

Abstract
Congenital heart defects, dysmorphic facial features and intellectual developmental disorders (CHDFIDD) syndrome in humans was recently associated with mutation in CDK13 gene. In order to assess the loss of function of Cdk13 during mouse development, we employed gene trap knock-out (KO) allele in Cdk13 gene. Embryonic lethality of Cdk13-deficient animals was observed by the embryonic day (E) 16.5, while live embryos were observed on E15.5. At this stage, improper development of multiple organs has been documented, partly resembling defects observed in patients with mutated CDK13. In particular, overall developmental delay, incomplete secondary palate formation with variability in severity among Cdk13-deficient animals or complete midline deficiency, kidney failure accompanied by congenital heart defects were detected. Based on further analyses, the lethality at this stage is a result of heart failure most likely due to multiple heart defects followed by insufficient blood circulation resulting in multiple organs dysfunctions. Thus, Cdk13 KO mice might be a very useful model for further studies focused on delineating signaling circuits and molecular mechanisms underlying CHDFIDD caused by mutation in CDK13 gene.
Keywords: cyclin-dependent kinase (CDK), cyclin, transcription regulation, development, mouse, cyclin-dependent kinase 13, cyclin K
Introduction
Recently, de novo missense variants in Cyclin-dependent kinase 13 (CDK13) gene have been identified as an emerging factor involved in the onset of congenial heart defects (CHD) in humans (Sifrim et al., 2016). Documented CHD cases were characterized by ventral and atrial septal defects accompanied by pulmonary valve abnormalities. CHD patients had syndromic facial gestalt, and two patients had agenesis of the corpus callosum. Additional mutations within the CDK13 gene were recognized in humans resembling at clinical level many symptoms previously associated with loss of function of CDK13 or newly described symptoms, such as autism spectrum disorder, seizures, feeding difficulties and craniofacial dysmorphism including short upslanting palpebral fissures, hypertelorism or telecanthus, medial epicathic folds, low-set, posteriorly rotated ears and small mouth with thin upper lip vermilion (Hamilton et al., 2018). In addition, genome wide search for de novo mutations responsible for developmental disorders in patients in Great Britain and Republic of Ireland led to identification of 14 genes that previously lacked compelling evidence of involvement in developmental disorders, among them the CDK13 gene (Deciphering Developmental Disorders Study, 2017). However, heart defects do not seem to be the key feature of this disorder since patients with heterozygous constitutional mutation in CDK13 lacking cardiac anomalies were reported by two groups (Carneiro et al., 2018; Uehara et al., 2018). Spectrum of clinical phenotypes of patients with CDK13 mutations varies from mild to severe with the ubiquitous intellectual disability and developmental delay (ID/DD) (van den Akker et al., 2018). Based on these observations, congenital heart defects, dysmorphic facial features and intellectual development disorder (CHDFIDD) have been recognized as novel syndrome caused by de novo variants of CDK13 gene (Sifrim et al., 2016; Bostwick et al., 2017; Carneiro et al., 2018; van den Akker et al., 2018).
Human CDK13 protein consists of 1512-amino acids with a conserved kinase domain surrounded by N- and C-terminal arms of undefined function (Kohoutek and Blazek, 2012). In order to be active, CDK13 binds cyclin K (CycK) and forms enzymatically active complex (Ko et al., 2001; Even et al., 2006; Bartkowiak et al., 2010; Blazek et al., 2011; Cheng et al., 2012; Dai et al., 2012; Kohoutek and Blazek, 2012; Liang et al., 2015). CDK13 belongs to the family of transcription-associated cyclin-dependent kinases phosphorylating the carboxyl-terminal domain (CTD) of RNA polymerase II (RNAPII). In particular, the CDK13 phosphorylates serine 2 (Ser2) and to a lesser extend also serine 5 (Ser5) within Y1S2P3T4S5P6S7 heptapeptides within the CTD of RNAPII in vitro (Greifenberg et al., 2016). Nevertheless, downregulation of CDK13 in tumor derived cell lines had a very small, if any, effect on level of Ser2 within CTD of RNAPII (Blazek et al., 2011; Greifenberg et al., 2016). In parallel to CDK13, there is CDK12 in mammalian cells able to associate with CycK as well (Blazek et al., 2011; Dai et al., 2012; Liang et al., 2015). Even though CDK13 shares high amino acid similarity with CDK12, both kinases appear to function in mutually exclusive complexes in mammalian cells (Blazek et al., 2011; Kohoutek and Blazek, 2012; Greenleaf, 2018).
In comparison to CDK12, there is a limited number of papers envisioning the likely function of CDK13 in various biological processes. For instance, the CDK13 was proposed to be involved in oncogenesis; yet, its precise function is still under examination (Kim et al., 2012; Pan et al., 2012). Even though factors involved in RNA processing, RNA splicing, polyadenylation and RNA cleavage were demonstrated to bind CDK13 as a result of global protein-protein interactions, truly associating partners of this kinase are still unknown (Davidson et al., 2014; Bartkowiak and Greenleaf, 2015; Liang et al., 2015). In addition to involvement of CDK13 in diverse cellular processes, this protein participates in regulation of alternative splicing of HIV-1 or influenza virus replication, thus suppressing viral production (Berro et al., 2008; Bakre et al., 2013). In developing mouse embryos and murine cells, CDK13 regulates hematopoiesis, stemness and axonal elongation, suggesting an important function in neuronal development (Pan et al., 2012; Chen et al., 2014).
To this date, the impact of complete loss of Cdk13 function during mammalian development has not been investigated. Therefore, we employed a Cdk13 knock-out (KO) mouse model to explore a novel role of Cdk13 during mouse embryonic development. We observed embryonic lethality of Cdk13 KO animals at the embryonic day 16.5. At this stage, improper development of multiple organs has been observed (heart, brain, kidney, liver, and palate formation) resembling phenotype of human patients with de novo missense variants of CDK13 gene. Therefore, our Cdk13-deficient mice may become an important model to study dysregulation of developmental processes occurring in human patients.
Results
Disruption of Cdk13 Gene Leads to Embryonic Lethality in Mice
To examine the role of CDK13 during mouse development, the mice carrying Cdk13tm1a allele were generated at the Transgenic and Archiving Module CCP (Institute of Molecular Genetics of the CAS, Prague). The Cdk13tm1a allele of Cdk13 gene enables cessation of transcription due to presence of two strong poly A sites leading to production of the aberrant transcript of Cdk13 mRNA resulting in non-functional truncated form of CDK13 protein harboring only N-terminal part of this protein, without kinase domain and C-terminal part (Figure 1A). Heterozygous Cdk13tm1a/+ mice were intercrossed to obtain Cdk13tm1a/tm1a offspring. Newborn mice were genotyped with specific sets of primers able to distinguish inserted cassette (257 bp PCR product, KO) and wild-type (179 bp PCR product, WT) alleles of Cdk13 gene (Figure 1B). Although the offspring with Cdk13 WT and heterozygous alleles was born at the expected Mendelian ratio, appeared normal and fertile, Cdk13tm1a/tm1a mice were not born at all. This finding suggested that a homozygous deficiency in Cdk13 gene leads to embryonic lethality in mice. To determine the precise stage, when the embryonic lethality occurs, mouse embryos were collected at various gestation time points (Table 1). There were no living Cdk13tm1a/tm1a embryos after the embryonic stage 15.5 (E15.5) judging by the lack of their heart beating. Moreover, from E13.5 to E16.5, we observed increased number of absorbed embryos as a reflection of empty decidua (Table 1). Based on these finding, we concluded that the deficiency of Cdk13 causes severe adverse developmental defects and consequent death from E14.5 resulting in total lethality before E16.5.
FIGURE 1.

Generation of Cdk13tm1a mice. (A) Scheme of Cdk13tm1a allele. The Cdk13tm1a allele consists of strong splicing acceptor (SA), β-galactosidase gene (lacZ) and neomycine resistance gene (neo) both with poly A sites (pA), being surrounded by two FRT sites within intron 2, having two and one lox P sites within intron 2 and 4. (B) PCR genotyping from E12.5 embryos using specific primers distinguishing Cdk13tm1a, mutant (KO, upper bands) and Cdk13+, wild-type, allele (WT, lower bands). (C) Brain extracts were prepared from E14.5 embryos of Cdk13+/+, Cdk13tm1a/+ and Cdk13tm1a/tm1a mice and protein levels of Ser2 and Ser5 of RNAPII, CDK13 and γ-catenin (CTNNG) were evaluated by Western blotting. The short and long expositions for CDK13 are presented to demonstrate residual expression of CDK13 in Cdk13tm1a/tm1a embryos.
TABLE 1.
Genotypes of offspring from Cdk13tm1a/+ intercross.
| Empty | |||||
| Stage |
Cdk13+/+ |
Cdk13tm1a/+ |
Cdk13tm1a/tm1a |
Litters | decidua |
| (25%) | (50%) | (25%) | |||
| E9.5 | 4 (21.1%) | 12 (63.2%) | 3 (15.8%)* | 3 | 2 |
| E10.5 | 4 (20%) | 10 (50%) | 6 (30%)* | 2 | 0 |
| E11.5 | 14 (31.8%) | 23 (52.3%) | 7 (15.9%)* | 6 | 4 |
| E12.5 | 31 (30.7%) | 52 (51.5%) | 18 (17.8%)*,# | 15 | 13 |
| E13.5 | 6 (16.2%) | 18 (48.6%) | 13 (35.1%)* | 5 | 5 |
| E14.5 | 48 (27.6%) | 95 (54.6%) | 31 (17.8%)*,# | 26 | 9 |
| E15.5 | 21 (27.5%) | 34 (58%) | 10 (14.5%)* | 11 | 5 |
| E16.5 | 8 (25%) | 18 (56.3%) | 6 (18.7%)*,# | 4 | 0 |
| P0 | 39 (33.1%) | 79 (66.9%) | 0 | 28 |
*Growth retardation, #dead embryo.
