

Abstract
Heterozygous missense mutations in lysyl oxidase (LOX) are associated with thoracic aortic aneurysms and dissections. To assess how LOX mutations modify protein function and lead to aortic disease, we studied the factors that influence the onset and progression of vascular aneurysms in mice bearing a Lox mutation (p.M292R) linked to aortic dilation in humans. We show that mice heterozygous for the M292R mutation did not develop aneurysmal disease unless challenged with increased hemodynamic stress. Vessel dilation was confined to the ascending aorta, although in both ascending and descending aortae, changes in vessel wall structure, smooth muscle cell number, and inflammatory cell recruitment differed between WT and mutant animals. Studies with isolated cells revealed that M292R-mutant LOX is retained in the endoplasmic reticulum and ultimately cleared through an autophagy/proteasome pathway. Because the mutant protein does not transit to the Golgi, where copper incorporation occurs, the protein is never catalytically active. These studies show that the M292R mutation results in LOX loss of function due to a secretion defect that predisposes the ascending aorta in mice (and by extension humans with similar mutations) to arterial dilation when exposed to risk factors that impart stress to the arterial wall.
Keywords: Cell Biology, Vascular Biology
Keywords: Collagens, Extracellular matrix, Mouse models
A missense mutation in lysyl oxidase results in intracellular retention of the mutant enzyme and predisposes the aorta to arterial dilation when exposed to hypertension.
Introduction
Thoracic aortic aneurysm and dissection (TAAD) is a major cardiovascular health problem that arises from mutations that alter vascular extracellular matrix (ECM), smooth muscle cell function, or growth factor signaling pathways (1). In contrast to abdominal aortic aneurysms, TAAD has a strong genetic component, with pathogenic variants typically inherited in an autosomal dominant manner (2). A single highly penetrant, pathogenic mutation is responsible for disease in only one-fourth of individuals with TAAD, indicating that other undiscovered genetic factors influence disease onset and progression. In addition to the known pathogenic gene mutations, low-penetrant risk variants interact with other genetic or environmental factors to trigger disease or to increase the risk for developing disease but are not themselves pathogenic (2). Significant risk factors for TAAD, primarily when occurring in the background of low-penetrant disease-predisposing variants, are hypertension and bicuspid aortic valve (1).
A common histopathological feature of TAAD in humans is fragmented elastin in the aortic medial region (3). Elastin is the most abundant ECM protein in the aorta, where it plays a central role in the vessel’s ability to expand and recoil in response to pulsatile blood flow. Smooth muscle cells (SMCs) synthesize and organize the protein into fenestrated sheets (lamellae) that separate smooth muscle cell layers. The secreted form of elastin (tropoelastin) undergoes extensive crosslinking outside the cell to form an elastic polymer, which is the functional form of the protein. The enzyme responsible for initiating crosslink formation is lysyl oxidase (LOX), a copper-binding amine oxidase that oxidizes lysine ε-amino groups in elastin and fibrillar collagens to aldehydes, which then spontaneously react to form covalent linkages between and within proteins (4, 5). Approximately 90% of the lysine residues in elastin undergo crosslink formation, which makes it one of the most highly crosslinked and most stable proteins in the body (4). Inactivation of the Lox gene in mice results in early postnatal death due to ruptured aortic aneurysms, emphasizing the critical role of fully crosslinked, functional elastin in maintaining aorta integrity and function (6, 7). The gene inactivation studies also show that other LOX isoforms cannot substitute for the loss of LOX in large, elastin-rich vessels.
LOX in vertebrates belongs to a family of 5 copper-dependent enzymes (LOX, LOXL1–4) that show a high degree of homology in their carboxy-terminal catalytic domain but differ in their amino-terminal sequences. LOXL2–4 form a subgroup that shares 4 amino-terminal scavenger receptor cysteine-rich domains that influence substrate binding but appear to have little role in regulating catalytic activity (8, 9). LOX and LOXL1, in contrast, are synthesized without scavenger receptor sequences but have a propeptide domain that keeps the enzymes in a latent state until proteolytically processed outside the cell by BMP1/Tolloid-like metalloproteinases (10, 11). Substrate specificity for each of the lysyl oxidases is still under investigation, but gene inactivation studies and phylogenetic analysis suggest that LOX and LOXL1 contribute to crosslinking of elastin and fibrillar collagens, whereas LOXL2–4 have different substrate specificities (12, 13). All 5 LOX isoforms are synthesized in the ER and traffic to the Golgi, where the inactive apoenzyme binds copper and forms the cofactor lysyl tyrosine quinone (LTQ); both copper and LTQ are essential active site components required for catalytic activity (14).
Mutations within LOX in families with TAAD were recently identified by whole exome sequencing (3, 15). Aortic disease in these families followed autosomal dominant inheritance, suggesting that 1 mutant LOX allele is sufficient to influence disease initiation or progression. Previously, we identified a LOX catalytic site mutation (p.Met298Arg) in a family with a history of TAAD (3). By introducing this human mutation into the homologous position in the mouse genome using CRISPR/Cas9 genome editing technology (c.857T>G encoding p.M292R; hereafter referred to as Mu), we confirmed that this amino acid change caused ruptured TAAD and perinatal death when both alleles were mutated (LoxMu/Mu) (3). In contrast to humans who developed TAAD when heterozygous for this LOX variant, aneurysms or other arterial disease were not observed in mice with a similar genotype (Lox+/Mu) unless they were challenged with hypertension. In this report, we show that the nature of the functional loss associated with the methionine-to-arginine change in LOX results from a secretion defect, where the protein is retained in the ER and degraded through an autophagy/proteasome pathway. We also show that the vessel wall alterations associated with LOX functional haploinsufficiency increase the likelihood of developing vascular disease in association with environmental factors that increase wall stress.
Results
Lox+/Mu adult animals are predisposed to aortic dilation in response to hypertension.
The generation and characterization of mice expressing the M292R mutation were described previously (3). Mice heterozygous for this Lox missense variant appear grossly normal, have normal blood pressure, and show no increased mortality through 2 years of age. Aortic diameter and circumferential wall stiffness at physiological blood pressure in Lox+/Mu animals are normal, but ascending aortic length measured from the aortic root to the brachiocephalic artery is 10% longer than in WT mice (3).
Hypertension is a known risk factor associated with aortic dilation (16). To test whether mice expressing mutant LOX are predisposed to aneurysm formation as a consequence of increased wall stress, Lox+/Mu mice were made hypertensive by subcutaneous delivery of angiotensin II (Ang II) via Alzet osmotic pumps. Pumps containing saline served as the control. Ang II infusion is a well-established model for inducing vessel wall changes in mice and other animals (17, 18). Blood pressure measurements using radiotelemetry in awake, freely moving Lox+/+ and Lox+/Mu mice showed an increase of approximately 30 mmHg in systolic pressure and 25–30 mmHg in mean blood pressure in both genotypes 7–10 days after pump implantation (data not shown), consistent with what others have found using this model (19). After 4 weeks of treatment, all animals receiving Ang II had cardiac hypertrophy, indicating altered cardiac physiology consistent with Ang II–induced hypertension (Figure 1A). However, only Lox+/Mu mice showed substantial dilation and elongation of the ascending aorta in response to Ang II; no change in aortic diameter was observed in Ang II–treated Lox+/+ mice or in Lox+/Mu mice infused with saline alone (Figure 1B). Biaxial pressure-diameter testing showed the ascending aorta in Ang II–treated Lox+/Mu animals to be approximately 25% larger than the aorta from WT or saline-treated controls at mean physiological blood pressure (Figure 1C). The shape of the pressure-diameter curves suggests that Ang II treatment did not affect aortic mechanical properties in either mutant or WT animals. Other than in the ascending aorta, no dilation or change in vessel mechanics occurred in the left common carotid artery for either Lox+/+ or Lox+/Mu animals with saline or Ang II treatment (Figure 1D), nor were dilations or areas of stenosis found in the aortic arch, descending aorta, or abdominal aorta.
Figure 1. The ascending aorta in Lox+/Mu animals undergoes aortic dilation in response to hypertension.

(A) Both Lox+/+ and Lox+/Mu animals developed significant cardiac hypertrophy following 4 weeks of treatment with 2 μg/kg/min Ang II compared with saline-treated controls. Two-way ANOVA with Tukey’s multiple-comparisons test, with the 2 variables being genotype and treatment, was used to assess differences. Data are presented as mean ± SD. ****P < 0.0001. (B) The ascending aorta in Lox+/Mu animals is longer than in Lox+/+ animals and undergoes substantial dilation following Ang II treatment. (C) Pressure-diameter measurements at intraluminal pressures ranging from 0 to 175 mmHg. The ascending aorta from Ang II–treated Lox+/Mu animals has a larger diameter over the entire range of pressures compared with Lox+/+ animals treated with Ang II or with saline controls. (D) There was no difference in the diameter or mechanical properties of the left common carotid artery in either genotype with or without Ang II treatment. Repeated measures 2-way ANOVA with Tukey’s multiple-comparisons test was used to assess differences between groups at each pressure level. Data are presented as mean ± SD. #P < 0.05, Lox+/+ with Ang II vs. Lox+/Mu with Ang II; *P < 0.05, **P < 0.01, Lox+/Mu with saline vs. Lox+/Mu with Ang II.
