

This article provides recommendations for the clinical management of key adverse reactions reported with lorlatinib.
Keywords: Lorlatinib, Non‐small cell lung cancer, Safety, Drug therapy management
Abstract
Lorlatinib is a novel, highly potent, brain‐penetrant, third‐generation ALK/ROS1 tyrosine kinase inhibitor (TKI), which has broad‐spectrum potency against most known resistance mutations that can develop during treatment with crizotinib and second‐generation ALK TKIs. The safety profile of lorlatinib was established based on 295 patients who had received the recommended dose of lorlatinib 100 mg once daily. Adverse events associated with lorlatinib are primarily mild to moderate in severity, with hypercholesterolemia (82.4%), hypertriglyceridemia (60.7%), edema (51.2%), peripheral neuropathy (43.7%), and central nervous system effects (39.7%) among the most frequently reported. These can be effectively managed with dose modification and/or standard supportive medical therapy, as indicated by a low incidence of permanent discontinuations due to adverse reactions. Most patients (81.0%) received at least one lipid‐lowering agent. Prescription of supportive therapy should also consider the potential for drug‐drug interactions with lorlatinib via engagement of specific CYP450 enzymes. This article summarizes the clinical experience from lorlatinib phase I investigators and was generated from discussion and review of the clinical study protocol and database to provide an expert consensus opinion on the management of the key adverse reactions reported with lorlatinib, including hyperlipidemia, central nervous system effects, weight increase, edema, peripheral neuropathy, and gastrointestinal effects. Overall, lorlatinib 100 mg once daily has a unique safety profile to be considered when prescribed, based on the recent U.S. Food and Drug Administration approval, for the treatment of patients with ALK‐positive metastatic non‐small cell lung cancer previously treated with a second‐generation ALK TKI.
Implications for Practice.
Despite the advancement of second‐generation anaplastic lymphoma kinase (ALK) tyrosine kinase inhibitors (TKIs), the emergence of resistance and progression of central nervous system metastases remain clinically significant problems in ALK‐positive non‐small cell lung cancer. Lorlatinib is a potent, brain‐penetrant, third‐generation, macrocyclic ALK/ROS1 TKI, with broad‐spectrum potency against most known resistance mutations that can develop during treatment with existing first‐ and second‐generation ALK TKIs. This article provides recommendations for the clinical management of key adverse reactions reported with lorlatinib.
摘要
劳拉替尼是一种新型、高效、具有脑渗透作用的第三代 ALK/ROS1 酪氨酸激酶抑制剂 (TKI),对克唑替尼和第二代 ALK TKI 治疗期间可能发生的大多数已知耐药突变具有广谱效力。295 名患者每日服用一次 100 mg 推荐剂量的劳拉替尼,在此基础上确立了劳拉替尼的安全谱。劳拉替尼相关不良事件的严重程度主要为轻度至中度,最常报告的不良事件是高胆固醇血症 (82.4%)、高甘油三酯血症 (60.7%)、水肿 (51.2%)、周围神经病变 (43.7%) 和中枢神经系统影响 (39.7%)。由于因不良事件引起的永久停药的发生率较低,因此可通过调整剂量和/或标准支持性药物疗法来有效管理上述不良事件。大多数患者 (81.0%) 服用至少一种降脂药。支持疗法的处方还应考虑劳拉替尼通过特定 CYP450 酶的参与发生药物相互作用的可能性。本文总结了劳拉替尼 I 期研究者的临床经验,并通过对临床研究方案和数据库的讨论和回顾,就报告的罗拉替尼主要不良事件(包括高脂血症、中枢神经系统影响、体重增加、水肿、周围神经病变和胃肠影响)的管理提供了专家共识意见。总体而言,根据美国食品药品监督管理局最新的批准,我们认为,凭处方每天服用一次 100 mg 的劳拉替尼对治疗 ALK 阳性转移性非小细胞肺癌患者(之前接受过第二代 ALK TKI 治疗)具有出色的安全性。
实践意义:尽管第二代间变性淋巴瘤激酶 (ALK) 酪氨酸激酶抑制剂 (TKI) 取得了发展,但中枢神经系统转移出现耐药性和进展仍是 ALK 阳性非小细胞肺癌面临的重要临床问题。劳拉替尼是一种高效、具有脑渗透作用的第三代大环 ALK/ROS1 TKI,对现有第一代和第二代 ALK TKI 治疗期间可能发生的大多数已知耐药突变具有广谱效力。本文为报告的劳拉替尼主要不良反应的临床管理提供了 建议。
Background
Rearrangements of the anaplastic lymphoma kinase (ALK) gene are found in 3%–5% of patients with non‐small cell lung cancer (NSCLC) [1], [2], [3], [4] and represent a clinically and molecularly distinct subtype of NSCLC. Standard therapy for patients with ALK‐positive NSCLC includes crizotinib, a multitargeted ALK/ROS1/MET tyrosine kinase inhibitor (TKI) [5] and, more recently, second‐generation ALK TKIs [6], [7], [8]. However, most patients treated with crizotinib relapse over time because of acquired (or secondary) resistance that occurs through several molecular mechanisms, including development of secondary mutations within the ALK kinase domain and activation of alternative signaling pathways [9]. More potent second‐generation ALK TKIs were developed to surmount the development of crizotinib resistance in ALK‐positive NSCLC and have demonstrated improved response rates in crizotinib‐refractory and treatment‐naïve patients [7], [8], [10], [11]. However, most patients will eventually develop resistance to second‐generation ALK TKIs [12].
Treatment options following the emergence of resistance to a second‐generation ALK TKI are limited, with platinum‐based chemotherapy as the current standard of care. However, the associated toxicities, consisting of adverse events such as nausea, vomiting, and neutropenia, can negatively impact patients’ quality of life. Although there are no published data on the antitumor activity of chemotherapy after second‐generation ALK TKIs, response rates of 27%–45% have been reported with platinum‐ and pemetrexed‐based chemotherapy in the first‐line treatment of ALK‐positive advanced NSCLC [5], [8], and response rates ranging from 7% to 11% have been reported with single‐agent chemotherapy in patients previously treated with a platinum doublet and crizotinib [13], [14], [15].