To confirm the loss of CDK13 protein in Cdk13tm1a/tm1a mice, developing brain from WT and Cdk13tm1a/tm1a homozygous embryos at E14.5 were collected and western blot analyses were carried out with specific antibodies recognizing CDK13 protein. As expected, the WT form of CDK13 was present in Cdk13+/+ and Cdk13tm1a/+ mice, but surprisingly, corroborated residual expression of CDK13 protein was detected in the embryonic brain of Cdk13tm1a/tm1a homozygous embryos (Figure 1C), suggesting that Cdk13tm1a/tm1a mice represent a hypomorphic mutant phenotype. Because the anti-Cdk13 antibody used in western blot recognizes its N-terminal part of CDK13, we were curious if the truncated form of CDK13, as a result of terminated transcription within intron 2, will be expressed in mice bearing the Cdk13tm1a allele. Indeed, the truncated form of CDK13 was detected in Cdk13tm1a/+ and Cdk13tm1a/tm1a animals (Supplementary Figure S1). Since CDK13 was demonstrated to phosphorylate CTD of RNAPII in vitro, we decided to evaluate phosphorylation status of CTD in hypomorphic mice. Thus, the effect of CDK13 downregulation on Ser2 was evaluated in the animal tissue. Even though expression of CDK13 was significantly lowered in Cdk13tm1a/tm1a mice, no effect on either Ser2 or Ser5 phosphorylation within CTD of RNAPII was detected (Figure 1C). The Ser5 phosphorylation was checked in parallel since it is phosphorylated by other CDK kinase, CDK7 in particular (Kohoutek, 2009).
Cdk13 Loss Causes Growth Retardation, Developmental Delay and Its Failure
To examine deficiency of CDK13 protein in Cdk13tm1a/tm1a mice, morphology at different embryonic stages compared to the WT animals was examined and various abnormalities were observed in Cdk13tm1a/tm1a embryos at each stage of gestation (Supplementary Figure S2). Observed growth retardation of Cdk13-deficient mice is presented in detail (Figure 2); however, the severity of the developmental delay was variable. On average, the Cdk13tm1a/tm1a embryos appeared to be one embryonic day behind in comparison to their littermate controls as evidenced by the shallow indentation of the footpad, which is characteristic of embryos at E13.5 (Figure 2F, bottom). In contrast, Cdk13+/+ littermates exhibited deep indentations between the developing toes (not yet separated), what is characteristic of embryos at 14.5 (Figure 2F, top). In addition, the retarded embryo exhibited nuchal edema (black arrow, Figure 2B), which correlates with the presence of cardiovascular phenotypes. Occasionally, the pericardial effusion were detected in developing Cdk13tm1a/tm1a embryos, most likely caused by dysfunction of the heart (Figure 2D). Summary of various developmental defects associated with hypomorphic Cdk13tm1a allele is presented in Supplementary Table S1.
FIGURE 2.

Comparison of gross morphology of wild-type, Cdk13tm1a/+ and Cdk13tm1a/tm1a embryos at various stages. Cdk13tm1a/tm1a embryos display significant growth retardation compared to wild-type and heterozygous embryos. (D–F) Detailed images of Cdk13tm1a/tm1a at relevant developmental stages. (A) Heterozygous and Cdk13tm1a/tm1a embryos at E12.5. (D) Occasional chest wall deformities manifest at hypomorphs. (B) Wild-type and Cdk13tm1a/tm1a embryos at E13.5. Cdk13tm1a/tm1a embryo exhibits nuchal edema (black arrow). (E) Hypervascularization of the peripheral vessels capillaries (black arrow). (C) Wild-type and Cdk13tm1a/tm1a embryos at E14.5. (F, top) Wild-type embryo demonstrates deep indentations between the developing fingers of embryos E14.5, although not yet separated. (F, bottom) In contrast, Cdk13tm1a/tm1a embryo appears to be 1 day delayed in development as evidenced by the shallow indentation of the footpad, which is characteristic of embryos E13.5.
Cdk13 Is Indispensable for the Development of Several Organs
To narrow down possible cause of embryonic lethality, embryos at E14.5 were contrasted with Lugol’s solution to visualize gross morphology of individual soft tissues by microCT (Figure 3). Indeed, several developmental abnormalities were detected within developing embryos. The heart wall of both ventricles in Cdk13tm1a/tm1a embryos (Figures 3B,D) appeared thinner in comparison to Cdk13+/+ littermate controls (Figures 3A,C). In addition, lung, liver and kidney in Cdk13tm1a/tm1a embryos were smaller and undeveloped (Figures 3D,F,H) in comparison to Cdk13+/+ littermates (Figures 3C,E,G). However, detailed 3D reconstruction of liver and kidney (Figures 3I–P) with movable display of E14.5 embryos of Cdk13tm1a/tm1a and Cdk13+/+ genotypes (Supplementary Figures S3A,B) revealed no defect in general gross morphology of these organs.
FIGURE 3.

MicroCT analysis of wild-type and Cdk13tm1a/tm1a embryos. High-contrast differentiation resolution by X-ray computed microtomography, where Lugol’s staining was used to visualize the soft tissues. Sagittal sections through body midline (A,B) and more lateral plane at E15.5 (C,D). Horizontal sections through lung (E,F) and liver (G,H). 3D reconstruction of kidney and liver in the right side view of embryo (I,J), left side view (K,L) and caudal view (M,N) with embryo outlined in gray where segmentation of serial sections was used for the liver and kidney reconstruction. (O,P) High power of 3D imaging for liver and kidney. Horizontal view (Q,R) and sagittal detailed view (S,T) on kidney and suprarenal gland. Abbreviation used for individual organs: ag, adrenal gland; d, diencephalon; h, heart; hb, hindbrain; k, kidney; l, lung; li, liver; mb, midbrain; t, tongue; te, telencephalon; s, stomach; sc, spinal cord. Scale bar = 1 mm.
To uncover possible discrepancies in developmental speed of individual organs, the volume analysis was performed with normalization to total body volume of given embryo. Liver size of Cdk13tm1a/tm1a embryos represented only about 46% in comparison to Cdk13+/+ littermates (Figures 3, 11). Similarly, kidney size of Cdk13tm1a/tm1a animals comprised only about 52% in comparison to Cdk13+/+. In parallel, histological sections of selected organs were analyzed at stages between E14.5 and E16 (Figure 4). Decelerated development of kidneys was identified in Cdk13tm1a/tm1a embryos at E14.5 and E16 (Figures 4D,F) including nephron differentiation as shown by altered proportional representation of individual nephron stages at E14.5 (Figure 4G). Moreover, statistically significant reduction in the number of S-shaped bodies and glomeruli was observed in Cdk13tm1a/tm1a embryos (Figure 4H). At lethality stage E16.5, kidney tissue exhibited tissue abrogation with only few, much reduced tubules visible (data not shown), very likely caused by general pre-necrotic changes.
FIGURE 11.

Comparison of Cdk13tm1a and Cdk13tm1d mice at E13.5 High-contrast differentiation resolution by X-ray computed microtomography where Lugol’s staining was used to visualize the soft tissues. The Cdk13tm1a/tm1a, Cdk13tm1d/tm1d and their littermate controls were analyzed at E13.5. (A–D) Overall view of Cdk13+/+, Cdk13tm1a/tm1a, and Cdk13tm1d/tm1d embryos. Both mutants exhibited smaller body size, with severe growth retardation in Cdk13tm1d/tm1d animals. (A’–D’) 3D reconstruction of heart (red color), kidney (violet color) and liver (light pink color) in right side view of embryo, with embryo outlined in gray where segmentation of serial sections was used for the reconstruction. Liver in Cdk13tm1a/tm1a and Cdk13tm1d/tm1d embryos were smaller than in Cdk13+/+ embryos. (E–H’) Frontal view of embryos with 3D reconstruction of heart, kidneys and liver. Clear deficiency in development of midfacial structures in Cdk13tm1d/tm1d embryos in contrast to Cdk13+/+ animals was observed. (I–L) 3D reconstruction of heart. The heart of Cdk13tm1a/tm1a and Cdk13tm1d/tm1d embryos were smaller in comparison to control mice with severe ventricle deficiency detected in Cdk13tm1d/tm1d animals. (M–P) 3D reconstruction of liver. Generally smaller liver were observed in Cdk13tm1a/tm1a and Cdk13tm1d/tm1d embryos in comparison to Cdk13+/+ littermate control mice. Liver in Cdk13tm1d/tm1d mice exhibit abrogated liver parts arrangements. (Q–T) 3D reconstruction of kidneys. Kidneys are smaller with abnormal shape in Cdk13tm1a/tm1a and Cdk13tm1d/tm1d embryos. Scale bar (A–H) = 1,5 mm, Scale bar (I–L) = 350 μm, scale bar (M,P) = 500 μm, and scale bar (Q–T) = 250 μm.
FIGURE 4.
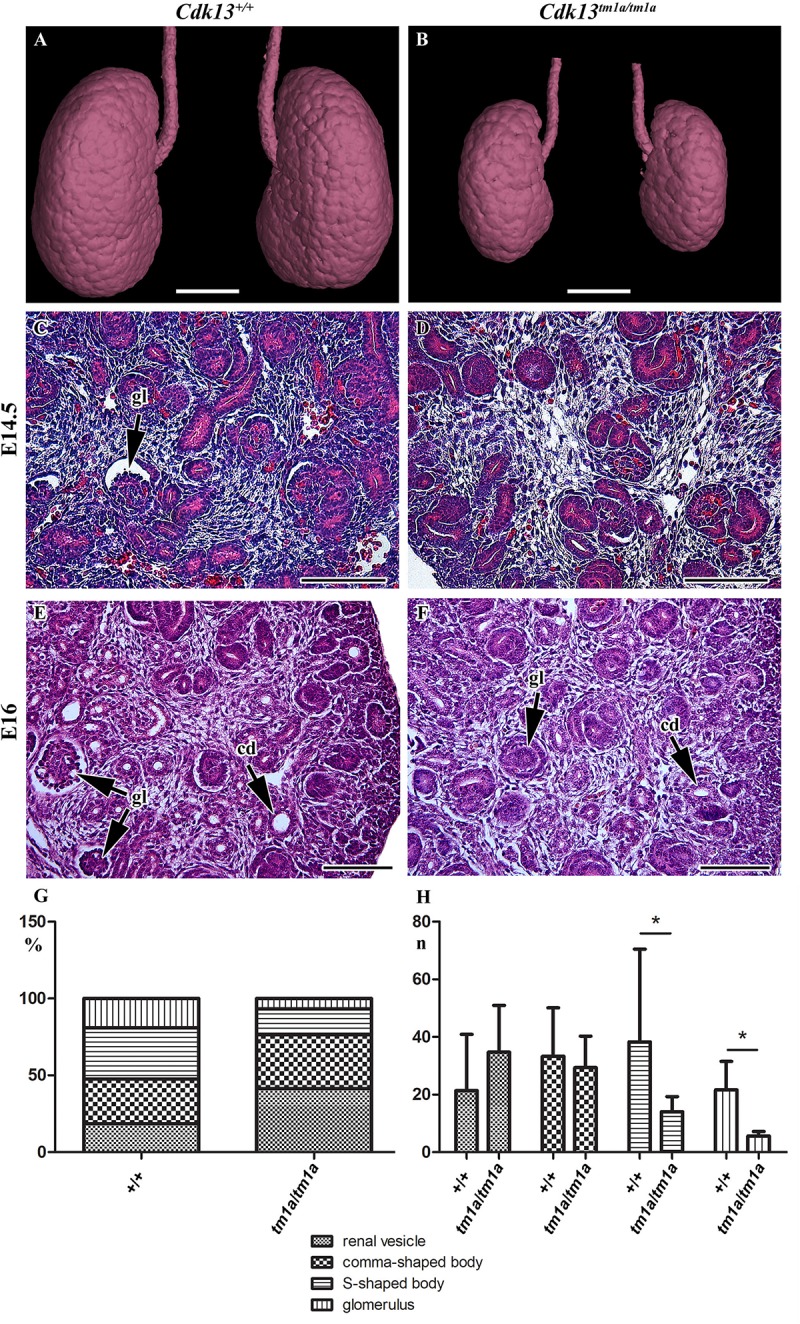
Gross anatomy and microscopic structure of kidney in wild-type and Cdk13tm1a/tm1a embryos. (A,B) High power view on segmented kidneys from Lugol’s stained sections and visualized by X-ray computed microtomography. Growth retardation of kidney is visible in Cdk13tm1a/tm1a embryos at E14.5 (D) and E16 (F) in contrast to littermate wild-type, Cdk13+/+, controls (C for E14.5 and E for E16). (E,F) Only very few just forming glomeruli (gl) were found in Cdk13tm1a/tm1a embryos. (G) Relative quantification of individual developmental stages of nephrogenesis in Cdk13+/+ and Cdk13tm1a/tm1a embryos. (H) Increased amount of renal vesicles together with the reduction of S-shaped bodies and glomeruli (gl) was found in Cdk13tm1a/tm1a embryos. The graph values denote median ± s.d., *p < 0.05, by unpaired t-test. Scale bar (A,B) = 0.3 mm, scale bar (C–H) = 100 μm.
