcinoma, as they contain the same mutations27.
Fig. 3 |. Proposed model for RB1 loss in transdifferentiation and drug resistance.

a | RB1 gene mutation is a frequent event in acquired resistance to targeted therapies in which the initial adenocarcinoma transdifferentiates into a neuroendocrine cancer. b | Initially, the adenocarcinoma cells express RB, which binds to DNA regions via recruitment factors (RFs) such as transcription factor E2F1, and use enhancer of zeste homologue 2 (EZH2) to establish histone H3 lysine 27 trimethylation (H3K27me3) and repress repeats, including the repeat-like gene paternally expressed 10 (PEG10) and stem cell-related factors such as transcription factor SOX2. Following treatment with a targeted agent that inhibits a driver oncogene in the adenocarcinoma, cells that mutate RB1 are able to change H3K27me3 patterns and express SOX2 and PEG10 to establish a new cell identity that is no longer dependent on the therapeutic target. Treatment of resistant neuroendocrine tumours with EZH2 inhibitors presumably reorganizes H3K27me3 and gene expression patterns, returning cells to an epithelial state and restoring expression and sensitivity to inhibition of the original driver oncogene. These molecular events in adenocarcinoma to neuroendocrine transdifferentiation were established by studying prostate cancer, while only RB1 loss and SOX2 transcription have been presently implicated in transdifferentiation of epidermal growth factor receptor (EGFR)-mutant lung adenocarcinoma. AR, androgen receptor.
RB loss is rare in prostate adenocarcinomas98 but frequent in treatment-acquired neuroendocrine prostate cancers (NEPCs)96, suggesting that RB loss plays a role in transdifferentiation or loss of lineage identity. Two recent studies have tested this hypothesis in engineered mouse models and human prostate cancer cell lines and found a causative relationship between RB loss and altered cell lineage99,100. Pten mutation in mouse prostate epithelium generates a weakly metastatic adenocarcinoma that does not transdifferentiate101, and Rb1 mutation in the same cells does not cause cancer101–103. Prostate cancer cells deficient in both Pten and Rb1 develop as adenocarcinoma then transition into a mixed tumour containing both luminal-like and neuroendocrine-like prostate cancer cells, demonstrating lineage plasticity100. Tumours are highly metastatic and remain sensitive to anti-androgen therapy but inevitably relapse in a form lacking AR expression, possessing neuroendocrine markers and often having acquired spontaneous Trp53 mutations100. Because Rb1 loss does not appreciably alter proliferation of tumour cells in this model100, the most logical conclusion is that Rb1 loss primarily contributes to lineage plasticity. The second study used a combination of RNAi and Cas9-mediated gene deletions to inactivate RB1 and TP53 in human anti-androgen-sensitive prostate cancer cell lines99. Combined RB1 and TP53 loss, but not single mutations, led to increased expression of the reprogramming transcription factor SOX2, and this conferred increased lineage plasticity, as indicated by upregulation of basal and neuroendocrine lineage markers at the expense of luminal markers, including AR99. Loss of AR expression was accompanied by acquired resistance to anti-androgens, and this could be reversed by blocking SOX2 expression99.
In both studies, RB loss drove lineage plasticity through de-repression of stem cell and epigenetic reprogramming factors SOX2 and EZH2 (REFS99,100). Analyses of human prostate cancer specimens also demonstrate increased expression of SOX2 and EZH2 in NEPC relative to prostate adenocarcinoma100. Silencing of SOX2 or EZH2 reversed NEPC transformation and restored sensitivity to anti-androgen therapy100. Small molecule EZH2 inhibitors can also reverse NEPC transformation and anti-androgen resistance100. These exciting data support a model of epigenetic instability fuelled by RB loss and lineage plasticity, which is detailed in FIG. 3b. Perhaps more importantly, they also indicate that acquired resistance to targeted therapies through lineage plasticity is reversible, opening the door to new therapeutic opportunities. Collectively, these studies offer a mechanistic link between RB loss, de-repression of epigenetic reprogramming factors, prostate cancer lineage plasticity and acquired anti-androgen resistance.
RB loss in EGFR inhibitor-resistant lung adenocarcinoma.
A very similar story of RB loss and transdifferentiation is emerging in the treatment of lung adenocarcinoma with epidermal growth factor receptor (EGFR) inhibitors. Specific activating mutations in EGFR drive tumorigenesis in some patients with lung adenocarcinoma, and these tumours are sensitive to EGFR tyrosine kinase inhibitors such as erlotinib104. Analogous to prostate cancer, mechanisms of acquired resistance to EGFR inhibitors involve alteration of EGFR itself105,106. For example, acquisition of a second site T790M substitution reduces erlotinib binding and reactivates EGFR signalling105. Newer EGFR inhibitors like osimertinib specifically inhibit EGFR-T790M107. Although osimertinib can extend survival in patients who have relapsed after treatment with erlotinib, beneficial therapeutic responses are often short-lived because target-independent resistance mechanisms arise108,109.
