

Drosophila l(3)mbt malignant brain tumors present sexual dimorphism.
Abstract
The notable male predominance across many human cancer types remains unexplained. Here, we show that Drosophila l(3)mbt brain tumors are more invasive and develop as malignant neoplasms more often in males than in females. By quantitative proteomics, we have identified a signature of proteins that are differentially expressed between male and female tumor samples. Prominent among them is the conserved chromatin reader PHD finger protein 7 (Phf7). We show that Phf7 depletion reduces sex-dependent differences in gene expression and suppresses the enhanced malignant traits of male tumors. Our results identify potential regulators of sex-linked tumor dimorphism and show that these genes may serve as targets to suppress sex-linked malignant traits.
INTRODUCTION
Cancer susceptibility and mortality rate are significantly higher in the male population, even after occupational and behavioral risk factors are taken into account (1–3). Male predominance is also observed in childhood malignancies that present themselves in very young infants before puberty (4–7). Studies covering a wide panel of cancer types show that the expression of many clinically relevant genes is strongly sex biased in malignant tumors (8, 9), hence suggesting molecular sexual dimorphism at the cellular level as a key determinant of sex-linked disparities in cancer (1, 2, 10). Understanding the molecular basis of sex-linked differences in cancer incidence and survival may pave the way for gender-specific, more efficient therapeutic strategies. However, the molecular cell biology for sex disparities in cancer remains very poorly understood.
Drosophila can be used to experimentally induce a wide range of tumors that affect a variety of organs in both adult flies and developing larvae (11). These tumor types range from hyperplasias to frankly malignant neoplasias that exhibit classic hallmarks of mammalian cancer. In addition, natural hyperplasias can develop in the adult fly testis and gut and are age dependent (12, 13). Some Drosophila tumors are being used as experimental models for leukemia, neuroblastoma, glioblastoma, ovarian cancer, and others [reviewed in (11, 14–16)].
Available information about sex-biased phenotypes in Drosophila tumors in organs with nonreproductive function is limited to nonmalignant, genetically induced hyperplastic tumors induced by altering Notch (N) or APC-ras signaling in the adult midgut (17) and natural hyperplasia formed in the aging gut (13), which are more frequent in females.
To determine whether Drosophila experimental models of malignant growth may serve to investigate the cell biological axes that control sex-linked tumor dimorphism, we have studied brain tumor (brat) (18) and lethal(3)malignant brain tumor [l(3)mbt] (19) malignant brain neoplasias (henceforth referred to as brat and mbt tumors, respectively). The TRIpartite Motif - NCL-1/HT2A/LIN-41 (TRIM-NHL) protein Brat inhibits translation, and its loss of function results in tumors that originate from type II intermediary neuronal progenitors in the larval brain (20, 21). Human Brat ortholog TRIM3 is a tumor suppressor that regulates neural stem cell equilibrium (22).
L(3)mbt harbors three MBT repeats and a zinc finger domain (23) and has been shown to repress the expression of dozens of genes, including germline genes, in somatic tissues as well as testis-specific and neuronal genes in the female germline (24–27). L(3)mbt interacts sub-stoichiometrically with the dREAM/Myb-Muv B complex (25, 28) and is a stoichiometric component of the L(3)mbt-interacting (LINT) complex (29). Loss-of-function conditions for l(3)mbt result in neoplastic growth that originates in the neuroepithelial regions in the larval brain lobes (30). Some of the germline genes that are ectopically expressed in l(3)mbt mutant brains are essential for mbt tumor growth (24, 31).
RESULTS
l(3)mbt tumors, but not brat tumors, show gender-dependent malignant traits
As a first step to investigate whether malignant growth is affected by the sex of the tumor bearer, we studied the anatomy of l(3)mbt and brat, male and female larval brain lobes stained with 4′,6-diamidino-2-phenylindole (DAPI) to label DNA and immunostained with anti-Miranda (Mira) (fig. S1) and anti–DE-cadherin antibodies (Fig. 1). Mira is highly expressed in brat tumor cells (21), and DE-cadherin labels adherens junctions that are enriched in the neuroepithelium (NE), which is the region from which mbt tumors derive (30).
Fig. 1. mbt tumors present sex-linked dimorphism.
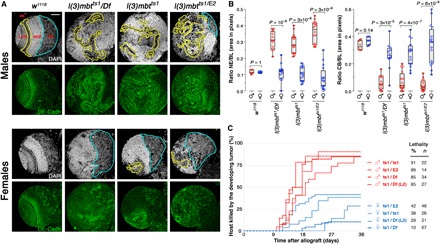
(A) Larval brain lobes from male and female control (w1118) and mbt mutant [l(3)mbtts1/Df, l(3)mbtts1, and l(3)mbtts1/E2] larvae stained with DAPI (gray) and anti–DE-cadherin antibody (green). Male mbt lobes present reduced central brains (CBs; blue) and overgrown neuroepithelia (NE; yellow) that invade medial regions. In contrast, in female mbt lobes, the NE do not overgrow and CBs remain as distinct as in wild-type larvae. Scale bar, 50 μm. (B) Relative sizes of NE and CB (as a fraction of brain lobe area) in male (red) and female (blue), control (w1118) and mbt mutant larvae. Differences in NE and CB sizes between mbt male and female brain lobes and between mbt males and control larvae of either sex are highly significant. (C) Tumor growth rate and host lethality caused by male (red) and female (blue) mbt larval brain lobes allografted to adult hosts. Allografted tissues were dissected from third instar larvae (132 ± 12 hours AEL; 29°C) except for ts1/Df (L2) samples that were dissected from second instar (36 ± 12 h AEL; 29°C). Male implants kill significantly more hosts and faster than do female implants. MED, medulla; LAM, lamina.
We found no sex-dependent differences in larval brain lobe anatomy in brat tumors (fig. S1). In contrast, brain lobes from three allelic combinations of l(3)mbt [l(3)mbtts1/Df(3R)ED10966, l(3)mbtts1, and l(3)mbtts1/l(3)mbtE2; henceforth referred to as ts1/Df, ts1, and ts1/E2, respectively] are all sexually dimorph. After staining with DAPI and anti–DE-cadherin antibody (Fig. 1A, gray and green, respectively), male mbt brain lobes present a long and convoluted NE (Fig. 1A, outlined in yellow) that reaches medially and is embedded in strongly DAPI-positive tissue (likely a tumorous version of the medulla) that invades most of the brain lobe including the central brain (CB) (Fig. 1A, outlined in blue). In female mbt brain lobes, however, the NE and the tissue that stains strongly with DAPI are restricted to the lateral half of the lobe, and the CB remains distinct (Fig. 1A). Differences in the fraction of the brain lobe area occupied by the NE and the CB are highly significant between mbt males and their female siblings (P < 10−4) and insignificant between mbt females and wild-type brains of either sex (Fig. 1B). Mean maximum Feret diameter of the brain lobe is also significantly larger (P < 10−10) in male than in female mbt individuals (fig. S2).
To determine growth potential, we allografted mbt tissue dissected from late third instar larvae [132 ± 12 hours at 29°C after egg laying (AEL)] and ts1/Df samples from early second instar larvae (36 ± 12 hours at 29°C AEL), a stage at which male and female mbt and wild-type brains are still indistinguishable. Under all four experimental conditions, male mbt implants (Fig. 1C, red lines) killed more than 85% of the hosts, half of them within 14 to 18 days after implantation, while female mbt implants (Fig. 1C, blue lines) took longer to develop and never reached the 50% host lethality mark. Notably, these results also apply to second instar male ts1/Df brains [Fig. 1C, ts1/Df(L2)] that grow faster and kill more hosts than the much larger late third instar female mbt implants. Male and female brat implants are equally aggressive in allograft tests (fig. S1). Thus, in summary, mbt tumors present sex-dependent dimorphism, while brat tumors do not.