Brains of Cdk13tm1a/tm1a embryos appeared developmentally delayed as demonstrated by reduced size as compared to littermate controls. Depicted in Figure 5 are E14.5 controls and Cdk13tm1a/tm1a mutant samples from two separate litters. The two litters were developmentally at different stages, pre-palatal fusion in the control embryo depicted (Figures 5A,B) and post-palatal fusion in the control embryo depicted (Figures 5E,F). In order to assess the developmental delay in Cdk13tm1a/tm1a embryos the cell proliferation was examined by Ki67 staining, a marker of proliferating cells (Figures 5A’–H’, embryos A and C, as well as E and G are littermates, representative pictures of two embryos are shown to display variability in mutant phenotype). As evident from performed quantification, there was decrease in number of proliferating cells in Cdk13tm1a/tm1a embryos in comparison to Cdk13+/+ littermate controls, but this decrease was not statistically significant (Figure 5I).
FIGURE 5.
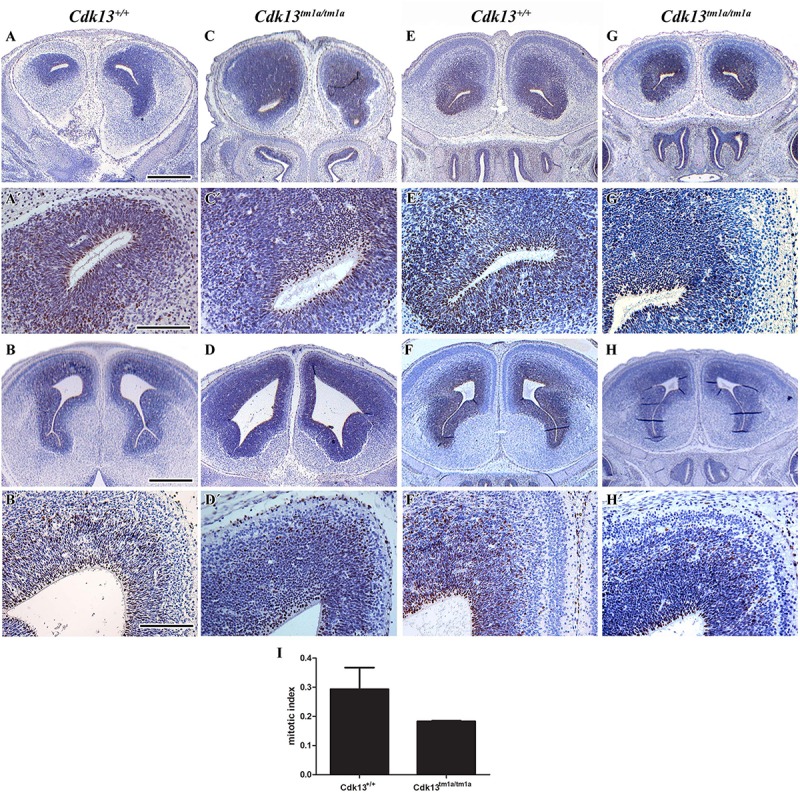
Cell proliferation in brain area in wild-type and Cdk13tm1a/tm1a embryos. (A,A’,B,B’) Cdk13+/+ and (C,C’,D,D’) Cdk13tm1a/tm1a embryos before palatal shelves fusion. (E,E’,F,F’) Cdk13+/+ and (G,G’,H,H’) Cdk13tm1a/tm1a embryos after palatal shelves fusion. Immunohistochemical nuclear labeling of Ki67-positive cells in the frontal head sections in the lower power view (A–H) and in detail (A’–H’). (I) Mitotic index was counted as the ratio between Ki67-positive cells and total amount of prosencephalon cells in three biological triplicates for each group. The graph values denote mean ± s.d, difference is not statistically significant according to unpaired t-test (p-value: 0.1047). Ki67-positive cells - brown nuclei, Ki67-negative cells - blue nuclei (hematoxylin). Scale bar (A–C) = 1 mm; scale bar (A’–C’) = 100 μm.
The analysis of craniofacial area revealed also defective palatal shelves development in several Cdk13tm1a/tm1a embryos resulting in their insufficient horizontal growth and the formation of the cleft palate at E15.5 (Figure 6) in Cdk13tm1a/tm1a mouse. Incomplete secondary palate formation exhibited variability in severity among Cdk13-deficient animals. Observed penetrance of secondary cleft palate was 2/4 animals at E15.5. In addition, Cdk13tm1a/tm1a mouse at E15.5 had smaller number of initiated nasal glands in comparison to the controls (Supplementary Figure S4, compare A–D and B–E).
FIGURE 6.

Transversal sections of the head in control Cdk13+/+ and Cdk13tm1a/tm1a embryos at E15.5. Rostro-caudal view in Cdk13+/+ animal (A) and four Cdk13tm1a/tm1a mutant mice to show variability in the secondary palate morphology (B–E). Palatal shelves do not meet each other in the midline (B”’,E”’) and cleft of secondary palate is visible. Abnormal shape of palatal shelves was observed also caudally with cleft expanding into the soft palate area. (A’–A””, B’–B””, C’–C””, D’–D””) are transversal sections of head in individual embryos in rostrocaudal direction. Scale bar = 100 μm.
Embryonic Lethality in Cdk13tm1a/tm1a Mice Is Due to Heart Failure
The heart is the one of the first organs to form during mammalian development. During heart development, significant changes in organ morphology and cardiomyocyte differentiation and organization reflect the increasing needs of growing embryos for nutrition and oxygen supply. Any of these developmental steps are critical for further development of whole embryos. Therefore, we analyzed the microscopic structure of the heart and found that the heart wall of Cdk13tm1a/tm1a mice embryos was less compact in comparison to the heart wall of Cdk13+/+ mice (Figures 7C,D). Further, apparent disruption of tissue architecture was detected in Cdk13tm1a/tm1a embryos with the reduction of myocardium (Figure 7, compare C, E and G to D, F and H). The heart volume was slightly increased to 103% (organ ratio to total body volume) in Cdk13tm1a/tm1a mice compared to Cdk13+/+ animals. However, the total volume of heart tissue to the organ volume was significantly lower in case of Cdk13tm1a/tm1a embryos (62.5%) in comparison to WT littermates (80%) suggestive of the thinner heart wall at E15.5 in Cdk13tm1a/tm1a embryos. Percent soft tissue volumes were measured by microCT using Bruker-microCT CT-analyzer, where the object volume representing soft tissues was divided by total VOI volume. The VOI area referring to the heart volume was selected from the dataset manually. Also, decreased expression of myosin was detected in ventricle myocardium of 14.5 hearts (Supplementary Figure S5, compare B and B’ to E and E’).
FIGURE 7.