Similar to observations in prostate cancer, an emerging mechanism of resistance in patients with lung adenocarcinoma who have relapsed after treatment with EGFR inhibitors involves transformation to histological variants lacking EGFR expression, including variants expressing neuroendocrine lineage markers and resembling small-cell lung cancer, a lung tumour subtype that is nearly universally RB1-mutant110 (FIG. 3a). Accordingly, a recent study has found that RB was lost universally in patients with EGFR-mutant lung adenocarcinoma who relapsed after treatment with EGFR inhibitors by neuroendocrine transformation111. In one particularly informative patient, three distinct metastatic tumours were sampled after relapse, and all shared the EGFR driver mutation of the original adenocarcinoma. RB1 was lost in both metastatic tumours exhibiting neuroendocrine transformation but not in a metastatic tumour retaining an adenocarcinoma phenotype that acquired resistance via the EGFR-T790M alteration. Thus, this study indicates that RB1 loss is correlated with neuroendocrine transformation of lung adenocarcinoma during relapse from EGFR inhibitors, even within the same patient111. Interestingly, cell culture models of EGFR resistance indicate that induction of SOX2 expression is associated with resistance to inhibitors112, suggesting that it shares molecular features with prostate lineage plasticity as well (FIG. 3b). However, whether RB1 loss can be an initiating or contributing factor for neuroendocrine transformation in lung cancer is unclear, as the described study only shows correlations and lacks any experimental data to document the direct role of RB1 loss in this transformation.
Taken together, while requiring further substantiation, these studies in prostate and lung cancer suggest two general pathways of acquired resistance to targeted therapies. One pathway alters the therapeutic target or engages a compensatory pathway while maintaining its original cancer phenotype. This is often caused by secondary mutations to the original therapeutic target, such as the T790M substitution in EGFR. Alternatively, some cancers acquire resistance through a fundamentally different route in which they lose lineage commitment, express neuroendocrine markers and no longer express the therapeutic target. The emerging evidence described above suggests that RB loss is key to lineage plasticity.
Genomic-repeat silencing and lineage plasticity.
Widespread de-repression of repetitive genomic sequences, such as satellites and LINE1 elements, has been shown to occur sporadically in a broad spectrum of epithelial cancers113. In particular, simultaneous expression of diverse repeat classes, including their genomic neighbours, often implicated in neural or stem cell pathways, has been observed113. This suggests that global alterations in repeat expression are linked to cell lineage. As described above, RB is uniquely capable of silencing broad classes of repetitive sequences such as LINEs and satellites7, and it occupies enhancers to silence expression of the pluripotency factors OCT4 and SOX2 through recruitment of histone deacetylases and EZH2 to deacetylate histones and deposit H3K27me38,114. Thus, its non-canonical functions suggest that RB loss is part of this lineage transformation mechanism (FIG. 3b). A prostate cancer study examining genes that are responsible for acquired therapeutic resistance to AR inhibitors further illustrates this concept. Using a patient-derived xenograft model of acquired resistance, loss of RB and expression of the gene paternally expressed 10 (PEG10) were identified as coincident with a shift in lineage commitment to NEPC115. PEG10 expression is normally restricted to the placenta, and its complex gene structure is derived from a retroviral repeat115. In AR-dependent prostate cancer cell lines, PEG10 is repressed by RB and E2F1, but when expressed in prostate cancer cells deficient in both RB1 and TP53, it is required for proliferation115.
RB loss is coincident with neuroendocrine transdifferentiation in prostate and lung cancer therapeutic resistance. In addition, the unique molecular mechanisms that RB uses to suppress expression of pluripotency factors and genomic repeats offer a direct mechanistic connection to transdifferentiation. At present, our understanding of the molecular events that underpin the conversion of adenocarcinomas to neuroendocrine tumours during the acquisition of therapeutic resistance remains in its infancy. However, we suggest that RB loss is critical to this process and that disruption of its non-canonical functions best explains its role in this emerging paradigm of drug resistance.
Conclusions
The studies described in this Opinion provide a number of scenarios for RB function outside its best-known role in cell cycle control. We suggest that chromatin-associated RB functions can be collectively organized into a non-canonical RB pathway, and FIG. 4 offers a visual summary of the features of this emerging pathway compared with the well known role for RB in cell cycle control.
