

Abstract
R‐loop disassembly by the human helicase Senataxin contributes to genome integrity and to proper transcription termination at a subset of RNA polymerase II genes. Whether Senataxin also contributes to transcription termination at other classes of genes has remained unclear. Here, we show that Sen1, one of two fission yeast homologues of Senataxin, promotes efficient termination of RNA polymerase III (RNAP3) transcription in vivo. In the absence of Sen1, RNAP3 accumulates downstream of RNAP3‐transcribed genes and produces long exosome‐sensitive 3′‐extended transcripts. Importantly, neither of these defects was affected by the removal of R‐loops. The finding that Sen1 acts as an ancillary factor for RNAP3 transcription termination in vivo challenges the pre‐existing view that RNAP3 terminates transcription autonomously. We propose that Sen1 is a cofactor for transcription termination that has been co‐opted by different RNA polymerases in the course of evolution.
Keywords: R‐loops, RNA polymerase III, Senataxin, transcription termination
Subject Categories: Protein Biosynthesis & Quality Control, RNA Biology, Transcription
Introduction
Senataxin is a conserved DNA/RNA helicase whose deficiency has been implicated in the neurological disorders amyotrophic lateral sclerosis type 4 (ALS4) and ataxia–ocular apraxia type 2 (AOA2; Chen et al, 2004; Moreira et al, 2004). How different Senataxin mutations contribute to the development of diseases with distinct pathologies remains unclear (Groh et al, 2017). Senataxin is the homologue of the yeast Sen1 helicase, and concordant observations have established that both human Senataxin and budding yeast Sen1 are important for transcription termination of at least a subset of RNAP2‐transcribed genes (Ursic et al, 1997; Steinmetz et al, 2006; Skourti‐Stathaki et al, 2011; Porrua & Libri, 2013). However, the mechanisms involved probably differ in both species as budding yeast Sen1 contributes to RNAP2 transcription termination as part of the Nrd1‐Nab3‐Sen1 (NNS) complex, which is not conserved in human cells. In addition, both budding yeast Sen1 and human Senataxin have been implicated in the repair of DNA damage (Li et al, 2016; Andrews et al, 2018; Cohen et al, 2018) and in the resolution of transcription–replication conflicts (Alzu et al, 2012; Richard et al, 2013; Yüce & West, 2013). Both budding and fission yeast Sen1 can translocate in a 5′–3′ direction on either single‐stranded DNA or RNA in vitro (Kim et al, 1999; Martin‐Tumasz & Brow, 2015; Han et al, 2017), and it is believed that long, co‐transcriptional RNA‐DNA hybrids (also known as R‐loops) represent a critical substrate of budding yeast Sen1 and human Senataxin in vivo (Mischo et al, 2011; Skourti‐Stathaki et al, 2011 and reviewed in Groh et al, 2017). Current models propose that the stabilization of R‐loops upon Senataxin inactivation underlies the associated transcription termination and DNA repair defects. This proposal however has been somewhat challenged by the observation that budding yeast Sen1 could directly dissociate pre‐assembled RNAP2 transcription elongation complexes in vitro by translocating on the nascent RNA, even in the absence of RNA‐DNA hybrids (Porrua & Libri, 2013; Han et al, 2017), suggesting that Sen1 has potentially R‐loop‐independent functions in the control of transcription. In addition, it has been suggested that the very low processivity of Sen1′s helicase activity might actually prevent it from unwinding long R‐loops in vivo (Han et al, 2017).
