

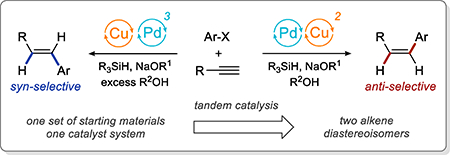
Abstract
A diastereodivergent hydroarylation of terminal alkynes is accomplished using tandem catalysis. The hydroarylation allows highly selective synthesis of both E and Z diastereoisomers of aryl alkenes, from the same set of starting materials, using the same combination of palladium and copper catalysts. The selectivity is controlled by simple changes in the stoichiometry of the alcohol additive. The hydroarylation has excellent substrate scope and can be accomplished in the presence of various classes of compounds, including esters, nitriles, alkyl halides, epoxides, carbamates, acetals, ethers, silyl ethers, and thioethers. The Z-selective hydroarylation is accomplished using a new approach based on tandem Sonogashira coupling and catalytic semireduction. The E-selective hydroarylation involves an additional catalytic isomerization of the Z-alkene. Our explorations of the reaction mechanism explain the role of individual reaction components and how the subtle changes in the reaction conditions influence the rates of specific steps of the hydroarylation. Our studies also show that although the Z- and E-selective hydroarylation reactions are mechanistically closely related, the roles of the palladium and copper catalysts in the two reactions are different.
Graphcial Abstract
INTRODUCTION
Alkenes play an important role in organic chemistry, both as common structural elements of organic molecules and as intermediates in organic synthesis. The preparation of the E and Z isomers of an alkene generally requires the synthesis of two different sets of precursors,1 often using different synthetic routes. For example, in extensively used cross-coupling reactions, the stereochemistry of the alkene product is determined by the stereochemistry of the starting material.1b As a result, the synthesis of E- and Z-aryl alkenes involves separate cross-coupling reactions of two diastereomeric alkene fragments with a functionalized arene (Scheme 1a).
Scheme 1.

Stereoselectivity in Cross Coupling
Although rare, stereodivergent methods that allow the synthesis of both alkene isomers from the same starting materials are known. For example, both isomers can be formed by the semireduction of an alkyne.2 Whereas this approach allows control over the double bond geometry and stereodivergence, no new carbon-carbon bond is formed. A more efficient approach is offered by alkene cross metathesis,3 which leads to fragment coupling through the formation of a new double bond and allows the control of the double bond geometry. The stereochemistry of the product is primarily determined by the catalyst used in the reaction.3b,4 The downside of this approach is that the selective formation of the desired cross metathesis product often necessitates the use of specific combinations of substrates and/or a large excess of one of the substrates.
Reductive cross-coupling reactions of alkynes also provide an excellent opportunity for stereodivergent alkene synthesis. Like the alkene cross metathesis reaction, these transformations are responsible both for the formation of a new C-C bond and for setting the stereochemistry of the alkene product. Therefore, in principle, a single hydroarylation reaction could provide access to both diastereoisomers of an aryl alkene from a single alkyne substrate (Scheme 1b). Despite the development of numerous reactions for the hydroarylation of alkynes, diastereodivergent methods remain rare.5
Recently, a diastereodivergent hydroalkylation of aryl alkynes (Scheme 2) was reported by Nishikata et al.6 demonstrating the feasibility of diastereodivergent reductive cross-coupling. Although no detailed mechanistic analysis was reported, the diastereodivergence seems to result from fundamentally different mechanisms for the two reactions. The Z-products are proposed to be formed through radical bromoalkylation, followed by the reduction of alkenyl bromide. The formation of E-alkenes, on the other hand, likely involves hydroboration followed by Suzuki cross-coupling.
Scheme 2.

Diastereodivergent Reductive Alkylation
Mechanistic analysis of known hydroarylation reactions suggests why stereodivergence has been difficult to achieve. Most current methods are based on syn-stereospecific carbo-7 or hydrometallation8 of alkynes, which forces the syn-selective hydroarylation and prevents the formation of the other isomer (Scheme 3a).
Scheme 3.

Methods for Hydroarylation of Alkynes
To our knowledge, the only approach to catalytic anti-selective hydroarylation was developed by Fujiwara in 2000 (Scheme 3b).9,10 Using a palladium or platinum catalyst, anti-selective hydroarylation is accomplished through electrophilic activation of alkynes.11 Unfortunately, the Friedel-Crafts mechanism12 of this reaction limits the scope to highly electron-rich arenes and electrophilic alkynes, such as propiolates. Furthermore, products of dialkenylation and diarylation are often formed. Finally, it is important to note that all three mechanistic approaches to hydroarylation shown in Scheme 3 (a and b) have been difficult to apply to reactions of terminal alkynes, making hydroarylation of this important class of substrates particularly challenging.13
Seeking to exploit the full potential of hydroarylation chemistry, we were interested in developing a diastereodivergent hydroarylation of terminal alkynes that would provide access to both Z- and E-aryl alkenes. Considering the diastereospecific nature of current hydroarylation methods we decided to explore a fundamentally different approach based on tandem catalysis.
Tandem catalysis has recently received a lot of attention as an efficient strategy for the development of new catalytic transformations.14 Performing two or more catalytic reactions in the same flask enables a more economical use of energy and time, and minimizes the use of reaction workup and product purification.14a At the same time, tandem catalysis allows the development of transformations that are difficult to accomplish relying on a single catalytic cycle.15
We reasoned that the Z-selective hydroarylation could be achieved using tandem Sonogashira coupling16 and Z-selective semireduction.2c Based on the seminal work by Sadighi et al.17 and the subsequent work by Tsuji18 and our group,19 we planned to use the same NHC copper catalyst to promote Sonogashira coupling and to achieve semireduction in the presence of a silane and an alcohol (Scheme 3c). E-Alkenes could be accessed using the same catalyst system through Sonogashira coupling and semireduction, followed by isomerization of the Z product. E-styrenes are known to be significantly more stable than Z isomers, and several distinct mechanisms for palladium-catalyzed isomerization have been established.20
RESULTS AND DISCUSSION
Z-Selective Hydroarylation of Alkynes
Based on our general strategy outlined in Scheme 3c, we initially explored Z-selective hydroarylation and found that Sonogashira coupling under a variety of known conditions followed by in situ copper-catalyzed semireduction was an ineffective method for hydroarylation. The best result (27% yield) was obtained using modified Fu-Buchwald Sonogashira conditions21 with conditions for the semireduction previously reported by our group19 (equation 1, See SI for details).
