

Collagen-binding ability improves serum albumin as an anti-cancer drug carrier, resulting in enhanced therapeutic efficacy.
Abstract
Serum albumin (SA) is used as a carrier to deliver cytotoxic agents to tumors via passive targeting. To further improve SA’s tumor targeting capacity, we sought to develop an approach to retain SA-drug conjugates within tumors through a combination of passive and active targeting. SA was recombinantly fused with a collagen-binding domain (CBD) of von Willebrand factor to bind within the tumor stroma after extravasation due to tumor vascular permeability. Doxorubicin (Dox) was conjugated to the CBD-SA via a pH-sensitive linker. Dox-CBD-SA treatment significantly suppressed tumor growth compared to both Dox-SA and aldoxorubicin treatment in a mouse model of breast cancer. Dox-CBD-SA efficiently stimulated host antitumor immunity, resulting in the complete eradication of MC38 colon carcinoma when used in combination with anti–PD-1 checkpoint inhibitor. Dox-CBD-SA decreased adverse events compared to aldoxorubicin. Thus, engineered CBD-SA could be a versatile and clinically relevant drug conjugate carrier protein for treatment of solid tumors.
INTRODUCTION
Serum albumin (SA) is the most abundant protein in blood (1). A number of compounds including small molecules, peptides, and cytokines have been fused to, conjugated to, or coformulated with SA for improved drug delivery to disease lesions. SA-binding fatty acid–modified insulin analog (2), SA-fused interferon-α (IFN-α) (3), and SA nanoparticle–formulated paclitaxel (Abraxane) have been developed (4). In these cases, the exceptionally long plasma half-life and/or hydrophilicity of SA contributes to improved pharmacokinetics, safety, and efficacy of the drugs (1). Moreover, SA can passively target tumors through the pathological permeability of the tumor vasculature (5), which is an advantage of SA-based drugs for cancer therapy.
On the basis of the rationale that combined passive and active targeting is beneficial in tumor drug delivery (6), molecular engineering approaches aiming for further improvement of SA-based drugs have been explored, such as incorporation of the targeting ligands cyclic arginylglycylaspartic acid peptide (7) or mannose-6-phosphate to SA (8). However, superior antitumor efficacy of modified SA has been shown compared to free drugs but not to drugs associated with unmodified SA in either case. Recently, we have shown the targeted delivery of checkpoint inhibitor (CPI) antibodies and the cytokine interleukin-2 (IL-2) using a collagen-binding domain (CBD), namely, the A3 domain of von Willebrand factor (VWF) (9). The A3 domain of VWF has the highest affinity for collagen type I and type III among reported nonbacterial origin proteins/peptides (10). Collagens are not accessible in most tissues due to the low permeability of the vasculature, yet are abnormally exposed to the bloodstream in the tumor microenvironment due to the hyperpermeability of the tumor vasculature (6). Thus, collagens are promising targets for cancer drug delivery. In addition, collagens are overexpressed in multiple types of cancers (11, 12). CBD-CPI and CBD–interleukin-2 (IL-2) showed significantly stronger antitumor effects compared with their unmodified forms in multiple murine cancer models (9). In addition, treatment-related adverse events of CPI and IL-2 were greatly suppressed by conjugating or fusing CBD, which demonstrates the usefulness of the CBD-based tumor-targeting strategy (9). Here, we hypothesized that the CBD would be compatible with SA-based drug delivery carriers, because both CBD and SA target vasculature permeability, and CBD fusion adds active targeting ability to SA.
Doxorubicin (Dox) is a small-molecule anticancer drug that is approved for treating a broad spectrum of cancers by the U.S. Food and Drug Administration. Dox internalizes within cells via passive transmembrane diffusion and interferes with DNA functions, leading to death of proliferating cells. Although Dox treatment prolongs survival of some populations of patients, antitumor efficacy is not notable partially due to acquired drug resistance. The poor therapeutic index of Dox also limits its therapeutic use. Considerable toxicity of Dox has been reported in the clinic, including bone marrow suppression, excessive inflammation, and cardiotoxicity (13, 14). To improve efficacy, Dox is often used in combination with other chemotherapeutic agents. Recently, Dox has been reported to facilitate immune cell infiltration into tumors through induction of immunogenic cell death (ICD) (15), suggesting the possibility of synergizing in combination treatment with CPI (16). Other approaches to improving efficacy and maximum tolerated dose of Dox are liposomal formulation (Doxil) (17) and use of a maleimide derivative of Dox with a pH-sensitive cleavable linker (aldoxorubicin), which was developed to achieve conjugation with cysteine-34 (in the human sequence) of circulating SA in situ (18). Aldoxorubicin displays extended blood half-life and accumulation within tumors through passive tumor targeting. The low pH within the tumor tissue (reportedly pH 6.5) allows Dox release from the conjugate with SA. Aldoxorubicin showed improved maximum tolerated dose and efficacy in mouse cancer models (18, 19) and in a clinical trial (20) compared to unmodified Dox.
Here, we designed recombinant mouse SA (CBD-SA) in which the N terminus is fused with the C terminus of the VWF A3 domain, and aldoxorubicin was conjugated to CBD-SA via a pH-dependent cleavable hydrazone linkage before injection (namely, Dox-CBD-SA) (21). We evaluated engineered CBD-SA as a tumor-targeted drug carrier, leading to improved antitumor efficacy by efficient Dox delivery to the tumor microenvironment.
RESULTS
CBD-SA binds to collagen and can be conjugated to Dox
We synthesized Dox-CBD-SA conjugates to target the tumor microenvironment (Fig. 1, A and B). We first investigated the binding abilities of CBD-SA to recombinant collagen protein in vitro. SA was expressed recombinantly with the CBD on the N terminus of mouse SA using a (GGGS)2 linker (table S1). The molecular weight of CBD-SA was analyzed by matrix-assisted laser desorption/ionization–time-of-flight mass spectrometry (MALDI-TOF MS) (fig. S1). We observed strong binding affinities [nanomolar range dissociation constant (Kd) values] of CBD-SA to collagen type I and type III (Fig. 1C and fig. S2). For Dox conjugation, we first thiolated the lysine residues of CBD-SA using 2-iminothiolane (also known as Traut’s reagent). Then, aldoxorubicin was covalently conjugated to CBD-SA. Unmodified SA was also conjugated with aldoxorubicin in the same way (Dox-SA). SDS–polyacrylamide gel electrophoresis (PAGE) under nonreducing conditions showed that purified Dox-SA and Dox-CBD-SA are monomeric (fig. S3). Before and after Dox conjugation, the hydrodynamic size of CBD-SA was measured (fig. S4). The results also showed that CBD-SA exists in a monomeric form, and Dox conjugation did not alter this character even after a lyophilization/reconstitution cycle. Approximately three Dox molecules were conjugated per SA molecule and per CBD-SA molecule (Fig. 1D). Notably, our conjugation method would not affect the binding ability of CBD-SA to collagens since there are no cysteine or lysine residues at the binding interface between the VWF A3 domain and human collagen III (Protein Data Bank: 4DMU; fig. S5) (22). This interface is also far from the C-terminal fusion site to the SA domain.
Fig. 1. Synthesis and characterization of Dox-CBD-SA.
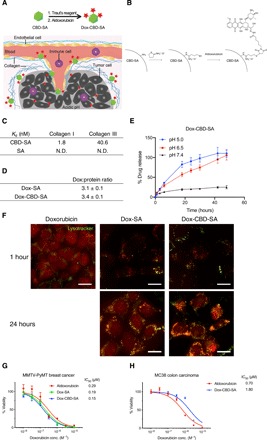
(A) Schematic of CBD-SA mediated drug delivery. (B) Synthesis scheme of Dox-CBD-SA. (C) Affinities [dissociation constant (Kd) values are shown] of CBD-SA and SA against collagen type I and collagen type III were measured by enzyme-linked immunosorbent assay (ELISA). N.D., not determined due to low signal. Graphs with [concentrations] versus [signals] are shown in fig. S2. Two experimental replicates. (D) Dox conjugation ratio per protein is presented. Values were calculated on the basis of the results of bicinchoninic acid assay protein quantification assay (proteins) and absorbance at 495 nm (Dox) (mean ± SD of three experimental replicates). (E) Dox release kinetics from Dox-CBD-SA under three different pH conditions was evaluated by fluorescence (excitation at 495 nm, emission at 590 nm) (n = 3, mean ± SD; two experimental replicates). (F) MMTV-PyMT cells were seeded and incubated overnight. Dox, Dox-SA, or Dox-CBD-SA was added (red). Cells were also stained with LysoTracker (green). Scale bars, 20 μm. Representative pictures are presented. Two experimental replicates. (G and H) Cytotoxicity of Dox variants against MMTV-PyMT cells or MC38 cells in vitro (n = 6, mean ± SEM). Two experimental replicates. IC50, half maximal inhibitory concentration.