The mbt proteome is sex dependent
To investigate the molecular basis of the sex-dependent dimorphism that we have observed in mbt tumors, we searched for proteins that present sex-biased expression levels in these tumors. We analyzed the proteome of male and female ts1/Df and w1118 brain lobes by Tandem Mass Tag (TMT) labeling and nanoliquid chromatography electrospray ionization tandem mass spectrometry (nanoLC-ESI-MS/MS). We identified a total of 7035 proteins and obtained reliable quantitated data for 5985. Among them, we found a group of 127 proteins that are expressed at significantly different levels in males and females in mbt tumors, but not in wild-type samples (Fig. 2A). This mbt proteome sex-linked dimorphic signature (pSDS) includes 66 proteins that are more expressed in males (M-pSDS) and 61 that are overexpressed in females (F-pSDS) (Fig. 2A and table S1). None of these proteins appears to be expressed in one sex only.
Fig. 2. The sex-linked mbt proteome.
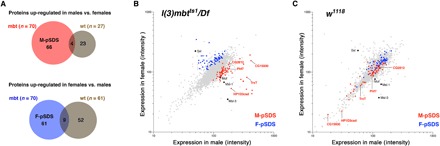
(A) Venn diagrams showing the number of proteins that present sex-biased expression in mbt (red and blue) and wild-type (brown) larval brains. Top and bottom diagrams show the number of proteins up-regulated in males compared to females and in females compared to males, respectively. (B and C) Plots showing the expression level of the proteins in l(3)mbtts1/Df (B) and control w1118 (C) brains lobes in male (x axis) and female (y axis) samples. Blue and red dots correspond to proteins that are significantly overexpressed in males (M-pSDS) and females (F-pSDS), respectively, in l(3)mbtts1/Df samples. As an example, the points corresponding to Phf7, TrxT, HP1D3csd, CG15930, and CG2812 are identified in the plot (red). Gray dots correspond to proteins that are expressed at levels that are not significantly different between male and female l(3)mbtts1/Df samples. Black dots correspond to the sex determination protein Sxl and to the dosage compensation proteins Msl-1, Msl-3, and Mof. The sex-biased expression of these proteins is unaffected in mbt tumors.
M-pSDS and F-pSDS proteins (Fig. 2, B and C, red and blue dots, respectively) appear as two distinct clouds that are well apart in plots showing the expression level of proteins in male versus female mbt tumor samples (Fig. 2B), but are mixed in plots showing sex-dependent expression levels in wild-type samples (Fig. 2C). Expression of the sex determination and Male-Specific Lethal (MSL)–complex proteins that we have unequivocally identified (i.e., Sxl, Msl-1, Msl-3, and Mof) is equally sex biased in mbt tumors and wild-type brains (Fig. 2, B and C, black dots), thus suggesting that the sexual identity of the XX and XY mbt tumors is not compromised.
Eight pSDS proteins [CG15930, CG31373, Agpat4, Cona, Fs(1)Yb, HP1D3csd, TrxT, and Vas] are encoded by genes that belong to the mbt signature (MBTS), identified in a previous transcriptome study in which male and female tissues were not examined separately (24), and four (CG15930, TrxT, Vas, and Pxn) are known to be required for mbt tumor growth (24, 31).
Enriched Gene Ontology (GO) terms refer to DNA replication and mitosis in the M-pSDS and to signaling, ecdysone, redox, and lipid transport in the F-pSDS (table S2). These results are fully consistent with the very different proliferative potential of mbt male and female samples and strongly substantiate the sex-dimorphic nature of mbt tumors. Differences in the expression level of L(3)mbt itself and other proteins of the LINT and Myb-MuvB/Dream complexes (25, 29) between male and female wild-type brains are not significant, and therefore unlikely to account for the different levels of transformation observed in male and female mbt mutants (table S3).
As a first step toward investigating the functional relevance of pSDS proteins, we focused our attention on those of the male signature (M-pSDS), whose contribution to the enhanced malignant traits observed in mbt male tumors can be tested by simple loss-of-function studies. Among them, the group that includes CG15930, CG2812, HP1D3csd, TrxT, and Phf7 stands out, first, because those genes are up-regulated in Drosophila snf148 ovarian tumors (henceforth referred to as snf tumors) that are driven by the unscheduled expression of PHD finger protein 7 (Phf7) (32–34) and, second, because the group includes Phf7 itself.
Phf7 encodes two transcripts: Phf7-RA and Phf7-RC. Phf7-RA transcripts have been found in a variety of tissues including adult ovaries and salivary glands, and larval central nervous system, trachea, and salivary glands (www.flyatlas2.org). Phf7-RC, which includes an additional small exon, is transcribed from an upstream testis-specific transcription start site (TSS), and consistently, has only been reported to be expressed in testis. These two mRNA isoforms have different 5′ untranslated regions that affect translation efficiency, and the PHF7 protein has only been detected in the male germline (33, 34, 35, 36). Ovarian snf tumors up-regulate Phf7-RC and therefore express the Phf7 protein, which is both necessary and sufficient for the tumor-forming pathway (34).
Visualization of our RNA-sequencing (RNA-seq) data in Integrated Genome Browser shows that Phf7-RC, estimated by the number of reads that map to the Phf7-RC–specific exon, is absent in wild-type samples, but is expressed in mbt tumor samples, at a higher level in males than in females (Fig. 3A). This conclusion is further substantiated by reverse transcription quantitative polymerase chain reaction (RT-qPCR) data (Fig. 3B).
Fig. 3. Phf7-RC is up-regulated in mbt tumors.

(A) Genome Browser view of RNA-seq reads aligned to the Phf7 locus. Reads that are unique to the Phf7-RC transcript are highlighted in gray. The arrows at the bottom correspond to the primers used to quantify Phf7-RC by RT-qPCR. (B) Quantification by RT-qPCR of the Phf7-RC transcript in l(3)mbtts1/Df, l(3)mbtts1 and w1118 larval brains. Error bars indicate SD of data derived from biological triplicates (two technical duplicates each). a.u., arbitrary units.
Phf7 depletion suppresses sex-linked dimorphism by inhibiting male-specific phenotypic traits
Because ectopic expression of Phf7 is a necessary step in the development of snf ovarian tumors, we wondered whether it may also have a function in the enhancement of malignant traits that we have observed in mbt male tumors. To test this hypothesis, we investigated the effect of Phf7 depletion on mbt tumor development in situ and on its behavior in allograft assays. We found that depletion of Phf7 has no effect on wild-type brain lobe development (Fig. 4A; compared to Fig. 1A), which is consistent with its reported strictly male germline function in flies (33). Depletion of Phf7 has little effect on the anatomy of female mbt tumors but significantly suppresses the two main anatomy traits that make male mbt tumors distinct: It brings about a significant reduction of the NE (P = 6 × 10−5) that no longer spreads over the brain lobe and the recovery of a well-defined CB region (P = 10−8) (Fig. 4, A and B). As a result, double-mutant Phf7ΔN2; l(3)mbtts1 brain lobes lack sex-linked dimorphism.
Fig. 4. Phf7 depletion inhibits male-specific mbt tumor traits.
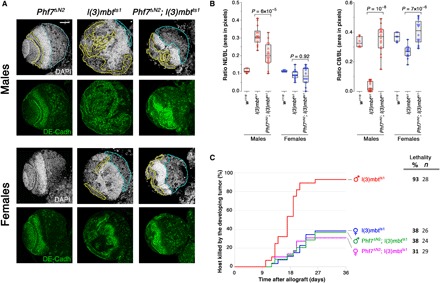
(A) Larval brain lobes from male and female, Phf7ΔN2 and l(3)mbtts1 single-mutant, and Phf7ΔN2; l(3)mbtts1 double-mutant larvae stained with DAPI (gray) and anti–DE-cadherin antibody (green). Phf7ΔN2 mutant lobes appear wild type. Compared to lobes from l(3)mbtts1 males, those from Phf7ΔN2; l(3)mbtts1 males present a much reduced NE (yellow) and a sizeable CB (blue). Scale bar, 50 μm. (B) Relative sizes of NE and CB (as a fraction of brain lobe area) in male (red) and female (blue) control (w1118), mbt [l(3)mbtts1], and Phf7-depleted mbt [Phf7ΔN2; l(3)mbtts1] larvae. Differences in NE and CB sizes between male l(3)mbtts1 and Phf7ΔN2; l(3)mbtts1 are highly significant. (C) Tumor growth rate and host lethality caused by allografted l(3)mbtts1 single-mutant and Phf7ΔN2; l(3)mbtts1 double-mutant, male (red and green, respectively) and female (blue and purple, respectively) brain lobes. Male Phf7ΔN2; l(3)mbtts1 implants develop and kill hosts at a much lower rate than do male l(3)mbtts1 implants.
