

Summary
Tumour necrosis factor receptor‐associated periodic syndrome (TRAPS) is a hereditary autoinflammatory disorder characterized by recurrent episodes of fever and inflammation. It is associated with autosomal dominant mutations in TNFRSF1A, which encodes tumour necrosis factor receptor 1 (TNF‐R1). Our aim was to understand the influence of TRAPS mutations on the response to stimulation of the pattern recognition Toll‐like receptor (TLR)‐9. Peripheral blood mononuclear cells (PBMCs) and serum were isolated from TRAPS patients and healthy controls: serum levels of 15 proinflammatory cytokines were measured to assess the initial inflammatory status. Interleukin (IL)‐1β, IL‐6, IL‐8, IL‐17, IL‐22, tumour necrosis factor (TNF)‐α, vascular endothelial growth factor (VEGF), interferon (IFN)‐γ, monocyte chemoattractant protein 1 (MCP‐1) and transforming growth factor (TGF)‐β were significantly elevated in TRAPS patients’ sera, consistent with constitutive inflammation. Stimulation of PBMCs with TLR‐9 ligand (ODN2006) triggered significantly greater up‐regulation of proinflammatory signalling intermediates [TNF receptor‐associated factor (TRAF 3), IL‐1 receptor‐associated kinase‐like 2 (IRAK2), Toll interacting protein (TOLLIP), TRAF6, phosphorylated transforming growth factor‐β‐activated kinase 1 (pTAK), transforming growth factor‐β‐activated kinase‐binding protein 2 (TAB2), phosphorylated TAK 2 (pTAB2), IFN‐regulatory factor 7 (IRF7), receptor interacting protein (RIP), nuclear factor kappa B (NF‐κB) p65, phosphorylated NF‐κB p65 (pNF‐κB p65) and mitogen‐activated protein kinase kinase (MEK1/2)] in TRAPS patients’ PBMCs. This up‐regulation of proinflammatory signalling intermediates and raised serum cytokines occurred despite concurrent anakinra treatment and no overt clinical symptoms at time of sampling. These novel findings further demonstrate the wide‐ranging nature of the dysregulation of innate immune responses underlying the pathology of TRAPS and highlights the need for novel pathway‐specific therapeutic treatments for this disease.
Keywords: autoinflammatory disease, cytokines, inflammation, toll like receptors (TLRs)
Introduction
The rare autoinflammatory disease, tumour necrosis factor receptor (TNF‐R)‐associated periodic syndrome (TRAPS) is caused by autosomal dominant missense mutations in the TNFRSF1A gene, encoding TNF‐R1 1. TRAPS patients demonstrate exaggerated innate immune responsiveness and are characterized clinically by recurrent prolonged episodes of fever, migratory rash, peritonitis, myalgia and arthralgia, with systemic amyloidosis occurring in a proportion of patients 2. Intracellular aggregation of mutant TNF‐R1 plays a major role in the pathogenesis of TRAPS through a variety of mechanisms, including misfolding and ligand‐independent signalling, an unfolded protein response, induction of reactive oxygen species (ROS) and impaired autophagy 3, 4. Both transfected cells and TRAPS patients’ PBMCs expressing a prototypical TRAPS‐related mutant TNF‐R1 (C33Y) reveal perturbations and activation of multiple proinflammatory signalling pathways 5. Thus, mutant TNF receptors create an intracellular environment conducive to drive chronic inflammation and synergy between mutant and wild‐type TNF‐R1 actions is required to generate the full TRAPS phenotype 6, 7.
An area of increasing interest is the cross‐talk between mutant TNFRs and pattern recognition receptors (PRRs), including the Toll‐like receptors (TLRs) involved in innate immune activation. It has recently been proposed that cells with TRAPS‐related mutant TNF‐R display greater sensitivity to PRR ligands such as the TLR‐4 ligand, bacterial lipopolysaccharide (LPS), due to excessive activation of downstream signalling through the mitogen‐activated protein kinases (MAPKs), resulting in enhanced production of proinflammatory cytokines 8.
TLR‐9 is an endosomal PRR which recognizes unmethylated CpG‐rich DNA motifs, frequently found in bacterial, mitochondrial and viral DNA. TLR‐9 activation initiates innate immune responses via the adaptor molecule myeloid differentiation primary response 88 (MyD88), and subsequently interleukin (IL)‐1 receptor‐associated kinase‐like (IRAK4) and TNF receptor‐associated factor (TRAF) 6, and ultimately activation of IFN‐regulatory factor 7 (IRF7), nuclear factor kappa B (NF‐κB), Janus kinase (JNK) and P38 MAPKs, ultimately leading to the production of inflammatory cytokines and type 1 IFNs 9.