Lorlatinib is a novel, highly potent, third‐generation, macrocyclic ALK/ROS1 TKI that competitively binds to the adenosine triphosphate‐binding pocket, blocking ALK‐dependent oncogenic signaling. Lorlatinib was also designed to penetrate the blood–brain barrier in part by minimizing p‐glycoprotein‐1‐mediated efflux, which can lead to poor blood–brain barrier penetration [16], [17]. Lorlatinib has broad‐spectrum potency against most known resistance mutations that can develop during treatment with crizotinib and second‐generation ALK TKIs, including the difficult‐to‐treat ALK G1202R mutation [18].
Lorlatinib: Pharmacology
In preclinical studies, advantageous pharmacokinetic properties have been reported for lorlatinib including low plasma clearance, 100% oral bioavailability, and a moderate volume of distribution [16]. Lorlatinib may be taken with or without food and is rapidly absorbed (peak plasma concentrations occurring 1–2 hours after dosing), with a terminal elimination half‐life of 19.0–28.8 hours (Pfizer, data on file). The high blood–brain barrier penetration of lorlatinib is supported by a mean ratio of cerebrospinal fluid/plasma (unbound) of 0.75 [19]. Lorlatinib is metabolized primarily by cytochrome P450 (CYP)3A, CYP2C19, and CYP2C8 and uridine 5’‐diphospho‐glucuronosyltransferase and has also exhibited time‐dependent inhibition of CYP3A4/5 as well as induction of CYP2B6 and CYP3A4 (Pfizer, data on file). Thus, lorlatinib may have the potential to alter the pharmacokinetics of other coadministered drugs that are metabolized by CYP2B6 or CYP3A. A phase I, open‐label, crossover study (ClinicalTrials.gov identifier: NCT02804399) found that concomitant administration of lorlatinib and rifampin led to elevated aspartate and alanine aminotransferase levels (Pfizer, data on file). Therefore, the use of strong CYP3A4/5 inducers with lorlatinib is contraindicated.
Lorlatinib: Clinical Experience
In an ongoing phase I/II study (NCT01970865), lorlatinib demonstrated clinically meaningful benefit, including substantial intracranial efficacy, among patients with ALK‐positive or ROS1‐positive advanced NSCLC who were treatment naïve or who had received a range of prior ALK inhibitors with or without chemotherapies [19], [20]. Patients in the phase II portion of this study were enrolled into six expansion cohorts based on prior treatment and rearrangement status (expansion cohorts 1–5, ALK‐positive; expansion cohort 6, ROS1‐positive), details of which have been previously presented [21]. In April 2017, lorlatinib was granted breakthrough therapy designation by the U.S. Food and Drug Administration (FDA) in patients with ALK‐positive advanced NSCLC previously treated with one or more ALK inhibitors and, in November 2018, the FDA granted lorlatinib an accelerated approval for the treatment of patients with ALK‐positive metastatic NSCLC who had disease progression on crizotinib and at least one other ALK TKI or who had disease progression on alectinib or ceritinib as the first ALK TKI. The phase III CROWN study (NCT03052608) comparing lorlatinib with crizotinib as first‐line treatment among patients with ALK‐positive advanced NSCLC is currently enrolling patients.
Lorlatinib: Safety and Tolerability
Overview
The safety profile of lorlatinib was established based on 295 patients who had received the recommended dose of lorlatinib 100 mg once daily (QD) in the fully recruited, ongoing phase I/II study (NCT01970865). This pooled group comprised 17 patients from the phase I portion of the study, 275 patients from the phase II portion, and 3 patients from a Japanese lead‐in cohort—an evaluation study of the safety and tolerability of lorlatinib in Japanese patients conducted before sites in Japan could participate in the phase II study because of lack of documented experience in this patient population. The data presented in this article are based on a cutoff date of March 15, 2017.
The incidence and management of the most frequent adverse reactions (i.e., adverse drug reactions) with lorlatinib, particularly those occurring in ≥10% of patients, are reported. Adverse drug reactions are defined as those adverse events that, after internal clinical and safety review applying medical judgment, were determined to be likely associated with the study treatment. This is in contrast to the broad category of all‐causality adverse events, which may or may not have an established causal relationship. It is further important to note that the definitions and reporting thresholds for adverse reactions can vary across different regulatory health authorities, and in certain circumstances, these concepts can be used interchangeably. In the phase I/II study (based on the data cutoff of March 15, 2017), adverse reactions consisted of adverse events including hypercholesterolemia, hypertriglyceridemia, central nervous system (CNS) effects (mood disorder, cognitive disorder, and speech disorder), weight increase, edema, peripheral neuropathy, diarrhea, constipation, fatigue, arthralgia, and vision disorder. Additional adverse events of clinical interest included lipase increase and atrioventricular (AV) block.
Adverse reactions in this pooled safety analysis population of 295 patients treated with lorlatinib 100 mg QD are shown in Table 1. Hyperlipidemia, edema, and peripheral neuropathy were among the most frequently reported adverse reactions with lorlatinib treatment. Most adverse reactions were grade 1 or 2 in severity, with few grade ≥ 3 adverse reactions reported (Table 1). Serious adverse reactions occurred in 9 patients (3.0%), and the most frequently reported were cognitive disorder (1.0%) and edema (0.7%).
Table 1. Adverse reactions in ≥10% of all patients or Grade ≥3 adverse reactions in any patient treated with lorlatinib 100 mg once daily.
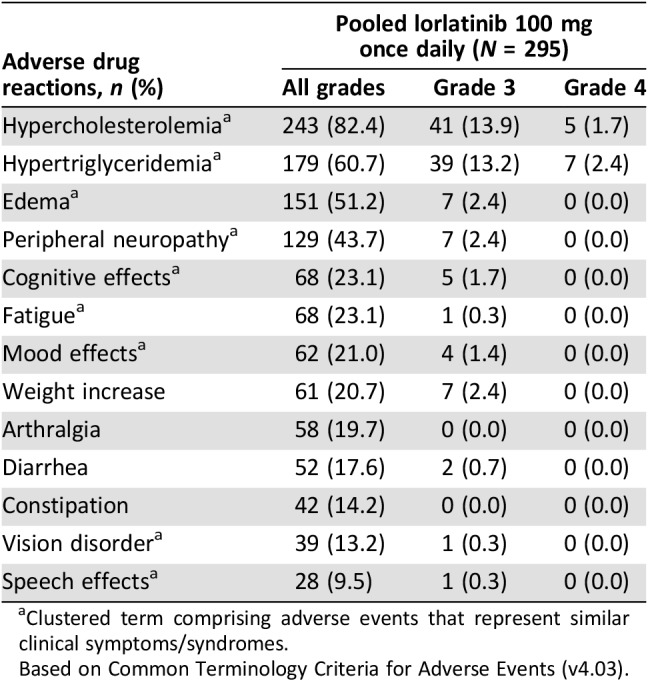
Clustered term comprising adverse events that represent similar clinical symptoms/syndromes.