Fig. 4 |. Features of the canonical and non-canonical RB pathways.

a | Positive growth signals activate cyclin-dependent kinases (CDKs) to phosphorylate and inactivate RB, whereas negative growth regulators (DNA damage and transforming growth factor-β (TGFβ)) augment CDK inhibitors. Inactivation of RB in the canonical pathway leads to E2F target gene transcription. b | Non-canonical RB functions are best defined by their ambivalence to CDK regulation. Signals that likely activate non-canonical RB include stresses that activate p38 MAPK phosphorylation and DNA damage that stimulates P300-associated factor (PCAF)-dependent acetylation and SET domain-containing protein 8 (SET8; also known as KMT5A) or SET and MYND domain-containing protein (SMYD)-dependent methylation. With the aid of recruitment factors (RFs), often transcription factor E2F1, RB is capable of maintaining genome stability by localizing to sites of DNA breaks and stimulating non-homologous end joining or homologous recombination repair in complex with the DNA binding proteins X-ray repair cross-complementing protein 5 (XRCC5), XRCC6 or transcription activator BRG1. RB also ensures fidelity of DNA replication and chromosome condensation through direct interactions with condensin II complexes and the indirect influence of cohesin recruitment. Lastly, RB recruits enhancer of zeste homologue 3 (EZH2) to trimethylate histone H3 lysine 27 (H3K27me3) and inhibit enhancers to control lineage commitment and to silence expression of repetitive sequences. Importantly, this diverse grouping of functions is facilitated by the association of RB with the genome over a much larger scale than proximal promoters. DP, dimeric partner.
Comparison of canonical and non-canonical pathways.
The regulatory inputs into the canonical RB pathway are known to be signals that contribute to proliferative control (FIG. 4a). Inputs such as p38 MAPK phosphorylation, as well as methylation or acetylation of RB, can serve to activate or preserve non-canonical aspects of RB function in proliferating cells (FIG. 4b). Stress responses that activate p38 MAPK and DNA damage signals are some of the most likely regulatory inputs that serve to mobilize RB to sites of DNA damage, to preserve genome stability and to maintain the epigenetic state of the cell. However, we know very little about these signals in relation to the non-canonical RB functions described in this article and suggest that the understanding of signals that activate the non-canonical RB pathway constitutes the most critical area of future work.
The output of canonical RB regulation is the release of E2F transcription factors from RB upon CDK-mediated phosphorylation to activate transcriptional targets that advance the cell cycle (FIG. 4a). The non-canonical pathway includes RB being distributed across the genome, often in complex with E2F1, in a sequence-independent manner, where it can engage numerous effectors such as cohesin, condensin II, BRG1, XRCC5 and XRCC6 (FIG. 4b). These serve to repair DNA, replicate DNA and ensure that chromosomes are faithfully distributed to daughter cells. In addition, the physical association of RB with repeat elements and enhancers allows it to silence repeat expression and control the expression of genes involved in pluripotency to regulate lineage commitment of the cell. The mechanism by which RB localizes to these regions and manages its many interactions and functions remains an important gap in our understanding.
Mutations that disable the two RB pathways.
We expect that upstream mutations affecting CDK regulation do not necessarily inactivate non-canonical RB functions. On the basis of present knowledge, we are unable to define a cascade of regulatory interactions analogous to the classic RB pathway (BOX 1). The best evidence for a non-canonical tumour suppressor pathway is a mutant mouse in which RB does not contact E2F1 through its marked box and yet the mouse is prone to develop spontaneous cancers without apparent cell cycle disruption7. A murine mutation in the serine protease 27 (Prss27) subunit of condensin II is also cancer prone74, as do Ezh2-mutant strains116. However, it is difficult to connect alterations in condensin II or Ezh2 that are contributing to cancer development directly to their impact on a non-canonical RB pathway owing to their multiple RB-independent functions. Our present understanding of non-canonical functions suggests that complete loss-of-function mutations in RB1 itself are the most reliable indicators of the lack of non-canonical RB pathway activity. Indeed, most RB1 mutations catalogued from comprehensive cancer genomic efforts are deletions or nonsense changes that are predicted to create null alleles (cBioPortal database). This suggests that specific interactions, or functions, unique to either canonical or non-canonical pathways are not being specifically selected for inactivation.