Dox is released under acidic pH conditions
Because Dox is linked to SA with a pH-sensitive cleavable linker, we examined the release kinetics of Dox from conjugates under different pH conditions (Fig. 1E). After 48 hours of incubation, Dox release from Dox-CBD-SA reached a maximum at pH 5.0 and 6.5 (reported tumor microenvironment condition). In contrast, only about 20% of Dox was released at pH 7.4 after 48 hours. Dox-SA showed similar release profiles (fig. S6). These data show the pH-dependent release of Dox from conjugates, consistent with previously reported release kinetics of small chemicals linked via a hydrazone linkage (21).
Dox conjugates are taken up by cancer cells and retain cytotoxicity
We compared the intracellular localization of Dox conjugates with free drug using confocal laser scanning microscopy by detecting the fluorescence of Dox. Because Dox is a major drug for breast cancer (23), here we chose mouse mammary tumor virus-polyomavirus middle T antigen (MMTV-PyMT) murine breast cancer as an experimental model. The MMTV-PyMT cells were cultured in the presence of Dox or Dox conjugates, and then their intracellular uptake was assessed (Fig. 1F). After 1 hour of incubation, free Dox was detected in the cytoplasm, intracellular acidic organelles, and preferentially in the nucleus, indicating that its delivery is mediated by passive transmembrane diffusion. In contrast, 1 hour after addition of either Dox-SA or Dox-CBD-SA, the cytoplasm did not show strong fluorescence compared to the unconjugated Dox. Rather, punctate fluorescence was observed, with some puncta colocalized with lysosomes, suggesting that Dox-SA and Dox-CBD-SA were both internalized via endocytosis. Twenty-four hours after the addition of Dox conjugates, we observed Dox-derived fluorescence in the nucleus as well, suggesting that the acidic pH in intracellular organelles induced drug liberation from the conjugates. We next examined the cytotoxicity of the different Dox forms in vitro. MMTV-PyMT cells or MC38 colon carcinoma cells were seeded and incubated in the presence of the Dox forms for 3 days. Viability tests showed that all three Dox forms have comparable cytotoxicity in vitro (Fig. 1, G and H).
Dox-CBD-SA demonstrates comparable blood plasma pharmacokinetics as aldoxorubicin and accumulates in tumors
Aldoxorubicin reacts with endogenous SA rapidly after intravenous administration; therefore it possesses substantially longer blood plasma half-life compared with Dox (1, 18, 19). We tested the plasma pharmacokinetics of aldoxorubicin with or without prior conjugation of SA and CBD-SA using tumor-free FVB mice. After intravenous injection, similar blood plasma half-lives of aldoxorubicin, Dox-SA, and Dox-CBD-SA were observed (Fig. 2, A and B). We also examined the plasma pharmacokinetics of fluorescently labeled SA and CBD-SA with a pH-insensitive linker (fig. S7). The result showed that the half-lives of each protein conjugated with either Dox or dye were similar, suggesting that Dox liberation from the conjugates does not occur in the blood circulation.
Fig. 2. Dox-CBD-SA shows comparable plasma pharmacokinetics with Dox-SA and higher tumor accumulation than aldoxorubicin and Dox-SA.
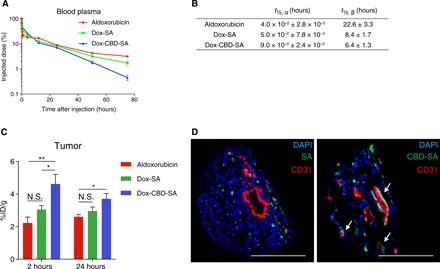
(A) Aldoxorubicin, Dox-SA, or Dox-CBD-SA (5 mg/kg on a Dox basis) was administered to tumor-free FVB mice via tail vein injection. Blood plasma was collected at the indicated time points. Plasma concentration of Dox was measured by fluorescence (mean ± SEM; n = 4 for aldoxorubicin, n = 5 for Dox-SA and Dox-CBD-SA). (B) Plasma half-lives of Dox were calculated using two-phase exponential decay: MFI (t) = Ae−αt + Be−βt. t½, α, fast clearance half-life; t½, β, slow clearance half-life (mean ± SEM; n = 4 for aldoxorubicin, n = 5 for Dox-SA and Dox-CBD-SA). (C) MMTV-PyMT tumor-bearing mice were treated with aldoxorubicin, Dox-SA, or Dox-CBD-SA (4.16 mg/kg on a Dox basis). At the indicated time points, tumors were harvested, and the amount of Dox within the tumors was quantified (mean ± SEM; n = 5 for 2 hours, n = 7 for 24 hours per group). (D) DyLight 488–labeled SA (100 μg) or equimolar amounts of DyLight 488–labeled CBD-SA were injected intravenously to MMTV-PyMT tumor-bearing mice. One hour after injection, tumors were harvested and fluorescence was analyzed by confocal microscopy. Tissues were also stained with 4′,6-diamidino-2-phenylindole (DAPI) and anti-CD31 antibody. Scale bars, 100 μm. Representative images of three tumors each. Two experimental replicates. Statistical analyses were done using analysis of variance (ANOVA) with Tukey’s test. *P < 0.05; **P < 0.01; N.S., not significant.
We next hypothesized that CBD fusion to SA would increase the amount of Dox within the tumor via active targeting against collagens within the tumor microenvironment. To test this hypothesis, we measured the amounts of Dox within tumor tissues after a single intravenous administration. Dox-CBD-SA showed significantly higher tumor accumulation of Dox compared to aldoxorubicin and Dox-SA at 2 hours after administration (Fig. 2C). Conjugation with CBD-SA achieved the highest tumor accumulation of Dox after 24 hours of injection as well, showing a significant increase compared to aldoxorubicin. Histological analysis revealed that fluorescently labeled CBD-SA colocalized with CD31 staining within tumor tissue, demonstrating that CBD-SA targets the tumor vasculature (Fig. 2D). These data demonstrate that CBD fusion to SA to which Dox is conjugated enables Dox to target tumors, resulting in enhanced tumor accumulation of Dox.
Dox-CBD-SA demonstrates superior efficacy in the MMTV-PyMT murine breast cancer model
Motivated by the plasma pharmacokinetics and tumor accumulation studies, we evaluated the antitumor effects of Dox-CBD-SA in vivo. MMTV-PyMT orthotopic tumor-bearing mice received a single intravenous injection of the Dox forms (5 mg/kg on a Dox basis) via the tail vein. Dox-SA and Dox-CBD-SA significantly suppressed tumor growth, whereas aldoxorubicin did not (Fig. 3, A and C to F). This suggests that preconjugation of Dox with SA would provide a higher therapeutic effect than in situ conjugation of aldoxorubicin with endogenous SA. Notably, Dox-CBD-SA showed a greater therapeutic effect compared to Dox-SA. Dox-CBD-SA treatment significantly extended the survival rate compared to all the other groups (Fig. 3B) and induced complete tumor remission in 2 of 12 mice. These data demonstrate that CBD-fused SA functions as a superior Dox carrier compared to unmodified SA in terms of antitumor efficacy.
Fig. 3. Dox-CBD-SA shows enhanced antitumor efficacy and infiltration of lymphocytes into tumor in the MMTV-PyMT breast cancer model.