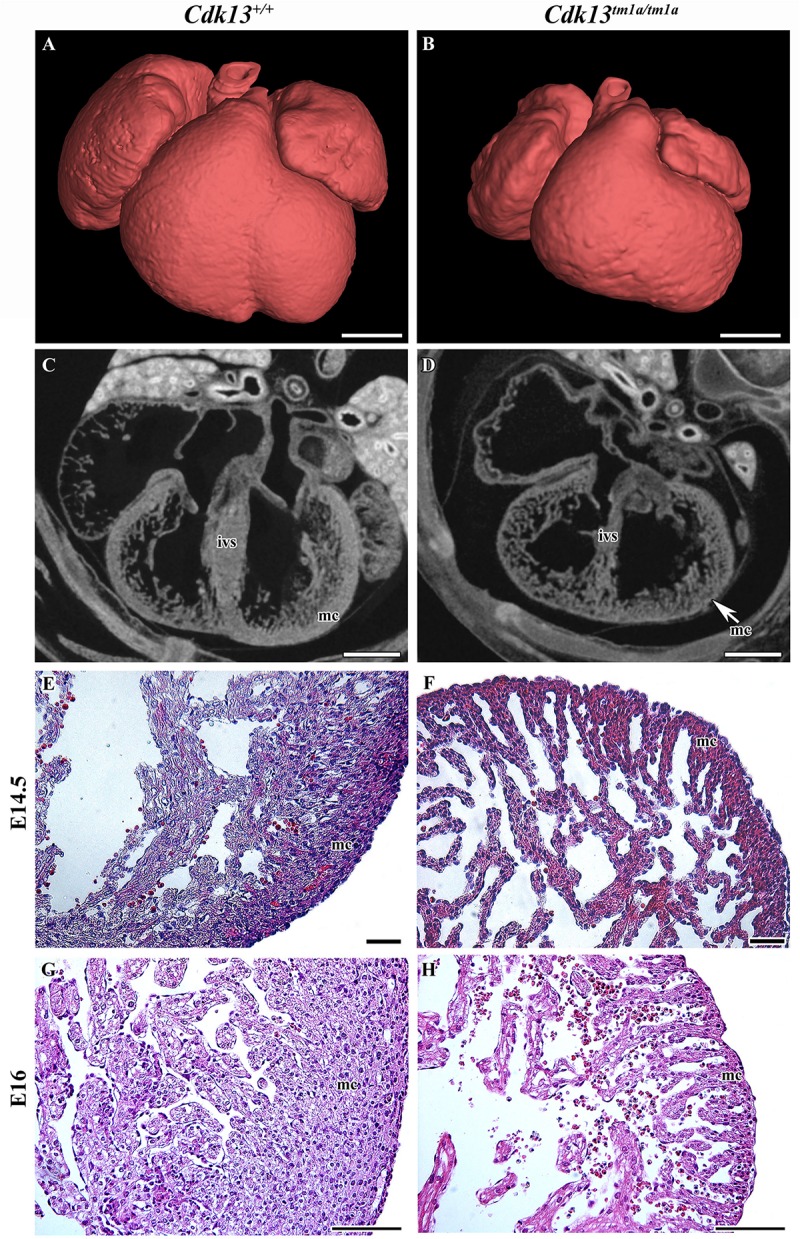
Gross anatomy and microscopic structure of heart in wild-type and Cdk13tm1a/tm1a embryos. (A,B) High power view on segmented heart from Lugol’s stained sections and visualized by X-ray computed microtomography. (C,D) Sagittal section of heart from Lugol’s staining visualized by X-ray computed microtomography. Smaller size of heart with broadening of ventricles was visible in Cdk13tm1a/tm1a animals. Abbreviations for the organ structures: ivs, interventricular septum; mc, myocardium. (E,F) At E14.5, myocardium (mc) of Cdk13tm1a/tm1a mice is already thinner in the ventricle area with small amount of cardiomyocytes layers, which is in contrast to thick wall of Cdk13+/+ littermate controls. Later, apparent disruption of tissue architecture with enlarged intercellular spaces and small amount of cardiomyocytes was observed in Cdk13tm1a/tm1a embryos at later stages at E16 (G,H). Scale bar (A,B) = 0.4 mm, scale bar (C,D) = 350 μm, and scale bar (E–H) = 100 μm.
Since the heart mass of Cdk13tm1a/tm1a E14.5 – E16 embryos is significantly reduced with hypomorphic muscular layers of myocardium compared with Cdk13+/+ mice (heart/body ratio), we presume that heart developmental defect is the cause of embryonic lethality. In order to evaluate cardiac circulatory physiology, we performed non-invasive ultrasound Doppler imaging to quantitatively assess the hemodynamic function in E14.5 and E15.5 embryos. Out of nine Cdk13tm1a/tm1a embryos dissected at E14.5 stage, only one was found dead (Table 2). All the other Cdk13tm1a/tm1a embryos at this stage exhibited comparable blood flow velocities and velocity-time integral (VTI) in dorsal aorta (Figures 8A,B). However, this situation changed dramatically at embryonic stage E15.5 (Figures 8C,D), where only few Cdk13tm1a/tm1a embryos retained normal blood flow parameters (5/16), while the rest of the embryos heart functions declined (standard measurements were not possible due to irregular or spare heart beating 11/16); moreover, an increased portion of embryos were already dead (Table 2).
TABLE 2.
Summary of ultrasound scanned embryos at E14.5 and E15.5.
| Stage |
Number of embryos |
Litters |
Number of Cdk13tm1a/tm1a |
|||
| Cdk13+/+ | Cdk13tm1a/+ | Cdk13tm1a/tm1a | Heart beating | Dying/dead | ||
| E14.5 | 11 | 27 | 9 | 6 | 8 | 1 |
| E15.5 | 14 | 36 | 17 | 8 | 6 | 11 |
FIGURE 8.

Hemodynamic analysis of wild-type, Cdk13tm1a/+ and Cdk13tm1a/tm1a embryos. The non-invasive ultrasound Doppler imaging to quantitatively assess the heart hemodynamic function in E14.5 and E15.5 embryos was carried out. (A,C) Velocity-time integral in dorsal aorta (DA VTI) and (B,D) Peak Doppler blood flow velocity measured on E14.5 (A,B) and E15.5 (C,D) day old embryos, respectively.
The assessment of cardiac function in developmental interval E14.5 – E15.5 embryos showed dramatic failure in heart function, probably corresponding to increasing needs of embryos at E15.5 for blood supply in growing organ systems, which is very challenging for the defective heart to achieve. The preserved blood flow in few Cdk13tm1a/tm1a embryos was probably due to the observed variability of the embryo size. Our findings suggest that the heart failure appears in most cases during the transition from E14.5 to E15.5.
Cdk13 Is Expressed in Affected Organs in the Prenatal and Also Postnatal Period
Currently, there is limited information about protein expression pattern of CDK13 either in developing or adult organs; therefore, we decided to evaluate expression of CDK13 protein in developing organs. First, the western blot of CDK13 protein was carried out in selected developing organs (Figure 9). As expected, expression of CDK13 was detected in organs with abnormal embryonic development. The highest protein level of CDK13 was detected in the brain, then lung, kidney and heart (Figure 9). In case of detection of CDK13 in the developing heart, four times concentrated protein lysates had to be used to detect any reproducible signal.
FIGURE 9.

Expression of CDK13 protein in embryonic mouse organs. Protein extracts were obtained from brain, heart, kidneys and lung of E14.5 Cdk13, Cdk13tm1a/+ and Cdk13tm1a/tm1a embryos and protein levels of CDK13, Cyclin T1 (CYCT1) and γ-catenin (CTNNG) were evaluated by Western blotting. The highest expression of CDK13 was observed in brain and kidney, with substantial expression in lung and heart in Cdk13+/+ embryos.
To explore gene expression of Cdk13 in adult tissues and organs, the particular organs were isolated and the expression of Cdk13 was examined by activity of β-galactosidase (Supplementary Figure S6). Cdk13 was strongly expressed in the retina of the eye, testes, ovary, uterus, gall bladder (Supplementary Figures S6A,F–I). To a lesser extent, the Cdk13 was detected in the urinary bladder (Supplementary Figure S6D). Interestingly, rather localized, yet strong expression within organ structure was observed in renal pelvis in kidney, thyroid gland and heart atrium, with substantial expression in the heart ventricle (Supplementary Figures S6B,C,E).
Cdk13tm1d/tm1d Mice Exhibits More Severe Phenotype and Earlier Lethality
As we observed residual expression of Cdk13 in analyzed organs of hypomorphic Cdk13tm1a/tm1a mice, we decided to cross Cdk13tm1a mice with Flp-deleter mice and Cre-deleter (detailed description of the utilized transgenic strains is provided at the Section “Experimental Procedure”) mice to generate Cdk13tm1d allele with deleted exons 3 and 4 (Figure 10A). As in case of Cdk13tm1a mice, the expression of CDK13 protein was investigated in the Cdk13tm1d mice. High expression of CDK13 was detected in the developing brain of WT Cdk13+/+ embryos (Figure 10B). Greatly downregulated expression of CDK13 was detected in heterozygous Cdk13tm1d/+ brain with undetectable expression of CDK13 protein in homozygous Cdk13tm1/tm1d brain (Figure 10B). As in the case of Cdk13tm1a mice, no significant reproducible downregulation of Ser2 phosphorylation was observed in Cdk13tm1d/tm1d brain extract (Figure 10B). Interestingly, expected Mendelian ratios were reflected in the portion of Cdk13tm1d/tm1d mice. High number of empty decidua at E12.5 were detected reflecting increased mortality before this stage (Table 3). Critically, only 19 litters out of 42 contained Cdk13tm1d/tm1d embryos suggesting homozygous mice carrying Cdk13/tm1d alleles exhibited more severe defects in phenotype than Cdk13tm1a/tm1a mice, especially in craniofacial area with midline facial cleft (Figure 10C). The prevalence of the midline orofacial deficiency and pericardial effusion was 60.5% in Cdk13tm1d homozygous mice at E12.5-E14.5. Out of 94 cases, only two cases of Cdk13tm1a/tm1a mice had externally visible orofacial clefting (Supplementary Table S1). The prevalence of the pericardial effusion (PE) in Cdk13tm1a/tm1a embryos at E12.5-E14.5 was less than 20% in comparison to high occurrence in Cdk13tm1d/tm1d embryos (Supplementary Table S1). No PE or orofacial cleft was recorded in any WT, though 6 embryos from 262 heterozygotes exhibited PE.
FIGURE 10.

Generation of Cdk13tm1d mice. (A) Scheme of Cdk13tm1d allele harbors deletion of critical exons 3 and 4, causing non-functional allele of Cdk13 gene. (B) Protein extract from the brain was prepared from E14.5 embryos and protein levels of CDK13 and γ-catenin (CTNNG) were evaluated by Western blotting. No residual expression of CDK13 was observed in Cdk13tm1d/tm1d embryos. Any significant downregulation of Ser2 and Ser5 phosphorylation was not observed in Cdk13tm1d/tm1d mice. (C) Cdk13tm1d/tm1d embryos exhibited distinct craniofacial phenotype with midfacial hypoplasia in comparison to wild-type embryos. Scale bar = 0.3 mm.
TABLE 3.
Genotypes of offspring from Cdk13tm1d/+ intercross.
| Empty | |||||
| Stage |
Cdk13 +/+ |
Cdk13tm1d/+ |
Cdk13 tm1d/tm1d |
Litter | decidua |
| (25%) | (50%) | (25%) | |||
| E12.5 | 33 (27.5%) | 69 (57.5%) | 18 (15%)*,# | 18 | 24 |
| E13.5 | 29 (31.5%) | 52 (56.5%) | 11 (12%)*,# | 14 | 5 |
| E14.5 | 24 (31.2%) | 44 (57.1%) | 9 (11.7%)*,# | 10 | 4 |
*Growth retardation. #Dead embryos.
Comparison of Cdk13tm1a/tm1a and Cdk13tm1d/tm1d Mice at E13.5
Finally, the phenotype of Cdk13tm1a/tm1a mice was compared to Cdk13tm1d/tm1d mice (Figure 11 and Supplementary Figures S7, S8). High-contrast differentiation resolution by X-ray computed microtomography was used to assess gross morphology of the Cdk13+/+, Cdk13tm1a/tm1a and Cdk13tm1d/tm1d embryos at E13.5. Both Cdk13 mutants exhibited smaller body size with severe growth retardation in Cdk13tm1d/tm1d animals (Figures 11A–D). Hypoplasia of midfacial structures was observed in Cdk13tm1d/tm1d embryos (Figure 11, compare H to G). The hearts were smaller in both Cdk13tm1a/tm1a as well as Cdk13tm1d/tm1d embryos in comparison to littermate control mice with severe ventricle deficiency detected in Cdk13tm1d/tm1d animals (Figure 11, compare J to I and L to K). Generally smaller liver were detected in Cdk13tm1a/tm1a and Cdk13tm1d/tm1d embryos in comparison to control Cdk13+/+ mice. Liver in Cdk13tm1d/tm1d mice exhibited abrogated liver lobes arrangement (Figure 11, compare P to O and N to M). Kidneys in Cdk13tm1d/tm1d mice do not follow right left kidney asymmetry (Figure 11, compare T to S).
