

Considering the limitations in understanding of how preclinical research and trial design influences clinical trial success, this article assesses factors that potentially may predict the success of phase II pediatric oncology clinical trials .
Keywords: Phase II, Pediatric, Oncology, Success, Predictors
Abstract
Background.
There are limited data to predict which novel childhood cancer therapies are likely to be successful. To help rectify this, we sought to identify the factors that impact the success of phase II clinical trials for pediatric malignancies.
Materials and Methods.
We examined the impact of 24 preclinical and trial design variables for their influence on 132 phase II pediatric oncology clinical trials. Success was determined by an objective assessment of patient response, with data analyzed using Fisher's exact test, Pearson's chi‐square test, and logistic regression models.
Results.
Trials that evaluated patients with a single histological cancer type were more successful than those that assessed multiple different cancer types (68% vs. 47%, 27%, and 17% for 1, 2–3, 4–7, and 8+; p < .005). Trials on liquid or extracranial solid tumors were more successful than central nervous system or combined trials (70%, 60%, 38%, and 24%; p < .005), and trials of combination therapies were more successful than single agents (71% vs. 28%; p < .005). Trials that added therapies to standard treatment backbones were more successful than trials testing novel therapies alone or those that incorporated novel agents (p < .005), and trials initiated based on the results of adult studies were less likely to succeed (p < .05). For 61% of trials (80/132), we were unable to locate any relevant preclinical findings to support the trial. When preclinical studies were carried out (52/132), there was no evidence that the conduct of any preclinical experiments made the trial more likely to succeed (p < .005).
Conclusion.
Phase II pediatric oncology clinical trials that examine a single cancer type and use combination therapies have the highest possibility of clinical success. Trials building upon a standard treatment regimen were also more successful. The conduct of preclinical experiments did not improve clinical success, emphasizing the need for a better understanding of the translational relevance of current preclinical testing paradigms.
Implications for Practice.
To improve the clinical outcomes of phase II childhood cancer trials, this study identified factors impacting clinical success. These results have the potential to impact not only the design of future clinical trials but also the assessment of preclinical studies moving forward. This work found that trials on one histological cancer type and trials testing combination therapies had the highest possibility of success. Incorporation of novel therapies into standard treatment backbones led to higher success rates than testing novel therapies alone. This study found that most trials had no preclinical evidence to support initiation, and even when preclinical studies were available, they did not result in improved success.
Introduction
The survival rates for childhood cancer have improved dramatically over the last 50 years, with approximately 80% of children now cured with standard therapy [1]. There has, however, been a recent plateau in the rate of increase in cure rates. This has occurred at the same time hundreds of novel anticancer compounds have been developed. The relative rarity of childhood cancer, and high cure rates, mean only a limited number of these novel therapies can be effectively tested in patients with relapsed or refractory disease [2]. Unfortunately, the majority of these novel therapeutic strategies fail in early‐phase trials and do not get incorporated into standard treatment protocols [3], [4]. Although every clinical trial increases the knowledge base, it is important to develop strategies to help maximize the success rate of future studies.
The ultimate goal of cancer research is the successful translation of preclinical findings into clinical applications [4]. However, anecdotally, there are conflicting data on the utility of preclinical studies to predict clinical success [5]. One of the earliest and most active therapies for pediatric leukemia, VAMP (vincristine, amethopterin, mercaptopurine, and prednisone), was not shown to be effective in in vivo models [6]. Despite this, the first 13 patients with leukemia treated with VAMP rapidly entered complete remission [7]. The VAMP protocol has since been successfully applied to other pediatric cancers with the same significant improvements in survival seen [7], [8], [9]. Conversely, cytarabine was shown to inhibit the therapeutic target in Ewing's sarcoma, with compelling preclinical activity both in vitro and in vivo [10], yet no activity was observed in a subsequent phase II trial [11]. Despite the specificity of cytarabine toward EWS‐FLI1 [10], subsequent preclinical studies in an extended in vivo panel inferred a lack of activity in this cancer type [12]. Although it has been suggested that more rigorous preclinical testing may have predicted this clinical outcome prior to trial initiation [11], there are no objective data to help guide preclinical researchers and trialists as to how much preclinical evidence is needed before a trial should be initiated, which preclinical studies best predict clinical success, or the translational relevance of current preclinical testing paradigms [5], [13], [14].
Similarly, there may be clinical factors that predict which trial may be successful. Phase I response rates have recently been found to significantly correlate with subsequent phase II response [15]. However, it is unknown whether a trial supported by prior adult or pediatric early‐phase studies will have a higher possibility of attaining clinical success compared with trials supported by preclinical research. Likewise, there is little understanding of the influence of trial design variables including single agents versus combination therapies, the number of cancer types included, or the type of cancer assessed [16].