Based on Common Terminology Criteria for Adverse Events (v4.03).
Temporary dose interruptions and dose reductions associated with adverse reactions were reported in 21.7% and 19.7% of patients, respectively. The most common cause for dose interruptions and dose modifications was edema (5.8% and 6.1% of patients, respectively). Overall, the incidence of permanent discontinuations due to adverse reactions was low (n = 6 [2.0%]). The adverse reactions that led to permanent treatment discontinuations were cognitive disorder (n = 2), mood disorder (n = 2), edema (n = 1), and fatigue (n = 1).
This article summarizes the clinical experience from lorlatinib phase I investigators and was generated from discussion and review of the clinical study protocol and database to provide an expert consensus opinion on the management of the key adverse reactions reported with lorlatinib, including hyperlipidemia, CNS effects, weight increase, edema, peripheral neuropathy and gastrointestinal effects. Lipase increase and AV block adverse events are also discussed.
Management of Specific Adverse Events
Hyperlipidemia.
Hyperlipidemia, comprising the cluster terms hypercholesterolemia and hypertriglyceridemia, was the most common adverse reaction reported with lorlatinib and usually occurred within the first few weeks of treatment (median time to onset, 15 days [range, 1–219]; supplemental online Fig. 1). Hypercholesterolemia and hypertriglyceridemia were reported in 82.4% and 60.7% of patients in the pooled safety group, respectively, and both events were mostly grade 1 or 2 in severity. Grade 3/4 hypercholesterolemia and hypertriglyceridemia both occurred at a frequency of 15.6%. However, hypercholesterolemia and hypertriglyceridemia were not common reasons for dose delay (3.4% and 4.7%, respectively) or dose reduction (0.7% and 1.7%, respectively) and did not result in any permanent discontinuations across the study. In most cases, these typically asymptomatic adverse events were easily managed with appropriate medical therapy (i.e., lipid‐lowering agents) and dose interruptions, coupled with dose modifications for more severe (grade ≥3) and difficult‐to‐treat adverse events (Table 2).
Table 2. Dose modification guidelines for lorlatinib‐related hyperlipidemias by severity.

Abbreviation: ULN, upper limit of normal.
Most patients (81.0%) received at least one lipid‐lowering agent and commenced treatment within 3 weeks of the first lorlatinib dose (median time to start of lipid‐lowering medication, 20 days [range, 1–190]). Among 226 patients who received a lipid‐lowering agent for hypercholesterolemia and 198 who received a lipid‐lowering agent for hypertriglyceridemia, 22.1% and 30.8% of patients required the addition of another lipid‐lowering agent, respectively. The most commonly prescribed lipid‐lowering agent was rosuvastatin (n = 124; 42.0% of patients). Treatment with a statin is recommended at the first sign of elevated cholesterol (upper limit of normal, 300 mg/dL [upper limit of normal, 7.75 mmol/L]) and/or triglyceride levels (150–300 mg/dL [1.71–3.42 mmol/L]). The choice and dosing of statins may be guided by information on the differential metabolism by the CYP450 pathway (supplemental online Table 1). Pitavastatin, pravastatin, or rosuvastatin should initially be considered based on their low involvement with specific CYP450 enzymes that can interact with lorlatinib (e.g., CYP3A4) [22]. The addition of fibrates or fish oils is considered effective in lowering triglyceride levels if treatment beyond statins is required [23]. Of these, fenofibrate can be administered first as it has the least involvement with the associated CYP450 enzymes, followed by fish oils and nicotinic acid [24], [25], [26]. Of note, the concomitant use of gemfibrozil and some statins is not recommended. In particular, coadministration of gemfibrozil with simvastatin is contraindicated [27]. Ezetimibe may also be considered if treatment with statins and fenofibrate is ineffective.
Dose interruptions are recommended if cholesterol levels reach >500 mg/dL and/or triglyceride levels reach >1,000 mg/dL despite the use of lipid‐lowering therapies. Lorlatinib should be withheld, while maximizing lipid‐lowering treatment, until severity is reduced before rechallenging at the same dose. Reducing the lorlatinib dose is suggested only if cholesterol levels >500 mg/dL and/or triglyceride levels >1,000 mg/dL recur (Table 2).
In light of the frequent occurrence of hyperlipidemia, patients should be informed of the likelihood of requiring lipid‐lowering therapy or that an increase in dose may be necessary for those patients already receiving lipid‐lowering agents. In addition, patients should be aware that monitoring of serum cholesterol and triglyceride levels is to be expected before and throughout the course of treatment with lorlatinib (supplemental online Table 2). However, patients should be advised that hyperlipidemia associated with lorlatinib treatment is easily managed and resolvable through lipid‐lowering therapy and/or dose modifications.
CNS Effects.
A broad spectrum of CNS effects can occur in patients receiving lorlatinib, and the following section summarizes the changes in cognitive function, mood, and speech that have been reported in patients receiving lorlatinib. These adverse reactions were reported at a frequency of 39.7%, with 11.9% of patients experiencing more than one type of CNS effect (supplemental online Fig. 2). Baseline CNS metastases were present in 84 (71.8%) of 117 patients with CNS effects (supplemental online Fig. 3). Of the 84 patients with CNS effects and baseline CNS metastases, 22 (26.2%) had received prior whole‐brain radiation therapy (supplemental online Fig. 4). CNS adverse reactions were generally mild in severity and intermittent and improved or resolved upon dose modifications. Out of 117 patients who experienced CNS effects, 24 required ≥1 dose modification (temporary discontinuation and/or dose reduction), with 15 (62.5%) of these patients experiencing resolution of their CNS effects. The median time to resolution of CNS effects was 12.5 days (range 2–112). Recurrence of CNS effects upon rechallenge at the same and reduced dose occurred in six (25.0%) and seven (29.2%) patients, respectively. General dose modification guidelines appropriate for any of the CNS effects are outlined in Table 3. Dose interruptions should be considered in the presence of grade 1 CNS effects and enforced until recovery to baseline before restarting lorlatinib at the same dose or, if required, at a lower dose. For patients with grade 2 or 3 CNS adverse events, lorlatinib should be withheld until recovery to grade ≤1 before rechallenging at a reduced dose. Permanent discontinuation from lorlatinib is recommended for grade 4 CNS effects. In addition, patients and their caregivers should be instructed to report any changes in cognitive function, mood, or speech to their health care provider.