In allograft assays, depletion of Phf7 results in a significant drop in host lethality, from 93% [male l(3)mbtts1, n = 28] down to 38% [malePhf7ΔN2; l(3)mbtts1, n = 24], a rate that is very similar to that of mbt female implants [female l(3)mbtts1, 38%, n = 26] (Fig. 4C). The same applies to the timing of tumor development and to the onset of host lethality that are delayed in male Phf7ΔN2; l(3)mbtts1 compared to male l(3)mbtts1 implants (Fig. 4C). Phf7 depletion has no significant effect on the parameters of growth and lethality of female mbt tissue in allograft assays. Thus, depletion of Phf7 suppresses both the anatomy traits and the greater growth potential that make mbt tumors more aggressive in males than in their female siblings.
Phf7 depletion erases phenotypic dimorphism by respectively down- and up-regulating the male and female sex-linked dimorphic signatures in males
Given Phf7 reported function as a histone code reader that binds lysine 4 di- and tri-methylated histone H3 (H3K4me2/me3) and controls gene expression programs (33–35), we decided to test whether Phf7 function in mbt sex-linked dimorphism could be due to a role for Phf7 in controlling sex-dependent differences in gene expression. To this end, we carried out RNA-seq to quantify the transcripts that present sex-biased expression in l(3)mbtts1, but not in w1118 larval brains [i.e., the male and female transcriptome sex-linked dimorphic signatures of l(3)mbtts1 tumors; M-tSDS and F-tSDS, respectively] and then determined whether such a bias is affected in Phf7ΔN2; l(3)mbtts1 double-mutant samples.
To analyze these data, we plotted the expression levels of transcripts in male and female brain samples from wild-type (w1118), mbt (l(3)mbtts1), and double-mutant Phf7ΔN2; l(3)mbtts1 larvae (Fig. 5A). As expected, dots corresponding to the expression levels of M-tSDS (red) and F-tSDS (blue) genes are mixed in control w1118 and form two distinct clouds that are well apart in l(3)mbtts1 samples. The red and blue clouds remain distinct in Phf7ΔN2; l(3)mbtts1 double mutant, but they are much closer than in l(3)mbtts1 alone, showing that sex-biased transcription of M-tSDS and F-tSDS genes is reduced in mbt tumors that lack Phf7 (Fig. 5A). Such a reduction of the difference in expression levels of the M-tSDS and F-tSDS gene sets in Phf7ΔN2; l(3)mbtts1 samples could be accounted for by changes in expression that affect either of the two signatures, or both, in either sex, or in both. To determine which are the actual changes that account for the observed results, we plotted the expression levels of each of the genes of M-tSDS and F-tSDS in mbt (l(3)mbtts1) and Phf7-depleted mbt [Phf7ΔN2; l(3)mbtts1] male and female tissues. For ease of understanding, signature genes were ordered from left to right along the x axis as a function of their expression level in l(3)mbtts1 (Fig. 5B).
Fig. 5. Phf7 depletion reduces sex-biased expression of the mbt sex-linked dimorphic signatures.
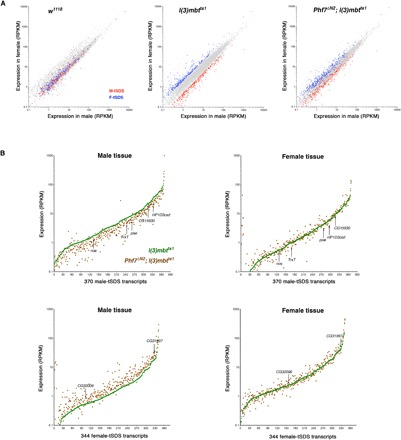
(A) Plots showing the expression level of transcripts in male (x axis) and female (y axis) samples from w1118 and l(3)mbtts1 single-mutant and Phf7ΔN2; l(3)mbtts1 double-mutant larvae. Blue and red dots correspond to genes that are significantly overexpressed in male versus female (M-tSDS) and female versus male (F-tSDS) mbt tumors, respectively. Gray dots correspond to genes that are expressed at levels that are not significantly different between males and females. (B) Expression levels of M-tSDS and F-tSDS transcripts in l(3)mbtts1 (green) and Phf7ΔN2; l(3)mbtts1 (brown) male and female tissues. The genes of each signature are ordered along the x axis as a function of their expression level in l(3)mbtts1 tissue. Arrows point at dots corresponding to selected M-tSDS (CG15930, HP1D3csd, TrxT, nos, and piwi) and F-tSDS (CG31997 and CG32006) genes.
We found that in male, but not in female, tissue, M-tSDS genes are on average expressed at a lower level in Phf7ΔN2; l(3)mbtts1 (Fig. 5B, brown dots) than in l(3)mbtts1 (Fig. 5B, green line). Wilcoxon tests confirm that differences between mean gene expression across the M-tSDS genes in the Phf7ΔN2; l(3)mbtts1, and l(3)mbtts1 samples are highly significant (P = 6.9 × 10−4) in males and not significant (P = 0.6) in females. Conversely, F-tSDS genes present a significantly higher (P = 2.5 × 10−6) level of expression in Phf7ΔN2; l(3)mbtts1 than in l(3)mbtts1 male samples, while differences are not significant (P = 0.7) in females (Fig. 5B). As an example, some of the genes that are up- and down-regulated in male samples are marked in Fig. 5, including five M-tSDS genes (CG15930, HP1D3csd, TrxT, nos, and piwi) and two F-tSDS genes (CG31997 and CG32006). All these seven genes belong to the MBTS, and four of them (CG15930, TrxT, nos, and piwi) are required for mbt tumor growth (24, 31).
The same conclusions are derived from plots showing the significance of the fold change in the expression level of M-tSDS and F-tSDS genes between Phf7ΔN2; l(3)mbtts1 and l(3)mbtts1 in male and female samples (volcano plots; fig. S3). In male tissues, most dots representing M-tSDS and F-tSDS transcripts are shifted to negative (i.e., down-regulated) and positive (i.e., up-regulated) values, respectively, while in female tissues fold-change value distribution is rather symmetric and most changes are not significant for both signatures.
In contrast to its effect in controlling gene expression differences in male and female tumors, Phf7 appears to have a minor role in controlling gene expression differences between tumor and wild-type samples. Thus, for instance, only six MBTS genes are down-regulated to any significant extent after the loss of Phf7 in mbt males and only one in females [Phf7ΔN2; l(3)mbtts1 compared to l(3)mbtts1]. None of these are germline genes (table S4).
These results strongly suggest that Phf7 contributes to bringing about sex-linked molecular disparities in mbt tumors by acting in male tissue both up-regulating M-tSDS genes and down-regulating F-tSDS genes while having little, if any, effect in the expression of either of these signatures in female samples.
DISCUSSION
Epidemiological studies show that in a wide range of cancer types unrelated to reproductive function, men have a worse prognosis than women (1, 2, 4, 6). The molecular basis for such disparities remains very poorly understood. We have found that the tumors that develop in Drosophila l(3)mbt mutant larvae are strongly dimorphic: Malignant traits are much more prominent in males than in females, to the extent that they can be used to objectively stratify mbt tumor samples into two populations that correlate tightly with the sex of the tumor bearer. Using mbt tumors as a genetically tractable experimental model to investigate the molecular basis of sex-linked disparities in malignant growth, we have identified two protein signatures that include those proteins that are significantly up-regulated in one sex compared to the other (male and female proteomic sex-linked dimorphic signatures; M-pSDS and F-pSDS, respectively). Many of the proteins that belong to these signatures have homologs in humans and are therefore promising leads for future research.