This study investigates how the host innate immune response to TLR‐9 stimulation is influenced by the TRAPS‐associated C33Y mutant TNF‐R1 (TNF‐R1(C33Y)). Venous blood from patients with TNF‐R1(C33Y) and controls were examined for serum cytokine levels to establish baseline levels of inflammatory mediators, and the TLR‐9 agonist (ODN2006) was used to stimulate isolated peripheral blood mononuclear cells (PBMCs) from both sample groups and the expression and activation of proinflammatory signalling pathways was subsequently evaluated using protein microarray technologies 10.
Material and methods
Patients and healthy controls
Four TRAPS patients carrying TNF‐R1(C33Y) [two males, two females; mean age 46·3 (range 33–66) years] and five healthy controls [two males, three females; mean age 43.5 (range 27–62) years] were included for TLR‐9 signalling pathway studies in PBMCs. For cytokine assays, seven controls’ sera [three males, four females; mean age 48 (range 27–62) years] were compared to the patients’ samples. All TRAPS patients were being treated with anakinra and none had any clinical symptoms of inflammation at the time of blood sampling.
Serum collection and PBMC separation
Blood samples were collected after ethical approval and with informed consent into citrate phosphate dextrose (ACD; Baxter Health Care, Norfolk, UK) and processed within 2 h of sampling. PBMCs were isolated by Ficoll‐density gradient centrifugation. PBMCs were resuspended in serum‐free RPMI‐1640, containing 25 mM HEPES and L‐glutamine, at a density of 2 × 105 cells/ml. Cells were exposed to 5 μM ODN 2006 (InvivoGen, Toulouse, France) for 2 h; cells were then washed with ice‐cold phosphate‐buffered saline (PBS), pelleted and lysed with 100 μl of RIPA lysis buffer containing 5 units/ml benzonase nuclease (Sigma‐Aldrich, Poole, UK) and 1 × Halt™ protease and phosphatase inhibitor cocktail (Thermo‐Fisher Scientific, Loughborough, UK) for 20 min with continuous agitation on ice.
TLR‐9 expression in PBMCs from TRAPS and healthy controls
Cells were stained for flow cytometry as described previously 11 to evaluate TLR‐9 expression. Following washing with PBA (PBS/1% BSA/0·05% NaN3), the PBMCs were permeabilized using saponin buffer and stained for 2 h in the dark with anti‐human CD289 (TLR‐9) phycoerythrin (PE) or appropriate isotype controls (rat IgG2a PE) (e‐Bioscience/Thermo‐Fisher Scientific). Cells were then washed twice in PBA, resuspended in 0·5% formaldehyde fixative and analysed on a Coulter FC500 flow cytometer. Analysis was performed using weasel software (Walter and Eliza Hall Institute, Parkville, Australia).
Reverse‐phase protein arrays (RPPA) analysis
RPPA was used as optimized and validated previously 5. Briefly, PBMC lysates were diluted 3 : 1 with × 4 sodium dodecyl sulphate (SDS) sample buffer (278 mM TrisCl, pH 6.8, 44% glycerol, 4·4% SDS, 8% 2‐mercaptoethanol) and heated (95°C for 5 min). Samples were spotted in quadruplicate onto nitrocellulose‐coated glass slides (Grace Bio‐labs, Roslin, UK) with a MicroGrid II robot (BioRobotics Inc., Redwood City, CA, USA). The printed slides were blocked overnight at 4°C in blocking solution (0·2% I‐block (Tropix, Bedford, MA, USA), 0·1% Tween‐20 in PBS) at 4°C. After washing three times for 5 min each in PBS + 0·05% Tween‐20 (PBST), the slides were incubated overnight at 4°C with the specific primary antibodies (Cell Signaling Technology, Danvers, MA, USA). Supporting information, Table S1 shows the details of the primary antibodies used for RPPA analysis. β‐actin antibody was used as a housekeeping protein to control protein loading. After washing in PBST (×3), primary antibody detection was achieved using infrared Licor secondary antibodies (800‐CW anti‐rabbit antibody and 700‐CW anti‐mouse antibody) for 30 min at room temperature in the dark with shaking. After washing in PBST × 3 and rinsing in water, arrays were centrifuged to dry and scanned with a Licor Odyssey SA scanner (LI‐COR Biosciences, UK) at 20 μm resolution. Fluorescence images were processed with Axon Genepix Pro‐6 Microarray Image Analysis software (Axon Instruments, Inc., Foster City, CA, USA). Target signals were finally determined after background subtraction and normalization to the internal housekeeping targets using RPPanalyzer, a module within the r statistical language 12. The specificity of all the primary antibodies was verified using gel electrophoresis and western blotting, as described previously 13. Only antibodies detecting single bands of the correct molecular weight target were used.
