

Summary
Increased peripheral levels of cytokines and central microglial activation have been reported in patients with psychiatric disorders. The degree of both innate and adaptive immune activation is also associated with worse clinical outcomes and poor treatment response in these patients. Understanding the possible causes and mechanisms leading to this immune activation is therefore an important and necessary step for the development of novel and more effective treatment strategies for these patients. In this work, we review the evidence of literature pointing to childhood trauma as one of the main causes behind the increased immune activation in patients with psychiatric disorders. We then discuss the potential mechanisms linking the experience of early life adversity (ELA) to innate immune activation. Specifically, we focus on the innervation of the bone marrow from sympathetic nervous system (SNS) as a new and emerging mechanism that has the potential to bridge the observed increases in both central and peripheral inflammatory markers in patients exposed to ELA. Experimental studies in laboratory rodents suggest that SNS activation following early life stress exposure causes a shift in the profile of innate immune cells, with an increase in proinflammatory monocytes. In turn, these cells traffic to the brain and influence neural circuitry, which manifests as increased anxiety and other relevant behavioural phenotypes. To date, however, very few studies have been conducted to explore this candidate mechanism in humans. Future research is also needed to clarify whether these pathways could be partially reversible to improve prevention and treatment strategies in the future.
Keywords: depression, early adversity, inflammation, psychosis, sympathetic nervous system
Introduction
In the past two decades, converging lines of evidence from several studies suggest a role for activation and/or dysfunction of the immune system in the pathogenesis of major psychiatric disorders such as depression and psychotic disorders. Specifically, there is evidence for increased peripheral immune activation, typically characterized by elevations in the levels of circulating proinflammatory cytokines such as interleukin (IL)‐6 or IL‐1β. In addition, other inflammatory markers, such as C reactive protein (CRP) are also commonly increased in these patients. Finally, in both depression and psychotic disorder there is evidence from post‐mortem brain tissue and in‐vivo neuroimaging studies to suggest central microglia activation, the brain’s resident innate immune cell, although these data are not unequivocal 1, 2. The increased innate immune activation in these patients may be of particular clinical relevance, based on the findings of our group and others that the degree of immune activation, at least in terms of peripheral cytokines and CRP, is associated with more severe clinical symptoms, suicidal ideation and poor response to treatment 2, 3, 4.
In order to develop novel treatment and/or preventative measures for these patients, we need to understand more clearly what are the causes and potential mechanisms underlying the immune activation in patients with psychiatric disorders. In this review we seek to address this question by first reviewing the evidence that links the experience of traumatic and stressful events during childhood to innate immune activation and development of psychopathology in later life 1. Childhood trauma is, unfortunately, highly prevalent in our society and one of the most widely recognized risk factors for development of psychiatric disorders later in life, including major depression, psychosis and post‐traumatic stress disorder (PTSD) 5, 6, 7. Research carried out in the past two decades has clearly identified a role for the activation of the immune system as a potential biological mechanism connecting childhood trauma and the subsequent development of psychopathology 8. In the second part of this review, we focus on the emerging evidence for a role of the sympathetic nervous system (SNS) as one of the main mechanisms behind the link between early life adversities (ELA) and peripheral and central inflammation. With this critical overview, the paper shows and identifies necessary next steps for research in this field, to enable the future development of clinical preventative and treatment strategies in victims of childhood trauma.
Evidence for the association between ELA and activation of the innate immune system
Increased circulating cytokines and acute‐phase proteins
The presence of increased immune activation in patients with psychiatric disorders across different diagnostic categories has generated increasing interest in unravelling causes behind this immune activation. In the context of peripheral immune activation, several clinical studies so far have shown that inflammatory markers, such as IL‐6 and CRP, are particularly high in patients who had experienced childhood traumatic events, including physical or sexual abuse, emotional abuse or neglect that occur in childhood; this has been demonstrated in patients suffering with psychosis, major depression or bipolar disorder 9, 10, 11, 12, 13. These primary research findings are supported by evidence from systematic reviews that provide evidence for prolonged peripheral inflammation following exposure to traumatic events in childhood 8, 11, 14. Increased levels of other peripheral circulating inflammatory markers, such as the acute‐phase protein CRP, are also widely reported in young children exposed to adverse psychosocial events 15, 16. Moreover, this initial early immune activation appears to persist and thus become chronic throughout adult life 14. In a previous meta‐analysis, we reported evidence to suggest that adult subjects with experience of childhood trauma have higher levels not only of CRP, but also of the major proinflammatory cytokines IL‐6 and tumour necrosis factor (TNF)‐α, when compared with adults without such experience 8.
One important point in this regard is that the magnitude of the increases in these inflammatory markers is not comparable to that found following acute infections. Rather, it more closely resembles a low‐threshold, chronic inflammation, which is broadly comparable to detectable levels of these markers in patients with cardiovascular disease or rheumatoid arthritis. Although modest in terms of intensity, this persistent immune activation appears to be relevant for development of psychiatric disorders later in life. Specifically, data derived from longitudinal cohorts of children provide evidence for statistically significant associations between experiencing childhood traumatic events and increases in peripheral CRP levels, which are particularly pronounced in those individuals who later developed depression in adult life 17, 18.