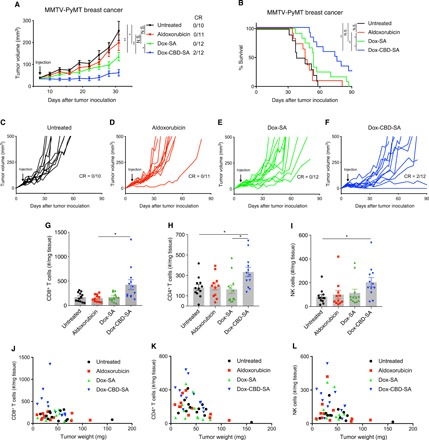
(A) MMTV-PyMT cells (5 × 105) were inoculated into FVB mice on day 0. Aldoxorubicin, Dox-SA, or Dox-CBD-SA (5 mg/kg on a Dox basis) was injected intravenously on day 7. Graphs depict tumor volume until the first mouse died (mean ± SEM). (B) Survival rate. (C to F) Individual tumor growth curves. CR indicates complete response frequency. Three experimental replicates. (G to L) MMTV-PyMT cells (5 × 105) were inoculated on day 0. Aldoxorubicin, Dox-SA, or Dox-CBD-SA (5 mg/kg on a Dox basis) was injected intravenously on day 7. Lymphocytes within tumors were extracted on day 14, followed by flow cytometric analysis. (G to I) Graphs depict the number of (G) CD45+CD8+CD3+ T cells, (H) CD45+CD4+CD3+ T cells, and (I) CD45+NK1.1+CD3− NK cells per tumor weight (in milligrams). Bars represent mean ± SEM. (J to L) Graph shows [CD45+CD8+CD3+ T cells per tumor weight (mg)] (J), [CD45+CD4+CD3+ T cells per tumor weight (mg)] (K), or [CD45+NK1.1+CD3− NK cells per tumor weight (mg)] (L) versus [tumor weight]. Two experimental replicates. Statistical analyses were done using (A, H, and I) ANOVA with Tukey’s test or (G) Kruskal-Wallis test followed by Dunn’s test or (B) log-rank (Mantel-Cox) test. *P < 0.05; **P < 0.01.
Dox-CBD-SA enhances tumor infiltration of lymphocytes
Dox reportedly induces ICD, which stimulates immune responses against antigens from necrotic cells (15). ICD increases the number of tumor-infiltrating lymphocytes (TILs), which is a marker of favorable prognosis in multiple types of cancers such as colorectal cancer and breast cancer (24, 25). We analyzed the TILs after Dox-CBD-SA treatment, particularly T cells and natural killer (NK) cells. Lymphocytes were extracted from the tumor and analyzed by flow cytometry 7 days after treatment with the various Dox forms. Dox-CBD-SA, but not aldoxorubicin or Dox-SA, significantly increased the numbers of CD8+ T cells, CD4+ T cells, and NK cells within the tumor per unit tumor mass (Fig. 3, G to I). In particular, Dox-CBD-SA treatment increased the number of CD8+ T cells more than twofold higher than the other treatment groups (Fig. 3G). Plots of individual tumor size and TIL cell number revealed that Dox-CBD-SA indeed induced a correlation between small tumor size and the number of tumor-infiltrated CD8+ T cells, CD4+ T cells, and NK cells (Fig. 3, J to L). These data suggest that enhanced infiltration of lymphocytes, particularly CD8+ cytotoxic T cells, may contribute to the superior antitumor effects of Dox-CBD-SA.
Dox-CBD-SA shows reduced toxicity
Because conjugated aldoxorubicin is only released very slowly from SA under physiological pH (Fig. 1E), we hypothesized that Dox-CBD-SA shows reduced toxicity compared to aldoxorubicin. We evaluated adverse events after a single injection of aldoxorubicin or Dox-CBD-SA (20 mg/kg on a Dox basis) using tumor-free FVB mice. Administration of aldoxorubicin increased the plasma concentration of inflammatory cytokines such as IFN-γ, TNF-α (tumor necrosis factor–α), IL-5, and IL-6, whereas Dox-CBD-SA did not (Fig. 4, A to D). Aldoxorubicin treatment also induced a significant decrease in red blood cell counts, white blood cell (WBC) counts, hematocrit, and hemoglobin concentration (Fig. 4, E and F, and fig. S8). In contrast, adverse effects of Dox-CBD-SA on hematological values were mild. Only a significant decrease in WBC counts compared to the untreated group was observed. Aldoxorubicin administration induced splenomegaly, whereas Dox-CBD-SA treatment did not (Fig. 4G). Histological analysis revealed that Dox-CBD-SA treatment provided no observable damage in heart, liver, kidney, or lung (fig. S9). These data suggest that preconjugation of Dox with CBD-SA reduced toxicity in various aspects.
Fig. 4. Dox-CBD-SA treatment shows reduced toxicity.
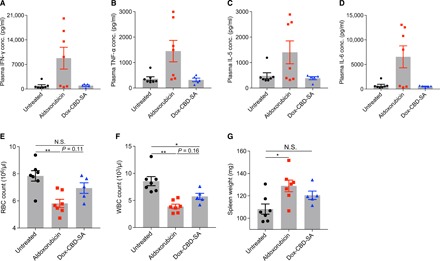
Aldoxorubicin or Dox-CBD-SA (20 mg/kg on a Dox basis) was administered to tumor-free FVB mice via tail vein injection on day 0. (A to D) Plasma cytokine concentrations on day 3. (E) Red blood cell (RBC) counts on day 6. (F) White blood cell (WBC) counts on day 3. (G) Spleen weights on day 16. Data represent mean ± SEM. Two experimental replicates. Statistical analyses were done using ANOVA with Tukey’s test. *P < 0.05; **P < 0.01.
Dox-CBD-SA in combination with anti–PD-1 antibody eradicates MC38 tumor
On the basis of the observation of increased TILs induced by Dox-CBD-SA treatment (Fig. 3, G to L), we hypothesized that Dox-CBD-SA combination therapy with CPI would show a greater therapeutic effect compared to aldoxorubicin combination therapy with CPI. To test this hypothesis, we selected anti–programmed cell death-1 antibody (αPD-1) as the most widely used CPI in the clinic (26). αPD-1 is used in combination with Dox in clinical trials (e.g., NCT02648477). We examined the antitumor effect of aldoxorubicin and Dox-CBD-SA in combination with αPD-1 using the MC38 colon carcinoma model, which is immunogenic (27) but not curable by Dox monotherapy (28). C57BL/6 mice were inoculated subcutaneously with 5 × 105 MC38 cells. The treatment schedule is shown in Fig. 5A. Aldoxorubicin or Dox-CBD-SA was administered to mice 6, 9, and 12 days after tumor inoculation. Since Dox-CBD-SA increases the number of TILs, we injected 100 μg of αPD-1 1 day after Dox treatment two times (on days 10 and 13). Dox-CBD-SA + αPD-1 therapy completely eradicated established MC38 tumors (average tumor volume was about 100 mm3 on day 6; Fig. 5, B and G) and significantly prolonged the survival of mice compared to all the other groups (Fig. 5C). In other treatment groups, a fraction of mice failed to show a complete response, and average tumor size increased gradually (Fig. 5, B and D to F). In Dox-CBD-SA + αPD-1–treated survivors, no mice rechallenged with MC38 cells without additional therapy developed palpable tumors, demonstrating that they had acquired strong immunologic antitumor memory (Fig. 5H and fig. S10A). During the treatments, no mouse showed more than 15% body weight loss (fig. S10B). These data show that Dox-CBD-SA, through induction of ICD, synergizes with αPD-1 to show further antitumor effects that could not be achieved by equivalent doses of aldoxorubicin + αPD-1.
Fig. 5. Dox-CBD-SA treatment completely eradicates established MC38 tumor in combination with anti-PD-1 CPI.
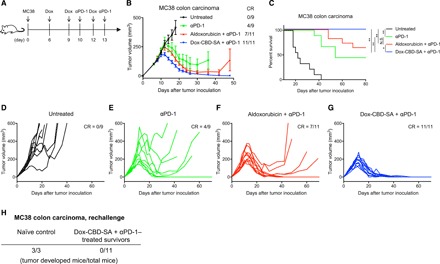
MC38 cells (5 × 105) were inoculated on day 0. Mice were injected intravenously with aldoxorubicin or Dox-CBD-SA (5 mg/kg on a Dox basis) on days 6, 9, and 12. αPD-1 was also injected intraperitoneally on days 10 and 13. (A) The experimental schedule. (B) Graphs depict tumor volume until the first mouse died (mean ± SEM). (C) Survival rate. (D to G) Individual tumor growth curves. (H) On day 60, Dox-CBD-SA + αPD-1–treated survivors were rechallenged by subcutaneous injection of 5 × 105 MC38 cells. Naïve mice were also challenged with the same amounts of cells as a control group. The number of mice that developed palpable tumors is shown. Two experimental replicates. Statistical analyses were done using log-rank (Mantel-Cox) test for survival curves. *P < 0.05; **P < 0.01.
