

A different route of administering GM-CSF may selectively manipulate tissue-resident macrophages to therapeutic benefit.
Abstract
Pneumonia poses profound health threats to preterm infants. Alveolar macrophages (AMs) eliminate inhaled pathogens while maintaining surfactant homeostasis. As AM development only occurs perinatally, therapies that accelerate AM maturation in preterms may improve outcomes. We tested therapeutic rescue of AM development in mice lacking the actin-bundling protein L-plastin (LPL), which exhibit impaired AM development and increased susceptibility to pneumococcal lung infection. Airway administration of recombinant granulocyte-macrophage colony-stimulating factor (GM-CSF) to LPL−/− neonates augmented AM production. Airway administration distinguishes the delivery route from prior human infant trials. Adult LPL−/− animals that received neonatal GM-CSF were protected from experimental pneumococcal challenge. No detrimental effects on surfactant metabolism or alveolarization were observed. Airway recombinant GM-CSF administration thus shows therapeutic promise to accelerate neonatal pulmonary immunity, protecting against bacterial pneumonia.
INTRODUCTION
Pneumonia causes considerable morbidity and mortality in premature infants and neonates (1, 2), yet remarkably little is known about host susceptibility to, and the pathophysiology of, lower respiratory tract infections in this age group. In particular, the development of innate immunity in preterm infant lungs is poorly understood. Given that alveolar macrophages (AMs) have unique roles as first responders to pathogenic infection and in maintaining the anti-inflammatory, homeostatic environment of the lungs conducive to appropriate lung development, understanding how the appearance of AMs is affected by preterm birth is essential to understanding how preterm infants may respond to pulmonary pathogen challenge (2–7).
A major paradigm shift occurred when Schulz et al. (8) demonstrated that tissue-resident macrophages arise during embryogenesis from yolk sac precursors and are not continuously rederived from circulating peripheral blood monocytes. The recognition that each tissue-resident macrophage lineage derives along a unique pathway, following distinct development and tissue-specific cues and finishing with specialized phenotypic markers and functions, revolutionized basic immunologists’ understanding and appreciation for the complex nature of the tissue-resident phagocytic system (9–19). AMs epitomize the highly specialized nature of tissue-resident macrophages, as they express CD11c (not CD11b) and depend on the growth factor granulocyte-macrophage colony-stimulating factor (GM-CSF) and the transcription factor peroxisome proliferator–activated receptor γ. They also arise during a temporally limited phase of development, which in mice begins on embryonic day 16 and closes on postnatal day 7 (PND7). They arise from monocyte precursor cells that seed the lungs of mice during embryonic days 15 and 16, coincident with the saccular phase of lung development (20). These monocytes develop to the intermediary pre-AM in the days before birth and then advance to fully mature AMs in mice by PND7 (9). Pre-AMs and AMs are phenotypically distinguished by surface markers; pre-AMs express CD11c, while AMs express both CD11c and SiglecF. The pre-AM, as a developmental intermediary, can only be found in the lungs from late embryonic or early neonatal mice (not adult). AM development during the perinatal period is driven by a burst of GM-CSF production, which subsides after birth (14). After the first week of life, no new precursor cells are generated, and in the absence of overwhelming inflammation, AMs self-renew over the lifetime of the host (9, 21).
In addition to serving as sentinel cells, engulfing pathogens, and preventing subsequent lung infection (13, 22), AMs are functionally distinct from interstitial lung macrophages in that AMs are programmed to tilt toward an anti-inflammatory, prohealing phenotype. AMs thus protect and preserve the delicate alveolar structure from inflammatory insults. In human infants, AMs can arise post-natally in the first 2 days (3, 4, 6, 23, 24); therefore, we posit that the exogenous augmentation of AM development may represent a new avenue to protect preterm infants from pulmonary disease. In our mouse model featuring the partial disruption of AM development, mice were rendered susceptible to pneumococcal challenge (22, 25). Mice lacking the actin-bundling protein L-plastin (LPL−/− mice) and CD11c.Cre+-LPLfl/fl mice exhibited cell intrinsic defects in AM production secondary to the diminished migration of AM precursors to, and retention within, the alveoli. LPL−/− and CD11c.Cre+-LPLfl/fl mice also exhibited impaired pneumococcal clearance from the lungs following intratracheal challenge, associated with increased dissemination to the bloodstream and decreased survival (22, 25). Because GM-CSF signaling is independent of LPL (22), we considered these mice to represent an ideal model system to test the therapeutic potential of GM-CSF delivery to neonates. We now show that the intranasal administration of GM-CSF to neonatal mice in the first week of life enhances AM development. We note that the systemic administration of GM-CSF did not enhance AM maturation as did direct airway administration, and therefore, these effects would not have been apparent in prior clinical trials of GM-CSF in preterm infants (26). Most notably, the augmented population of AMs persists to adulthood, and animals treated in the neonatal period are protected as adults from mortality during pneumococcal lung infection.
RESULTS
Neonatal administration of rGM-CSF enhances AM population in LPL−/− mice
We have previously shown that LPL−/− mice exhibit defective AM development due to the impaired migration of pre-AM precursors into the alveolar space (22). Impaired migration results in reduced numbers of mature AMs and subsequent susceptibility to pneumococcal lung infection. Notably, AM precursors in LPL−/− mice retain responsiveness to the required growth factor GM-CSF (22). We undertook the present study to determine whether perinatal AM development could be augmented by the exogenous intranasal administration of GM-CSF and provide durable protection against pneumococcal lung infection.
To test whether the airway administration of GM-CSF would augment AM maturation during the normal physiological window of development, we intranasally administered recombinant GM-CSF (rGM-CSF) to wild-type (WT) and LPL−/− mouse pups or phosphate-buffered saline (PBS) to control mice on the day of birth (DOB), PND1, and PND2 (Fig. 1A). Mice were euthanized on PND3, and populations of fetal monocytes, pre-AMs, and AMs in whole-lung homogenates were determined by flow cytometry (Fig. 1B) (9). As previously observed (22), untreated LPL−/− pups showed no deficiencies in fetal monocyte or pre-AM populations compared to WT pups but did exhibit a reduced percentage of mature AMs. The administration of rGM-CSF to LPL−/− pups increased the proportion of CD11c+ maturing cells (combined pre-AMs and AMs; Fig. 1C), while these proportions were unchanged by rGM-CSF administration to WT pups. Administering the same amount of rGM-CSF (20 ng) to LPL−/− pups via subcutaneous injection (systemic administration) on DOB, PND1, and PND2 did not enhance AM maturation, as assessed on PND6 (fig. S1).
