

Abstract
Mutated RNA splicing machinery drives many human diseases and is a promising therapeutic target for engineering and small molecule therapy. In the case of mutations in individual genes that cause them to be incorrectly spliced, engineered splicing factors can be introduced to correct splicing of these aberrant transcripts and reduce the disease phenotype. Mutations that occur in certain splicing factor genes themselves have been implicated in many cancers, particularly myelodysplastic syndromes. Small molecules that target splicing factors have been developed as therapies to preferentially induce apoptosis in these cancer cells. Specifically, drugs targeting the splicing factor SF3B1 have led to recent clinical trials. Here we review the role of alternative splicing in disease, approaches to rescue incorrect splicing using engineered splicing factors, and small molecule splicing inhibitors developed to treat hematological cancers.
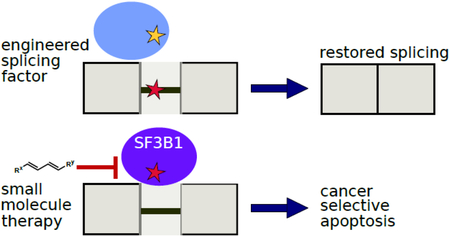
Graphical_Abstract
Introduction
Alternative splicing (AS) of precursor messenger RNA is responsible for the precise regulation of gene expression and AS dysregulation is the cause of various human genetic diseases [1]. The final spliced fate of an RNA transcript depends on which potential splice sites in a transcript are chosen by the spliceosome, a megadalton ribonucleoprotein complex [2]. Aberrant splicing can occur when 1) mutations arise in the splicing signals of a gene or 2) mutations arise in the spliceosomal genes themselves. A splicing signal mutation can prevent proper association of the spliceosome at an intron/exon boundary or activate usage of a cryptic splice site in another location of the gene [3]. Mutations in splicing factor genes change alternative splicing patterns by altering the binding affinities of the splicing factor proteins for their targets in the RNA [4]. These two mechanisms lead to human disease. In this review, we will address therapeutic strategies utilized against both of these mutation modes of AS. AS also contributes to mechanisms of cancer resistance by modulating drug targets, which is well reviewed by Siegfried and Karni [5] and not further discussed here.
Spliceosomes dynamically assemble across introns and exons
The excision of introns and ligation of exons to process a pre-mRNA into a protein-coding transcript is completed by a multitude of trans-acting factors that comprise the spliceosome. The main building blocks of the major spliceosome are 5 small nuclear ribonucleoproteins (snRNPs) composed of small nuclear RNAs (U1, U2, U4, U5, and U6) that interact with a variety of associated protein factors [6]. The spliceosome dynamically assembles on the pre-mRNA via stepwise recruitment of the snRNPs and additional protein factors. Introns in pre-mRNA contain four sequence motifs: 5’ splice site, branchpoint site (BS), polypyrimidine tract, and 3’ splice site. These motifs are among the splice signals that are recognized multiple times by different splicing factors during splicing to ensure fidelity and to determine alternative splicing patterns of the pre-mRNA. The subunits of the spliceosome undergo extensive remodeling during splicing as the RNA active site components are initially sequestered as inactive conformations through a network of RNA-RNA interactions [7]. These subunits become active through conformational changes in the spliceosome initiated by RNA helicases [8]. The spliceosome operates through two general principles: 1) splicing reactions are catalyzed by the snRNAs bound to intron splice sites; and 2) proteins maneuver these snRNAs into correct position for splicing, forming snRNPs and various other protein complexes in the spliceosome. The therapeutic strategies discussed below primarily focus on engineering or interfering with the components of the spliceosome in the A complex, particularly the U1 and U2 snRNPs (Figure 1).
Figure 1.

A spliceosome assembled on a pre-mRNA transcript and an overview of the two approaches to treating spliceosomal diseases. On the left-hand side, the modifications of the U1 snRNP are shown to improve binding of the splicing factor to the transcript. Various proteins in the spliceosome can be targeted for inhibition using small molecules, which leads to cancer-selective apoptosis in certain cancers. The SR protein on the far right is a stand-in for the family of proteins targeted by that list of molecules. The spliceosome depicted is beginning to transition out of the A complex, as detailed by the arrival of the tri-snRNP (U4/U6.U5). Brr2, a protein target of several inhibitors discussed in this review, is one component of the U5 snRNP. N-palmitoyl-L-leucine inhibits late-stage splicing assembly.
Recovering splicing of mutated splice signals
If a disease-causing mutation is in a splicing signal of a gene (i.e., splice sites and intronic/exonic splicing enhancer/silencer sequences), then engineered splicing factors can be introduced to the host to correct the erroneous splicing at the level of the transcript (Figure 2). This precision engineering of splicing factors has therapeutic potential for many RNA diseases, according to promising preclinical data from experiments on mammalian cell lines, patient-derived cells, and mouse models. For example, exon-specific U1 snRNAs (ExSpeU1s) have been engineered to correct splicing defects in genes causing spinal muscular atrophy (SMA) [9,10],[11•], propionic acidemia [12], Netherton syndrome [13], blood coagulation disorders [14-18], and familial dysautonomia [19]. In the studies discussed here, gene constructs containing ExSpeU1s have been virally transfected into human and mammalian cell lines or microinjected into mouse models for expression of the engineered splicing factor without modification to the wild-type copy of U1 snRNA. Corrected splicing of disease-relevant transcripts was detected using RT-PCR and RNA sequencing. Protein expression arising from these properly processed transcripts has been demonstrated via protein blotting or functional assays [11•, 13,15-19]. ExSpeU1s have been particularly effective when combined with antisense oligonucleotide molecules [20], generating more spliced transcripts than application of either strategy alone. The engineered U1 snRNAs provide more stable binding to a mutated splicing signal and promote correct splicing of these transcripts. These approaches take advantage of the positive effect on human splicing observed by hyperstabilization of extended U1 snRNA on 5’ss [21].
Figure 2.

Recovering splicing of mutated and inefficiently spliced transcripts. (A) In this example diagram, the wild-type U1 snRNP binds poorly to a mutated splice signal. The transcript is broken into exons (depicted as solid blocks) and an intron with a mutated splice signal (line with a star). This results in an aberrantly spliced mature mRNA and potentially a disease phenotype. (B) An engineered U1 snRNP can be introduced to allow for improved binding to a mutated splice signal. This restores splicing of the correct mature mRNA. (C) Upon addition of the compound NVS-SM2, wild-type U1 snRNP is stabilized against the inefficiently spliced SMN2 transcript, allowing production of full length SMN mRNA [26].