Ang II treatment leads to vessel wall thickening and changes in vascular cellularity.
On a structural level, Ang II treatment resulted in ascending and descending aortic wall thickening in Lox+/Mu animals, but there were no changes in elastic lamellar number or lamellar organization, quantified by measurement of medial area (Figure 2A and Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.127748DS1). Elastic lamellar fragmentation, already elevated in Lox+/Mu compared with Lox+/+ animals (3), did not increase in either genotype following Ang II treatment (data not shown). We analyzed elastin crosslinking using an ELISA assay for desmosine, a major crosslink in elastin. We found no difference in desmosine levels in aortic tissue from Lox+/+ and Lox+/Mu mice, confirming normal elastin levels and that elastin crosslinking is not affected in animals heterozygous for the Lox mutation (Supplemental Figure 1C). Hydroxyproline levels (as a measure of total collagen) trended higher in vessels from the mutant animals but were not significantly different from WT when normalized to the change in total protein (Supplemental Figure 1D). Higher protein quantities are consistent with the increase in wall thickness that occurred with Ang II treatment (Supplemental Figure 1B).
Figure 2. Ang II treatment leads to changes in vascular cellularity and infiltration of immune cells.

(A) Frozen sections of descending aorta from Lox+/+ and Lox+/Mu animals following 28 days of treatment with Ang II or saline (asterisks indicate vessel lumen; scale bars: 5 μm). Macrophages (stained green, arrowheads) were detected with an antibody to CD68, and cell nuclei were visualized with DAPI. Red is elastin autofluorescence. The vessel wall was thicker in both Lox+/+ and Lox+/Mu animals following Ang II treatment, with more DAPI+ and CD68+ cells in the adventitia of both genotypes. (B) In the medial layer, there were fewer SMCs in the aorta of WT animals treated with Ang II but more cells in Lox+/Mu aorta. Shown are the mean ± SD for cell number counts between the internal and external elastic lamina on 7 tissue sections from each group. Two-way ANOVA with Tukey’s multiple-comparisons test, with the 2 variables being genotype and treatment, was used to assess differences. Data are presented as mean ± SD. *P < 0.05, **P < 0.01. (C) Medial CD68+ cells were mostly located on the luminal side of the media in the Lox+/+ aorta but nearer the adventitia in Lox+/Mu aorta after Ang II treatment.
Changes in the number and types of cells were observed in the thickened aortic wall of the hypertensive animals. The most substantial change occurred in the adventitia of both the ascending and descending aorta, which underwent considerable expansion in the Lox+/+ and Lox+/Mu animals as a result of Ang II exposure (Figure 2A). In the medial layer, Ang II infusion led to a decrease in the number of SMCs in Lox+/+ animals but an increase in SMCs in Lox+/Mu mice (Figure 2B). There was also an increase in intralamellar area in both genotypes, although the change was most notable in Lox+/Mu mice. The nuclei of the intralamellar cells in the mutant media were rounder than cells in Lox+/+ vessels, suggesting a less polarized phenotype and noncircumferential orientation. Immunostaining for smooth muscle α-actin demonstrated uniform reactivity throughout the medial intralamellar spaces, whereas cell staining in the adventitia was heterogeneous (not shown).
Staining for the monocyte/macrophage marker CD68 in descending aorta showed an increased number of CD68+ cells in the media and adventitia of both Lox+/+ and Lox+/Mu descending aorta following Ang II treatment (Figure 2C). These results are consistent with previous studies showing that infusion of Ang II promotes macrophage accumulation within the aortic wall (20). While the adventitia showed the highest level of immune cell infiltration, macrophage accretion in the medial layer was also observed, but the spatial localization of the CD68+ cells was different for the 2 genotypes. In Ang II–treated Lox+/+ vessels, medial CD68+ cells were associated with the first 2 elastic layers nearest to the intima. Medial CD68+ cells in the aorta of Lox+/Mu animals treated with Ang II, however, were in the outer layers of the media closest to the adventitia (Figure 2C).
Evaluation of factors that adversely affect vessel wall integrity and SMC function.
An explanation for why Lox+/Mu mice are more sensitive than WT animals to Ang II infusion was explored by first checking for possible differences in Ang II receptor expression using quantitative PCR (qPCR). In ascending aorta from Lox+/Mu mice, all 3 Ang II receptors (Agtr1a, Agtr1b, and Agtr2) were detected, and there was no difference in their expression levels compared with WT controls (Supplemental Figure 2A). Thus, the potential for Ang II receptor–mediated signaling is similar in both genotypes.
Many Ang II–induced responses on SMCs are attributable to stimulation of NADPH oxidase with the subsequent generation of ROS (21, 22). We used dihydroethidium, a cell-permeable compound that interacts with superoxide, to assess oxidant levels in sections of WT and mutant ascending aorta and found no difference between the 2 genotypes (Supplemental Figure 2B). Thus, differences in levels of oxidative stress do not appear to be a factor in the Ang II susceptibility of Lox+/Mu mice.
In many models of aortic disease, the presence of proteases that degrade elastin can initiate and propagate disease by degrading the elastic fiber system in the vessel wall. We therefore investigated whether 3 elastolytic MMPs commonly linked to vascular remodeling — MMP2, MMP9, and MMP12 — were differentially expressed in mutant versus WT aorta. There were no differences in Mmp2 and Mmp9 mRNA expression levels, while expression of Mmp12 was diminished in the mutant aorta (Supplemental Figure 2C).
Active LOX is absent in LoxMu/Mu mouse embryonic fibroblast conditioned medium.
To elucidate how the M292R modification affects LOX function, we used mouse embryonic fibroblasts (MEFs) from WT, KO, and mutant animals to characterize LOX secretion and activity. Mutant cells were indistinguishable from WT cells in morphology and in their rate of proliferation (data not shown). LOX activity (Figure 3A), assessed using an Amplex Red assay, was not detected in cell layer extracts of any MEF genotype (Lox+/+, Lox+/Mu, LoxMu/Mu, and Lox–/–), which was expected, since the full-length intracellular form of LOX (pro-LOX) is an inactive zymogen. LOX enzymatic activity was readily detected in the conditioned medium from Lox+/+ MEFs and showed linear enzyme kinetics over the period of the Amplex Red assay. Conditioned medium from LoxMu/Mu MEFs, however, had no detectable activity, similar to conditioned medium from cells with inactivated Lox alleles (Lox–/–) (Figure 3B). Activity was detected in conditioned medium from Lox+/Mu cells, where the linearity and rate of substrate oxidation were comparable to those seen in Lox+/+ cells, indicating that the mutant form of the protein does not adversely affect the enzymatic activity of WT LOX and hence does not function in a dominant negative fashion.
Figure 3. LOX enzymatic activity is absent in LoxMu/Mu cells.

(A) LOX activity in cell lysates from Lox+/+, Lox+/Mu, LoxMu/Mu, and Lox–/– mouse embryonic fibroblasts measured using the Amplex Red assay over 150 minutes and expressed as relative fluorescence units (RFU). As expected, LOX activity was not detected in any of the cell extracts, as intracellular LOX is catalytically inactive until proteolytically processed outside of cells. (B) LOX activity in conditioned medium from the same cells as in A. LOX activity was detected in Lox+/+ and Lox+/Mu cells, but was absent in conditioned medium from LoxMu/Mu MEFs. Cells from Lox–/– animals served as a negative control. Repeated measures 2-way ANOVA with Tukey’s multiple-comparisons test was used to assess differences between groups at each time point. Values are mean ± SD. *P < 0.05, ****P < 0.0001, Lox+/+ vs. LoxMu/Mu.
M292R LOX is poorly secreted and is retained in the ER.
LOX is maintained as an inactive 50-kDa proenzyme through every stage of the synthetic and secretory pathway. Activation requires secretion into the extracellular space, where proteolytic removal of the N-terminal propeptide portion of the protein generates the 30-kDa active catalytic domain. To determine whether LOX activity is absent in M292R MEF conditioned medium because mutant LOX is not secreted, we performed Western blots using an anti-LOX antibody on Lox+/+, Lox+/Mu, and LoxMu/Mu MEF cell layers and conditioned medium. Immunoblotting showed increased levels of 50-kDa intracellular pro-LOX protein in cell extracts from LoxMu/Mu MEFs compared with Lox+/+ cells (Figure 4A). Conversely, there was abundant 30-kDa mature (processed) LOX in conditioned medium from WT cells but barely detectable levels in medium from LoxMu/Mu MEFs (Figure 4B). These findings indicate that the mutant protein is poorly secreted and is retained intracellularly as the proenzyme.
Figure 4. Mutant LOX is retained inside the cell.

(A) Immunoblotting of MEF lysate with an antibody to LOX shows increased amounts of pro-LOX (50 kDa) in LoxMu/Mu cells compared with Lox+/+ MEFs. (B) Immunoblots of conditioned medium demonstrate less processed LOX (30 kDa) associated with LoxMu/Mu cells, which is consistent with LOX retention within the cell. β-Actin– and Ponceau S–stained gel blots served as loading controls.