Fig. 1. Administration of rGM-CSF increases AMs in LPL−/− mice.
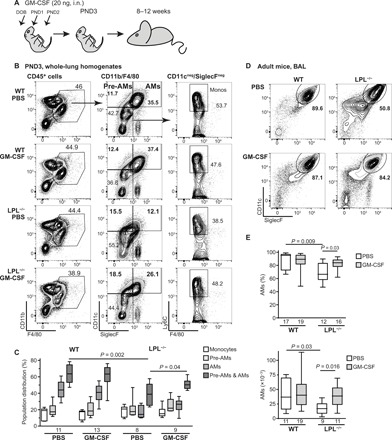
(A) rGM-CSF (20 ng) in 6 μl of PBS was administered via intranasal (i.n.) instillation to neonatal pups on DOB, PND1, and PND2. Mice were evaluated at indicated times after rGM-CSF administration. (B) Representative flow cytometry of whole-lung homogenates from PND3 WT and LPL−/− pups treated intranasally with PBS or rGM-CSF. (C) Quantification of the distribution of monocytes, pre-AMs, AMs, and total CD11c+ (maturing) cells in PND3 WT and LPL−/− pups. (D) Representative flow cytometry from BAL fluid obtained from adult WT and LPL−/− mice that had received neonatal rGM-CSF therapy. (E) Percentage and number of AMs recovered from BAL fluid from adult WT and LPL−/− mice that had received neonatal rGM-CSF treatment. (C and E) n of each group is provided below x axes. Data were obtained from four independent cohorts of animals. P values are determined using the Mann-Whitney U test. Kruskal-Wallis test comparing four groups revealed P = 0.0014 for AM % (top) and P = 0.015 for AM numbers (bottom). The “n” of AM numbers (bottom) in some groups is lower than AM % (top) because cell numbers in one experiment were counted manually rather than by cytometer acquisition. Only cell counts obtained by the same method (cytometer acquisition) are included in data shown here.
To determine whether neonatal rGM-CSF administration elicited durable change in AM numbers, we analyzed the percentage and numbers of AMs recovered from adult (defined as ≥8 week old) animals that had received intranasally either PBS or rGM-CSF as neonatal pups. As previously observed (22), untreated adult LPL−/− mice exhibited reduced AMs compared with WT mice (Fig. 1, D and E). However, LPL−/− mice that had received rGM-CSF as pups harbored significantly higher numbers of AMs in bronchoalveolar lavage (BAL) fluid (Fig. 1E), while similarly treated WT mice demonstrated no alteration in AMs. Thus, exogenous rGM-CSF enhanced AM production under conditions where maturation was impaired but did not alter normal AM development.
GM-CSF has been considered pro-inflammatory in the lung (27), and excess inflammation has been associated with the disruption of alveolarization in neonatal mice (5). To test whether the exogenous administration of rGM-CSF to neonatal pups interfered with alveolarization in the developing lungs, histological sections of WT and LPL−/− pups (PND3) and adults that received either PBS or rGM-CSF were examined by a veterinary pathologist. No disruption of alveolarization was observed (fig. S2).
Neonatal rGM-CSF administration protected adult LPL−/− mice from infection
To determine whether enhanced neonatal AM development protected mice from subsequent infection, adult LPL−/− mice that received neonatal rGM-CSF were challenged intratracheally with pneumococcus (Fig. 2A). We selected the same inoculum used in our prior studies, in which about 10 to 20% of WT animals would be expected to succumb, while up to 70 to 80% of LPL−/− mice might succumb. This sizable differential in susceptibility would provide the optimal conditions for observing an effect of neonatal rGM-CSF therapy, if any (22, 25). As anticipated, untreated LPL−/− mice were more susceptible to pneumococcal pneumonia than were WT mice (Fig. 2B). However, adult LPL−/− mice that had received neonatal rGM-CSF therapy were significantly protected from mortality, while the already low mortality in infected WT mice was unaltered by neonatal rGM-CSF therapy (Fig. 2B). Quantitative blood cultures obtained 24 hours after pneumococcal inoculation revealed that untreated LPL−/− mice suffered from augmented dissemination (Fig. 2C), as reported previously (25) and aligned with their increased mortality. The neonatal rGM-CSF treatment of LPL−/− mice reduced pneumococcal bloodstream dissemination in infected adults to match that in WT animals, showing that the neonatal administration of inhaled rGM-CSF provided sustained rescue of pulmonary host defense. An analysis of peripheral blood 24 hours after infection revealed no significant difference in the percentages of circulating neutrophils or monocytes in infected WT or LPL−/− mice treated with neonatal PBS or rGM-CSF. LPL−/− mice exhibit reduced B cell maturation (28), which was expectedly unchanged by neonatal rGM-CSF intranasal administration (Fig. 2D). Thus, we did not find evidence that intranasal neonatal rGM-CSF treatment altered systemic host immune responses (Fig. 2D) while protecting against pulmonary infection (Fig. 2, B and C).
Fig. 2. Adult LPL−/− mice that received neonatal rGM-CSF therapy are protected from pneumococcal infection.