Table 3. Dose modification guidelines for lorlatinib‐related central nervous system effects by CTCAE gradea .
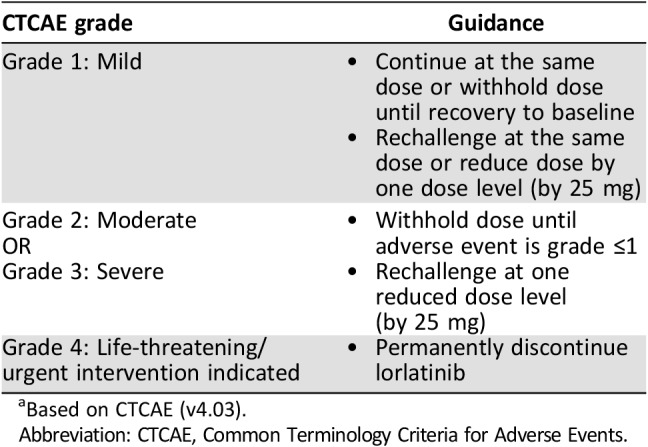
Based on CTCAE (v4.03).
Abbreviation: CTCAE, Common Terminology Criteria for Adverse Events.
Mood Effects.
Behavioral alterations and mood changes associated with lorlatinib in clinical studies were collected under the cluster term “mood effects” and are shown in Table 4. These effects occurred at a frequency of 21.0%, with a median time to onset of 43 days (range, 1–452 days). Irritability, anxiety, depression, and affect lability were the most common mood adverse events reported. Mood changes observed with lorlatinib were mostly mild in severity (19.7%), with no grade 4 adverse events reported. The overall frequency of mood effects was comparable between patients <65 years of age (21.6%) and those ≥65 years (18.5%); however, they were reported more frequently in non‐Asian (27.3%) compared with Asian patients (10.2%). Most patients with mood effects will describe feeling more irritable, more prone to impatience or anger, or sometimes more anxious. When patients first start treatment, some report feeling a little “energized” or even “buzzed.” Patients have also reported “feeling flat” and/or “feeling less excited about things.” Additional assessments of mood and suicidal ideation and behavior were a requirement of the phase II study and were administered to patients at the beginning of each cycle up to cycle 6 and then every other cycle thereafter. Based on the phase II portion (n = 275), there were no trends in the Beck Depression Inventory‐II summary data to suggest worsening of symptoms during treatment with lorlatinib. Likewise, there were no trends in the Columbia‐Suicide Severity Rating Scale group summary data to suggest a notable shift in suicidal ideation or behavior during treatment with lorlatinib (Pfizer, data on file). Mood effects related to lorlatinib were associated with dose interruptions and reductions in eight (2.7%) and seven (2.4%) patients, respectively. Out of the 12 mood events that required dose modification (i.e., dose reduction, temporary discontinuation, or both), 8 (66.7%) events resolved. The median time to resolution was 14 days (range 2–112). Recurrence upon rechallenge at a reduced dose was reported with four (33.3%) mood events; no recurrence following rechallenge at the same dose was reported. Affect lability and anxiety resulted in permanent treatment discontinuation in one patient each.
Table 4. Mood disorder adverse events with lorlatinib.
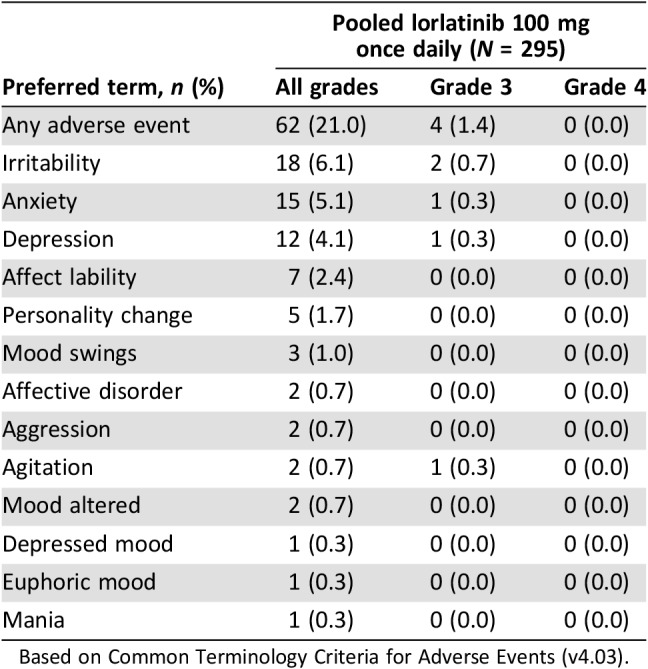
Based on Common Terminology Criteria for Adverse Events (v4.03).
Effects on mood should be discussed with patients before lorlatinib is administered. It is particularly important to review these effects with patients who have pre‐existing psychiatric conditions. Caregivers and family members, as well as patients, should also be encouraged to report any changes in patients’ mood during treatment with lorlatinib in the event that patients are unaware of such changes. These adverse events are temporary and reversible upon dose interruption or reduction.
Cognitive Effects.
