

Abstract
Giant cell arteritis and Takayasu arteritis are autoimmune vasculitides that cause aneurysm formation and tissue infarction. Extravascular inflammation consists of an intense acute phase response. Deeper understanding of pathogenic events in the vessel wall has highlighted the loss of tissue protective mechanisms, the intrusion of immune cells into “forbidden territory”, and the autonomy of self-renewing vasculitic infiltrates. Adventitial vasa vasora critically control vessel wall access and drive differentiation of tissue-invasive T cells. Selected T cells establish tissue residency and build autonomous, self-sufficient inflammatory lesions. Pathogenic effector T cells intrude and survive due to failed immune checkpoint inhibition. Vasculitis-sustaining T cells and macrophages provide a broad portfolio of effector functions, involving heterogeneous populations of pro-inflammatory T cells and diverse macrophage subsets that ultimately induce wall capillarization and intimal hyperplasia. Redirecting diagnostic and therapeutic strategies from control of extravascular inflammatory markers to suppression of vascular inflammation will improve disease management.
Keywords: giant cell arteritis and Takayasu arteritis, inflammation, blood vessel (or artery), cytokines, proteases
1. Introduction
In humans, medium-sized and large arteries are organ systems with defined structural features. Due to abundance of medial collagen and elastin filaments, the large elastic arteries (aorta and primary branches), have Windkessel function and are distinguished from the smaller muscular arteries (2nd- 5th aortic branches) that distribute blood to dependent tissues [1, 2]. The wall thickness of medium and large arteries requires a dedicated blood supply system localized in the adventitial layer, the vasa vasorum network [3, 4]. Together with the heart, medium and large arteries are critical to survival and are protected from inflammatory attack; described as arterial wall immunoprivilege [5, 6]. A breakdown of this immunoprivilege results in vasculitis; Takayasu arteritis (TAK) in the elastic arteries including the aorta and the proximal segments of its major branches and pulmonary arteries and Giant Cell Arteritis (GCA) in the muscular arteries and the aorta [7, 8].
TAK and GCA are granulomatous arteritides [9]. Inflammatory infiltrates composed of T cells and macrophages accumulated in the vessel wall, predominantly in the medial layer [10]. Histomorphologic hallmarks of TAK and GCA provide clues towards disease mechanisms of relevance (Fig.1A). Tissue damage patterns include fragmentation of the elastic laminae, indicative of abundant elastolytic activity [11, 12]. Accordingly, multinucleated giant cells are often aligned along the digested lamina elastic interna. Disruption of elastic tissue is associated with loss of vascular smooth muscle cells, producing medial thinning. The inflammation-induced wall remodeling process leads to pronounced intimal thickening, with hyperplastic intima occluding the lumen [10]. In the large elastic arteries, arteritis causes aneurysm formation, in the muscular arteries luminal stenosis gives rise to tissue ischemia.
Figure 1. Histology and Cellular Players in Giant Cell Arteritis.
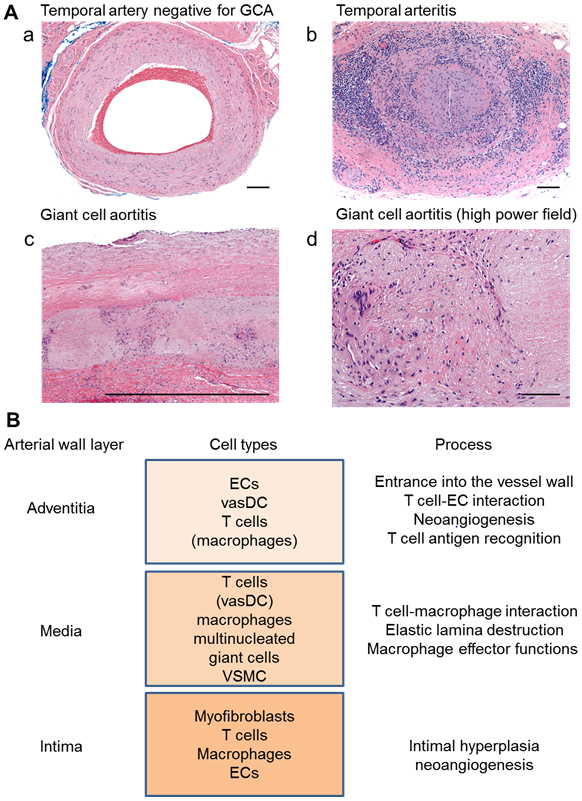
(A) Histological findings in giant cell arteritis and aortitis. Hematoxylin& Eosin (H&E) staining of temporal artery and aortic tissues. (a) Temporal artery showing normal “aging” changes with mild concentric intimal thickening. Scale bar indicates 100 μm. (b) Temporal artery showing severe inflammation especially in the medial and adventitial layers. Note the slit-like lumen of the artery due to marked intimal hyperplasia. Scale bar, 100 μm. (c) Scanning magnification of aorta showing medial disruption by alternating cellular and necrotic zones. Scale bar, 100 μm. (d) High power showing granulomatous lesion containing a multinucleate giant cell admixed with chronic inflammatory cells and a zone of medial necrosis to the right side. Scale bar, 100 μm. (B) Multiple cell types and multiple pathways participate in the pathogenic events of GCA. Specific cell types and pathways depend upon the tissue microenvironment. The arterial adventitia provides access for inflammatory cells and is the home of antigen-presenting vascular dendritic cells. Sprouting of adventitial vasa vasora leads to wall capillarization. Granulomatous infiltrates composed of macrophages, multinucleated giant cells and T cells accumulate in the medial layer, eventually causing medial thinning. Conversely, the intimal layer expands from 1-3 layers to multiple layers and acquires a microvascular network. Luminal occlusion by newly formed intima causes the ischemic complications of GCA. Wall-infiltrating T cells and macrophages may invade all wall layers, but granulomatous arrangements are most typically encountered in the media. EC, endothelial cells; vasDC, vascular dendritic cells; VSMC, vascular smooth muscle cells.
In GCA and TAK patients, the vasculitis is frequently associated with a systemic inflammatory syndrome, easily detected by an intense acute phase response [13–15]. While previous disease models have suggested that the disease begins extravascularly and progresses to the arterial wall, recent data strongly support the concept that the extravascular and vascular components have independent trajectories and the acute phase response may lie downstream of arterial wall inflammation [16–18]. This conceptual shift has implications not only for the understanding of pathomechanisms, but profoundly affects the diagnostic and therapeutic approach to GCA and TAK patients.
This review will discuss recent progress in the identification of cells and tissue niches actively involved in the disruption of the arterial wall immunoprivilege, will draw attention to inflammatory amplification processes damaging the vessel wall, and will present data demonstrating the autonomy of the vascular lesion. Together, these studies call for a reassessment of therapeutic goals in GCA; defining the suppression of vessel wall inflammation as the ultimate target of optimized immunotherapy.
2. Vascular GCA
2.1. Tissue niches in the vessel wall (Fig. 1B)
2.1.1. The arterial adventitia
A healthy and functional arterial wall is free of inflammatory cells and a combination of mechanisms prevents influx of inflammatory cells to minimize the risk for unintended collateral damage in a life-sustaining organ. Infiltration of innate and adaptive immune cells requires a loss of this immunoprivilege [5, 6]. All three wall layers, the adventitia, the media and the intima, play a disease-relevant role. In established GCA, innate and adaptive immune cells are encountered in a panartertic distribution, but packed lymphocyte infiltrates are often seen in the adventitia and granulomatous arrangements prefer the tunica media. It is now clear that GCA results from an active partnership between immune cells and indigenous vascular cells. Endothelial cells (EC) and stromal cells building the wall are instrumental in forming permissive tissue niches and are the drivers of the remodeling process, including angiogenesis within the wall and the formation of hyperplastic intima. Immune cells are highly selected; they must be able to actively communicate with vascular cells and must be able to survive and prosper in a hostile environment.