Discussion
Our analysis revealed a new function of Cdk13 in the context of the mouse development and found that this protein is critical for proper development at later stages of embryogenesis. Loss of Cdk13 affects negatively several organs and tissue structures during mouse development and its inactivation leads to late embryonic lethality.
Animals with hypomorphic allele (Cdk13tm1a) retained low residual expression of CDK13 and they were lethal by E16.5 most due to heart failure and delayed development of several organs was caused likely due to insufficient supply of oxygen and nutrients. Interestingly, several miRNAs able to target Cdk13 mRNA for degradation were recently recognized during the acute myocardial infarctions (Wang et al., 2015) supporting the idea of CDK13 as a strategic molecule for optimal heart function. In addition, the expression of CDK13 was detectable not just in the heart, but also in other organs during embryonic development, such as craniofacial area or brain. However, the expression only cannot explain the complexity of phenotype exclusively in tissue autonomous manner or by insufficient nutrients and oxygen distribution. Thus, we assume that the organs with higher expression might be affected either by non-functional CDK13 in cell autonomous manner and/or by underdevelopment of cardiovascular system and lack of nutrients, mostly by insufficient heart function. Importantly, the affected cardiovascular system is superior to development of other organ systems and can drive systemic developmental phenotype; the example of such phenotype is growth retardation in the whole embryo with no respect to local CDK13 expression levels, which is affecting size of all organs within the embryo body. Therefore, in organs where the expression of CDK13 is under detection limit, we suggest that the phenotype is caused mostly by failure of cardiovascular system development and its systemic influence.
It is also interesting why the Cdk13tm1a allele exhibits the hypomorphic mutant phenotype. One possible explanation might be due to the insertion of neomycin selection cassette in non-coding region that has been shown to affect gene expression, both at the DNA and RNA levels (Pham et al., 1996; Meyers et al., 1998; Scacheri et al., 2001). Moreover, in silico analyses of post-transcriptional exon shuffling (PTES) events in humans revealed high PTES frequencies in CDK13 gene enabling the formation of aberrant functional CDK13 transcripts (Al-Balool et al., 2011).
In contrast to our observations, genetic depletion of Cdk12, a kinase with high amino acid similarity to Cdk13, resulted in early developmental lethality at the blastocyst stage due to deregulated expression of DNA-damage repair genes leading to enhanced genomic instability (Juan et al., 2016). Importantly, KO of CycK resembled the same lethal phenotype at the blastocyst stage as Cdk12 animals (Blazek et al., 2011). Besides clear effect of CycK during preimplantation stage, CycK is highly expressed in mouse embryonic stem cells and testes in a developmentally regulated manner (Dai et al., 2012). During neonatal spermatogenesis, CycK is highly expressed in gonocytes, spermatogonial stem cells and is absent in differentiating spermatogonia, spermatids and spermatozoa (Xiang et al., 2014). Similar expression in testes, ovary and uterus was also documented for CDK13 in our heterozygous mice.
Downregulation of Cdk13 had also significant impact on brain development. It is known from previous studies that CycK, a Cdk13 binding partner, has been identified in a genome-wide screen as one of the factors involved in the formation of the nervous system in Drosophila (Neumuller et al., 2011). In parallel, function of CDK12 in process of embryonic neural development was described in the conditional KO mouse model for this gene (Chen et al., 2014, 2017). When neuronal differentiation model of mouse embryonic carcinoma cell (P19) was employed, CDK12 and CDK13 kinases participated in the axonal elongation through a common signaling pathway that modulates protein expression of CDK5 (Chen et al., 2014). Recently, CDK13 was found remarkably dysregulated in hippocampus and suggested as one of the hub genes useful to elucidate Alzheimer’s disease (Pang et al., 2017). Importantly, studies of patients with CDK13 mutations suggested their essential role in brain development. As demonstrated in Figure 5, we were able to observe alteration of proliferation in neural tissue in Cdk13tm1a mice (as analyzed by Ki67 expression) however overall changes in brain morphology were not determined. On the other hand, observed proliferation changes can be also associated with developmental delay of Cdk13tm1a animals and subventricular zone in brains may significantly expands later in development. However, it is also possible that a prominent role of CDK13 may be apparent in later stages of brain development, for instance, in the axon pathfinding, synapse formation and etc. Confirmation of this statement, however, has to be proven in future by employing the nestin-Cre or CreErt-Flox systems.
Importantly, developmental defects within craniofacial formation was documented for Cdk13tm1a mice followed by complete loss of frontonasal part in Cdk13tm1d mice. It is probably the most surprising observation in respect to patients with heterozygous mutation of CDK13 gene (Sifrim et al., 2016; Bostwick et al., 2017; Carneiro et al., 2018; Hamilton et al., 2018). Moreover, gradual deregulation of CDK13 in mice resulted in enhanced craniofacial phenotype. The hypomorphic Cdk13tm1a mice exhibited in the rostral area, altered shape of nasal septa and later at E15.5, incomplete formation of the secondary palate was observed in several animals, where palatal shelves did not reach the midline, while control animals exhibited complete fusion of palatal processes. Nevertheless, complete depletion of CDK13 in the Cdk13tm1d/tm1d mice led to ablation of the midline area of the head. There is a limited information regarding the role of CDK13 in this developmental process at the moment, yet. The association between SNP polymorphism rs373711932 located within CDK13 gene and cranial base width was recently documented (Lee et al., 2017). Importantly, heterozygous copy number loss of CCNK gene in humans caused a syndromic neurodevelopmental disorder with distinctive facial dysmorphism (Fan et al., 2018). Therefore, it seems highly probable that CDK13 is indeed associated with craniofacial development, however, with unclear function, which will be necessary to uncover in the future.
From the functional point of view, it is surprising that only heterozygous mutation in one allele of the CDK13 gene was sufficient to cause the CHDFIDD in human patients. Common feature in all studies depicting deleterious effect of pathogenic CDK13 in patients was presence of missense substitutions in the protein kinase domain around ATP-binding and magnesium binding sites (Sifrim et al., 2016; Bostwick et al., 2017; Hamilton et al., 2018). In addition, two nonsense variants and a frame-shift variant accompanying production of aberrant CDK13 transcripts have been identified (van den Akker et al., 2018). Based on crystal structure of CDK13/CycK complex, it is very possible that all these mutations most likely caused inactivation of catalytic activity of CDK13 or disrupted binding of CycK, its associating partner, leading to abrogated function of CDK13 (Kohoutek and Blazek, 2012; Greifenberg et al., 2016; Hamilton et al., 2018). Nevertheless, some of identified mutations in patients were predicted to retain binding to cyclin K with affected catalytic activity. If this is the case, then CDK13/CycK complex represents a dominant negative complex in cells (Hamilton et al., 2018). It is of note that two patients carrying stop codons at the end of the kinase domain represented milder phenotypes, although obvious genotype-phenotype correlation has not been recorded (van den Akker et al., 2018). From all these results, we can postulate that even loss of function of one CDK13 allele is enough to interfere with developmental pathways and cause the CHDFIDD in humans, while complete abrogation of CDK13 gene would lead to embryonic lethality as documented in our mouse model.
Currently, we can only speculate how CDK13 contributes to the developmental processes: if it participates in phosphorylation of Ser2 within CTD of RNAPII, it is highly probable that inhibition of CDK13 will block or dramatically affect ability of RNAPII to effectively synthesize nascent RNA. CDK13 was demonstrated to bind various splicing factors or proteins involved in splicing (Davidson et al., 2014; Bartkowiak and Greenleaf, 2015; Liang et al., 2015). Moreover, CDK13 was found to associate with ASF/SF2 factor, a member of the spliceosome complex (Even et al., 2006). CDK13 was also identified in perinucleolar compartments enriched for proteins primarily implicated in pre-mRNA processing; therefore, lack of CDK13 could abrogate also RNA processing of given metazoan genes (Even et al., 2016). Thus, it is possible that a loss of kinase activity or associating potential of CDK13 will cripple proper splicing of transcribed RNA by RNAPII resulting in production of improperly spliced mRNA of proteins engaged in development processes.
It has been postulated that CDK13 phosphorylates Ser2 and Ser5 within CTD of RNAPII, if Ser7 is prephosphorylated at the C terminus of heptapeptide (Greifenberg et al., 2016). However, we were not able to detect any effect on Ser2 or Ser5, either in Cdk13tm1a/tm1a or Cdk13tm1d/tm1d animals with substantial decreased level of CDK13 in developing brain, where physiologically high level of CDK13 is present. Even though the global effect on Ser2 or Ser5 was not documented in Cdk13tm1a/tm1a developing brains, CDK13 could still orchestrate phosphorylation of RNAPII at the specific sets of genes as earlier described for the CDK9/cyclin T2 or CDK12/cyclin K complexes in mouse and human cells (Zhu et al., 2009; Blazek et al., 2011; Johnson et al., 2016). When CDK13 was downregulated in HCT116 cells, gene ontology analysis revealed enrichment of functions connected to various extracellular and growth signaling pathways (Liang et al., 2015; Greifenberg et al., 2016). Similar modus operandi was implicated for CDK9 activation by MEK-1 and MSK1 in response to extracellular cues (Fujita et al., 2008; Smerdova et al., 2014). In addition, transcriptional kinase CDK8 was described to affect canonical Notch signaling by targeting activated Notch intracellular domain for proteasomal degradation (Fryer et al., 2004). In addition, CDK8 was suggested to stabilize β-catenin interaction with the promoter of WNT targets to achieve regulatory control (Firestein et al., 2008). Therefore, it is possible to envision that CDK13 regulates a specific set of genes at a specific developmental stage of embryonic development or in specialized cell types. Therefore, it will be critical in the near future to identify unique genes under control of CDK13 at given tissue or organ by employment of exome RNA-sequencing analyses in WT and Cdk13-deficient mice, which will be our next goal.
We believe that the Cdk13tm1a hypomorphic mouse can be a very instrumental animal model to delineate cellular pathways or mechanisms participating in the onset or propagation of severe developmental cues in humans with mutated form of CDK13. Moreover, our Cdk13tm1d mouse model with enhanced craniofacial phenotype may be invaluable for further clarification of molecular processes, which are responsible for clinical symptoms of human patients.