Biallelic loss of RB1 is relatively rare from a pan-cancer perspective (cBioPortal database); however, we note that some aspects of the non-canonical pathway have been shown to be sensitive to single-copy RB1 loss24,59,78,117, so it may be possible to specifically inactivate non-canonical functions. Lastly, canonical and non-canonical roles may be targeted individually in a way that is more cancer type-specific or related to disease stage or treatment choice. On the basis of the prostate and lung cancer studies highlighted in this article, we suggest that genes encoding components of the canonical RB pathway are mutated early to contribute to proliferation (for example, activation of CDK4), while later mutation of RB1 proves advantageous for transdifferentiation or other epigenetic alterations that contribute to cancer progression or treatment resistance. Understanding these intricacies is a critical challenge for future research.
Future outlook for research on the role of RB in cancer.
As mentioned above, our intention with this Opinion is to create a starting point for the grouping of chromatin-associated, non-cell-cycle-regulated functions of RB. We expect that future investigation will determine the boundaries of this non-canonical RB pathway. Exciting work on metabolism30,31 and related processes32,35 has established clearcut roles for RB and E2F1 that are highly cancer relevant. Connections to the non-canonical functions discussed here may be discovered in the future. Conceptually, the recent reports of RB functions in the cytoplasm51,118,119 suggest that there are more cancer-relevant functions that cannot easily be placed in either of the pathways described here (FIG. 4). This suggests that additional RB-dependent pathways exist but remain to be fully elucidated.
Lastly, the multifunctional properties of RB make it a critical target in many cancerrelevant contexts. Mutations in the canonical RB pathway can trigger inappropriate cell proliferation while leaving the RB1 gene intact and preserving the non-canonical pathway at relatively early stages in tumorigenesis. Later in disease progression, loss of RB may fuel metastatic dissemination through lineage alterations and genome instability, or loss of RB may allow escape from targeted therapies. Together, the loss of both RB regulatory pathways in cancer likely creates a powerful and synergistic cancer-promoting combination. It is exciting to see how an improved understanding of the molecular mechanisms of action of RB is leading to a new and better appreciation of the reasons why RB1 loss plays such a central role in human cancer.
Acknowledgements
Research in the authors’ laboratories is supported by the US National Institutes of Health (R01 CA207757 and R21 CA179907 to D.W.G., R21 AG050296 and R01 CA114102 to J.S. and R01 GM117413 to N.J.D.) and the Canadian Institutes of Health Research (MOP-89765 and MOP-64253 to F.A.D.). F.A.D. is the Wolfe Senior Fellow in Tumour Suppressor Genes at Western University. J.S. is the Harriet and Mary Zelencik Scientist in Children’s Cancer and Blood Diseases.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Competing interests statement
The authors declare no competing interests.
Reviewer information
Nature Reviews Cancer thanks E. Knudsen, N. La Thangue and S. Mittnacht for their contribution to the peer review of this work.
References
- 1.Dyson N The regulation of E2F by pRB-family proteins. Genes Dev. 12, 2245–2262 (1998). [DOI] [PubMed] [Google Scholar]
- 2.Classon M & Harlow E The retinoblastoma tumour suppressor in development and cancer. Nat. Rev. Cancer 2, 910–917 (2002). [DOI] [PubMed] [Google Scholar]
- 3.Knudsen ES & Knudsen KE Tailoring to RB: tumour suppressor status and therapeutic response. Nat. Rev. Cancer 8, 714–724 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sherr CJ Cancer cell cycles. Science 274, 1672–1677 (1996). [DOI] [PubMed] [Google Scholar]
- 5.Sherr CJ & McCormick F The RB and p53 pathways in cancer. Cancer Cell 2, 103–112 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Hanahan D & Weinberg RA The hallmarks of cancer. Cell 100, 57–70 (2000). [DOI] [PubMed] [Google Scholar]
- 7.Ishak CA et al. An RB-EZH2 complex mediates silencing of repetitive DNA sequences. Mol. Cell 64, 1074–1087 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kareta MS et al. Inhibition of pluripotency networks by the Rb tumor suppressor restricts reprogramming and tumorigenesis. Cell Stem Cell 16, 39–50 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ferrari R et al. Adenovirus small E1A employs the lysine acetylases p300/CBP and tumor suppressor Rb to repress select host genes and promote productive virus infection. Cell Host Microbe 16, 663–676 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Avni D et al. Active localization of the retinoblastoma protein in chromatin and its response to S phase DNA damage. Mol. Cell 12, 735–746 (2003). [DOI] [PubMed] [Google Scholar]