To restore correct splicing of a gene causing retinitis pigmentosa, researchers transfected mutant U1 snRNAs into patient-derived primary skin fibroblasts and demonstrated improved splicing of the target transcript [22,23]. Another study focused on another engineered splicing protein, U2AF, to promote the wild-type splicing isoform [24]. These engineered U1 snRNAs have also been studied as HIV-1 inhibitors. Targeting the U1 snRNP to the 3' end of HIV-1 mRNA substantially reduced viral protein expression in cell lines by blocking pre-mRNA polyadenylation and targeting the HIV-1 mRNA for degradation [25]. These studies highlight the wide variety of diseases that can potentially be treated using this approach, but also show that current research has been primarily focused on using U1 snRNA as a splicing modulator in cell lines or mice expressing human minigenes.
In addition to engineering splicing factor genes, small molecules have been identified that can stabilize the spliceosome on mutated splicing signals. A small molecule stabilizer of SMN2 pre-mRNA and the U1 snRNP complex elevates levels of full-length SMN protein and extends the survival of SMA mice [26] (Figure 2C). These small molecule therapies have been proven to increase motor function and longevity in these mice models [27]. Despite the low toxicities and therapeutic successes observed in mouse studies, there have not been any clinical trials attempted yet using this engineered splicing factor approach. Further studies will be needed to determine the specificity of this technology and any potential off-target effects before application to human patients.
Common splicing factor mutations that lead to cancer
Mutations in the spliceosome drive many cancers [28], as a disease-relevant mutation in a spliceosomal gene induces widespread changes to intron/exon recognition in the cell. Given the relationship between spliceosome mutations and cancer progression, the spliceosome also represents a therapeutic vulnerability [29]. As there are a limiting number of spliceosomes in the cell, spliceosome substrate competition will have strong global splicing affects [30,31]. Cancers with spliceosome mutations depend on wild-type spliceosome functionality for survival [32,33•]. High-throughput sequencing experiments have implicated four splicing factor mutations in various cancers. These mutations alter splicing factor functionality with far-reaching impacts on downstream genes and pathways that enable cancerous growth. Change/gain-of-function mutations in the genes for splicing factor 3B subunit 1 (SF3B1) [34], U2 small nuclear RNA auxiliary factor 1 (U2AF1) [35,36], and serine/arginine rich splicing factor 2 (SRSF2) [4,37] are frequent in patients with myelodysplastic syndromes (MDS) [38], chronic myelomonocytic leukemia (CMML), and chronic lymphocytic leukemia (CLL) [39••]. For a cell with splicing factor mutations, knocking down function of the limited wild-type spliceosomes will create a synthetic lethality phenotype. Therefore, malignancies with mutated spliceosomal genes are more sensitive to pharmacological intervention [40], which has been exploited in the development of new therapeutic compounds. This is highly crucial as a report by Seiler et al. detailed the frequency of somatic mutations in splicing factor genes across 33 tumor types and found that 119 splicing factor genes have recurring mutations in various cancers [41••].
Small molecule spliceosome inhibitors
Chemical compounds can modulate aberrant splicing in a variety of human RNA diseases [42]. Natural products derived primarily from bacteria have provided many leads for therapeutic compounds targeting splicing [43]. Medicinal chemistry has been useful to optimize tumor-selective spliceosome modulators and enhance potency [44]. The recent determination of the different stages of the spliceosome through cryo-electron microscopy is ushering in a new era in understanding spliceosome interactions by capturing splicing factors bound to their inhibitors [45••,46••,47••], and will undoubtedly lead to rational design of new small molecule inhibitors.
Shared pharmacophore
The natural products FR901464, pladienolide B, and herboxidiene are the three major splicing inhibitor classes that have given rise to a plethora of more potent derivatives. Spliceostatin A (a derivative of FR901464), pladienolide B, herboxidiene, as well as the sudemycins (structurally similar synthetic compounds) have been demonstrated to target SF3B1, one of six components of the SF3b subunit of the U2 snRNP and a highly influential target in splicing (Figure 3). These compounds inhibit the spliceosome at multiple stages, both at early steps and at exon ligation, indicating that the SF3B1 subunit is involved at multiple stages in splicing [48]. To point toward a common mechanism of action, spliceostatin A, pladienolide B, herboxidiene, and a synthetic cyclopropane analog [49] all share similar structures, mainly a central diene unit flanked by varying functional groups. In a recent study by Teng et al., spliceostatin A, pladienolide B, and herboxidiene have been shown to target the PHF5A-SF3B1 complex, as mutations in PHF5A and SF3B1 can cause cells to become resistant to the inhibitors [45••]. This target was conclusively visualized via cryo-EM structures of the SF3b complex with pladienolide B [47••] and a potent derivative E7107 [46••].
Figure 3.

Five chemical scaffolds used to target the SF3b complex in cancer therapies. The compounds prevent the SF3B1-PHF5A complex from transitioning to a closed form and thus inhibit the splicing process. All compounds share a similar structure, featuring a central diene flanked by large functional groups, depicted in a general form inside the red box.
FR901464, spliceostatin A, and derivatives
Antitumor compound FR901464 was first isolated from fermentation broth containing Pseudomonas spp. 2663 [50-52], but the native producer was later identified to be Burkholderia spp. [53]. The 1-O-methylated derivative spliceostatin A (SSA) has increased potency [54] and displays strong effects on intron retention [55••]. SSA induces apoptosis at nanomolar concentrations in CLL cells through altered MCL-1 RNA splicing [56]. SSA has demonstrated the promise of targeting the spliceosome for combinatorial treatments in cancer. Combining SSA with Bcl-2/Bcl-xL antagonists has increased potential for CLL therapy as an approach to target residual disease in the lymph nodes [56]. The stimulation of CD40L/interleukin-4 (IL-4) occurs in the tumor microenvironment of CLL and reduces the effectiveness of monotherapies. Larrayoz et al. found that treatment with Bcl-2/Bcl-xL antagonists along with SSA induced greater apoptosis of CLL cells compared to the monotherapies in the presence of CD40L/IL-4 [56]. Despite its promising use in research studies, SSA has not been attempted as a therapy in any clinical trials. This has likely been due to the lack of a reliable biomarker of efficacy as SSA has widespread effects on transcripts [57] and the demonstrated toxicity of similar compound E7107 in the clinic (described in the next section). Another compound related to FR901464 and SSA, Thailanstatin A, was first isolated from bacteria Burkholderia thailandensis [53]. It was recently used as the bioactive component of antibody-drug conjugates and found to be highly potent against gastric cancer xenograft models [58]. Furthermore, derivatives of Thailanstatin A have been generated through total synthesis with more potent antitumor activities [59], highlighting the possibility of improved payloads in future studies.