The fission yeast Schizosaccharomyces pombe expresses two non‐essential homologues of Senataxin, Sen1 and Dbl8. Surprisingly, transcription termination at RNAP2‐transcribed genes is largely unaffected by lack of either or both homologues (Lemay et al, 2016; Larochelle et al, 2018) and to date, the roles of the fission yeast Senataxin enzymes have remained largely unknown. Dbl8 was shown to localize at sites of double‐strand breaks (Yu et al, 2013), and we reported previously that fission yeast Sen1 physically associates with RNAP3 and is recruited to specific tRNA genes (Legros et al, 2014). The function of Sen1 at RNAP3‐transcribed genes has remained unclear, however. RNAP3 is predominantly implicated in the transcription of the short and abundant tRNA and 5S rRNA species. Internal promoter sequences called A‐ and B‐box recruit the TFIIIC complex, which helps to position TFIIIB upstream of the transcription start site (TSS). TFIIIB in turn recruits the 17 subunits of RNAP3 complex at the TSS to initiate transcription (reviewed in Schramm and Hernandez 2002). In fission yeast, an upstream TATA box assists TFIIIC in recruiting TFIIIB and is essential for the proper recruitment of RNAP3 (Hamada et al, 2001). In vitro transcription assays have indicated that once loaded, RNAP3 can terminate transcription autonomously upon reaching a transcription termination signal, which is constituted by a simple stretch of five thymine residues on the non‐template strand (Mishra & Maraia, 2019), although this number may vary for different genes and organisms (reviewed in Arimbasseri et al 2013). The C37/53/11 subcomplex of RNAP3 is particularly important for this intrinsic transcription termination mechanism (Arimbasseri et al, 2013). Interestingly, however, low levels of read‐through transcription are observed at many tRNA genes in vivo in budding yeast, especially at tRNA genes with weaker terminator sequences (Turowski et al, 2016). The resulting 3′‐extended transcripts are degraded by specific mechanisms involving the RNA exosome and the poly(A)‐binding protein Nab2 (Turowski et al, 2016). In addition, human RNAP3 was also found in the 3′ regions of many tRNA genes (Orioli et al, 2011), suggesting that in distantly related systems, RNAP3 frequently overrides primary termination signals. These observations suggest that efficient RNAP3 transcription termination might be more challenging in vivo than suggested by in vitro studies. Yet, to what extent robust RNAP3 transcription termination requires the support of ancillary factors in vivo remains elusive.
We previously showed that unstable R‐loops form at tRNA genes in fission yeast (Legros et al, 2014). Similar observations have been made in budding yeast (El Hage et al, 2014), humans (Chen et al, 2017) and the plant Arabidopsis thaliana (Xu et al, 2017), suggesting that R‐loop formation is a conserved feature of RNAP3 transcription. The genome‐wide stabilization of R‐loops by the deletion of RNase H had a mild impact on pre‐tRNA processing in budding yeast (El Hage et al, 2014). However, whether these mild perturbations were a direct consequence of R‐loop stabilization in cis at tRNA genes was not addressed. Thus, the contribution of R‐loops to RNAP3 transcription still remains largely unknown.
Our previous work revealed that fission yeast tRNA genes form R‐loops and recruit Sen1. As both R‐loops and Senataxin were previously proposed to facilitate transcription termination of RNAP2 in humans (Skourti‐Stathaki et al, 2011), we investigated the possibility that Sen1 together with R‐loops might participate in transcription termination of RNAP3 in fission yeast. We find that Sen1 primarily associates with RNAP3 transcription units at the genome‐wide level and does indeed promote proper RNAP3 transcription termination, albeit in a manner that is insensitive to the presence of R‐loops. Thus, in contrast to conclusions drawn from in vitro studies, our work reveals the need for a protein cofactor to ensure robust RNAP3 transcription termination in vivo.
Results
Sen1 but not Dbl8 associates primarily with RNAP3‐transcribed genes
There are two homologues of Senataxin in fission yeast, Sen1 and Dbl8. Our previous mass spectrometry (MS) analysis of the protein partners of GFP‐tagged Sen1 indicated that Sen1 associates physically with RNAP3 but not with RNAP1 or RNAP2 (Legros et al, 2014). Using affinity purification coupled to MS with Flag‐tagged proteins expressed from their endogenous chromosomal loci, we show here that Sen1 and Dbl8 associate with different sets of proteins (Appendix Table S1). Sen1 primarily associated with RNAP3 (Fig EV1A), whereas Dbl8 associated with RNAP1 subunits but not with RNAP3 (Fig EV1A and B). The association between Sen1 and RNAP3 was resistant to a benzonase treatment, indicating that it is mediated by direct protein contacts rather than by DNA or RNA (Appendix Fig S1). Taken together, these data suggested that Sen1 but not Dbl8 could play a direct role at RNAP3‐transcribed genes. Accordingly, the current study will focus on the role of Sen1 in RNAP3 transcription.
Figure EV1. Sen1 and Dbl8 associate with different RNA polymerases.