Cytokine analysis
Cytokines were analysed in sera from TRAPS patients and controls using antibody microarray‐based sandwich enzyme‐linked immunosorbent assay (ELISA). DuoSet paired antibody kits (R&D Systems, Minneapolis, MN, USA) were used for the detection of 16 cytokines: eotaxin‐1, eotaxin‐2, IFN‐γ, IL‐1β, IL‐4, IL‐6, IL‐8, IL‐10, IL‐17, IL‐22, monocyte chemoattractant protein (MCP)‐1, receptor for advanced glycation end‐products (RAGE), transforming growth factor (TGF)‐β, TNF‐α and vascular endothelial growth factor (VEGF) 10. In brief, capture antibodies were printed at 100 μg/ml in quadruplicate as regular arrays (16 per slide) onto poly‐L‐lysine‐coated glass slides (Thermo‐Fisher Scientific) using a MicroGrid II robot. The array slides were blocked with I‐Block (0·2% in PBS) for 1 h and washed three times with PBST. A cocktail of recombinant cytokine standards was prepared with each cytokine at the maximum recommended standard curve concentration, and then used to generate eight doubling dilutions. Standard dilutions and samples were added to independent arrays for 1 h at room temperature with frequent shaking followed by three washes with PBST. A mixture of the manufacturer’s biotinylated detection antibodies was added at the recommended concentrations to each array for 1 h with shaking. After washing with PBST as before, A 1 : 1 : 300 mixture of Genisphere streptavidin 3DNA Oyster 650‐40: Nucleic Acid Blocker (Genisphere LLC, Hatfield, PA, USA) antibody diluent (Dako, Ely, UK) was added and incubated for 30 min at room temperature with frequent shaking in the dark. Slides were then washed three times with PBST, rinsed with ultra‐pure water and dried by centrifugation. Slides were scanned at 635 nm with a Genepix 4200AL scanner (Axon Instruments) and fluorescence signals quantified using Axon Genepix Pro‐6 software. The concentration of each cytokine in samples was subsequently extrapolated from the standard curve values.
Statistical analysis
Statistical analyses were performed using GraphPad Prism version 6 software. One‐way analysis of variance (ANOVA) and Bonferroni’s multiple comparisons test was undertaken to compare signalling molecules expression in different conditions. The statistical differences in cytokine expression and TLR‐9 expression between patient and control samples were determined by Mann–Whitney U‐test.
Results
In‐vivo constitutive inflammatory stimulation in TRAPS patients
Fifteen inflammatory cytokines and chemokines were measured in the sera of TRAPS patients with TNF‐R1(C33Y) and age‐ and sex‐matched healthy controls to determine the underlying inflammatory status of each patient at the time of PBMC collection (Fig. 1). Significantly higher levels of the following cytokines were observed in TRAPS patients compared to healthy controls: IL‐1β, IL‐6, IL‐8, IL‐17, IL‐22, TNF‐α, VEGF, IFN‐γ, MCP‐1 and TGF‐β (Fig. 1a,c,d,f,g,h,i,m,o). Eotaxin‐1, RAGE, IL‐10 and IL‐4 were not significantly different between the groups (Fig. 1b,e,n) and eotaxin‐2 was significantly higher in the controls (Fig. 1k). These findings support and extend previous studies 14, 15, 16, 17 by revealing the presence of additional proinflammatory serum cytokines/chemokines (IFN‐γ, IL‐17, IL‐22 and MCP‐1) in TRAPS patients and also increased serum TGF‐β and VEGF.
Figure 1.
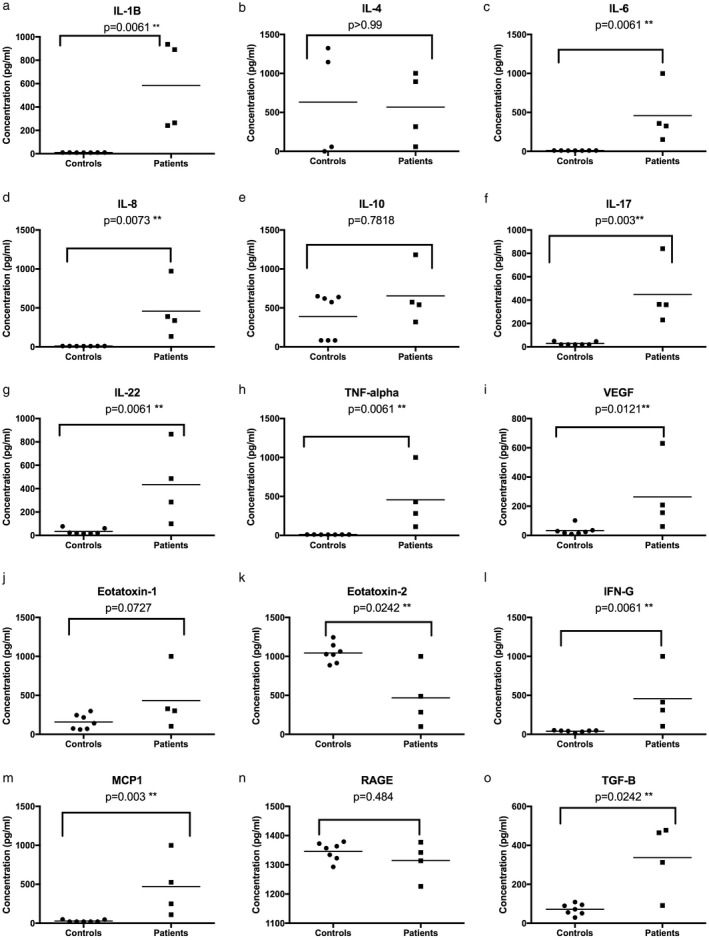
Serum cytokine levels in tumour necrosis factor receptor‐associated periodic syndrome (TRAPS) patients and healthy controls. The data are shown for individual serum samples (four TRAPS patients and seven healthy controls, with three technical replicates per sample). Horizontal bars indicate the mean values. The Mann–Whitney test was performed and P < 0·05 was considered statistically significant.