Pladienolide B and derivatives
Pladienolide B has a verified target in the SF3B1 subunit of the spliceosomal SF3b subcomplex [60]. Pladienolide B inhibits splicing by forcing SF3b to maintain an open complex, inhibiting stable BS-pocket formation for first step splicing, as identified by cryo-EM [47••]. It is used widely as a tool for studying the sensitivity of cancer cells after exposure to genetic or environmental changes. Expression of wild-type p53 promotes pladienolide B-induced apoptosis in cutaneous squamous cell carcinoma cells [61•]. Pladienolide B and a related macrolide FD-895 induce intron retention and selectively induce apoptosis in CLL cells versus normal lymphocytes [62]. However, FD-895 is not stable [49] and more stable analogs have been synthesized by decorating the central scaffold with carbohydrate motifs [63].
E7107 has been a highly studied derivative of pladienolide B, including clinical data. A structure of E7107 bound to the SF3b spliceosome subcomplex was generated at 3.95 Å through cryo-EM [46••]. This structure suggests the mechanism of action of this inhibitor is to interfere with branch point adenosine recognition to prevent first step splicing. E7107 displays dose-dependent reversible inhibition of pre-mRNA processing in patients, proving that splicing can be modulated with this drug in humans [64]. A phase I clinical trial, which involved testing the pharmacokinetic profile of E7107 in patients with advanced solid tumors, unfortunately resulted in two patients undergoing vision loss, likely due to global inhibition of the spliceosome in the optic nerve. The limited efficacy observed in this study indicates that more study should be done into which tumor types would be best treated by this inhibitor [65].
Splicing inhibitor E7107 is also well-studied in treating acute myeloid leukemia in mouse models [66]. Mouse models were built with a mutant allele of SRSF2(P95H) involved with MDS and acute myeloid leukemia (AML). Treatment of these models with E7107 resulted in greater reduction of leukemia burden in systems containing the spliceosome mutation versus wild-type leukemia [66]. The most common SF3B1 mutation in MDS (K700E) was constructed in a mouse model and E7107 was shown to selectively induce apoptosis in cells expressing this mutant [67]. E7107 also demonstrates the utility of targeting the spliceosome in combination therapy. When combined with the proteasome inhibitor bortezomib, E7107 enhanced antitumor response [68•].
Sudemycins share a pharmacophore with FR901464 and pladienolide B
Sudemycins (C, D1, D6, E, F1, F2, and K) are a class of synthetic analogs of FR901464. Cells treated with sudemycin exhibit differential activity on exon skipping [55••,69•]. Upon reanalysis of published RNA-seq data sets, exon skipping was also demonstrated in HeLa cells treated with SSA [69•], providing additional evidence for the effects of this shared pharmacophore.
Sudemycins have proven effective for treating hematological cancers. A study by Xargay-Torrent et al. demonstrates that sudemycin D1 has greater toxicity in CLL patient cells compared to healthy lymphocytes or tumors from different B-lymphoid malignancies. CLL cells with mutations in SF3B1 or other splicing and RNA processing genes were more sensitive to sudemycin D1 than CLL cells without these mutations. Sudemycin efficacy was also demonstrated in vivo using immunodeficient mice engrafted with peripheral blood cells from CLL patients [70]. Sudemycin D1 and D6 show comparable cytotoxicity but D6 was optimized for in vivo stability [70]. Sudemycin D6 has dose- and time-dependent anti-tumor activity and changes to alternative splicing patterns were observed with RT-PCR [71]. In a recent study, sudemycin D6 shows effects on exon skipping in DUSP11 and SRRM1 pre-mRNAs, which serve as pharmacodynamic markers at 9 hours after treatment of blood lymphocytes extracted from donors [72].
In MDS, patient-derived hematopoietic cells expressing mutant U2AF1 are more sensitive to sudemycin [73•]. Tests performed in transgenic mice with mutant U2AF1(S34F) found that sudemycin D6 increases exon skipping and intron retention, which when coupled with splicing changes caused by the mutant U2AF1, creates negative cumulative synergy on the cell and can offer an explanation for the increased sensitivity to sudemycin.
Novel sudemycins developed from the shared pharmacophore show improved potency, specifically sudemycin K, which has an amide group in place of an oxycarbonyl [74] (Figure 3). Similar to the other splicing inhibitors, there is positive combinatorial benefit when combining sudemycins with other cancer therapies. Sudemycin combined with ibrutinib shows an enhanced antitumor effect, likely due to induction of alternative splicing of IBTK which promotes BtK expression, leading to ibrutinib sensitivity [70].
Additional SF3b complex inhibitors
Herboxidiene is a polyketide natural product first isolated from fermentation broth of the bacteria Streptomyces chromofuscus [75]. Given its potent inhibition of the SF3b protein complex in the spliceosome, it was targeted for synthesis of improved derivatives [76]. The study showed an efficient synthesis and that derivatives were functional in an in vitro splicing assay. Jerantinine A is an indole alkaloid which inhibits tubulin polymerization, inducing G2/M cell cycle arrest and tumor-specific cell death through upregulation of SF3B1 and SF3B3 and inducing dissociation of SF3B1 in the nucleosome [77].
A highly promising new inhibitor is H3B-8800, which is effective against epithelial and hematologic tumor cells. This compound was identified using medicinal chemistry. H3B-8800 is also an inhibitor of the SF3b complex (Figure 3) and demonstrated efficacy against a xenograft system for CMML [39••]. Cells with SF3B1(K700E) or SRSF2(P95H) mutations were preferentially affected and tumor growth was halted. Seiler et al. found that upon exposure to H3B-8800, a large number of genes that encode splicing factors had increased rates of intron retention. However, SF3B1(R1074H) or PHF5A(Y36C) mutants conferred resistance to H3B-8800 [39••], indicating that this splicing factor complex is targeted by H3B-8800, similar to the shared pharmacophore of FR901464, pladienolide B, and herboxidiene. Notably, H3B-8800 also shares the central diene unit identified in the other inhibitors. Unlike E7107, H3B-8800 activity increases the retention of short, GC-rich introns, which are enriched in splicing factor genes [39••]. A clinical trial is currently recruiting to study the preliminary activity of H3B-8800 in patients with MDS, AML, or CMML (NCT02841540).