Circulating immune cell populations – evidence for changes in both innate and adaptive immunity following ELA exposure
In contrast to the wealth of data on circulating cytokines following early life trauma exposure, proportionally far fewer studies have directly investigated the effects of exposure to childhood adversities on circulating immune cell populations using, for example, fluorescence activated cell sorting (FACS). In adolescents who experienced early life stress compared to those without such experience, do Prado and colleagues reported increases in circulating T cells expressing the activation markers CD2+/CD4+/CD25+ and CD3+/CD69+, but also an increase in senescent T cells (CD8+/CD28– and CD4+/CD28– cells) 19. These changes were further accompanied by decreased percentages of natural killer (NK; CD3–/CD56+) and NK T cells (CD3+/CD56+) 19. Comparing individuals separated from their mothers in early life to those reared by their parents, Elwenspoek and colleagues also reported increased T cell senescence (CD57+ cells) 20. In another FACS study from this group, comparing individuals with a history of ELA to those without, a reduction in the numbers of CD69+/CD8+ T cells and increased numbers of human leucocyte antigen D‐related HLA‐DR+/CD4+, HLA‐DR+/CD8+ and CD25+/CD8+ T cells was reported 21. Collectively, these results suggest that ELA exposure is associated with a sustained activation of adaptive immunity and accelerated immune ageing, as indexed by the increase in T cell senescence. These effects appeared to be independent of either stress or health‐risk behaviours, suggestive of a primary effect of early life programming on specific immune cell populations 20, 21, 22. Exploring how social adversity may affect peripheral immune cells, Counotte et al. observed that individuals with high clinical risk for psychosis who were exposed to ELA were characterized by an increased number of CD4+ T helper type 17 (Th17) cells when compared with those at lower risk 23. There were also significant predictive relationships between increased numbers of T regulatory (Treg) cells and decreased NK cell numbers, with the increased degree of stress experienced during exposure to virtual social stressors 23. If and how these different associations fit together still needs to be elucidated. Other studies characterizing the effects of social adversities on peripheral leucocyte populations have mainly identified increases in circulating monocytes and dendritic cells (DCs) following exposure to traumatic experiences in early life 24, 25. Furthermore, Schwaiger and colleagues identified an altered stress‐induced gene expression in CD14+ monocytes of adults with history of childhood trauma; the differences were particularly linked to the expression of genes involved in steroid hormone activity and signal transduction which were, in turn, associated with increased activity of proinflammatory signalling upstream 26.
These clinical data are supported by a large body of evidence from preclinical studies in adult mice exposed to repeated social defeat (RSD), which results in a significant accumulation of neutrophils and CD11b+LyC6high monocytes in both the circulation and spleen 27, 28. Splenic DCs from mice exposed to RSD also have increased surface expression of major histocompatibility complex (MHC) class I, CD80 and CD44, indicative of an activated state 29. It should also be noted, however, that CD44 is commonly used as a T cell activation marker 29. Furthermore, following in‐vitro stimulation, these cells display an up‐regulated proinflammatory gene expression signature and produce comparatively higher levels of pro‐ and anti‐inflammatory cytokines, including IL‐6, TNF‐α and IL‐10 29. Extending these findings, exposure to RSD in mice and low socio‐economic status (SES) in humans results in a relative expansion of a transcriptional profile associated with immature proinflammatory monocytes in peripheral blood mononuclear cells (PMBCs) 30. This is characterized (at least in the mouse) by increased activity of transcription factors that regulate both differentiation to the myeloid lineage and also the proinflammatory effector functions of these cells, such as PU.1 30. In addition, exposure to RSD in mice results in increased formation of Ly6Chigh monocytes, which could be prevented using β‐adrenoreceptor antagonists 30. Collectively, these data suggest a role for the sympathetic nervous system driving, at least in part, up‐regulation of myelopoiesis, characterized by a proinflammatory state which we will return to later in this review 30. Data on the effects of social stress on adaptive immunity in preclinical models are less developed, although there are data suggesting a reduction in the numbers of T cells in the circulation and spleen 27, 28, which is at odds with some of the aforementioned clinical findings. Conversely, RSD is reported to result in enhanced CD4+ and CD8+ memory T cell responses when this stress is experienced before a viral infection 31, 32, 33. These data suggest a view that exposure to RSD in adult mice may significantly enhance virus‐specific immune memory through the augmentation of specific memory T cell populations 31, 32, 33. Interestingly, using a mediation analysis, Elwenspoek and colleagues found that cytomegalovirus (CMV) infection largely explained the increase in T cell senescence markers (CD57+) following ELA exposure, suggesting that viral infections may affect T cell populations in humans following ELA exposure 20. When comparing these data it should be remembered that the great majority of preclinical studies that have characterized the peripheral immune response to stress exposure have primarily focused on the effects of repeated RSD exposure in adult animals. Detailed peripheral immunophenotyping data regarding the specific effects of early life stress in preclinical models is, however, currently lacking. Another important point is that the effects of any experimental stress paradigm in animals may have both common and distinct effects on the immune system, depending on the nature and duration of the stressor 34, 35, 36. In this context, preclinical studies could be immensely valuable, as they allow the experimenter to probe the effects of different types of early life stress with a precise control of genetic and other environmental variables, something that is very challenging in humans. Preclinical studies therefore have the clear potential to help dissect the specific peripheral immune cell responses associated with exposure to different stressors at different vulnerable periods from the prenatal period, throughout early postnatal life and into adolescence.
Overall, the evidence from the few studies available suggests that ELA exposure is associated with: (1) direct programming of innate and adaptive immune cell function; (2) increased numbers of circulating innate and adaptive immune cells with a proinflammatory phenotype, a finding consistent with the elevation in circulating proinflammatory cytokines and acute‐phase proteins in those exposed to ELA; and (3) accelerated immune ageing, as indicated by T cell senescence. What remains unclear from these data is whether these peripheral changes are related to central immune activation. Here again, studies of preclinical models of stress exposure are vital, as they permit invasive assessments in both the brain and periphery to aid dissection of the mechanisms governing immune‐to‐brain communication and vice versa 37, 38. However, in addressing this question, we must first consider the clinical evidence base for central immune activation as a function of ELA exposure.