DISCUSSION
Because small-molecule anticancer drugs broadly distribute to tissues and induce systemic side effects, modifications of drugs to improve their pharmacokinetics and biodistribution have been attempted. Nanoparticle-formulated (17) or SA-reactive (18, 19) Dox exhibits improved pharmacokinetics and accumulation within tumors based in part on their pathologically abnormal vasculature (5). However, this effect may not always be effective in human cancers because of their heterogeneity (29). Thus, drugs that are dependent on passive targeting alone may have room for improvement. Active targeting of tumor-specific or tumor-associated antigens for drug delivery is another therapeutic strategy. However, this intrinsically limits the applicable range of cancers and may also lead to acquired drug resistance due to antigen-selective cell targeting and killing, in which antigen may be lost by mutation (30). Here, we engineered CBD-SA to overcome these issues. Unlike other active targeting strategies, CBD-SA does not require the prior investigation of tumor-associated antigen expression because collagen is nearly ubiquitously expressed in tumors, and the CBD gains access to the tumor stroma via the abnormal blood vessel structure within the tumor microenvironment (6). Subsequently, the CBD-SA binds to exposed collagen (Figs. 1C and 2D; fig. S2) and converts the tumor stroma into a reservoir for chemotherapeutics. Dox conjugation to CBD-SA showed significantly higher accumulation of Dox within tumor tissue compared to aldoxorubicin and Dox-SA (Fig. 2C). After accumulation of Dox-CBD-SA within the tumor tissue, the hydrazone linkage, which can be cleaved under the slightly acidic conditions in the tumor microenvironment (Fig. 1E) (21), enables the sustained release of Dox from CBD-SA. At the same time, it is known that tumor cells uptake SA (1). Notably, CBD fusion did not alter the cellular uptake of SA (Fig. 1F), indicating that Dox-CBD-SA can also be delivered intracellularly as efficient as Dox-SA. Thus, part of the Dox release may occur in the tumor stroma, while the Dox-CBD-SA is still matrix-bound, and part may occur in the endolysosomal compartment following endocytosis. Incidentally, cancer stromal targeting (CAST) therapy using stroma-targeting antibodies has been proposed recently (31). The relatively low molecular mass of CBD-SA (88 kDa; fig. S1) may be a benefit compared to stroma-targeting antibodies in terms of diffusion into tumor tissues (32).
Regarding the payload of Dox, we conjugated about three Dox molecules per CBD-SA (Fig. 1D). The drug-to-protein ratio of antibody-drug conjugates is typically close to 4 (33). Considering that CBD-SA is 59% of the molecular weight of an immunoglobulin G (IgG) antibody, CBD-SA at a ratio of 3 achieved a high payload of Dox compared to typical antibody-drug conjugates. In addition, SA fusion increases the solubility of the CBD, which is important for hydrophobic drugs (4).
In terms of antitumor efficacy, Dox-CBD-SA significantly suppressed the growth of MMTV-PyMT breast cancer and extended the survival of mice compared to aldoxorubicin and Dox-SA (Fig. 3, A to F). Because Dox-CBD-SA showed the highest accumulation into tumor tissue in vivo, Dox-CBD-SA should induce tumor cell death more efficiently via inhibition of tumor cell proliferation. In addition to this effect, a single injection of Dox-CBD-SA brought a long-lasting therapeutic effect in spite of its faster plasma clearance half-life (Fig. 2, A and B). This could be explained by our observation that Dox-CBD-SA treatment induces a higher number and density of TILs compared to Dox-SA and aldoxorubicin treatments (Fig. 3, G to L). Therefore, the antitumor mechanism of action of Dox-CBD-SA may be not only direct cell killing but also the stimulation of host antitumor immunity. Since Dox-CBD-SA efficiently accumulates within tumors, it may induce ICD and tumor antigen exposure to the immune system more efficiently than aldoxorubicin and Dox-SA. As a consequence, Dox-CBD-SA synergistically eradicated MC38 colon carcinoma when administered in combination with αPD-1 (Fig. 5, B and G). Improved therapeutic efficacy of Dox-SA and Dox-CBD-SA in comparison with aldoxorubicin (Fig. 3, A to F) also indicates that preconjugation of Dox before injection would provide higher antitumor efficacy. In addition to rapid clearance from blood circulation, in situ conjugation of aldoxorubicin with other sulfhydryl compounds such as cysteine, glutathione, fibronectin, or α1-antitrypsin in plasma (18) is also a possible cause of inefficient therapeutic efficacy of aldoxorubicin.
Cardiac toxicity is a major drawback of Dox, which limits the lifetime cumulative dose of Dox (13). Histological analysis revealed that even Dox-CBD-SA administration (20 mg/kg) did not show any signs of cardiac damage (fig. S9). This suggests that Dox preconjugated with CBD-SA is less cardiotoxic than free Dox, which irreversibly damages cardiac tissue at a cumulative dose of 15 mg/kg in mouse (34). A cumulative dose of 15 mg/kg is nearly equivalent to the maximum cumulative dose in human (35). We hypothesize that unconjugated Dox could penetrate into and damage cardiac tissue, whereas its preconjugated form would not penetrate into tissues or release Dox under physiological pH (i.e., within nonmalignant tissue or blood), contributing to reduced adverse events.
In terms of the manufacturing process, we conjugated Dox using Traut’s reagent, which allows precise control of the drug conjugation ratio (36). This method has little risk to abrogate binding between the CBD and collagen since there are no lysine residues at the binding interface between the VWF A3 domain and collagen (fig. S5) (22). Moreover, SA contains approximately sevenfold the number of lysine residues as the CBD sequence (table S1), also suggesting the low risk of unfavorable conformational changes in the CBD due to conjugation. Traut’s reagent is also used for an ADC targeting CD70 (MDX-1203, Bristol-Myers Squibb) (37), indicating its translational applicability. As CBD-SA is produced with high yield [human embryonic kidney 293 (HEK293) cell culture (~70 to 100 mg/liter)], we propose that preconjugation of Dox to CBD-SA produces high antitumor efficacy with a simple and translatable production method.
As a potential limitation, CBD-SA might accumulate in undesirable sites in the body such as liver, kidney, and wounds, where collagens may be exposed via a fenestrated or leaky endothelium, although Dox would not be released from the CBD-SA if such locations are at neutral pH. At least, we did not observe pathological damage in the liver and kidney after Dox-CBD-SA administration (20 mg/kg) (fig. S9). As another limitation, chemical conjugation may decrease the half-life of SA in general. Methotrexate conjugation reportedly accelerated the clearance of methotrexate-SA conjugates from circulation in a drug-to-protein ratio–dependent manner (38). In this study, the half-lives of Dox-SA and Dox-CBD-SA were shorter than the reported half-life of native mouse SA [t½, β = 35 (hours)] (39). The reason why aldoxorubicin showed a relatively longer half-life than Dox-SA conjugates is probably that it reacted with endogenous SA at a 1:1 ratio due to the abundance of SA in circulation.
In conclusion, Dox-CBD-SA accumulated into tumors and activated host antitumor immunity. As a consequence, monotherapy of Dox-CBD-SA suppressed orthotopic MMTV-PyMT breast tumor growth and prolonged survival. Combination therapy of Dox-CBD-SA with immune checkpoint inhibition via αPD-1 completely eradicated tumors in the immunogenic MC38 model. CBD fusion provided an active targeting ability to SA, which is classically used as a passively targeted drug carrier, enabling effective drug delivery to tumors from the systemic circulation. CBD-SA is expected to be nonimmunogenic and biologically acceptable because it is composed of two proteins (VWF A3 domain and SA) that naturally exist in the blood. Furthermore, CBD-SA acts independently of tumor type–specific antigens and thus provides broad applicability to various types of solid tumors as a drug carrier. Therefore, CBD-SA may hold potential for clinical translation to cancer therapy as an antitumor drug carrier.