Ex‐vivo constitutive activation of inflammatory pathways in TRAPS patients
TRAPS patients’ PBMCs in the absence of external stimulation have already been shown to exhibit enhanced activation of proinflammatory signalling pathways 13. In this study, we therefore investigated the influence of TNF‐R1(C33Y) on responses to TLR‐9 stimulation to isolate PBMCs. Binding of the synthetic agonist ODN2006 to TLR‐9 triggers a complex series of intracellular signal transduction events 9. Concurrent evaluation of target protein expression and activation‐related post‐translational modifications were measured using reverse‐phase protein arrays. PBMCs from TRAPS patients with TNF‐R1(C33Y) and healthy controls were cultured with or without ODN2006, lysed and 49 signalling intermediates, including TLR‐9 pathway‐associated molecules, were subsequently measured. β‐actin abundance was measured as a normalizing protein control. Supporting information, Table S1 shows the list of the primary antibodies used in this study. Comparisons were then made between the TNF‐R1(C33Y) patient samples and control samples, both before and after ODN2006 stimulation (Fig. 2). Table 1 (column A) shows that 36 of the 49 signalling molecules measured revealed significantly higher baseline levels in untreated TRAPs patients’ PBMCs compared to untreated PBMCs from healthy controls. Similarly, comparison of ODN2006‐stimulated PBMCs revealed 42 of 49 signalling molecules detected at significantly higher levels in patients’ PBMCs compared to healthy controls (Table 1, column B). However, the most important finding derives from comparisons between ODN2006‐stimulated PBMCs and unstimulated cells: for the TRAPS patients, 17 of 49 signalling molecules were significantly raised by stimulation with ODN2006 (Table 1, column C), but only four of 49 signalling molecules were significantly raised in the healthy control PBMCs, only one of which (IRAK4) was also not up‐regulated in the patients’ cells (Table 1, column D).
Figure 2.

Inflammatory signalling pathway intermediates measured by reverse‐phase protein arrays (RPPA) in peripheral blood mononuclear cells (PBMCs) of tumour necrosis factor receptor‐associated periodic syndrome (TRAPS) patients and controls. (1) Heat‐map columns represent control samples (C1–5) and patient samples (P1–4) either unstimulated (US) or ODN2006‐stimulated (ODN). Individual rows represent the signalling molecules studied. Antibodies used and working dilutions are described in full in Supporting information, Table S1. The white to black heat‐map colour range represents RPPA signal data normalized to represent percentages of the signal range for each molecule examined (0–100%). Letters a–r at the left‐hand side refer to the scatter‐plots in section 2 of the figure. (2) Scatter‐plots representing actin‐normalized relative abundance of selected target proteins which show statistically significant differences in columns C and D of Table 1. Data are shown for individual PBMC lysates (mean of four technical replicates per sample) of four patient samples and five control samples. Signals are presented as arbitrary fluorescence units after background subtraction and normalization to β‐actin. The four conditions were: unstimulated healthy controls’ PBMCs; Con (US), unstimulated patients’ PBMCs; Pat (US), ODN2006‐stimulated healthy controls’ PBMCs; Con (ODN) and ODN2006‐stimulated patients’ PBMCs; Pat (US). Significant differences between conditions for each signalling molecule were determined by one‐way analysis of variance (ANOVA) and Bonferroni’s multiple comparisons test; these are presented in Table 1.
Table 1.