Cognitive effects (cluster term) were reported in 23.1% of patients treated with lorlatinib in the pooled safety analysis. These adverse events, which most commonly included memory impairment, cognitive disorder, and amnesia, usually presented within the first 2 months of treatment (median time to onset, 53 days [range, 1–423]) and were mainly grade ≤ 2 in severity (21.4%; Table 5). The frequency of cognitive effects was comparable between age groups (<65 years [22.4%] or ≥ 65 years [25.9%]); however, they were reported at a higher frequency in non‐Asian (28.0%) compared with Asian patients (12.0%). Five grade 3 cognitive effects were reported: cognitive disorder (n = 2), confusional state (n = 2), and delirium (n = 1). Patients have reported experiences described as “sluggish thought,” “fogginess,” “trouble connecting the dots,” difficulty multitasking, difficulty finding the right words, issues with short‐term memory or recall, and confusion and hallucinations in severe cases. An assessment of cognitive function, which tested verbal learning, psychomotor function, delayed recall, attention, and working memory, was conducted in the phase II portion and validated by a central vendor (Cogstate, Inc., New Haven, CT). From this analysis, there was little evidence of any systematic decline in cognition associated with lorlatinib (Pfizer, data on file). Cognitive effects were among the most frequently reported adverse events associated with dose interruptions (3.7%) and dose reductions (2.7%). Out of the 33 cognitive effects that required dose modification(s), 22 (66.7%) resolved. The median time to resolution was 10 days (range, 3–39). Seven (21.2%) cognitive effects recurred upon rechallenge with the same dose of lorlatinib and four (12.1%) recurred with a reduced dose. Two patients permanently discontinued lorlatinib as a result of cognitive disorder (n = 1) and confusional state (n = 1).
Table 5. Cognitive disorder adverse events with lorlatinib.
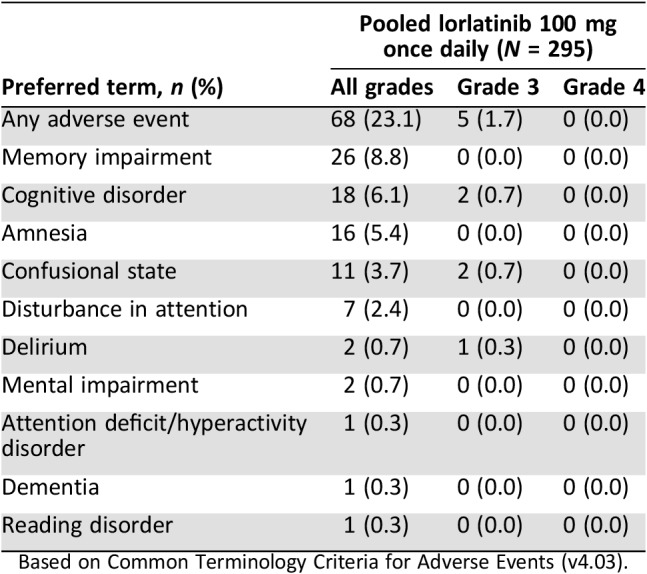
Based on Common Terminology Criteria for Adverse Events (v4.03).
The possibility of cognitive‐related adverse reactions should be discussed with patients and caregivers before starting lorlatinib treatment, along with advice on how to minimize the impact on daily activities (e.g., setting reminders). Patients should be advised to inform their health care provider if they experience any changes in cognitive functioning. As with all CNS effects, dose modifications, imposed at low thresholds, are generally effective in reversing these adverse events (Table 3), and the importance of dose reduction should be emphasized if patients’ usual activities or relationships are impaired by cognitive or mood effects.
Speech Effects.
Speech effects (cluster term) occurred in 9.5% of patients, with a median time to onset of 42 days (range, 1–404). Dysarthria, slow speech, and speech disorder were reported in 3.7%, 3.4%, and 2.4% of patients, respectively (supplemental online Table 3). Overall, these adverse events were predominantly mild in severity (grade 1, 8.5%). One patient required a dose reduction and interruption as a result of dysarthria. Of the three speech events that required dose modification(s), two (66.7%) events resolved. The median time to resolution was 38.5 days (range, 35–42). One (33.3%) out of three speech events with dose modifications recurred upon rechallenge at the same dose of lorlatinib; no recurrence was reported with rechallenge at a reduced dose. No patients permanently discontinued treatment because of speech effects.
Speech effects have been reported by patients as a perception of slowed speech or difficulty in word finding. Although the altered speech experienced is typically mild, the ability to tolerate such adverse events can vary between patients. Therefore, patients should be counseled on the potential for speech effects at the time of treatment initiation and should be reassured that these adverse events are reversible following dose modifications or treatment cessation, if required. Patients should also notify their health care provider if they experience any changes in their speech.
Weight Increase.
Reports of weight increases among patients treated with lorlatinib usually presented within 2 months of treatment initiation (median time to onset, 64 days [range, 1–519]). Of 282 evaluable patients in the pooled safety group, 87 (30.9%) had a 10%–20% increase of their baseline body weight and 38 (13.5%) had a >20% increase of their baseline body weight. The median of maximum percent change from baseline was 11.4% (range, 0.2–55.2). However, weight increase was only reported as an adverse event in 61 patients (20.7%), most of which were grade ≤2 in severity (18.3%). An increase in weight was reported as an adverse event in 4.1% of patients at Cycle 1, 13.3% of patients by Cycle 4, and 23.4% of patients by Cycle 8. An increase in appetite has been reported by some patients, suggesting that body weight increase may potentially be associated with increased caloric intake and a heightened desire to eat; however, causality has not been determined. Additionally, cases of weight gain (≥10% increase from baseline) reported concurrently with edema were limited and are shown in supplemental online Figure 5.
Two patients required dose interruptions and two required dose reductions as a result of weight increase associated with lorlatinib treatment. Although relatively uncommon, dose modifications (Table 6) should be applied in more severe cases of weight gain.
Table 6. General dose modification guidelines for lorlatinib‐related adverse reactions by CTCAE gradea .
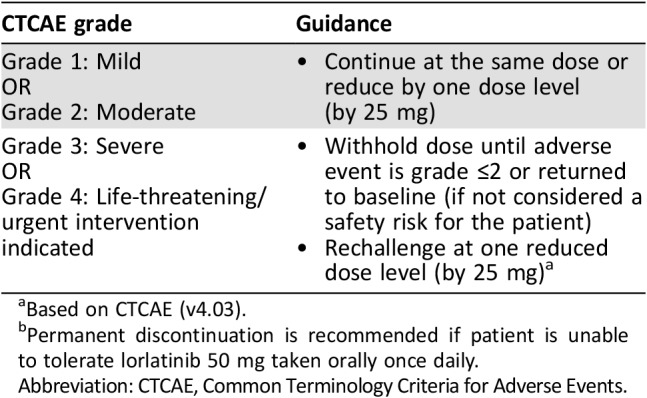
Based on CTCAE (v4.03).
Permanent discontinuation is recommended if patient is unable to tolerate lorlatinib 50 mg taken orally once daily.