Given the limitations in our understanding of how preclinical research and trial design influences trial success, we assessed potential factors that may predict the success of phase II pediatric oncology clinical trials. We systematically identified published phase II pediatric oncology clinical trials, sought and analyzed the preclinical data supporting each trial, and assessed each trial to measure the impact of 24 preclinical research and trial design parameters on success.
Materials and Methods
Study Design
We conducted database searches of PubMed and Google Scholar using the search terms “Phase 2,” “cancer,” “pediatric,” “tumor,” and “clinical trial” and their spelling variants for phase II pediatric oncology clinical trials. Manuscripts meeting the inclusion criteria were reviewed in full (n = 169). Thirty‐seven trials were excluded because of insufficient information provided to allow an objective assessment of response rates, or because the trial assessed the activity of a chemo‐protectant rather than antitumor activity.
Variable Assessment
For each trial that met the inclusion criteria (n = 132), we assessed 24 preclinical and trial design variables for their impact on trial success (supplemental online Table 1).
To identify relevant preclinical studies, we obtained all references to preclinical findings within each phase II manuscript. We also conducted PubMed and Google Scholar searches to identify preclinical, phase I, and clinical manuscripts relevant to each of the 132 trials. To find these manuscripts, we searched for each individual agent and/or combination therapy assessed in the trial against each cancer type enrolled in the trial along with the search terms “in vitro” or “in vivo” or “Phase 1.” To account for publication delays [17], database searches were carried out until December 31, 2015.
Preclinical data were classified as “relevant” to the clinical trial if the preclinical studies were performed on models representing the same tumor type(s) included in the trial using the same therapeutic drug(s) as the trial. To limit the number of references, we have not cited these publications. Each trial manuscript and all acquired publications were then reviewed and relevant information extracted to answer each variable. Where available, we also reviewed the trial protocol for references, unpublished preclinical data, and trial design parameters.
Establishing Success
Each trial was then categorized as “successful” or “unsuccessful” based on an independent assessment of response. For extracranial solid and central nervous system (CNS) trials, an overall response rate (complete + partial response) ≥20% was defined as successful, based on the 19.6% response rate observed in Pediatric Oncology Group clinical trials [18]. For liquid tumor trials, a response rate ≥40% was defined as successful, based on the Therapeutic Advances in Childhood Leukemia guidelines reported by Ko et al. [19]. Secondary measures of success included whether the trial investigators (the authors) concluded the trial was successful and whether the phase II results led to the conduct of a phase III trial.
Statistical Analysis
Upon completion of data collection, each categorical variable was transformed to a numerical format and examined independently to determine its individual influence on successful trial outcome. Statistical analysis was performed using IBM‐SPSS Statistics version‐22 (IBM, Armonk, NY). Pearson's chi‐square and Fisher's exact tests were used to determine the individual association between each variable and successful trial outcome, whereas multivariate logistic regression models were used to determine the grouped influence on trial outcome. All variables with a p < .2 were included in the regression models. p values and 95% confidence intervals were extracted for each identified predictor.
Results
Using a search of the keywords, we identified 33,641 manuscripts (Fig. 1). Of these, 5,921 were outside the inclusion timeframe, not performed on humans, or not available in English. We then excluded trials of noncancer diagnoses, trials on adult patients, and manuscripts that discussed the findings of phase I or III results alone. A detailed review of the eligible manuscripts (n = 169) led to the exclusion of 37 trials that contained insufficient data for an objective assessment of response rates or that assessed a chemo‐protectant.
Figure 1.

Consolidated Standards of Reporting Trials (CONSORT) diagram depicting the process of pediatric oncology trial identification.
In total, 132 trials met all criteria for inclusion in this study (supplemental online data). The clinical trial characteristics of each trial are summarized in Table 1. Sixty‐nine trials (52%) included patients with a single histological cancer type only, whereas 19 (14%), 26 (20%), and 18 trials (14%) included 2–3, 4–7, and ≥8 cancer diagnoses, respectively. Patients with liquid tumors were included in 23 trials (17%), extracranial solid tumors in 50 trials (38%), and CNS tumors in 42 trials (32%). Seventeen trials (13%) included patients from at least two of these tumor categories. Sixty‐four trials (48%) studied the activity of single agents compared with 68 trials (52%) that examined combination therapies (two or more agents). Antitumor activity was demonstrated in a corresponding phase I trial in 71 cases (54%). Seventeen trials tested targeted therapies (13%), and the remainder tested conventional cytotoxic agents. Only three trials (2%) selected patients based on the presence or absence of a biomarker. Ninety‐three trials (71%) studied the activity of a novel agent(s), whereas 23 trials (17%) tested a modification of an established standard therapy through the addition of one or more agents. Sixteen trials (12%) examined the activity of an established drug or regimen through a dose or schedule change.
Table 1. Characteristics of the 132 phase II pediatric oncology trials.