- 11.Wells J, Yan PS, Cechvala M, Huang T & Farnham PJ Identification of novel pRb binding sites using CpG microarrays suggests that E2F recruits pRb to specific genomic sites during S phase. Oncogene 22, 1445–1460 (2003). [DOI] [PubMed] [Google Scholar]
- 12.Ianari A et al. Proapoptotic function of the retinoblastoma tumor suppressor protein. Cancer Cell 15, 184–194 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cecchini MJ & Dick FA The biochemical basis of CDK phosphorylation-independent regulation of E2F1 by the retinoblastoma protein. Biochem. J 434, 297–308 (2011). [DOI] [PubMed] [Google Scholar]
- 14.Cecchini MJ et al. Loss of the retinoblastoma tumor suppressor correlates with improved outcome in patients with lung adenocarcinoma treated with surgery and chemotherapy. Hum. Pathol 46, 1922–1934 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Bosco EE et al. The retinoblastoma tumor suppressor modifies the therapeutic response of breast cancer. J. Clin. Invest 117, 218–228 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Witkiewicz AK et al. RB-pathway disruption is associated with improved response to neoadjuvant chemotherapy in breast cancer. Clin. Cancer Res 18, 5110–5122 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kommoss S et al. Independent prognostic significance of cell cycle regulator proteins p16(INK4a) and pRb in advanced-stage ovarian carcinoma including optimally debulked patients: a translational research subprotocol of a randomised study of the Arbeitsgemeinschaft Gynaekologische Onkologie Ovarian Cancer Study Group. Br. J. Cancer 96, 306–313 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ludovini V et al. Vascular endothelial growth factor, p53, Rb, Bcl-2 expression and response to chemotherapy in advanced non-small cell lung cancer. Lung Cancer 46, 77–85 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Zhao W et al. Altered p16(INK4) and RB1 expressions are associated with poor prognosis in patients with nonsmall cell lung cancer. J. Oncol 2012, 957437 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garsed DW et al. Homologous recombination DNA repair pathway disruption and retinoblastoma protein loss are associated with exceptional survival in high-grade serous ovarian cancer. Clin. Cancer Res 24, 569–580 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Knudsen ES et al. Retinoblastoma and phosphate and tensin homolog tumor suppressors: impact on ductal carcinoma in situ progression. J. Natl Cancer Inst 104, 1825–1836 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharma A et al. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J. Clin. Invest 120, 4478–4492 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Burkhart DL & Sage J Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat. Rev. Cancer 8, 671–682 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McNair C et al. Differential impact of RB status on E2F1 reprogramming in human cancer. J. Clin. Invest 128, 341–358 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robinson DR et al. Integrative clinical genomics of metastatic cancer. Nature 548, 297–303 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson D et al. Integrative clinical genomics of advanced prostate cancer. Cell 162, 454 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Beltran H et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med 22, 298–305 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dick FA & Rubin SM Molecular mechanisms underlying RB protein function. Nat. Rev. Mol. Cell Biol 14, 297–306 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kitajima S & Takahashi C Intersection of retinoblastoma tumor suppressor function, stem cells, metabolism, and inflammation. Cancer Sci 108, 1726–1731 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nicolay BN & Dyson NJ The multiple connections between pRB and cell metabolism. Curr. Opin. Cell Biol 25, 735–740 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benevolenskaya EV & Frolov MV Emerging links between E2F control and mitochondrial function. Cancer Res 75, 619–623 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ciavarra G & Zacksenhaus E Direct and indirect effects of the pRb tumor suppressor on autophagy. Autophagy 7, 544–546 (2011). [DOI] [PubMed] [Google Scholar]
- 33.Indovina P, Pentimalli F, Casini N, Vocca I & Giordano A RB1 dual role in proliferation and apoptosis: cell fate control and implications for cancer therapy. Oncotarget 6, 17873–17890 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sage J The retinoblastoma tumor suppressor and stem cell biology. Genes Dev 26, 1409–1420 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dyson NJ RB1: a prototype tumor suppressor and an enigma. Genes Dev 30, 1492–1502 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Blanchet E et al. E2F transcription factor-1 regulates oxidative metabolism. Nat. Cell Biol 13, 1146–1152 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones RA et al. RB1 deficiency in triple-negative breast cancer induces mitochondrial protein translation. J. Clin. Invest 126, 3739–3757 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee WH et al. The retinoblastoma susceptibility gene encodes a nuclear phosphoprotein associated with DNA binding activity. Nature 329, 642–645 (1987). [DOI] [PubMed] [Google Scholar]