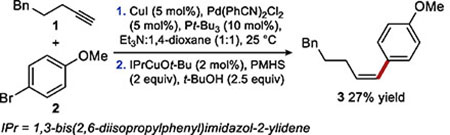 |
(1) |
These results demonstrate the major challenge in tandem catalysis of ensuring that the catalytic reactions involved in the process are mutually compatible. Cross-talk between components of different catalytic cycles often leads to side reactions and catalyst deactivation or decomposition.14 In our case, the major concern was the ability of palladium catalysts to react with silanes and unsaturated compounds in a variety of ways. For example, previously, when the product of a Sonogashira reaction was treated with a silane in situ, exclusive formation of the alkane product was observed,22 consistent with the general propensity of palladium complexes to promote over-reduction of aryl alkynes.2c, 23 Palladium catalysts have also been shown to promote isomerization of Z-alkenes24 in the presence of hydride donors, such as tin hydrides and silanes.25 Furthermore, palladium-catalyzed hydrosilylation of alkenes26 and alkynes27 is also known. Overall, even though both Sonogashira and semireduction reactions are well-established, merging them into a single process presents significant challenges, and requires a catalyst system that will selectively promote the desired combination of reactions and suppress other well-established reaction pathways.
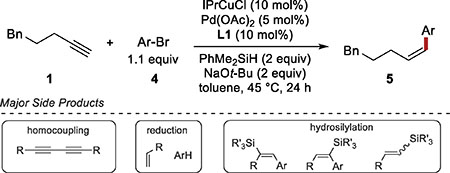
Based on this analysis, we focused on finding a combination of a palladium catalyst and a silane that would minimize the possible side reactions. Considering our previous work on the Z-selective semireduction of alkynes, we focused on exploring various combinations of IPrCuOt-Bu and palladium catalysts in the presence of a silane and an alkoxide (Table 1). We observed a wide range of reactions promoted by various Cu/Pd catalyst combinations, including alkyne homocoupling, reduction of the aryl bromide22, and hydrosilylation of the terminal and internal alkynes (see SI for details).26a
Table 1.
Reaction Development
 | |||
|---|---|---|---|
| entry | deviation from above | yielda | conversion |
| 1. | BINAP | 14% | 71% |
| 2. | PCy3 | 2% | 44% |
| 3. | XPhos | 8% | 100% |
| 4. | SPhos | 26% | 100% |
| 5. | none | 47% | 100% |
| 6. | Me2i-PrSiH | 43%(87%)b | 100% |
| 7. | Me2i-PrSiH, MeOHc | 94% | 100% |
Ar = 4-butylbenzene
GC yields are reported.
Yield after 72 h reported in parenthesis.
MeOH added after the consumption of 1 (2 h). Total reaction time 4 h.
The performance of various phosphine ligands in the hydroarylation reaction revealed some general trends that guided further reaction development. Homocoupling of the alkyne was a major product with common bidentate ligands, such as BINAP (Table 1, entry 1). Monodentate trialkyl phosphine ligands, such as PCy3, fully suppressed the homocoupling, but greatly decreased conversion (entry 2).
With dialkylbiaryl phosphine ligands,28 such as XPhos and SPhos, full consumption of the starting material was achieved, although hydrosilylation and reduction of the starting materials dominated (entries 3 and 4). Interestingly, ligand L129 showed significantly higher selectivity for the desired alkene product than the closely related SPhos (entries 4 vs 5). The use of L1 eliminated the aryl bromide reduction and suppressed hydrosilylation of the alkynes, resulting in increased yield of the hydroarylation product (entry 5). Hydrosilylation was further suppressed by switching to dimethylisopropylsilane (Me2i-PrSiH), resulting in clean formation of the Sonogashira product and a slow semireduction (entry 6). Full conversion of the Sonogashira product was achieved only after 72 h, and the product was obtained in 87% yield and 28:1 Z:E selectivity. The semireduction was dramatically faster when 1.5 equivalents of MeOH was added after the Sonogashira coupling was completed (entry 7). Within two hours of the MeOH addition we observed the formation of the desired Z alkene in 94% yield and 19:1 Z:E selectivity. Once the terminal alkyne (1) has been consumed, the timing of the MeOH addition is not critical. The same results were obtained with MeOH added two or twenty-four hours after the start of the reaction.
In a control experiment, we discovered that adding the silane at the beginning of the reaction was beneficial. Under standard conditions described in entry 7 (Table 1), 99% of the Sonogashira product was formed after 2 h, while in the absence of the silane, only 24% of the coupling product was formed at the same time point.30 Another interesting aspect of our Sonogashira reaction is the tolerance of a copper cocatalyst. Previously, Buchwald et al. have documented deleterious effect of copper co-catalysts in a Sonogashira reaction promoted by a closely related palladium catalyst stabilized by XPhos ligand.31
Having established the reactions conditions for the Z-selective hydroarylation of terminal alkynes (Table 1, entry 7), we explored the scope of the reaction (Table 2). Alkynes containing reductively labile functional groups such as an alkyl chloride (12), alkyl bromide (11), ester (9), and nitrile (8) were compatible with this reaction. In addition, the reaction could be successfully performed in the presence of epoxides (16), silyl ethers (15), and protected amines (10). Notably, both electron-rich and electron-deficient aryl alkynes were competent coupling partners in this reaction (17 and 18).
Table 2.