MATERIALS AND METHODS
Study design
This study was designed to verify the strategy for anticancer drug delivery to tumors by engineered collagen-binding SA as a drug conjugation carrier. Specifically, we tested whether antitumor efficacy of Dox-CBD-SA against mouse models of breast cancer and colon carcinoma is improved compared to their unmodified forms. The adverse effects of Dox-CBD-SA were also tested using tumor-free mice. We measured tumor growth, anticancer immune responses, and multiple aspects of toxicity after treatment. Statistical methods were not used to predetermine required sample size, but sample sizes were determined on the basis of estimates from pilot experiments and previously published results such that appropriate statistical tests could yield significant results. CBD-SA was produced by multiple individuals to ensure reproducibility. All experiments were replicated at least twice except for fig. S7 (once). For animal studies, mice were randomized into treatment groups within a cage immediately before the first Dox-CBD-SA injection and treated in the same way. Samples were excluded from analysis only when an animal developed a health problem for a non–treatment-related reason, according to the animal care guidelines. The survival endpoint was reached when the tumor volume reached more than 500 mm3 for the MMTV-PyMT model and 600 mm3 for the MC38 model. The n values used to calculate statistics are indicated in the figures or in the figure legends. Drug administration and pathological analyses were performed in a blinded fashion. Statistical methods are described in the “Statistical analysis” section.
Cell culture
MMTV-PyMT cells were obtained from spontaneously developed breast cancer in FVB-Tg (MMTV-PyMT) transgenic mice as described previously (9). The MC38 colon carcinoma cell line was provided by the R. Weichselbaum laboratory (University of Chicago). Dulbecco’s modified Eagle’s medium (DMEM; Gibco) supplemented with sodium pyruvate (110 mg/liter), 10% heat-inactivated fetal bovine serum (FBS), and 1% penicillin-streptomycin was used for both cell lines. The cell lines were checked for mycoplasma contamination by an IMPACT I pathogen test (IDEXX BioResearch).
Mice
Female FVB mice, ages 8 to 12 weeks, were obtained from Charles River and the Jackson Laboratory. Female C57BL/6 mice, ages 8 to 12 weeks, were obtained from the Jackson Laboratory. All the animal experiments performed in this work were approved by the Institutional Animal Care and Use Committee of the University of Chicago.
Production and purification of CBD-SA
CBD-SA protein was designed, produced, and purified similarly to previously reported CBD proteins (9). The sequences encoding for the fusion of human VWF A3 domain residues Cys1670-Gly1874 (907-1111 of mature VWF) and mouse SA without pro-peptide (25 to 608 amino acids of whole SA) were synthesized and subcloned into the mammalian expression vector pcDNA3.1(+) by GenScript. A sequence encoding for a His-tag (6-His) was inserted at the C terminus for further purification of the recombinant protein. Suspension-adapted HEK293F cells were routinely maintained in serum-free FreeStyle 293 Expression Medium (Gibco). On the day of transfection, cells were diluted into fresh medium at a density of 1 × 106 cells/ml. Plasmid DNA (2 μg/ml), linear 25-kDa polyethylenimine (2 μg/ml; Polysciences), and OptiPRO SFM medium (4% final concentration; Thermo fisher Scientific) were added. The culture flask was agitated by orbital shaking at 135 rpm at 37°C in the presence of 5% CO2. Seven days after transfection, the cell culture medium was collected by centrifugation and filtered through a 0.22-μm filter. Culture medium was loaded into a HisTrap HP 5-ml column (GE Healthcare), using an ÄKTA pure 25 (GE Healthcare). After washing of the column with wash buffer [20 mM imidazole, 20 mM NaH2PO4, and 0.5 M NaCl (pH 7.4)], protein was eluted with a gradient of 500 mM imidazole [in 20 mM NaH2PO4 and 0.5 M NaCl (pH 7.4)]. The eluent was further purified with size exclusion chromatography using a HiLoad Superdex 200PG column (GE Healthcare). All purification steps were carried out at 4°C. The protein was verified as >90% pure by SDS-PAGE.
Matrix-assisted laser desorption/ionization–time-of-flight mass spectrometry
Purified CBD-SA was analyzed by MALDI-TOF MS (Bruker Ultraflextreme MALDI TOF/TOF) as described previously (9). Bruker flexControl was used for data acquisition, and Bruker flexAnalysis was used for data processing. First, a saturated solution of α-cyano-4-hydroxycinnamic acid (Sigma-Aldrich) was prepared in 50:50 acetonitrile:1% trifluoroacetic acid in water as a solvent. CBD-SA in phosphate-buffered saline (PBS; 5 μl, 0.1 mg/ml) and the matrix solution (25 μl) were mixed, and 1 μl of that mixture was dropped on the MTP 384 ground steel target plate. The drop was dried in a nitrogen gas flow. All samples were analyzed using high mass linear positive mode method with 2500 laser shots at a laser intensity of 75%. The measurements were externally calibrated at three points with a mix of carbonic anhydrase, phosphorylase B, and bovine SA (BSA).
Binding affinity assay
The binding affinity of CBD-SA to collagens was tested as described previously (9). Ninety-six–well enzyme-linked immunosorbent assay (ELISA) plates (Greiner Bio-One) were coated with collagen I or collagen III (10 μg/ml each in PBS) overnight at 37°C, followed by blocking with 2% BSA in PBS with 0.05% Tween 20 (PBS-T) for 1 hour at room temperature. Then, wells were washed with PBS-T and further incubated with CBD-SA at increasing concentrations for 2 hours at room temperature. After three washes with PBS-T, wells were incubated for 1 hour at room temperature with biotin-conjugated antibodies against mouse SA. After washes, bound CBD-SA was detected with tetramethylbenzidine substrate by measurement of the absorbance at 450 nm with subtraction of the absorbance at 570 nm. The apparent Kd values were obtained by nonlinear regression analysis in Prism software (version 7, GraphPad) assuming one-site-specific binding.
Synthesis of Dox conjugates
Mouse SA or CBD-SA was solubilized in PBS containing 2 mM EDTA. Four molar equivalents of Traut’s reagents solved in PBS containing 2 mM EDTA were added and incubated for 1 hour at room temperature in the dark. Excess Traut’s reagents were removed by a Zeba spin desalting column (Thermo Fisher Scientific). Fifteen molar equivalents of aldoxorubicin (MedChemExpress) dissolved in 10 mM sodium phosphate buffer (pH 5.9) was added and incubated for 1 hour at room temperature and overnight at 4°C in the dark. To quench the reaction, 20 molar equivalents of l-cysteine [dissolved in PBS containing 2 mM EDTA; Sigma-Aldrich (pharma-grade)] against aldoxorubicin was added. Unreacted Dox precipitates were removed by centrifugation (10,000g, 5 min). Supernatant was further purified by a Zeba spin desalting column, followed by ultrafiltration using Amicon Ultra (Merck, 10 kDa molecular mass cutoff). Concentration of Dox in the final product was quantitated by absorbance at 495 nm, using a molar extinction coefficient of 10,650 (L mol−1 cm−1). The concentration of protein content was measured by Pierce BCA (bicinchoninic acid assay) Protein Assay Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Dynamic light scattering
The hydrodynamic size of Dox conjugates in PBS was measured using Zetasizer Nano ZS (Malvern Panalytical). Conjugates were analyzed immediately after synthesis or lyophilized and stored at −20°C until use.
SDS–polyacrylamide gel electrophoresis
SDS-PAGE was performed as described in our previous work (9). A 4 to 20% gradient gel (Bio-Rad) was used, and 0.5 μg of each Dox conjugate was loaded with or without reduction with 10 mM dithiothreitol. After electrophoresis, SimplyBlue SafeStain (Thermo Fisher Scientific) was used for gel staining according to the manufacturer’s instruction. ChemiDoc XRS+ system (Bio-Rad) was used for image acquisition.
pH-dependent release of Dox from conjugates
Slide-A-Lyzer MINI Dialysis Device (Thermo Fisher Scientific, 10 kDa molecular mass cutoff) was used to determine the release profile of Dox from conjugates. Dox conjugates were diluted in either PBS (pH 6.5 or 7.4) or 0.1 M acetate buffer (pH 5.0) to a final concentration of 100 μM (Dox basis). Each sample (150 μl) was loaded into the device and dialyzed with buffer (50 ml). Dialysis was performed using a magnetic stirrer. The temperature of the stage was set to 37°C, and samples were protected from light during dialysis. Dialysate was collected at various time points and stored at 4°C until the sample collection was finished. Dialysate was loaded onto a 96-well black plate in duplicate (90 μl/well). Fluorescence was determined using excitation at 495 nm and emission at 590 nm. Serial dilution of Dox hydrochloride was prepared in the same buffer to create a standard curve.