A conspicuous group of the proteins that we have found to be expressed at a higher rate in male mbt tumors are also ectopically expressed in the tumors that develop in the ovaries of flies homozygous for the viable allele of sans fille, snf148. One of these common proteins is Phf7, which is both necessary and sufficient for the snf tumor–forming pathway (33, 34). Phf7 is a histone code reader that bears three PHD domains and binds histone H3 N-terminal tails with a preference for dimethyl lysine 4 (H3K4me2) (33). In wild-type flies, expression of Phf7 is restricted to male germline stem cells and spermatogonia (33, 35). Loss of Phf7 impairs the ability of male germline cells to transit through the different stages of spermatogenesis and results in reduced fertility (33, 35).
There are notable similarities as well as differences in Phf7 function, regulation of expression, and targets in wild-type testis, snf ovarian tumors, and mbt tumors.
As far as function is concerned, similarly to its oncogenic effect in ovaries, we have found that Phf7 has a key role in enhancing malignant traits in male mbt brain tumors both in situ and in allograft test. Our transcriptomics data and the little, if any, phenotypic consequences brought about by the loss of Phf7 in female mbt tumors strongly suggest that Phf7 exerts this role by contributing to the dysregulation of dozens of genes mostly in male mbt tumors. We do not know the reason for such sex-linked differential impact of Phf7 depletion. It could be quantitative (i.e., the PHF7 level in female tumors is not sufficient enough), qualitative [i.e., dependent upon male-specific (or male-enriched) factors], or both.
Unscheduled expression of Phf7-RC in wild-type female germ cells is prevented by deposition of the H3K9me3 repressive mark over the testis-specific TSS of Phf7-RC through a process that is controlled by the female sex determination protein Sxl and depends on the eggless/SETDB1 methyltransferase and other members of the H3K9me3 pathway (36). Phf7-RC is expressed in snf tumors because the homozygous condition for snf148 interferes with the splicing of Sxl in germ cells (32). The mechanisms that repress Phf7-RC transcription in wild-type somatic cells of both sexes remain unknown, but certainly in males, cannot depend on Sxl. Ectopic Phf7-RC expression in mbt tumors and the overlap of the TSS of Phf7-RC with a L(3)mbt binding site identified in cephalic complex samples from third instar larvae (30) suggest that repression of Phf7-RC in the soma may depend on L(3)mbt itself. Something similar may apply to another M-pSDS protein, CG15930, also known as Tudor-domain–containing protein 5-prime (Tdrd5p), that is also normally highly expressed in male germ cells and is repressed by Sxl in female germ cells (34, 37). Expression of CG15930 is strongly up-regulated in mbt tumors of both sexes and, like Phf7-RC, more so in males than in females. The TSS of CG15930 overlaps with both L(3)mbt (30) and LINT binding sites (29).
A common theme between ovarian snf and larval brain mbt tumors is the unscheduled expression of hundreds of genes including many testis genes. However, the extent of overlap between the genes dysregulated in both tumors is low: Only 10% of the MBTS genes (11) are up-regulated in snf compared to wild-type ovaries. Included among these is the CG15930 gene referred to above, which we have previously shown to be essential for mbt tumor growth (31). In wild-type flies, CG15930 localizes to cytoplasmic granules with some characteristics of RNA processing (P-) bodies and has been shown to promote proper male fertility and germline differentiation (37). Likewise, the overlap between genes controlled by Phf7 in testis and those dysregulated in mbt is negligible. Transcriptomic comparisons between single-mutant bag-of-marbles (bam) testis and double-mutant Phf7; bam testis identified 45 genes that are dysregulated upon Phf7 loss (35). Only one of those (EbpIII) is also dysregulated in male mbt tumors that lack Phf7 [Phf7ΔN2; l(3)mbtts1 compared to l(3)mbtts1]. These results reflect a fundamental difference in the targets of Phf7 function in snf tumors and wild-type testis, and those that we have identified in mbt tumors, and suggest possible differences in the distribution of H3K4 methylation marks.
A distinct feature of mbt tumors is the up-regulation of a signature of transcripts, the MBTS, that can be used to unequivocally tell these tumors apart not only from wild-type brains but also from other malignant brain neoplasms, like those caused by loss-of-function conditions for lethal giant larvae (lgl), miranda (mira), prospero (pros), or brat. Our new RNA-seq data obtained from male and female samples provide an opportunity to determine the extent to which up-regulation of the MBTS, which was identified in a study where male and female tissues were not examined separately, is sex dependent. Our data show that the majority of the MBTS genes (80) are up-regulated in both sexes: 8 only in males and 1 only in females. These results show that L(3)mbt safeguards larval brain cells against unscheduled gene expression in both sexes. This includes the germline genes that account for a quarter of the MBTS genes, many of which are necessary for mbt tumor growth (24, 31). Notably, however, 19 of the 80 MBTS genes up-regulated in both sexes are significantly more expressed in male than in female mbt tumors (i.e., belong to the M-tSDS). Whether such quantitative—not qualitative—differences reflect a sex-dependent efficiency in the role of L(3)mbt safeguarding against ectopic gene expression remains unclear.
L(3)mbt’s function as a repressor of unscheduled gene programs is not limited to the somatic cells of the larval brains. Loss of l(3)mbt in some Drosophila cell lines and in the somatic cells of the ovary leads to the ectopic activation of germline genes, including components of the PIWI ping-pong cycle, vas, nos, and others (26, 27). Loss of l(3)mbt in the female germline results in the ectopic activation of testis and neuronal genes (26).
The human genome contains three orthologs to Drosophila l(3)mbt—L3MBTL1, L3MBTL3, and L3MBTL4—that, like the fly gene, encode chromatin-interacting transcriptional repressors (23). L3MBTL3 maps to chromosomal region 6q23 that is frequently altered in acute leukemia cells, and homozygous deletion of this gene has been observed in human patients with medulloblastoma (38, 39). Moreover, reexpression of L3MBTL3 attenuates malignancy in human medulloblastoma cell lines that are deleted for L3MBTL3 (39). Medulloblastoma groups 3 and 4, which are very frequently metastatic, exhibit a 2:1 male to female incidence ratio (40, 41). The cause for such gender disparities remains unknown. Homologs of Drosophila Phf7 have been identified in vertebrates including mammals, and human Phf7 expression is also highly enriched in the testis (42, 43, but there is currently no evidence suggesting a role for Phf7 in sex dimorphism in human cancer. Despite substantial sequence homology, human Phf7 and Drosophila Phf7 did not evolve from a common Phf7 ancestor, but rather, both genes evolved in parallel through independent duplication events from an ancestral G2-M phase–specific E3 ubiquitin protein ligase (G2E3) (42). Functional overlap between both homologs is high, but not full: Human Phf7 can rescue the fertility defects brought about by the loss of Phf7 in Drosophila males, but does not have the deleterious effect brought about by the fly protein when expressed in the female germline (33, 42).
A significant number of clinically actionable genes show strong sex-biased signatures in different cancer types, but the functional relevance of such disparities remains to be determined (8). Our results show that proteins that belong to sex-biased tumor signatures can be targeted to eliminate sex-linked enhanced malignancy.
MATERIALS AND METHODS
Fly stocks
The following mutant alleles were used in this study: l(3)mbtts1 (19), l(3)mbtE2 (44), Df(3R)ED10966 (DGRC#150208 from Kyoto Stock Center), bratK06028 (45), and Phf7ΔN2 (33). The wild-type strain used was w1118. To distinguish male from female mbt mutant larvae, the strains Dp(1:Y)y+ and an X insertion of pUbq-αtub84B-GFP were used (46). Because of the temperature-sensitive condition of mbt, all crosses, including controls, were maintained at 29°C. Double-mutant Phf7ΔN2; l(3)mbtts1 was generated using standard genetic techniques.