Immunofluorescence imaging with an antibody to LOX showed minimal intracellular staining in Lox+/+ cells, confirming that WT LOX is secreted efficiently (Figure 5A). Staining of LoxMu/Mu cells, however, showed abundant intracellular staining, consistent with the immunoblot results described above. The staining pattern for intracellular LOX was typical of accumulation within the ER, which was confirmed by colocalization of LOX with calnexin, an ER-resident protein (Figure 5, B and C).
Figure 5. Intracellular mutant LOX interacts with ER-resident protein calnexin.

(A) Immunofluorescence imaging of Lox+/+, Lox+/Mu, and LoxMu/Mu mouse embryonic fibroblasts (MEFs) using an anti-LOX antibody showed intracellular accumulation of mutant but not WT LOX protein. (B) Imaging using an antibody to calnexin showed a uniform distribution of intracellular calnexin staining in all cells. (C) Merged images of LOX and calnexin staining established colocalization of intracellular mutant LOX with calnexin. Scale bars: 1 μm (D) Cell lysates from Lox+/+, Lox+/Mu, LoxMu/Mu, and Lox–/– MEFs were immunoprecipitated with anti-calnexin antibody, separated by SDS-PAGE, then immunoblotted with an anti-LOX antibody. Both WT and mutant LOX were immunoprecipitated with calnexin, but the level of calnexin-bound mutant LOX was much higher than that of WT LOX. Cell lysate from Lox–/– cells served as a negative control.
A direct interaction between LOX and calnexin was shown using coimmunoprecipitation from Lox+/+, Lox+/Mu, and LoxMu/Mu MEF lysates. Lox–/– cells served as the negative control. Cell lysates were immunoprecipitated with an anti-calnexin antibody, followed by immunoblotting using an antibody to LOX. LOX protein was detected in calnexin immunoprecipitates from all genotypes except Lox–/–, confirming an interaction between the 2 proteins (Figure 5D).
M292R LOX has an altered protein conformation.
The high level of LOX detected in LoxMu/Mu MEF calnexin immunoprecipitates suggests that M292R LOX is misfolded and trapped in the ER bound to calnexin. To determine whether M292R LOX is conformationally different from the WT enzyme, we explored possible structural changes by assessing differential sensitivity to proteolytic degradation. Changes in protein structure frequently expose or mask protease binding sites, leading to altered degradation products and a different degradation fingerprint. Immunoblotting of cell lysate from Lox+/+, Lox+/Mu, and LoxMu/Mu MEFs incubated at 37°C for 6 hours to exploit protein degradation by endogenous enzymes within the cell lysate showed time-dependent degradation of WT but not M292R LOX (Figure 6). These results suggest that WT and M292R pro-LOX have different susceptibilities to degradation arising from cell-associated proteases. Differential degradation was also observed when cell lysates were incubated with pancreatic elastase. In digests of Lox+/+ cell lysate, there was a 25-kDa fragment evident with all doses of elastase that was absent in LoxMu/Mu samples (Supplemental Figure 3, arrow). When treated with the highest dose of elastase, pro-LOX from Lox+/+ and Lox+/Mu samples degraded predominantly to a 30-kDa fragment. LOX from LoxMu/Mu samples, in contrast, degraded further to mainly a 10-kDa fragment. It is important to note that pancreatic elastase does not cleave after methionine or arginine, so the Met-to-Arg mutation does not create or negate a protease cleavage site for this enzyme. Together, the conformational difference between the WT and mutant proteins suggests that M292R LOX is misfolded, which is consistent with its protracted interaction with calnexin in the ER.
Figure 6. M292R Lox mutation leads to altered protein conformation.

(A) Cell lysates from Lox+/+ and LoxMu/Mu mouse embryonic fibroblasts were incubated at 37°C, and samples were collected every hour for 6 hours. Immunoblotting with an anti-LOX antibody showed more extensive degradation of WT LOX compared with the mutant protein. (B) Quantitation of band density from immunoblot in A.
M292R LOX does not elicit an ER stress response.
Accumulation of misfolded proteins in the ER generally leads to ER stress, which subsequently activates the unfolded protein response (UPR) (23). UPR triggers 3 primary stress sensors, PERK, IRE1, and ATF6. Under resting conditions, BIP/Grp78 maintains these sensors in an inactive state. Upon ER stress, BIP binds to misfolded proteins, thereby allowing for sensor activation. To determine whether the continuous presence of misfolded mutant LOX induces ER stress, we assessed Bip transcript levels by qPCR. We also measured mRNA levels of Atf4 and Chop, downstream transcriptional activators of the PERK pathway. Atf4 and Chop were not elevated in LoxMu/Mu MEFs compared with Lox+/+ cells (Figure 7, A and B). While there was a statistically significant increase in Bip mRNA in mutant cells (Figure 7C), the ~20% change was modest compared with the ~2-fold and ~4-fold increase in Atf4 and Chop, respectively, when ER stress was stimulated by treatment of MEFs with lopinavir/ritonavir, which served as a positive control (data not shown). Electron microscopic analysis of WT and mutant cells showed that ER volume was not markedly different in the 2 cell types (Supplemental Figure 4), suggesting that mutant protein does not accumulate to levels that would produce dilated saccules. Together, these results suggest that LOX protein containing the M292R mutation does not induce substantial ER stress.
Figure 7. M292R LOX does not elicit an ER stress response.
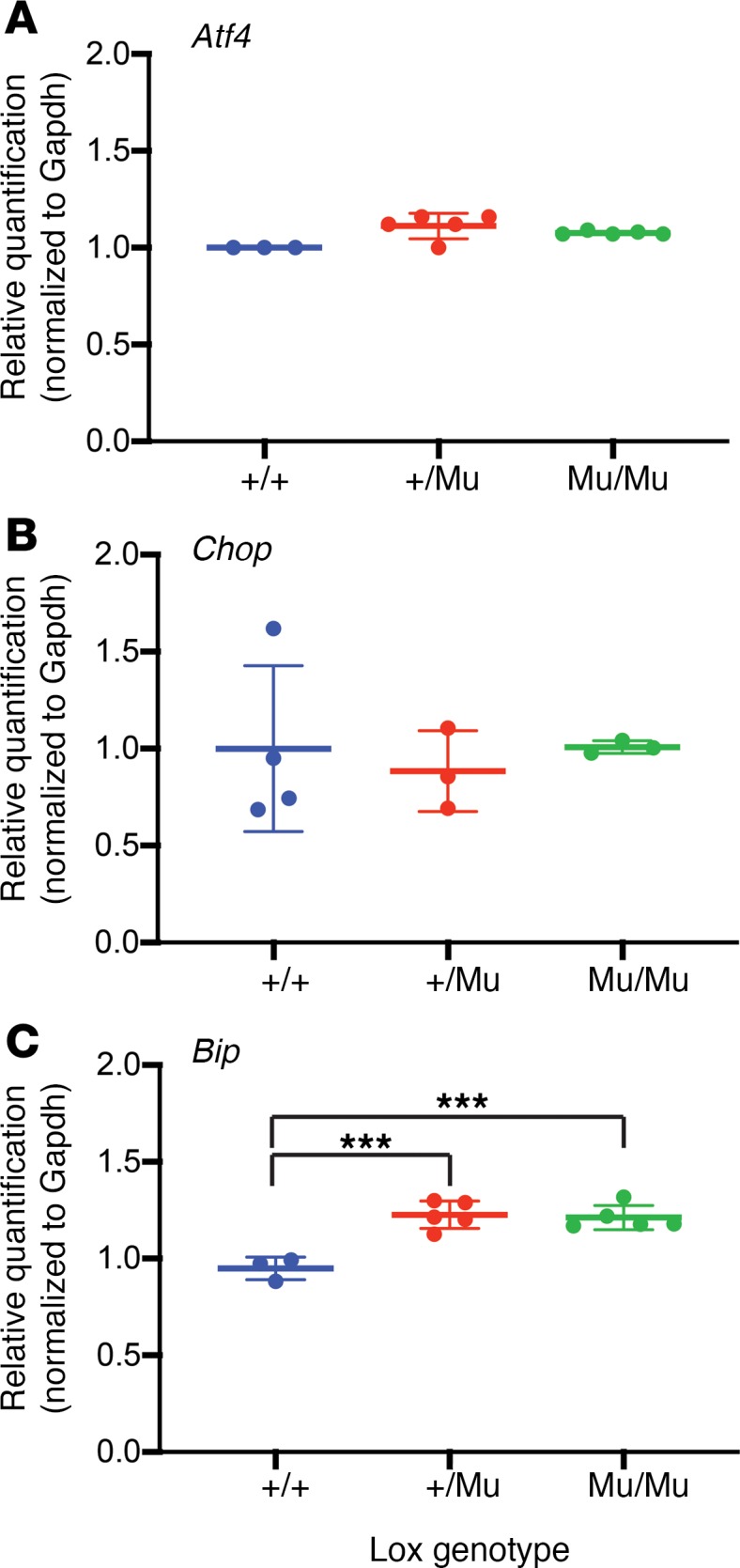
qPCR showing transcript levels of the ER stress markers (A) Atf4, (B) Chop, and (C) Bip normalized to Gapdh expression in mouse embryonic fibroblasts from Lox+/+, Lox+/Mu, and LoxMu/Mu mice. One-way ANOVA with Tukey’s multiple-comparisons test was used to assess differences, which were minimal between the 3 genotypes. Data are presented as mean ± SD. ***P < 0.001.