Cellular uptake of Dox conjugates
To evaluate the cellular uptake of Dox conjugates, MMTV-PyMT cells were seeded in a 96-well high-content imaging plate (Corning) at 5000 cells per well and incubated overnight. Cells were washed with PBS and treated with free Dox, Dox-SA, or Dox-CBD-SA dissolved in DMEM [sodium pyruvate (110 mg/liter), 10% heat-inactivated FBS, 1% penicillin-streptomycin, and Phenol red (−)] at a concentration of 50 μM equivalent of Dox. After incubation, cells were washed twice, treated with 75 nM LysoTracker Deep Red, and further incubated for 30 min at 37°C. Cells were washed twice and observed by an IX83 microscope (Olympus) with ×60 magnification. Images were processed using ImageJ software (National Institutes of Health, NIH). Scale bar, 20 μm.
In vitro cytotoxicity
MMTV-PyMT cells or MC38 cells were seeded in a 96-well tissue culture plate (BD Falcon) at 3000 cells per well and incubated overnight. Cells were washed with DMEM [sodium pyruvate (110 mg/liter), 10% heat-inactivated FBS, 1% penicillin-streptomycin, and Phenol red (−)], and DMEM (80 μl/well) was added. Then, serial dilutions of aldoxorubicin, Dox-SA, or Dox-CBD-SA in PBS were added (20 μl/well). Cells were incubated 3 days at 37°C, and the viability was determined using CellTiter 96 AQueous One Solution Cell Proliferation Assay Kit (Promega) according to the manufacturer’s instructions. Cells treated with DMEM (80 μl/well) and PBS (20 μl/well) were defined as 100% viable, whereas the cell-free wells with the same mixture were defined as 0% viable. Half maximal inhibitory concentration (IC50) values were obtained by nonlinear regression analysis in Prism software ([inhibitor] versus normalized response).
Plasma pharmacokinetics of Dox conjugates
A previous report about polypeptide-Dox nanoparticles was referred (40). To measure pharmacokinetics of Dox, 5 mg/kg Dox equivalent of aldoxorubicin, Dox-SA, or Dox-CBD-SA was injected intravenously into female FVB mice. Blood samples were collected in EDTA-coated tubes at 5 min, 30 min, 1 hour, 4 hours, 12 hours, 25 hours, 50 hours, and 75 hours after injection. Blood samples were stored at 4°C until the end of sample collection. The samples were centrifuged (2000g, 5 min), and plasma was collected. Plasma samples diluted in acidified isopropanol (75 mM HCl, 10% water, 90% isopropanol) were loaded onto a 96-well black plate (100 μl/well). Fluorescence was measured as described above. Plasma samples were also collected from mice, which received no injections, diluted in acidified isopropanol, and measured to create the standard curve of background fluorescence. Exponential two-phase decay (Y = Ae−αt + Be−βt) fitting was used to calculate the plasma half-life. Fast clearance half-life, t1/2,α; slow clearance half-life, t1/2,β. Data were analyzed using Prism software (v7, GraphPad).
Plasma pharmacokinetics of SA and CBD-SA
SA and CBD-SA were labeled with DyLight 800 NHS ester (Thermo Fisher Scientific) according to the manufacturer’s instructions. Unreacted dye was removed by a Zeba spin desalting column as described above. After labeling, 200 μg of each protein was injected intravenously into female FVB mice. Blood samples were collected in EDTA-coated tubes at 1 min, 1 hour, 4 hours, 24 hours, 74 hours, and 120 hours after injection. Blood samples were stored at 4°C until the end of sample collection. The samples were centrifuged (2000g, 5 min), and plasma was collected. Plasma samples were diluted in PBS and loaded into a 96-well black plate (100 μl/well). The concentration of each protein in plasma was measured with a LI-COR Infrared Odyssey Imager (Li-COR Biosciences). The method of curve fitting and calculation of plasma half-life was described above.
MMTV-PyMT tumor inoculation and treatments
The MMTV-PyMT murine breast cancer model was prepared as described previously (9). A total of 5 × 105 MMTV-PyMT cells suspended in 50 μl of PBS were injected subcutaneously into the mammary gland on the right side of each mouse. Mice were treated with aldoxorubicin, Dox-SA, or Dox-CBD-SA on day 7 (5 mg/kg) via tail vein injection. Tumors were measured with a digital caliper at the indicated time points, and volumes were calculated as ellipsoids, where V = 4/3 × 3.14 × depth/2 × width/2 × height/2. Mice were euthanized when tumor volume had reached more than 500 mm3 or when active ulceration was observed. For the therapeutic experiment, FVB mice that originated from Charles River were used. For the TIL analysis, FVB mice that originated from both the Jackson Laboratory and Charles River were used. The proportion of mice from different providers was equalized among all groups.
MC38 tumor inoculation and treatments
The MC38 murine colon carcinoma model was prepared similarly to the B16F10 melanoma model as described previously (9). A total of 5 × 105 MC38 cells suspended in 50 μl of PBS were injected intradermally on the left side of the back of each C57BL/6 mouse. Mice were injected intravenously on days 6, 9, and 12 with aldoxorubicin, Dox-SA, or Dox-CBD-SA (5 mg/kg). Mice were also treated intraperitoneally with 100 μg of anti–PD-1 (clone 29F.1A12, Bio X Cell) on days 10 and 13. Tumor growth was monitored as described above. Mice were euthanized when tumor volume had reached more than 600 mm3 or when active ulceration was observed. On day 60, naïve C57BL/6 mice or tumor-free survivors were rechallenged by intradermal injection of 5 × 105 MC38 cells.
Tumor accumulation study
A previous report about polypeptide-Dox nanoparticles was referred (40). Aldoxorubicin, Dox-SA, or Dox-CBD-SA was injected into FVB mice with established tumors at 4.16 mg/kg via tail vein. Tumor was collected, weighed, and put on ice for 2 or 24 hours after injection. Tumor tissues were suspended in 1 ml of acidified isopropanol and homogenized using Lysing Matrix D and FastPrep-24 5G (MP Biomedical) for 40 s at 5000 beats/min. After homogenization, samples were protected from light and incubated overnight at 4°C. Samples were centrifuged (5000g, 5 min), and the supernatants were loaded onto a 96-well black plate (100 μl/well, triplicate). Fluorescence was measured to quantify the amount of Dox in tissue extracts as described above. Tumors from untreated mice were also processed, and serial dilutions of tissue extracts were measured to obtain the standard curves of tissue-derived autofluorescence.
Histological analysis of injected CBD-SA within tumor
Mouse SA (Sigma-Aldrich) and CBD-SA were conjugated with NHS-DyLight 488 according to the manufacturer’s instructions. Unreacted dye was removed by a Zeba spin desalting column and then fluorescent protein solution was stored at 4°C until use. Fluorescent-labeled SA (100 μg) or CBD-SA labeled with equimolar amounts of dye was intravenously injected into MMTV-PyMT tumor-bearing mice. One hour after injection, tumors were harvested and frozen in dry ice with optimal cutting temperature compound. Tissue slices (10 μm) were obtained by cryo-sectioning. The tissues were fixed with 2% paraformaldehyde in PBS for 15 min at room temperature. After washing with PBS-T, the tissues were blocked with 2% BSA in PBS-T for 1 hour at room temperature. The tissues were stained with biotin-labeled anti-mouse CD31 antibody (1:100, BioLegend) and Alexa Fluor 647 streptavidin (1:1000, BioLegend). The tissues were washed three times and then covered with ProLong gold antifade mountant with 4′,6-diamidino-2-phenylindole (DAPI; Thermo Fisher Scientific). An IX83 microscope (Olympus) was used for imaging with ×60 magnification. Images were processed using ImageJ software (NIH).
Flow cytometry and antibodies
The MMTV-PyMT model was prepared as described above. Mice were treated on day 7 with aldoxorubicin, Dox-SA, or Dox-CBD-SA (5 mg/kg). Mice were euthanized on day 14. Cell suspensions were obtained from each tumor as described previously (9). Tumors were harvested and digested in DMEM supplemented with 2% FBS, collagenase D (2 mg/ml), and DNase I (40 μg/ml) (Roche) for 30 min at 37°C. Single-cell suspensions were obtained by gently disrupting the organs through a 70-μm cell strainer. Red blood cells were lysed with ACK lysing buffer (Quality Biological). Fixable live/dead cell discrimination was performed using Fixable Viability Dye eFluor 455 (eBioscience) according to the manufacturer’s instructions. Following a washing step, cells were stained with specific antibodies for 20 min on ice before fixation. The following antibodies were used to stain the cells: CD3 (145-2C11, BD Biosciences), CD4 (RM4-5, BD Biosciences), CD8α (53-6.7, BD Biosciences), CD45 (30-F11, BD Biosciences), and NK1.1 (PK136, BD Biosciences). All flow cytometric analyses were done using a Fortessa flow cytometer (BD Biosciences) and analyzed using FlowJo software (Tree Star).