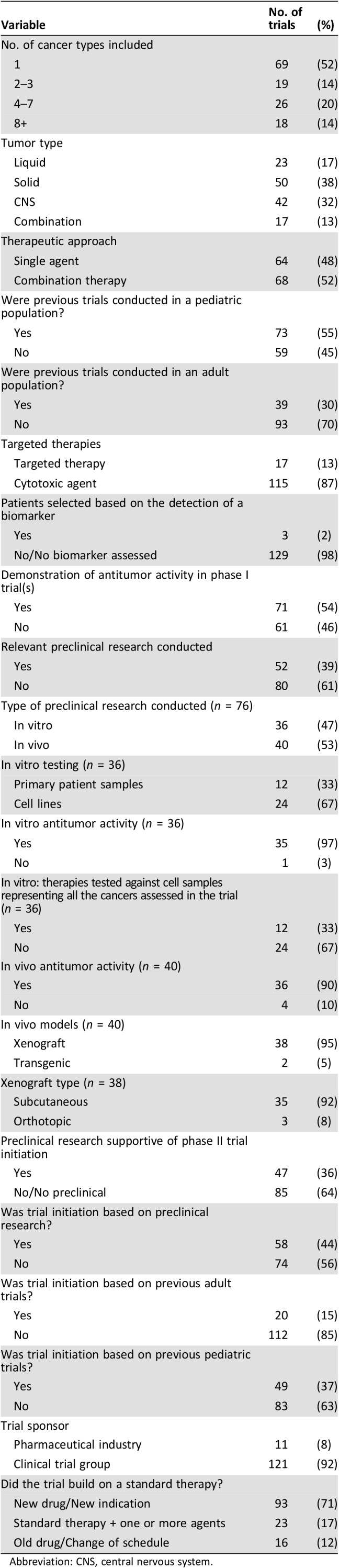
Abbreviation: CNS, central nervous system.
We found 52 trials (39%) that had preclinical research available to support trial initiation. For 36 of these trials, the in vitro studies used the same therapy or therapies against one or more of the same target cancer(s) as the phase II trial. In 12 of these cases, the in vitro experiments used primary patient‐derived samples; the remainder used established tumor cell lines. Of the 36 studies with in vitro data, in 35 cases, the treatment regimen was shown to be active against one or more of the tumor type(s) tested in the phase II trial. In the remaining case, the treatment was inactive in vitro. In 12 of the 36 trials (33%), the treatment regimen included in the trial had been tested against cell samples that represented all the tumor types included in the phase II trial.
Relevant in vivo studies were identified for 40 trials. In 36 cases, the treatment was shown to have activity in vivo. In four cases, the treatment regimen was concluded to be inactive against at minimum one of the same tumor types later assessed in the phase II trial. The types of in vivo models used included xenografts in 38 trials (28%) and transgenic models in 2 trials (2%). Subcutaneous xenografts were used for 35 trials (26%) compared with orthotopic models in 3 trials (2%).
Trial Justification
We examined the reasons stated by the investigators for initiating each of the trials. In total, 90 of the trial manuscripts (67%) referred to preclinical data to support trial initiation. Of these, 47 manuscripts (52%) discussed preclinical data that either used different drugs than those used in the trial or discussed preclinical studies using the same drug(s) but on different tumor types than those included in the trial.
Trial investigators stated that their decision to launch the trial was based on preclinical data (published or unpublished in‐house testing) in 58 cases (44%) and/or the results of previous pediatric trials and/or the results of previous adult trials. In total, 73 trials (55%) used therapies that had previously been examined in other early‐phase pediatric clinical trials (e.g., phase I or smaller phase II trials in subgroups of patients); however, only 49 trials (37%) stated that the trial was initiated based on the conduct of prior pediatric trials. We found that 39 trials (30%) involved therapies previously tested in adult patients with the same cancer type(s); however, only 20 trials (15%) stated that their decision to launch was based on supporting data in an adult population.
Trial Success Rates
Based on an objective assessment of response rates, 66/132 trials (50%) were classed as successful. Trial investigators were significantly more likely to rate the trial as being successful compared with the independent assessment of response rates (63% vs. 50%: p = .037). Conversely, eight trials (6%) appeared successful based on response rate but were deemed unsuccessful by the investigators because of significant toxicities or no observed benefit above standard therapy. Only 8/132 trials (6%) had progressed to phase III studies at the time of analysis, making statistical analysis of this measure not feasible.