The adventitia provides the gateway to the “forbidden tissue site”, whereby T cells and monocytes/macrophages enter through vasa vasora. Recent studies have revealed that the digestion of the basal lamina by matrix metalloprotease (MMP)-9–producing monocytes is a critical step in the invasion process [12]. T cells cannot penetrate through collagen IV-rich matrix, unless they are accompanied by proteinase-releasing monocytes. Monocytes from GCA patients spontaneously produce MMP-9 and assist T cells in the infiltrative process. A second enabling event is the communication between CD4 T cells and microvascular endothelial cells [17]. In GCA-affected temporal arteries as well as GCA-affected aortas, adventitial microvessels express Jagged-1, a ligand for the NOTCH1 receptor. Circulating CD4 T cells in GCA patients express NOTCH1 [19] and receive stimulatory signals from Jagged-1+ microvessel EC; shifting the differentiation program of such T cells towards the production of IFN-γ and IL-17. EC-mediated T cell activation involves activation of the mTOR pathway, identifying possible therapeutic targets for novel immunosuppressive strategies [17]. The study also identified upstream signals leading to aberrant Jagged-1 expression on adventitial microvascular cells. Vascular endothelial growth factor (VEGF), abundantly present in the serum of GCA patients [20], functions as a Jagged-1 inducer, priming the EC for immunostimulatory functions. These findings emphasize the role of the adventitia in serving as a protective shield for the artery under normal conditions.
After the successful invasion into the wall, incoming monocytes/macrophages and T cells will encounter strategically positioned vascular dendritic cells (vasDC) [21]. These tissue-residing DC have been implicated in the production of cytokines and chemokines. They possess the machinery to present antigen and thus elicit an antigen-selected immune response [21]. Their major abnormalities lie in the deficient expression of the immunoinhibitory ligand PD-L1 [22]. Instead of sending negative signals to incoming T cells and preventing local immune reactivity, they predominantly provide co-stimulatory signals, thus promoting activation and survival of T cells in the tissue niche. This immune checkpoint failure classifies GCA as an autoimmune disease in which principal threshold settings are defective. Analogous to malignancies, where excessive activity of the PD-1/PD-L1 immune checkpoint leads to dismantling of anti-tumor immunity [23], immune protection of the arterial wall fails with insufficiency of negative signaling. Accordingly, the wall lesions in GCA are filled with PD-1+ T cells that are unopposed due to the loss of inhibitory signals. PD-L1 loss-of-function extends beyond the vessel wall and affects circulating monocytes/macrophages and monocyte-derived dendritic cells [16, 22]. Checkpoint dependency of the pathogenic immune responses in GCA has multiple implications for patient care. Firstly, GCA patients potentially have the benefit of robust anti-pathogen and anti-cancer immune responses. Secondly, given that the checkpoint failure is antigen-nonspecific, T cells with many specificities can survive in the wall lesions. Thirdly, overcoming the deficient checkpoint may render the patients susceptible to malignancies. And fourthly, therapeutic weakening of checkpoint function in cancer patients may lead to vasculitis as an immune-mediated side effect [24].
Recent studies have demonstrated that vasculitogenic immunity is highly sensitive to blocking CD28 function, emphasizing that professional antigen-presenting cells deliver strong co-stimulatory signals [25]. Metabolic fitness, specifically glycolytic breakdown of glucose fueling T cell effector functions, was identified as a CD28-dependent pathway. Together, these data support a novel disease paradigm, drawing attention to the balance of activating and inhibitory signals rather than to specific antigens driving the aberrant immunity underlying vascular GCA.
2.1.2. The arterial media
Arrangements of T cells, highly activated macrophages (so-called histocytes) and multinucleated giant cells are often encountered in the medial layer, a part of the artery composed of vascular smooth muscle cells (VSMC), collagen and elastic fibers. GCA is associated with VSMC loss, in a diffuse pattern in muscular arteries and in a patchy distribution in the aorta. How VSMC and elastic matrix components contribute to the disease process is not well understood. Matrix breakdown products are known to exhibit proinflammatory functions, as captured by the term “matrikines” [26–29]. Degradation products of extracellular matrix proteins, including elastin and collagens, reportedly have chemotactic activity and promote cellular activation and production of proteolytic enzymes. Matrikines, specifically N-acetyl-proline-glycine-proline (acPGP), have mild inflammatory activity in a GCA model system [12], suggesting that they participate in inflammatory amplification loops.
2.1.3. The arterial intima
In young healthy arteries, the intima consists of 1 to 3 endothelial cell layers and matrix. Intimal thickness increases with age, as exemplified by concentric intimal thickening encountered in temporal artery biopsies of older individuals (Fig.1A). In GCA-affected muscular arteries, the intima expands to more than 10 layers, often causing complete luminal occlusion. There is agreement that the cell type driving intimal hyperplasia is the myofibroblast. Myofibroblasts express α-smooth muscle actin (α-SMA) as a signature molecule, and have high migratory and proliferative capacities [18]. The cellular origin and their heterogeneity are insufficiently understood, but over the last decade several mechanisms have emerged that can supply myofibroblasts. Transdifferentiation of endothelial cells (endothelial-mesenchymal transition) is believed to give rise to myofibroblasts. De-differentiation of VSMC and their directed migration towards the intima is often proposed as the major pathway leading to intimal hyperplasia. Also, adventitial sources have been proposed, specifically, adventitial fibroblasts, mesenchymal stem cells and pericytes [30, 31]. Lineage tracing studies would need to be applied to answer this question, but contribution of several cellular origins is a likely scenario.
Precise molecular mechanisms underlying the disease-critical process of intimal hyperplasia in vasculitis are undefined. Older studies have suggested that intimal hyperplasia is a coordinated process with neoangiogenesis [32], a predictable element in supporting the outgrowth of matrix-producing myofibroblasts. As the major lumen of the artery occludes, oxygen and nutrients can no longer be supplied by diffusion and supporting microvascular networks need to be build. Mechanistic studies suggest that intimal hyperplasia is a T-cell–-dependent pathway [22]. Specifically, unleashing of T cell immunity through antibody-mediated checkpoint paralysis is sufficient to enhance intimal thickness and the diameter of the tunica intima doubled after anti-PD-1 treatment.
3. Heterogeneity of the vasculitogenic immune response
3.1. Disease-inducing T cell populations in GCA
Early studies in temporal arteries affected by GCA supported the disease model that the tissue-infiltrating T cells are selected for specificity, strongly supportive for antigen-driven wall inflammation [33]. Recent data have uncovered the role of antigen-nonspecific events as disease risk and amplification factors. The breakdown of physical barriers, the signals provided by the tissue microenvironment, activation threshold setting through co-stimulation and co-inhibition all appear to be critical in inducing and promoting vessel wall inflammation. Accordingly, functional analysis of wall-residing T cells has revealed tremendous heterogeneity, with the common denominator being heightened effector function. Analysis of lineage-defining T cell cytokines in inflamed arteries have provided evidence for a multitude of T cell populations, producing IL-2, IFN-γ, IL-17, IL-21, IL-9, and IL-22 [22, 34–37] (Fig. 2A). Quantitative approaches have demonstrated that IFN-γ–producing Th1 cells are the dominant population in the arteries and the circulation of GCA patients. IFN-γ is instrumental in stimulating dendritic cells, macrophages, endothelial cells, and stromal cells to produce cytokines, chemokines, growth factors and proteases and IFN-γ is now considered as one of the major drivers of autoimmunity [38, 39]. Commitment to IFN-γ production is an identifying characteristic of circulating CD4 T cells in GCA patients, possibly resulting from interaction with Jagged-1-expressing endothelial cells [17]. Binding of IFN-γ to its receptor triggers activation of the Janus kinase (JAK) and signal transducer and activator of transcription (STAT) signaling pathway [40, 41]. Notably, IFN-γ production by Th1 cells is also regulated by the JAK-STAT pathway [18]. Transcriptome analysis in GCA-affected arteries identified STAT1, STAT2 and the STAT target genes Tbet, CXCL9, ISG15 and OAS1 as strongly upregulated, indicative for active ongoing IFN-γ-dependent signaling in the tissue lesion [18]. Therapeutic targeting of JAK-STAT signaling with a small molecule inhibitor that targets JAK3/1 was highly successful in suppressing vasculitis [18], reinforcing the concept that JAK1/3-dependent cytokine signaling has mechanistic relevance in GCA.
Figure 2. Pathogenic Effector Cells in Giant Cell Arteritis.