Central immune activation following ELA exposure
Assessment of central immune activation in clinical samples is typically achieved through post‐mortem neuropathological assessment at either the cellular or molecular level or in vivo, using positron emission tomography (PET) combined with selective radioligands for the translocator protein (TSPO) comparing patient groups with healthy age‐ and sex‐matched controls 37. Searching the extant literature, we found only one TSPO PET study which had tested for an association between psychosocial risk factors for psychosis, including childhood trauma and central TSPO radioligand binding. Dahoun and colleagues 39 measured TSPO binding in a small sample of healthy individuals who were classified into two groups on the basis of exposure to psychosocial risk (high‐risk group; n = 12) or no significant exposure to psychosocial risk (low‐risk group; n = 12). However, there were no significant group differences in TSPO volume of distribution (VT) in either total grey matter or the frontal, parietal, temporal and occipital lobes 39. At first glance, these data are not consistent with the hypothesis that exposure to stress and/or ELA is associated with increased central microglial activation. It should be noted however, that these were healthy individuals without a psychiatric diagnosis, raising the possibility that the ‘high‐risk’ group in this case are a resilient group in whom putative central microglial activation may not be seen 39. Moreover, although some of the stressors assessed in these individuals occurred recently, there was a temporal separation between stress exposure and TSPO PET imaging. Hence, it remains possible that acute stress exposure could be associated with increased TSPO, which may normalize over time 39. Further studies are clearly required to address these issues and explore in more detail the effect of ELA on central TSPO binding in both preclinical models and clinical populations. In contrast, several TSPO PET studies have been conducted to search for any evidence of central immune activation in the psychiatric disorders for which ELA is a known risk factor, such as schizophrenia and depression. The data published so far suggest that there is evidence for putative central innate immune activation associated with psychiatric disorders for which ELA exposure is a known risk factor 37, but evidence for specific effects of ELA on central immune cells in humans is currently lacking. Hence, to address this question, we must consider the evidence from preclinical rodent models of ELA exposure.
Exposing rodents or non‐human primates to early life stress increases anxiety‐like behaviours and impairs cognitive function in adulthood, suggesting that animal models may provide important insights into the effects of ELA in humans 40, 41, 42. As already mentioned, it should be remembered when comparing data from preclinical models to clinical studies that the effects of any experimental stress paradigm in animals may have both common and distinct effects on the immune system, depending on the nature and duration of the stressor. This is no less true for central immune activation following stress exposure in rodents 36, 37. Interestingly, genetically controlled depletion of brain microglia in healthy rodents in early life is associated with abnormal social and motivated behaviour in a sex‐specific manner, strongly implying that microglia play a role in the early life programming of behaviour 43, 44. With regard to the evidence that early life stress exposure influences central microglia, several preclinical studies in different early life stress models provide converging lines of evidence for increased innate immune activation in the central nervous system (CNS). Prenatal stress exposure, induced by exposing pregnant rat dams to restraint or other physical stressors, is reported to increase the number of Iba1+/CD68+ microglia, coupled with a shift in their morphology to an activated, amoeboid state and decreased process motility, as well as increasing proinflammatory cytokine expression in the brains of the affected offspring early in postnatal life primarily (but not exclusively) in the hippocampus, amygdala and prefrontal cortex 45, 46, 47, 48, 49, 50, 51, 52, 53, 54. None of these studies, however, have investigated the effects of ELA exposure on TSPO binding, limiting cross‐species comparisons to humans. At the molecular level, Delpech and colleagues have examined the effect of brief daily separation (BDS) stress on the microglial transcriptome in the affected offspring, providing evidence that this early life stress actually perturbs the normal functional development of microglia in the developing hippocampus, with these cells displaying increased phagocytic activity and reduced expression of genes that normally increase across development 55. It is increasingly appreciated that microglia cells play critical roles in shaping the processes of synaptic maturation and axonal guidance in the developing and maturing rodent brain 56, 57, 58, 59, 60, 61, 62, 63, 64. Hence, any stress‐induced perturbation of microglial physiology during these critical developmental windows would be predicted to result in the defective maturation of neural circuits 56, 57, 65. This could have long‐lasting consequences on synaptic function and may be another mechanism whereby activation of central innate immunity after ELA exposure may directly increase the vulnerability to develop psychopathology in later life. These cells may also play a role in central to peripheral immune communication, as we discuss in the next section.
How does ELA lead to immune activation?
Although both clinical and preclinical studies provide converging lines of evidence for both central and peripheral immune activation following ELA exposure (notably for both innate and adaptive immunity), a critical question remains as to how ELA exposure specifically triggers both central and peripheral inflammation. Although the precise mechanisms by which peripheral immunity may influence central immunity and neuronal function (and vice versa) remain largely unknown, a number of interesting candidate mechanisms have been advanced. These include: (1) the development of glucocorticoid resistance and therefore an impairment in the anti‐inflammatory action of glucocorticoids; (2) activation of the SNS; (3) a change in the composition of gut microbiota; (4) exposure to physical injuries or infection as part of the early life trauma; and (5) behavioural mechanisms, including obesity, alcohol consumption and poor sleep, which may themselves promote immune activation 66. For the purposes of this concise review, we will focus on the link between SNS activation in response to stress exposure and immune activation, for which there is a recent increasing body of evidence 67.
The role of the SNS in the link between early adversities and immune activation
The SNS is one of the main biological systems involved in the stress response. Its activation also results in a cascade of signals that affect the regulation of the innate immune system, which occurs in a cell‐state and organ‐specific manner. Of particular interest is the bone marrow (BM), which is densely innervated by the SNS 68. Activation of the SNS in response to stress, for example, leads to the production and release of noradrenaline (NA), which binds to β‐adrenergic (βAR) and α‐adrenergic (αAR) receptors expressed on peripheral immune cell populations in both rodents and humans 68, 69. Preclinical studies in animals show that SNS activation and NA production triggers the release of monocytes into the circulation, which can be modulated pharmacologically by systemic administration of either a β2AR agonist (increase) or βAR antagonist (decrease) 27, 30, 32. Increased numbers of circulating monocytes is also correlated with oscillations in SNS tone as a function of circadian rhythm 70 and can occur following other forms of SNS stimuli, such as exercise 71. These observations are also true for lymphocytes, with exposure to acute stress in humans resulting in mobilization of total monocytes and lymphocytes and CD8+ T lymphocytes 71. NA released during SNS activation may, however, lead to different immunomodulatory effects, depending on the cell type, state and organ involved. For example, Case and Zimmerman 72 propose a model in which NA release can have a suppressive effect on naive T lymphocytes in lymphoid organs such as the spleen, but in other regions, such as the vasculature and renal system, NA may drive the activation of proinflammatory Th17 T lymphocytes.