Increased activation of autophagy in LoxMu/Mu cells.
The absence of ER stress in LoxMu/Mu cells was surprising given that M292R LOX does not leave the ER. To determine whether mutant LOX is being removed from the ER for degradation through the lysosomal pathway, we costained cells for LOX and the lysosomal marker LAMP-2. No colocalization of the 2 proteins was observed, suggesting that the mutant protein was not trafficked through lysosomes (Supplemental Figure 5).
Autophagy is a general mechanism for clearing proteins accumulating in cells. Clearing of misfolded proteins from the ER, a process termed ER-phagy, plays a critical role in ER homeostasis (24). To determine whether mutant LOX directly activates autophagy-mediated protein clearance from the ER, we measured 2 autophagy activation markers, p62 and microtubule-associated protein 1A/1B, light chain 3 (LC3), in WT and LoxMu/Mu MEFs. Following pretreatment with bafilomycin A1 (BafA1) to inhibit organelle acidification–induced degradation of p62 and LC3, only LoxMu/Mu cells with and without BafA1 pretreatment had significantly increased p62 expression compared with Lox+/+ cells without BafA1 pretreatment (Figure 8, A and B). The autophagy activation markers LC3-I and -II were also elevated in mutant compared with WT cells (Figure 8, A, C, and D), providing more evidence for increased autophagy.
Figure 8. Intracellular mutant LOX activates the autophagy markers p62 and LC3.

(A) Mouse embryonic fibroblasts (MEFs) were pretreated with bafilomycin A1 (BafA1) to prevent organelle acidification–induced degradation of autophagy activation markers. Immunoblots were then used to detect P62 and LC3 isoforms in MEF lysates. (B) Quantification of the detected proteins showed a significant increase in p62 only in cell lysates of LoxMu/Mu MEFs with and without BafA1 treatment compared with Lox+/+ MEFs without BafA1. (C and D) Also, LC3 was elevated in LoxMu/Mu cells compared with Lox+/+ cells. Two-way ANOVA with Tukey’s multiple-comparisons test, with the 2 variables being genotype and treatment, was used to assess differences. Data are presented as mean ± SD. *P < 0.05, **P < 0.001.
We also used electron microscopy to screen for the presence of autophagosome-like vesicles in WT and mutant cells (Supplemental Figure 4). We observed that multi- and single-membrane vesicles typical of autophagosomes were particularly abundant in the perinuclear region but were also present throughout the cytoplasm of LoxMu/Mu cells. Such structures were rare in WT cells. Together, the existence of autophagic vesicles along with increased expression of autophagy activation markers in mutant cells is consistent with the clearance of misfolded mutant LOX from the ER through an autophagy/proteasome pathway.
Mutant LOX does not influence the secretion of the WT protein.
To assess whether the secretion of WT LOX is affected by the presence of M292R mutant protein, we transiently expressed plasmids containing WT LOX, and M292R LOX fused to the fluorescent protein mApple, in Lox–/– MEFs. Successful transfection was confirmed by intracellular mApple expression using fluorescence imaging (data not shown). In the conditioned medium of Lox–/– cells transfected with empty plasmid, no LOX was observed by Western blot analysis (Figure 9A). In Lox–/– MEFs transfected with 2.5 μg WT Lox-mApple plasmid, an immunoreactive protein band at approximately 60 kDa was detected in the conditioned media, corresponding to the size of active LOX (30 kDa) fused to mApple (27 kDa). In Lox–/– MEFs transfected with 2.5 μg of M292R Lox-mApple plasmid, LOX protein was observed inside the cell by fluorescence microscopy, but no LOX protein was detected in conditioned medium by Western blotting, consistent with the inability of M292R LOX to be secreted. To determine whether mutant LOX alters the secretion of the WT protein, or if the WT protein acts as a chaperone to facilitate the secretion of the mutant protein, we cotransfected cells with 1.25 μg each of WT and mutant plasmid, which is half as much as was used for single plasmid transfection. If mutant LOX does not affect the secretion of the WT protein, then LOX should be detected in conditioned medium, but at half the level of the single plasmid transfection. If the mutant protein has an adverse effect on WT LOX secretion, then levels will be lower than 50%. If WT LOX acts as a chaperone to facilitate secretion of the mutant protein, then the combination of both secreted proteins would be higher than 50% and possibly equivalent to the WT transfection (1.25 + 1.25 = 2.5 μg). Figure 9 shows that LOX protein was detected in conditioned medium of doubly transfected cells at half the level of singly transfected cells, confirming that the mutant protein does not affect the secretion of WT LOX.
Figure 9. Mutant LOX does not interfere with secretion of the WT protein.

Lox–/– mouse embryonic fibroblasts were transiently transfected with WT or mutant LOX fused to the fluorescent reporter mApple, and protein in conditioned medium from transfected cells was separated by SDS-PAGE. (A) Immunoblotting using an antibody to LOX showed that protein from the WT LOX construct is properly secreted, while mutant (Mu) LOX is not. When the WT and mutant LOX constructs were cotransfected into Lox–/– cells at half the level of the single transfections, the amount of secreted protein that was detected was equivalent to that expected from the WT allele, suggesting that the presence of mutant LOX does not alter the secretion of the WT protein. The bottom panel is an image of the stained gel to document equal sample loading. (B) Quantification of the LOX bands in A.
M292R propeptide processing.
Our experimental results are consistent with the M292R mutation being a loss-of-function phenotype due to a secretion defect. The final activation step of the proenzyme occurs outside the cell through proteolytic separation of the propeptide and catalytic domains. To determine whether M292R pro-LOX could be processed to the 30-kDa mature form if it were secreted, we incubated cell lysates from WT and mutant MEFs with recombinant BMP1 (rBMP1), the enzyme responsible for propeptide cleavage. Western blot analysis showed that BMP1 appropriately processed both WT and M292R pro-LOX to the 30-kDa mature form (Supplemental Figure 6). Thus, the mutation did not inhibit propeptide processing. The processed mutant protein was not assayed for oxidase activity because intracellular LOX in M292R MEFs does not have bound copper and cannot be enzymatically active.
Discussion
LOX plays a critical role in vascular ECM maturation by catalyzing the crosslinking of elastin and fibrillar collagens, the 2 major ECM protein classes in the aortic wall. Phylogenic studies suggest that the current form of vertebrate LOX coevolved with elastin and helped facilitate the appearance of a closed circulatory system (12, 25). Therefore, it is no surprise that mutations in LOX give rise to vascular insufficiency and disease. All LOX mutations associated with TAAD in humans show autosomal dominant inheritance, and most result in loss of function. We know from mouse studies that inactivation of both Lox alleles is lethal and that other members of the LOX family cannot assume LOX’s critical role in vascular development and maintenance of the vessel wall. In this report, we utilize a mouse model of a well-characterized human mutation to explore how LOX haploinsufficiency leads to vascular dilation and aneurysm formation.
Mice heterozygous for the Lox M292R variant (Lox+/Mu) have increased elastic lamellar fragmentation in the wall of the ascending aorta (3), but do not develop aortic dilation under normal conditions. The absence of aneurysmal disease was surprising given that humans heterozygous for this same mutation are prone to aneurysm formation and vessel wall changes (3). Variability in the age of disease onset and disease severity in humans, however, suggested that a secondary challenge is a factor in disease initiation and progression. Here we show that hypertension induced by Ang II infusion resulted in vessel wall changes in both Lox+/+ and Lox+/Mu mice, but only Lox+/Mu animals showed aortic dilation localized to the ascending aorta. Several groups have reported that the effects of Ang II are most pronounced in the ascending aorta region in this model (26–28), but the underlying factors governing this vessel specificity are unknown. Differences in the embryonic origin of the cells that populate the aortic wall are one explanation (29, 30), but the composition and maturation of the aortic ECM are also factors. In Lox+/Mu mice, changes in ECM crosslinking associated with LOX insufficiency could have more negative consequences for this vessel segment because of high collagen and elastin levels and the high mechanical stress experienced by the aortic wall during the cardiac cycle.
While several Ang II–induced vascular changes were expected (19, 27) and common to WT and mutant animals, there were also significant differences. For instance, medial expansion was evident in both genotypes, but the effect was most notable in mutant mice, in which there was an increase in the number of smooth muscle cells between the elastic lamellae. This response is particularly unusual, since the intralamellar space is typically populated by a single smooth muscle cell layer, as can be seen in vessels from saline-treated Lox+/+ and Lox+/Mu animals (Figure 2). The opposite change occurred in WT vessels, where there were fewer smooth muscle cells in response to Ang II, suggesting Ang II–related apoptosis or growth suppression (26), which we did not assess.
Another cellular change in the ascending and descending aorta following Ang II treatment was the accumulation of macrophages in the aortic wall of both WT and Lox+/Mu mice. CD68+ cells were found in the adventitia of both genotypes, with fewer cells in the medial layer. In Ang II–treated WT mice, CD68+ cells accumulated in the intima and inner elastic layers of the media and did not localize to breaks in elastic lamella, as has been reported in other models (18, 20). Their location within the wall, however, shows that some cells were able to penetrate past the internal elastic lamina into the first or second layer of smooth muscle cells. Medial CD68+ cells in Lox+/Mu mice exposed to Ang II, in contrast, were localized to the outer aspect of the media and occurred at higher numbers than in WT vessels. These results suggest that the cellular origin of the CD68+ cells or the route of inflammatory cell infiltration is different in Lox+/+ and Lox+/Mu vessels as a response to wall stress.