Abbreviation: CTCAE, Common Terminology Criteria for Adverse Events.
Food intake counseling, dietary advice from a nutritionist, and the addition of exercise also appear to be effective weight management strategies, although nonadherence can be an issue. Patients should be advised from the outset that they are likely to experience some degree of weight gain. This will ensure that patients are prepared and can expect to implement some lifestyle adjustments.
Edema.
Treatment with lorlatinib was associated with edema (51.2%), with peripheral edema arising as the most frequently reported event (41.7%). Other adverse events reported within the edema cluster term were edema (7.5%), peripheral swelling (6.1%), generalized edema (0.7%), and swelling (0.7%). The median time to onset of edema was 42 days (range, 1–232), with a median duration of 163 days. Most adverse events were mild in severity (grade ≤2, 48.8%), and only 2.4% of patients experienced grade 3 events. Edema was the most common cause of dose interruptions (5.8%) and dose reductions (6.1%) in the pooled safety analysis group.
In practice, compression stockings, leg elevations, and lifestyle modifications, such as increased exercise and limiting dietary salt, should initially be considered in patients with low‐grade edema before commencing with dose modifications. These conservative measures, in combination with diuretics (usually furosemide), have shown to be effective in the management of edema. The addition of spironolactone can also be beneficial in the treatment of edema refractory to furosemide monotherapy. However, if edema persists or worsens, general dose modification guidance can be used with lorlatinib treatment and held until improvement to grade ≤2 (if not a safety risk) or returned to baseline and rechallenged at a reduced dose (Table 6).
Peripheral Neuropathy.
Peripheral neuropathy (cluster term) associated with lorlatinib was reported at a frequency of 43.7%, with a median time to onset of 77 days (range, 1–723). Within the cluster term, the most common adverse events reported were paresthesia (13.2%) and neuropathy peripheral (11.2%). Other peripheral neuropathy adverse events seen with lorlatinib included peripheral sensory neuropathy and muscular weakness (Table 7). In general, peripheral neuropathy adverse events were mild in severity (grade ≤2, 41.4%) and reversible following dose modifications or standard medical therapy. Symptoms were often described as tingling, numbness, and pain at night in extremities (especially in patients experiencing symptoms similar to carpal tunnel syndrome). In patients with pre‐existing carpal tunnel syndrome, symptoms often worsened when they received lorlatinib. Patients with peripheral neuropathy often also reported adverse events of weight gain and/or edema (71.3%); overlap of these three adverse events is shown in supplemental online Figure 6.
Table 7. Peripheral neuropathy adverse events with lorlatinib.
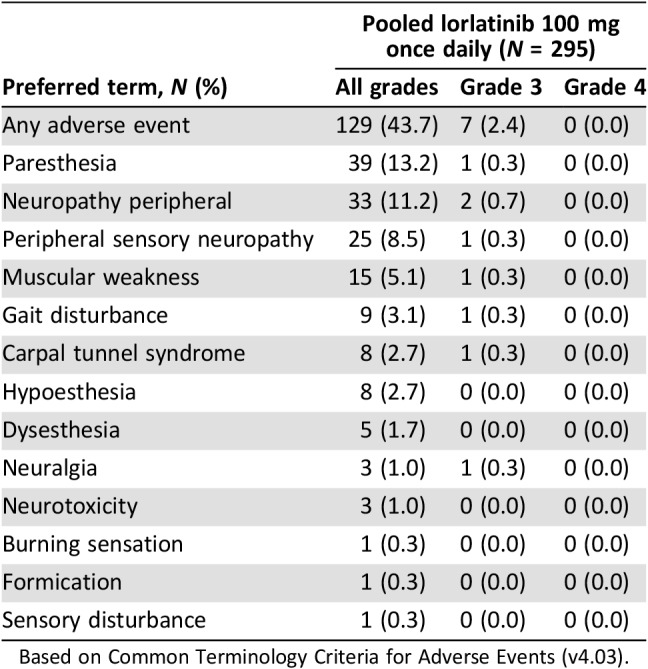
Based on Common Terminology Criteria for Adverse Events (v4.03).
Peripheral neuropathy was among the most frequently reported adverse events associated with dose interruptions (4.1%) and dose reductions (4.1%) but responded well to these dose modifications. General guidance on dose modifications should be considered in higher‐severity cases (Table 6). Thus, if grade ≥3 peripheral neuropathy occurs following administration of lorlatinib, treatment should be temporarily discontinued until adverse events are reduced to grade ≤2 (if not a safety risk) or returned to baseline and the patient rechallenged at a reduced lorlatinib dose. Treatment with vitamin B1 and vitamin B6 and medications for pain associated with peripheral neuropathy (e.g., gabapentin or pregabalin) may also provide symptom relief in some cases. For carpal tunnel syndrome, the use of a night splint has been found to provide improvement in some patients. However, in more significant cases, patients may opt for release surgery.
In addition, peripheral neuropathy may be associated with peripheral edema (primarily of the upper extremities). In such instances, treatment with diuretics might improve peripheral neuropathy symptoms as well as reduce edema.
Increase in Lipase.
In the pooled safety analysis group, an increase in lipase was reported as an adverse event in 10.8% of patients treated with lorlatinib. Of 290 evaluable patients, lipase increases were reported as a laboratory abnormality (any grade) in 77 patients (26.6%), most of which were grade ≤2 (17.6%). Pancreatitis was rare and reported in 1 of 295 patients; however, other potential confounding factors were noted with this patient. Lipase elevations were often sporadic throughout the course of treatment and episodic in some patients. Based on laboratory data, increases in lipase generally occurred within 2 months of starting lorlatinib treatment (median time to onset, 62 days [range, 7–619]). Distributions of lipase levels across treatment cycles are shown in supplemental online Figure 7.
Dose interruptions and dose reductions due to lipase elevations were reported in nine (3.1%) and three (1.0%) patients, respectively. General guidance on dose modifications should be applied to grade ≥3 adverse events (Table 6).
Gastrointestinal Effects.
Gastrointestinal side effects such as constipation and diarrhea occurred in 14.2% and 17.6% of patients treated with lorlatinib 100 mg QD, respectively, and were predominantly mild in severity. No grade ≥3 constipation adverse events were reported, and only two patients (0.7%) experienced grade 3 diarrhea. Dose modifications were rarely required, and no patients discontinued lorlatinib as a result of gastrointestinal adverse events. General guidance should be followed in more severe cases (Table 6), and constipation and diarrhea can be managed with standard medical therapy.