Taken together, the aforementioned lines of evidence clearly show that SNS activation is well placed to mediate both innate and adaptive immunity and brain–immune communication across species. There are no studies in healthy humans, either alone or in comparison to clinical populations, that provide direct evidence for a link between ELA exposure, SNS activation and elevated peripheral and central immunity, beyond the observations that stress‐exposed individuals who have developed psychopathology have elevated numbers of circulating proinflammatory monocytes and lymphocytes – as already evidenced in this review. Here, again, we must therefore turn to preclinical studies in rodent models to dissect this mechanism.
As already stated, the SNS innervates peripheral lymphoid tissues including the spleen and bone marrow. Preclinical data suggest that chronic stress exposure is associated with a decrease in expression of the chemokine CXCL12 in the haematopoietic stem cell niche which, in turn, accelerates proliferation of haematopoietic stem cells resulting in enhanced production of neutrophils and monocytes 73. Increases in SNS outflow and NA release following RDS exposure also drives mobilization of lymphocytes from the murine spleen and bone marrow, which then enter the circulation 27, 28, 30, 32, 71, 74, 75. These data are consistent with the aforementioned findings in humans of increased number of leucocytes in the circulation of individuals with stress‐related psychiatric disorders 76, and the aforementioned studies reported increases in these cells in the circulation of individuals exposed to ELA. The specific contribution of the rodent models, however, has been to elegantly demonstrate a potential connection between central and peripheral immunity, which highlights the involvement of the SNS. Specifically, RDS in adult mice increases neural activation (indexed by increased immunostaining for the immediate early gene c‐Fos) in mouse brain regions that regulate fear and threat sensing, such as the amygdala which, in turn, is associated with both social avoidance and anxiety‐like behaviour that is sensitive to βAR antagonism, suggesting SNS involvement downstream of the activation of neural stress circuitry 75. In parallel to this increased neural activity, RDS results in central microglial activation, with increased density of amoeboid microglia in the amygdala, PFC and hippocampus, with higher surface expression of CD68, CD14 and Toll‐like receptor (TLR)‐4 and increased expression of both IL‐1β and glucocorticoid‐responsive genes (e.g. FKBP5) in these microglia, indicative of activation 67, 74, 75, 77, 78. Moreover, CD11b+CD45highLy6Chigh macrophages are increased in the circulation, but also trafficked into the mouse brain, where they express higher amounts of IL‐1β and adopt a proinflammatory phenotype (see Fig. 1) 67, 74, 75, 77, 78. These effects are dependent on βAR, as they may be blocked by propranolol (a βAR antagonist), suggestive of a role for SNS activation in the initial mobilization of the monocytes 75. Consistent with this view, the trafficking of monocytes from the spleen to the brain is necessary for the re‐establishment of anxiety in stress‐sensitized mice, and this also occurs downstream of SNS activation 74, 79. Depletion of central microglia also prevents monocyte trafficking to the brain and blocks the development of anxiety behaviour following repeated RDS exposure 77. These data suggest a model in which stress exposure results in elevated neural activity in stress circuits, leading to increased SNS outflow. The former triggers a microglial response, the latter triggers the mobilization of proinflammatory monocyte‐derived macrophages, which subsequently traffic to the brain in a CCR2‐ and IL‐1β‐dependent manner, involving communication between central microglia and the endothelial cells of the blood–brain barrier 38, 67, 80, 81. These elegant studies demonstrate a potential pathway by which SNS activation is a key part of a larger multi‐faceted interaction between central neural activity in specific circuits, central microglial activation and mobilization of peripheral immunity, which then traffics to the brain to close the loop by influencing neural dynamics in the same circuits, promoting the emergence of anxiety‐like phenotypes.
Figure 1.
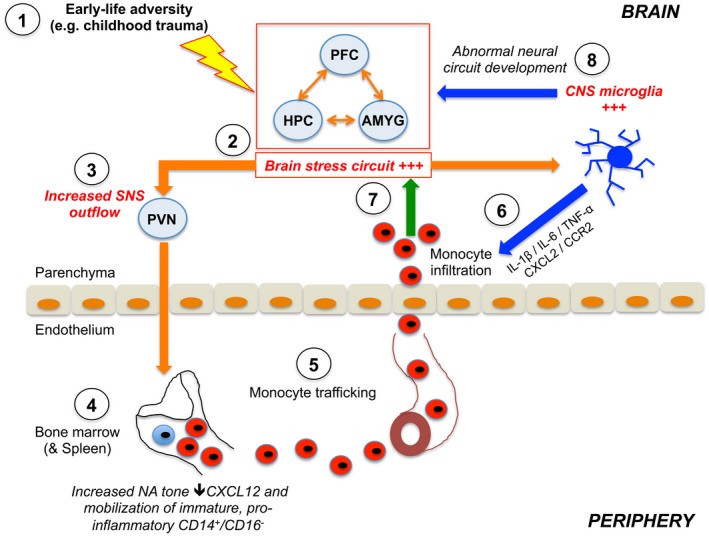
A schematic representation of how early life adversities may contribute to peripheral and central immune activation through activation of the sympathetic nervous system (SNS). Orange arrows indicate neural pathways, blue indicate central microglial effects and green the influence of peripheral monocytes. (1) Exposure to early life adversity (ELA) such as childhood trauma results in (2) activation of the brain stress circuitry and (3) increased outflow of the sympathetic nervous system to the bone marrow with increased noradrenaline (NA) release. (4) This decreases the expression of the chemokine CXCL12 in the bone marrow haematopoietic stem cell niche, which accelerates proliferation of haematopoietic stem cells resulting in enhanced production of proinflammatory CD14+/CD16– monocytes, which are released into circulation and (5) traffic to the blood–brain barrier through the vasculature. (6) At the same time, central microglial cells are also activated in response to neural cues as a result of stress circuit activation. These cells facilitate the infiltration of the peripheral monocytes through secretion of specific cytokines and chemokines, which modulate endothelial cell permeability and allow ingress of the monocytes from the vasculature to the perivascular space and the brain parenchyma. (7) The infiltrating proinflammatory monocytes lead to further sustained activation of neural stress circuits, resulting in the emergence of abnormal behaviour. (8) In parallel, ELA exposure may also influence the development and function of central microglia. Converging lines of evidence suggest that microglia play key roles in shaping the development of the brain including the formation and pruning of dendritic spines and synapses. Hence, any disruption of this process following stress exposure in early life may result in the abnormal neural circuit development which, by itself, may predispose an individual to psychopathology in later life. PFC = prefrontal cortex; AMYG = amygdala; HPC = hippocampus; SNS = sympathetic nervous system.