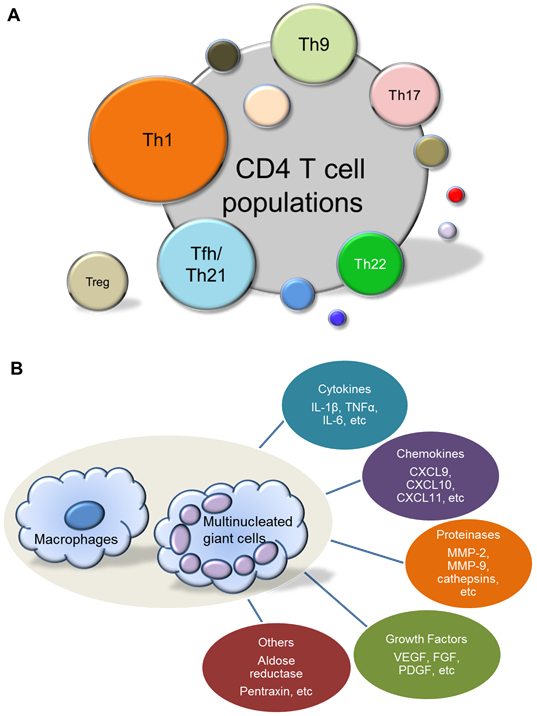
(A) Heterogeneity of wall-residing T effector cells in giant cell arteritis. The T cell infiltrate in the vessel wall is composed of multiple functional subsets. T helper 1 (Th1) cells are the dominant population and produce interferon-γ, activating macrophages, endothelial cells and vascular smooth muscle cells. IL-17 derived from Th17 cells acts upon endothelial and stromal cells. The marker cytokine of follicular helper T cells (Tfh) is IL-21, which promotes local T cell differentiation and amplifies tissue inflammation. Precise effector functions of IL-9–producing Th9 cells and IL-22–producing Th22 cells are unknown. Anti-inflammatory regulatory T cells (Treg) are distinctly infrequent in the vasculitic lesions. (B) Macrophage effector functions in giant cell arteritis. Lesional T cells produce regulatory molecules that drive macrophage activation and differentiation. Macrophages and multinucleated giant cells mediate tissue damage and contribute to maladaptive reparative responses, vessel wall remodeling, through the production of a large portfolio of effector molecules: cytokines, chemokines, proteinases, growth factors, etc. CXCL, C-X-C motif ligand; FGF, fibroblast growth factor; IL, interleukin; MMP, matrix metalloprotease; PDGF, platelet-derived growth factor; TNF-α, tumor necrosis factor α; VEGF, vascular endothelial growth factor.
Based on examination of tissue-resident T cells, IL-21–producing follicular helper T (Tfh) cells account for the second most prominent population [12]. IL-21 is known for its role in germinal centers, where it promotes B cell differentiation towards plasma cells and has a role in Tfh and Th17 cell differentiation [42–44]. B cells are distinctly rare in the vasculitic lesions and germinal center like structures have not been identified. IL-21 activates multiple cellular signaling cascades, including the JAK-STAT, MAPK, and PI3K/Akt pathway and is considered disease promoting in several autoimmune diseases [45]. Understanding IL-21’s role in the inflamed wall will require further investigations. IL-21 is an abundant cytokine expressed in the arterial lesions [12] and in the blood of GCA patients and appears to be sensitive to glucocorticoid treatment GCA [35].
Several smaller populations of committed T cells contribute to the vasculitic infiltrates (Fig. 2A). IL-17–producing T cells are represented in the blood and the arteries of GCA patients [46]. IL-17 is involved in the host defense against extracellular pathogens and contributes to inflammatory disease [47, 48]. IL-17 acts primarily upon epithelial, endothelial and stromal cells [49] and anti-IL-17 therapy is highly effective in psoriasis [50]. However, given the relatively low frequencies of Th17 cells in GCA and the high sensitivity of IL-17+ effector T cells to glucocorticoid-mediated immunosuppression [46], IL-17 might not be a major target for the effective treatment in GCA. IL-9, originally discovered as a T cell growth factor [51], is a pleiotropic cytokine regulating pro-inflammatory and anti-inflammatory processes [52, 53]. Comparison of non-inflamed and vasculitic temporal arteries by immunostaining has yielded a strong signal for IL-9 in the inflamed vessels [34]. However, which particular effector functions are perpetuated by IL-9 is currently unknown. Similarly, IL-22 production has been reported in GCA-affected arteries [37], yet functional studies linking this cytokine to disease mechanisms are lacking. IL-22 is closely linked to epithelial barrier function and augments IL-17 function [54, 55]. IL-22 is mainly produced by Th22 cells, a novel subset of helper T cells that secret IL-22 and TNF-α, but not IL-17, IFN-γ, and IL-4 [56]. In humans, IL-6 and TNF-α are the two major cytokines inducing Th22 differentiation, signified by the expression of the lineage-defining transcription factor aryl hydrocarbon receptor [57].
Due to the longevity of T cells, they are particularly susceptible to the aging process and it has been proposed that the almost exclusive risk of individuals over 50 years of age to develop GCA may reflect abnormalities in immune aging [58]. While available data are limited, evidence has been provided that GCA patients have a defect in anti-inflammatory CD8 T cells, rendering them susceptible to unopposed immune reactivity [59]. Defective CD8 Treg cells in GCA patients lose the ability to package NADPH oxidase into immunosuppressive exosomes; a defect even more pronounced in GCA patients than in healthy older individuals. CD8 T cells are known to age faster than CD4 T cells and a hallmark of immune aging is the loss of naïve CD8 T cells [60], suggesting acceleration of T cell aging in GCA with direct pathogenic relevance.
3.2. Disease-relevant effector macrophages in GCA (Fig. 2B)
Recent studies have connected monocytes to very early steps in the disease process. Specifically, MMP-9–producing monocytes are required to enable the transmigration of monocytes and T cells through the basal lamina [12]. Gene expression data evaluating the transcription of 8 metalloproteinases in monocytes and ex vivo differentiated macrophages established a “GCA-specific profile” of spontaneously upregulated transcription of MMP-2, MMP-7 and MMP-9. Disease specificity of this pattern was established by comparative studies in patients with a diagnosis of GCA versus Takayasu arteritis (Fig. 3). Equipped with the three major proteases, GCA monocytes have high potential to invade into tissue and move through extracellular matrix. Collagen and elastin fibrils are highly resistant to proteolysis, a resistance that can be broken by MMPs [61]. Given the elastolytic and gelatinolytic activities of MMP-9, this protease appears particularly important in the disease process of GCA, which requires the invasion into the arterial wall and the mobility of wall-infiltrating cells to reach the medial layer. Studies in temporal arteritis and in GCA aortitis assigned MMP-9 production exclusively to macrophages [12]. Considering the structural barriers in the vessel wall, specific blockade of MMP-9 essentially made the artery inaccessible and effectively prevented vasculitis [12]. Conversely, systemically delivered MMP-9 was sufficient to render arteries highly susceptible to infiltrating macrophages, T cells, inflammation and wall remodeling.
Figure 3. Macrophage Matrix metalloprotease production in large vessel vasculitides.
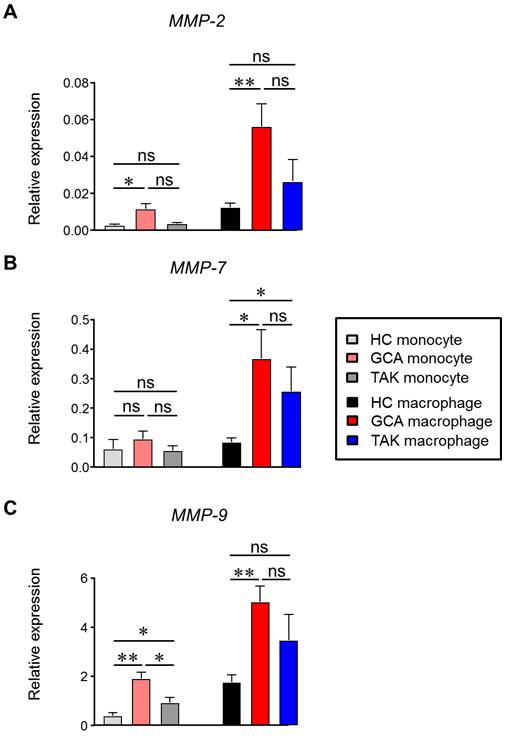
Peripheral blood mononuclear cells (PBMCs) were isolated from healthy individuals, patients with GCA and patients with TAK. Monocytes were purified and stimulated with interferon-γ (IFN-γ, 100 U/ml) and lipopolysaccharide (LPS, 100 ng/ml) for 24 hrs. Macrophages were differentiated from monocyte with macrophage-colony stimulating factor (M-CSF, 20 ng/ml)-contained RPMI 1640 medium for 5 days and then stimulated with IFN-γ and LPS for 24 hrs. Transcripts for MMP-2 (A), MMP-7 (B) and MMP-9 (C) were measured by RT-PCR. Primer sequences for MMPs were previously described [12]. HC, healthy control; GCA, giant cell arteritis; MMP, matrix metalloprotease; TAK, Takayasu arteritis. One-way ANOVA. ns, not significant. *P<0.05, **P<0.01.