AV Block and Other Electrocardiogram Findings.
In a study of healthy volunteers who were administered lorlatinib, analysis of electrocardiogram (ECG) data revealed some evidence of PR prolongation, although no adverse events associated with AV block were reported. However, of 295 patients treated with lorlatinib, asymptomatic first‐degree AV block (grade 1 in severity) was reported in 2 patients (0.7%). Complete AV block was reported for one patient (0.3%; grade 3) and led to temporary discontinuation from treatment, although it should be noted that this patient had pre‐existing second‐degree AV block. There were no other dose modifications or discontinuations as a result of AV block.
ECG QT prolongation was reported in 19 patients (6.4%), most of which were grade ≤2, although one event was grade 3 in severity and required temporary discontinuation. None of these events resulted in permanent treatment discontinuation. Before initiating treatment with lorlatinib, patients should be informed of the potential risks of AV block and advised to contact their health care provider immediately if they experience any new chest pain or discomfort, changes in heartbeat, palpitations, dizziness, lightheadedness, or fainting, and changes in or new use of heart or blood pressure medication. For patients with pre‐existing PR prolongation, ECG monitoring should be conducted throughout the course of treatment (supplemental online Table 2). For patients who develop AV block, dose modification may be required depending upon the degree of AV block and whether the patient exhibits symptoms. Dose modification guidelines for patients with AV block are described in supplemental online Table 4.
Summary
Lorlatinib has an unique safety profile, distinct from those of other ALK TKIs, and is generally well tolerated with a low incidence of permanent discontinuations due to adverse reactions (n = 6 [2.0%]). Temporary dose interruptions and dose reductions associated with adverse reactions were reported in 21.7% and 19.7% of patients, respectively.
Hyperlipidemia is the most common adverse drug reaction associated with lorlatinib and is largely manageable with lipid‐lowering therapy. In our analysis, most patients (81.0%) received at least one lipid‐lowering agent, and 22.1% and 30.8% required two or more agents for hypercholesterolemia and hypertriglyceridemia, respectively. Therefore, patients should be informed that they may experience these adverse events and be monitored throughout treatment. The CNS effects associated with lorlatinib generally improved or resolved following dose modifications. Patients, and importantly patients’ family members and/or caregivers, should be advised to alert health care providers if any CNS‐related symptoms arise. The proactive counseling of patients on how to manage adverse events, as well as pre‐emptive monitoring and treatment, is an integral component of patient care when initiating lorlatinib or any new treatment regimen. Other adverse drug reactions with lorlatinib are primarily mild to moderate in severity and can also be effectively managed with dose modifications and/or standard supportive medical therapy.
Conclusion
Lorlatinib has a unique safety profile to be considered when prescribed for the treatment of patients with ALK‐positive, metastatic, advanced NSCLC previously treated with a second‐generation ALK TKI. A phase III study comparing lorlatinib with crizotinib in treatment‐naïve, ALK‐positive, advanced NSCLC is underway.
See http://www.TheOncologist.com for supplemental material available online.
Acknowledgments
Medical writing support was provided by Jade Drummond and Brian Szente of inScience Communications, Springer Healthcare (Chester, U.K., and Philadelphia, PA), and was funded by Pfizer Inc. We thank Leonard P. James, formerly of Pfizer Inc., for insightful discussion and review of the manuscript.
Author Contributions
Conception/design: Todd M. Bauer, Enriqueta Felip, Benjamin J. Solomon, Holger Thurm, Gerson Peltz, Marc D. Chioda, Alice T. Shaw
Provision of study material or patients: Todd M. Bauer, Enriqueta Felip, Benjamin J. Solomon, Alice T. Shaw
Collection and/or assembly of data: Todd M. Bauer, Enriqueta Felip, Benjamin J. Solomon, Holger Thurm, Gerson Peltz, Marc D. Chioda, Alice T. Shaw
Data analysis and interpretation: Todd M. Bauer, Enriqueta Felip, Benjamin J. Solomon, Holger Thurm, Gerson Peltz, Marc D. Chioda, Alice T. Shaw
Manuscript writing: Todd M. Bauer, Enriqueta Felip, Benjamin J. Solomon, Holger Thurm, Gerson Peltz, Marc D. Chioda, Alice T. Shaw
Final approval of manuscript: Todd M. Bauer, Enriqueta Felip, Benjamin J. Solomon, Holger Thurm, Gerson Peltz, Marc D. Chioda, Alice T. Shaw
Disclosures
Todd M. Bauer: Guardant Health, Ignyta, Loxo, Moderna Therapeutics, Pfizer (C/A); Enriqueta Felip: Celgene, Eli Lilly, Guardant Health, Takeda, AstraZeneca, Boehringer Ingelheim, Bristol‐Myers Squibb, Merck Sharp & Dohme, Novartis, Pfizer, Roche (C/A), AstraZeneca, Boehringer Ingelheim, Bristol‐Myers Squibb, Merck Sharp & Dohme, Novartis, Pfizer, Roche (H); Benjamin J. Solomon: AstraZeneca, Eisai (C/A), AstraZeneca, Bristol‐Myers Squibb, Merck, Novartis, Roche (other: travel expenses), Biodesix (VeriStrat) (IP), AstraZeneca, Bristol‐Myers Squibb (H); Holger Thurm: Pfizer (E, OI); Gerson Peltz: Pfizer (E, OI); Marc D. Chioda: Pfizer (E, OI); Alice T. Shaw: Ariad, Blueprint Medicines, Daiichi Sankyo, EMD Serono, Genentech, Ignyta, KSQ Therapeutics, Natera, Novartis, Pfizer, Roche and Taiho Pharmaceutical (C/A), Novartis, Pfizer, Roche/Genentech (RF), Novartis, Pfizer, Roche/Genentech, Foundation Medicine (H).