Statistical evaluation of intracellular signalling molecule expression in peripheral blood mononuclear cells (PBMCs) from tumour necrosis factor receptor‐associated periodic syndrome (TRAPS) patients and healthy controls without and with ODN2006 treatment.
| Signalling molecule | (A) Patients’ US versus controls’ US | (B) Patients’ ODN versus controls’ ODN | (C) Patients’ ODN versus patients’ US | (D) Controls’ ODN versus controls’ US |
|---|---|---|---|---|
| MyD88 | ||||
| TRAF3 | 0·0001 | <0·0001 | 0·002 | |
| IRAK‐4 | 0·0052 | 0·0273 | ||
| pIRAK‐4 | ||||
| IRAK‐1 | <0·0001 | <0·0001 | ||
| IRAK‐2 | 0·0048 | <0·0001 | 0·0003 | 0·0425 |
| IRAK‐M | ||||
| TOLLIP | <0·0001 | <0·0001 | <0·0001 | <0·0001 |
| TRAF6 | <0·0001 | <0·0001 | 0·0132 | |
| pTAK | <0·0001 | <0·0001 | 0·0115 | |
| TAB2 | 0·0007 | 0·0336 | ||
| pTAB2 | 0·0011 | <0·0001 | 0·0150 | |
| TBK‐NAK | 0·0009 | |||
| pTBK‐NAK | ||||
| IRF7 | 0·0028 | <0·0001 | 0·0028 | |
| pIRF7 | 0·0222 | 0·0054 | ||
| IRF3 | 0·0037 | 0·0002 | 0·0428 | |
| pIRF3 | <0·0001 | <0·0001 | ||
| TRAF2 | 0·0123 | 0·0006 | ||
| RIP | 0·0003 | <0·0001 | 0·0107 | |
| pRIP | 0·0106 | |||
| IKB‐α | 0·0003 | <0·0001 | ||
| pIKK‐α | 0·0001 | <0·0001 | ||
| NF‐κB p65 | 0·0004 | <0·0001 | 0·0216 | |
| pNF‐κB p65 | <0·0001 | <0·0001 | 0·0455 | |
| PIKK‐γ | 0·0178 | 0·0014 | ||
| PIKK‐β | 0·0006 | 0·0005 | ||
| NF‐KB p50 | 0·0013 | 0·002 | ||
| A20 | 0·0105 | |||
| MEK1/2 | 0·0276 | 0·0063 | ||
| pMEK1/2 | <0·0001 | <0·0001 | ||
| pP38 | 0·0057 | |||
| pERK1/2 | 0·0311 | |||
| MKK7 | 0·0028 | |||
| pSEK1/MKK4 | 0·0022 | |||
| pHSP27 | <0·0001 | <0·0001 | ||
| pROS | 0·0016 | <0·0001 | ||
| pATF | 0·0169 | <0·0001 | 0·0109 | |
| pCRAF | 0·0071 | 0·0015 | ||
| pAKT Thr | 0·0002 | <0·0001 | 0·0078 | |
| pAKT Serine | <0·0001 | <0·0001 | ||
| pPDK1 | 0·0035 | 0·0001 | ||
| pGSK‐3B | <0·0001 | <0·0001 | ||
| PI3K110 | 0·0039 | 0·0014 | ||
| PI3KP85 | 0·0004 | <0·0001 | ||
| CIAP | 0·0041 | 0·025 | ||
| ICAM | 0·0014 | <0·0001 | 0·0174 | |
| MMP12 | <0·0001 | <0·0001 | ||
| Caspase 8 | 0·0001 | <0·0001 |
Table shows statistical comparison [one‐way analysis of variance (ANOVA) with Bonferroni’s multiple comparison test] of patient and control group normalized signal intensities of 49 signalling molecules, measured by reverse‐phase protein arrays (RPPA), with and without ODN2006 stimulation. Significant differences between the patient and control group are reported (P‐values < 0·05). Detailed data are shown in Fig. 2. (A) Patients’ US (unstimulated patients’ PBMCs) versus controls US (unstimulated healthy controls’ PBMCs). (B) Patients’ ODN (ODN2006‐stimulated patients’ PBMCs) versus controls’ ODN (ODN2006‐stimulated healthy controls’ PBMCs). (C) Patients’ ODN versus patients’ US. (D) Controls ODN versus controls US.
TLR‐9 expression in PBMCs from TRAPS patients and healthy controls
To establish whether the hypersensitivity of the TLR‐9‐stimulated response seen in the TNF‐R1(C33Y) patients PBMCs might be due to differential TLR‐9 expression, we compared TLR‐9 expression in PBMCs of patients and healthy controls by intracellular staining and flow cytometry. There was no significant difference in TLR‐9 expression between the two groups as measured by the mean fluorescence intensity (MFI) or the percentage of TLR‐9‐positive cells (Supporting information, Fig. S1). Thus, the hypersensitivity of the TRAPS patients’ PBMCs to ODN2006 is not due to increased levels of TLR‐9.
Discussion
TRAPS and the other autoinflammatory diseases reflect primary dysfunctions of innate immunity, with increasing support suggesting a complex interplay of inflammatory signals and inflammasome involvement resulting in the clinical presentations seen 18, 19. We hypothesized that the effects of the TRAPS mutant TNF‐R1 and any subsequent TLR activation (TLR‐9 used here), may synergize to exacerbate proinflammatory responses in TRAPS.