Genotypes and crossing schemes
mbt experiments
Crosses for ts1/Df experiments were done with females yw; Df(3R)ED10966/TM6B,Tb and males yw/Dp(1:Y)y+; l(3)mbtts1/TM6B,Tb. The progeny used for the experiments were males yw/Dp(1:Y)y+; l(3)mbtts1/ Df(3R)ED10966 and females yw; l(3)mbtts1/ Df(3R)ED10966. Similarly, for ts1 experiments, females yw; l(3)mbtts1/TM6B,Tb were crossed with males yw/Dp(1:Y)y+; l(3)mbtts1/TM6B,Tb. The progeny used were males yw/Dp(1:Y)y+; l(3)mbtts1 and females yw; l(3)mbtts1. For ts1/E2 experiments, females yw; l(3)mbtts1/TM6B,Tb were crossed with males l(3)mbtE2/TM6B,Tb. The progeny used were males yw; l(3)mbtts1/l(3)mbtE2 and females yw/+; l(3)mbtts1/l(3)mbtE2. In addition, sexing of larvae in some allograft assays was achieved by using the X insertion pUbq-αtub84B-GFP. A further unbiased method for sexing larvae was to allograft the lobes of unmarked larvae (i.e., the same genotype for male and female progeny) and to subsequently assign the sex by immunostaining of the corresponding salivary glands with an H4K16ac-specific antibody (17). The actual method used for sexing larvae did not alter the dimorphic outcome of independent allograft assays done with the same allelic combination.
brat experiments
Crosses for brat experiments were done with females w; bratk08026/CyO,Tb and males pUbq-αtub84B-GFP; bratK08026/CyO,Tb. The progeny used for experiments were males w; bratk06028 and females pUbq-αtub84B-GFP /+; bratk06028.
Phf7 experiments
Crosses for the double-mutant condition were done crossing females ywPhf7ΔN2; l(3)mbtts1/TM6B,Tb with males ywPhf7ΔN2/Dp(1:Y)y+; l(3)mbtts1/TM6B,Tb. The progeny selected were males ywPhf7ΔN2/Dp(1:Y)y+; l(3)mbtts1 and females ywPhf7ΔN2; l(3)mbtts1.
Immunohistochemistry
Immunostaining of whole larval brains was performed as described (47). Briefly, brains were dissected in phosphate-buffered saline (PBS), fixed in 4% formaldehyde, rinsed in PBS–0.3% Triton X-100 (PBST), and blocked for 60 min in PBST with 10% fetal calf serum (PBSTF). Primary and secondary antibodies were incubated in PBSTF overnight at 4°C. Primary antibodies used in this study include rat anti–DE-cadherin [DCAD2, 1:100, Developmental Studies Hybridoma Bank (DSHB)], mouse anti-Dac (mAbdac1-1, 1:100, DSHB), and rabbit anti-H4K16ac (1:1000, Active Motif). We used Alexa Fluor secondary antibodies (1:1000, Life Technologies). DNA was stained with DAPI. Larval brains were mounted in Vectashield (Molecular Probes). Images were acquired with an SP8 Leica confocal image microscope and processed in Adobe Photoshop CS6 and ImageJ.
Allograft assays
Larval brain lobe grafts were carried out in female hosts as described in (48). Tumor lethality (%) was calculated by the number of hosts killed by the developing tumor over the total of allografted adults. Implanted hosts were kept at 29°C.
Quantification and characterization of phenotypes
Eggs were collected for 24 hours and allowed to develop for up to 7 days (156 ± 12 hours AEL) for anatomy analysis and up to 8 days (180 ± 12 h AEL) for Feret diameter measurement, except for control w1118 larvae that were dissected at 5 days AEL. The ratios area of NE/area of the brain lobe (ratio NE/BL) and area of CB/area of the brain lobe (ratio CB/BL) were calculated by using images acquired with an SP8 Leica confocal image microscope and by measuring the areas corresponding to the NE, the CB, and the brain lobe using ImageJ software. The Feret diameter measurement of the brain lobe pairs was performed by analyzing the images of brains taken using a Leica EC3 camera coupled to a Nikon SMZ800 stereoscope. The images were analyzed by a purpose-made macro (31) written in ImageJ software to measure the maximum brain Feret diameter of the brain lobe pair. Ventral ganglions were digitally masked before measurement. The results were represented in boxplots, and P values were calculated by nonparametric Mann-Whitney U tests using GraphPad Prism 7.00 for MacOS X (GraphPad Software, La Jolla, CA, USA) (www.graphpad.com). All genotypes and crosses were done as described above.
Proteomics
Protein extraction
Larval brains were dissected in PBS, transferred to a 1.5-ml tube, and frozen in liquid nitrogen after removing the buffer. One hundred fifty brains of each genotype were homogenized in 150 μl of a buffer containing 4% SDS, 100 mM tris-HCl (pH 7.6), and 0.1 M dithiothreitol and incubated at 95°C for 3 min. The samples were sonicated to shear the DNA to reduce the viscosity of the sample. Before starting sample processing, the lysate was clarified by centrifugation at 16,000g for 5 min.
Sample preparation, TMT labeling, and basic reversed-phase prefractionation
Protein extracts were quantified using the Pierce 660 Protein Assay Kit (#22662) and Ionic Detergent Compatibility Reagent (#22663). They were alkylated with 2-iodoacetamide and digested with trypsin following the Filter Aided Sample Preparation (FASP) protocol (49). After digestion and requantification at the peptide level by the Colorimetric Peptide Assay (Pierce Thermo, #23275), samples were isotopically labeled with the corresponding TMT10plex reagent (Thermo Fisher Scientific) according to the experimental design (labels 126 to 131). Validation for correct isotopic labeling was performed by LC-MS/MS, and samples were then mixed in two different batches (taking into account peptide quantification) and desalted using PolyLC C18 and PolyLC SCX strong cationic exchange tips. Each of the two TMT10plex experiments was fractionated offline by high-pH reversed-phase peptide chromatography using Pierce columns (ref. 84868). Ten fractions were collected for each batch (F0 to F9), dried, and reconstituted in 1% formic acid, 3% acetonitrile for nanoLC-ESI-MS/MS analysis (600 ng of protein on column).
Nanoliquid chromatography electrospray ionization tandem mass spectrometry
Peptides from basic reversed-phase prefractionation (20 fractions from two TMT10plex experiments) were analyzed using an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher Scientific) equipped with a Thermo Scientific Dionex Ultimate 3000 ultrahigh-pressure chromatographic system (Thermo Fisher Scientific) and Advion TriVersa NanoMate (Advion Biosciences Inc.) as the nanospray interface. Peptide mixtures were loaded to a μ-precolumn (300 μm inside diameter × 5 mm, C18 PepMap100, 5 μm, 100 Å, C18 Trap column; Thermo Fisher Scientific) at a flow rate of 15 μl/min and separated using a C18 analytical column (Acclaim PepMap TM RSLC: 75 μm × 75 cm, C18 2 μm, nanoViper) with a flow rate of 200 nl/min and a 300-min run, comprising three consecutive steps with linear gradients from 1 to 35% B in 262 min, from 35 to 50% B in 5 min, and from 50 to 85% B in 2 min, followed by isocratic elution at 85% B in 5 min and stabilization to initial conditions (A = 0.1% formic acid in water, B = 0.1% formic acid in acetonitrile).
The mass spectrometer was operated in a data-dependent acquisition mode. In each data collection cycle, one full MS scan (400 to 1600 m/z) was acquired in the Orbitrap [1.2 × 105 resolution setting and automatic gain control (AGC) of 2 × 105]. The following MS2-MS3 analysis was conducted with a top speed approach. The most abundant ions were selected for fragmentation by collision-induced dissociation (CID). CID was performed with a collision energy of 35%, 0.25 activation Q, an AGC target of 1 × 104, an isolation window of 0.7 Da, a maximum ion accumulation time of 50 ms, and turbo ion scan rate. Previously analyzed precursor ions were dynamically excluded for 30 s. For the MS3 analyses for TMT quantification, multiple fragment ions from the previous MS2 scan (SPS ions; synchronous precursor selection) were co-selected and fragmented by HCD using a 65% collision energy and a precursor isolation window of 2 Da. Reporter ions were detected using the Orbitrap with a resolution of 60,000, an AGC of 1 × 105, and a maximum ion accumulation time of 120 ms.
Spray voltage in the NanoMate source was set to 1.60 kV. Radio frequency lenses were tuned to 30%. The spectrometer was working in positive polarity mode, and singly charge state precursors were rejected for fragmentation.