Predictors of Success
Trials that included patients with a single histological cancer type were significantly more likely to achieve success than trials examining multiple tumor types (Table 2; Fig. 2A). Of the 69 trials that examined patients with a single cancer type, 68% were successful. As the number of cancers included in each trial increased to 2–3, 4–7, and 8+, the success rate decreased to 47%, 27%, and 17%, respectively (p < .0005). The type of cancer assessed also significantly influenced success. Trials in patients with liquid tumors had the highest success rate at 70%, compared with 60%, 38%, and 24% for trials of extracranial solid tumors, CNS tumors, or combined trials (p < .0005; Fig. 2B). The type of therapeutic approach used also significantly influenced success rates. The 68 trials that tested combination therapies had a higher likelihood of achieving success (71%) compared with single‐agent studies with a success rate of 28% (p < .0005; Fig. 2C). For each of these variables, similar results were observed when the investigators’ assessment of success was used as the marker of success (supplemental online Table 3).
Table 2. The influence of clinical trial design characteristics on pediatric oncology clinical trial success rates.

Fisher's exact or Pearson's chi‐square tests.
Abbreviation: CNS, central nervous system.
Figure 2.

The influence of trial design factors on clinical success. Success rates were assessed according to the number of cancer types included in each trial (A; p < .0005), the category of tumors assessed in each trial (B; p < .0005), the therapeutic approach used, that is, single agent versus combination therapy (C; p < .0005), and the type of therapy used (D; ** = p < .005). Statistical analysis was carried out using Fisher's exact or Pearson's chi‐square tests.
Trials sponsored by a pharmaceutical company were no more successful than trials sponsored by cooperative groups; however, the investigators on pharmaceutical‐sponsored trials were more likely to conclude that the trial was successful (91% vs. 60%; p = .05). There was no significant difference in trial success when prior relevant pediatric trials had been conducted (Table 2); however, when prior adult trials had been conducted using the same agent(s) on the same cancer type(s), the pediatric trial was significantly less likely to be successful (36% vs. 56%; p = .036).
For 52/132 trials (39%), relevant in vitro and/or in vivo had been conducted, and in 47/52 cases (90%), the preclinical results were positive. However, we found that trials with supportive preclinical results were significantly less likely to be successful than those with unsupportive or no preclinical findings (p < .001; Table 3). To further understand this finding, we examined multiple preclinical factors but could not identify any preclinical experiments that positively influenced trial success (Table 3). For example, experiments on primary patient‐derived cultures were no more predictive of success than experiments on established cells lines. Similarly, the conduct of in vivo studies in any model type did not influence trial success rates. The reasons stated by investigators for initiating the trial were also assessed for their influence on success rates. Trials in which investigators used preclinical data or adult trials as their rationale were less likely to succeed (supplemental online Table 2).
Table 3. The influence of preclinical research characteristics on pediatric oncology phase II trial success rates.

Fisher's exact or Pearson's chi‐square tests.
To understand why trials with preclinical data were less likely to succeed, we examined the relationship between the types of therapies tested and trial success. We found that trials that examined the activity of a completely novel therapeutic regimen were significantly less likely to be successful compared with trials that added treatments to a standard therapy backbone (40% vs. 83%; p < .0006; Fig. 2D). Importantly, trials of novel regimens were 5 times more likely to have preclinical data prior to initiation compared with trials that built on a standard therapy or trials that did not incorporate novel agents (49% vs. 9% and 12%, respectively). We also found that 67% of novel‐agent trials tested the activity of single agents, compared with 12% for trials of old drugs. Given the lower clinical success rates for single novel‐agent studies (27%) versus combination studies (65%; p < .0005), this may partly explain the low success rate of both novel‐agent trials and trials with supportive preclinical data. However, trials of novel agents that had supportive preclinical findings were still less likely to be successful (28%) compared with those with no preclinical or negative findings (52%; p = .02). To further understand these findings, we looked in depth into the preclinical studies for each of the trials that had supportive preclinical data but were unsuccessful in trial (n = 35). We found that eight of these trials that assessed novel agents had preclinical indicators that may have suggested the trial was unlikely to succeed. In six cases, we found that the drug(s) used in the trial had little to no efficacy in vitro or in vivo against at least one of the cancer types included in the trial [20], [21], [22], [23], [24], [25]. In one trial, in vivo doses resulted in high mouse mortality rates [26], whereas for two trials, the preclinical studies concluded that the treatment was less effective than existing standard therapies [27], [28].
Multivariate Analysis
Using logistic regression models, four clinical variables were predictive of trial success (Table 4): The number of cancers assessed during the trial influenced success, with an identified decrease in the likelihood of achieving success as the number of cancer types assessed increased; the type of therapeutic approach (trials using combination therapies were more successful than single‐agent trials); trials that did not base initiation on prior clinical data in adults; and investigators using previous pediatric trials as their rationale for trial initiation.
Table 4. Logistic regression analysis for the variables predictive of phase II success.