Inhibitors of additional splicing targets
Brr2 is an RNA helicase involved in the activation of the spliceosome’s catalytic center by unwinding the U4/U6 duplex (Figure 1). The Brr2 inhibitor spiro[indole-3,20-pyrrolidin]-2(1H)-one has superior activity (inhibition at submicromolar concentrations) to the previously reported 4,6-dihydropyrido[4,3-d]pyrimidine-2,7(1H,3H)-dione series [78]. These potent and selective Brr2 inhibitors with helicase inhibition activity were uncovered using ATPase activity-based screening [79]. N-palmitoyl-L-leucine inhibits late stage assembly of human spliceosomes and was discovered in a high-throughput in vitro splicing screening platform for extracts from marine natural products [80]. Lynamicin D has been shown to alter the levels of SRPK1, which encodes a kinase involved in constitutive and alternative splicing [81]. A library containing cyclic and linear peptides was screened for interactions that inhibit the interaction between the UHM domain of splicing factor 45 (SPF45) and the ULM motifs found in constitutive splicing factors [82] (Figure 1). Additionally, a small molecule has been shown to prevent the transition of human spliceosomal B complexes into activated B complex, revealing the new insight into spliceosome assembly. Specifically, U4/U6 snRNP proteins are released during activation before incorporation of the Prp19.CDC5L complex [83].
Spliceosome phosphorylation is another intriguing target for cancer therapies. TG003 targets the spliceosome phosphatase family of proteins, specifically inhibiting CDC2-like kinase 1. TG003 induces exon-skipping in shorter exons with fewer splicing factor binding sites than insensitive-exons, according to RNA-seq data from human and mouse skeletal cells [84], but it is unstable for clinical applications [85]. A more stable molecule, TG693, promotes the skipping of a mutated exon in Duchenne muscular dystrophy (DMD) patients and creates a functional protein. Administration to mice alters the pre-mRNA splicing in skeletal muscle [85]. T-025 also targets CDC-like kinases (CLK) and administration of the compound decreased CLK-dependent phosphorylation which increased the number of skipped exons. It shows in vivo antitumor efficacy in an allograft model of MYC-driven breast cancer [86]. Small molecules Cpd-1, Cpd-2, Cpd-3 all show inhibition of CLKs and serine/arginine protein kinases (SRPKs), resulting in modulation of pre-mRNA splicing and growth inhibition of breast cancer MDA-MB-468 cells [87].
Another promising therapeutic approach is to promote selective protein degradation in cancer cell lines using anticancer sulfonamides, which target RBM39/CAPERα (U2AF related splicing cofactor activator) [88]. Compounds including indisulam, tasisulam, and chloroquinoxaline sulfonamide share the same mechanism of action of inactivation of RBM39. The sensitivity of cancer cell lines to these drugs correlates to their levels of DCAF15 expression, which could prove a useful biomarker [89].
Summary and conclusions
Spliceosomes dynamically assemble across introns and exons in order to process pre-mRNA as a crucial step toward protein synthesis. The multitude of splicing isoforms that can be created due to splicing logic is termed alternative splicing and plays a large role in human disease. There are two types of splicing mutations: 1) those in splicing signals which alter splicing of that specific transcript; and 2) splicing mutations in splicing factor genes, which can have global effects on transcripts. In order to recover aberrant splicing of mutated/inefficiently splice signals, engineered splicing factors with specific recognition of the mutated splicing signal can be introduced into cells. Most research has focused on applications of U1 snRNA modifications for applications in blood diseases and spinal muscular atrophy. Mutations in splicing factor genes are prominent in many cancers, particularly hematological cancers such as MDS and leukemia; new research is unveiling an ever-increasing role of these splicing mutations in cancer. Small molecule spliceosome inhibitors have demonstrated high potential as novel therapies. Three widely studied natural product-based inhibitors have a shared pharmacophore and target the essential splicing factor SF3B1, as demonstrated by elegant genetic studies and the advent of cyro-EM. Inhibitors of additional splicing targets are in development and it is likely more therapeutic approaches will be determined. Increasingly, researchers are studying the positive synergistic effects observed when combining splicing inhibitors with other treatments for cancer therapy. Overall, learning how to control alternative splicing is crucial to designing new therapies for human spliceosomal diseases.
Highlights.
Engineered U1 splicing factors rescue splicing of disease-relevant transcripts with mutated splice signals.
Mutated splicing factor SF3B1 is an increasingly well-studied therapeutic target in certain blood cancers.
Small molecule H3B-8800 targeting SF3B1 has begun clinical trials as a cancer therapy.
Cryogenic electron microscopy is enabling a deeper understanding of how small molecule therapies interact with the spliceosome.
Additional small molecules targeting different spliceosomal components have the potential to be new anticancer drugs.
Acknowledgements
Due to space constraint, we apologize for the omission of other relevant reports. ABD was supported by National Institutes of Health (NIH) 1U01GM110706.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Recommended Reading
Papers of particular interest, published within the period of the review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Scotti MM, Swanson MS: RNA mis-splicing in disease. Nat Rev Genet 2016, 17:19–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Black DL: Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem 2003, 72:291–336. [DOI] [PubMed] [Google Scholar]
- 3.Pagani F, Baralle FE: Genomic variants in exons and introns: identifying the splicing spoilers. Nat Rev Genet 2004, 5:389–396. [DOI] [PubMed] [Google Scholar]
- 4.Zhang J, Lieu YK, AN AM, Penson A, Reggio KS, Rabadan R, Raza A, Mukherjee S, Manley JL: Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc Natl Acad Sci U S A 2015, 112:E4726–4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Siegfried Z, Karni R: The role of alternative splicing in cancer drug resistance. Curr Opin Genet Dev 2018, 48:16–21. [DOI] [PubMed] [Google Scholar]
- 6.Shi Y: The Spliceosome: A Protein-Directed Metalloribozyme. J Mol Biol 2017, 429:2640–2653. [DOI] [PubMed] [Google Scholar]
- 7.Will CL, Luhrmann R: Spliceosome structure and function. Cold Spring Harb Perspect Biol 2011, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cordin O, Hahn D, Beggs JD: Structure, function and regulation of spliceosomal RNA helicases. Curr Opin Cell Biol 2012, 24:431–438. [DOI] [PubMed] [Google Scholar]
- 9.Fernandez Alanis E, Pinotti M, Dal Mas A, Balestra D, Cavallari N, Rogalska ME, Bernardi F, Pagani F: An exon-specific U1 small nuclear RNA (snRNA) strategy to correct splicing defects. Hum Mol Genet 2012, 21:2389–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Singh NN, Del Rio-Malewski JB, Luo D, Ottesen EW, Howell MD, Singh RN: Activation of a cryptic 5' splice site reverses the impact of pathogenic splice site mutations in the spinal muscular atrophy gene. Nucleic Acids Res 2017, 45:12214–12240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •11.Rogalska ME, Tajnik M, Licastro D, Bussani E, Camparini L, Mattioli C, Pagani F: Therapeutic activity of modified U1 core spliceosomal particles. Nat Commun 2016, 7:11168. [DOI] [PMC free article] [PubMed] [Google Scholar]; Exon-specific U1 snRNAs are developed to increase SMN2 exon 7 inclusion and improve full-length SMN protein production. Treatment with this engineered splicing factor significantly improved the life span of the SMA mouse model.