The location of macrophages in the outer region of the wall provides a mechanistic explanation for why Lox+/Mu, but not WT, aorta dilates following Ang II treatment. ECM degradation is a primary factor leading to vessel wall dilation. Elastases and other matrix-degrading enzymes secreted by macrophages destabilize the vessel wall, particularly when matrix degradation occurs in or near the adventitia — the vessel compartment essential for maintaining the structural integrity of the artery (31). The medial cell hypertrophy and inflammatory cell accumulation in the Lox+/Mu aorta in response to hemodynamic stress are similar in several ways to vascular changes in mouse models of Marfan syndrome with mutations in fibrillin-1, a key elastic fiber protein. In Marfan mice, inflammatory cell accumulation at the medial-adventitial interface correlates with increased elastin degradation and a loss of structural integrity that leads to vessel wall dilation (32). As in the Ang II–treated Lox+/Mu animals, only the ascending aorta is affected in Marfan mice.
Our previous studies showed that mice with the Lox M292R mutation share numerous phenotypic traits with Lox+/– animals (3), suggesting that M292R is a loss-of-function mutation in mice and humans. Even though the amino acid change is in the catalytic domain of LOX close to the copper binding site, it was not clear whether the mutation results in a catalytically dead enzyme or if some other aspect of LOX processing is negatively affected by the mutation. Our data show that the answer fits both possibilities: the mutant protein is retained in the ER and not secreted, and the enzyme is catalytically dead because of its inability to traffic to the Golgi, where it needs to bind copper and form the redox quinone cofactor LTQ required for catalytic activity (33). Even though we found that the propeptide of mutant pro-LOX could be cleaved by rBMP1 similarly to WT LOX, full-length LOX in the ER cannot be catalytically active even if propeptide processing does occur.
Why mutant LOX is retained in the ER is not clear, but our data point to protein misfolding as a probable cause. ER retention is characteristic of incompletely folded proteins. Folding intermediates of most secreted proteins interact with the calnexin/calreticulin protein complex in the ER to achieve the proper native conformation required for secretion (34). The calnexin/calreticulin protein complex recognizes N-linked glycan structures (LOX has 3 N-linked glycosylation sites in the propeptide region; ref. 11) that undergo cycles of sugar residue removal and addition. Once folded correctly, calnexin and calreticulin dissociate, and the folded proteins are transported from the ER to the Golgi apparatus. Mutant LOX colocalized intracellularly with calnexin and was isolated bound to calnexin in cellular extracts, suggesting that the protein remains misfolded despite repeated folding interactions with calnexin. Furthermore, the differential susceptibility to proteolytic degradation of mutant compared with WT LOX is consistent with structural differences between the 2 proteins.
It was interesting that mutant LOX accumulation in the ER did not induce an appreciable UPR. We saw no upregulation in mutant cells of the ER stress–related markers Bip (an ER luminal chaperone protein and critical ER stress sensor), Atf4 (a transcription factor downstream of PERK that plays a crucial role in the adaptation to ER stress), or Chop (a transcription factor regulated by ATF4 that drives proapoptotic gene transcription). Nor could we detect in mutant cells an increase in ER volume frequently associated with ER stress. One pathway whereby ER stress is mitigated is removal from the ER of mutant protein and degradation at the lysosome (ERAD-II) or proteasome (ERAD-I) (24). We were unable to localize LOX within lysosomes but did find increased expression of the autophagy markers p62 and LC3. Electron microscopy also identified an increased number of autophagic vesicles in LoxMu/Mu cells. These results suggest that misfolded mutant LOX is being cleared from the ER through an autophagy/proteasome pathway and would explain why mutant protein levels never reached a critical threshold to sustain an ER stress response. Furthermore, there is mounting evidence that the autophagy and secretory pathways are coregulated and intimately linked (35), which may represent a mechanistic connection into the LOX trafficking defect that we identified. It should be noted that all the experiments probing the fate of the mutant protein were done in cultured cells. It is possible, but unlikely, that in vivo, in tissues, and at developmental stages relevant to the TAAD phenotype, LOX folding, secretion, ER retention, and stress occur differently than in MEFs.
Guo et al. (15) described 2 other LOX active site variants (S280R and S348R) that segregate with disease in families with TAAD. When expressed from transgenes in HeLa cells and examined in cell lysates, a portion of the pro-LOX protein was processed to its active form and was able to oxidize substrate at rates approximately equivalent to the WT enzyme (S348R) or with approximately 50% lower activity (S280R) (2, 15). Enzymatic activity in conditioned medium from the transfected cells was not evaluated, so it is not known whether these LOX variants are efficiently secreted. However, the fact that the proteins are catalytically active implies trafficking through the Golgi and proteolytic removal of the propeptide domain. Thus, unlike the M292R mutation, which is retained in the ER, the S348R and S280R variants appear capable of traversing the secretory pathway. How these proteins, which have full (or somewhat reduced) catalytic activity, cause thoracic aortic disease is an interesting question. An intriguing possibility is that the mutant proteins contribute to disease pathogenesis through mechanisms that do not involve their amine oxidase activity. Noncatalytic functions for LOX are emerging (5, 36, 37), and much remains to be learned about how mutations in this multifunctional enzyme lead to aortic disease.
Methods
Antibodies.
Antibodies used in this study are described in Supplemental Table 1.
Mouse models and cell isolation.
A knockin mouse strain bearing the Lox M292R substitution was generated in the C57BL/6 background using CRISPR/Cas9 genome editing technology as previously described (3). Lox+/– mice in the same background are described in ref. 6. Primary MEFs were isolated from the various mouse lines by digestion of E14.5 mouse embryos with trypsin/EDTA at 37°C for 30 minutes in DMEM (38). Cells were collected by centrifugation and grown in DMEM, 10% FBS, 1% penicillin/streptomycin, and 1 mM l-glutamine.
Ang II treatment and blood pressure measurements.
Telemetry transmitters (Data Sciences International, model PA-C10) were implanted surgically in 3- to 4-month-old mice under isoflurane anesthesia. The catheter was inserted in the left common carotid artery and advanced to the aortic arch, and the transmitter was placed subcutaneously along the abdomen. Animals were allowed to recover for 7 days prior to activation of transmitters, and continuous recording of heart rate and arterial pressure was obtained, with sampling every 5 minutes for 15-second intervals for 3 days. Data were collected and stored using Dataquest ART (Data Sciences International).
After baseline blood pressure measurements were obtained, mice were implanted with osmotic pumps (Alzet model 1004, DURECT Corp.) that delivered Ang II (MilliporeSigma) subcutaneously at a dose of 2 μg/kg/min in the interscapular region, according to published procedures (39, 40). Mice infused with saline were used as controls. After a week of Ang II treatment, arterial pressure and heart rate were again continuously recorded using the telemeters, with sampling every 5 minutes for 15-second intervals for 3 days. Animals were fed a standard laboratory chow diet. After 28 days, animals were sacrificed under isoflurane anesthesia, and vessels were collected for compliance studies and histological characterization.
Vessel compliance measurement.
For vessel compliance measurements, the ascending aorta and left common carotid artery were excised and placed in a physiological saline solution. Vessels were then cleaned of surrounding fat and connective tissue and mounted onto a pressure arteriograph (Danish Myo Technology). Experiments were done at 37°C in an organ bath containing physiological saline. The mounted vessels were visualized using a computerized imaging system, allowing continuous recording of vessel diameters. Vessel outer diameters were recorded while intravascular pressure was increased from 0 to 175 mmHg by increments of 25 mmHg (12 seconds per step). The average of 3 outer diameter measurements was taken at each pressure (40, 41).
Protein quantification, hydroxyproline, and desmosine assays.
Each ascending aorta was hydrolyzed at 105°C overnight with 20 μL of 6 N hydrochloric acid (Thermo Fisher Scientific) and dried under vacuum in a SpeedVac. The dried pellet was dissolved in 400 μL water and filtered with a 0.45-μm filter. Total protein, hydroxyproline, and desmosine levels were determined as described previously (42).
Dihydroethidium staining for superoxide detection in aortic sections.
Superoxide in aortic sections was detected using dihydroethidum (DHE) staining. In the presence of superoxide, DHE is oxidized to 2-hydroxyethidium, which is highly fluorescent and intercalates within the DNA, staining the nucleus a fluorescent red (43, 44). After euthanasia and thoracotomy, the right atrium was clipped, and 5 mL cold (4°C) 1× PBS was flushed through the left ventricle to clear the blood. Ascending aorta was then dissected, placed in Tissue-Tek O.C.T. Compound (Sakura Finetek), and flash frozen in liquid nitrogen. Four-micrometer cryosections were obtained using a Leica CM1850 cryostat and placed on charged microscope slides (Thermo Fisher Scientific). A 10-mM stock solution of DHE (Life Technologies) in DMSO was diluted to 10 μM using 1× PBS. The 10-μM DHE solution was applied to unfixed aortic sections and incubated at 37°C for 30 minutes in the dark. After staining with DHE, sections were then rinsed with 1× PBS for 5 minutes at room temperature (RT) and mounted with a coverslip using ProLong Diamond Antifade Mountant with DAPI (Invitrogen). Images were obtained on the Zeiss Axioscope fluorescence microscope with a QImaging MicroPublisher 3.3 RTV camera using QCapture software (QImaging). The entire procedure (from mouse euthanasia to image acquisition) occurred within 8 hours.