Database search
Database searches were performed with Proteome Discoverer v2.1.0.81 software (Thermo Fisher Scientific) using Sequest HT search engine and UniProt Canonical and Isoforms DROME_2017_06 with contaminants. Search was run against targeted and decoy database to determine the false discovery rate (FDR). Search parameters included trypsin, allowing for two missed cleavage sites, carbamidomethyl in cysteine and TMT peptide N terminus as static modification and TMT in K, methionine oxidation, and acetylation in protein N terminus as dynamic modifications. Peptide mass tolerance was 10 parts per million (ppm), and the MS/MS tolerance was 0.6 Da in MS2 and 20 ppm in MS3. Peptides with a q value lower than 0.1 and an FDR of <1% were considered as positive identifications with a high confidence level.
Quantitative analysis
TMT reporter ion intensities were used for protein quantification. Unique peptides (peptides that are not shared between different protein groups) were considered for further quantitative and statistical analysis. Within each TMT experiment, peptide quantitation was normalized by summing the abundance values for each channel over all peptides identified within an experiment, and then the channel with the highest total abundance was taken as a reference and all abundance values were corrected in all other channels by a constant factor per channel so that, at the end, the total abundance is the same for all channels. Protein quantitation was done by summing all peptide normalized intensities for a given protein. Protein intensities were scaled so that, for every protein in an experiment, the average of all channels is 100. Proteins were only considered quantifiable if all quan channels have abundance values.
DanteR (50), by Pacific Northwest National Laboratory, was used to preprocess, visualize data (boxplots and principal components analysis), and perform relative quantification of proteins labeled with TMT. Protein quantitative measurements were log2-transformed, and normalization across the four TMT10plex experiments was performed using quantile normalization (51). Two-way analysis of variance (ANOVA) was performed at the protein level using a linear model. Conditions were considered as the principal factor and TMT batch as the second factor. Weighting function was used to allow data variability to depend on data value. Comparisons considering condition or age were performed. Last, P values were adjusted for multiple testing using the Benjamini-Hochberg FDR correction. Data were also processed by performing a one-way ANOVA statistical analysis to take also into account those proteins found only in one batch. Differential expressed proteins were determined using an adjusted P value cutoff of 0.05 and a fold change lower than 0.67 (down) or higher than 1.5 (up).
Venn diagrams
Venn diagrams were done using the web application BioVenn (http://www.biovenn.nl/) (52).
GO analysis
Functional annotation of GO terms was performed using the online tool Database for Annotation, Visualization and Integrated Discovery (DAVID 6.8; http://david.abcc.ncifcrf.gov/). GO terms for biological process (GOTERM_BP_DIRECT) with a P value of <0.05 were accepted as a significant enrichment.
Transcriptomics
RNA-seq sample preparation and sequencing
RNA was isolated using magnetic beads (RNAClean XP, Beckman Coulter, A63987) from 10 Drosophila larval brains following the protocol described in (24). RNA concentration was determined with a Qubit fluorometer (Thermo Fisher Scientific), and integrity was assessed on the Agilent 2100 Bioanalyzer.
RNA poly(A) purification was performed from 0.8 to 1.2 μg of total RNA using the NEBNext Poly(A) mRNA Magnetic Isolation Module Kit (NEB, E7490). Then, complementary DNA (cDNA) generation, adaptor ligation, and library amplification were done with NEBNext Ultra RNA II Library Prep Kit for Illumina and NEBNext Multiplex Oligos for Illumina Set 1, 2, and 3 (NEB E7770, E7335, E7500, and E7710, respectively) following the manufacturer’s instructions. Library amplification was performed with SYBR Green (Sigma, S9430) to establish the necessary number of cycles to quantify (Qubit fluorometer) and to check size distribution (2100 Bioanalyzer) and sequencing. Libraries were sequenced in 125-nucleotide paired-end lanes of an Illumina HiSeq 2500 system, obtaining between 27 million and 56 million of reads per sample.
RNA-seq data processing
Data were processed with the Grape RNA-seq pipeline (https://github.com/guigolab/grape-nf). Raw reads were aligned to the fly genome (dmel6 assembly from http://hgdownload.soe.ucsc.edu/goldenPath/dm6/bigZips/dm6.fa.gz) and transcriptome (dmel6-05 from ftp://ftp.flybase.net/genomes/Drosophila_melanogaster/dmel_r6.05_FB2015_02/gff/dmel-all-no-analysis-r6.05.gff.gz) using STAR (https://doi.org/10.1093/bioinformatics/bts635, v2.4.0j). A maximum of four mismatches per sequence was used, and only reads with at most 10 multiple mappings were retained. Genes and transcripts were quantified using RNA-seq by Expectation Maximization (RSEM) (https://doi.org/10.1186/1471-2105-12-323, v1.2.21) with default parameters. RPKM (reads per kilobase of exon per million fragments mapped) was used as a measure of gene and transcript abundances. The library size for each sample was scaled according to the TMM (trimmed mean of M values) normalization method (https://doi.org/10.1186/gb-2010-11-3-r25). Several filters were applied to the gene expression matrix to obtain a stable gene set for the analysis. We removed all ribosomal RNA (rRNA) and transfer RNA (tRNA) genes as well as messenger RNA (mRNA) genes coding for ribosomal proteins. Lowly expressed genes with less than 10 reads in all replicates were discarded. To ensure reproducibility across replicates, we applied nonparametric Irreproducible Discovery Rate (IDR) (53) on read counts for all pairwise combinations of three replicates: If IDR < 0.01 for any comparison, then the read counts in all replicates are set to 0. Last, we only kept genes with RPKM > 1 in at least three samples. This resulted in 9340 genes out of 17,159.
Expression coverage files in BigWig format (https://genome.ucsc.edu/goldenPath/help/bigWig.html) were generated using STAR (v2.4.0j) and later normalized according to the scaling factors obtained by the TMM normalization method. The resulting files were visually inspected with the Integrated Genome Browser (http://igb.bioviz.org/) software.
RNA-seq data analysis
Batch effect correction was applied to the gene expression matrix using Limma (https://doi.org/10.1093/nar/gkv007, v3.30.13). The gender was used as the fixed factor for batch correction.
Differential expression analysis
Pairwise comparisons were performed to identify differentially expressed genes between females and males and genotypes using edgeR. Genes with fold change > 2 and FDR < 0.05 were considered differentially expressed.
To define the transcriptomics signatures (M-tSDS and F-tSDS), we filtered out genes with a coefficient of variation (CV%) greater than 50% for all replicates.
Wilcoxon test and generation of volcano plots
Two-sample Wilcoxon tests were performed using the function wilcox.test from R standard library (default parameters).
Volcano plots represent log2 fold change of the expression of M-tSDS and F-tSDS genes (x axis) versus negative log10 of the P values (y axis), as resulting from the differential expression analysis between Phf7ΔN2; l(3)mbtts1 and l(3)mbtts1 from male and female samples.
Gene expression analysis by RT-qPCR
Total RNA was isolated from dissected larval brains using proteinase K and deoxyribonuclease (DNase) (Invitrogen) and purified using magnetic beads. RNA yield and quality were assessed with Qubit, followed by reverse transcription using random hexamers. Transcript levels were measured with PowerUp SYBR Green Master Mix in QuantStudio 6 Flex System (Applied Biosystems). Initial activation was performed at 95°C for 20 s, followed by 40 cycles of 95°C for 5 s and 60°C for 15 s. The melting curve was generated ranging from 50° to 95°C with an increment of 0.5°C each 5 s. Primers specific for the transcript Phf7-RC used for RT-qPCR are as follows: AGTTCGGGAATTCAACGCTT (forward) and GAGATAGCCCTGCAGCCA (reverse) (34). Measurements were performed on biological triplicates, with technical duplicates of each biological sample. RNA levels were normalized to rp49. The relative transcript levels were calculated using the 2−ΔΔCT method (54).