Once embedded in the tissue, circulating monocytes respond to microenvironmental cues and differentiate into tissue macrophages. A shared feature of pathogenic macrophages and T cells is the involvement of many different effector functions (Fig. 2B). For macrophages even more than T cells, the disease lesions in patient-derived tissue sections demonstrate geographic distribution of select macrophage functions. Adventitial macrophages have been connected to cytokine production, although it may be difficult to distinguish dendritic cells and macrophages in the tissue. The tunica media is most impenetrable, requiring matrix digestion. Besides producing MMPs, medial macrophages have also been described to produce reactive oxygen species (ROS) [62], contribute to nitrosative stress [63] and upregulate the molecular machinery to deal with oxidative attack [64]. As an example, induction of aldose reductase may serve as a protective shield [64], permitting survival of macrophages in a very hostile environment.
The GCA-specific destruction pattern is a combination of tissue damage with a maladaptive repair response, including the mobilization of myofibroblasts that expand and deposit matrix to form the hyperplastic intima. Cellular growth depends on growth factors that promote proliferative activity in the vasculitic lesions and build neointima to remodel the wall. A critical growth factor seems to be platelet-derived growth factor (PDGF), supplied by macrophages and giant cells placed at the media-intima junction [65]. Multiple mesenchymal cell types, such as fibroblasts, VSMC and mesenchymal stem cells express PDGF receptors and respond with proliferation and directed migration to PDGF. Fibroblast growth factors (FGF), in particular FGF-2 may partner with PDGF to enhance healing responses to local injury and may contribute to maladaptive arterial remodeling [32].
A critical growth factor, both systemically and locally, is VEGF. Circulating VEGF primes adventitial endothelial cells to “open the door” for incoming T cells [17]. VEGF-producing macrophages and multinucleated giant cells are positioned in the proximal media, next to the fragmented lamina elastica [32], where they can create a local tissue gradient to support sprouting of new microvessels. A circumferential ring of capillaries typically forms in the distal neointima.
These data identify diverse populations of protease and growth factor-producing macrophages as a coordinated force in tissue injury and repair.
The most recent studies have attempted to provide insights into regulatory abnormalities that deviate monocytes and macrophages from tissue protection to excessive tissue digestion, wall destruction and inappropriate neotissue formation. Stimulation of myeloid cells with microbial and non-microbial stimuli can induce an inflammation-prone phenotype in a process called trained immunity. Both, metabolic and epigenetic reprogramming are core events in training monocytes to become inflammatory macrophages [66–68]. Intracellular metabolism distinguishes and supports discrete functional states and the question has been raised whether the metabolic machinery of patient-derived cells allows for distinction between “healthy” and vasculitogenic macrophages. These studies relied on comparing monocytes and macrophages from patients with GCA and patients with coronary artery disease (CAD), another condition manifesting with inflammation in the vessel wall [16]. Transcriptome analysis in monocytes/macrophages defined a metabolic profile of high glucose import, high glycolytic activity and high mitochondrial activity exclusively in CAD patients [16]. These data predict a prime state in CAD cells, but not in GCA cells and points towards fundamentally different activation conditions in these two inflammatory vasculopathies. Excess glucose uptake and utilization was linked to several functional domains, particularly cytokine production and expression of the co-inhibitory ligand PD-L1 [69, 70]. Macrophages differentiated from CAD patients consistently were high expressers for IL-6 and PD-L1, a function dependent on glucose availability in the microenvironment [16].
Taken together monocytes and macrophages isolated from GCA patients have an unprimed phenotype.
4. Extravascular GCA
Granulomatous lesions mimicking those formed in the vessel wall are extremely rare, if not non-existent in extravascular tissues. Instead, patients frequently present with constitutional symptoms, such as fever, malaise, anorexia, weight loss, night sweats and depression. Laboratory abnormalities include anemia and thrombocytosis, all manifestations of an acute phase response (APR). Acute phase proteins (APP) are typically abundant and easily detectable in serum [71, 72]. Trauma, infection, neoplasia, inflammation and tissue necrosis trigger a rapid early-response program meant to protect, defend and heal. The core process of this acute phase response is the production of APPs by hepatocytes. The most prominent APP is C-reactive protein, first described in the early 1930s, which was discovered in the “acute phase” of pneumococcal infection in monkeys and humans [73]. CRP can be easily measured and is considered an informative marker in malignancy, infection, trauma and tissue breakdown. The erythrocyte sedimentation rate (ESR) is strongly dependent on the APR, and changes in ESR and CRP are often synchronized. The APR is a complex program characterized by the rapid increase of many and the decline of few serum proteins [74, 75]. Known APPs are listed in Table 1, with the most prominent members being CRP, serum amyloid A, fibrinogen and pentraxin 3. APPs are consistently elevated in GCA and many have been studied as possible disease amplifiers [76, 77]. CRP induces downstream targets, most prominently IL-6 [78]. About 50% of GCA patients develop a syndrome of muscle pain and stiffness, named polymyalgia rheumatica (PMR) [79]. PMR symptoms are closely related to excess APPs.
Table 1.
Acute Phase Proteins in Giant Cell Arteritis
| Increased | Decreased |
|---|---|
| C-reactive protein (CRP) | Serum albumin |
| Serum amyloid A (SAA) | Transferrin |
| a1-antitrypsin (Serpin) | Transthyretin |
| a2-macroglobulin | |
| Complement (C3, CD4) | |
| Ferritin | |
| Fibrinogen | |
| Haptoglobin | |
| Hepcidin | |
| Pentraxin 3 |
Most untreated GCA patients have highly elevated ESR, CRP and IL-6 levels. The acute phase response is exquisitely sensitive to glucocorticoids and thus APPs decline rapidly when patients are treated with corticosteroid therapy [8]. Of all innate cytokines involved in the acute phase response, blocking IL-6 should be particularly effective in reducing APPs.
5. Therapeutic targeting of vascular GCA
In almost all patients, glucocorticoids are highly effective in suppressing extravascular GCA [8, 80]. Glucocorticoids eliminate vascular inflammation in about 50% of patients treated for one year [81]. To assess the remission-inducing potential of glucocorticoid therapy, 40 patients with a positive temporal artery biopsy received standard doses of prednisone and were re-biopsied on the collateral side after 3, 6, 9 or 12 months [81]. Even patients with a positive second biopsy responded clinically and had normal laboratory findings, emphasizing that vascular and extravascular GCA follow distinct trajectories.
Currently, little information is available on whether steroid-sparing agents can successfully suppress vessel wall inflammation. Tocilizumab, a recently approved antibody that blocks the IL-6 receptor, is highly effective in suppressing ESR and CRP [82], but it is not known whether diminishing IL-6 signaling has beneficial effects on the arterial wall inflammatory lesion. Data from patients with TAK question whether IL-6 is an important contributor to vascular disease. In a randomized, double-blind, placebo-controlled trial testing the efficacy of tocilizumab in refractory TAK the primary endpoint of time to relapse was not met [83]. And disease progression has been described in TAK patients on tocilizumab therapy [84, 85].
As disease mechanisms in the vessel wall lesions are further elucidated, it may be possible to design therapeutic strategies that exploit key events in vasculitis. A major hurdle is the autonomy of the lesion, with repopulation of inflammatory cells from tissue-resident populations. Also, niche effects provided by the affected arteries, creating ideal conditions for T cells and macrophages to survive and function in a non-lymphoid tissue site complicate simplified anti-proliferative and anti-inflammatory approaches.