Immunofluorescence on aortic sections and area measurements.
Ascending and descending aortas were dissected as above and placed in O.C.T. compound, and the blocks were frozen on dry ice. Four-micrometer-thick cryosections were placed on glass slides and stored at –80°C. The sections were fixed in 4% paraformaldehyde (Electron Microscopy Sciences) in 1× PBS at 4°C for 10 minutes, washed twice with 1× PBS (5 minutes each) at RT, and blocked with a solution containing 1% BSA (MilliporeSigma), 1% fish gelatin (MilliporeSigma), and 0.05% Triton-X (Acros Organics) in 1× PBS for 1 hour at RT. The aortic sections were then incubated with rat anti–mouse CD68 antibody (Bio-Rad) at a 1:1000 dilution in blocking solution at 4°C overnight. The next day, sections were washed 3 times with 1× PBS (5 minutes each) and incubated in goat anti-rat IgG Alexa Fluor 488 secondary antibody (Thermo Fisher Scientific) diluted 1:2000 and DAPI diluted 1:3000 in blocking solution at RT for 1 hour. The sections were washed twice with 1× PBS (5 minutes each) at RT, mounted with a coverslip using ProLong Diamond Antifade Mountant, and imaged using the Axioscope fluorescence microscope equipped with the QImaging MicroPublisher 3.3 RTV camera using QCapture software as above. For area measurements, frozen sections from the descending aorta from 3-month-old mice were prepared as described above. The area between the internal and external elastic laminae was traced in each ×40 image using ImageJ software (NIH) and measured in pixels2.
LOX activity assay.
MEFs were grown in 10-cm dishes and maintained as above until fully confluent. Twenty-four hours before collection, culture medium was replaced with DMEM lacking phenol red and FBS, supplemented with 50 μg/mL ascorbic acid and 0.1% BSA. Conditioned medium from each dish was concentrated using Amicon Ultra-15 Centrifugal Filter Units (Millipore) to a final volume of 1 mL. Cell lysates were collected by scraping in 1× PBS containing protease inhibitors. LOX activity in cell lysate and concentrated culture medium, incubated with and without the LOX inhibitor β-aminopropionitrile (BAPN), was quantified using the Amplex Red assay as previously described (3, 45). Resorufin fluorescence, the product of Amplex Red oxidation, was measured at excitation and emission wavelengths (540 and 600 nm, respectively) every 30 minutes for 150 minutes using a BioTek H4 Hybrid Reader. LOX activity was calculated as the difference between total activity and activity in the presence of BAPN. Differences in relative fluorescent units were tested using 2-way ANOVA with Tukey’s multiple-comparisons test.
Immunofluorescence imaging of primary cells.
Primary MEFs were seeded in 4-chamber glass slides at 3 × 105 cells per/well. After 3 days in culture, the cells were fixed with cold methanol (2 washes, 5 seconds each) followed by 3 washes with blocking solution containing 1× PBS, 1% BSA, 1% fish gelatin (MilliporeSigma), and 0.05% Triton-X100 at pH 7.4, then incubated in blocking solution for 30 minutes at RT. The cells were then incubated with a primary antibody to LOX at 1:1000 in blocking solution overnight at 4°C. After washing the cells with blocking solution to remove the primary antibody, the cells were incubated with a goat anti-rabbit IgG Alexa Fluor 568 secondary antibody at 1:2000 (Thermo Fisher Scientific) and anti-calnexin primary antibody directly labeled with Alexa Fluor 488 at 1:4000 in blocking solution for 30 minutes at RT. The cells were washed with blocking solution 3 times, then mounted using ProLong Diamond Antifade Mountant (Invitrogen) and coverslipped. The samples were imaged using the Axioscope fluorescence microscope and QCapture Pro software.
Protein extraction from cell culture and aortic tissue.
Cultured MEFs were switched to serum-free medium 24 hours before protein extraction. MEFs in 10-cm dishes were incubated in 1 mL lysis (RIPA) buffer (MilliporeSigma) containing protease inhibitors for 30 minutes at 4°C. The lysates were scraped from the dish into microfuge tubes and centrifuged at 17,500 g for 5 minutes, and the supernatant was collected. Conditioned medium from each dish was concentrated using Amicon Ultra-15 Centrifugal Filter Units (Millipore) to a final volume of 1 mL. The protein concentration in cell lysates and concentrated media was measured using bicinchoninic acid (BCA) assay (Thermo Fisher Scientific). Laemmli buffer with and without DTT was added to samples for SDS-PAGE and native gel electrophoresis, respectively. Only the samples with DTT were boiled at 100°C for 5 minutes.
RNA extraction and qPCR.
RNA was extracted from confluent cell cultures using TRIzol (Thermo Fisher Scientific) following the manufacturer’s protocol. For RNA isolation from aortic tissue, ascending aorta from adult Lox+/+ and Lox+/Mu mice were each homogenized in 500 μl TRIzol using QIAGEN TissueLyser. The manufacturer’s protocol was followed for RNA isolation. At the time of RNA precipitation, 10 μg glycogen (MilliporeSigma) was added as a carrier to each sample. Each RNA pellet was dissolved in 10 μL molecular-grade water. The RNA was quantified using the NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific). One microgram of the isolated RNA was treated with DNase I (Invitrogen) according to the manufacturer’s instructions. Reverse transcription was then carried out using the High-Capacity RNA-to-cDNA Kit (Applied Biosystems). One microliter of cDNA was used for qPCR using TaqMan Fast Universal PCR Master Mix and TaqMan assay primer/probes (Life Technologies) for Bip (Mm00517691_m1), Atf4 (Mm00515325_g1), Chop (Mm01135937_g1), Agtr1a (Mm01957722_s1), Agtr1b (Mm02620758_s1), Agtr2 (Mm00431727_91), Mmp2 (Mm00439498_m1), Mmp9 (Mm00442991_m1), Mmp12 (Mm00500554_m1), and Gapdh (Mm9999999915_g1), which was used as an internal control. Ten-microliter reactions were performed in duplicate using a ViiA 7 Real-Time PCR System for Bip, Atf4, and Chop and QuantStudio 3 Real-Time PCR system (Applied Biosystems) for Agtr1a, Agtr1b, Agtr2, Mmp2, Mmp9, and Mmp12. All mRNA levels were normalized to Gapdh expression.
rBMP1 treatment.
Protein was extracted from Lox+/+, Lox+/Mu, and LoxMu/Mu MEFs as described above. Fifty microliters of cell lysates from cultured MEFs containing 50 μg total protein were incubated with 30, 60, and 90 ng rBMP1 (R&D Systems) for 45 minutes at 37°C in reaction buffer (50 mM Tris, 150 mM NaCl, 5 mM CaCl2, pH 7.5). The enzyme reaction was stopped by adding Laemmli buffer with DTT and incubated at 100°C for 3 minutes before analysis by SDS-PAGE and immunoblotting.
Western blotting.
Protein extracts from cells, concentrated condition medium, or tissues (generally 50 μg) were analyzed using 10% Mini-PROTEAN TGX Stain-Free Protein Gels (Bio-Rad) run at 100 mV for 1 hour. The protein was then transferred to ProBlott Membrane (Applied Biosystems). The blots were incubated in 5% nonfat dried milk in 0.1% 1× PBS-Tween for 1 hour at RT, then incubated with a primary antibody overnight at 4°C. The next day, the blots were washed with 0.1% 1× PBS-Tween and incubated in ECL anti-rabbit or anti-mouse IgG HRP-Linked Secondary Antibody (GE Healthcare) for 1 hour at RT. The blots were then washed and the immunoreactive bands detected using Immobilon Western Chemiluminescent HRP Substrate (Millipore). The blots were stripped by incubating in 2% SDS, 75 mM Tris-HCl, and 100 mM β-mercaptoethanol for 1 hour at 65°C while shaking. The blot was then incubated in 0.1% 1× PBS-Tween wash buffer for 1 hour at RT before immunodetection of β-actin (MilliporeSigma) at 1:20,000 in wash buffer, which was used as a loading control. For protein samples from concentrated conditioned media, membranes were stained with Ponceau S solution (MilliporeSigma) before blocking and used as a loading control. Alternatively, stain-free images of Mini-PROTEAN TGX Stain-Free Protein Gels (Bio-Rad) taken following the manufacturer’s protocol served as a loading control.
Coimmunoprecipitation.
Cell lysates were prepared from MEFs 5 days after confluency as described above. Immobilized protein A anti-rabbit IgG beads (50 μL), preequilibrated by washing with lysis buffer, were then added to 250 μL cell extract. After incubation on a rocking platform for 60 minutes at 4°C, the sample was centrifuged at 2500 g for 3 minutes, and the supernatant was incubated with 8 μg immunoprecipitation antibody for 1 hour at 4°C. Fifty microliters of precleared protein A beads was then added, and the samples incubated overnight at 4°C with rocking. Immune complexes bound to the protein A beads were collected by centrifugation and washed with lysis buffer prior to boiling at 100°C for 10 minutes with Laemmli buffer containing DTT. The supernatant was collected by centrifugation, and proteins were separated by SDS-PAGE and immunoblotted following the same protocol described above.