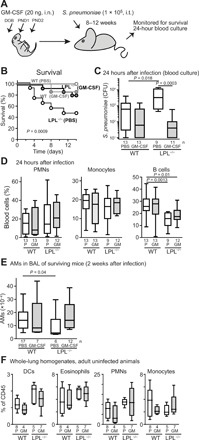
(A) Experimental design: Neonatal WT and LPL−/− pups were intranasally given 20 ng of rGM-CSF or PBS (control) on DOB, PND1, and PND2. After reaching adulthood, mice were challenged intratracheally (i.t.) with pneumococcus. (B) Adult WT (gray) or LPL−/− (black) mice that had received neonatal rGM-CSF therapy (open symbols) or PBS (control; closed symbols) were infected intratracheally with pneumococcus and monitored for survival. Survival curves show data combined from three independent experiments (WT + PBS, n = 19; WT + rGM-CSF, n = 7; LPL−/− + PBS, n = 11; LPL−/− + rGM-CSF, n = 16) and were compared using Mantel-Cox log-rank test. (C) Quantitative blood culture obtained from adult mice 24 hours after intratracheal instillation of pneumococci. Comparison of all groups by one-way analysis of variance (ANOVA) (Kruskal-Wallis test), P = 0.0007. (D) Distribution of neutrophils [polymorphonuclear leukocytes (PMNs)], monocytes, and B cells in the peripheral blood of WT and LPL−/− mice challenged with pneumococcus. Mice had received neonatal rGM-CSF therapy (GM) or PBS (P; control). (E) Number of AMs recovered from BAL fluid of WT or LPL−/− adult mice that survived 2 weeks after intratracheal pneumococcal infection. Grossly bloody BAL fluids were excluded from analysis (clotted). (F) Distribution of DCs, eosinophils, neutrophils (PMNs), and monocytes in whole-lung homogenates from uninfected adult WT and LPL−/− mice that had received neonatal rGM-CSF therapy or PBS (control). Percentages of total CD45+ cells are given. (C to F) Data are combined from three (C to E) or two (F) independent experiments. n of each group is given below x axes. P values of comparisons between two groups are determined using the Mann-Whitney U test.
We also quantified AMs in the BAL of adult animals that survived 2 weeks after intratracheal pneumococcal inoculation. AM numbers in control and rGM-CSF–treated WT animals were equivalent. Untreated LPL−/− mice again exhibited reduced AM numbers compared to WT animals, while rGM-CSF–treated LPL−/− mice harbored AM numbers equivalent to those of WT animals (Fig. 2E). We did not observe any changes in the proportions of other pulmonary innate immune cell types [dendritic cells (DCs), eosinophils, neutrophils, or monocytes] preceding infection (Fig. 2F). Protection from pneumococcal infection thus correlated specifically with increased AM populations.
Neonatal rGM-CSF administration enhances AM and AM precursor proliferation
We next sought to define a mechanism by which exogenous rGM-CSF therapy enhanced AM neonatal development. We have previously shown that the migration of AM precursors into the alveoli is impaired in the absence of LPL. While it is not obvious how rGM-CSF could overcome this migration defect, we hypothesized that exogenous rGM-CSF could increase the proliferation of precursor cells that do successfully reach the alveoli in LPL−/− pups—in other words, acting on cells “already there.” Furthermore, the contribution of proliferation to the increase in lung-resident monocyte, pre-AM, and AM populations during neonatal development has not been previously assessed. We therefore quantified 5-bromo-2′-deoxyuridine (BrdU) incorporation into monocytes, pre-AMs, and AMs in PND3 pups treated intranasally with PBS or rGM-CSF (Fig. 3A). AMs and precursors were identified using surface expression of CD11b, F4/80, CD11c, SiglecF, and Ly6C as previously defined [Fig. 1B and (9)]. In untreated WT pups, we noted proliferation in all three subpopulations, with substantial BrdU incorporation into fetal monocytes and AMs; meanwhile, BrdU incorporation into all three cell types was diminished in LPL−/− pups (Fig. 3, A and B). Notably, neonatal rGM-CSF treatment significantly increased pre-AM proliferation by threefold in LPL−/− pups (Fig. 3B); thus, one mechanism by which rGM-CSF restores mature AM numbers in LPL−/− mice is by enhancing the proliferation of precursors that do succeed in reaching the alveolar space. Intranasal rGM-CSF therapy had no effect on the proliferation of pre-AMs or AMs in WT mice, again indicating that rGM-CSF therapy exerts little, if any, effect on AM development that is proceeding normally (Fig. 3B).
Fig. 3. rGM-CSF administration increased pre-AM proliferation in LPL−/− pups.
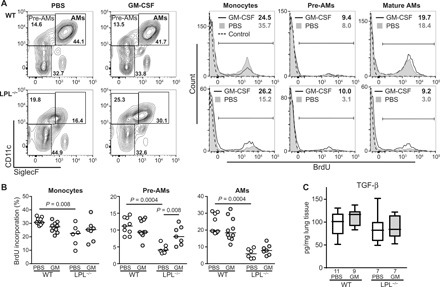
(A) Representative flow cytometry of BrdU incorporation into AMs and precursors (monocytes and pre-AMs, defined as shown) in whole-lung homogenates from PND3 WT and LPL−/− pups receiving rGM-CSF therapy or PBS (control). Percentage of cells in each gate incorporating BrdU listed in the top right-hand corner. (B) Quantification of the percentage of monocytes, pre-AMs, or AMs incorporating BrdU in PND3 WT or LPL−/− pups receiving neonatal rGM-CSF therapy (gray symbols) or PBS (control; open symbols). Each symbol represents one animal. Data are combined from three independent experiments. Line shows median value. P values are determined using the Mann-Whitney U test. (C) TGF-β concentration in whole-lung homogenates from PND3 WT and LPL−/− pups receiving neonatal rGM-CSF therapy (gray bars) or PBS (open bars). Line shows the median value. n of each group is listed below the x axis. Data are from two independent cohorts of animals.
Recently, the autocrine production of transforming growth factor–β (TGF-β) has been shown to be essential for AM development and maintenance (29). We had not previously evaluated whether TGF-β production by AMs was altered by LPL deficiency. Furthermore, if TGF-β were dependent on LPL for generation, then a second mechanism of AM rescue by rGM-CSF could be by augmenting TGF-β production. Refuting this possibility, we found no difference in the whole-lung TGF-β concentration in PND3 WT or LPL−/− pups that had received PBS or rGM-CSF (Fig. 3C).