5.1. JAK-STAT pathway inhibition
Transcriptome analysis in inflamed temporal arteries has identified STAT signaling target genes, with a bias towards IFN type I and type II-dependent responses. Treatment of human arteries -SCID chimeras with tofacitinib, a selective JAK1 and JAK3 inhibitor effectively suppressed vasculitis, including expansion of the intima and wall capillarization [18].
5.2. Blocking T cell co-stimulation and enhancing T cell co-inhibition
T cells sustaining vasculitis require co-stimulatory signals and are highly sensitive to anti-CD28 antibody treatment [25]. Specifically, the metabolic fitness of lesional T cells, characterized by strong upregulation of glycolytic activity, depended on CD28-derived co-stimulation. Curbing fuel consumption of pathogenic T cells would be a novel approach in suppressing wall inflammation.
Conversely, the PD-1/PD-L1 inhibitory checkpoint is defective in GCA [22]. While cancer immunotherapy attempts to unleash T cell immunity by blocking the pathway, it is conceivable that agonist anti-PD-1 antibodies could supply re-introduce negative signaling. Alternatively, soluble PD-L1, e.g. fused to an Fc domain, could be applied to activate PD-1-dependent inhibitory signaling.
5.3. Blocking activity of metalloproteinases
Invasion into the vessel wall, particularly into the elastin-rich tunica media, is only possible after digestion of matrix and elastic laminae. MMP-9–producing monocytes and macrophages are particularly abundant in GCA, are present in the circulation and occupy the inflamed medial layer [11, 12]. Preclinical studies in human artery-SCID chimeras have identified two critical steps in the disease process that are MMP-9 dependent: breakdown of the basal lamina to permit penetration of inflammatory cells form the circulation into the tissue; and destruction of elastic barriers in the media. Antibody-mediated blockade of MMP-9 enzyme activity inhibited the accumulation of T cells in the arterial wall and suppressed wall remodeling [12]. This approach may avoid non-specific immunosuppression as MMP-9 has no role in immune cell production.
5.4. Targeting the VEGF-Notch pathway
Besides the acute phase proteins, VEGF is also highly elevated in the sera of GCA patients and this growth factor has been directly implicated in pathogenic events [17, 20]. Most significantly, VEGF mediates the activation of adventitial microvessels, opening the door to “forbidden tissue”. Anti-VEGF therapy is widely applied in cancer therapy and has become a routine treatment to arrest aberrant neovascularization in ocular structures [86, 87]. Since work described above has identified ligands within the NOTCH pathway as downstream targets of VEGF signaling, there is conceptual support to explore interference with the NOTCH pathway. This pathway too is implicated in driving uncontrolled growth of malignant cells [88, 89] and efforts are underway to develop clinically applicable reagents that can prevent NOTCH pathway activation.
5.5. Inhibiting the mTOR pathway
Several lines of evidence point towards mTOR as an integrator of disease-inducing effector functions in GCA. Active signaling through the mTOR pathway is a hallmark of vasculitogenic T cells [17, 90]. Such T cells are metabolically reprogrammed, offering an alternative target for therapeutic interference. mTOR is a central nutrient sensor in the cell, which coordinates energy needs with cellular proliferative activity [91–93]. mTOR inhibitors can thus disrupt the fueling of pathogenic effector functions by curbing energy supply [90].
5.6. Other potential therapies
Pilot studies in patients with GCA and Takayasu arteritis have shown that some patients may benefit from targeting p40, a shared molecule in the IL-12 and IL23 signaling pathway [94–96]. Rationale for this approach stems from the critical role of IL-12 in polarizing Th1 cell lineage commitment and a role of IL-23 in promoting Th17 cell development.
6. Conclusions
Pathogenesis-oriented studies have yielded important insights into disease-inducing and disease-sustaining pathways in large vessel vasculitis. Key concepts include a separation of vascular and extravascular GCA, recognition that the vessel wall has unique structural and functional features that make it “forbidden territory” for routine immune responses, autonomy of established vasculitis lesions and the unexpected heterogeneity of pathogenic effector cells. Extravascular manifestations of GCA, mostly an intense acute phase response, constitutional symptoms and proximal myalgias, have previously been considered to be upstream of vasculitis. In an updated disease model the APR is conceptualized as consequence and not a cause of vasculitis; calling into question the current practice of relying on ESR and CRP monitoring to diagnose and manage vasculitis. ESR and CRP are easily measured but they should not replace a tissue biopsy nor can normalization of downstream laboratory findings be considered a hallmark of therapeutic success. As demonstrated in a recent study of patients that were biopsied before and 3–12 months after initiation of therapy [81], vascular inflammation persists in most patients, despite normal CRP and ESR results. The lack of reliable biomarkers capturing the load of vascular inflammation is challenging for practicing physicians, particularly in patients without a large vessel component that can be assessed by imaging.
Deeper understanding of how inflammatory cells enter the vessel wall, how they survive and flourish and how they mediate tissue injury has emphasized that the wall of medium and large arteries is primarily unaccessible and self-protective. In patients, this natural protection breaks down; facilitated by MMP-9–producing monocytes that digest the basal lamina and the failure of wall-residing dendritic cells to express the immune-inhibitory ligand PD-L1. Established vasculitic lesions are self-sustained for the production of tissue-resident T cells and thus can persist for extended periods. Macrophages are end-differentiated cells and their precursors may have to be continuously recruited from the circulation. Therapeutic targeting of tissue-resident memory T cells will need to be a requirement of successful patient management. Injury-inducing effector cells, both T cells as well as macrophages, have a high degree of heterogeneity, strongly suggestive for abnormalities in antigen-nonspecific pathways. A broad spectrum of cytokines, enzymes and growth factors actively participate in wall damage and wall remodeling, providing a rich array of potential therapeutic targets.
Table 2.
Current and Emerging Therapies in Large Vessel Vasculitis
| A. Currently used therapies | ||
|---|---|---|
| Vascular GCA | Extravascular GCA | |
| Glucocorticoids | ++ | +++ |
| Methotrexate | ? | ? |
| Leflunomide | ? | ? |
| Cyclophosphamide | ? | ? |
| Azathioprine | ? | ? |
| Anti-TNF inhibitor | ? | ? |
| Anti-IL-6 therapy | ? | ++ |
| B. Emerging therapies | ||
| Target | Function | Example |
| JAK-STAT pathway | Cytokine responses | Tofacitinib Baricitinib |
| T cell co-stimulation | Adaptive immunity | Abatacept Anti-CD28 |
| T cell co-inhibition | Adaptive immunity | PD-L1 Fc |
| IL-12/IL-23 pathway | T cell differentiation | Ustekinumab |
| Proteases | Matrix degradation | Blocking MMP-9 |
| VEGF | Angiogenesis | Bevacizumab |
| mTOR pathway | Cellular nutrient sensor | Rapamycin |
Acknowledgments
This work was supported by the National Institutes of Health (R01 AR042547, R01 HL117913, R01 AI108906, R01 HL142068 and P01 HL129941 to CMW and R01 AI108891, R01 AG045779, U19 AI057266, R01 AI129191, and I01 BX001669 to JJG). RW received fellowship support from the Cahill Discovery Fund.
Footnotes
Conflict of interest statement
The authors have declared that no conflict of interest exists.