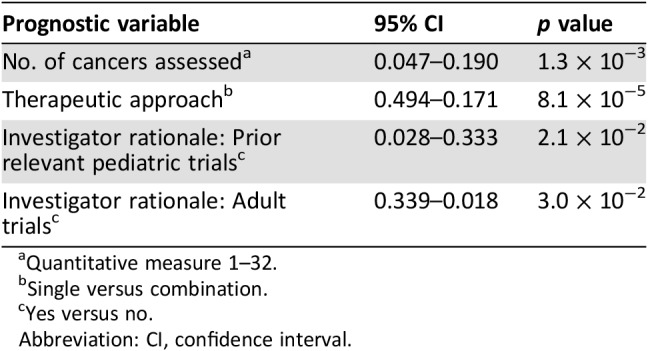
Quantitative measure 1–32.
Single versus combination.
Yes versus no.
Abbreviation: CI, confidence interval.
Discussion
Although the majority of children with cancer are cured, there remains an urgent need to develop novel treatment strategies for patients with tumors refractory to standard therapies. There are, however, enormous challenges around conducting trials in this patient population, given their low numbers, heterogeneity of tumor types, high clinical trial costs, lengthy patient accrual timeframes, and the reluctance of pharmaceutical companies to support pediatric trials [16], [29], [30]. Thus, despite the development of hundreds of new anticancer agents, only a small number can be effectively tested in children with cancer. Although many of these trials are successful, and cure rates continue to increase, a large proportion of phase II trials remain unsuccessful, showing insufficient antitumor activity, excessive toxicity, or no benefit above current treatment strategies [31], [32].
To our knowledge, this is the first study to examine the factors that predict the success of phase II pediatric oncology trials, and until now, much has remained unknown. What preclinical information is needed before starting a trial? Do different preclinical models better predict clinical activity? How should phase II trials be designed to maximize the chance of success? Should drugs be tested as single agents or in combination? The results here provide a first step in answering these questions. Our findings suggest that studies using combination therapies and focusing on a limited number of tumor types were significantly more likely to succeed.
Given that preclinical studies of drug activity are meant to guide the inclusion of novel treatment strategies in clinical trials [4], it is concerning that for the majority of trials initiated (61%), we were unable to locate any preclinical data to support the trial. Surprisingly, even when supporting preclinical findings were available, we found no positive association between the conduct of preclinical experiments and trial success. Although the reasons for this remain unclear, the results emphasize the pressing need for a better understanding of the reliability and clinical relevance of preclinical research. We identified several cases in which even when there was supportive clinical data, there were indications that the clinical trials were unlikely to succeed because of, for example, tumor types being included where little to no activity was seen preclinically.
It is also possible that a lack of control for tumor heterogeneity and limitations of molecular subtyping may have influenced the reliability of preclinical data in the past. Preclinical studies sometimes use poorly characterized xenograft models [33]. Another potential confounder is the inability to model the impact of immune responses or mimic the human condition, as preclinical testing usually occurs in immunocompromised animals. Doses used in vitro, and sometimes in vivo, are often not achievable in the clinic, or drugs may not penetrate into the tumor or past biological barriers such as the blood‐brain barrier [34], [35]. Negative preclinical results are usually not published, and it has also been proposed that up to 50% of preclinical findings cannot be reproduced [31], [36], [37]. Preclinical target validation may not be considered robust enough to warrant further investigation, or the results may not consider each potential variable that may be causing the result including off‐target effects [38].
Although our results suggest that the gap between the laboratory and the clinic was wider than expected, there are current attempts to strengthen the bridge between scientific justification and patient care [39]. It has been suggested that clinical translation can be enhanced through pharmacokinetic and pharmacodynamics studies, randomization and double blinding of preclinical analysis, the use of clinically relevant drug dosages, scheduling in vivo treatment regimens to mimic clinically relevant regimes, and the application of early‐phase studies in mice directly comparable to the clinical environment [33], [39]. The aim of this new preclinical archetype is to increase the reliability of research findings and ease the transition of novel therapies from the laboratory to the clinic. Whether this will increase the successful translation of laboratory experiments to the clinic will hopefully be established in time.
An additional concern may be the relevance of prior clinical trials to support phase II initiation in a pediatric population. The rationale for trial initiation is often supported by results of prior early‐phase studies in both adult and pediatric populations. Given that the approval for a new therapy in a pediatric population generally occurs following approval for its use in adults [29], prior adult trials are often used as the rationale for trial initiation in pediatric patients. Our study, however, showed that trials initiated based on activity data from adult trials were less likely to succeed. This may be because most pediatric malignancies are biologically distinct from adult cancers, even if histologically they bear similarities [29], [40]. For example, glioblastoma (GBM) in adults and children is histologically identical; however, molecular aberrations found in adult tumors (e.g., IDH1) are rarely found in childhood GBM, and mutations identified in pediatric GBM (e.g., H3‐K27 M mutations) are rarely found in adult tumors [40], [41]. In agreement, logistic regression models identified both prior activity in a pediatric population and not using proof of principal in an adult population as rationale for trial initiation as predictors of clinical success.