Protein degradation analysis.
Confluent cultures of WT and mutant MEFs were treated with 5 μg/mL Brefeldin A (MilliporeSigma) to inhibit protein transport from ER to the Golgi apparatus. This ensured that WT LOX remained in the cells so that the starting amount of protein was the same across genotypes. After 4 hours, the cell lysate was extracted using lysis buffer placed at 37°C to initiate protein degradation. Every hour, 50 μL aliquot of cell lysate was collected, then immediately boiled with Laemmli sample buffer at 100°C for 5 minutes prior to SDS-PAGE analysis. For exogenous protease-mediated degradation, human pancreatic elastase in increasing concentrations (0.625, 1.25, 2.5, 5 ng/μL) was added to cell lysates containing 1 mg/mL protein at 4°C for 30 minutes. After treatment, elastase activity was inhibited with protease inhibitor cocktail, and samples were boiled with Laemmli sample buffer at 100°C for 5 minutes for SDS-PAGE analysis.
Autophagy markers.
Confluent MEFs in 6-well dishes were pretreated with 100 mM BafA1 (MilliporeSigma) for 3 hours before cell lysates were collected. The protein samples were separated on 4%–15% gradient gels for SDS-PAGE analysis. Immunoblotting was performed following the same procedure as above.
Lox-mApple plasmid cloning and expression.
The mouse Lox cDNA in a mammalian expression vector driven by a pCMV promoter was purchased from GE Healthcare. The Lox open reading frame was PCR amplified using a 5′ primer bearing an XhoI restriction enzyme site (5′-CAGATCTCGAGAGCGTTTCGCCTGGGCTGTGC-3′) and a 3′ primer designed to remove the Lox stop codon bearing a BamHI restriction enzyme site (5′-GGATCCACATACGGTGAAATTGTGCAGCCTGAG-3′). The PCR product and the pGEMT shuttle vector (Promega) were digested with XhoI and BamHI, then ligated together using T4 ligase. The mutant Lox plasmid bearing the 893 T>G base pair change was generated using the Lox-pGEMT plasmid as the template, using the Phusion Site-Directed Mutagenesis Kit (Thermo Fisher Scientific), following the manufacturer’s protocol (5′-TTACCACAGCAGGGACGAATTCAGCCACTATG-3′ and 5′-TGTTGGTGACAGCTGTGCCACTCCC-3′). The WT and mutant Lox-pGEMT plasmid sequences were confirmed using Sanger sequencing performed at the Washington University Protein and Nucleic Acid Chemistry Laboratory. To generate the Lox-mApple plasmids, WT and mutant Lox pGEMT plasmids and the mApple plasmid (46), provided by David W. Piston (Washington University School of Medicine in St. Louis), were digested with XhoI and BamHI restriction enzymes. The WT and mutant mLox cDNA inserts were then ligated into mApple vectors to generate the mLox-mApple fusion plasmids and sequenced to confirm. To express the WT and mutant Lox–mApple plasmids in MEFs, Lipofectamine 3000 reagents (Thermo Fisher Scientific) were used following the manufacturer’s protocol.
Transmission electron microscopy.
MEFs from Lox+/+, Lox+/Mu, and LoxMu/Mu mice, cultured on glass coverslips, were washed in cacodylate buffer (0.15 M cacodylate in 2 mM CaCl2, pH 7.4) and fixed for 5 minutes with 2.5% glutaraldehyde and 2% paraformaldehyde in cacodylate buffer prewarmed to 37°C. The samples were then incubated at RT for an additional hour, washed with cacodylate buffer, and treated with 1% OsO4/1.5% potassium ferrocyanide for 1 hour in the dark. After washing with ultrapure water, en bloc staining was performed in 2% uranyl acetate for 1 hour in the dark. The samples were washed again in ultrapure water before dehydration in a graded acetone series. Finally, the samples were embedded in Epon resin and cured in a 60°C oven for 48 hours. The processed samples were imaged using a JEOL 1400 electron microscope.
Statistics.
One-way ANOVA with Tukey’s multiple-comparisons test was used to determine the significance, if any, between different groups, particularly genotypes. When 2 variables were present, e.g., genotype and treatment or genotype and gene, 2-way ANOVA with Tukey’s multiple-comparisons test was used to assess differences. If the same sample was measured repeatedly (e.g., at different pressures in the pressure-diameter curves in Figure 1, C and D, or at different time points in Figure 3), then repeated measures 2-way ANOVA with Tukey’s multiple-comparisons test was performed. Prism 8 for Mac OS X (GraphPad Software) was used to run statistical analyses. In all numerical figures, data are presented as mean ± SD. P < 0.05 was considered statistically significant. The statistical test used and significant differences are noted in each figure legend.
Study approval.
All animal experiments were carried out following protocols approved by Washington University School of Medicine Institutional Animal Care and Use Committee.
Author contributions
VSL, CMH, TJB, and PCT participated in experimental design and acquired and analyzed primary data. NOS helped with data analysis, and RPM designed the research study and analyzed primary data. VSL wrote the initial draft of the manuscript, to which all authors contributed edits.
Supplementary Material
Acknowledgments
This work was supported by NIH grants R01HL53325, HL105314 (RPM), R21AR072748 (PCT), and R01HL131961 (NOS). Funds were also provided to RPM by the Ines Mandl Research Foundation. VSL was supported by training grant T32 HL125241 and a Predoctoral Individual National Research Service Award (F31HL136073). CMH was supported by NIH grant K08 HL135400. We thank Robyn Roth for assistance with electron microscopy and gratefully acknowledge assistance from James Fitzpatrick and staff at the Washington University Center for Cellular Imaging (WUCCI), which is supported by Washington University School of Medicine, the Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital (CDI-CORE-2015-505), and the Foundation for Barnes-Jewish Hospital (grant no. 3770). Additionally, we thank Michael Wallendorf of the Washington University Division of Biostatistics for assistance with statistical data analysis.
Version 1. 06/18/2019
In-Press Preview
Version 2. 08/08/2019
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2019, American Society for Clinical Investigation.
Reference information: JCI Insight. 2019;4(15):e127748.https://doi.org/10.1172/jci.insight.127748.
Contributor Information
Vivian S. Lee, Email: V.lee@wustl.edu.
Carmen M. Halabi, Email: halabi_c@kids.wustl.edu.
Thomas J. Broekelmann, Email: tbroekel@wustl.edu.
Philip C. Trackman, Email: trackman@bu.edu.
Nathan O. Stitziel, Email: nstitziel@wustl.edu.
Robert P. Mecham, Email: bmecham@wustl.edu.