Neonatal rGM-CSF improves surfactant homeostasis in LPL−/− mice
In addition to their critical role in host defense, AMs are essential to surfactant metabolism. Surfactant is produced by alveolar epithelial type II cells but taken up and catabolized in part by AMs, a process stimulated by GM-CSF. Animals deficient in GM-CSF or the GM-CSF receptor suffer from primary alveolar proteinosis, a progressive and ultimately fatal accumulation of surfactant protein in the alveoli (30–32). Although LPL−/− animals have not exhibited overt evidence of progressive lung disease (22, 25), we have not previously formally assessed surfactant protein in these mice. In addition, we wished to determine whether neonatal rGM-CSF treatment would alter surfactant catabolism in WT or LPL−/− mice. We therefore quantified surfactant protein D (SP-D) and SP-A in WT and LPL−/− mice that had received intranasally either PBS or rGM-CSF as pups (Fig. 4). In PND3 and adult LPL−/− mice, the amount of SP-D per milligram of lung tissue was increased in LPL−/− whole-lung homogenates compared to untreated WT mice (Fig. 4A). Neonatal rGM-CSF therapy in LPL−/− mice partially ameliorated the increase in SP-D by PND3 and normalized SP-D concentrations by adulthood while not altering SP-D levels in WT mice (Fig. 4A). SP-A concentrations were not altered in the lungs of adult untreated LPL−/− mice and were not affected by neonatal rGM-CSF therapy (Fig. 4B). An analysis of SP-D and SP-A protein amounts compared to total lung protein revealed similar trends (fig. S3). Therefore, neonatal rGM-CSF therapy did not alter surfactant metabolism in WT mice but ameliorated defects in LPL−/− mice associated with reduced AM production.
Fig. 4. Neonatal rGM-CSF corrects increased SP-D in LPL−/− mice.
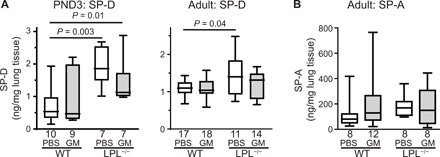
(A) SP-D or (B) SP-A concentrations in whole-lung homogenates obtained from PND3 pups or adult WT or LPL−/− mice that had received neonatal rGM-CSF therapy (gray bars) or PBS (control; open bars). Line shows the median value. P values are determined using the Mann-Whitney U test. n of each group is listed below x axes. Data are combined from two independent cohorts of animals.
DISCUSSION
Here, we used a preclinical model of impaired AM development to show that GM-CSF, a readily available, U.S. Food and Drug Administration–approved therapy, can be administered safely to the airways of newborn mice to accelerate AM maturation (Fig. 1) and protect otherwise susceptible animals from bacterial pneumonia (Fig. 2). One mechanism by which rGM-CSF rescues AM development in LPL−/− mice is by significantly increasing pre-AM proliferation (Fig. 3B) without affecting levels of TGF-β or disrupting alveolarization or normal surfactant homeostasis (Fig. 4 and fig. S2). We propose that the translation of neonatal GM-CSF therapy to human infants could have a direct impact on improving outcomes of respiratory disease in preterm neonates, a benefit that would not have been apparent in prior studies that either subcutaneously or intravenously administered systemic GM-CSF (26, 33). Our work shows that the route and timing of GM-CSF administration—directly to the airways, in the first 24 hours of life, to mimic the physiological burst of GM-CSF production that naturally occurs (9)—could exert effects very different from those previously observed when GM-CSF was administered systemically and, later, postnatally. In addition, the present studies expand on our prior work revealing that LPL is an essential regulator of AM development (22, 25); we now show that beyond supporting the migration and adhesion of AMs and AM precursors into the alveoli (22), LPL also facilitates the proliferation of AM precursors and developing AMs (Fig. 3, A and B).
The observation that tissue-resident, long-lived macrophages arise during prenatal development and then self-renew (8) overturned the long-standing presumption that tissue-resident macrophages are continually repopulated from circulating blood monocytes. This finding prompted new work demonstrating the different ontogeny and function of varied macrophage lineages (9, 11, 14–18, 21). AMs represent a unique phagocytic lineage with a limited temporal window for development (9), occurring in the perinatal period and driven by a “burst” of GM-CSF occurring shortly after birth. The maturation pathway from fetal liver monocyte to mature AM has been well documented in mice (9, 14), and several key regulators have been identified (29). Translation to humans, which requires mapping of ontology and functional phenotypes of the multiple interstitial and AMs in mice to corresponding lineages in humans, will inform the role of each lineage in both lung infection and chronic lung disease (CLD) in preterm infants. One major obstacle to clinical translation has recently been overcome, as markers for clear differentiation of human AMs from interstitial lung macrophages have recently been defined (24, 33). The recent characterization of distinct lineages (34, 35) now enables the re-evaluation of prior links between CLD outcome and lung macrophages; for instance, the macrophages studied in a landmark paper examining alveolarization inhibition by macrophage-specific pro-inflammatory signaling (5) were, in retrospect, likely fetal interstitial lung macrophages rather than AMs. The pathogenesis of CLD (including inhibition of airway maturation) might be greatly clarified by additional studies leveraging new knowledge of macrophage lineages and AM development.
Existing clinical data, examined in light of the new murine paradigm, support the consideration of AM maturation as a therapeutic target. First, a similar monocyte-to-AM maturation may occur in human infants (3, 6). While preterm lungs have been shown to bear monocytes (6), very few, if any, mature AMs are present in the lungs of stillborn infants (3, 24), strongly suggesting that AMs are not present in the lungs of infants in utero. In a cohort of live-born infants who died in the early neonatal period, AMs were present in 33 of 46 (72%) infants who died before 48 hours of age but were apparent in the lungs of 50 of 54 (93%) infants who survived longer than 48 hours (3). Thus, the emergence of AMs in the lungs of neonates correlated with time since birth, consistent with the murine model in which AM precursors differentiate to mature AM shortly after birth.
Moreover, the developmentally regulated increase of GM-CSF noted in newborn mice has also been observed in humans (23). An analysis of GM-CSF concentrations in amniotic fluid revealed gradually increasing GM-CSF concentrations, beginning at 28 weeks of gestation (23). After birth, tracheal aspirates demonstrated a sharp and significant increase in GM-CSF concentrations in samples between 12 and 48 hours of age, after which concentrations did not change appreciably (23). This study provides evidence that GM-CSF may also increase at the time of birth in human infants, concurrently with the appearance of mature AMs.
Because AMs are phenotypically and functionally distinct from the interstitial macrophage lineages, delayed or disturbed AM maturation in preterm and term neonates could be pathophysiologically linked to either impaired immunity or increased pro-inflammatory signaling associated with bronchopulmonary dysplasia or CLD. For instance, the analysis of BAL fluid from preterm infants revealed a significant increase in nonclassical, pro-inflammatory CD14+/CD16+ monocyte-macrophages and a decrease in the anticipated mature, anti-inflammatory CD14+/CD36+ macrophages compared to term infants. Furthermore, preterm infants that developed CLD had significantly fewer mature anti-inflammatory CD14+/CD36+ macrophages (AMs) recovered from BAL fluid than did preterm infants who did not develop CLD. Results from this study thus correlate with mouse ontogeny models, in that a higher proportion of immature (monocytic) cells is observed in preterm infants, and suggest that this perturbation is associated with CLD (6).