References
- [1].Belz GG, Elastic properties and Windkessel function of the human aorta, Cardiovasc Drugs Ther. 9 (1) (1995) 73–83. [DOI] [PubMed] [Google Scholar]
- [2].Westerhof N, Lankhaar JW, Westerhof BE, The arterial Windkessel, Med Biol Eng Comput. 47 (2) (2009) 131–141. [DOI] [PubMed] [Google Scholar]
- [3].Mulligan-Kehoe MJ, The vasa vasorum in diseased and nondiseased arteries, Am J Physiol Heart Circ Physiol. 298 (2) (2010) H295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Mulligan-Kehoe MJ, Simons M, Vasa vasorum in normal and diseased arteries, Circulation. 129 (24) (2014) 2557–2566. [DOI] [PubMed] [Google Scholar]
- [5].Galea I, Bechmann I, Perry VH, What is immune privilege (not)?, Trends Immunol. 28 (1) (2007) 12–18. [DOI] [PubMed] [Google Scholar]
- [6].Tellides G, Pober JS, Inflammatory and immune responses in the arterial media, Circ Res. 116 (2) (2015) 312–322. [DOI] [PubMed] [Google Scholar]
- [7].Weyand CM, Goronzy JJ, Medium- and large-vessel vasculitis, N Engl J Med. 349 (2) (2003) 160–169. [DOI] [PubMed] [Google Scholar]
- [8].Weyand CM, Goronzy JJ, Clinical practice. Giant-cell arteritis and polymyalgia rheumatica, N Engl J Med. 371 (1) (2014) 50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, Flores-Suarez LF, Gross WL, Guillevin L, Hagen EC, Hoffman GS, Jayne DR, Kallenberg CG, Lamprecht P, Langford CA, Luqmani RA, Mahr AD, Matteson EL, Merkel PA, Ozen S, Pusey CD, Rasmussen N, Rees AJ, Scott DG, Specks U, Stone JH, Takahashi K, Watts RA, 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides, Arthritis Rheum. 65 (1) (2013) 1–11. [DOI] [PubMed] [Google Scholar]
- [10].Weyand CM, Goronzy JJ, Immune mechanisms in medium and large-vessel vasculitis, Nat Rev Rheumatol. 9 (12) (2013) 731–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rodriguez-Pla A, Bosch-Gil JA, Rossello-Urgell J, Huguet-Redecilla P, Stone JH, Vilardell-Tarres M, Metalloproteinase-2 and −9 in giant cell arteritis: involvement in vascular remodeling, Circulation. 112 (2) (2005) 264–269. [DOI] [PubMed] [Google Scholar]
- [12].Watanabe R, Maeda T, Zhang H, Berry GJ, Zeisbrich M, Brockett R, Greenstein AE, Tian L, Goronzy JJ, Weyand CM, MMP (Matrix Metalloprotease)-9-Producing Monocytes Enable T Cells to Invade the Vessel Wall and Cause Vasculitis, Circ Res. 123 (6) (2018) 700–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Buttgereit F, Dejaco C, Matteson EL, Dasgupta B, Polymyalgia Rheumatica and Giant Cell Arteritis: A Systematic Review, JAMA. 315 (22) (2016) 2442–2458. [DOI] [PubMed] [Google Scholar]
- [14].Numano F, Okawara M, Inomata H, Kobayashi Y, Takayasu’s arteritis, Lancet. 356 (9234) (2000) 1023–1025. [DOI] [PubMed] [Google Scholar]
- [15].Yoshida M, Watanabe R, Ishii T, Machiyama T, Akita K, Fujita Y, Shirota Y, Sugimura K, Fujii H, Shimokawa H, Harigae H, Retrospective analysis of 95 patients with large vessel vasculitis: a single center experience, Int J Rheum Dis. 19 (1) (2016) 87–94. [DOI] [PubMed] [Google Scholar]
- [16].Watanabe R, Hilhorst M, Zhang H, Zeisbrich M, Berry GJ, Wallis BB, Harrison DG, Giacomini JC, Goronzy JJ, Weyand CM, Glucose metabolism controls disease-specific signatures of macrophage effector functions, JCI Insight. 3 (20) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wen Z, Shen Y, Berry G, Shahram F, Li Y, Watanabe R, Liao YJ, Goronzy JJ, Weyand CM, The microvascular niche instructs T cells in large vessel vasculitis via the VEGF-Jagged1-Notch pathway, Sci Transl Med. 9 (399) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Zhang H, Watanabe R, Berry GJ, Tian L, Goronzy JJ, Weyand CM, Inhibition of JAK-STAT Signaling Suppresses Pathogenic Immune Responses in Medium and Large Vessel Vasculitis, Circulation. 137 (18) (2018) 1934–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Piggott K, Deng J, Warrington K, Younge B, Kubo JT, Desai M, Goronzy JJ, Weyand CM, Blocking the NOTCH pathway inhibits vascular inflammation in large-vessel vasculitis, Circulation. 123 (3) (2011) 309–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Baldini M, Maugeri N, Ramirez GA, Giacomassi C, Castiglioni A, Prieto-Gonzalez S, Corbera-Bellalta M, Di Comite G, Papa I, Dell’antonio G, Ammirati E, Cuccovillo I, Vecchio V, Mantovani A, Rovere-Querini P, Sabbadini MG, Cid MC, Manfredi AA, Selective up-regulation of the soluble pattern-recognition receptor pentraxin 3 and of vascular endothelial growth factor in giant cell arteritis: relevance for recent optic nerve ischemia, Arthritis Rheum. 64 (3) (2012) 854–865. [DOI] [PubMed] [Google Scholar]
- [21].Ma-Krupa W, Jeon MS, Spoerl S, Tedder TF, Goronzy JJ, Weyand CM, Activation of arterial wall dendritic cells and breakdown of self-tolerance in giant cell arteritis, J Exp Med. 199 (2) (2004) 173–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhang H, Watanabe R, Berry GJ, Vaglio A, Liao YJ, Warrington KJ, Goronzy JJ, Weyand CM, Immunoinhibitory checkpoint deficiency in medium and large vessel vasculitis, Proc Natl Acad Sci U S A. 114 (6) (2017) E970–E979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sun C, Mezzadra R, Schumacher TN, Regulation and Function of the PD-L1 Checkpoint, Immunity. 48 (3) (2018) 434–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Watanabe R, Zhang H, Berry G, Goronzy JJ, Weyand CM, Immune checkpoint dysfunction in large and medium vessel vasculitis, Am J Physiol Heart Circ Physiol. 312 (5) (2017) H1052–H1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Zhang H, Watanabe R, Berry GJ, Nadler SG, Goronzy JJ, Weyand CM, CD28 signaling controls metabolic fitness of pathogenic T cells in medium and large vessel vasculitis J Am Coll Cardiol. In press (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Duca L, Floquet N, Alix AJ, Haye B, Debelle L, Elastin as a matrikine, Crit Rev Oncol Hematol. 49 (3) (2004) 235–244. [DOI] [PubMed] [Google Scholar]
- [27].Grahovac J, Wells A, Matrikine and matricellular regulators of EGF receptor signaling on cancer cell migration and invasion, Lab Invest. 94 (1) (2014) 31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Sofeu Feugaing DD, Gotte M, Viola M, More than matrix: the multifaceted role of decorin in cancer, Eur J Cell Biol. 92 (1) (2013) 1–11. [DOI] [PubMed] [Google Scholar]
- [29].Wells A, Nuschke A, Yates CC, Skin tissue repair: Matrix microenvironmental influences, Matrix Biol. 49 (2016) 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Falke LL, Gholizadeh S, Goldschmeding R, Kok RJ, Nguyen TQ, Diverse origins of the myofibroblast-implications for kidney fibrosis, Nat Rev Nephrol. 11 (4) (2015) 233–244. [DOI] [PubMed] [Google Scholar]
- [31].Wang N, Deng Y, Liu A, Shen N, Wang W, Du X, Tang Q, Li S, Odeh Z, Wu T, Lin H, Novel Mechanism of the Pericyte-Myofibroblast Transition in Renal Interstitial Fibrosis: Core Fucosylation Regulation, Sci Rep. 7 (1) (2017) 16914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kaiser M, Younge B, Bjornsson J, Goronzy JJ, Weyand CM, Formation of new vasa vasorum in vasculitis. Production of angiogenic cytokines by multinucleated giant cells, Am J Pathol. 155 (3) (1999) 765–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Weyand CM, Schonberger J, Oppitz U, Hunder NN, Hicok KC, Goronzy JJ, Distinct vascular lesions in giant cell arteritis share identical T cell clonotypes, J Exp Med. 179 (3) (1994) 951–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ciccia F, Rizzo A, Guggino G, Cavazza A, Alessandro R, Maugeri R, Cannizzaro A, Boiardi L, Iacopino DG, Salvarani C, Triolo G, Difference in the expression of IL-9 and IL-17 correlates with different histological pattern of vascular wall injury in giant cell arteritis, Rheumatology (Oxford). 54 (9) (2015) 1596–1604. [DOI] [PubMed] [Google Scholar]