Scope and Applications of Z-Selective Hydroarylation
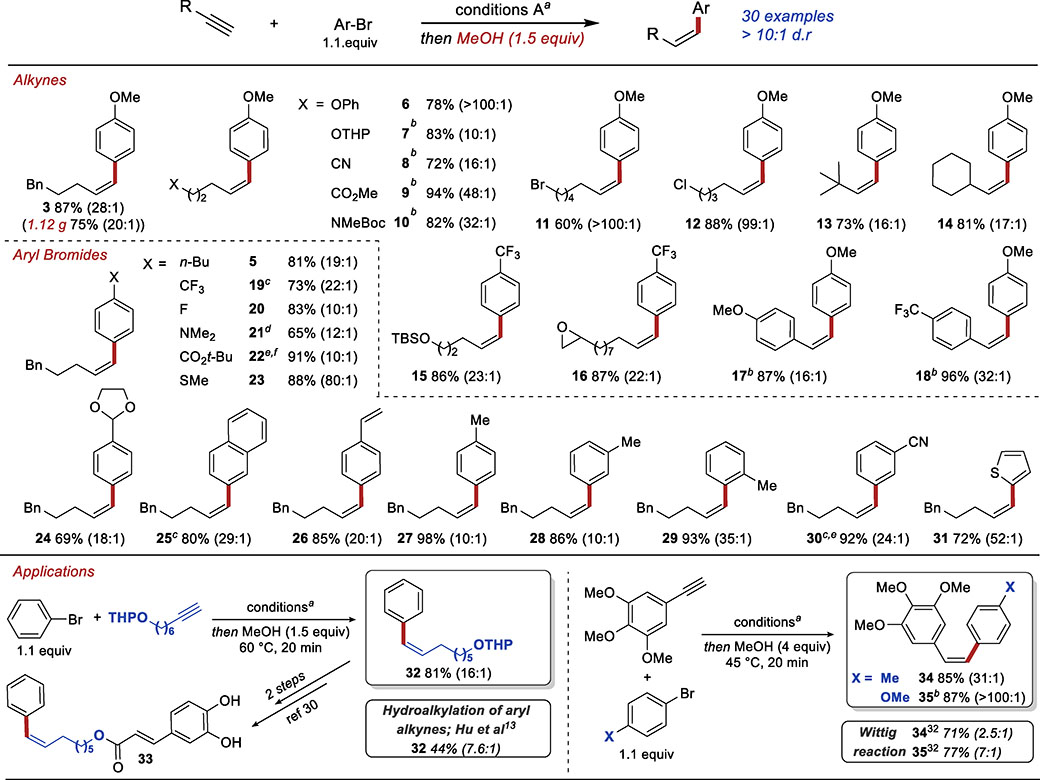 |
Reactions performed on 0.5-mmol scale. Reported are isolated yields of purified mixtures of product diastereoisomers. Z:E ratios of products determined by GC analysis of crude reaction mixtures are reported in paranthesis.
Conditions: IPrCuCl (10 mol%), Pd(OAc)2 (5 mol%), L1 (10 mol%), Mc2i-PrSiH (2 equiv), NaOt-Bu (2 cquiv), toluene, 45 °C, 2 h.
Reaction conditions: IPrCuCl (10 mol%), Pd(OAc)2 (5 mol%), L1 (10 mol%), Me2i-PrSiH (2 equiv), LiOt-Bu (2 equiv), toluene, 45 °C, 3 h, then MeOH (4 equiv) and NaOt-Bu (2 equiv), 60 °C.
i-BuOH was used instead of MeOH.
Reaction performed at 0 °C.
t-BuOH was used instead of MeOH and the semireduction was performed at 60 °C after addition of alcohol.
Reaction was monitored by GC.
A wide range of aryl bromides could also be used as substrates (Table 2). Both electron-rich (3 and 21) and electron-poor (19, 22 and 30) aryl bromides were viable substrates, as were heteroaryl bromides. A variety of functional groups were tolerated on the arene substrate, including thioethers (23), acetals (24), and alkenes (26). Finally, products derived from ortho-, meta-, and para-substituted aryl bromides were isolated in high yields and good to excellent diastereoselectivities (27-29).
To demonstrate the utility of the new method, we prepared 3 on a gram scale. We also used the hydroarylation reaction in the synthesis of biologically relevant compounds shown in Table 2. These applications allow us to make a direct comparison to other methods previously used to accomplish the synthesis of aryl alkenes. Compound 32 was used in the synthesis of caffeic acid derivative 33, which showed selective antiproliferative activity in certain highly metastatic carcinoma cell lines.32 The compound was originally prepared using a Wittig reaction (25% yield and 3.3:1 Z:E selectivity).32 More recently, Hu et al. prepared 32 by Z-selective hydroalkylation of aryl acetylenes using 3 equiv of the alkyl iodide (44% yield and 7.6:1 Z:E selectivity).13a The hydroarylation of terminal alkyne shown in Table 2 provided 32 in 81% yield and 16:1 Z:E selectivity. Compounds 34 and 35 are among the most active analogues of natural product Combretastatin A4, which is a potent inhibitor of tubulin polymerization.33 Like many other analogues, these have been prepared with relatively low Z-selectivity using Wittig reaction.34 Using our hydroarylation reaction both compounds were prepared in high yield (>80%) and with excellent Z-selectivity (>30:1).
To ensure high Z-selectivity in synthesis of compounds shown in Table 2, we monitored the reaction progress by TLC, and stopped the reactions when the products of the Sonogashira coupling were consumed. Careful monitoring of the reaction progress by gas chromatography (GC) established that in several representative cases, high Z selectivity (10:1) can be achieved in a twenty-minute window within the first hour after the addition of MeOH (Table 3). In the absence of MeOH, isomerization is significantly slower and good Z selectivity is observed even 24 h after the complete consumption of the Sonogashira product (Table 1, entry 6: after 96 h, we obtained the Z-alkene in 87% yield and Z:E = 11:1).
Table 3.