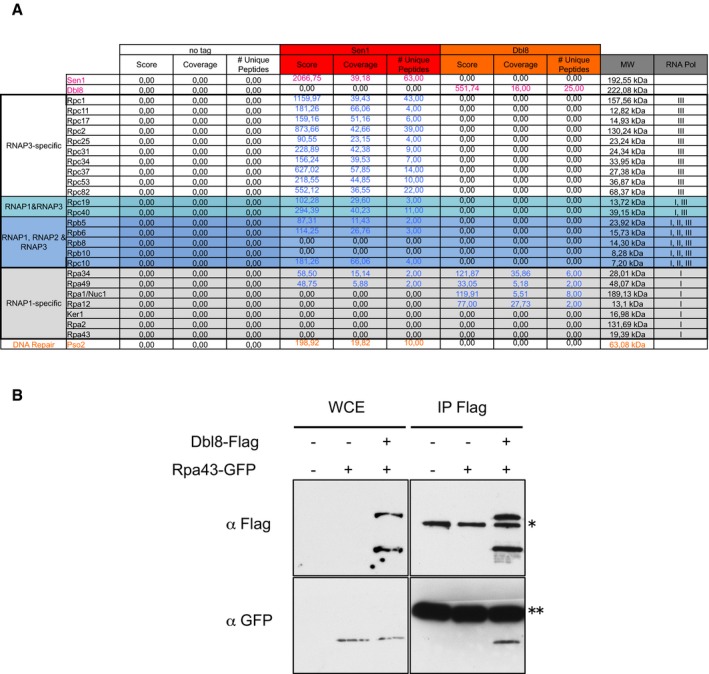
- Flag‐tagged Sen1 and Dbl8 were affinity‐purified and their associated proteins identified using mass spectrometry analysis. The table lists the RNA polymerase components that were recovered with either Sen1 or Dbl8. The full list of proteins identified is shown in Appendix Table S1.
- Co‐immunoprecipitation between Flag‐tagged Dbl8 and the GFP‐tagged RNAP1 subunit Rpa43. * Aspecific protein recognized by the anti‐Flag antibody; ** IgG.
Consistent with the idea that Sen1 acts at RNAP3‐transcribed genes, we reported previously that Sen1 associates with specific tRNA genes in an R‐loop‐independent manner (Legros et al, 2014). However, the extent of Sen1 recruitment to all RNAP3‐transcribed genes (tRNA, 5S rRNA, srp7 and U6 snRNA) had remained unclear. We therefore used chromatin immunoprecipitation (ChIP) assays coupled to high‐throughput sequencing (ChIP‐seq) to analyse the genome‐wide distribution of Sen1 relative to RNAP2 and RNAP3 occupancy. Analysis of ChIP‐seq data confirmed that Sen1 is primarily enriched at tRNA and 5S rRNA genes, which also showed specific and robust RNAP3 binding (Fig 1A). In contrast, RNAP2‐transcribed genes, as exemplified by the strongly expressed tef3 gene in Fig 1A, showed only background Sen1 signal. Sen1 was also enriched at the RNAP3‐transcribed U6 snRNA snu6 (Fig 1B) and srp7 (Fig 1C) loci. A breakdown of all aligned ChIP‐seq reads from two independent experiments revealed that more than 85% of Sen1‐associated regions correspond to RNAP3‐transcribed genes (Fig 1D, tRNA and 5S rRNA). This contrasts to Seb1 (Lemay et al, 2016), the fission yeast homologue of the NNS component Nrd1, that primarily associates with RNAP2‐transcribed genes (Fig 1D). This is consistent with previous observations that Sen1 and Seb1 do not form a functional complex in fission yeast (Legros et al, 2014; Lemay et al, 2016; Larochelle et al, 2018). Overall, we observed a positive genome‐wide correlation between ChIP‐seq signals of Sen1 and two independent subunits of RNAP3 but not with RNAP2 at all loci (Fig 1E and Appendix Fig S2). We confirmed the enrichment of Sen1 at RNAP3‐transcribed sites using ChIP‐qPCR (Fig 1F). Consistent with the observation that Dbl8 does not form a complex with RNAP3, Dbl8 was not enriched at RNAP3‐transcribed genes (Fig 1F) and lack of Dbl8 did not affect the recruitment of Sen1 at RNAP3‐transcribed genes (Appendix Fig S3). Interestingly, chromosome‐organizing clamp (COC) sites, which recruit TFIIIC but not RNAP3 (Noma et al, 2006), were not enriched for Sen1 (Fig 1F). This is consistent with our observation that Sen1 physically associates with RNAP3 but not with TFIIIC (Legros et al, 2014) and suggests that the RNAP3 transcription complex might recruit Sen1 to target genes. To test this possibility, we mutated the upstream TATA box of a model tRNA gene, SPCTRNAARG.10 (tRNAARG UCG), in order to interfere with the recruitment of RNAP3 specifically at this locus. The mutated TATA‐less SPCTRNAARG.10 locus showed reduced levels of both RNAP3 and Sen1 (Fig 1G), indicating that the TATA box‐dependent recruitment of RNAP3 is important for Sen1 occupancy at RNAP3‐transcribed genes. Collectively, these data indicate that fission yeast Sen1 is primarily enriched at RNAP3‐transcribed genes, where it is likely to perform a function that is not shared with Dbl8.
Figure 1. Sen1 associates predominantly with RNAP3‐transcribed genes.