- 12.Sanchez-Alcudia R, Perez B, Perez-Cerda C, Ugarte M, Desviat LR: Overexpression of adapted U1snRNA in patients' cells to correct a 5' splice site mutation in propionic acidemia. Mol Genet Metab 2011, 102:134–138. [DOI] [PubMed] [Google Scholar]
- 13.Dal Mas A, Fortugno P, Donadon I, Levati L, Castiglia D, Pagani F: Exon-Specific U1s Correct SPINK5 Exon 11 Skipping Caused by a Synonymous Substitution that Affects a Bifunctional Splicing Regulatory Element. Hum Mutat 2015, 36:504–512. [DOI] [PubMed] [Google Scholar]
- 14.Pinotti M, Rizzotto L, Balestra D, Lewandowska MA, Cavallari N, Marchetti G, Bernardi F, Pagani F: U1-snRNA-mediated rescue of mRNA processing in severe factor VII deficiency. Blood 2008, 111:2681–2684. [DOI] [PubMed] [Google Scholar]
- 15.Pinotti M, Balestra D, Rizzotto L, Maestri I, Pagani F, Bernardi F: Rescue of coagulation factor VII function by the U1+5A snRNA. Blood 2009, 113:6461–6464. [DOI] [PubMed] [Google Scholar]
- 16.Balestra D, Faella A, Margaritis P, Cavallari N, Pagani F, Bernardi F, Arruda VR, Pinotti M: An engineered U1 small nuclear RNA rescues splicing defective coagulation F7 gene expression in mice. J Thromb Haemost 2014, 12:177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balestra D, Scalet D, Pagani F, Rogalska ME, Mari R, Bernardi F, Pinotti M: An Exon-Specific U1snRNA Induces a Robust Factor IX Activity in Mice Expressing Multiple Human FIX Splicing Mutants. Mol Ther Nucleic Acids 2016, 5:e370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tajnik M, Rogalska ME, Bussani E, Barbon E, Balestra D, Pinotti M, Pagani F: Molecular Basis and Therapeutic Strategies to Rescue Factor IX Variants That Affect Splicing and Protein Function. PLoS Genet 2016, 12:e1006082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Donadon I, Pinotti M, Rajkowska K, Pianigiani G, Barbon E, Morini E, Motaln H, Rogelj B, Mingozzi F, Slaugenhaupt SA, et al. : Exon-specific U1 snRNAs improve ELP1 exon 20 definition and rescue ELP1 protein expression in a familial dysautonomia mouse model. Hum Mol Genet 2018, 27:2466–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balestra D, Barbon E, Scalet D, Cavallari N, Perrone D, Zanibellato S, Bernardi F, Pinotti M: Regulation of a strong F9 cryptic 5'ss by intrinsic elements and by combination of tailored U1snRNAs with antisense oligonucleotides. Hum Mol Genet 2015, 24:4809–4816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Freund M, Hicks MJ, Konermann C, Otte M, Hertel KJ, Schaal H: Extended base pair complementarity between U1 snRNA and the 5' splice site does not inhibit splicing in higher eukaryotes, but rather increases 5' splice site recognition. Nucleic Acids Res 2005, 33:5112–5119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glaus E, Schmid F, Da Costa R, Berger W, Neidhardt J: Gene therapeutic approach using mutation-adapted U1 snRNA to correct a RPGR splice defect in patient-derived cells. Mol Ther 2011, 19:936–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmid F, Glaus E, Barthelmes D, Fliegauf M, Gaspar H, Nurnberg G, Nurnberg P, Omran H, Berger W, Neidhardt J: U1 snRNA-mediated gene therapeutic correction of splice defects caused by an exceptionally mild BBS mutation. Hum Mutat 2011, 32:815–824. [DOI] [PubMed] [Google Scholar]
- 24.Agrawal AA, McLaughlin KJ, Jenkins JL, Kielkopf CL: Structure-guided U2AF65 variant improves recognition and splicing of a defective pre-mRNA. Proc Natl Acad Sci U S A 2014, 111:17420–17425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knoepfel SA, Abad A, Abad X, Fortes P, Berkhout B: Design of modified U1i molecules against HIV-1 RNA. Antiviral Res 2012, 94:208–216. [DOI] [PubMed] [Google Scholar]
- 26.Palacino J, Swalley SE, Song C, Cheung AK, Shu L, Zhang X, Van Hoosear M, Shin Y, Chin DN, Keller CG, et al. : SMN2 splice modulators enhance U1-pre-mRNA association and rescue SMA mice. Nat Chem Biol 2015, 11:511–517. [DOI] [PubMed] [Google Scholar]
- 27.Naryshkin NA, Weetall M, Dakka A, Narasimhan J, Zhao X, Feng Z, Ling KK, Karp GM, Qi H, Woll MG, et al. : Motor neuron disease. SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science 2014, 345:688–693. [DOI] [PubMed] [Google Scholar]
- 28.Read A, Natrajan R: Splicing dysregulation as a driver of breast cancer. Endocr Relat Cancer 2018, 25:R467–R478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hsu TY, Simon LM, Neill NJ, Marcotte R, Sayad A, Bland CS, Echeverria GV, Sun T, Kurley SJ, Tyagi S, et al. : The spliceosome is a therapeutic vulnerability in MYC-driven cancer. Nature 2015, 525:384–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Munding EM, Shiue L, Katzman S, Donohue JP, Ares M Jr., : Competition between pre-mRNAs for the splicing machinery drives global regulation of splicing. Mol Cell 2013, 51:338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Effenberger KA, Urabe VK, Jurica MS: Modulating splicing with small molecular inhibitors of the spliceosome. Wiley Interdiscip Rev RNA 2017, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fei DL, Motowski H, Chatrikhi R, Prasad S, Yu J, Gao S, Kielkopf CL, Bradley RK, Varmus H: Wild-Type U2AF1 Antagonizes the Splicing Program Characteristic of U2AF1-Mutant Tumors and Is Required for Cell Survival. PLoS Genet 2016, 12:e1006384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •33.Paolella BR, Gibson WJ, Urbanski LM, Alberta JA, Zack TI, Bandopadhayay P, Nichols CA, Agarwalla PK, Brown MS, Lamothe R, et al. : Copy-number and gene dependency analysis reveals partial copy loss of wild-type SF3B1 as a novel cancer vulnerability. Elite 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study confirmed SF3B1 as a CYCLOPS gene (Copy-number alterations Yield Cancer Liabilities Owing to Partial losS). Cancer cells with partial copy-loss of SF3B1 lack a reservoir of SF3B1 to protect them from the effects of the splicing inhibitors.