- [35].Terrier B, Geri G, Chaara W, Allenbach Y, Rosenzwajg M, Costedoat-Chalumeau N, Fouret P, Musset L, Benveniste O, Six A, Klatzmann D, Saadoun D, Cacoub P, Interleukin-21 modulates Th1 and Th17 responses in giant cell arteritis, Arthritis Rheum. 64 (6) (2012) 2001–2011. [DOI] [PubMed] [Google Scholar]
- [36].Watanabe R, Hosgur E, Zhang H, Wen Z, Berry G, Goronzy JJ, Weyand CM, Pro-inflammatory and anti-inflammatory T cells in giant cell arteritis, Joint Bone Spine. 84 (4) (2017) 421–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zerbini A, Muratore F, Boiardi L, Ciccia F, Bonacini M, Belloni L, Cavazza A, Cimino L, Moramarco A, Alessandro R, Rizzo A, Parmeggiani M, Salvarani C, Croci S, Increased expression of interleukin-22 in patients with giant cell arteritis, Rheumatology (Oxford). 57 (1) (2018) 64–72. [DOI] [PubMed] [Google Scholar]
- [38].Green DS, Young HA, Valencia JC, Current prospects of type II interferon gamma signaling and autoimmunity, J Biol Chem. 292 (34) (2017) 13925–13933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Lees JR, Interferon gamma in autoimmunity: A complicated player on a complex stage, Cytokine. 74 (1) (2015) 18–26. [DOI] [PubMed] [Google Scholar]
- [40].O’Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A, The JAK-STAT pathway: impact on human disease and therapeutic intervention, Annu Rev Med. 66 (2015) 311–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Schwartz DM, Bonelli M, Gadina M, O’Shea JJ, Type I/II cytokines, JAKs, and new strategies for treating autoimmune diseases, Nat Rev Rheumatol. 12 (1) (2016) 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Ettinger R, Sims GP, Fairhurst AM, Robbins R, da Silva YS, Spolski R, Leonard WJ, Lipsky PE, IL-21 induces differentiation of human naive and memory B cells into antibody-secreting plasma cells, J Immunol. 175 (12) (2005) 7867–7879. [DOI] [PubMed] [Google Scholar]
- [43].Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK, IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells, Nature. 448 (7152) (2007) 484–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Vogelzang A, McGuire HM, Yu D, Sprent J, Mackay CR, King C, A fundamental role for interleukin-21 in the generation of T follicular helper cells, Immunity. 29 (1) (2008) 127–137. [DOI] [PubMed] [Google Scholar]
- [45].Dinesh P, Rasool M, Multifaceted role of IL-21 in rheumatoid arthritis: Current understanding and future perspectives, J Cell Physiol. 233 (5) (2018) 3918–3928. [DOI] [PubMed] [Google Scholar]
- [46].Deng J, Younge BR, Olshen RA, Goronzy JJ, Weyand CM, Th17 and Th1 T-cell responses in giant cell arteritis, Circulation. 121 (7) (2010) 906–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Amatya N, Garg AV, Gaffen SL, IL-17 Signaling: The Yin and the Yang, Trends Immunol. 38 (5) (2017) 310–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Iwakura Y, Ishigame H, Saijo S, Nakae S, Functional specialization of interleukin-17 family members, Immunity. 34 (2) (2011) 149–162. [DOI] [PubMed] [Google Scholar]
- [49].Gaffen SL, Recent advances in the IL-17 cytokine family, Curr Opin Immunol. 23 (5) (2011) 613–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Langley RG, Elewski BE, Lebwohl M, Reich K, Griffiths CE, Papp K, Puig L, Nakagawa H, Spelman L, Sigurgeirsson B, Rivas E, Tsai TF, Wasel N, Tyring S, Salko T, Hampele I, Notter M, Karpov A, Helou S, Papavassilis C, E.S. Group, F.S. Group, Secukinumab in plaque psoriasis--results of two phase 3 trials, N Engl J Med. 371 (4) (2014) 326–338. [DOI] [PubMed] [Google Scholar]
- [51].Uyttenhove C, Simpson RJ, Van Snick J, Functional and structural characterization of P40, a mouse glycoprotein with T-cell growth factor activity, Proc Natl Acad Sci U S A. 85 (18) (1988) 6934–6938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kaplan MH, Th9 cells: differentiation and disease, Immunol Rev. 252 (1) (2013) 104–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Rauber S, Luber M, Weber S, Maul L, Soare A, Wohlfahrt T, Lin NY, Dietel K, Bozec A, Herrmann M, Kaplan MH, Weigmann B, Zaiss MM, Fearon U, Veale DJ, Canete JD, Distler O, Rivellese F, Pitzalis C, Neurath MF, McKenzie ANJ, Wirtz S, Schett G, Distler JHW, Ramming A, Resolution of inflammation by interleukin-9-producing type 2 innate lymphoid cells, Nat Med. 23 (8) (2017) 938–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Duhen T, Geiger R, Jarrossay D, Lanzavecchia A, Sallusto F, Production of interleukin 22 but not interleukin 17 by a subset of human skin-homing memory T cells, Nat Immunol. 10 (8) (2009) 857–863. [DOI] [PubMed] [Google Scholar]
- [55].Eyerich K, Dimartino V, Cavani A, IL-17 and IL-22 in immunity: Driving protection and pathology, Eur J Immunol. 47 (4) (2017) 607–614. [DOI] [PubMed] [Google Scholar]
- [56].Eyerich S, Eyerich K, Pennino D, Carbone T, Nasorri F, Pallotta S, Cianfarani F, Odorisio T, Traidl-Hoffmann C, Behrendt H, Durham SR, Schmidt-Weber CB, Cavani A, Th22 cells represent a distinct human T cell subset involved in epidermal immunity and remodeling, J Clin Invest. 119 (12) (2009) 3573–3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Ramirez JM, Brembilla NC, Sorg O, Chicheportiche R, Matthes T, Dayer JM, Saurat JH, Roosnek E, Chizzolini C, Activation of the aryl hydrocarbon receptor reveals distinct requirements for IL-22 and IL-17 production by human T helper cells, Eur J Immunol. 40 (9) (2010) 2450–2459. [DOI] [PubMed] [Google Scholar]
- [58].Goronzy JJ, Weyand CM, Successful and Maladaptive T Cell Aging, Immunity. 46 (3) (2017) 364–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wen Z, Shimojima Y, Shirai T, Li Y, Ju J, Yang Z, Tian L, Goronzy JJ, Weyand CM, NADPH oxidase deficiency underlies dysfunction of aged CD8+ Tregs, J Clin Invest. 126 (5) (2016) 1953–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Moskowitz DM, Zhang DW, Hu B, Le Saux S, Yanes RE, Ye Z, Buenrostro JD, Weyand CM, Greenleaf WJ, Goronzy JJ, Epigenomics of human CD8 T cell differentiation and aging, Sci Immunol. 2 (8) (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Van Doren SR, Matrix metalloproteinase interactions with collagen and elastin, Matrix Biol. 44–46 (2015) 224–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Rittner HL, Kaiser M, Brack A, Szweda LI, Goronzy JJ, Weyand CM, Tissue-destructive macrophages in giant cell arteritis, Circ Res. 84 (9) (1999) 1050–1058. [DOI] [PubMed] [Google Scholar]
- [63].Borkowski A, Younge BR, Szweda L, Mock B, Bjornsson J, Moeller K, Goronzy JJ, Weyand CM, Reactive nitrogen intermediates in giant cell arteritis: selective nitration of neocapillaries, Am J Pathol. 161 (1) (2002) 115–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Rittner HL, Hafner V, Klimiuk PA, Szweda LI, Goronzy JJ, Weyand CM, Aldose reductase functions as a detoxification system for lipid peroxidation products in vasculitis, J Clin Invest. 103 (7) (1999) 1007–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Kaiser M, Weyand CM, Bjornsson J, Goronzy JJ, Platelet-derived growth factor, intimal hyperplasia, and ischemic complications in giant cell arteritis, Arthritis Rheum. 41 (4) (1998) 623–633. [DOI] [PubMed] [Google Scholar]