Overall, clinical trial design factors were the only variables that best predicted clinical success. The number of cancer types examined was one of the most important factors identified. Although widening patient enrollment to include different cancer types may allow for more rapid patient enrollment, our data suggest this reduces response rates and the possibility of attaining success. Similarly, the type of cancer examined significantly impacted success rates, with trials involving liquid or extracranial solid tumors having the highest possibility of success. CNS tumors had the lowest response rates, possibly related to poor penetration of the blood‐brain barrier [34], [35].
Combination therapies were also almost 3 times more likely to be active compared with single agents. Notably, half the studies included in this review tested drugs as single agents. The necessity to use combination therapies to improve cure rates in both child and adult cancers has been theorized for decades [15]. There may, however, be reasons trialists opt for single‐agent studies, including the reticence of pharmaceutical companies to work with competitors and difficulty in understanding which agents are active when using combination therapies. Novel trial designs, such as randomized phase II trials testing, for example, a novel agent added to a standard treatment backbone can address the latter concern while facilitating a more rapid progression to a combination therapeutic approach.
Importantly, trials that added therapies to standard treatment backbones were more successful than trials testing novel therapies alone or trials that did not incorporate novel agents. This in part may relate to the fact that trials of novel therapeutic strategies were more often tested as single agents, which we found were less active than combination therapies. This finding suggests that future trials should focus on this strategy of building on successful therapies in order to lift success rates. It is also possible that clinicians who used therapies containing standard therapies were more likely to use these treatments at earlier timepoints, when response rates may be expected to be higher.
Although these results have the potential to impact clinical success moving forward, there are limitations to this study. Most importantly, our analysis should not be interpreted to imply that preclinical research is unnecessary or that treatment strategies should enter clinical development without a solid foundation of supportive preclinical findings. The majority of trials did not have any supportive preclinical data, and when preclinical studies had been performed, the methodology was variable, often with significant differences in dose and tumor types compared with the clinical trial that was performed. Greater emphasis should be placed on preclinical studies that take into account clinically achievable concentrations and treatment schedules.
We also recognize that because of the retrospective nature of the review there are factors we could not control, or assess adequately, including the percentage of trial responders, exposure to prior therapies, and the tumor types assessed. Despite our best attempts, literature searches may not have uncovered all of the preclinical manuscripts available, and we may not have been able to account for the presence of in‐house or unpublished preclinical testing if the data were not readily available. The small number of studies that involved the use of targeted therapies, transgenic models, and biomarkers may have also influenced the ability to detect the impact of these variables on trial outcome. Therefore, the lack of influence of these variables on clinical outcome may not be a reflection of their lack of impact on clinical success but rather the small sample sizes. Further investigation into the impact of these factors on clinical outcome is warranted. Finally, only published clinical findings were reviewed. The number of trials in the timeframe assessed may be greater than that identified [17], [42]. The small number of agents progressing to phase III trials was surprising. This may reflect the difficulty in conducting large randomized phase III studies in many pediatric cancers, because of the relatively small number of patients with what is essentially a rare disease. Activity in phase II trials may be a surrogate that is being used to guide the development of treatment regimens, especially for those cancers with poor outcomes.
Conclusion
Overall, we identified key variables that significantly influence and/or enhance the success rate of phase II trials for patients with childhood cancer. The results suggest that phase II trials should focus on one cancer type and on the development of combination therapies. Preclinical studies should be focused on drug doses that are achievable in patients, and trialists should rigorously assess preclinical data to help generate the best and most effective trials.
See http://www.TheOncologist.com for supplemental material available online.
Acknowledgments
This work was supported by the Kids Cancer Alliance and the Cancer Institute NSW.
Author Contributions
Conception/design: Jennifer Byrne, Glenn M. Marshall, David S. Ziegler
Provision of study material or patients: Laura Franshaw, Maria Tsoli, David S. Ziegler
Collection and/or assembly of data: Laura Franshaw, Maria Tsoli, Jennifer Byrne, Chelsea Mayoh, Siva Sivarajasingam, Murray Norris, Glenn M. Marshall, David S. Ziegler
Data analysis and interpretation: Laura Franshaw, Maria Tsoli, Jennifer Byrne, Chelsea Mayoh, Siva Sivarajasingam, Murray Norris, Glenn M. Marshall, David S. Ziegler
Manuscript writing: Laura Franshaw, Maria Tsoli, Jennifer Byrne, Chelsea Mayoh, Siva Sivarajasingam, Murray Norris, Glenn M. Marshall, David S. Ziegler
Final approval of manuscript: Laura Franshaw, Maria Tsoli, Jennifer Byrne, Chelsea Mayoh, Siva Sivarajasingam, Murray Norris, Glenn M. Marshall, David S. Ziegler
Disclosures
The authors indicated no financial relationships.