Experimental Procedures
Mice, Genotyping, Breeding of Cdk13tm1a Mice
Mouse embryos (Cdk13tm1a(EUCOM)Hmgu) bearing splicing acceptor, β-galactosidase and neomycine resistance gene both with poly A signals (Figure 1A) were obtained from the Infrafrontier Research Infrastructure–Mouse disease Models1. The acquired embryos carrying Cdk13tm1a allele were transferred into pseudopregnant females of C57BL/6N mouse strain and the heterozygous Cdk13tm1a mice were obtained at the Transgenic and Archiving Module, CCP (IMG, Prague, Czechia). Heterozygous mice for Cdk13tm1a allele were bred and newborn mice were genotyped by PCR amplification (94°C, 30 s; 62°C/65°C, 20 s; 72°C, 30 s; 35 cycles) of Cdk13 gene to distinguish WT (Cdk13+/+) and Cdk13tm1a/tm1a alleles. For PCR analysis, the following sets of primers were used for KO and WT alleles: (KOf, 5′-CCA GCT TTA AAG GCG CAT AAC-3′; KOr, 5′-TGA CCT TGG GCA AGA ACA TAA-3′; WTf, 5′-TAG CTG GCC AAT GAG CTT TC-3′; WTr, 5′-AGT CTA GGA AGC TGG GAA GAT- 3′). Primers KOf and KOr amplify a 257-bp fragment from Cdk13tm1a allele while primers WTf and WTr amplify a 179-bp fragment from WT allele. Genomic DNA was isolated from mouse ears by NaOH extraction (Truett et al., 2000). Heterozygous mice carrying Cdk13tm1a allele were bred and after mating, females were examined for vaginal plugs, embryonic day 0.5 (E0.5) was determined at noon at the day of vaginal plaque record. Embryos at various stages were obtained from uteri of pregnant heterozygous female mice, genotyped and the Mendelian ratios were defined.
To generate mouse carrying the Cdk13tm1d allele, the Cdk13tm1a/tm1a mice were crossed with the Flp-deleter mouse strain Gt(ROSA)26Sortm2(CAG–flpo,–EYFP)Ics (MGI: 5285396, Philippe Soriano) and Cre-deleter strain Gt(ROSA)26Sortm1(ACTB–cre,–EGFP)Ics (MGI: 5285392, Philippe Soriano) in order to delete critical exons 3 and 4 within Cdk13 gene. All animal procedures were performed in strict accordance to the Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee (Veterinary Research Institute, Brno, Czechia).
Western Blotting
Particular organ from Cdk13tm1a/tm1a, Cdk13tm1a/+ and Cdk13+/+ embryos at E14.5 were lysed in lysis buffer (100 mM Tris, pH 7.4; 1% SDS; 10% glycerol) and sonicated. The brain from Cdk13tm1d/tm1d, Cdk13tm1d/+ and Cdk13+/+ were lysed in lysis buffer (100 mM Tris, pH 7.4; 1% SDS; 10% glycerol) and sonicated. Concentrations of total proteins were determined with bicinchoninic acid (BCA). Lysates were diluted to the same concentration, mixed with the 3xLaemmli sample buffer and boiled for 5 min. Western blotting was performed using anti-mouse CDK13 polyclonal antibody (SAB1302350, Sigma-Aldrich), γ-catenin (CTNNG) antibody (2309, Cell Signaling Technology), cyclin T1 (CYCT1) (sc-10750, Santa Cruz Biotechnology) and anti-serine 2 (Ser2) and serine 5 (Ser5) rat monoclonal antibodies (clones 3E10 and 3E8 respectively, ChromoTek GmbH, Germany).
Histological Analysis
Wild-type and homozygous Cdk13tm1a/tm1a embryos at E14.5 to E16.5 were fixed in 4% paraformaldehyde and dehydrated. After that hearts, brains, kidneys and livers were dissected, embedded in paraffin, and sectioned at 5 μm. Hematoxylin-eosin (H&E) staining was performed. Images were taken under bright field using a Leica compound microscope (DMLB2) with a Leica camera (DFC480) attached (Leica Microsystems, Wetzlar, Germany).
Immunohistochemistry
Proliferating cells were visualized on E14.5 brain sections by labeling of Ki67 (positive cells are brown). Sections were pretreated in Citrate buffer, pH6, 10 mM, 20 min/97°C in water bath and were labeled with Ki67 primary antibody (RBK027-05, Zytomed Systems). For primary antibody detection, specific secondary antibody and avidin-biotin complex were used (Vectastain kit, PK-6101, Vector laboratories). The signal was developed by DAB chromogen system (K3468, DAKO). Nuclei were counterstained by hematoxylin (blue). Mitotic index was counted as the ratio between Ki67-positive cells and total amount of cells in three biological samples for each group (three Cdk13+/+ embryos, three Cdk13tm1a/tm1a embryos). Statistical significance in cell number differences between control and deficient mice were evaluated by unpaired t-test.
E14.5 heart sections were pretreated in DAKO Target Retrieval (32367, DAKO) solution for 15 min/97°C in water bath and labeled using primary antibodies against actin (sc-1615-R, Santa Cruz Biotechnology) and myosin (sc-32732, Santa Cruz Biotechnology). To detect primary antibodies, secondary Alexa Fluor antibodies (A11004, A11008, Invitrogen) were used. Nuclei were counterstained by DRAQ5TM (62251, Thermo Scientific). Images were captured on fluorescence confocal microscope Leica SP8 (Leica).
Micro-Computed Tomography (micro-CT)
The embryos at E13.5 or E15.5 were contrasted with Lugol’s solution to visualize gross morphology of individual soft tissues by microCT. For the purpose of motion stabilization during the micro CT scan, mouse embryo was embedded in 1% agarose gel in Falcon conical centrifuge tube. The micro-CT scan was performed using - laboratory system GE Phoenix v| tome| x L 240 (GE Sensing & InspectionTechnologies GmbH, Germany), equipped with a 180 kV/15W maximum power nanofocus X-ray tube and high contrast flat panel detector DXR250 2048 px × 2048 px with 200 μm × 200 μm pixel size. The measurement was carried out in the air-conditioned cabinet (21°C) at acceleration voltage of 60 kV and X-ray tube current of 200 μA. Thousand nine hundred projections were taken over 360° with exposure time 900 ms resulting in 5.5 μm voxel resolution. The tomographic reconstruction was realized by software GE phoenix datos| x 2.0 (GE Sensing & Inspection Technologies GmbH, Germany). Reconstructed slice data were processed using VG Studio MAX 3.1 software (Volume Graphics GmbH, Germany) and segmentation of liver, kidneys and heart was completed manually.
Embryonic Ultrasound Imaging and Doppler Echocardiography
Pregnant females were anesthetized on gestational day 14.5 or 15.5 (before noon) with an isoflurane/oxygen mixture (anesthesia was initiated with 3–4%, and maintained with 1–2% isoflurane), and maintained on a temperature-controlled mouse platform (with sensors for monitoring of maternal electrocardiogram, respiration and core body temperature). Maternal temperature was maintained at 34–37°C, and maternal heart beat at ∼400 beats/minute by adjusting the level of anesthesia. An incision of about 2–3 cm in the lower abdomen was made, and a uterine horn was externalized through the incision to provide imaging access. The number of fetuses in right and left uterine horns was counted from the mother’s bladder and later during echocardiographic imaging, labeled as R1, R2, R3, etc (right side) and L1, L2, L3, etc (left side). Pre-warmed ultrasound gel (37°C) without bubbles was applied between the individual fetuses and the transducer. B-mode imaging scanning of the whole embryo with the focus on the heart and Color Doppler and PW Doppler measurements for heart aorta and dorsal aorta were obtained using a high-frequency ultrasound system (Vevo 2100, FUJIFILM VisualSonics, Inc., Toronto, ON, Canada) equipped with a MS-550S transducer operating at a center frequency of 44 MHz. At the end of imaging of all fetuses, the mother was sacrificed by cervical dislocation and a small piece of yolk sac of each fetus was taken for genotyping.
Data Availability
All datasets generated for this study are included in the manuscript and/or the Supplementary Files.
Ethics Statement
All animal procedures were performed in strict accordance to the Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee (Veterinary Research Institute, Brno, Czechia).
Author Contributions
JKo conceptualized and supervised the project, acquired the funding, wrote the original draft, and administrated the project. MN conceptualized the project, contributed to formal analysis, investigation, and visualization, and wrote, review, and edited the manuscript. DV contributed to investigation and visualization. MH contributed to formal analysis, investigation, and visualization. JP contributed to conceptualization, formal analysis, supervision, and investigation and wrote, review, and edited the manuscript. SP and MP contributed to investigation and visualization. RS supervised the project. MK, TZ, and JKa contributed to software development and visualization. H-CJ contributed to investigation, and wrote, review, and edited the manuscript. M-JF and MB contributed to formal analysis, supervision, writing, review, and editing.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Assoc. Prof. S. Sevcikova for a critical comments to the manuscript and other members of the laboratory for helpful inputs.
Funding. This research was supported by grants n. 16-24043J of the Czech Science Foundation to JKo, MN, and DV. The research for this manuscript was financially supported by the Ministry of Agriculture n. MZE-RO0518, by the Ministry of Education, Youth and Sports, n. LM2015040 and LQ1604 by, and by the Ministry of Education, Youth and Sports and European Fund for Regional Development n. OP RDE CZ.02.1.01/0.0/0.0/16_013/0001789 and n. OP RDI CZ.1.05/1.1.00/02.0109 and OP RDI CZ.1.05/2.1.00/19.0395 to JP, MP, SP, and RS. Work by MB and MH was supported by the Ministry of Education, Youth and Sports of the Czech Republic (CZ.02.1.01/0.0/0.0/15_003/0000460). The cooperation between MB and TZ labs is supported by the Czech Science Foundation (17-14886S). MicroCT analyses performed by MK, TZ, and JKa were carried out under the project CEITEC 2020 (LQ1601) with financial support from the Ministry of Education, Youth and Sports of the Czech Republic under the National Sustainability Program II. Work by H-CJ and M-JF was supported by Ministry of Science and Technology, Taiwan (MOST 106-2811-B-010-046 and MOST 105-2923-B-010 -002 -MY3).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2019.00155/full#supplementary-material
Truncated form of CDK13 protein. (A)Predicted amino acid sequence of truncated CDK13 protein due totranscriptional block mediated by two Poly A sites introduced intothe intron 2 as part of trap vector in mice bearing Cdk13tm1a allele.Black and Blue capital letters represent amino acids encoded within Exon 1 and Exon 2 of Cdk13 gene, respectively. Predicted Molecular weight of truncated CDK13 protein is 66.22 KDa (https://www.bioinformatics.org/sms/ prot_mw.html). (B) Brain extracts were prepared from E14.5 embryos of given genotype and protein levels of wild-type and truncated form of CDK13 was evaluated by Western Blotting. The antibody used throughout our study recognizes N-terminal part of CDK13, which is expressed in both forms of Cdk13, wild-type as well as truncated one. The Cdk13+/+ mice contained only the wild-type CDK13 protein (WT-CDK13). The heterozygous Cdk13tm1a/+ mice included wild-type (WT-CDK13) and truncated (TR-CDK13) forms of CDK13 protein. The Cdk13tm1a expressed the truncated form of CDK13 and minor wild-type CDK13 as demonstrated by immunoblot.