- [66].Leentjens J, Bekkering S, Joosten LAB, Netea MG, Burgner DP, Riksen NP, Trained Innate Immunity as a Novel Mechanism Linking Infection and the Development of Atherosclerosis, Circ Res. 122 (5) (2018) 664–669. [DOI] [PubMed] [Google Scholar]
- [67].Quintin J, Cheng SC, van der Meer JW, Netea MG, Innate immune memory: towards a better understanding of host defense mechanisms, Curr Opin Immunol. 29 (2014) 1–7. [DOI] [PubMed] [Google Scholar]
- [68].Rivera A, Siracusa MC, Yap GS, Gause WC, Innate cell communication kick-starts pathogen-specific immunity, Nat Immunol. 17 (4) (2016) 356–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Shirai T, Nazarewicz RR, Wallis BB, Yanes RE, Watanabe R, Hilhorst M, Tian L, Harrison DG, Giacomini JC, Assimes TL, Goronzy JJ, Weyand CM, The glycolytic enzyme PKM2 bridges metabolic and inflammatory dysfunction in coronary artery disease, J Exp Med. 213 (3) (2016) 337–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Watanabe R, Shirai T, Namkoong H, Zhang H, Berry GJ, Wallis BB, Schaefgen B, Harrison DG, Tremmel JA, Giacomini JC, Goronzy JJ, Weyand CM, Pyruvate controls the checkpoint inhibitor PD-L1 and suppresses T cell immunity, J Clin Invest. 127 (7) (2017) 2725–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Goronzy JJ, Weyand CM, Cytokines in giant-cell arteritis, Cleve Clin J Med. 69 Suppl 2 (2002) SII91–94. [DOI] [PubMed] [Google Scholar]
- [72].Hazleman B, Laboratory investigations useful in the evaluation of polymyalgia rheumatica (PMR) and giant cell arteritis (GCA), Clin Exp Rheumatol. 18 (4 Suppl 20) (2000) S29–31. [PubMed] [Google Scholar]
- [73].Pepys MB, Baltz ML, Acute phase proteins with special reference to C-reactive protein and related proteins (pentaxins) and serum amyloid A protein, Adv Immunol. 34 (1983) 141–212. [DOI] [PubMed] [Google Scholar]
- [74].Ahmed MS, Jadhav AB, Hassan A, Meng QH, Acute phase reactants as novel predictors of cardiovascular disease, ISRN Inflamm. 2012 (2012) 953461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.[] Gabay C, Kushner I, Acute-phase proteins and other systemic responses to inflammation, N Engl J Med. 340 (6) (1999) 448–454. [DOI] [PubMed] [Google Scholar]
- [76].O’Neill L, Rooney P, Molloy D, Connolly M, McCormick J, McCarthy G, Veale DJ, Murphy CC, Fearon U, Molloy E, Regulation of Inflammation and Angiogenesis in Giant Cell Arteritis by Acute-Phase Serum Amyloid A, Arthritis Rheumatol. 67 (9) (2015) 2447–2456. [DOI] [PubMed] [Google Scholar]
- [77].Roche NE, Fulbright JW, Wagner AD, Hunder GG, Goronzy JJ, Weyand CM, Correlation of interleukin-6 production and disease activity in polymyalgia rheumatica and giant cell arteritis, Arthritis Rheum. 36 (9) (1993) 1286–1294. [DOI] [PubMed] [Google Scholar]
- [78].Ballou SP, Lozanski G, Induction of inflammatory cytokine release from cultured human monocytes by C-reactive protein, Cytokine. 4 (5) (1992) 361–368. [DOI] [PubMed] [Google Scholar]
- [79].Kermani TA, Warrington KJ, Polymyalgia rheumatica, Lancet. 381 (9860) (2013) 63–72. [DOI] [PubMed] [Google Scholar]
- [80].Salvarani C, Hatemi G, Management of large-vessel vasculitis, Curr Opin Rheumatol. 31 (1) (2019) 25–31. [DOI] [PubMed] [Google Scholar]
- [81].Maleszewski JJ, Younge BR, Fritzlen JT, Hunder GG, Goronzy JJ, Warrington KJ, Weyand CM, Clinical and pathological evolution of giant cell arteritis: a prospective study of follow-up temporal artery biopsies in 40 treated patients, Mod Pathol. 30 (6) (2017) 788–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Stone JH, Tuckwell K, Dimonaco S, Klearman M, Aringer M, Blockmans D, Brouwer E, Cid MC, Dasgupta B, Rech J, Salvarani C, Schett G, Schulze-Koops H, Spiera R, Unizony SH, Collinson N, Trial of Tocilizumab in Giant-Cell Arteritis, N Engl J Med. 377 (4) (2017) 317–328. [DOI] [PubMed] [Google Scholar]
- [83].Nakaoka Y, Isobe M, Takei S, Tanaka Y, Ishii T, Yokota S, Nomura A, Yoshida S, Nishimoto N, Efficacy and safety of tocilizumab in patients with refractory Takayasu arteritis: results from a randomised, double-blind, placebo-controlled, phase 3 trial in Japan (the TAKT study), Ann Rheum Dis. 77 (3) (2018) 348–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Czihal M, Lottspeich C, Schrottle A, Treitl KM, Treitl M, Leipe J, Schulze-Koops H, Hoffmann U, Dechant C, Relapses in three patients with Takayasu arteritis under tocilizumab treatment detected by contrast enhanced ultrasound, Vasa. 47 (2) (2018) 149–152. [DOI] [PubMed] [Google Scholar]
- [85].Sanchez-Alvarez C, Koster M, Duarte-Garcia A, Warrington KJ, Disease progression of Takayasu arteritis in two patients treated with tocilizumab, Ann Rheum Dis. (2018). [DOI] [PubMed] [Google Scholar]
- [86].Loupakis F, Cremolini C, Masi G, Lonardi S, Zagonel V, Salvatore L, Cortesi E, Tomasello G, Ronzoni M, Spadi R, Zaniboni A, Tonini G, Buonadonna A, Amoroso D, Chiara S, Carlomagno C, Boni C, Allegrini G, Boni L, Falcone A, Initial therapy with FOLFOXIRI and bevacizumab for metastatic colorectal cancer, N Engl J Med. 371 (17) (2014) 1609–1618. [DOI] [PubMed] [Google Scholar]
- [87].Yang JC, Haworth L, Sherry RM, Hwu P, Schwartzentruber DJ, Topalian SL, Steinberg SM, Chen HX, Rosenberg SA, A randomized trial of bevacizumab, an anti-vascular endothelial growth factor antibody, for metastatic renal cancer, N Engl J Med. 349 (5) (2003) 427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Chiang MY, Radojcic V, Maillard I, Oncogenic Notch signaling in T-cell and B-cell lymphoproliferative disorders, Curr Opin Hematol. 23 (4) (2016) 362–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Sanchez-Martin M, Ferrando A, The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia, Blood. 129 (9) (2017) 1124–1133. [DOI] [PubMed] [Google Scholar]
- [90].Maciejewski-Duval A, Comarmond C, Leroyer A, Zaidan M, Le Joncour A, Desbois AC, Fouret JP, Koskas F, Cluzel P, Garrido M, Cacoub P, Saadoun D, mTOR pathway activation in large vessel vasculitis, J Autoimmun. 94 (2018) 99–109. [DOI] [PubMed] [Google Scholar]
- [91].Hardie DG, AMPK--sensing energy while talking to other signaling pathways, Cell Metab. 20 (6) (2014) 939–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Kim J, Guan KL, mTOR as a central hub of nutrient signalling and cell growth, Nat Cell Biol. 21 (1) (2019) 63–71. [DOI] [PubMed] [Google Scholar]
- [93].Mossmann D, Park S, Hall MN, mTOR signalling and cellular metabolism are mutual determinants in cancer, Nat Rev Cancer. 18 (12) (2018) 744–757. [DOI] [PubMed] [Google Scholar]
- [94].Conway R, O’Neill L, Gallagher P, McCarthy GM, Murphy CC, Veale DJ, Fearon U, Molloy ES, Ustekinumab for refractory giant cell arteritis: A prospective 52-week trial, Semin Arthritis Rheum. (2018). [DOI] [PubMed] [Google Scholar]
- [95].Conway R, O’Neill L, McCarthy GM, Murphy CC, Fabre A, Kennedy S, Veale DJ, Wade SM, Fearon U, Molloy ES, Interleukin 12 and interleukin 23 play key pathogenic roles in inflammatory and proliferative pathways in giant cell arteritis, Ann Rheum Dis. 77 (12) (2018) 1815–1824. [DOI] [PubMed] [Google Scholar]
- [96].Terao C, Yoshifuji H, Nakajima T, Yukawa N, Matsuda F, Mimori T, Ustekinumab as a therapeutic option for Takayasu arteritis: from genetic findings to clinical application, Scand J Rheumatol. 45 (1) (2016) 80–82. [DOI] [PubMed] [Google Scholar]