- 34.Alsafadi S, Houy A, Battistella A, Popova T, Wassef M, Henry E, Tirade F, Constantinou A, Piperno-Neumann S, Roman-Roman S, et al. : Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun 2016, 7:10615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shirai CL, Ley JN, White BS, Kim S, Tibbitts J, Shao J, Ndonwi M, Wadugu B, Duncavage EJ, Okeyo-Owuor T, et al. : Mutant U2AF1 Expression Alters Hematopoiesis and Pre-mRNA Splicing In Vivo. Cancer Cell 2015, 27:631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li B, Liu J, Jia Y, Wang J, Xu Z, Qin T, Shi Z, Song Z, Peng S, Huang H, et al. : Clinical features and biological implications of different U2AF1 mutation types in myelodysplastic syndromes. Genes Chromosomes Cancer 2018, 57:80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, Li Y, Chung YR, Micol JB, Murphy ME, et al. : SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 2015, 27:617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thol F, Kade S, Schlarmann C, Loffeld P, Morgan M, Krauter J, Wlodarski MW, Kolking B, Wichmann M, Gorlich K, et al. : Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes. Blood 2012, 119:3578–3584. [DOI] [PubMed] [Google Scholar]
- ••39.Seiler M, Yoshimi A, Darman R, Chan B, Keaney G, Thomas M, Agrawal AA, Caleb B, Csibi A, Sean E, et al. : H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat Med 2018, 24:497–504. [DOI] [PMC free article] [PubMed] [Google Scholar]; A new orally available splicing inhibitor is described in this study. H3B-8800 targets the SF3b complex, but induces retention of short, GC-rich introns which are enriched in spliceosome genes. A Phase 1 clinical trial is currently recruiting patients with certain hematological cancers.
- 40.Bonnal S, Vigevani L, Valcarcel J: The spliceosome as a target of novel antitumour drugs. Nat Rev Drug Discov 2012, 11:847–859. [DOI] [PubMed] [Google Scholar]
- ••41.Seiler M, Peng S, Agrawal AA, Palacino J, Teng T, Zhu P, Smith PG, Cancer Genome Atlas Research N, Buonamici S, Yu L: Somatic Mutational Landscape of Splicing Factor Genes and Their Functional Consequences across 33 Cancer Types. Cell Rep 2018, 23:282–296 e284. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors utilize sequencing data from The Cancer Genome Atlas to identify 119 splicing factor genes that are enriched for mutations across 33 tumor types. Analysis of RNA-seq data revealed the extent of alternative splicing dysregulation due to these mutations, underscoring the importance of studying spliceosome mutations in cancer development and progression.
- 42.Kataoka N: Modulation of aberrant splicing in human RNA diseases by chemical compounds. Hum Genet 2017, 136:1237–1245. [DOI] [PubMed] [Google Scholar]
- 43.Leon B, Kashyap MK, Chan WC, Krug KA, Castro JE, La Clair JJ, Burkart MD: A Challenging Pie to Splice: Drugging the Spliceosome. Angew Chem Int Ed Engl 2017, 56:12052–12063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lagisetti C, Palacios G, Goronga T, Freeman B, Caufield W, Webb TR: Optimization of antitumor modulators of pre-mRNA splicing. J Med Chem 2013, 56:10033–10044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••45.Teng T, Tsai JH, Puyang X, Seiler M, Peng S, Prajapati S, Aird D, Buonamici S, Caleb B, Chan B, et al. : Splicing modulators act at the branch point adenosine binding pocket defined by the PHF5A-SF3b complex. Nat Commun 2017, 8:15522. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors identify the SF3B1-PHF5A complex as the target for the splicing inhibitors. Mutations in PHF5A and SF3B1 confer resistance to the inhibitors and reduce the effect of the inhibitors on global splicing patterns.
- ••46.Finci LI, Zhang X, Huang X, Zhou Q, Tsai J, Teng T, Agrawal A, Chan B, Irwin S, Karr C, et al. : The cryo-EM structure of the SF3b spliceosome complex bound to a splicing modulator reveals a pre-mRNA substrate competitive mechanism of action. Genes Dev 2018, 32:309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study utilized cryo-EM to solve crystal structures of E7107 bound to the SF3b complex. The results support a model of substrate competitive inhibition where the inhibitor molecule competes with the branch point site of the intron.
- ••47.Cretu C, Agrawal AA, Cook A, Will CL, Fekkes P, Smith PG, Luhrmann R, Larsen N, Buonamici S, Pena V: Structural Basis of Splicing Modulation by Antitumor Macrolide Compounds. Mol Cell 2018, 70:265–273 e268. [DOI] [PubMed] [Google Scholar]; The investigators utilized cryo-EM to study how the splicing inhibitor pladienolide B binds a hinge of SF3B1, thus locking the complex in an open conformation and preventing splicing. These crystal structures will enable further studies into drug design.