Comparison of gross morphology of Cdk13-deficent mice phenotype. (A–H) Wild-type Cdk13+/+, (I–P) heterozygous Cdk13tm1a/+, and (Q–X) Cdk13tm1a embryos at various stages. Growth retardation of Cdk13-deficient mice was apparent from early developmental stages (Q,R). Some embryos exhibited nuchal edema and peripheral hypervascularization (U) and abnormal craniofacial shape (S,T). Developmental delay was observed at later stages (V,W). At E16.5, each of the Cdk13tm1a/tm1a embryos was undergoing resorption (X). No morphological differences between wild-type and heterozygous embryos were observed. Scale bar = 0.1 mm.
The 3D reconstruction of selected organs in micro-CT of control Cdk13+/+ and Cdk13tm1a/tm1a embryos. Overall view on embryos of Cdk13+/+ (A) and Cdk13tm1a/tm1a (B) with 3D reconstruction of heart, liver and kidney (left). Gross morphology is well visible on movable model on the right side. To visualize only one organ, click on the lower row and select heart (orange), liver (pink) or kidney (purple) organ only. Scale bar = 1 mm.
Transversal sections of the head of control Cdk13+/+ and Cdk13tm1a/tm1a embryos. Transversal sections through the mouse head with details of the nasal cavity area. At E14.5, no obvious differences were observed and only one epithelial protrusion (marked by arrow) were developed close to each nasal cavity (nc) (compare A to D). Later at E15.5, the number of nasal glands was reproducibly smaller in Cdk13tm1a/tm1a mice in comparison to control animals (compare B to E). (C,F) Coronal section through the head of embryos at the level of the eyes. White arrows depict different complexity of the nasal cavity in wild-type, Cdk13+/+, and Cdk13tm1a/tm1a animals. Abbreviations nc – nasal cavity, arrowhead – nasal gland. Scale bar (A,B,D,F) = 100 μm and scale bar (C,F) = 1 mm.
Immunohistochemical detection of myosin and actin in the heart of Cdk13+/+ and Cdk13tm1a/tm1a embryos. Sagittal sections through the mouse heart were prepared and immunohistochemical detection of myosin and actin was carried out with specific antibodies. Decreased expression of myosin was detected in ventricle myocardium, compare B and B’ to E and E’. Scale bar (A,D) = 500 μm; Scale bar (B–F, without D) − 100 μm.
The expression of Cdk13 in adult mouse organs visualized by β-galactosidase activity. The expression of Cdk13 was examined by activity of β-galactosidase in adult mouse organs (A) eye, (B) heart, (C) thyroid gland, (D) urinary bladder, (E) kidney, (F) uterus, (G) gall bladder, (H) testes, and (I) ovary.
Sagittal sections through heads of Cdk13tma1 and Cdk13tm1d and Cdk13tma1 and Cdk13tm1d mice. High-contrast differentiation resolution by X-ray computed microtomography where Lugol’s staining was used to visualize the soft tissues. The Cdk13tm1a/tm1a, Cdk13tm1d/tm1d and their littermate controls are displayed at E13.5 to show overall developmental delay.
Horizontal sections through heads of Cdk13tma1 and Cdk13tm1d mice. High-contrast differentiation resolution by X-ray computed microtomography where Lugol’s staining was used to visualize the soft tissues. The Cdk13tm1a/tm1a, Cdk13tm1d/tm1d and their littermate controls are displayed at E13.5.
The prevalence of developmental defects in Cdk13tma1 and Cdk13tm1d transgenic mice.
References
- Al-Balool H. H., Weber D., Liu Y., Wade M., Guleria K., Nam P. L., et al. (2011). Post-transcriptional exon shuffling events in humans can be evolutionarily conserved and abundant. Genome Res. 21 1788–1799. 10.1101/gr.116442.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakre A., Andersen L. E., Meliopoulos V., Coleman K., Yan X., Brooks P., et al. (2013). Identification of host kinase genes required for Influenza virus replication and the regulatory role of MicroRNAs. PLoS One 8:e66796. 10.1371/journal.pone.0066796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkowiak B., Greenleaf A. L. (2015). Expression, purification, and identification of associated proteins of the full-length hCDK12/CyclinK complex. J. Biol. Chem. 290 1786–1795. 10.1074/jbc.M114.612226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkowiak B., Liu P., Phatnani H. P., Fuda N. J., Cooper J. J., Price D. H., et al. (2010). CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev. 24 2303–2316. 10.1101/gad.1968210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berro R., Pedati C., Kehn-Hall K., Wu W., Klase Z., Even Y., et al. (2008). CDK13, a new potential human immunodeficiency virus type 1 inhibitory factor regulating viral mRNA splicing. J. Virol. 82 7155–7166. 10.1128/JVI.02543-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazek D., Kohoutek J., Bartholomeeusen K., Johansen E., Hulinkova P., Luo Z., et al. (2011). The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev. 25 2158–2172. 10.1101/gad.16962311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostwick B. L., McLean S., Posey J. E., Streff H. E., Gripp K. W., Blesson A., et al. (2017). Phenotypic and molecular characterisation of CDK13-related congenital heart defects, dysmorphic facial features and intellectual developmental disorders. Genome Med. 9:73. 10.1186/s13073-017-0463-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carneiro T. N., Krepischi A. C., Costa S. S., Tojal da Silva I., Vianna-Morgante A. M., Valieris R., et al. (2018). Utility of trio-based exome sequencing in the elucidation of the genetic basis of isolated syndromic intellectual disability: illustrative cases. Appl. Clin. Genet. 11 93–98. 10.2147/TACG.S165799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H. R., Juan H. C., Wong Y. H., Tsai J. W., Fann M. J. (2017). Cdk12 Regulates Neurogenesis and Late-Arising Neuronal Migration in the Developing Cerebral Cortex. Cereb. Cortex 27 2289–2302. 10.1093/cercor/bhw081 [DOI] [PubMed] [Google Scholar]
- Chen H. R., Lin G. T., Huang C. K., Fann M. J. (2014). Cdk12 and Cdk13 regulate axonal elongation through a common signaling pathway that modulates Cdk5 expression. Exp. Neurol. 261 10–21. 10.1016/j.expneurol.2014.06.024 [DOI] [PubMed] [Google Scholar]
- Cheng S. W., Kuzyk M. A., Moradian A., Ichu T. A., Chang V. C., Tien J. F., et al. (2012). Interaction of cyclin-dependent kinase 12/CrkRS with cyclin K1 is required for the phosphorylation of the C-terminal domain of RNA polymerase II. Mol. Cell. Biol. 32 4691–4704. 10.1128/MCB.06267-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Q., Lei T., Zhao C., Zhong J., Tang Y. Z., Chen B., et al. (2012). Cyclin K-containing kinase complexes maintain self-renewal in murine embryonic stem cells. J. Biol. Chem. 287 25344–25352. 10.1074/jbc.M111.321760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson L., Muniz L., West S. (2014). 3′ end formation of pre-mRNA and phosphorylation of Ser2 on the RNA polymerase II CTD are reciprocally coupled in human cells. Genes Dev. 28 342–356. 10.1101/gad.231274.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deciphering Developmental Disorders Study (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature 542 433–438. 10.1038/nature21062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Even Y., Durieux S., Escande M. L., Lozano J. C., Peaucellier G., Weil D., et al. (2006). CDC2L5, a Cdk-like kinase with RS domain, interacts with the ASF/SF2-associated protein p32 and affects splicing in vivo. J. Cell. Biochem. 99 890–904. 10.1002/jcb.20986 [DOI] [PubMed] [Google Scholar]
- Even Y., Escande M. L., Fayet C., Geneviere A. M. (2016). CDK13, a Kinase Involved in Pre-mRNA Splicing, Is a Component of the Perinucleolar Compartment. PLoS One 11:e0149184. 10.1371/journal.pone.0149184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y., Yin W., Hu B., Kline A. D., Zhang V. W., Liang D., et al. (2018). De Novo mutations of CCNK cause a syndromic neurodevelopmental disorder with distinctive facial dysmorphism. Am. J. Hum. Genet. 103 448–455. 10.1016/j.ajhg.2018.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestein R., Bass A. J., Kim S. Y., Dunn I. F., Silver S. J., Guney I., et al. (2008). CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature 455 547–551. 10.1038/nature07179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryer C. J., White J. B., Jones K. A. (2004). Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Mol. Cell. 16 509–520. 10.1016/j.molcel.2004.10.014 [DOI] [PubMed] [Google Scholar]
- Fujita T., Ryser S., Piuz I., Schlegel W. (2008). Up-regulation of P-TEFb by the MEK1-extracellular signal-regulated kinase signaling pathway contributes to stimulated transcription elongation of immediate early genes in neuroendocrine cells. Mol. Cell. Biol. 28 1630–1643. 10.1128/mcb.01767-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenleaf A. L. (2018). Human CDK12 and CDK13, multi-tasking CTD kinases for the new millennium. Transcription 10 91–110. 10.1080/21541264.2018.1535211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greifenberg A. K., Honig D., Pilarova K., Duster R., Bartholomeeusen K., Bosken C. A., et al. (2016). Structural and functional analysis of the Cdk13/Cyclin K complex. Cell Rep. 14 320–331. 10.1016/j.celrep.2015.12.025 [DOI] [PubMed] [Google Scholar]