Change in Z Selectivity Over Time
 | ||||||
|---|---|---|---|---|---|---|
| Ar | 20 min | 25 min | 30 min | 35 min | 40 min | Yield d |
 |
20:1 | 18:1 | 13:1 | 10:1 | 10:1 | 92 – 95 |
 |
>100:1c | 18:1 | 18:1 | 17:1 | 13:1 | 70 – 79 |
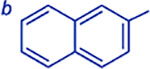 |
28:1 | 28:1 | 27:1 | 26:1 | 25:1 | 84 – 89 |
Reported selectivities of crude reaction mixture.
i-BuOH used instead of MeOH
Reaction mixture contains <10% of alkyne.
Combined yileds of Z and E isomers.
We found it convenient that the rate of the semireduction can be adjusted by varying the temperature and the alcohol additive. The semireduction of internal alkynes containing an electron-deficient arene was slow in the presence of MeOH and significantly faster in the presence of isobutanol (i-BuOH). For example, with MeOH as an additive, the syntheses of compounds 19 and 25 required more than 24 h, while with i-BuOH the reductions were completed in less than 2 h. Conversely, under the standard reaction conditions, the reduction of the Sonogashira products with electron-rich aryl bromides (21) was completed in several minutes, which made monitoring the reaction progress difficult and resulting in low Z selectivity. At a lower temperature, the reduction was completed in 6 h. Finally, in syntheses of several products shown in Table 2, we used LiOt-Bu instead of NaOt-Bu. We noticed that this was required with more acidic alkynes that contain electron-withdrawing groups. In these reactions, NaOt-Bu was completely ineffective in promoting Sonogashira coupling and we could recover the starting materials. Monitoring reactions between different alkynes and the two bases by in situ 1H NMR did not reveal any clear differences in these reactions.
E-Selective Hydroarylation of Alkynes
Next, we explored the development of E-selective hydroarylation of alkynes. The formation of the E isomer would involve the execution of the following three catalytic processes in tandem: Sonogashira coupling, semireduction, and alkene isomerization (Scheme 3c). The feasibility of this approach was supported by the strong thermodynamic preference for the E isomer of aryl alkenes. For example, the equilibrium constant for isomerization of Z-1-phenyl-1-propene to the E isomer is 32.2.20 Furthermore, several mechanisms for palladium-catalyzed alkene isomerization have been established through detailed mechanistic studies.20
Despite numerous documented mechanisms for palladium-catalyzed isomerization of aryl alkenes, previous efforts to develop preparatively useful method for this transformation encountered significant problems. Spencer et al. have shown that (MeCN)2PdCl2 promotes alkene isomerization through a cationic intermediate.24 Good yields and selectivities are obtained under mild reaction conditions. However, the reaction is limited to electron-rich aryl alkenes that can support the formation of the carbenium intermediate. A more general method based on reversible hydropalladation of alkenes was later developed by Jung et al.25 However, they found that if silane is used as a hydride source, a significant amount of alkane product is formed well before the thermodynamic E:Z ratio of alkenes can be reached. To avoid the reduction of alkenes, Jung et al. used n-Bu3SnH (2.2 equivalents) as a hydride source.
Based on Jung’s report and our observation that isomerization of the Z-alkene occurs after the Sonogashira product is completely consumed, we explored the formation of E-alkenes using the standard reaction conditions and a range of phosphine ligands (Table 4). QPhos and DavePhos provided promising initial results and showed fast isomerization to the E-isomer. Unfortunately, after 24 h, the reaction with QPhos ligand afforded the alkene mixture with E:Z ratio of only 5.2:1.
Table 4.
Isomerization of the Aryl Alkene Product
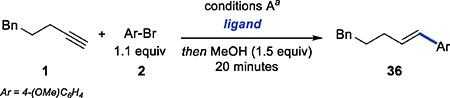 | |||
|---|---|---|---|
| entry | ligand | yieldb | E:Z ratio |
| 1. | QPhos | 100% | 1.9:1 |
| 2. | DavePhos | 81% | 1:2.5 |
| 3. | SPhos | 69% | 1:2.8 |
| 4. | RuPhos | 81% | 1:1.2 |
| 5. | L1 | 90% | 1:29 |
| 6. | L1 (6 days) | 95% | 2:1 |
| 7. | L1 (5 equiv of MeOH, 24 h) | 94% | >100:1 |
See Table 2.
Combined yields of E- and Z-alkenes determined by GC.
In the reaction with DavePhos, we observed a significant amount of hydrosilylation products before E-alkene was the dominant component in the mixture. L1, which was initially optimized for the formation of Z isomer, led to the slowest isomerization of the Z-alkene, but with no side reactions. Even after 6 days, 95% of the alkene was present as a mixture of the two isomers (E:Z =2:1) (entry 6).
Prompted by the excellent combined yield of the alkene obtained with L1, we explored the effect of other reaction parameters on isomerization hoping to identify a perturbation of the standard reaction conditions that would allow us to achieve full isomerization of Z-alkenes. Surprisingly, we found that E-alkenes could be obtained using the standard conditions for Z-selective hydroarylation with one simple change in reaction stoichiometry. With a larger excess of MeOH additive (5 equiv), isomerization proceeds within 24 h to provide E-alkenes in excellent yield and high selectivity (E:Z >100:1)(entry 7). In contrast to the Z-selective hydroarylation, strict monitoring of the reaction progress was not necessary, as we generally observed <5% yield of the alkane over-reduction product after 24 h.
The addition of excess MeOH allowed the synthesis of a wide range of E-aryl alkenes in yields and diastereoselectivities that were comparable to those observed in the synthesis of Z-alkenes (Table 5) We observed a similar functional group compatibility and general scope of the reaction. Nitriles, esters, epoxides, alkyl halides, tertiary amines, acetals, and silyl ethers were all compatible with the reaction conditions. Both aryl and alkyl alkynes were, again, viable substrates.
Table 5.