Our data support the concept that the augmentation of AM number and function, if perturbed in preterm infants, could confer translational benefit to the preterm lung, both by protecting against pulmonary infection (a common complication of prematurity) and by promoting surfactant function and metabolism [as reduced AM number impairs surfactant catabolism (36–38)]. In our mouse model of deficient AM maturation, intranasal rGM-CSF restored AM numbers and provided durable protection against pneumococcal challenge. Similarly, in preterm rabbits, reduced AM number correlated with increased susceptibility to group B streptococcal (GBS) lung infection (4). Specifically, preterm rabbits harbored barely detectable AMs and permitted the proliferation of GBS in the lungs, while term rabbits had 17-fold higher numbers of AMs than preterms and easily cleared a similar pulmonary GBS challenge (4). We also show an increase in SP-D concentrations in LPL−/− neonatal pups (Fig. 4A), consistent with reduced surfactant catabolism that would be anticipated with diminished numbers of AMs [Fig. 1B and (22)]. Exogenous neonatal rGM-CSF therapy reduced SP-D concentrations in PND3 LPL−/− pup lungs concordant with the acceleration in AM maturation, suggesting that the airway administration of GM-CSF to augment AM maturation may promote surfactant homeostasis in preterm neonates.
Our data show that administering three doses of intranasal GM-CSF to neonatal mice is feasible and was effective in promoting AM maturation and pneumococcal clearance. The airway administration of GM-CSF has not been previously studied in clinical trials of human infants (26). One major clinical trial of GM-CSF administered subcutaneous (systemic) GM-CSF to preterm infants within 72 hours of birth, with a primary end point of sepsis-free survival at 14 days of life and with a goal of increasing peripheral neutrophil counts. No survival benefit of systemic GM-CSF was observed in this study nor were any differences in oxygen requirements observed at 28 days from enrollment (26). Follow-up evaluations of 2- and 5-year outcomes revealed no differences in neurodevelopmental, growth, or infectious outcomes with systemic GM-CSF administration (39, 40). We found that the subcutaneous administration of GM-CSF did not increase AM maturation as did direct airway administration (fig. S1). We propose that direct airway administration results in higher alveolar concentrations of rGM-CSF than does systemic injection, as systemic injection would distribute rGM-CSF throughout all tissues, while direct airway administration would provide a high alveolar concentration before systemic absorption. Further pharmacokinetic studies could define optimal dosing and timing of neonatal rGM-CSF to modify AM development. A meta-analysis revealed a higher response rate and greater improvements in PaO2 in subjects with autoimmune pulmonary alveolar proteinosis treated with inhaled GM-CSF than with subcutaneous GM-CSF (41). Inhaled GM-CSF has recently been used to successfully treat Mycobacterium abscessus infections in two patients with cystic fibrosis (42). We further note that we did not find any evidence of adverse effects of neonatal rGM-CSF airway administration to WT animals, in which AM maturation would be expected to be proceeding normally, as assessed by alveolarization, surfactant protein concentrations, and infectious susceptibility. We therefore suggest that revisiting GM-CSF therapy in neonates via this alternate route of administration would be warranted.
Furthermore, administering rGM-CSF to neonates with perturbed AM development produced a longer-lasting effect (8 to 12 weeks) than prior techniques using GM-CSF to protect against lung infections (43). For instance, adenoviral-mediated expression continuously delivered GM-CSF to the airway and protected mice from pneumococcal pneumonia 14 days later, but the effect of GM-CSF on leukocyte recruitment waned after 28 days of administration (43). The airway administration of GM-CSF also improved clearance of pneumococcus when given to adult mice from 12 hours before 6 hours after infection, but clinical translation of this application was limited by the narrow window of time for effective GM-CSF administration in relation to pneumococcal infection (43). The airway overexpression of GM-CSF driven by a surfactant promoter was also protective in a model of influenza infection, as was the administration of GM-CSF 1 week before infection (44). However, the sustained expression of GM-CSF by either adenoviral or transgenic expression is not clinically translatable, and the effect of GM-CSF administered to adults was not durable. Our approach of administering GM-CSF during the neonatal window of AM development is therefore unique and clinically adaptable. We note that we did not challenge WT mice with a pneumococcal inoculum anticipated to induce notable mortality, as our intention was to test whether neonatal rGM-CSF therapy of LPL−/− mice rescued susceptibility (Fig. 2A). While we can conclude that neonatal rGM-CSF therapy did not increase susceptibility of WT animals to lung pneumococcal infection, further studies at higher inocula are required to test whether neonatal rGM-CSF therapy also boosts antipneumococcal immunity in WT mice. Last, also supporting the translatability of this approach, GM-CSF has been administered to the airways of a small cohort of adults with refractory acute respiratory distress syndrome, providing benefit in oxygenation and lung compliance compared with control patients (45). The successful use of rGM-CSF to provide sustained protection against bacterial pneumonia in susceptible neonatal mice, without perturbing airway immunity and lung development in the normal host, illuminates how the airway administration of GM-CSF might be used in preterm infants to improve clinical outcomes in this highly vulnerable patient population.
MATERIALS AND METHODS
Study design
Research objective
The primary objective of this research was to test the hypothesis that administering rGM-CSF to neonatal LPL−/− pups (in the temporal window of AM development) would protect LPL−/− mice during subsequent bacterial lung infection. This objective was defined before initiation of the experiments and data analysis.
Experimental design
The experimental design was a controlled laboratory experiment using genetically modified mice.
Randomization
Pups within each litter of mice were randomly assigned to receive intranasal rGM-CSF or PBS (control). In one set of experiments, pups within litters were randomized to receive subcutaneous rGM-CSF or PBS (control).
Blinding
Histology sections were read by a veterinary pathologist who was not associated with the study outcomes and was blinded to mouse genotype and treatment group (slides were provided with coded labels). Because of the technical challenges associated with working with timed pregnancies and neonatal pups, and the necessity of ensuring that pups receiving PBS could be consistently distinguished from the pups receiving rGM-CSF (pups were marked with permanent ink, but this would wear off if not reapplied), most of the studies could not be blinded. However, the outcomes are all objectively quantifiable [flow cytometry, colony forming units (CFU), etc.].