- Hamilton M. J., Caswell R. C., Canham N., Cole T., Firth H. V., Foulds N., et al. (2018). Heterozygous mutations affecting the protein kinase domain of CDK13 cause a syndromic form of developmental delay and intellectual disability. J. Med. Genet. 55 28–38. 10.1136/jmedgenet-2017-104620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson S. F., Cruz C., Greifenberg A. K., Dust S., Stover D. G., Chi D., et al. (2016). CDK12 inhibition reverses de novo and acquired PARP inhibitor resistance in BRCA wild-type and mutated models of triple-negative breast cancer. Cell Rep. 17 2367–2381. 10.1016/j.celrep.2016.10.077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juan H. C., Lin Y., Chen H. R., Fann M. J. (2016). Cdk12 is essential for embryonic development and the maintenance of genomic stability. Cell Death Differ. 23 1038–1048. 10.1038/cdd.2015.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H. E., Kim D. G., Lee K. J., Son J. G., Song M. Y., Park Y. M., et al. (2012). Frequent amplification of CENPF, GMNN and CDK13 genes in hepatocellular carcinomas. PLoS One 7:e43223. 10.1371/journal.pone.0043223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko T. K., Kelly E., Pines J. (2001). CrkRS: a novel conserved Cdc2-related protein kinase that colocalises with SC35 speckles. J. Cell Sci. 114 2591–2603. [DOI] [PubMed] [Google Scholar]
- Kohoutek J. (2009). P-TEFb- the final frontier. Cell Div. 4:19. 10.1186/1747-1028-4-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohoutek J., Blazek D. (2012). Cyclin K goes with Cdk12 and Cdk13. Cell Div. 7:12. 10.1186/1747-1028-7-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M. K., Shaffer J. R., Leslie E. J., Orlova E., Carlson J. C., Feingold E., et al. (2017). Genome-wide association study of facial morphology reveals novel associations with FREM1 and PARK2. PLoS One 12:e0176566. 10.1371/journal.pone.0176566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang K., Gao X., Gilmore J. M., Florens L., Washburn M. P., Smith E., et al. (2015). Characterization of human cyclin-dependent kinase 12 (CDK12) and CDK13 complexes in C-terminal domain phosphorylation, gene transcription, and RNA processing. Mol. Cell. Biol 35 928–938. 10.1128/MCB.01426-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers E. N., Lewandoski M., Martin G. R. (1998). An Fgf8 mutant allelic series generated by Cre- and Flp-mediated recombination. Nat. Genet. 18 136–141. 10.1038/ng0298-136 [DOI] [PubMed] [Google Scholar]
- Neumuller R. A., Richter C., Fischer A., Novatchkova M., Neumuller K. G., Knoblich J. A. (2011). Genome-wide analysis of self-renewal in Drosophila neural stem cells by transgenic RNAi. Cell Stem Cell 8 580–593. 10.1016/j.stem.2011.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan J., Xue Y., Chen S., Qiu H., Wu C., Jiang H., et al. (2012). Establishment and characterization of a new human acute myelomonocytic leukemia cell line JIH-3. Leuk. Res. 36 889–894. 10.1016/j.leukres.2012.01.012 [DOI] [PubMed] [Google Scholar]
- Pang X., Zhao Y., Wang J., Zhou Q., Xu L., Kang, et al. (2017). The bioinformatic analysis of the dysregulated genes and microRNAs in entorhinal cortex, hippocampus, and blood for Alzheimer’s disease. Biomed. Res. Int. 2017:9084507. 10.1155/2017/9084507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham C. T., MacIvor D. M., Hug B. A., Heusel J. W., Ley T. J. (1996). Long-range disruption of gene expression by a selectable marker cassette. Proc. Natl. Acad. Sci. U.S.A. 93 13090–13095. 10.1073/pnas.93.23.13090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scacheri P. C., Crabtree J. S., Novotny E. A., Garrett-Beal L., Chen A., Edgemon K. A., et al. (2001). Bidirectional transcriptional activity of PGK-neomycin and unexpected embryonic lethality in heterozygote chimeric knockout mice. Genesis 30 259–263. 10.1002/gene.1072 [DOI] [PubMed] [Google Scholar]
- Sifrim A., Hitz M. P., Wilsdon A., Breckpot J., Turki S. H., Thienpont B., et al. (2016). Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat. Genet. 48 1060–1065. 10.1038/ng.3627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smerdova L., Smerdova J., Kabatkova M., Kohoutek J., Blazek D., Machala M., et al. (2014). Upregulation of CYP1B1 expression by inflammatory cytokines is mediated by the p38 MAP kinase signal transduction pathway. Carcinogenesis 35 2534–2543. 10.1093/carcin/bgu190 [DOI] [PubMed] [Google Scholar]
- Truett G. E., Heeger P., Mynatt R. L., Truett A. A., Walker J. A., Warman M. L. (2000). Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT). Biotechniques 29 52, 54. 10.2144/00291bm09 [DOI] [PubMed] [Google Scholar]
- Uehara T., Takenouchi T., Kosaki R., Kurosawa K., Mizuno S., Kosaki K. (2018). Redefining the phenotypic spectrum of de novo heterozygous CDK13 variants: three patients without cardiac defects. Eur. J. Med. Genet. 61 243–247. 10.1016/j.ejmg.2017.12.004 [DOI] [PubMed] [Google Scholar]
- van den Akker W. M. R., Brummelman I., Martis L. M., Timmermans R. N., Pfundt R., Kleefstra T., et al. (2018). De novo variants in CDK13 associated with syndromic ID/DD: molecular and clinical delineation of 15 individuals and a further review. Clin. Genet. 93 1000–1007. 10.1111/cge.13225 [DOI] [PubMed] [Google Scholar]
- Wang Y., Pan X., Fan Y., Hu X., Liu X., Xiang M., et al. (2015). Dysregulated expression of microRNAs and mRNAs in myocardial infarction. Am. J. Transl. Res. 7 2291–2304. [PMC free article] [PubMed] [Google Scholar]
- Xiang X., Deng L., Zhang J., Zhang X., Lei T., Luan G., et al. (2014). A distinct expression pattern of cyclin K in mammalian testes suggests a functional role in spermatogenesis. PLoS One 9:e101539. 10.1371/journal.pone.0101539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H., Doherty J. R., Kuliyev E., Mead P. E. (2009). CDK9/cyclin complexes modulate endoderm induction by direct interaction with Mix.3/mixer. Dev. Dyn. 238 1346–1357. 10.1002/dvdy.21920 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Truncated form of CDK13 protein. (A)Predicted amino acid sequence of truncated CDK13 protein due totranscriptional block mediated by two Poly A sites introduced intothe intron 2 as part of trap vector in mice bearing Cdk13tm1a allele.Black and Blue capital letters represent amino acids encoded within Exon 1 and Exon 2 of Cdk13 gene, respectively. Predicted Molecular weight of truncated CDK13 protein is 66.22 KDa (https://www.bioinformatics.org/sms/ prot_mw.html). (B) Brain extracts were prepared from E14.5 embryos of given genotype and protein levels of wild-type and truncated form of CDK13 was evaluated by Western Blotting. The antibody used throughout our study recognizes N-terminal part of CDK13, which is expressed in both forms of Cdk13, wild-type as well as truncated one. The Cdk13+/+ mice contained only the wild-type CDK13 protein (WT-CDK13). The heterozygous Cdk13tm1a/+ mice included wild-type (WT-CDK13) and truncated (TR-CDK13) forms of CDK13 protein. The Cdk13tm1a expressed the truncated form of CDK13 and minor wild-type CDK13 as demonstrated by immunoblot.
Comparison of gross morphology of Cdk13-deficent mice phenotype. (A–H) Wild-type Cdk13+/+, (I–P) heterozygous Cdk13tm1a/+, and (Q–X) Cdk13tm1a embryos at various stages. Growth retardation of Cdk13-deficient mice was apparent from early developmental stages (Q,R). Some embryos exhibited nuchal edema and peripheral hypervascularization (U) and abnormal craniofacial shape (S,T). Developmental delay was observed at later stages (V,W). At E16.5, each of the Cdk13tm1a/tm1a embryos was undergoing resorption (X). No morphological differences between wild-type and heterozygous embryos were observed. Scale bar = 0.1 mm.
The 3D reconstruction of selected organs in micro-CT of control Cdk13+/+ and Cdk13tm1a/tm1a embryos. Overall view on embryos of Cdk13+/+ (A) and Cdk13tm1a/tm1a (B) with 3D reconstruction of heart, liver and kidney (left). Gross morphology is well visible on movable model on the right side. To visualize only one organ, click on the lower row and select heart (orange), liver (pink) or kidney (purple) organ only. Scale bar = 1 mm.
Transversal sections of the head of control Cdk13+/+ and Cdk13tm1a/tm1a embryos. Transversal sections through the mouse head with details of the nasal cavity area. At E14.5, no obvious differences were observed and only one epithelial protrusion (marked by arrow) were developed close to each nasal cavity (nc) (compare A to D). Later at E15.5, the number of nasal glands was reproducibly smaller in Cdk13tm1a/tm1a mice in comparison to control animals (compare B to E). (C,F) Coronal section through the head of embryos at the level of the eyes. White arrows depict different complexity of the nasal cavity in wild-type, Cdk13+/+, and Cdk13tm1a/tm1a animals. Abbreviations nc – nasal cavity, arrowhead – nasal gland. Scale bar (A,B,D,F) = 100 μm and scale bar (C,F) = 1 mm.
Immunohistochemical detection of myosin and actin in the heart of Cdk13+/+ and Cdk13tm1a/tm1a embryos. Sagittal sections through the mouse heart were prepared and immunohistochemical detection of myosin and actin was carried out with specific antibodies. Decreased expression of myosin was detected in ventricle myocardium, compare B and B’ to E and E’. Scale bar (A,D) = 500 μm; Scale bar (B–F, without D) − 100 μm.
The expression of Cdk13 in adult mouse organs visualized by β-galactosidase activity. The expression of Cdk13 was examined by activity of β-galactosidase in adult mouse organs (A) eye, (B) heart, (C) thyroid gland, (D) urinary bladder, (E) kidney, (F) uterus, (G) gall bladder, (H) testes, and (I) ovary.
Sagittal sections through heads of Cdk13tma1 and Cdk13tm1d and Cdk13tma1 and Cdk13tm1d mice. High-contrast differentiation resolution by X-ray computed microtomography where Lugol’s staining was used to visualize the soft tissues. The Cdk13tm1a/tm1a, Cdk13tm1d/tm1d and their littermate controls are displayed at E13.5 to show overall developmental delay.
Horizontal sections through heads of Cdk13tma1 and Cdk13tm1d mice. High-contrast differentiation resolution by X-ray computed microtomography where Lugol’s staining was used to visualize the soft tissues. The Cdk13tm1a/tm1a, Cdk13tm1d/tm1d and their littermate controls are displayed at E13.5.
The prevalence of developmental defects in Cdk13tma1 and Cdk13tm1d transgenic mice.
Data Availability Statement
All datasets generated for this study are included in the manuscript and/or the Supplementary Files.