It is worth emphasizing again that this model has been developed based on experiments conducted in adult animals and in the context of the RDS paradigm. Hence, two important preclinical extensions of this work are necessary: first, to confirm whether this mechanism is necessary and sufficient for the development of abnormal behaviour (such as anxiety) following exposure to other types of stressor, including social isolation (SI), repeated social defeat stress (RSDS) or chronic unpredictable mild stress (CUMS). In this context, exposure to chronic variable stress is reported to increase Lin−Sca‐1+c‐Kit+ haematopoietic stem cells in the BM 73. Moreover, in mice susceptible to RSDS, elevated peripheral IL‐6 and increased circulating proinflammatory Ly6Chigh monocytes are reported, which also traffic to the perivascular space to increase central IL‐6 leading to depressive‐like behaviour 80. Lastly, as already described herein, there is also evidence for central microglial activation in several animal models of early life stress, although forward translation of this evidence to humans using TSPO binding for example is currently lacking 39. These data may suggest that there are indeed common mechanisms across different stress exposures, but further work is needed in this respect to clarify this mechanistically. Secondly, studies in the pre‐, perinatal and adolescent period are necessary to directly test if the model proposed from adult animals contributes to the association between ELA exposure and psychopathology in later life. It is worth recalling here that microglia depletion by itself in this period (without stress exposure) was sufficient to induce long‐term emotional behavioural changes in mice 43. Moreover, molecular studies suggest that microglia development may be abnormal following ELA exposure, perhaps indicative of later loss‐of‐function or reduced flexibility of these cells 55, 82. Hence, the relationships between neural circuit activity central and peripheral immunity may differ in the developing compared to the adult brain, which requires investigation 82, 83.
From the clinical perspective, direct evidence for involvement of the SNS per se following ELA exposure still needs to be provided. Indeed, the evidence supporting the link between childhood trauma, for example, and hyperactivation of the SNS in humans, is conflicting, with some studies reporting an increased reactivity of SNS following high levels of family adversities, while others find no such association 84. For example, no study has so far investigated immune markers together with markers of SNS activation in individuals with experience of childhood trauma compared with relevant controls. This would be an extremely important step to understand if the immune activation is found mainly in those who present an enhanced reactivity of the SNS.
Conclusions
In conclusion, increasing evidence points towards a fundamental role of ELA exposure in the activation of both innate and adaptive arms of the immune system in patients with psychiatric disorders. The SNS appears to play a central role in this link by increasing the activation of T cells and shifting the differentiation of haematopoietic stem cells in the bone marrow to proinflammatory monocytes, with elevated expression of proinflammatory molecules. Preclinical studies in adult animals suggest that trafficking of peripheral immune cells to the brain is an important mechanism for the development of anxiety following stress exposure. We highlight an important lack of studies addressing these issues, insofar as it is possible to do so in human samples, but also the lack of detailed work on these potential mechanisms which link central and peripheral immunity following early life stress exposure in preclinical models. Future studies to address these questions have the potential to identify molecular targets and time windows of opportunity for both prevention and treatment strategies to ameliorate the devastating long‐term effects of ELA on mental health.
Disclosures
V. M. has received research funding from Johnson & Johnson as part of a research programme on depression and inflammation. A. C. V. has received research funding from F. Hoffman La Roche Ltd and UBC Biopharma SPRL as part of a research programme on early life immune activation. The views expressed are those of the authors and not necessarily those of Johnson & Johnson, F. Hoffman La Roche or UCB Biopharma SPRL. These funders had no influence on the decision to publish this work.
Acknowledgements
V. M. is supported by MQ: Transforming Mental Health (Grant: MQBF1), the Medical Research Foundation (Grant: MRF‐160‐0005‐ELP‐MONDE), and the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health. A. C. V. acknowledges the financial support of the Medical Research Council (New Investigator Research Grant MR/N025377/1 and Centre Grant MR/N026063/1). The authors are also supported by the King’s Together Fund of King’s College London.
OTHER ARTICLES PUBLISHED IN THIS REVIEW SERIES
Neuroimmune interactions: how the nervous and immune systems influence each other. Clinical and Experimental Immunology 2019, 197: 276–277.
Neuroimmunology – the past, present and future. Clinical and Experimental Immunology 2019, 197: 278–293.
The immune system and psychiatric disease: a basic science perspective. Clinical and Experimental Immunology 2019, 197: 294–307
Depressive symptoms in inflammatory bowel disease: an extraintestinal manifestation of inflammation? Clinical and Experimental Immunology 2019, 197: 308–318.