- 48.Effenberger KA, Urabe VK, Prichard BE, Ghosh AK, Jurica MS: Interchangeable SF3B1 inhibitors interfere with pre-mRNA splicing at multiple stages. RNA 2016, 22:350–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kumar D, Kashyap MK, La Clair JJ, Villa R, Spaanderman I, Chien S, Rassenti LZ, Kipps TJ, Burkart MD, Castro JE: Selectivity in Small Molecule Splicing Modulation. ACS Chem Biol 2016, 11:2716–2723. [DOI] [PubMed] [Google Scholar]
- 50.Nakajima H, Sato B, Fujita T, Takase S, Terano H, Okuhara M: New antitumor substances, FR901463, FR901464 and FR901465. I. Taxonomy, fermentation, isolation, physico-chemical properties and biological activities. J Antibiot (Tokyo) 1996, 49:1196–1203. [DOI] [PubMed] [Google Scholar]
- 51.Nakajima H, Hori Y, Terano H, Okuhara M, Manda T, Matsumoto S, Shimomura K: New antitumor substances, FR901463, FR901464 and FR901465. II. Activities against experimental tumors in mice and mechanism of action. J Antibiot (Tokyo) 1996, 49:1204–1211. [DOI] [PubMed] [Google Scholar]
- 52.Nakajima H, Takase S, Terano H, Tanaka H: New antitumor substances, FR901463, FR901464 and FR901465. III. Structures of FR901463, FR901464 and FR901465. J Antibiot (Tokyo) 1997, 50:96–99. [DOI] [PubMed] [Google Scholar]
- 53.He H, Ratnayake AS, Janso JE, He M, Yang HY, Loganzo F, Shor B, O'Donnell CJ, Koehn FE: Cytotoxic Spliceostatins from Burkholderia sp. and Their Semisynthetic Analogues. J Nat Prod 2014, 77:1864–1870. [DOI] [PubMed] [Google Scholar]
- 54.Kaida D, Motoyoshi H, Tashiro E, Nojima T, Hagiwara M, Ishigami K, Watanabe H, Kitahara T, Yoshida T, Nakajima H, et al. : Spliceostatin A targets SF3b and inhibits both splicing and nuclear retention of pre-mRNA. Nat Chem Biol 2007, 3:576–583. [DOI] [PubMed] [Google Scholar]
- ••55.Vigevani L, Gohr A, Webb T, Irimia M, Valcarcel J: Molecular basis of differential 3' splice site sensitivity to anti-tumor drugs targeting U2 snRNP. Nat Commun 2017, 8:2100. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study identifies the sequence determinants of the sensitivity of the 3’ splice sites to splicing inhibitors. Introns retained by drug effects display higher GC-content. The authors determine that sudemycins primarily affect exon skipping while spliceostatin A affects intron retention.
- 56.Larrayoz M, Blakemore SJ, Dobson RC, Blunt MD, Rose-Zerilli MJ, Walewska R, Duncombe A, Oscier D, Koide K, Forconi F, et al. : The SF3B1 inhibitor spliceostatin A (SSA) elicits apoptosis in chronic lymphocytic leukaemia cells through downregulation of Mcl-1. Leukemia 2016, 30:351–360. [DOI] [PubMed] [Google Scholar]
- 57.Bates DO, Morris JC, Oltean S, Donaldson LF: Pharmacology of Modulators of Alternative Splicing. Pharmacol Rev 2017, 69:63–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Puthenveetil S, Loganzo F, He H, Dirico K, Green M, Teske J, Musto S, Clark T, Rago B, Koehn F, et al. : Natural Product Splicing Inhibitors: A New Class of Antibody-Drug Conjugate (ADC) Payloads. Bioconjug Chem 2016, 27:1880–1888. [DOI] [PubMed] [Google Scholar]
- 59.Nicolaou KC, Rhoades D, Kumar SM: Total Syntheses of Thailanstatins A-C, Spliceostatin D, and Analogues Thereof. Stereodivergent Synthesis of Tetrasubstituted Dihydro- and Tetrahydropyrans and Design, Synthesis, Biological Evaluation, and Discovery of Potent Antitumor Agents. J Am Chem Soc 2018, 140:8303–8320. [DOI] [PubMed] [Google Scholar]
- 60.Aouida M, Eid A, Mahfouz MM: CRISPR/Cas9-mediated target validation of the splicing inhibitor Pladienolide B. Biochim Open 2016, 3:72–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •61.Hepburn LA, McHugh A, Fernandes K, Boag G, Proby CM, Leigh IM, Saville MK: Targeting the spliceosome for cutaneous squamous cell carcinoma therapy: a role for c-MYC and wild-type p53 in determining the degree of tumour selectivity. Oncotarget 2018, 9:23029–23046. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors demonstrate the potential of SF3B1 inhibitors to treat cutaneous squamous cell carcinoma. However, they determined that the inactivation of p53 (common to this cancer) will limit the therapeutic effect of pladienolide B in treatment.
- 62.Kashyap MK, Kumar D, Villa R, La Clair JJ, Benner C, Sasik R, Jones H, Ghia EM, Rassenti LZ, Kipps TJ, et al. : Targeting the spliceosome in chronic lymphocytic leukemia with the macrolides FD-895 and pladienolide-B. Haematologica 2015, 100:945–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dhar S, La Clair JJ, Leon B, Hammons JC, Yu Z, Kashyap MK, Castro JE, Burkart MD: A Carbohydrate-Derived Splice Modulator. J Am Chem Soc 2016, 138:5063–5068. [DOI] [PubMed] [Google Scholar]
- 64.Eskens FA, Ramos FJ, Burger H, O'Brien JP, Piera A, de Jonge MJ, Mizui Y, Wiemer EA, Carreras MJ, Baselga J, et al. : Phase I pharmacokinetic and pharmacodynamic study of the first-in-class spliceosome inhibitor E7107 in patients with advanced solid tumors. Clin Cancer Res 2013, 19:6296–6304. [DOI] [PubMed] [Google Scholar]
- 65.Hong DS, Kurzrock R, Naing A, Wheler JJ, Falchook GS, Schiffman JS, Faulkner N, Pilat MJ, O'Brien J, LoRusso P: A phase I, open-label, single-arm, dose-escalation study of E7107, a precursor messenger ribonucleic acid (pre-mRNA) splicesome inhibitor administered intravenously on days 1 and 8 every 21 days to patients with solid tumors. Invest New Drugs 2014, 32:436–444. [DOI] [PubMed] [Google Scholar]
- 66.Lee SC, Dvinge H, Kim E, Cho H, Micol JB, Chung YR, Durham BH, Yoshimi A, Kim YJ, Thomas M, et al. : Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat Med 2016, 22:672–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Obeng EA, Chappell RJ, Seiler M, Chen MC, Campagna DR, Schmidt PJ, Schneider RK, Lord AM, Wang L, Gambe RG, et al. : Physiologic Expression of Sf3b1(K700E) Causes Impaired Erythropoiesis, Aberrant Splicing, and Sensitivity to Therapeutic Spliceosome Modulation. Cancer Cell 2016, 30:404–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •68.Chan S, Sridhar P, Kirchner R, Lock YJ, Herbert Z, Buonamici S, Smith P, Lieberman J, Petrocca F: Basal-A Triple-Negative Breast Cancer Cells Selectively Rely on RNA Splicing for Survival. Mol Cancer Ther 2017, 16:2849–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]; E7107 treatment prevents tumor growth in triple-negative breast cancer xenografts. This response was improved by the addition of the proteasome inhibitor bortezomib, indicating the utility of combinatorial therapy.
- •69.Wu G, Fan L, Edmonson MN, Shaw T, Boggs K, Easton J, Rusch MC, Webb TR, Zhang J, Potter PM: Inhibition of SF3B1 by molecules targeting the spliceosome results in massive aberrant exon skipping. RNA 2018, 24:1056–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]; The authors analyze RNA-seq data on Hela cell lines treated with sudemycin D1 and spliceostatin A and reveal exon skipping to be the dominant change to alternative splicing patterns. This study contrasts with previous reports suggesting that intron retention was the major change to splicing.