Supplementary Material
Acknowledgments
We are very grateful to R. Guigó, J. Pampalona, and D. Garrido-Martín for their contribution to this study; M. V. Doren, the Bloomington Stock Center, the Kyoto Stock Center, and the Developmental Studies of Hybridoma Bank for fly strains and antibodies; and the Functional Genomics Facility of IRB Barcelona for their technical assistance. Funding: Part of this study was funded by ERC AdG 2011 294603 and BFU2015-66304-P from the MINECO, Spain. Author contributions: C.G. conceived and supervised the project and wrote the manuscript. C.M., J.P.H., J.R., and S.L. executed the experiments. C.G., C.M., G.P., J.P.H., J.R., and S.L. analyzed the data. C.M. prepared the figures. A.B. designed the transcriptome analysis. E.P. processed the RNA-seq data and performed the corresponding bioinformatic analysis. M.G., L.V., and M.V. performed the proteomic studies and the corresponding bioinformatics analysis. All authors read and approved the final manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaaw7965/DC1
Fig. S1. brat tumors do not present sex-dependent dimorphism.
Fig. S2. mbt tumor size is sex dependent.
Fig. S3. Loss of Phf7 reduces the expression of mbt M-tSDS genes and increases the expression of mbt F-tSDS genes in male tissue.
Table S1. Proteomic sex-linked dimorphic signatures.
Table S2. GO terms enriched in the M-pSDS and the F-pSDS.
Table S3. Relative expression levels of L(3)mbt and other components of the LINT and Myb/Muv/Dream complexes between male and female wild-type brain lobes.
Table S4. Sex-dependent dysregulation of MBTS genes in l(3)mbtts1 tumors versus wild-type and in Phf7ΔN2; l(3)mbtts1 versus l(3)mbtts1.
REFERENCES AND NOTES
- 1.Clocchiatti A., Cora E., Zhang Y., Dotto G. P., Sexual dimorphism in cancer. Nat. Rev. Cancer 16, 330–339 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Siegel R. L., Miller K. D., Jemal A., Cancer statistics, 2018. CA Cancer J. Clin. 68, 7–30 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Dunford A., Weinstock D. M., Savova V., Schumacher S. E., Cleary J. P., Yoda A., Sullivan T. J., Hess J. M., Gimelbrant A. A., Beroukhim R., Lawrence M. S., Getz G., Lane A. A., Tumor-suppressor genes that escape from X-inactivation contribute to cancer sex bias. Nat. Genet. 49, 10–16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dorak M. T., Karpuzoglu E., Gender differences in cancer susceptibility: An inadequately addressed issue. Front. Genet. 3, 268 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zannoni G. F., Ciucci A., Marucci G., Travaglia D., Stigliano E., Foschini M. P., Scambia G., Gallo D., Sexual dimorphism in medulloblastoma features. Histopathology 68, 541–548 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Khanna V., Achey R. L., Ostrom Q. T., Block-Beach H., Kruchko C., Barnholtz-Sloan J. S., de Blank P. M., Incidence and survival trends for medulloblastomas in the United States from 2001 to 2013. J. Neurooncol. 135, 433–441 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Pearce M. S., Parker L., Childhood cancer registrations in the developing world: Still more boys than girls. Int. J. Cancer 91, 402–406 (2001). [DOI] [PubMed] [Google Scholar]
- 8.Yuan Y., Liu L., Chen H., Wang Y., Xu Y., Mao H., Li J., Mills G. B., Shu Y., Li L., Liang H., Comprehensive characterization of molecular differences in cancer between male and female patients. Cancer Cell 29, 711–722 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lopes-Ramos C. M., Kuijjer M. L., Ogino S., Fuchs C., DeMeo D. L., Glass K., Quackenbush J., Gene regulatory network analysis identifies sex-linked differences in colon cancer drug metabolism. Cancer Res. 78, 5538–5547 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mokdad A. H., Dwyer-Lindgren L., Fitzmaurice C., Stubbs R. W., Bertozzi-Villa A., Morozoff C., Charara R., Allen C., Naghavi M., Murray C. J. L., Trends and patterns of disparities in cancer mortality among US counties, 1980-2014. JAMA 317, 388–406 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gonzalez C., Drosophila melanogaster: A model and a tool to investigate malignancy and identify new therapeutics. Nat. Rev. Cancer 13, 172–183 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Salomon R. N., Jackson F. R., Tumors of testis and midgut in aging flies. Fly 2, 265–268 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Regan J. C., Khericha M., Dobson A. J., Bolukbasi E., Rattanavirotkul N., Partridge L., Sex difference in pathology of the ageing gut mediates the greater response of female lifespan to dietary restriction. eLife 5, e10956 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tipping M., Perrimon N., Drosophila as a model for context-dependent tumorigenesis. J. Cell. Physiol. 229, 27–33 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sonoshita M., Cagan R. L., Modeling human cancers in Drosophila. Curr. Top. Dev. Biol. 121, 287–309 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Mirzoyan Z., Sollazzo M., Allocca M., Valenza A. M., Grifoni D., Bellosta P., Drosophila melanogaster: A model organism to study cancer. Front. Genet. 10, 51 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hudry B., Khadayate S., Miguel-Aliaga I., The sexual identity of adult intestinal stem cells controls organ size and plasticity. Nature 530, 344–348 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kurzik-Dumke U., Phannavong B., Gundacker D., Gateff E., Genetic, cytogenetic and developmental analysis of the Drosophila melanogaster tumor suppressor gene lethal(2)tumorous imaginal discs (1(2)tid). Differentiation 51, 91–104 (1992). [DOI] [PubMed] [Google Scholar]
- 19.Gateff E., Löffler T., Wismar J., A temperature-sensitive brain tumor suppressor mutation of Drosophila melanogaster: Developmental studies and molecular localization of the gene. Mech. Dev. 41, 15–31 (1993). [DOI] [PubMed] [Google Scholar]
- 20.Bowman S. K., Rolland V., Betschinger J., Kinsey K. A., Emery G., Knoblich J. A., The tumor suppressors Brat and Numb regulate transit-amplifying neuroblast lineages in Drosophila. Dev. Cell 14, 535–546 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Betschinger J., Mechtler K., Knoblich J. A., Asymmetric segregation of the tumor suppressor Brat regulates self-renewal in Drosophila neural stem cells. Cell 124, 1241–1253 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Mukherjee S., Tucker-Burden C., Zhang C., Moberg K., Read R., Hadjipanayis C., Brat D. J., Drosophila Brat and human ortholog TRIM3 maintain stem cell equilibrium and suppress brain tumorigenesis by attenuating Notch nuclear transport. Cancer Res. 76, 2443–2452 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bonasio R., Lecona E., Reinberg D., MBT domain proteins in development and disease. Semin. Cell Dev. Biol. 21, 221–230 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Janic A., Mendizabal L., Llamazares S., Rossell D., Gonzalez C., Ectopic expression of germline genes drives malignant brain tumor growth in Drosophila. Science 330, 1824–1827 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Georlette D., Ahn S., MacAlpine D. M., Cheung E., Lewis P. W., Beall E. L., Bell S. P., Speed T., Manak J. R., Botchan M. R., Genomic profiling and expression studies reveal both positive and negative activities for the Drosophila Myb MuvB/dREAM complex in proliferating cells. Genes Dev. 21, 2880–2896 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coux R.-X., Teixeira F. K., Lehmann R., L(3)mbt and the LINT complex safeguard cellular identity in the Drosophila ovary. Development 145, dev160721 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sumiyoshi T., Sato K., Yamamoto H., Iwasaki Y. W., Siomi H., Siomi M. C., Loss of l(3)mbt leads to acquisition of the ping-pong cycle in Drosophila ovarian somatic cells. Genes Dev. 30, 1617–1622 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lewis P. W., Beall E. L., Fleischer T. C., Georlette D., Link A. J., Botchan M. R., Identification of a Drosophila Myb-E2F2/RBF transcriptional repressor complex. Genes Dev. 18, 2929–2940 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meier K., Mathieu E.