Scope and Applications of E-Selective Hydroarylation
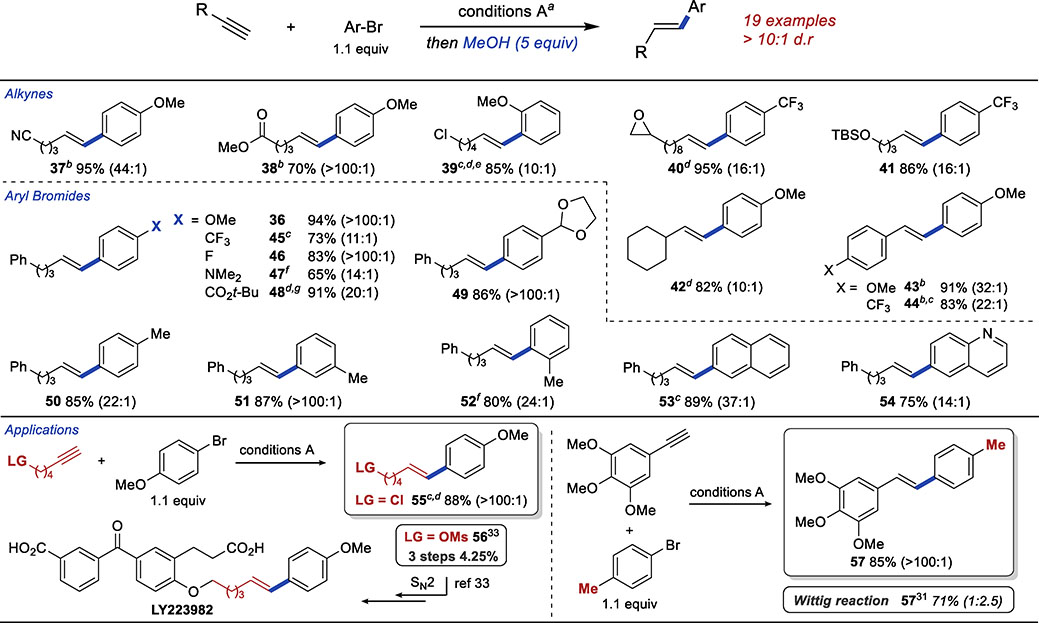 |
Reactions performed on 0.5-mmol scale. Reported are isolated yields of purified mixtures of product diastereoisomers. E:Z ratios reported in paranthesis, were determined by GC analysis of crude reaction mixtures.
See Table 2.
Reaction conditions: Same as A, except LiOt-Bu instead of NaOt-Bu, then after 3h, NaOt-Bu (2 equiv), MeOH, 60 °C.
i-BuOH used instead of MeOH
Reaction placed at 60 °C after addition of alcohol.
Pd(OAc)2 (10 mmol%), L1 (20 mol%)
0.5 equiv Me2i-PrSiH added after 2 h in addition to 5.0 equiv MeOH
t-BuOH used instead of MeOH.
We have also used the E-selective hydroarylation in the synthesis of biologically active compounds or their precursors. Electrophile 56 was previously used in the synthesis of LY223982, a Leukotriene B4 antagonist developed by Ely Lilly & Co.35 56 was originally prepared in 3 steps and 5% yield and was used in alkylation of a phenol en route to the target molecule.35 We have prepared an alternative electrophile 55 in one step from commercially available materials, in 88% yield and in excellent selectivity (>100:1). We have also prepared 57 in 85% yield and >100:1 selectivity. In the context of an SAR study of Combretastatin A4,33 57 was previously prepared using Wittig reaction in 71% yield as a mixture of E and Z isomers (E:Z = 1:2.5).
Overall, the two hydroarylation reactions shown in Tables 2 and 5 allow us to access both Z- and E-aryl alkenes, using one set of starting materials and reagents, and one catalyst system.
Mechanism of Hydroarylation Reactions
Considering the established mechanisms of the Sonogashira coupling16b and the copper-catalyzed semireduction,18–19 a plausible mechanism for the Z-selective hydroarylation is presented in Scheme 4. Monitoring the reaction progress confirmed that the starting materials (I and II) are fully converted to the Sonogashira product III before the semireduction.
Scheme 4.

Plausible Mechanism of Z-Selective Hydroarylation
In the context of this general mechanism, we were interested in understanding the relative contributions of the two catalysts to the individual steps of the reaction. First, we explored the role of the palladium catalyst in the semireduction and found evidence that under certain reaction conditions the semireduction of the Sonogashira product is more complicated than Scheme 4 suggests. Experiments presented in Scheme 5a show that both palladium and copper catalysts independently promote selective semireduction. These results suggest that in the catalytic hydroarylation both catalysts likely contribute to the semireduction of intermediate III.36 However, in the absence of the copper catalyst, palladium-catalyzed semireduction does not result in complete conversion and the maximum yield of the semireduction product was 41% after 24 h (see SI). Furthermore, the relative contributions of the palladium and copper catalysts to the semireduction depend on the alcohol additive used in the reaction.
Scheme 5.

Role of Pd and Cu Catalysts in the Semireduction
When i-BuOH is used instead of MeOH, the contribution of the palladium catalyst is significantly reduced, and the semireduction is mostly effected by the copper catalyst (Scheme 5b). Control experiments presented in the SI (Table S8) confirm that palladium contribution depends on the alcohol additive and not the substrate of the reaction.
We were also interested in understanding the role of the alcohol additive and the reasons for the changes to the standard conditions that were necessary with certain substrates. Initially, we explored the mechanism behind the significant acceleration of the semireduction in the presence of MeOH additive, which we observed in both E- and Z-selective hydroarylation reaction. We found that the rate of the stoichiometric hydrocupration is significantly higher using IPrCuOMe vs IPrCuOt-Bu (Scheme 6a) (See SI for complete kinetics data), although IPrCuOMe is only partially soluble in toluene. Considering that the alkoxide group plays no role in the hydrocupration of the alkyne, this observation implies significant difference in the rate of IPrCuH formation from IPrCuOR and a silane. These results are consistent with a computational study, which attributed a high activation barrier for the reaction between NHCCuOR and trialkylsilanes to significant steric repulsion in the transition state.37 Surprisingly, this effect has not been experimentally observed in reactions mediated by NHCCuH.