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1.Koivunen JP, Mermel C, Zejnullahu K et al. EML4‐ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res 2008;14:4275–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kris MG, Johnson BE, Berry LD et al. Using multiplexed assays of oncogenic drivers in lung cancers to select targeted drugs. JAMA 2014;311:1998–2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takeuchi K, Choi YL, Soda M et al. Multiplex reverse transcription‐PCR screening for EML4‐ALK fusion transcripts. Clin Cancer Res 2008;14:6618–6624. [DOI] [PubMed] [Google Scholar]
- 4.Takeuchi K, Soda M, Togashi Y et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med 2012;18:378–381. [DOI] [PubMed] [Google Scholar]
- 5.Solomon BJ, Mok T, Kim DW et al. First‐line crizotinib versus chemotherapy in ALK‐positive lung cancer. N Engl J Med 2014;371:2167–2177. [DOI] [PubMed] [Google Scholar]
- 6.Kim DW, Tiseo M, Ahn MJ et al. Brigatinib in patients with crizotinib‐refractory anaplastic lymphoma kinase‐positive non‐small‐cell lung cancer: A randomized, multicenter phase II trial. J Clin Oncol 2017;35:2490–2498. [DOI] [PubMed] [Google Scholar]
- 7.Peters S, Camidge DR, Shaw AT et al. Alectinib versus crizotinib in untreated ALK‐positive non‐small‐cell lung cancer. N Engl J Med 2017;377:829–838. [DOI] [PubMed] [Google Scholar]
- 8.Soria JC, Tan DSW, Chiari R, et al. First‐line ceritinib versus platinum‐based chemotherapy in advanced ALK‐rearranged non‐small‐cell lung cancer (ASCEND‐4): A randomised, open‐label, phase 3 study. Lancet 2017;389:917–929. [DOI] [PubMed] [Google Scholar]
- 9.Katayama R, Shaw AT, Khan TM et al. Mechanisms of acquired crizotinib resistance in ALK‐rearranged lung cancers. Sci Transl Med 2012;4:120ra117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shaw AT, Gandhi L, Gadgeel S et al. Alectinib in ALK‐positive, crizotinib‐resistant, non‐small‐cell lung cancer: A single‐group, multicentre, phase 2 trial. Lancet Oncol 2016;17:234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shaw AT, Kim DW, Mehra R et al. Ceritinib in ALK‐rearranged non‐small‐cell lung cancer. N Engl J Med 2014;370:1189–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gainor JF, Dardaei L, Yoda S et al. Molecular mechanisms of resistance to first‐ and second‐generation ALK inhibitors in ALK‐rearranged lung cancer. Cancer Discov 2016;6:1118–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Novello S, Mazieres J, Oh IJ et al. Primary results from the phase III ALUR study of alectinib versus chemotherapy in previously treated ALK+ non‐small‐cell lung cancer (NSCLC). Ann Oncol 2017;28(suppl 5):1299O_PRa. [Google Scholar]
- 14.Shaw AT, Kim DW, Nakagawa K et al. Crizotinib versus chemotherapy in advanced ALK‐positive lung cancer. N Engl J Med 2013;368:2385–2394. [DOI] [PubMed] [Google Scholar]
- 15.Shaw AT, Kim TM, Crino L et al. Ceritinib versus chemotherapy in patients with ALK‐rearranged non‐small‐cell lung cancer previously given chemotherapy and crizotinib (ASCEND‐5): A randomised, controlled, open‐label, phase 3 trial. Lancet Oncol 2017;18:874–886. [DOI] [PubMed] [Google Scholar]
- 16.Johnson TW, Richardson PF, Bailey S et al. Discovery of (10R)‐7‐amino‐12‐fluoro‐2,10,16‐trimethyl‐15‐oxo‐10,15,16,17‐tetrahydro‐2h‐8,4‐(m etheno)pyrazolo[4,3‐h][2,5,11]‐benzoxadiazacyclotetradecine‐3‐carbonitrile (PF‐06463922), a macrocyclic inhibitor of anaplastic lymphoma kinase (ALK) and c‐ros oncogene 1 (ROS1) with preclinical brain exposure and broad‐spectrum potency against ALK‐resistant mutations. J Med Chem 2014;57:4720–4744. [DOI] [PubMed] [Google Scholar]
- 17.Schinkel AH. P‐glycoprotein, a gatekeeper in the blood‐brain barrier. Adv Drug Deliv Rev 1999;36:179‐194. [DOI] [PubMed] [Google Scholar]
- 18.Zou HY, Friboulet L, Kodack DP et al. PF‐06463922, an ALK/ROS1 inhibitor, overcomes resistance to first and second generation ALK inhibitors in preclinical models. Cancer Cell 2015;28:70–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shaw AT, Felip E, Bauer TM et al. Lorlatinib in non‐small‐cell lung cancer with ALK or ROS1 rearrangement: An international, multicentre, open‐label, single‐arm first‐in‐man phase 1 trial. Lancet Oncol 2017;18:1590–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Solomon BJ, Shaw AT, Ou S‐HI et al. Phase 2 study of lorlatinib in patients with advanced ALK+/ROS1+ non‐small‐cell lung cancer. Paper presented at: International Association for the Study of Lung Cancer 18th World Conference on Lung Cancer; October 15–18, 2017; Yokohama, Japan.
- 21.Solomon BJ, Besse B, Bauer TM et al. Lorlatinib in patients with ALK‐positive non‐small‐cell lung cancer: Results from a global phase 2 study. Lancet Oncol 2018;19:1654–1667. [DOI] [PubMed] [Google Scholar]
- 22.Neuvonen PJ, Niemi M, Backman JT. Drug interactions with lipid‐lowering drugs: Mechanisms and clinical relevance. Clin Pharmacol Ther 2006;80:565–581. [DOI] [PubMed] [Google Scholar]
- 23.Reiner Z, Catapano AL, De Backer G et al. ESC/EAS guidelines for the management of dyslipidaemias: The task force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Eur Heart J 2011;32:1769–1818. [DOI] [PubMed] [Google Scholar]
- 24.Niaspan (niacin extended‐release): U.S. prescribing information. North Chicago, IL: Abbvie Inc., 2016. [Google Scholar]
- 25.Fenoglide (fenofibrate): U.S. prescribing information. San Diego, CA: Santarus, Inc; 2012. [Google Scholar]
- 26.Lovaza (omega‐3‐acid ethyl esters): U.S. prescribing information. Research Triangle Park, NC: GlaxoSmithKline, 2015. [Google Scholar]
- 27.Lopid (gemfibrozil): Summary of product characteristics. Kent, U.K.: Pfizer Ltd, 2016. [Google Scholar]