- 39.Adams PD et al. Retinoblastoma protein contains a C-terminal motif that targets it for phosphorylation by cyclin-cdk complexes. Mol. Cell. Biol 19, 1068–1080 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carr SM, Munro S, Kessler B, Oppermann U & La Thangue NB Interplay between lysine methylation and Cdk phosphorylation in growth control by the retinoblastoma protein. EMBO J 30, 317–327 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Munro S, Khaire N, Inche A, Carr S & La Thangue NB Lysine methylation regulates the pRb tumour suppressor protein. Oncogene 29, 2357–2367 (2010). [DOI] [PubMed] [Google Scholar]
- 42.Chan HM, Krstic-Demonacos M, Smith L, Demonacos C & La Thangue NB Acetylation control of the retinoblastoma tumour-suppressor protein. Nat. Genet 3, 667–674 (2001). [DOI] [PubMed] [Google Scholar]
- 43.Hirschi A et al. An overlapping kinase and phosphatase docking site regulates activity of the retinoblastoma protein. Nat. Struct. Mol. Biol 17, 1051–1057 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Julian LM, Palander O, Seifried LA, Foster JE & Dick FA Characterization of an E2F1-specific binding domain in pRB and its implications for apoptotic regulation. Oncogene 27, 1572–1579 (2008). [DOI] [PubMed] [Google Scholar]
- 45.Rubin SM, Gall AL, Zheng N & Pavletich NP Structure of the Rb C-terminal domain bound to E2F1-DP1: a mechanism for phosphorylation-induced E2F release. Cell 123, 1093–1106 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Calbo J et al. G1 cyclin/cyclin-dependent kinase-coordinated phosphorylation of endogenous pocket proteins differentially regulates their interactions with E2F4 and E2F1 and gene expression. J. Biol. Chem 277, 50263–50274 (2002). [DOI] [PubMed] [Google Scholar]
- 47.Liban TJ et al. Conservation and divergence of C-terminal domain structure in the retinoblastoma protein family. Proc. Natl Acad. Sci. USA 114, 4942–4947 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cecchini MJ et al. A retinoblastoma allele that is mutated at its common E2F interaction site inhibits cell proliferation in gene targeted mice. Mol. Cell. Biol 34, 2029–2045 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dick FA & Dyson N pRB contains an E2F1-specific binding domain that allows E2F1-induced apoptosis to be regulated separately from other E2F activities. Mol. Cell 12, 639–649 (2003). [DOI] [PubMed] [Google Scholar]
- 50.Gubern A et al. The N-terminal phosphorylation of RB by p38 bypasses its inactivation by CDKs and prevents proliferation in cancer cells. Mol. Cell 64, 25–36 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Zhang J et al. Inhibition of Rb phosphorylation leads to mTORC2-mediated activation of Akt. Mol. Cell 62, 929–942 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Julian LM et al. Opposing regulation of Sox2 by cell-cycle effectors E2f3a and E2f3b in neural stem cells. Cell Stem Cell 12, 440–452 (2013). [DOI] [PubMed] [Google Scholar]
- 53.Alabert C & Groth A Chromatin replication and epigenome maintenance. Nat. Rev. Mol. Cell Biol 13, 153–167 (2012). [DOI] [PubMed] [Google Scholar]
- 54.Alabert C et al. Two distinct modes for propagation of histone PTMs across the cell cycle. Genes Dev 29, 585–590 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Calo E et al. Rb regulates fate choice and lineage commitment in vivo. Nature 466, 1110–1114 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Blais A & Dynlacht BD E2F-associated chromatin modifiers and cell cycle control. Curr. Opin. Cell Biol 19, 658–662 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cook R et al. Direct involvement of retinoblastoma family proteins in DNA repair by non-homologous end-joining. Cell Rep 10, 2006–2018 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Velez-Cruz R et al. RB localizes to DNA double-strand breaks and promotes DNA end resection and homologous recombination through the recruitment of BRG1. Genes Dev 30, 2500–2512 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Coschi C et al. Haploinsufficiency of an RB-E2F1-Condensin II complex leads to aberrant replication and aneuploidy. Cancer Discov 4, 840–853 (2014). [DOI] [PubMed] [Google Scholar]
- 60.Montoya-Durango DE et al. Epigenetic control of mammalian LINE-1 retrotransposon by retinoblastoma proteins. Mutat. Res 665, 20–28 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen J et al. E2F1 promotes the recruitment of DNA repair factors to sites of DNA double-strand breaks. Cell Cycle 10, 1287–1294 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saddic LA et al. Methylation of the retinoblastoma tumor suppressor by SMYD2. J. Biol. Chem 285, 37733–37740 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Carnevale J, Palander O, Seifried LA & Dick FA DNA damage signals through differentially modified E2F1 molecules to induce apoptosis. Mol. Cell. Biol 32, 900–912 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chong JL et al. E2f1–3 switch from activators in progenitor cells to repressors in differentiating cells. Nature 462, 930–934 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yamasaki L et al. Tumor induction and tissue atrophy in mice lacking E2F-1. Cell 85, 537–548 (1996). [DOI] [PubMed] [Google Scholar]