Scheme 6.

Effect of MeOH on Semireduction
The rate of protonation of the alkenyl copper intermediate 60 was also significantly higher with MeOH than with t-BuOH, as demonstrated by the stoichiometric experiment shown in Scheme 6b. Consistent with stoichiometric experiments, we found that the catalytic semireduction of 58 was significantly faster in the presence of MeOH than in the presence of t-BuOH (Scheme 6c). Significant concentration of the alkenyl copper intermediate 60 was observed during copper-catalyzed semireduction in the presence of either alcohol. This result indicates that the protonation of the intermediate is at least partially rate limiting. Overall, it is likely that MeOH accelerates the semireduction by facilitating both the formation of the IPrCuH and the protonation of the alkenyl copper intermediate.
To understand the effect of i-BuOH additive in reactions with electron-deficient aryl bromides, we did initial rate measurements for the semireduction performed in the presence of MeOH and i-BuOH (Scheme 7). The experiment confirmed that the reaction is faster in the presence of i-BuOH. In both reactions, we observed a significant amount of the alkenyl copper intermediate (61) throughout the course of the reaction (18–20% of alkenyl copper in the reaction with 20 mol% loading of IPrCuOt-Bu, see SI for details). These results suggest that the resting state of the catalyst in the reaction of electron-deficient alkynes is the alkenyl copper intermediate and that the turnover limiting step is the protonation of this intermediate. Somewhat surprisingly, these results indicate that the protonation of the alkenyl copper intermediate is significantly faster with i-BuOH than with MeOH.
Scheme 7.

i-BuOH in Semireduction
Finally, we also explored aspects of the reaction mechanism specific to the conditions employed in the synthesis of E-aryl alkenes. In this case, the relative contributions of palladium and copper catalysts to the semireduction was dramatically different from their contributions in the Z-selective reaction. With 5 equivalents of MeOH, copper-catalyzed semireduction of the Sonogashira product provides only 10% of the desired product and 10% conversion. The data in Scheme 8a shows the impact the increasing amounts of methanol have on the copper-catalyzed semireduction. These results are consistent with competitive protonation of IPrCuH by methanol we previously observed in the semireduction reaction.19
Scheme 8.

Role of Pd Catalyst and MeOH in E-selective Hydroarylation.
The major implication of these results is that under conditions used in E-selective hydroarylation, the semireduction is predominantly mediated by the palladium catalyst and is promoted by excess MeOH. Experiments shown in Scheme 8b confirm the more prominent role of the palladium catalyst and the key role of MeOH in E-selective hydroarylation.
We were also interested in understanding the mechanism of alkene isomerization in the E-selective hydroarylation. Based on available precedents, isomerization through reversible hydrocupration of aryl alkenes seems unlikely under the conditions employed in our reaction.2b,38 Control experiments shown in Scheme 8b and 9a demonstrate that isomerization requires both the palladium catalyst and the silane. Furthermore, we found that isomerization can be effectively accomplished using only the palladium catalyst and a substoichiometric amount of a silane (Scheme 9b). Interestingly, in contrast to the results reported by Jung,25 we did not observe the formation of the alkane products. Based on observations we made during the development of the reaction, we believe that both the ligand and the silane used in the reaction are responsible for this difference.
Scheme 9.

Palladium-Catalyzed Alkene Isomerization
Considering the conditions necessary for isomerization, it seems likely that the formation of E-aryl alkene proceeds though a reversible hydropalladation of the alkene, one of the established mechanisms for alkene isomerization. Further evidence for this mechanism was provided by isotope incorporation experiments (Scheme 9c). With 10 mol% of silane, we observed selective and full incorporation of a proton into d2-3. The position of the proton incorporation is consistent with the established regioselectivity of styrene hydropalladation.39 In the presence of MeOH, we observed the complete exchange of the same deuterium label, suggesting that the mechanism remains the same in the presence of methanol. The extent of deuterium exchange in the presence of MeOH suggests fast exchange between Pd-H and MeOH.40
In summary, our exploration of the reaction mechanism provided insight into the roles of the two catalysts and the effects that alcohol additives and various changes from the standard reaction conditions have on the reaction. The most interesting finding is that the alcohol additive changes the role of each of the two catalysts in the hydroarylation. i-BuOH inhibits palladium catalyzed semireduction, while excess MeOH, suppresses copper-catalyzed semireduction and promotes both the palladium-catalyzed semireduction and alkene isomerization. This complementary reactivity of the two metal catalysts is essential for the success of the hydroarylation.