Mice
LPL−/− mice fully back-crossed to the C57BL/6 background have been described (22, 25). WT and LPL−/− mice were bred and cohoused in the same specific pathogen–free barrier animal facility. Human rGM-CSF (20 ng in 6 μl of PBS per inoculation) was administered intranasally to pups in all experiments except for the data presented in fig. S1. In fig. S1, some neonatal pups received 20 ng of rGM-CSF in 10 μl of PBS via subcutaneous injection. Litters of WT and LPL−/− pups were divided such that approximately half received PBS and half received rGM-CSF and such that littermate controls were used to discern the effects of rGM-CSF. Mice matched for sex and age were used in all experiments, which were conducted in accordance with a protocol approved by the Institutional Animal Care and Use Committee at Washington University School of Medicine (WUSM).
Cell isolation and media
BAL was performed as described (25), and cells were quantified by flow cytometry (22). Lungs were homogenized using collagenase D (2.5 mg/ml) in Hanks’ balanced salt solution and 3% fetal calf serum (22).
Flow cytometry
Commercial antibodies to the indicated murine antigens were used: CD11c-phycoerythrin (PE)/Cy7, CD11c-allophycocyanin (APC) (N418), CD11c-APCCy7 (N418), CD64-PE (X54-5/7.1), F4/80-PerCP/Cy5.5, F4/80-APC (BM8), I-Ak-PE (10-3.6), Ly-6C-PacBlue (HK1.4), Ly-6C-BV510 (HK1.4), CD45-PacBlue (30-F11), CD45-BV785 (30-F11), and Ly-6G-PacBlue (1A8) (all from BioLegend San Diego, CA); CD11b-fluorescein isothiocyanate (M1/70), CD11b-PeCy7 (M1/70), and CD11c-PE (N418) (all from eBioscience, San Diego, CA); CD45-BV510 (104), SiglecF-PE, SiglecF–Alexa Fluor 647, SiglecF-APC700, and (E50-2440) Ly6G-BUV395 (1A8) (all from BD Biosciences, San Jose, CA); and MerTK-PE/Cy7 (DS5MMER) (from Invitrogen, Carlsbad, CA). BrdU labeling was performed using the BrdU-APC labeling Kit (BD Biosciences) according to the manufacturer’s protocol. Cells were acquired either on the BD Biosciences LSRFortessa or with a BD FACScan flow cytometer with DxP multicolor upgrades by Cytek Development Inc. (Woodland Park, NJ) and then analyzed using FlowJo software (FlowJo LLC, Ashland, OR). Samples were preincubated with Fc-block [Hybridoma 2.4G2, American Type Culture Collection (ATCC)].
Infection
Streptococcus pneumoniae [ATCC 6303, serotype 3; 5 × 104 CFU per animal in 20 μl of Dulbecco’s PBS) was instilled intratracheally as before (22, 25). Blood was obtained for quantitative culture 24 hours after inoculation. Mice were monitored at least twice daily using a “humane end points” scoring system that included daily weight and temperature monitoring and observation of animals’ grooming, activity, behavior, and respirations. Animals that lost >20% of starting weight or that scored >5 on the clinical observation score were euthanized.
Tissues were harvested for either flow cytometry or for histology. Histological specimens were preserved in formalin (10%) and then embedded in paraffin. Sections were prepared and stained with hematoxylin and eosin by the Division of Comparative Medicine core facility at WUSM. Histological sections were reviewed by an independent veterinary pathologist.
Surfactant protein analysis
Concentrations of SP-A (Biotang Inc.) and SP-D (R&D Systems) in whole-lung homogenates were determined by enzyme-linked immunosorbent assay according to the manufacturer’s protocol.
Statistics
Nonparametric tests were used to compare non-Gaussian data. All quantitative data are fully displayed in graphs, as they are represented either with box and whisker plots (line shows the median value, box shows 25th to 75th percentiles, and whiskers show minimum and maximum values) or with symbols indicating each value. Comparisons of two groups were made with the Mann-Whitney U test, and comparisons of multiple groups used the Kruskal-Wallis test. Survival curves were compared using the log-rank Mantel-Cox test. A P value of <0.05 was considered statistically significant.
As these studies were performed to generate preliminary data and demonstrate feasibility, power calculations could not be performed in advance; the number of experiments to be performed was determined on the basis of the number of experiments required in prior reports to demonstrate differences between groups of animals (22, 25). End points of all experiments were determined in advance of performing the experiment. All results of all experiments that were technically interpretable were included. No outliers from any experiment were excluded. The number of animals and replicates for each experiment is provided in the figure or legend.
Supplementary Material
Acknowledgments
We thank D. Kreamalmeyer for technical assistance with support of the mouse colony and D. A. Hunstad for critical review of the manuscript. Funding: S.C.M. was supported by the National Institutes of Health (R01-AI104732 and R21-AI142723). This work was supported by the Hope Center Transgenic Vectors Core at WUSM. Experimental support was also provided by the Speed Congenics Facility of the Rheumatic Diseases Core Center. Research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases, part of the National Institutes of Health, under award no. P30-AR048335. Author contributions: Conceptualization: E.M.T. and S.C.M. Methodology: E.M.T., R.R., and S.C.M. Investigation: E.M.T., R.R., and T.P.S. Writing (original draft), funding acquisition, and supervision: S.C.M. Writing (review and editing): T.P.S., R.R., E.M.T., and S.C.M. Competing interests: E.T. and S.C.M. have a pending provisional patent application entitled “Inhaled GM-CSF in Neonatal Mice Provides Durable Protection Against Bacterial Pneumonia” (application no. 62/831,318, filed 9 April 9 2019), which might be converted to a patent application within a year from filing. T.P.S. and R.R. declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaax3387/DC1
Fig. S1. No increase in mature AMs following the subcutaneous administration of rGM-CSF (20 ng) to LPL−/− neonatal pups on DOB, PND1, and PND2.
Fig. S2. No disruption of alveolarization observed after intranasal neonatal rGM-CSF therapy.
Fig. S3. Increased SP-D in LPL−/− PND3 neonatal pups.