Toxicity profiles
Tumor-free FVB mice received aldoxorubicin or Dox-CBD-SA (20 mg/kg) by intravenous injection. Blood samples were collected from each mouse in an EDTA-coated tube by submandibular bleeding on days 3 and 6 after injection for plasma cytokine analysis and hematological analysis. Body weight of each mouse was measured at the indicated time points. On day 16, mice were euthanized and organs were harvested. Spleens were weighed, and the other organs were used for histological analysis. Mice were euthanized when more than 15% decrease of initial body weight was observed.
Hematological analysis
Blood samples were analyzed using a COULTER Ac•T 5diff CP hematology analyzer (Beckman Coulter) according to the manufacturer’s instructions.
Measurement of plasma cytokines
Blood plasma was collected from whole-blood sample as described above and stored at −20°C until use. Cytokine concentrations in plasma were measured using Ready-SET-Go! ELISA kits (eBioscience) and Can Get Signal solution (TOYOBO) according to the manufacturer’s instructions.
Histological analysis of heart, liver, kidney, and lung
Organs were fixed with 2% paraformaldehyde in PBS overnight. After embedding in paraffin, blocks were cut into 5-μm sections, followed by hematoxylin and eosin staining.
Statistical analysis
Statistically significant differences between experimental groups were determined using Prism software (v7, GraphPad) as described previously (9). Where one-way analysis of variance (ANOVA) followed by Tukey’s honestly significant difference post hoc test was used, variance between groups was found to be similar by Brown-Forsythe test. For nonparametric data (Fig. 3G), Kruskal-Wallis test followed by Dunn’s multiple comparison test was used. Survival curves were analyzed by using the log-rank (Mantel-Cox) test. The symbols * and ** indicate P values less than 0.05 and 0.01, respectively; N.S., not significant.
Supplementary Material
Acknowledgments
We thank the Human Tissue Resource Center of the University of Chicago for histology analysis. Molecular graphics and analyses were performed with the UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41-GM103311. We thank A. Solanki for assistance in tail vein injections, P. Hosseinchi and J. Reda for assistance in imaging analysis, M. Raczy for assistance in MALDI-TOF MS measurement, L.T. Gray, S. Gomes, and K. Katsumata for experimental advice and helpful discussions, and J.-M. Williford for careful reading of the manuscript. Funding: This work was supported by the European Research Council grant Cytrix. K.S. and R.M were supported by the JSPS Research Fellowship for Young Scientists (JP17J05032 and JP17J09905). K.S. was supported by the Advanced Graduate Course on Molecular Systems for Devices (Kyushu University). R.M. was supported by the Training Program of Leaders for Integrated Medical System (Kyoto University). Author contributions: K.S., J.I., K.F., and J.A.H. designed the project. K.S., J.I., A.I., R.M., and A.M. performed the experiments. K.S., J.I., A.I., R.M., and J.A.H. analyzed the data. K.S., J.I., and J.A.H. wrote the paper. Competing interests: K.S., J.I., and J.A.H. are inventors on U.S. Provisional Patent applications 62/856,468 filed on 3 June 2019, which cover the technology reported in this article. J.I., A.I., and J.A.H. are shareholders in Arrow Immune, Inc., which is developing the technology presented in this report. The other authors declare that they have no competing interests. Data and materials availability: Correspondence and requests for materials should be addressed to J.A.H. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaaw6081/DC1
Fig. S1. Confirmation of CBD fusion to SA by MALDI-TOF MS analysis.
Fig. S2. Binding affinities of CBD-SA to collagen type I and type III.
Fig. S3. SDS-PAGE analysis of mouse SA and CBD-SA conjugated with Dox.
Fig. S4. Hydrodynamic sizes.
Fig. S5. The binding interface between collagen type III and A3 domain of VWF.
Fig. S6. In vitro release kinetics of Dox from Dox-SA.
Fig. S7. Plasma pharmacokinetics of DyLight 800–labeled SA and CBD-SA.
Fig. S8. Changes of hematological values in mice receiving aldoxorubicin or Dox-CBD-SA (20 mg/kg).
Fig. S9. Histological analysis of major organs after Dox-CBD-SA treatment.
Fig. S10. MC38 tumor rechallenge and body weight changes of MC38 tumor-bearing mice during the treatment.
Table S1. Amino acid sequence of CBD-SA.
REFERENCES AND NOTES
- 1.Kratz F., Albumin as a drug carrier: Design of prodrugs, drug conjugates and nanoparticles. J. Control. Release 132, 171–183 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Morales J., Defining the role of insulin detemir in basal insulin therapy. Drugs 67, 2557–2584 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Flisiak R., Flisiak I., Albinterferon-alfa 2b: A new treatment option for hepatitis C. Expert. Opin. Biol. Ther. 10, 1509–1515 (2010). [DOI] [PubMed] [Google Scholar]
- 4.Green M. R., Manikhas G. M., Orlov S., Afanasyev B., Makhson A. M., Bhar P., Hawkins M. J., Abraxane®, a novel Cremophor®-free, albumin-bound particle form of paclitaxel for the treatment of advanced non-small-cell lung cancer. Ann. Oncol. 17, 1263–1268 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Matsumura Y., Maeda H., A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 46, 6387–6392 (1986). [PubMed] [Google Scholar]
- 6.Danhier F., Feron O., Préat V., To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J. Control. Release 148, 135–146 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Temming K., Meyer D. L., Zabinski R., Dijkers E. C., Poelstra K., Molema G., Kok R. J., Evaluation of RGD-targeted albumin carriers for specific delivery of auristatin E to tumor blood vessels. Bioconjug. Chem. 17, 1385–1394 (2006). [DOI] [PubMed] [Google Scholar]
- 8.Prakash J., Beljaars L., Harapanahalli A. K., Zeinstra-Smith M., de Jager-Krikken A., Hessing M., Steen H., Poelstra K., Tumor-targeted intracellular delivery of anticancer drugs through the mannose-6-phosphate/insulin-like growth factor II receptor. Int. J. Cancer 126, 1966–1981 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Ishihara J., Ishihara A., Sasaki K., Lee S. S.-Y., Yasui M., Abe H., Potin L., Hosseinchi P., Fukunaga K., Raczy M. M., Gray L. T., Williford J.-M., Fukayama M., Kron S. J., Swartz M. A., Hubbell J. A., Targeted antibody and cytokine cancer immunotherapies through collagen affinity. Sci. Transl. Med. 11, eaau3259 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Addi C., Murschel F., De Crescenzo G., Design and use of chimeric proteins containing a collagen-binding domain for wound healing and bone regeneration. Tissue Eng. Part B Rev. 23, 163–182 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Provenzano P. P., Inman D. R., Eliceiri K. W., Knittel J. G., Yan L., Rueden C. T., White J. G., Keely P. J., Collagen density promotes mammary tumor initiation and progression. BMC Med. 6, 11 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou Z.-H., Ji C.-D., Xiao H.-L., Zhao H.-B., Cui Y.-H., Bian X.-W., Reorganized collagen in the tumor microenvironment of gastric cancer and its association with prognosis. J. Cancer 8, 1466–1476 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Young R. C., Ozols R. F., Myers C. E., The anthracycline antineoplastic drugs. N. Engl. J. Med. 305, 139–153 (1981). [DOI] [PubMed] [Google Scholar]