Contributor Information
V. Mondelli, Email: valeria.mondelli@kcl.ac.uk.
A. C. Vernon, Email: anthony.vernon@kcl.ac.uk
References
- 1. Baumeister D, Russell A, Pariante CM, Mondelli V. Inflammatory biomarker profiles of mental disorders and their relation to clinical, social and lifestyle factors. Soc Psychiatry Psychiatr Epidemiol 2014; 49:841–9. [DOI] [PubMed] [Google Scholar]
- 2. Mondelli V, Vernon AC, Turkheimer F, Dazzan P, Pariante CM. Brain microglia in psychiatric disorders. Lancet Psychiatry 2017. [DOI] [PubMed] [Google Scholar]
- 3. Mondelli V, Ciufolini S, Belvederi Murri M et al Cortisol and inflammatory biomarkers predict poor treatment response in first episode psychosis. Schizophr Bull 2015; 41:1162–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cattaneo A, Gennarelli M, Uher R et al Candidate genes expression profile associated with antidepressants response in the GENDEP study: differentiating between baseline ‘predictors’ and longitudinal ‘targets’. Neuropsychopharmacology 2013; 38:377–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schafer I, Fisher HL. Childhood trauma and psychosis – what is the evidence? Dialogues Clin Neurosci 2011; 13:360–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Infurna MR, Reichl C, Parzer P, Schimmenti A, Bifulco A, Kaess M. Associations between depression and specific childhood experiences of abuse and neglect: a meta‐analysis. J Affect Disord 2016; 190:47–55. [DOI] [PubMed] [Google Scholar]
- 7. Yehuda R, Hoge CW, McFarlane AC et al Post‐traumatic stress disorder. Nat Rev Dis Primers 2015; 1:15057. [DOI] [PubMed] [Google Scholar]
- 8. Baumeister D, Akhtar R, Ciufolini S, Pariante CM, Mondelli V. Childhood trauma and adulthood inflammation: a meta‐analysis of peripheral C‐reactive protein, interleukin‐6 and tumour necrosis factor‐alpha. Mol Psychiatry 2016; 21:642–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aas M, Dieset I, Hope S et al Childhood maltreatment severity is associated with elevated C‐reactive protein and body mass index in adults with schizophrenia and bipolar diagnoses. Brain Behav Immun 2017; 65:342–9. [DOI] [PubMed] [Google Scholar]
- 10. Di Nicola M, Cattaneo A, Hepgul N et al Serum and gene expression profile of cytokines in first‐episode psychosis. Brain Behav Immun 2013; 31:90–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hepgul N, Pariante CM, Dipasquale S et al Childhood maltreatment is associated with increased body mass index and increased C‐reactive protein levels in first‐episode psychosis patients. Psychol Med 2012:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grosse L, Ambree O, Jorgens S et al Cytokine levels in major depression are related to childhood trauma but not to recent stressors. Psychoneuroendocrinology 2016; 73:24–31. [DOI] [PubMed] [Google Scholar]
- 13. Munjiza A, Kostic M, Pesic D, Gajic M, Markovic I, Tosevski DL. Higher concentration of interleukin 6 – a possible link between major depressive disorder and childhood abuse. Psychiatry Res 2018; 264:26–30. [DOI] [PubMed] [Google Scholar]
- 14. Danese A, Pariante CM, Caspi A, Taylor A, Poulton R. Childhood maltreatment predicts adult inflammation in a life‐course study. Proc Natl Acad Sci USA 2007; 104:1319–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Danese A, McEwen BS. Adverse childhood experiences, allostasis, allostatic load, and age‐related disease. Physiol Behav 2012; 106:29–39. [DOI] [PubMed] [Google Scholar]
- 16. Schmeer KK, Yoon A. Socioeconomic status inequalities in low‐grade inflammation during childhood. Arch Dis Child 2016; 101:1043–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Danese A, Moffitt TE, Pariante CM, Ambler A, Poulton R, Caspi A. Elevated inflammation levels in depressed adults with a history of childhood maltreatment. Arch Gen Psychiatry 2008; 65:409–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Khandaker GM, Zuber V, Rees JMB et al Shared mechanisms between coronary heart disease and depression: findings from a large UK general population‐based cohort. Mol Psychiatry 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. do Prado CH, Grassi‐Oliveira R, Daruy‐Filho L, Wieck A, Bauer ME. Evidence for immune activation and resistance to glucocorticoids following childhood maltreatment in adolescents without psychopathology. Neuropsychopharmacol 2017; 42:2272–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Elwenspoek MMC, Sias K, Hengesch X et al T cell immunosenescence after early life adversity: association with cytomegalovirus infection. Front Immunol 2017; 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Elwenspoek MMC, Hengesch X, Leenen FAD et al Proinflammatory T cell status associated with early life adversity. J Immunol 2017; 199:4046–55. [DOI] [PubMed] [Google Scholar]
- 22. Elwenspoek MMC, Kuehn A, Muller CP, Turner JD. The effects of early life adversity on the immune system. Psychoneuroendocrinology 2017; 82:140–54. [DOI] [PubMed] [Google Scholar]
- 23. Counotte J, Drexhage HA, Wijkhuijs JM et al Th17/T regulator cell balance and NK cell numbers in relation to psychosis liability and social stress reactivity. Brain Behav Immun 2018; 69:408–17. [DOI] [PubMed] [Google Scholar]
- 24. Cole SW, Hawkley LC, Arevalo JM, Cacioppo JT. Transcript origin analysis identifies antigen‐presenting cells as primary targets of socially regulated gene expression in leukocytes. Proc Natl Acad Sci USA 2011; 108:3080–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. O'Donovan A, Sun B, Cole S et al Transcriptional control of monocyte gene expression in post‐traumatic stress disorder. Dis Markers 2011; 30:123–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schwaiger M, Grinberg M, Moser D et al Altered stress‐induced regulation of genes in monocytes in adults with a history of childhood adversity. Neuropsychopharmacology 2016; 41:2530–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Engler H, Dawils L, Hoves S et al Effects of social stress on blood leukocyte distribution: the role of alpha‐ and beta‐adrenergic mechanisms. J Neuroimmunol 2004; 156:153–62. [DOI] [PubMed] [Google Scholar]