References
- 1.Smith MA, Altekruse SF, Adamson PC et al. Declining childhood and adolescent cancer mortality. Cancer 2014;120:2497–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bond MC, Pritchard S. Understanding clinical trials in childhood cancer. Paediatr Child Health 2006;11:148–150. [PMC free article] [PubMed] [Google Scholar]
- 3.Hay M, Thomas DW, Craighead JL et al. Clinical development success rates for investigational drugs. Nat Biotechnol 2014;32:40–51. [DOI] [PubMed] [Google Scholar]
- 4.Mak IW, Evaniew N, Ghert M. Lost in translation: Animal models and clinical trials in cancer treatment. Am J Transl Res 2014;6:114–118. [PMC free article] [PubMed] [Google Scholar]
- 5.Voskoglou‐Nomikos T, Pater JL, Seymour L. Clinical predictive value of the in vitro cell line, human xenograft, and mouse allograft preclinical cancer models. Clin Cancer Res 2003;9:4227–4239. [PubMed] [Google Scholar]
- 6.Freireich EJ, Karon M, Frei E III. Quadruple combination therapy (VAMP) for acute lymphocytic leukemia of childhood. Proc Amer Assoc Cancer Res 1964:260. [Google Scholar]
- 7.Unguru Y. The successful integration of research and care: How pediatric oncology became the subspecialty in which research defines the standard of care. Pediatr Blood Cancer 2011;56:1019–1025. [DOI] [PubMed] [Google Scholar]
- 8.Donaldson SS, Hudson MM, Lamborn KR et al. VAMP and low‐dose, involved‐field radiation for children and adolescents with favorable, early‐stage Hodgkin's disease: Results of a prospective clinical trial. J Clin Oncol 2002;20:3081–3087. [DOI] [PubMed] [Google Scholar]
- 9.Kavan P, Kabickova E, Koutechy J et al. Treatment of Hodgkin's disease in children with VAMP (vinblastine, adriamycin, methotrexate, prednisone) and VEPA (vinblastine, etoposide, prednisone, adriamycin). Pediatr Hematol Oncol 1999;16:141–148. [DOI] [PubMed] [Google Scholar]
- 10.Stegmaier K, Wong JS, Ross KN et al. Signature‐based small molecule screening identifies cytosine arabinoside as an EWS/FLI modulator in Ewing sarcoma. PLoS Med 20017;4:e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DuBois SG, Krailo MD, Lessnick SL et al. Phase II study of intermediate‐dose cytarabine in patients with relapsed or refractory Ewing sarcoma: A report from the Children's Oncology Group. J Pediatr Blood Cancer 2009;52:324–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Houghton PJ, Morton CL, Kang M et al. Evaluation of cytarabine against Ewing sarcoma xenografts by the pediatric preclinical testing program. Pediatr Blood Cancer 2010;55:1224–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kerbel RS. Human tumor xenografts as predictive preclinical models for anticancer drug activity in humans: Better than commonly perceived‐but they can be improved. Cancer Biol Ther 2003;2(4 suppl 1):S134–S139. [PubMed] [Google Scholar]
- 14.Scarlett UK, Chang DC, Murtagh TJ et al. High‐throughput testing of novel‐novel combination therapies for cancer: An idea whose time has come. Cancer Discov 2016;6:956–962. [DOI] [PubMed] [Google Scholar]
- 15.Yeh JC, Huang P, Cohen KJ. Phase I and phase II objective response rates are correlated in pediatric cancer trials: An argument for better clinical trial efficiency. J Pediatr Hematol Oncol 2016;38:360–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.El‐Maraghi RH, Eisenhauer EA. Review of phase II trial designs used in studies of molecular targeted agents: Outcomes and predictors of success in phase III. J Clin Oncol 2008;26:1346–1354. [DOI] [PubMed] [Google Scholar]
- 17.Chan JK, Ueda SM, Sugiyama VE et al. Analysis of phase II studies on targeted agents and subsequent phase III trials: What are the predictors for success? J Clin Oncol 2008;26:1511–1518. [DOI] [PubMed] [Google Scholar]
- 18.Weitman S, Ochoa S, Sullivan J et al. Pediatric phase II cancer chemotherapy trials: A Pediatric Oncology Group study. J Pediatr Hematol Oncol 1997;19:187–191. [DOI] [PubMed] [Google Scholar]
- 19.Ko RH, Ji L, Barnette P et al. Outcome of patients treated for relapsed or refractory acute lymphoblastic leukemia: A Therapeutic Advances in Childhood Leukemia Consortium study. J Clin Oncol 2010;28:648–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Atkinson JM, Shelat AA, Carcaboso AM et al. An integrated in vitro and in vivo high‐throughput screen identifies treatment leads for ependymoma. Cancer Cell 2011;20:384–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kubo T, Piperdi S, Rosenblum J et al. Platelet‐derived growth factor receptor as a prognostic marker and a therapeutic target for imatinib mesylate therapy in osteosarcoma. Cancer 2008;112:2119–2129. [DOI] [PubMed] [Google Scholar]