REFERENCES AND NOTES
- 1.O’Brien K. L., Wolfson L. J., Watt J. P., Henkle E., Deloria-Knoll M., McCall N., Lee E., Mulholland K., Levine O. S., Cherian T., Hib and Pneumococcal Global Burden of Disease Study Team , Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: Global estimates. Lancet 374, 893–902 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Pryhuber G. S., Postnatal infections and immunology affecting chronic lung disease of prematurity. Clin. Perinatol. 42, 697–718 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alenghat E., Esterly J. R., Alveolar macrophages in perinatal infants. Pediatrics 74, 221–223 (1984). [PubMed] [Google Scholar]
- 4.Sherman M. P., Johnson J. T., Rothlein R., Hughes B. J., Smith C. W., Anderson D. C., Role of pulmonary phagocytes in host defense against group B streptococci in preterm versus term rabbit lung. J. Infect. Dis. 166, 818–826 (1992). [DOI] [PubMed] [Google Scholar]
- 5.Blackwell T. S., Hipps A. N., Yamamoto Y., Han W., Barham W. J., Ostrowski M. C., Yull F. E., Prince L. S., NF-κB signaling in fetal lung macrophages disrupts airway morphogenesis. J. Immunol. 187, 2740–2747 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prince L. R., Maxwell N. C., Gill S. K., Dockrell D. H., Sabroe I., McGreal E. P., Kotecha S., Whyte M. K., Macrophage phenotype is associated with disease severity in preterm infants with chronic lung disease. PLOS ONE 9, e103059 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Georgountzou A., Papadopoulos N. G., Postnatal innate immune development: From birth to adulthood. Front. Immunol. 8, 957 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schulz C., Gomez Perdiguero E., Chorro L., Szabo-Rogers H., Cagnard N., Kierdorf K., Prinz M., Wu B., Jacobsen S. E., Pollard J. W., Frampton J., Liu K. J., Geissmann F., A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336, 86–90 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Guilliams M., De Kleer I., Henri S., Post S., Vanhoutte L., De Prijck S., Deswarte K., Malissen B., Hammad H., Lambrecht B. N., Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J. Exp. Med. 210, 1977–1992 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Plantinga M., Guilliams M., Vanheerswynghels M., Deswarte K., Branco-Madeira F., Toussaint W., Vanhoutte L., Neyt K., Killeen N., Malissen B., Hammad H., Lambrecht B. N., Conventional and monocyte-derived CD11b+ dendritic cells initiate and maintain T helper 2 cell-mediated immunity to house dust mite allergen. Immunity 38, 322–335 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Yona S., Kim K. W., Wolf Y., Mildner A., Varol D., Breker M., Strauss-Ayali D., Viukov S., Guilliams M., Misharin A., Hume D. A., Perlman H., Malissen B., Zelzer E., Jung S., Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gautier E. L., Ivanov S., Williams J. W., Huang S. C., Marcelin G., Fairfax K., Wang P. L., Francis J. S., Leone P., Wilson D. B., Artyomov M. N., Pearce E. J., Randolph G. J., Gata6 regulates aspartoacylase expression in resident peritoneal macrophages and controls their survival. J. Exp. Med. 211, 1525–1531 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schneider C., Nobs S. P., Heer A. K., Kurrer M., Klinke G., van Rooijen N., Vogel J., Kopf M., Alveolar macrophages are essential for protection from respiratory failure and associated morbidity following influenza virus infection. PLOS Pathog. 10, e1004053 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schneider C., Nobs S. P., Kurrer M., Rehrauer H., Thiele C., Kopf M., Induction of the nuclear receptor PPAR-γ by the cytokine GM-CSF is critical for the differentiation of fetal monocytes into alveolar macrophages. Nat. Immunol. 15, 1026–1037 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Gomez Perdiguero E., Klapproth K., Schulz C., Busch K., Azzoni E., Crozet L., Garner H., Trouillet C., de Bruijn M. F., Geissmann F., Rodewald H. R., Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518, 547–551 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kopf M., Schneider C., Nobs S. P., The development and function of lung-resident macrophages and dendritic cells. Nat. Immunol. 16, 36–44 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Mass E., Ballesteros I., Farlik M., Halbritter F., Gunther P., Crozet L., Jacome-Galarza C. E., Handler K., Klughammer J., Kobayashi Y., Gomez-Perdiguero E., Schultze J. L., Beyer M., Bock C., Geissmann F., Specification of tissue-resident macrophages during organogenesis. Science 353, aaf4238 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van de Laar L., Saelens W., De Prijck S., Martens L., Scott C. L., Van Isterdael G., Hoffmann E., Beyaert R., Saeys Y., Lambrecht B. N., Guilliams M., Yolk sac macrophages, fetal liver, and adult monocytes can colonize an empty niche and develop into functional tissue-resident macrophages. Immunity 44, 755–768 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Jakubzick C. V., Randolph G. J., Henson P. M., Monocyte differentiation and antigen-presenting functions. Nat. Rev. Immunol. 17, 349–362 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Whitsett J. A., Weaver T. E., Alveolar development and disease. Am. J. Respir. Cell Mol. Biol. 53, 1–7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hashimoto D., Chow A., Noizat C., Teo P., Beasley M. B., Leboeuf M., Becker C. D., See P., Price J., Lucas D., Greter M., Mortha A., Boyer S. W., Forsberg E. C., Tanaka M., van Rooijen N., Garcia-Sastre A., Stanley E. R., Ginhoux F., Frenette P. S., Merad M., Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 38, 792–804 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Todd E. M., Zhou J. Y., Szasz T. P., Deady L. E., D’Angelo J. A., Cheung M. D., Kim A. H., Morley S. C., Alveolar macrophage development in mice requires L-plastin for cellular localization in alveoli. Blood 128, 2785–2796 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bry K., Hallman M., Teramo K., Waffarn F., Lappalainen U., Granulocyte-macrophage colony-stimulating factor in amniotic fluid and in airway specimens of newborn infants. Pediatr. Res. 41, 105–109 (1997). [DOI] [PubMed] [Google Scholar]