- 66.Chen HZ, Tsai SY & Leone G Emerging roles of E2Fs in cancer: an exit from cell cycle control. Nat. Rev. Cancer 9, 785–797 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Biedermann S et al. The retinoblastoma homolog RBR1 mediates localization of the repair protein RAD51 to DNA lesions in Arabidopsis. EMBO J 36, 1279–1297 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Horvath BM et al. Arabidopsis RETINOBLASTOMA RELATED directly regulates DNA damage responses through functions beyond cell cycle control. EMBO J 36, 1261–1278 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Montoya-Durango DE et al. LINE-1 silencing by retinoblastoma proteins is effected through the nucleosomal and remodeling deacetylase multiprotein complex. BMC Cancer 16, 38 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Manning AL et al. Suppression of genome instability in pRB-deficient cells by enhancement of chromosome cohesion. Mol. Cell 53, 993–1004 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gonzalo S et al. Role of the RB1 family in stabilizing histone methylation at constitutive heterochromatin. Nat. Cell Biol 7, 420–428 (2005). [DOI] [PubMed] [Google Scholar]
- 72.Isaac CE et al. The retinoblastoma protein regulates pericentric heterochromatin. Mol. Cell. Biol 26, 3659–3671 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Longworth MS, Herr A, Ji JY & Dyson NJ RBF1 promotes chromatin condensation through a conserved interaction with the Condensin II protein dCAP-D3. Genes Dev 22, 1011–1024 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Woodward J et al. Condensin II mutation causes T-cell lymphoma through tissue-specific genome instability. Genes Dev 30, 2173–2186 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lukas J, Lukas C & Bartek J More than just a focus: the chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell Biol 13, 1161–1169 (2011). [DOI] [PubMed] [Google Scholar]
- 76.Mankouri HW, Huttner D & Hickson ID How unfinished business from S-phase affects mitosis and beyond. EMBO J 32, 2661–2671 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Munro S et al. Linker histone H1.2 directs genome-wide chromatin association of the retinoblastoma tumor suppressor protein and facilitates its function. Cell Rep 19, 2193–2201 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zheng L, Flesken-Nikitin A, Chen PL & Lee WH Deficiency of Retinoblastoma gene in mouse embryonic stem cells leads to genetic instability. Cancer Res 62, 2498–2502 (2002). [PubMed] [Google Scholar]
- 79.Coschi CH et al. Mitotic chromosome condensation mediated by the retinoblastoma protein is tumor-suppressive. Genes Dev 24, 1351–1363 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Conklin JF, Baker J & Sage J The RB family is required for the self-renewal and survival of human embryonic stem cells. Nat. Commun 3, 1244 (2012). [DOI] [PubMed] [Google Scholar]
- 81.Schvartzman JM, Sotillo R & Benezra R Mitotic chromosomal instability and cancer: mouse modelling of the human disease. Nat. Rev. Cancer 10, 102–115 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sharma A et al. Retinoblastoma tumor suppressor status is a critical determinant of therapeutic response in prostate cancer cells. Cancer Res 67, 6192–6203 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Varma H & Conrad SE Reversal of an antiestrogen-mediated cell cycle arrest of MCF-7 cells by viral tumor antigens requires the retinoblastoma protein-binding domain. Oncogene 19, 4746–4753 (2000). [DOI] [PubMed] [Google Scholar]
- 84.Mayhew CN et al. Discrete signaling pathways participate in RB-dependent responses to chemotherapeutic agents. Oncogene 23, 4107–4120 (2004). [DOI] [PubMed] [Google Scholar]
- 85.Zagorski WA, Knudsen ES & Reed MF Retinoblastoma deficiency increases chemosensitivity in lung cancer. Cancer Res 67, 8264–8273 (2007). [DOI] [PubMed] [Google Scholar]
- 86.Bourgo RJ et al. RB restricts DNA damage-initiated tumorigenesis through an LXCXE-dependent mechanism of transcriptional control. Mol. Cell 43, 663–672 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xiao H & Goodrich DW The retinoblastoma tumor suppressor protein is required for efficient processing and repair of trapped topoisomerase II-DNA-cleavable complexes. Oncogene 24, 8105–8113 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Branzei D & Foiani M Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol 9, 297–308 (2008). [DOI] [PubMed] [Google Scholar]
- 90.Sherr CJ, Beach D & Shapiro GI Targeting CDK4 and CDK6: from discovery to therapy. Cancer Discov 6, 353–367 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Koh J, Enders GH, Dynlacht BD & Harlow E Tumor-derived p16 alleles encoding proteins defective in cell cycle inhibition. Nature 375, 506–510 (1995). [DOI] [PubMed] [Google Scholar]