-L., Finkernagel F., Reuter L. M., Scharfe M., Doehlemann G., Jarek M., Brehm A., LINT, a novel dL(3)mbt-containing complex, represses malignant brain tumour signature genes. PLOS Genet. 8, e1002676 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Richter C., Oktaba K., Steinmann J., Müller J., Knoblich J. A., The tumour suppressor L(3)mbt inhibits neuroepithelial proliferation and acts on insulator elements. Nat. Cell Biol. 13, 1029–1039 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rossi F., Molnar C., Hashiyama K., Heinen J. P., Pampalona J., Llamazares S., Reina J., Hashiyama T., Rai M., Pollarolo G., Fernández-Hernández I., Gonzalez C., An in vivo genetic screen in Drosophila identifies the orthologue of human cancer/testis gene SPO11 among a network of targets to inhibit lethal(3)malignant brain tumour growth. Open Biol. 7, 170156 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chau J., Kulnane L. S., Salz H. K., Sex-lethal facilitates the transition from germline stem cell to committed daughter cell in the Drosophila ovary. Genetics 182, 121–132 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang S. Y., Baxter E. M., Van Doren M., Phf7 controls male sex determination in the Drosophila germline. Dev. Cell 22, 1041–1051 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shapiro-Kulnane L., Smolko A. E., Salz H. K., Maintenance of Drosophila germline stem cell sexual identity in oogenesis and tumorigenesis. Development 142, 1073–1082 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang S. Y., Chang Y. C., Wan Y. H., Whitworth C., Baxter E. M., Primus S., Pi H., van Doren M., Control of a novel spermatocyte-promoting factor by the male germline sex determination factor PHF7 of Drosophila melanogaster. Genetics 206, 1939–1949 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smolko A. E., Shapiro-Kulnane L., Salz H. K., The H3K9 methyltransferase SETDB1 maintains female identity in Drosophila germ cells. Nat. Commun. 9, 4155 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.S. Primus, C. Pozmanter, K. Baxter, M. Van Doren, tudor-domain containing protein 5-prime promotes male sexual identity in the Drosophila germline and is repressed in females by Sex lethal bioRxiv 388850 [Preprint]. 9 August 2018. 10.1101/388850, (2018). [DOI] [PMC free article] [PubMed]
- 38.Sundareshan T. S., Prabhash K., Bapsy P. P., Deletion of 6q23 as sole abnormality in acute myelocytic leukemia. Cancer Genet. Cytogenet. 143, 87–88 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Northcott P. A., Nakahara Y., Wu X., Feuk L., Ellison D. W., Croul S., Mack S., Kongkham P. N., Peacock J., Dubuc A., Ra Y.-S., Zilberberg K., Mcleod J., Scherer S. W., Sunil Rao J., Eberhart C. G., Grajkowska W., Gillespie Y., Lach B., Grundy R., Pollack I. F., Hamilton R. L., van Meter T., Carlotti C. G., Boop F., Bigner D., Gilbertson R. J., Rutka J. T., Taylor M. D., Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat. Genet. 41, 465–472 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Northcott P. A., Korshunov A., Witt H., Hielscher T., Eberhart C. G., Mack S., Bouffet E., Clifford S. C., Hawkins C. E., French P., Rutka J. T., Pfister S., Taylor M. D., Medulloblastoma comprises four distinct molecular variants. J. Clin. Oncol. 29, 1408–1414 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Taylor M. D., Northcott P. A., Korshunov A., Remke M., Cho Y.-J., Clifford S. C., Eberhart C. G., Parsons D. W., Rutkowski S., Gajjar A., Ellison D. W., Lichter P., Gilbertson R. J., Pomeroy S. L., Kool M., Pfister S. M., Molecular subgroups of medulloblastoma: The current consensus. Acta Neuropathol. 123, 465–472 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang X. R., Ling L. B., Huang H. H., Lin J. J., Fugmann S. D., Yang S. Y., Evidence for parallel evolution of a gene involved in the regulation of spermatogenesis. Proc. Biol. Sci. 284, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xiao J., Xu M., Li J., Chang Chan H., Lin M., Zhu H., Zhang W., Zhou Z., Zhao B., Sha J., NYD-SP6, a novel gene potentially involved in regulating testicular development/spermatogenesis. Biochem. Biophys. Res. Commun. 291, 101–110 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Wismar J., Löffler T., Habtemichael N., Vef O., Geißen M., Zirwes R., Altmeyer W., Sass H., Gateff E., The Drosophila melanogaster tumor suppressor gene lethal(3)malignant brain tumor encodes a proline-rich protein with a novel zinc finger. Mech. Dev. 53, 141–154 (1995). [DOI] [PubMed] [Google Scholar]
- 45.Gateff E., Malignant neoplasms of genetic origin in Drosophila melanogaster. Science 200, 1448–1459 (1978). [DOI] [PubMed] [Google Scholar]
- 46.Rebollo E., Llamazares S., Reina J., Gonzalez C., Contribution of noncentrosomal microtubules to spindle assembly in Drosophila spermatocytes. PLOS Biol. 2, E8 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.C. Gonzalez, D. M. Glover, Techniques for studying mitosis in Drosophila, in The Cell Cycle, P. Fantes, R. Brooks, Eds. (Oxford Univ. Press, 1993), pp. 143–174. [Google Scholar]
- 48.Rossi F., Gonzalez C., Studying tumor growth in Drosophila using the tissue allograft method. Nat. Protoc. 10, 1525–1534 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Wiśniewski J. R., Zougman A., Nagaraj N., Mann M., Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 (2009). [DOI] [PubMed] [Google Scholar]
- 50.Taverner T., Karpievitch Y. V., Polpitiya A. D., Brown J. N., Dabney A. R., Anderson G. A., Smith R. D., DanteR: An extensible R-based tool for quantitative analysis of -omics data. Bioinformatics 28, 2404–2406 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chick J. M., Munger S. C., Simecek P., Huttlin E. L., Choi K., Gatti D. M., Raghupathy N., Svenson K. L., Churchill G. A., Gygi S. P., Defining the consequences of genetic variation on a proteome-wide scale. Nature 534, 500–505 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hulsen T., de Vlieg J., Alkema W., BioVenn – a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics 9, 488 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Djebali S., Davis C. A., Merkel A., Dobin A., Lassmann T., Mortazavi A., Tanzer A., Lagarde J., Lin W., Schlesinger F., Xue C., Marinov G. K., Khatun J., Williams B. A., Zaleski C., Rozowsky J., Röder M., Kokocinski F., Abdelhamid R. F., Alioto T., Antoshechkin I., Baer M. T., Bar N. S., Batut P., Bell K., Bell I., Chakrabortty S., Chen X., Chrast J., Curado J., Derrien T., Drenkow J., Dumais E., Dumais J., Duttagupta R., Falconnet E., Fastuca M., Fejes-Toth K., Ferreira P., Foissac S., Fullwood M. J., Gao H., Gonzalez D., Gordon A., Gunawardena H., Howald C., Jha S., Johnson R., Kapranov P., King B., Kingswood C., Luo O. J., Park E., Persaud K., Preall J. B., Ribeca P., Risk B., Robyr D., Sammeth M., Schaffer L., See L.-H., Shahab A., Skancke J., Suzuki A. M., Takahashi H., Tilgner H., Trout D., Walters N., Wang H., Wrobel J., Yu Y., Ruan X., Hayashizaki Y., Harrow J., Gerstein M., Hubbard T., Reymond A., Antonarakis S. E., Hannon G., Giddings M. C., Ruan Y., Wold B., Carninci P., Guigó R., Gingeras T. R., Landscape of transcription in human cells. Nature 489, 101–108 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Livak K. J., Schmittgen T. D., Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408 (2001). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaaw7965/DC1
Fig. S1. brat tumors do not present sex-dependent dimorphism.
Fig. S2. mbt tumor size is sex dependent.
Fig. S3. Loss of Phf7 reduces the expression of mbt M-tSDS genes and increases the expression of mbt F-tSDS genes in male tissue.
Table S1. Proteomic sex-linked dimorphic signatures.
Table S2. GO terms enriched in the M-pSDS and the F-pSDS.
Table S3. Relative expression levels of L(3)mbt and other components of the LINT and Myb/Muv/Dream complexes between male and female wild-type brain lobes.
Table S4. Sex-dependent dysregulation of MBTS genes in l(3)mbtts1 tumors versus wild-type and in Phf7ΔN2; l(3)mbtts1 versus l(3)mbtts1.