References
- 1.Milewicz DM, Prakash SK, Ramirez F. Therapeutics targeting drivers of thoracic aortic aneurysms and acute aortic dissections: insights from predisposing genes and mouse models. Annu Rev Med. 2017;68:51–67. doi: 10.1146/annurev-med-100415-022956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kwartler CS, et al. Variants of unknown significance in genes associated with heritable thoracic aortic disease can be low penetrant “risk variants.”. Am J Hum Genet. 2018;103(1):138–143. doi: 10.1016/j.ajhg.2018.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee VS, et al. Loss of function mutation in LOX causes thoracic aortic aneurysm and dissection in humans. Proc Natl Acad Sci U S A. 2016;113(31):8759–8764. doi: 10.1073/pnas.1601442113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kozel BA, Mecham RP, Rosenbloom J. Elastin. In: Mecham RP, ed. Biology of Extracellular Matrix: An Overview. Berlin-Heidelberg, Germany: Springer-Verlag; 2011:267-301. [Google Scholar]
- 5.Trackman PC. Enzymatic and non-enzymatic functions of the lysyl oxidase family in bone. Matrix Biol. 2016;52-54:7–18. doi: 10.1016/j.matbio.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hornstra IK, Birge S, Starcher B, Bailey AJ, Mecham RP, Shapiro SD. Lysyl oxidase is required for vascular and diaphragmatic development in mice. J Biol Chem. 2003;278(16):14387–14393. doi: 10.1074/jbc.M210144200. [DOI] [PubMed] [Google Scholar]
- 7.Mäki JM, et al. Inactivation of the lysyl oxidase gene Lox leads to aortic aneurysms, cardiovascular dysfunction, and perinatal death in mice. Circulation. 2002;106(19):2503–2509. doi: 10.1161/01.CIR.0000038109.84500.1E. [DOI] [PubMed] [Google Scholar]
- 8.López-Jiménez AJ, Basak T, Vanacore RM. Proteolytic processing of lysyl oxidase-like-2 in the extracellular matrix is required for crosslinking of basement membrane collagen IV. J Biol Chem. 2017;292(41):16970–16982. doi: 10.1074/jbc.M117.798603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mäki JM, Tikkanen H, Kivirikko KI. Cloning and characterization of a fifth human lysyl oxidase isoenzyme: the third member of the lysyl oxidase-related subfamily with four scavenger receptor cysteine-rich domains. Matrix Biol. 2001;20(7):493–496. doi: 10.1016/S0945-053X(01)00157-3. [DOI] [PubMed] [Google Scholar]
- 10.Borel A, et al. Lysyl oxidase-like protein from bovine aorta. Isolation and maturation to an active form by bone morphogenetic protein-1. J Biol Chem. 2001;276(52):48944–48949. doi: 10.1074/jbc.M109499200. [DOI] [PubMed] [Google Scholar]
- 11.Trackman PC, Bedell-Hogan D, Tang J, Kagan HM. Post-translational glycosylation and proteolytic processing of a lysyl oxidase precursor. J Biol Chem. 1992;267(12):8666–8671. [PubMed] [Google Scholar]
- 12.Grau-Bové X, Ruiz-Trillo I, Rodriguez-Pascual F. Origin and evolution of lysyl oxidases. Sci Rep. 2015;5:10568. doi: 10.1038/srep10568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bignon M, et al. Lysyl oxidase-like protein-2 regulates sprouting angiogenesis and type IV collagen assembly in the endothelial basement membrane. Blood. 2011;118(14):3979–3989. doi: 10.1182/blood-2010-10-313296. [DOI] [PubMed] [Google Scholar]
- 14. Kagan HM, Ryvkin F. Lysyl oxidase and lysyl oxidase-like enzymes. In: Mecham RP, ed. The Extracellular Matrix: An Overview. Berlin Heidelberg, Germany: Springer-Verlag; 2011:303–335. [Google Scholar]
- 15.Guo DC, et al. LOX mutations predispose to thoracic aortic aneurysms and dissections. Circ Res. 2016;118(6):928–934. doi: 10.1161/CIRCRESAHA.115.307130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Humphrey JD, Tellides G. Central artery stiffness and thoracic aortopathy. Am J Physiol Heart Circ Physiol. 2019;316(1):H169–H182. doi: 10.1152/ajpheart.00205.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu H, Daugherty A. Aortic aneurysms. Arterioscler Thromb Vasc Biol. 2017;37(6):e59–e65. doi: 10.1161/ATVBAHA.117.309578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Daugherty A, Cassis L. Angiotensin II-mediated development of vascular diseases. Trends Cardiovasc Med. 2004;14(3):117–120. doi: 10.1016/j.tcm.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 19.Wu J, et al. Origin of Matrix-producing cells that contribute to aortic fibrosis in hypertension. Hypertension. 2016;67(2):461–468. doi: 10.1161/HYPERTENSIONAHA.115.06123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saraff K, Babamusta F, Cassis LA, Daugherty A. Aortic dissection precedes formation of aneurysms and atherosclerosis in angiotensin II-infused, apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2003;23(9):1621–1626. doi: 10.1161/01.ATV.0000085631.76095.64. [DOI] [PubMed] [Google Scholar]
- 21.Dikalova AE, Góngora MC, Harrison DG, Lambeth JD, Dikalov S, Griendling KK. Upregulation of Nox1 in vascular smooth muscle leads to impaired endothelium-dependent relaxation via eNOS uncoupling. Am J Physiol Heart Circ Physiol. 2010;299(3):H673–H679. doi: 10.1152/ajpheart.00242.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jiménez-Altayó F, et al. Redox stress in Marfan syndrome: dissecting the role of the NADPH oxidase NOX4 in aortic aneurysm. Free Radic Biol Med. 2018;118:44–58. doi: 10.1016/j.freeradbiomed.2018.02.023. [DOI] [PubMed] [Google Scholar]
- 23.Hetz C, Papa FR. The unfolded protein response and cell fate control. Mol Cell. 2018;69(2):169–181. doi: 10.1016/j.molcel.2017.06.017. [DOI] [PubMed] [Google Scholar]
- 24.Smith M, Wilkinson S. ER homeostasis and autophagy. Essays Biochem. 2017;61(6):625–635. doi: 10.1042/EBC20170092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wagenseil JE, Mecham RP. Vascular extracellular matrix and arterial mechanics. Physiol Rev. 2009;89(3):957–989. doi: 10.1152/physrev.00041.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Owens AP, et al. Angiotensin II induces a region-specific hyperplasia of the ascending aorta through regulation of inhibitor of differentiation 3. Circ Res. 2010;106(3):611–619. doi: 10.1161/CIRCRESAHA.109.212837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Daugherty A, Rateri DL, Charo IF, Owens AP, Howatt DA, Cassis LA. Angiotensin II infusion promotes ascending aortic aneurysms: attenuation by CCR2 deficiency in apoE-/- mice. Clin Sci. 2010;118(11):681–689. doi: 10.1042/CS20090372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trachet B, et al. Ascending aortic aneurysm in angiotensin II-infused mice: formation, progression, and the role of focal dissections. Arterioscler Thromb Vasc Biol. 2016;36(4):673–681. doi: 10.1161/ATVBAHA.116.307211. [DOI] [PubMed] [Google Scholar]
- 29.Hungerford JE, Little CD. Developmental biology of the vascular smooth muscle cell: building a multilayered vessel wall. J Vasc Res. 1999;36(1):2–27. doi: 10.1159/000025622. [DOI] [PubMed] [Google Scholar]
- 30.Majesky MW. Developmental basis of vascular smooth muscle diversity. Arterioscler Thromb Vasc Biol. 2007;27(6):1248–1258. doi: 10.1161/ATVBAHA.107.141069. [DOI] [PubMed] [Google Scholar]
- 31.Pyo R, et al. Targeted gene disruption of matrix metalloproteinase-9 (gelatinase B) suppresses development of experimental abdominal aortic aneurysms. J Clin Invest. 2000;105(11):1641–1649. doi: 10.1172/JCI8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pereira L, et al. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. Proc Natl Acad Sci U S A. 1999;96(7):3819–3823. doi: 10.1073/pnas.96.7.3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klinman JP, Bonnot F. Intrigues and intricacies of the biosynthetic pathways for the enzymatic quinocofactors: PQQ, TTQ, CTQ, TPQ, and LTQ. Chem Rev. 2014;114(8):4343–4365. doi: 10.1021/cr400475g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang JX, Braakman I, Matlack KE, Helenius A. Quality control in the secretory pathway: the role of calreticulin, calnexin and BiP in the retention of glycoproteins with C-terminal truncations. Mol Biol Cell. 1997;8(10):1943–1954. doi: 10.1091/mbc.8.10.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Farhan H, Kundu M, Ferro-Novick S. The link between autophagy and secretion: a story of multitasking proteins. Mol Biol Cell. 2017;28(9):1161–1164. doi: 10.1091/mbc.e16-11-0762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trackman PC. Functional importance of lysyl oxidase family propeptide regions. J Cell Commun Signal. 2018;12(1):45–53. doi: 10.1007/s12079-017-0424-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vallet SD, et al. Insights into the structure and dynamics of lysyl oxidase propeptide, a flexible protein with numerous partners. Sci Rep. 2018;8(1):11768. doi: 10.1038/s41598-018-30190-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jain K, Verma PJ, Liu J. Isolation and handling of mouse embryonic fibroblasts. Methods Mol Biol. 2014;1194:247–252. doi: 10.1007/978-1-4939-1215-5_13. [DOI] [PubMed] [Google Scholar]
- 39.Daugherty A, Manning MW, Cassis LA. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J Clin Invest. 2000;105(11):1605–1612. doi: 10.1172/JCI7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Halabi CM, Broekelmann TJ, Knutsen RH, Ye L, Mecham RP, Kozel BA. Chronic antihypertensive treatment improves pulse pressure but not large artery mechanics in a mouse model of congenital vascular stiffness. Am J Physiol Heart Circ Physiol. 2015;309(5):H1008–H1016. doi: 10.1152/ajpheart.00288.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Faury G, et al. Developmental adaptation of the mouse cardiovascular system to elastin haploinsufficiency. J Clin Invest. 2003;112(9):1419–1428. doi: 10.1172/JCI19028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stoilov I, Starcher BC, Mecham RP, Broekelmann TJ. Measurement of elastin, collagen, and total protein levels in tissues. Methods Cell Biol. 2018;143:133–146. doi: 10.1016/bs.mcb.2017.08.008. [DOI] [PubMed] [Google Scholar]
- 43.Cai H, Dikalov S, Griendling KK, Harrison DG. Detection of reactive oxygen species and nitric oxide in vascular cells and tissues: comparison of sensitivity and specificity. Methods Mol Med. 2007;139:293–311. doi: 10.1007/978-1-59745-571-8_20. [DOI] [PubMed] [Google Scholar]
- 44.Fink B, Laude K, McCann L, Doughan A, Harrison DG, Dikalov S. Detection of intracellular superoxide formation in endothelial cells and intact tissues using dihydroethidium and an HPLC-based assay. Am J Physiol Cell Physiol. 2004;287(4):C895–C902. doi: 10.1152/ajpcell.00028.2004. [DOI] [PubMed] [Google Scholar]
- 45.Trackman PC, Bais MV. Measurement of lysyl oxidase activity from small tissue samples and cell cultures. Methods Cell Biol. 2018;143:147–156. doi: 10.1016/bs.mcb.2017.08.009. [DOI] [PubMed] [Google Scholar]
- 46.Kremers GJ, Hazelwood KL, Murphy CS, Davidson MW, Piston DW. Photoconversion in orange and red fluorescent proteins. Nat Methods. 2009;6(5):355–358. doi: 10.1038/nmeth.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.