- 14.Wang L., Chen Q., Qi H., Wang C., Wang C., Zhang J., Dong L., Doxorubicin-induced systemic inflammation is driven by upregulation of toll-like receptor TLR4 and endotoxin leakage. Cancer Res. 76, 6631–6642 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Kroemer G., Galluzzi L., Kepp O., Zitvogel L., Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 31, 51–72 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Rios-Doria J., Durham N., Wetzel L., Rothstein R., Chesebrough J., Holoweckyj N., Zhao W., Leow C. C., Hollingsworth R., Doxil synergizes with cancer immunotherapies to enhance antitumor responses in syngeneic mouse models. Neoplasia 17, 661–670 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O’Brien M. E., Wigler N., Inbar M., Rosso R., Grischke E., Santoro A., Catane R., Kieback D., Tomczak P., Ackland S., Orlandi F., Mellars L., Alland L., Tendler C., Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYX™/Doxil®) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Ann. Oncol. 15, 440–449 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Kratz F., Warnecke A., Scheuermann K., Stockmar C., Schwab J., Lazar P., Drückes P., Esser N., Drevs J., Rognan D., Bissantz C., Hinderling C., Folkers G., Fichtner I., Unger C., Probing the cysteine-34 position of endogenous serum albumin with thiol-binding doxorubicin derivatives. Improved efficacy of an acid-sensitive doxorubicin derivative with specific albumin-binding properties compared to that of the parent compound. J. Med. Chem. 45, 5523–5533 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Graeser R., Esser N., Unger H., Fichtner I., Zhu A., Unger C., Kratz F., INNO-206, the (6-maleimidocaproyl hydrazone derivative of doxorubicin), shows superior antitumor efficacy compared to doxorubicin in different tumor xenograft models and in an orthotopic pancreas carcinoma model. Investig. New Drugs 28, 14–19 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Chawla S. P., Papai Z., Mukhametshina G., Sankhala K., Vasylyev L., Fedenko A., Khamly K., Ganjoo K., Nagarkar R., Wieland S., Levitt D. J., First-line aldoxorubicin vs doxorubicin in metastatic or locally advanced unresectable soft-tissue sarcoma: A phase 2b randomized clinical trial. JAMA Oncol. 1, 1272–1280 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Prabaharan M., Grailer J. J., Pilla S., Steeber D. A., Gong S., Amphiphilic multi-arm-block copolymer conjugated with doxorubicin via pH-sensitive hydrazone bond for tumor-targeted drug delivery. Biomaterials 30, 5757–5766 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Brondijk T. H. C., Bihan D., Farndale R. W., Huizinga E. G., Implications for collagen I chain registry from the structure of the collagen von Willebrand factor A3 domain complex. Proc. Natl. Acad. Sci. U.S.A. 109, 5253–5258 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuerer H. M., Newman L. A., Smith T. L., Ames F. C., Hunt K. K., Dhingra K., Theriault R. L., Singh G., Binkley S. M., Sneige N., Buchholz T. A., Ross M. I., McNeese M. D., Buzdar A. U., Hortobagyi G. N., Singletary S. E., Clinical course of breast cancer patients with complete pathologic primary tumor and axillary lymph node response to doxorubicin-based neoadjuvant chemotherapy. J. Clin. Oncol. 17, 460–469 (1999). [DOI] [PubMed] [Google Scholar]
- 24.Ropponen K. M., Eskelinen M. J., Lipponen P. K., Alhava E., Kosma V.-M., Prognostic value of tumour-infiltrating lymphocytes (TILs) in colorectal cancer. J. Pathol. 182, 318–324 (1997). [DOI] [PubMed] [Google Scholar]
- 25.Loi S., Sirtaine N., Piette F., Salgado R., Viale G., Van Eenoo F., Rouas G., Francis P., Crown J. P. A., Hitre E., de Azambuja E., Quinaux E., Leo A. D., Michiels S., Piccart M. J., Sotiriou C., Prognostic and predictive value of tumor-infiltrating lymphocytes in a phase III randomized adjuvant breast cancer trial in node-positive breast cancer comparing the addition of docetaxel to doxorubicin with doxorubicin-based chemotherapy: BIG 02-98. J. Clin. Oncol. 31, 860–867 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Hargadon K. M., Johnson C. E., Williams C. J., Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. Int. Immunopharmacol. 62, 29–39 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Yadav M., Jhunjhunwala S., Phung Q. T., Lupardus P., Tanguay J., Bumbaca S., Franci C., Cheung T. K., Fritsche J., Weinschenk T., Modrusan Z., Mellman I., Lill J. R., Delamarre L., Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 515, 572–576 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Kuai R., Yuan W., Son S., Nam J., Xu Y., Fan Y., Schwendeman A., Moon J. J., Elimination of established tumors with nanodisc-based combination chemoimmunotherapy. Sci. Adv. 4, eaao1736 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prabhakar U., Maeda H., Jain R. K., Sevick-Muraca E. M., Zamboni W., Farokhzad O. C., Barry S. T., Gabizon A., Grodzinski P., Blakey D. C., Challenges and key considerations of the enhanced permeability and retention effect for nanomedicine drug delivery in oncology. Cancer Res. 73, 2412–2417 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Holohan C., Van Schaeybroeck S., Longley D. B., Johnston P. G., Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 13, 714–726 (2013). [DOI] [PubMed] [Google Scholar]
- 31.Yasunaga M., Manabe S., Tarin D., Matsumura Y., Cancer-stroma targeting therapy by cytotoxic immunoconjugate bound to the collagen 4 network in the tumor tissue. Bioconjug. Chem. 22, 1776–1783 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Pluen A., Boucher Y., Ramanujan S., McKee T. D., Gohongi T., di Tomaso E., Brown E. B., Izumi Y., Campbell R. B., Berk D. A., Jain R. K., Role of tumor–host interactions in interstitial diffusion ofmacromolecules: Cranial vs. subcutaneous tumors. Proc. Natl. Acad. Sci. U.S.A. 98, 4628–4633 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Diamantis N., Banerji U., Antibody-drug conjugates—An emerging class of cancer treatment. Br. J. Cancer 114, 362–367 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fisher P. W., Salloum F., Das A., Hyder H., Kukreja R. C., Phosphodiesterase-5 inhibition with sildenafil attenuates cardiomyocyte apoptosis and left ventricular dysfunction in a chronic model of doxorubicin cardiotoxicity. Circulation 111, 1601–1610 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Takemura G., Fujiwara H., Doxorubicin-induced cardiomyopathy: From the cardiotoxic mechanisms to management. Prog. Cardiovasc. Dis. 49, 330–352 (2007). [DOI] [PubMed] [Google Scholar]
- 36.McCall M. J., Diril H., Meares C. F., Simplified method for conjugating macrocyclic bifunctional chelating agents to antibodies via 2-iminothiolane. Bioconjug. Chem. 1, 222–226 (1990). [DOI] [PubMed] [Google Scholar]
- 37.Owonikoko T. K., Hussain A., Stadler W. M., Smith D. C., Kluger H., Molina A. M., Gulati P., Shah A., Ahlers C. M., Cardarelli P. M., Cohen L. J., First-in-human multicenter phase I study of BMS-936561 (MDX-1203), an antibody-drug conjugate targeting CD70. Cancer Chemother. Pharmacol. 77, 155–162 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Stehle G., Sinn H., Wunder A., Schrenk H. H., Schütt S., Maier-Borst W., Heene D. L., The loading rate determines tumor targeting properties of methotrexate-albumin conjugates in rats. Anticancer Drugs 8, 677–685 (1997). [PubMed] [Google Scholar]
- 39.Chaudhury C., Mehnaz S., Robinson J. M., Hayton W. L., Pearl D. K., Roopenian D. C., Anderson C. L., The major histocompatibility complex-related Fc receptor for IgG (FcRn) binds albumin and prolongs its lifespan. J. Exp. Med. 197, 315–322 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.MacKay J. A., Chen M., McDaniel J. R., Liu W., Simnick A. J., Chilkoti A., Self-assembling chimeric polypeptide-doxorubicin conjugate nanoparticles that abolish tumours after a single injection. Nat. Mater. 8, 993–999 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaaw6081/DC1
Fig. S1. Confirmation of CBD fusion to SA by MALDI-TOF MS analysis.
Fig. S2. Binding affinities of CBD-SA to collagen type I and type III.
Fig. S3. SDS-PAGE analysis of mouse SA and CBD-SA conjugated with Dox.
Fig. S4. Hydrodynamic sizes.
Fig. S5. The binding interface between collagen type III and A3 domain of VWF.
Fig. S6. In vitro release kinetics of Dox from Dox-SA.
Fig. S7. Plasma pharmacokinetics of DyLight 800–labeled SA and CBD-SA.
Fig. S8. Changes of hematological values in mice receiving aldoxorubicin or Dox-CBD-SA (20 mg/kg).
Fig. S9. Histological analysis of major organs after Dox-CBD-SA treatment.
Fig. S10. MC38 tumor rechallenge and body weight changes of MC38 tumor-bearing mice during the treatment.
Table S1. Amino acid sequence of CBD-SA.