The TRAPS patients studied, all heterozygous for TNF‐R1(C33Y) were receiving anakinra (IL‐1 receptor antagonist) therapy at the time of sampling and were without clinical symptoms of inflammation. These patients nevertheless demonstrated increased serum levels of multiple proinflammatory cytokines, including IL‐1β, IL6, IL‐8 and TNF‐α relative to healthy controls samples. The elevated levels of IL‐1β seen also give further support to the role of inflammasome activation and IL‐1β release in TRAPS, as with many other autoinflammatory diseases 18, 20, and emphasizes the usefulness of therapeutics targeting IL‐1 responses, despite TRAPS being a TNF‐R1 mutation‐associated disease 21.
In addition, our results also revealed increased serum levels of IFN‐γ, IL‐17 and IL‐22, suggestive of additional proinflammatory responses, most probably from NK and T cell populations 22 and of VEGF and the pleotrophic cytokine TGF‐β, although their relevance in TRAPS disease pathology is currently unclear. The raised levels of MCP‐1 seen in patients may be a consequence of the elevated IL‐1‐β, IL‐6 and TNF‐α and may have some bearing on muscle inflammation in TRAPS 23, 24. Our findings from patient sera therefore highlight the complexity of the constitutive inflammatory status of TRAPS patients, even when clinically well and maintained on a cytokine neutralization therapy that does not target the central disease mechanisms 16.
TLR‐9 ligation by appropriate DNA ligands leads to stimulation of IRF7, NF‐κB, JNK and P38 MAPK pathways, and can also prime the NLRP3 inflammasome by increasing expression of both NLRP3 and pro‐IL‐1β. Similar expression levels of TLR‐9 in PBMCs from TRAPS patients and healthy controls were observed; however, heightened expression and activation of intracellular signaling molecules are seen in PBMCs of the TNF‐R1(C33Y) patients. These include, as expected, molecules involved in the TLR‐9 signal transduction, including key adaptors such as TRAF6 and, as we have reported previously, many elements of the NF‐κB associated with TNF‐R1 signalling (Fig. 2, 1 and Fig 2, 2k,i,o). Phosphorylation of many of these molecules is linked with activation status. The increased levels of phosphorylated IRF7, TAK and TAB2, the IKKs and NFκB P65 (examples shown in Fig. 2 (2i,d,g,j,o) indicates that TLR‐9 stimulation can further increase activation of the associated pathways from an already above‐supranormal level of activity in PBMCs of TRAPS patients. The evidence that numerous other signalling intermediates are also further stimulated suggests that the TNF‐R1(C33Y) mutation has a wide‐ranging influence on cross‐talk and activation of proinflammatory pathways (Fig. 3).
Figure 3.
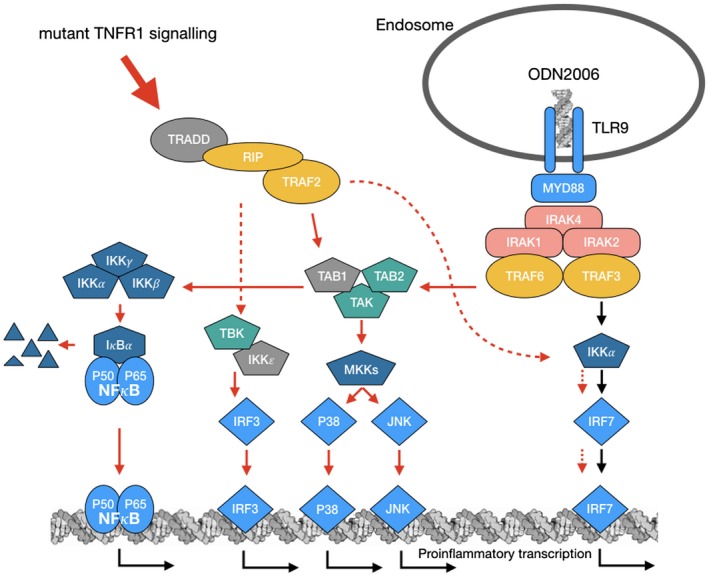
Visual depiction of findings: the graphic represents elements of the common pathways active by both tumour necrosis factor receptor 1 (TNF‐R1) signalling and signalling through Toll‐like receptor (TLR)‐9. Arrows indicate activation route of molecules in the pathways. Red arrows indicate those pathways which are found to be both more abundant and more activated as a result of the C33Y tumour necrosis factor receptor‐associated periodic syndrome (TRAPS) mutation and also further stimulated by TLR‐9 ligation as ODN2006 as an artificial activator. Molecules depicted in grey were not studied.