- 24.Bharat A., Bhorade S. M., Morales-Nebreda L., McQuattie-Pimentel A. C., Soberanes S., Ridge K., DeCamp M. M., Mestan K. K., Perlman H., Budinger G. R., Misharin A. V., Flow cytometry reveals similarities between lung macrophages in humans and mice. Am. J. Respir. Cell Mol. Biol. 54, 147–149 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deady L. E., Todd E. M., Davis C. G., Zhou J. Y., Topcagic N., Edelson B. T., Ferkol T. W., Cooper M. A., Muenzer J. T., Morley S. C., L-plastin is essential for alveolar macrophage production and control of pulmonary pneumococcal infection. Infect. Immun. 82, 1982–1993 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carr R., Brocklehurst P., Dore C. J., Modi N., Granulocyte-macrophage colony stimulating factor administered as prophylaxis for reduction of sepsis in extremely preterm, small for gestational age neonates (the PROGRAMS trial): A single-blind, multicentre, randomised controlled trial. Lancet 373, 226–233 (2009). [DOI] [PubMed] [Google Scholar]
- 27.Puljic R., Benediktus E., Plater-Zyberk C., Baeuerle P. A., Szelenyi S., Brune K., Pahl A., Lipopolysaccharide-induced lung inflammation is inhibited by neutralization of GM-CSF. Eur. J. Pharmacol. 557, 230–235 (2007). [DOI] [PubMed] [Google Scholar]
- 28.Todd E. M., Deady L. E., Morley S. C., The actin-bundling protein L-plastin is essential for marginal zone B cell development. J. Immunol. 187, 3015–3025 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu X., Buttgereit A., Lelios I., Utz S. G., Cansever D., Becher B., Greter M., The cytokine TGF-β promotes the development and homeostasis of alveolar macrophages. Immunity 47, 903–912.e4 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Trapnell B. C., Whitsett J. A., Gm-CSF regulates pulmonary surfactant homeostasis and alveolar macrophage-mediated innate host defense. Annu. Rev. Physiol. 64, 775–802 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Trapnell B. C., Carey B. C., Uchida K., Suzuki T., Pulmonary alveolar proteinosis, a primary immunodeficiency of impaired GM-CSF stimulation of macrophages. Curr. Opin. Immunol. 21, 514–521 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suzuki T., Arumugam P., Sakagami T., Lachmann N., Chalk C., Sallese A., Abe S., Trapnell C., Carey B., Moritz T., Malik P., Lutzko C., Wood R. E., Trapnell B. C., Pulmonary macrophage transplantation therapy. Nature 514, 450–454 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carr R., Modi N., Doré C., G-CSF and GM-CSF for treating or preventing neonatal infections. Cochrane Database Syst. Rev. 2003, CD003066 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mould K. J., Barthel L., Mohning M. P., Thomas S. M., McCubbrey A. L., Danhorn T., Leach S. M., Fingerlin T. E., O'Connor B. P., Reisz J. A., D'Alessandro A., Bratton D. L., Jakubzick C. V., Janssen W. J., Cell origin dictates programming of resident versus recruited macrophages during acute lung injury. Am. J. Respir. Cell Mol. Biol. 57, 294–306 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gibbings S. L., Thomas S. M., Atif S. M., McCubbrey A. L., Desch A. N., Danhorn T., Leach S. M., Bratton D. L., Henson P. M., Janssen W. J., Jakubzick C. V., Three unique interstitial macrophages in the murine lung at steady state. Am. J. Respir. Cell Mol. Biol. 57, 66–76 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Andreeva A. V., Kutuzov M. A., Voyno-Yasenetskaya T. A., Regulation of surfactant secretion in alveolar type II cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 293, L259–L271 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Wright J. R., Youmans D. C., Degradation of surfactant lipids and surfactant protein A by alveolar macrophages in vitro. Am. J. Physiol. 268, L772–L780 (1995). [DOI] [PubMed] [Google Scholar]
- 38.Dong Q., Wright J. R., Degradation of surfactant protein D by alveolar macrophages. Am. J. Physiol. 274, L97–L105 (1998). [DOI] [PubMed] [Google Scholar]
- 39.Marlow N., Morris T., Brocklehurst P., Carr R., Cowan F. M., Patel N., Petrou S., Redshaw M. E., Modi N., Dore C., A randomised trial of granulocyte-macrophage colony-stimulating factor for neonatal sepsis: Outcomes at 2 years. Arch. Dis. Child. Fetal Neonatal Ed. 98, F46–F53 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marlow N., Morris T., Brocklehurst P., Carr R., Cowan F., Patel N., Petrou S., Redshaw M., Modi N., Dore C. J., A randomised trial of granulocyte-macrophage colony-stimulating factor for neonatal sepsis: Childhood outcomes at 5 years. Arch. Dis. Child. Fetal Neonatal Ed. 100, F320–F326 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sheng G., Chen P., Wei Y., Chu J., Cao X., Zhang H. L., Better approach for autoimmune pulmonary alveolar proteinosis treatment: Inhaled or subcutaneous granulocyte-macrophage colony-stimulating factor: A meta-analyses. Respir. Res. 19, 163 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scott J. P., Ji Y., Kannan M., Wylam M. E., Inhaled granulocyte-macrophage colony-stimulating factor for Mycobacterium abscessus in cystic fibrosis. Eur. Respir. J. 51, 1702127 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Steinwede K., Tempelhof O., Bolte K., Maus R., Bohling J., Ueberberg B., Langer F., Christman J. W., Paton J. C., Ask K., Maharaj S., Kolb M., Gauldie J., Welte T., Maus U. A., Local delivery of GM-CSF protects mice from lethal pneumococcal pneumonia. J. Immunol. 187, 5346–5356 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Huang F. F., Barnes P. F., Feng Y., Donis R., Chroneos Z. C., Idell S., Allen T., Perez D. R., Whitsett J. A., Dunussi-Joannopoulos K., Shams H., GM-CSF in the lung protects against lethal influenza infection. Am. J. Respir. Crit. Care Med. 184, 259–268 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Herold S., Hoegner K., Vadasz I., Gessler T., Wilhelm J., Mayer K., Morty R. E., Walmrath H. D., Seeger W., Lohmeyer J., Inhaled granulocyte/macrophage colony-stimulating factor as treatment of pneumonia-associated acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 189, 609–611 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/8/eaax3387/DC1
Fig. S1. No increase in mature AMs following the subcutaneous administration of rGM-CSF (20 ng) to LPL−/− neonatal pups on DOB, PND1, and PND2.
Fig. S2. No disruption of alveolarization observed after intranasal neonatal rGM-CSF therapy.
Fig. S3. Increased SP-D in LPL−/− PND3 neonatal pups.