CONCLUSION
In conclusion, we have developed a diastereodivergent method for hydroarylation of terminal alkynes. The new method involves a sequence of catalytic reactions promoted by a combination of palladium and copper catalysts operating in tandem. The Z-selective hydroarylation is achieved through Sonogashira coupling of alkynes and aryl bromides, followed by semireduction. The E-selective hydroarylation involves an additional isomerization of the Z-aryl alkene. The new hydroarylation reactions allow access to both isomers of aryl alkenes using the same set of starting materials, and the same combination of catalysts. The hydroarylation reactions have excellent substrate scopes and functional group compatibility and provide the desired products in high yields and with high diastereoselectivity. Mechanistic experiments indicate different roles of palladium and copper catalysts in Z- and E-selective hydroarylation reactions. While both are involved in the Sonogashira coupling, the semireduction is predominantly promoted by a copper catalyst in the Z-selective reaction, while the palladium catalyst is necessary for both the semireduction and isomerization in the E-selective reaction.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH and by the University of Washington.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Experimental procedure and product characterization. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).(a) Maryanoff BE; Reitz AB Chem. Rev 1989, 89, 863 [Google Scholar]; (b) Meijere A; Brase S; Oestreich M Metal-Catalyzed Cross-Coupling Reactions and More; Wiley-VCH Verlag GmbH & Co. KGaA, 2014. [Google Scholar]
- (2).(a) Radkowski K; Sundararaju B; Fürstner A Angew. Chem., Int. Ed 2013, 52, 355. [DOI] [PubMed] [Google Scholar]; (b) Karunananda MK; Mankad NP J. Am. Chem. Soc 2015, 137, 14598. [DOI] [PubMed] [Google Scholar]; (c) Oger C; Balas L; Durand T; Galano J-M Chem. Rev 2013, 113, 1313. [DOI] [PubMed] [Google Scholar]; (d) Fu S; Chen N-Y; Liu X; Shao Z; Luo S-P; Liu Q J. Am. Chem. Soc 2016, 138, 8588. [DOI] [PubMed] [Google Scholar]; (e) Richmond E; Moran J J. Org. Chem 2015, 80, 6922. [DOI] [PubMed] [Google Scholar]
- (3).(a) Crowe WE; Goldberg DR J. Am. Chem. Soc 1995, 117, 5162 [Google Scholar]; (b) Chatterjee AK; Choi T-L; Sanders DP; Grubbs RH J. Am. Chem. Soc 2003, 125, 11360. [DOI] [PubMed] [Google Scholar]; (c) Cossy J; BouzBouz S; Hoveyda AH J. Organomet. Chem 2001, 634, 216. [Google Scholar]
- (4).(a) Meek SJ; O’Brien RV; Llaveria J; Schrock RR; Hoveyda AH Nature 2011, 471, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Herbert MB; Grubbs RH Angew. Chem., Int. Ed 2015, 54, 5018. [DOI] [PubMed] [Google Scholar]; (c) Koh MJ; Nguyen TT; Lam JK; Torker S; Hyvl J; Schrock RR; Hoveyda AH Nature 2017, 542, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Xu C; Shen X; Hoveyda AH J. Am. Chem. Soc 2017, 139, 10919. [DOI] [PubMed] [Google Scholar]
- (5).Diastereodivergence has been reported in hydroarylation of two symmetrical diarylalkynes using chromones as coupling partners. See:; Min M; Kim D; Hong S Chem. Commun 2014, 50, 8028. [DOI] [PubMed] [Google Scholar]
- (6).Nakamura K; Nishikata T ACS Catal. 2017, 7, 1049. [Google Scholar]
- (7).(a) Schipper DJ; Hutchinson M; Fagnou K J. Am. Chem. Soc 2010, 132, 6910. [DOI] [PubMed] [Google Scholar]; (b) Kim N; Kim KS; Gupta AK; Oh CH Chem. Commun 2004, 618. [DOI] [PubMed] [Google Scholar]; (c) Lautens M; Yoshida M Org. Lett 2002, 4, 123. [DOI] [PubMed] [Google Scholar]; (d) Liu Z; Derosa J; Engle KM J. Am. Chem. Soc 2016, 138, 13076. [DOI] [PubMed] [Google Scholar]; (e) Shirakawa E; Takahashi G; Tsuchimoto T; Kawakami Y Chem. Commun 2001, 2688. [DOI] [PubMed] [Google Scholar]
- (8).(a) Nakao Y; Kanyiva KS; Oda S; Hiyama T J. Am. Chem. Soc 2006, 128, 8146. [DOI] [PubMed] [Google Scholar]; (b) Nakao Y; Kanyiva KS; Hiyama T J. Am. Chem. Soc 2008, 130, 2448. [DOI] [PubMed] [Google Scholar]; (c) Kortman GD; Hull KL ACS Catal. 2017, 7, 6220. [Google Scholar]
- (9).(a) Jia C; Lu W; Oyamada J; Kitamura T; Matsuda K; Irie M; Fujiwara Y J. Am. Chem. Soc 2000, 122, 7252 [Google Scholar]; (b) Jia C; Piao D; Oyamada J; Lu W; Kitamura T; Fujiwara Y Science 2000, 287, 1992. [DOI] [PubMed] [Google Scholar]
- (10).Currently, the best practical approach to Z-selective hydroarylation of alkynes relies on the stoichiometric preparation of a Z-alkenyl metal intermediate and its use in palladium-catalyzed cross coupling with aryl halides. See,; Takami K; Yorimitsu H; Oshima K Org. Lett 2002, 4, 2993. [DOI] [PubMed] [Google Scholar]
- (11).(a) Reetz MT; Sommer K Eur. J. Org. Chem 2003, 2003, 3485 [Google Scholar]; (b) Tsuchimoto T; Maeda T; Shirakawa E; Kawakami Y Chem. Commun 2000, 1573 [Google Scholar]; (c) Oyamada J; Kitamura T Tetrahedron 2007, 63, 12754. [Google Scholar]
- (12).Tunge JA; Foresee LN Organometallics 2005, 24, 6440. [Google Scholar]