This study therefore highlights the effect of TLR‐9 activation in cells expressing the TNF‐R1(C33Y) mutation. The TRAPS patients’ PBMCs demonstrate a constitutive proinflammatory state of activation, but are also much more sensitive than those of healthy controls to activation of proinflammatory signalling pathways via stimulation of TLR‐9. This hypersensitivity, like that reported for the TLR‐4 ligand LPS 7, could partly explain the seemingly spontaneous inflammatory attacks seen in TRAPS, and may be triggered by subclinical infections and TLR or possibly other PRR stimulation that would go unnoticed in normal individuals.
Despite being subject to IL‐1‐neutralizing therapy (anakinra) prior to and at the time of sampling, patients showed considerable serum levels of multiple proinflammatory mediators, including IL‐1β, IL‐6, IL‐8, IL‐17, IL‐22 and TNF‐α, indicating that the underlying effects of TNF‐R1(C33Y)‐mediated dysregulation are largely unchecked as a result of treatment 13. The extensive signalling activation seen intracellularly in patients’ PMBCs, in both the absence and presence of TLR‐9 stimulation, however, suggests that suppressing such activity may require direct targeting of pathway components which lie at convergent points, such as has been suggested for TAB1 and 2/TAK1 complex 25, inhibition of the NLRP3 inflammasome 18, 26 or further novel targets 27, 28.
Disclosures
The authors declare that they have no conflicts of interest.
Supporting information
Fig. S1. Flow cytometric analysis of TLR9 expression in TRAPS and healthy control PBMCs. Representative dot plots of forward scatter vs. side scatter showing gating used for flow cytometric analysis of control PBMCs (a) and TRAPS patient PBMCs (b). Representative histograms of TLR9 expression by control PBMCs (c) and TRAPS patient PBMCs (d). Black lines denote isotype controls and red lines denote TLR9 expression. No significant differences were observed by unpaired Mann whitney test between TLR9 expression (MFI median fluorescence intensity) (e) in controls and patients.
Table S1. List of primary antibodies, and dilutions used in the RPPA signalling pathway analysis. All primary antibodies were obtained from Cell Signalling Technologies Inc.
Acknowledgments
This work was supported by The Jones 1986 Charitable Trust. We would like to thank Dr Luisa Martinez‐Pomares (University of Nottingham) for helpful discussions regarding the PBMC stimulations.
References
- 1. McDermott MF, Aksentijevich I, Galon J et al Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 1999; 97:133–44. [DOI] [PubMed] [Google Scholar]
- 2. Magnotti F, Vitale A, Rigante D et al The most recent advances in pathophysiology and management of tumour necrosis factor receptor‐associated periodic syndrome (TRAPS): personal experience and literature review. Clin Exp Rheumatol 2013; 31:141–9. [PubMed] [Google Scholar]
- 3. Todd I, Radford PM, Draper‐Morgan KA et al Mutant forms of tumour necrosis factor receptor I that occur in TNF‐receptor‐associated periodic syndrome retain signalling functions but show abnormal behaviour. Immunology 2004; 113:65–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bulua AC, Simon A, Maddipati R et al Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1‐associated periodic syndrome (TRAPS). J Exp Med 2011; 208:519–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Negm OH, Mannsperger HA, McDermott EM et al A pro‐inflammatory signalome is constitutively activated by C33Y mutant TNF receptor 1 in TNF receptor‐associated periodic syndrome (TRAPS). Eur J Immunol 2014; 44:2096–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fairclough LC, Stoop AA, Negm OH, Radford PM, Tighe PJ, Todd I. Tumour necrosis factor receptor I blockade shows that TNF‐dependent and TNF‐independent mechanisms synergise in TNF receptor associated periodic syndrome. Eur J Immunol 2015; 45:2937–44. [DOI] [PubMed] [Google Scholar]
- 7. Simon A, Park H, Maddipati R et al Concerted action of wild‐type and mutant TNF receptors enhances inflammation in TNF receptor 1‐associated periodic fever syndrome. Proc Natl Acad Sci USA 2010; 107:9801–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rigante D, Lopalco G, Vitale A et al Key facts and hot spots on tumor necrosis factor receptor‐associated periodic syndrome. Clin Rheumatol 2014; 33:1197–207. [DOI] [PubMed] [Google Scholar]
- 9. O'Neill LA, Golenbock D, Bowie AG. The history of Toll‐like receptors – redefining innate immunity. Nat Rev Immunol 2013; 13:453–60. [DOI] [PubMed] [Google Scholar]
- 10. Selvarajah S, Negm OH, Hamed MR et al Development and validation of protein microarray technology for simultaneous inflammatory mediator detection in human sera. Mediators Inflamm 2014; 2014:820304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Huggins ML, Radford PM, McIntosh RS et al Shedding of mutant tumor necrosis factor receptor superfamily 1A associated with tumor necrosis factor receptor‐associated periodic syndrome: differences between cell types. Arthritis Rheum 2004; 50:2651–9. [DOI] [PubMed] [Google Scholar]