- 28. Engler H, Bailey MT, Engler A, Sheridan JF. Effects of repeated social stress on leukocyte distribution in bone marrow, peripheral blood and spleen. J Neuroimmunol 2004; 148:106–15. [DOI] [PubMed] [Google Scholar]
- 29. Powell ND, Bailey MT, Mays JW et al Repeated social defeat activates dendritic cells and enhances Toll‐like receptor dependent cytokine secretion. Brain Behav Immun 2009; 23:225–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Powell ND, Sloan EK, Bailey MT et al Social stress up‐regulates inflammatory gene expression in the leukocyte transcriptome via beta‐adrenergic induction of myelopoiesis. Proc Natl Acad Sci USA 2013; 110:16574–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mays JW, Bailey MT, Hunzeker JT et al Influenza virus‐specific immunological memory is enhanced by repeated social defeat. J Immunol 2010; 184:2014–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Powell ND, Mays JW, Bailey MT, Hanke ML, Sheridan JF. Immunogenic dendritic cells primed by social defeat enhance adaptive immunity to influenza A virus. Brain Behav Immun 2011; 25:46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mays JW, Powell ND, Hunzeker JT, Hanke ML, Bailey MT, Sheridan JF. Stress and the anti‐influenza immune response: Repeated social defeat augments clonal expansion of CD8(+)T cells during primary influenza A viral infection. J Neuroimmunol 2012; 243:34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Alleva E, Santucci D. Psychosocial vs. ‘physical’ stress situations in rodents and humans – role of neurotrophins. Physiol Behav 2001; 73:313–20. [DOI] [PubMed] [Google Scholar]
- 35. Sheridan JF, Padgett DA, Avitsur R, Marucha PT. Experimental models of stress and wound healing. World J Surg 2004; 28:327–30. [DOI] [PubMed] [Google Scholar]
- 36. Calcia MA, Bonsall DR, Bloomfield PS, Selvaraj S, Barichello T, Howes OD. Stress and neuroinflammation: a systematic review of the effects of stress on microglia and the implications for mental illness. Psychopharmacology 2016; 233:1637–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mondelli V, Vernon AC, Turkheimer F, Dazzan P, Pariante CM. Brain microglia in psychiatric disorders. Lancet Psychiatry 2017; 4:563–72. [DOI] [PubMed] [Google Scholar]
- 38. Weber MD, Godbout JP, Sheridan JF. Repeated social defeat, neuroinflammation, and behavior: monocytes carry the signal. Neuropsychopharmacology 2017; 42:46–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dahoun T, Calcia MA, Veronese M et al The association of psychosocial risk factors for mental health with a brain marker altered by inflammation: a translocator protein (TSPO) PET imaging study. Brain Behav Immun 2019. doi: 10.1016/j.bbi.2019.05.023. [DOI] [PubMed] [Google Scholar]
- 40. Wei L, Hao J, Kaffman A. Early Life stress inhibits expression of ribosomal RNA in the developing hippocampus. PLOS ONE 2014; 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wei L, Hao J, Lacher RK et al Early‐life stress perturbs key cellular programs in the developing mouse hippocampus. Dev Neurosci (Basel) 2015; 37:476–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wei L, Simen A, Mane S, Kaffman A. Early life stress inhibits expression of a novel innate immune pathway in the developing hippocampus. Neuropsychopharmacology 2012; 37:567–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nelson LH, Lenz KM. Microglia depletion in early life programs persistent changes in social, mood‐related, and locomotor behavior in male and female rats. Behav Brain Res 2017; 316:279–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bilbo SD, Schwarz JM. The immune system and developmental programming of brain and behavior. Front Neuroendocrinol 2012; 33:267–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gomez‐Gonzalez B, Escobar A. Prenatal stress alters microglial development and distribution in postnatal rat brain. Acta Neuropathol 2010; 119:303–15. [DOI] [PubMed] [Google Scholar]
- 46. Roque A, Ochoa‐Zarzosa A, Torner L. Maternal separation activates microglial cells and induces an inflammatory response in the hippocampus of male rat pups, independently of hypothalamic and peripheral cytokine levels. Brain Behav Immun 2016; 55:39–48. [DOI] [PubMed] [Google Scholar]
- 47. Delpech JC, Wei L, Hao J et al Early life stress perturbs the maturation of microglia in the developing hippocampus. Brain Behav Immun 2016; 57:79–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Giovanoli S, Engler H, Engler A et al Stress in puberty unmasks latent neuropathological consequences of prenatal immune activation in mice. Science 2013; 339:1095–9. [DOI] [PubMed] [Google Scholar]
- 49. Giovanoli S, Engler H, Engler A et al Preventive effects of minocycline in a neurodevelopmental two‐hit model with relevance to schizophrenia. Transl Psychiatry 2016; 6:e772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cohen S, Ke X, Liu Q, Fu Q, Majnik A, Lane R. Adverse early life environment increases hippocampal microglia abundance in conjunction with decreased neural stem cells in juvenile mice. Int J Dev Neurosci 2016; 55:56–65. [DOI] [PubMed] [Google Scholar]
- 51. Wang HT, Huang FL, Hu ZL et al Early‐life social isolation‐induced depressive‐like behavior in rats results in microglial activation and neuronal histone methylation that are mitigated by minocycline. Neurotox Res 2017; 31:505–20. [DOI] [PubMed] [Google Scholar]
- 52. Gong Y, Tong LJ, Yang RR et al Dynamic changes in hippocampal microglia contribute to depressive like behavior induced by early social isolation. Neuropharmacology 2018; 135:223–33. [DOI] [PubMed] [Google Scholar]
- 53. Reus GZ, Silva RH, de Moura AB et al Early maternal deprivation induces microglial activation, alters glial fibrillary acidic protein immunoreactivity and indoleamine 2,3‐dioxygenase during the development of offspring rats. Mol Neurobiol 2019; 56:1096–108. [DOI] [PubMed] [Google Scholar]