- (13).For a complementary approach based on hydroalkylation of aryl alkynes, see:; (a) Cheung CW; Zhurkin FE; Hu X J. Am. Chem. Soc 2015, 137, 4932. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kambe N; Moriwaki Y; Fujii Y; Iwasaki T; Terao J Org. Lett 2011, 13, 4656. [DOI] [PubMed] [Google Scholar]
- (14).(a) Lohr TL; Marks TJ Nat. Chem 2015, 7, 477. [DOI] [PubMed] [Google Scholar]; (b) Wasilke J-C; Obrey SJ; Baker RT; Bazan GC Chem. Rev 2005, 105, 1001. [DOI] [PubMed] [Google Scholar]
- (15).(a) Li L; Herzon SB Nat. Chem 2014, 6, 22. [DOI] [PubMed] [Google Scholar]; (b) Trost BM; Machacek MR; Faulk BD J. Am. Chem. Soc 2006, 128, 6745. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Goldman AS; Roy AH; Huang Z; Ahuja R; Schinski W; Brookhart M Science 2006, 312, 257. [DOI] [PubMed] [Google Scholar]; (d) Persson BA; Larsson ALE; Le Ray M; Backvall J-E J. Am. Chem. Soc 1999, 121, 1645 [Google Scholar]; (e) Yuki Y; Takahashi K; Tanaka Y; Nozaki K J. Am. Chem. Soc 2013, 135, 17393. [DOI] [PubMed] [Google Scholar]; (f) Li Z; Assary RS; Atesin AC; Curtiss LA; Marks TJ J. Am. Chem. Soc 2014, 136, 104. [DOI] [PubMed] [Google Scholar]; (g) Tjutrins J; Arndtsen BA Chem. Sci 2017, 8, 1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).(a) Sonogashira K; Tohda Y; Hagihara N Tetrahedron Lett. 1975, 16, 4467 [Google Scholar]; (b) Chinchilla R; Nájera C Chem. Rev 2007, 107, 874. [DOI] [PubMed] [Google Scholar]
- (17).Mankad NP; Laitar DS; Sadighi JP Organometallics 2004, 23, 3369. [Google Scholar]
- (18).Semba K; Fujihara T; Xu T; Terao J; Tsuji Y Adv. Synth. Catal 2012, 354, 1542. [Google Scholar]
- (19).Whittaker AM; Lalic G Org. Lett 2013, 15, 1112. [DOI] [PubMed] [Google Scholar]
- (20).Tan EHP; Lloyd-Jones GC; Harvey JN; Lennox AJJ; Mills BM Angew. Chem., Int. Ed 2011, 50, 9602. [DOI] [PubMed] [Google Scholar]
- (21).(a) Hundertmark T; Littke AF; Buchwald SL; Fu GC Org. Lett 2000, 2, 1729. [DOI] [PubMed] [Google Scholar]; (b) Mohamed RK; Mondal S; Gold B; Evoniuk CJ; Banerjee T; Hanson K; Alabugin IV J. Am. Chem. Soc 2015, 137, 6335. [DOI] [PubMed] [Google Scholar]
- (22).Bhattacharjya A; Klumphu P; Lipshutz BH Org. Lett 2015, 17, 1122. [DOI] [PubMed] [Google Scholar]
- (23).Laren M. W. v.; Elsevier CJ Angew. Chem. Int. Ed. Engl 1999, 38, 3715. [DOI] [PubMed] [Google Scholar]
- (24).Yu J; Gaunt MJ; Spencer JB J. Org. Chem 2002, 67, 4627. [DOI] [PubMed] [Google Scholar]
- (25).Kim IS; Dong GR; Jung YH J. Org. Chem 2007, 72, 5424. [DOI] [PubMed] [Google Scholar]
- (26).(a) Motoda D; Shinokubo H; Oshima K Synlett 2002, 2002, 1529 [Google Scholar]; (b) Uozumi Y; Kitayama K; Hayashi T Tetrahedron: Asymmetry 1993, 4, 2419. [Google Scholar]
- (27).(a) Planellas M; Guo W; Alonso F; Yus M; Shafir A; Pleixats R; Parella T Adv. Synth. Catal 2014, 356, 179 [Google Scholar]; (b) Bal Reddy C; Shil AK; Guha NR; Sharma D; Das P Catal. Lett 2014, 144, 1530 [Google Scholar]; (c) Zhang J.-w.; Lu G.-p.; Cai C Green Chem. 2017, 19, 2535. [Google Scholar]
- (28).(a) Surry DS; Buchwald SL Angew. Chem., Int. Ed 2008, 47, 6338. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Martin R; Buchwald SL Acc. Chem. Res 2008, 41, 1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Ligand L1 has not been previously used in catalysis but has been prepared as an intermediate in the synthesis of a ligand immobilized on polystyrene beads. See:; Parrish CA; Buchwald SL J. Org. Chem 2001, 66, 3820. [DOI] [PubMed] [Google Scholar]
- (30).We suspect that under standard reaction conditions the silane is responsible for the formation of the active palladium catalyst by facilitating the reduction of Pd(OAc)2 to palladium(0). The same silane effect was not observed with Pd2(dba)3 as the palladium source, although with this catalyst the reaction was significantly slower than under our standard conditions (29% yield of Sonogashira product after 8 hours, and >99% after 24 hours). See SI for details.
- (31).Gelman D; Buchwald Stephen L Angew. Chem., Int. Ed. 2003, 42, 5993. [DOI] [PubMed] [Google Scholar]
- (32).Nagaoka T; Banskota AH; Tezuka Y; Saiki I; Kadota S Bioorg. Med. Chem. Lett 2002, 10, 3351. [DOI] [PubMed] [Google Scholar]
- (33).Tron GC; Pirali T; Sorba G; Pagliai F; Busacca S; Genazzani AA J. Med. Chem 2006, 49, 3033. [DOI] [PubMed] [Google Scholar]
- (34).(a) Cushman M; Nagarathnam D; Gopal D; Chakraborti AK; Lin CM; Hamel E J. Med. Chem 1991, 34, 2579. [DOI] [PubMed] [Google Scholar]; (b) Cushman M; Nagarathnam D; Gopal D; He HM; Lin CM; Hamel E J. Med. Chem 1992, 35, 2293. [DOI] [PubMed] [Google Scholar]
- (35).Gapinski DM; Mallett BE; Froelich LL; Jackson WT J. Med. Chem 1990, 33, 2807. [DOI] [PubMed] [Google Scholar]
- (36).For complete data see SI.
- (37).Vergote T; Nahra F; Merschaert A; Riant O; Peeters D; Leyssens T Organometallics 2014, 33, 1953. [Google Scholar]
- (38).Mazzacano TJ; Mankad NP ACS Catal. 2017, 7, 146. [Google Scholar]
- (39).Hayashi T; Hirate S; Kitayama K; Tsuji H; Torii A; Uozumi Y J. Org. Chem 2001, 66, 1441. [DOI] [PubMed] [Google Scholar]
- (40).Lipshutz BH; Servesko JM; Taft BR J. Am. Chem. Soc 2004, 126, 8352. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.