- 12. Mannsperger HA, Gade S, Henjes F, Beissbarth T, Korf U. RPPanalyzer: analysis of reverse‐phase protein array data. Bioinformatics 2010; 26:2202–3. [DOI] [PubMed] [Google Scholar]
- 13. Akrem A, Yousef N, Begum A et al Preliminary crystallographic analysis of a cruciferin protein from seeds of Moringa oleifera . Protein J 2014; 33:253–7. [DOI] [PubMed] [Google Scholar]
- 14. Rebelo SL, Amel‐Kashipaz MR, Radford PM et al Novel markers of inflammation identified in tumor necrosis factor receptor‐associated periodic syndrome (TRAPS) by transcriptomic analysis of effects of TRAPS‐associated tumor necrosis factor receptor type I mutations in an endothelial cell line. Arthritis Rheum 2009; 60:269–80. [DOI] [PubMed] [Google Scholar]
- 15. La Torre F, Muratore M, Vitale A, Moramarco F, Quarta L, Cantarini L. Canakinumab efficacy and long‐term tocilizumab administration in tumor necrosis factor receptor‐associated periodic syndrome (TRAPS). Rheumatol Int 2015; 35:1943–7. [DOI] [PubMed] [Google Scholar]
- 16. Vaitla PM, Radford PM, Tighe PJ et al Role of interleukin‐6 in a patient with tumor necrosis factor receptor‐associated periodic syndrome: assessment of outcomes following treatment with the anti‐interleukin‐6 receptor monoclonal antibody tocilizumab. Arthritis Rheum 2011; 63:1151–5. [DOI] [PubMed] [Google Scholar]
- 17. Nedjai B, Hitman GA, Church LD et al Differential cytokine secretion results from p65 and c‐Rel NF‐kappaB subunit signaling in peripheral blood mononuclear cells of TNF receptor‐associated periodic syndrome patients. Cell Immunol 2011; 268:55–9. [DOI] [PubMed] [Google Scholar]
- 18. Hoffman HM, Broderick L. The role of the inflammasome in patients with autoinflammatory diseases. J Allergy Clin Immunol 2016; 138:3–14. [DOI] [PubMed] [Google Scholar]
- 19. Peckham D, Scambler T, Savic S, McDermott MF. The burgeoning field of innate immune‐mediated disease and autoinflammation. J Pathol 2017; 241:123–39. [DOI] [PubMed] [Google Scholar]
- 20. Carta S, Semino C, Sitia R, Rubartelli A. Dysregulated IL‐1beta secretion in autoinflammatory diseases: a matter of stress? Front Immunol 2017; 8:345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jesus AA, Goldbach‐Mansky R. IL‐1 blockade in autoinflammatory syndromes. Annu Rev Med 2014; 65:223–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sabat R, Ouyang W, Wolk K. Therapeutic opportunities of the IL‐22‐IL‐22R1 system. Nat Rev Drug Discov 2014; 13:21–38. [DOI] [PubMed] [Google Scholar]
- 23. Bostrom EA, Kindstedt E, Sulniute R et al Increased eotaxin and MCP‐1 levels in serum from individuals with periodontitis and in human gingival fibroblasts exposed to pro‐inflammatory cytokines. PLOS ONE 2015; 10:e0134608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Patsouris D, Cao JJ, Vial G et al Insulin resistance is associated with MCP1‐mediated macrophage accumulation in skeletal muscle in mice and humans. PLOS ONE 2014; 9:e110653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fechtner S, Fox DA, Ahmed S. Transforming growth factor beta activated kinase 1: a potential therapeutic target for rheumatic diseases. Rheumatology (Oxf) 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Moll M, Kuemmerle‐Deschner JB. Inflammasome and cytokine blocking strategies in autoinflammatory disorders. Clin Immunol 2013; 147:242–75. [DOI] [PubMed] [Google Scholar]
- 27. Marcuzzi A, Piscianz E, Valencic E, Monasta L, Vecchi Brumatti L, Tommasini A. To extinguish the fire from outside the cell or to shutdown the gas valve inside? Novel trends in anti‐inflammatory therapies. Int J Mol Sci 2015; 16:21277–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Feltham R, Vince JE, Lawlor KE. Caspase‐8: not so silently deadly. Clin Transl Immunology 2017; 6:e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Flow cytometric analysis of TLR9 expression in TRAPS and healthy control PBMCs. Representative dot plots of forward scatter vs. side scatter showing gating used for flow cytometric analysis of control PBMCs (a) and TRAPS patient PBMCs (b). Representative histograms of TLR9 expression by control PBMCs (c) and TRAPS patient PBMCs (d). Black lines denote isotype controls and red lines denote TLR9 expression. No significant differences were observed by unpaired Mann whitney test between TLR9 expression (MFI median fluorescence intensity) (e) in controls and patients.
Table S1. List of primary antibodies, and dilutions used in the RPPA signalling pathway analysis. All primary antibodies were obtained from Cell Signalling Technologies Inc.