- 70.Xargay-Torrent S, Lopez-Guerra M, Rosich L, Montraveta A, Roldan J, Rodriguez V, Villamor N, Aymerich M, Lagisetti C, Webb TR, et al. : The splicing modulator sudemycin induces a specific antitumor response and cooperates with ibrutinib in chronic lymphocytic leukemia. Oncotarget 2015, 6:22734–22749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shi Y, Joyner AS, Shadrick W, Palacios G, Lagisetti C, Potter PM, Sambucetti LC, Stamm S, Webb TR: Pharmacodynamic assays to facilitate preclinical and clinical development of pre-mRNA splicing modulatory drug candidates. Pharmacol Res Perspect 2015, 3:e00158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Thurman M, van Doorn J, Danzer B, Webb TR, Stamm S: Changes in Alternative Splicing as Pharmacodynamic Markers for Sudemycin D6. Biomark Insights 2017, 12:1177271917730557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •73.Shirai CL, White BS, Tripathi M, Tapia R, Ley JN, Ndonwi M, Kim S, Shao J, Carver A, Saez B, et al. : Mutant U2AF1-expressing cells are sensitive to pharmacological modulation of the spliceosome. Nat Commun 2017, 8:14060. [DOI] [PMC free article] [PubMed] [Google Scholar]; Hematopoietic cells that express U2AF1(S34F) mutants have an increased sensitivity to treatment with sudemycin and exhibit differential gene expression.
- 74.Makowski K, Vigevani L, Albericio F, Valcarcel J, Alvarez M: Sudemycin K: A Synthetic Antitumor Splicing Inhibitor Variant with Improved Activity and Versatile Chemistry. ACS Chem Biol 2017, 12:163–173. [DOI] [PubMed] [Google Scholar]
- 75.Miller-Wideman M, Makkar N, Tran M, Isaac B, Biest N, Stonard R: Herboxidiene, a new herbicidal substance from Streptomyces chromofuscus A7847. Taxonomy, fermentation, isolation, physico-chemical and biological properties. J Antibiot (Tokyo) 1992, 45:914–921. [DOI] [PubMed] [Google Scholar]
- 76.Ghosh AK, Lv K, Ma N, Cardenas EL, Effenberger KA, Jurica MS: Design, synthesis and in vitro splicing inhibition of desmethyl and carba-derivatives of herboxidiene. Org Biomol Chem 2016, 14:5263–5271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chung FF, Tan PF, Raja VJ, Tan BS, Lim KH, Kam TS, Hii LW, Tan SH, See SJ, Tan YF, et al. : Jerantinine A induces tumor-specific cell death through modulation of splicing factor 3b subunit 1 (SF3B1). Sci Rep 2017, 7:42504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ito M, Iwatani M, Yamamoto T, Tanaka T, Kawamoto T, Morishita D, Nakanishi A, Maezaki H: Discovery of spiro[indole-3,2'-pyrrolidin]-2(1H)-one based inhibitors targeting Brr2, a core component of the U5 snRNP. Bioorg Med Chem 2017, 25:4753–4767. [DOI] [PubMed] [Google Scholar]
- 79.Iwatani-Yoshihara M, Ito M, Klein MG, Yamamoto T, Yonemori K, Tanaka T, Miwa M, Morishita D, Endo S, Tjhen R, et al. : Discovery of Allosteric Inhibitors Targeting the Spliceosomal RNA Helicase Brr2. J Med Chem 2017, 60:5759–5771. [DOI] [PubMed] [Google Scholar]
- 80.Effenberger KA, James RC, Urabe VK, Dickey BJ, Linington RG, Jurica MS: The Natural Product N-Palmitoyl-l-leucine Selectively Inhibits Late Assembly of Human Spliceosomes. J Biol Chem 2015, 290:27524–27531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sigala I, Ganidis G, Thysiadis S, Zografos AL, Giannakouros T, Sarli V, Nikolakaki E: Lynamicin D an antimicrobial natural product affects splicing by inducing the expression of SR protein kinase 1. Bioorg Med Chem 2017, 25:1622–1629. [DOI] [PubMed] [Google Scholar]
- 82.Jagtap PK, Garg D, Kapp TG, Will CL, Demmer O, Luhrmann R, Kessler H, Sattler M: Rational Design of Cyclic Peptide Inhibitors of U2AF Homology Motif (UHM) Domains To Modulate Pre-mRNA Splicing. J Med Chem 2016, 59:10190–10197. [DOI] [PubMed] [Google Scholar]
- 83.Sidarovich A, Will CL, Anokhina MM, Ceballos J, Sievers S, Agafonov DE, Samatov T, Bao P, Kastner B, Urlaub H, et al. : Identification of a small molecule inhibitor that stalls splicing at an early step of spliceosome activation. Elife 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sakuma M, Iida K, Hagiwara M: Deciphering targeting rules of splicing modulator compounds: case of TG003. BMC Mol Biol 2015, 16:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sako Y, Ninomiya K, Okuno Y, Toyomoto M, Nishida A, Koike Y, Ohe K, Kii I, Yoshida S, Hashimoto N, et al. : Development of an orally available inhibitor of CLK1 for skipping a mutated dystrophin exon in Duchenne muscular dystrophy. Sci Rep 2017, 7:46126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Iwai K, Yaguchi M, Nishimura K, Yamamoto Y, Tamura T, Nakata D, Dairiki R, Kawakita Y, Mizojiri R, Ito Y, et al. : Anti-tumor efficacy of a novel CLK inhibitor via targeting RNA splicing and MYC-dependent vulnerability. EMBO Mol Med 2018, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Araki S, Dairiki R, Nakayama Y, Murai A, Miyashita R, Iwatani M, Nomura T, Nakanishi O: Inhibitors of CLK protein kinases suppress cell growth and induce apoptosis by modulating pre-mRNA splicing. PLoS One 2015, 10:e0116929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Uehara T, Minoshima Y, Sagane K, Sugi NH, Mitsuhashi KO, Yamamoto N, Kamiyama H, Takahashi K, Kotake Y, Uesugi M, et al. : Selective degradation of splicing factor CAPERalpha by anticancer sulfonamides. Nat Chem Biol 2017, 13:675–680. [DOI] [PubMed] [Google Scholar]
- 89.Han T, Goralski M, Gaskill N, Capota E, Kim J, Ting TC, Xie Y, Williams NS, Nijhawan D: Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Science 2017, 356. [DOI] [PubMed] [Google Scholar]