- 92.Lukas J et al. Retinoblastoma-protein-dependent inhibition by the tumor-suppressor p16. Nature 375, 503–506 (1995). [DOI] [PubMed] [Google Scholar]
- 93.Bruce JL, Hurford RKJ, Classon M, Koh J & Dyson N Requirements for cell cycle arrest by p16INK4a. Mol. Cell 6, 737–742 (2000). [DOI] [PubMed] [Google Scholar]
- 94.Watson PA, Arora VK & Sawyers CL Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 15, 701–711 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tran C et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science 324, 787–790 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rickman DS, Beltran H, Demichelis F & Rubin MA Biology and evolution of poorly differentiated neuroendocrine tumors. Nat. Med 23, 1–10 (2017). [DOI] [PubMed] [Google Scholar]
- 97.Bluemn EG et al. Androgen receptor pathway-independent prostate cancer is sustained through FGF signaling. Cancer Cell 32, 474–489 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.The Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell 163, 1011–1025 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mu P et al. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 355, 84–88 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ku SY et al. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 355, 78–83 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang S et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 4, 209–221 (2003). [DOI] [PubMed] [Google Scholar]
- 102.Zhou Z et al. Synergy of p53 and Rb deficiency in a conditional mouse model for metastatic prostate cancer. Cancer Res 66, 7889–7898 (2006). [DOI] [PubMed] [Google Scholar]
- 103.Maddison LA, Sutherland BW, Barrios RJ & Greenberg NM Conditional deletion of Rb causes early stage prostate cancer. Cancer Res 64, 6018–6025 (2004). [DOI] [PubMed] [Google Scholar]
- 104.Lynch TJ et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med 350, 2129–2139 (2004). [DOI] [PubMed] [Google Scholar]
- 105.Kobayashi S et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med 352, 786–792 (2005). [DOI] [PubMed] [Google Scholar]
- 106.Yun CH et al. Structures of lung cancer-derived EGFR mutants and inhibitor complexes: mechanism of activation and insights into differential inhibitor sensitivity. Cancer Cell 11, 217–227 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pirker R Third-generation epidermal growth factor receptor tyrosine kinase inhibitors in advanced nonsmall cell lung cancer. Curr. Opin. Oncol 28, 115–121 (2016). [DOI] [PubMed] [Google Scholar]
- 108.Sequist LV et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl Med 3, 75ra26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Oser MG, Niederst MJ, Sequist LV & Engelman JA Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol 16, e165–e172 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.George J et al. Comprehensive genomic profiles of small cell lung cancer. Nature 524, 47–53 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Niederst MJ et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat. Commun 6, 6377 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rothenberg SM et al. Inhibition of mutant EGFR in lung cancer cells triggers SOX2-FOXO6-dependent survival pathways. eLife 4, e06132 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ting DT et al. Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science 331, 593–596 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liu Y et al. Mouse fibroblasts lacking RB1 function form spheres and undergo reprogramming to a cancer stem cell phenotype. Cell Stem Cell 4, 336–347 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Akamatsu S et al. The placental gene PEG10 promotes progression of neuroendocrine prostate cancer. Cell Rep 12, 922–936 (2015). [DOI] [PubMed] [Google Scholar]
- 116.Herrera-Merchan A et al. Ectopic expression of the histone methyltransferase Ezh2 in haematopoietic stem cells causes myeloproliferative disease. Nat. Commun 3, 623 (2012). [DOI] [PubMed] [Google Scholar]
- 117.Gonzalez-Vasconcellos I et al. Rb1 haploinsufficiency promotes telomere attrition and radiation-induced genomic instability. Cancer Res 73, 4247–4255 (2013). [DOI] [PubMed] [Google Scholar]
- 118.Hilgendorf KI et al. The retinoblastoma protein induces apoptosis directly at the mitochondria. Genes Dev 27, 1003–1015 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Araki K, Kawauchi K, Hirata H, Yamamoto M & Taya Y Cytoplasmic translocation of the retinoblastoma protein disrupts sarcomeric organization. eLife 2, e01228 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Weinberg RA The retinoblastoma protein and cell cycle control. Cell 81, 323–330 (1995). [DOI] [PubMed] [Google Scholar]
- 121.The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ciriello G et al. Comprehensive molecular portraits of invasive lobular breast cancer. Cell 163, 506–519 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]