- 22.Lefranc F, James S, Camby I et al. Combined cimetidine and temozolomide, compared with temozolomide alone: Significant increases in survival in nude mice bearing U373 human glioblastoma multiforme orthotopic xenografts. J Neurosurg 2005;102:706–714. [DOI] [PubMed] [Google Scholar]
- 23.Mathe G, Kidani Y, Noji M et al. Antitumor activity of l‐OHP in mice. Cancer Lett 1985;27:135–143. [DOI] [PubMed] [Google Scholar]
- 24.McGary EC, Weber K, Mills L et al. Inhibition of platelet‐derived growth factor‐mediated proliferation of osteosarcoma cells by the novel tyrosine kinase inhibitor STI571. Clin Cancer Res 2002;8:3584–3591. [PubMed] [Google Scholar]
- 25.Ponthan F, Lindskog M, Karnehed N et al. Evaluation of anti‐tumour effects of oral fenretinide (4‐HPR) in rats with human neuroblastoma xenografts. Oncol Rep 2003;10:1587–1592. [PubMed] [Google Scholar]
- 26.Fouladi M, Pollack IF. A phase II study of R115777 (ZARNESTRA) (NSC# 702818, IND# 58359) in children with recurrent or progressive: High grade glioma, medulloblastoma/PNET or brainstem glioma. Children's Oncology Group 2003;55. [Google Scholar]
- 27.Bodmer N, Walters D, Fuchs B. Pemetrexed, a multitargeted antifolate drug, demonstrates lower efficacy in comparison to methotrexate against osteosarcoma cell lines. Pediatr Blood Cancer 2008;50:905–908. [DOI] [PubMed] [Google Scholar]
- 28.Riccardi A, Ferlini C, Meco D et al. Antitumour activity of oxaliplatin in neuroblastoma cell lines. Eur J Cancer 1999;35:86–90. [DOI] [PubMed] [Google Scholar]
- 29.Adamson PC, Houghton PJ, Perilongo G et al. Drug discovery in paediatric oncology: Roadblocks to progress. Nat Rev Clin Oncol 2014;11:732–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Caldwell PH, Murphy SB, Butow PN et al. Clinical trials in children. Lancet 2004;364:803–811. [DOI] [PubMed] [Google Scholar]
- 31.Begley CG, Ellis LM. Drug development: Raise standards for preclinical cancer research. Nature 2012;483:531–533. [DOI] [PubMed] [Google Scholar]
- 32.Rubin EH, Gilliland DG. Drug development and clinical trials‐‐The path to an approved cancer drug. Nat Rev Clin Oncol 2012;9:215–222. [DOI] [PubMed] [Google Scholar]
- 33.Langenau DM, Sweet‐Cordero A, Wechsler‐Reya R et al. Preclinical models provide scientific justification and translational relevance for moving novel therapeutics into clinical trials for pediatric cancer. Cancer Res 2015;75:5176–5186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gabathuler R. Approaches to transport therapeutic drugs across the blood‐brain barrier to treat brain diseases. Neurobiol Dis 2010;37:48–57. [DOI] [PubMed] [Google Scholar]
- 35.Wohlfart S, Gelperina S, Kreuter J. Transport of drugs across the blood‐brain barrier by nanoparticles. J Control Release 2012;161:264–273. [DOI] [PubMed] [Google Scholar]
- 36.Freedman LP, Cockburn IM, Simcoe TS. The economics of reproducibility in preclinical research. PLoS Biol 2015;13:e1002165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prinz F, Schlange T, Asadullah K. Believe it or not: How much can we rely on published data on potential drug targets? Nat Rev Drug Discov 2011;10:712. [DOI] [PubMed] [Google Scholar]
- 38.Kaelin WG Jr. Common pitfalls in preclinical cancer target validation. Nat Rev Cancer 2017;17:425–440. [DOI] [PubMed] [Google Scholar]
- 39.Stewart E, Goshorn R, Bradley C et al. Targeting the DNA repair pathway in Ewing sarcoma. Cell Rep 2014;9:829–841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Paugh BS, Qu C, Jones C et al. Integrated molecular genetic profiling of pediatric high‐grade gliomas reveals key differences with the adult disease. J Clin Oncol 2010;28:3061–3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pollack IF, Hamilton RL, Sobol RW et al. IDH1 mutations are common in malignant gliomas arising in adolescents: A report from the Children's Oncology Group. Childs Nerv Syst 2011;27:87–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stern JM, Simes RJ. Publication bias: Evidence of delayed publication in a cohort study of clinical research projects. BMJ 1997;315:640–645. [DOI] [PMC free article] [PubMed] [Google Scholar]