- 54. Chastain LG, Franklin T, Gangisetty O et al life alcohol exposure primes hypothalamic microglia to later‐life hypersensitivity to immune stress: possible epigenetic mechanism. Neuropsychopharmacology 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Delpech JC, Wei L, Hao J et al Early life stress perturbs the maturation of microglia in the developing hippocampus. Brain Behav Immun 2016; 57:79–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Paolicelli RC, Ferretti MT. Function and dysfunction of microglia during brain development: consequences for synapses and neural circuits. Front Synaptic Neurosci 2017; 9:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhan Y, Paolicelli RC, Sforazzini F et al Deficient neuron–microglia signaling results in impaired functional brain connectivity and social behavior. Nat Neurosci 2014; 17:400–6. [DOI] [PubMed] [Google Scholar]
- 58. Paolicelli RC, Bolasco G, Pagani F et al Synaptic pruning by microglia is necessary for normal brain development. Science 2011; 333:1456–8. [DOI] [PubMed] [Google Scholar]
- 59. Paolicelli RC, Gross CT. Microglia in development: linking brain wiring to brain environment. Neuron Glia Biol 2011; 7:77–83. [DOI] [PubMed] [Google Scholar]
- 60. Schafer DP, Lehrman EK, Stevens B. The ‘quad‐partite’ synapse: microglia–synapse interactions in the developing and mature CNS. Glia 2013; 61:24–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Schafer DP, Lehrman EK, Kautzman AG et al Microglia sculpt postnatal neural circuits in an activity and complement‐dependent manner. Neuron 2012; 74:691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Basilico B, Pagani F, Grimaldi A et al Microglia shape presynaptic properties at developing glutamatergic synapses. Glia 2019; 67:53–67. [DOI] [PubMed] [Google Scholar]
- 63. Weinhard L, di Bartolomei G, Bolasco G et al Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat Commun 2018; 9:1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Weinhard L, Neniskyte U, Vadisiute A et al Sexual dimorphism of microglia and synapses during mouse postnatal development. Dev Neurobiol 2018; 78:618–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Paolicelli RC, Gross CT. Microglia in development: linking brain wiring to brain environment. Neuron Glia Biol 2011; 7:77–83. [DOI] [PubMed] [Google Scholar]
- 66. Danese A, Lewis SJ. Psychoneuroimmunology of early‐life stress: the hidden wounds of childhood trauma? Neuropsychopharmacology 2017; 42:99–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wohleb ES, Delpech JC. Dynamic cross‐talk between microglia and peripheral monocytes underlies stress‐induced neuroinflammation and behavioral consequences. Prog Neuro‐Psychopharmacol Biol Psychiatry 2017; 79:40–8. [DOI] [PubMed] [Google Scholar]
- 68. Elenkov IJ, Wilder RL, Chrousos GP, Vizi ES. The sympathetic nerve–an integrative interface between two supersystems: the brain and the immune system. Pharmacol Rev 2000; 52:595–638. [PubMed] [Google Scholar]
- 69. Gosain A, Muthu K, Gamelli RL, DiPietro LA. Norepinephrine suppresses wound macrophage phagocytic efficiency through alpha‐ and beta‐adrenoreceptor dependent pathways. Surgery 2007; 142:170–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Curtis AM, Bellet MM, Sassone‐Corsi P, O’Neill LA. Circadian clock proteins and immunity. Immunity 2014; 40:178–86. [DOI] [PubMed] [Google Scholar]
- 71. Riddell NE, Burns VE, Wallace GR et al Progenitor cells are mobilized by acute psychological stress but not beta‐adrenergic receptor agonist infusion. Brain Behav Immun 2015; 49:49–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Case AJ, Zimmerman MC. Sympathetic‐mediated activation versus suppression of the immune system: consequences for hypertension. J Physiol 2016; 594:527–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Heidt T, Sager HB, Courties G et al Chronic variable stress activates hematopoietic stem cells. Nat Med 2014; 20:754–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. McKim DB, Patterson JM, Wohleb ES et al Sympathetic release of splenic monocytes promotes recurring anxiety following repeated social defeat. Biol Psychiatry 2016; 79:803–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wohleb ES, Powell ND, Godbout JP, Sheridan JF. Stress‐induced recruitment of bone marrow‐derived monocytes to the brain promotes anxiety‐like behavior. J Neurosci 2013; 33:13820– 13833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Beydoun MA, Beydoun HA, Dore GA et al White blood cell inflammatory markers are associated with depressive symptoms in a longitudinal study of urban adults. Transl Psychiatry 2016; 6:e895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. McKim DB, Weber MD, Niraula A et al Microglial recruitment of IL‐1 beta‐producing monocytes to brain endothelium causes stress‐induced anxiety. Mol Psychiatry 2018; 23:1421–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Wohleb ES, Mckim DB, Sheridan JF, Godbout JP. Monocyte trafficking to the brain with stress and inflammation: a novel axis of immune‐to‐brain communication that influences mood and behavior. Front Neurosci‐Switz 2015; 8:447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Wohleb ES, McKim DB, Shea DT et al Re‐establishment of anxiety in stress‐sensitized mice is caused by monocyte trafficking from the spleen to the brain. Biol Psychiatry 2014; 75:970–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Menard C, Pfau ML, Hodes GE et al Social stress induces neurovascular pathology promoting depression. Nat Neurosci 2017; 20:1752‐+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Weber M, McKim D, Sawicki C, Niraula A, Godbout J, Sheridan J. Microglial Recruitment of monocytes to the brain causes stress‐induced anxiety. Neuropsychopharmacology 2016; 41:S461–S462. [Google Scholar]
- 82. Johnson FK, Kaffman A. Early life stress perturbs the function of microglia in the developing rodent brain: new insights and future challenges. Brain Behav Immun 2018; 69:18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Johnson FK, Delpech JC, Thompson GJ et al Amygdala hyper‐connectivity in a mouse model of unpredictable early life stress. Transl Psychiatry 2018; 8:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. McLaughlin KA, Sheridan MA, Alves S, Mendes WB. Child maltreatment and autonomic nervous system reactivity: identifying dysregulated stress reactivity patterns by using the biopsychosocial model of challenge and threat. Psychosom Med 2014; 76:538–46. [DOI] [PMC free article] [PubMed] [Google Scholar]