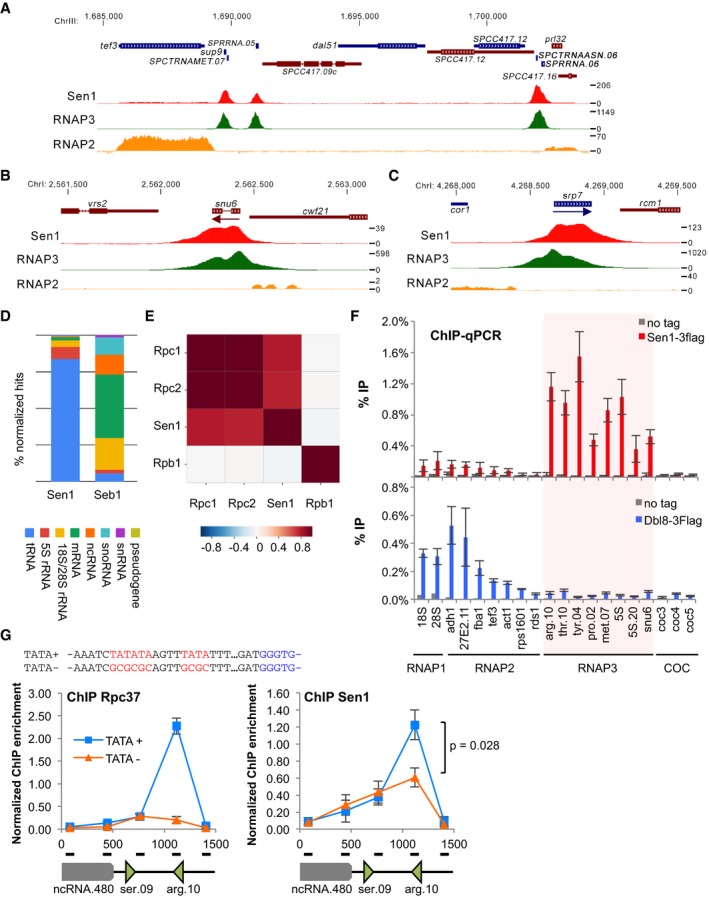
-
A–CSnapshots of ChIP‐seq signals of Sen1 (this study), RNAP3 (this study) and RNAP2 (data from Larochelle et al 2018) across (A) a 25‐kb region of chromosome 3 containing several tRNA and 5S rRNA genes, (B) the U6 snRNA snu6 and (C) the srp7 loci.
-
DComparison of the distribution of ChIP‐seq reads across the indicated categories of genes for Sen1 (this study) and Seb1 (data from Lemay et al 2016).
-
EGenome‐wide pairwise Pearson's correlation coefficient matrix at a resolution of 10 bp.
-
FChIP‐qPCR analysis of Flag‐tagged Sen1 at the indicated loci in a population of cycling cells (mean ± SD from 4 biological replicates).
-
GChIP‐qPCR analysis of the Flag‐tagged RNAP3 subunit Rpc37 and Flag‐tagged Sen1 across the SPCTRNAARG.10 tRNA locus, whose TATA box was either mutated (TATA−) or not (TATA+). The mutations introduced to disrupt the putative TATA boxes are indicated in red above the graphs. The enrichment values were normalized to SPCTRNATHR.10 (mean ± SD from four biological replicates; P‐value obtained using the Wilcoxon–Mann–Whitney statistical test).
Sen1 is required for normal RNAP3 transcription
Next, we investigated whether lack of Sen1 affects RNAP3 transcription. Using ChIP‐qPCR, we detected increased amount of RNAP3 at all sites tested in the absence of Sen1 (Fig 2A). Strikingly, this accumulation of RNAP3 over its target genes was not associated with an increased amount of RNAP3 transcripts in the cell (Fig 2B, compare lanes 1–2). The steady state level of 5S RNAs remained unchanged, and the overall levels of tRNA detected in the absence of Sen1 were even slightly reduced (Fig 2B), as confirmed using gene‐specific RT–qPCR (Fig 2C). Interestingly, this reduction was even more apparent when Dis3‐mediated tRNA degradation (Gudipati et al, 2012; Schneider et al, 2012) was impaired (Fig 2B, compare lanes 3–4). Since the steady state levels of tRNAs are controlled by the equilibrium between synthesis and Dis3‐mediated degradation (Gudipati et al, 2012; Schneider et al, 2012), the reduction in nuclear exosome activity associated with the depletion of Dis3 is expected to uncover the direct impact that lack of Sen1 has on RNAP3 transcription. Taken together, these results therefore indicate that lack of Sen1 impairs RNAP3 transcription.
Figure 2. Sen1 is required for normal RNAP3 transcription.
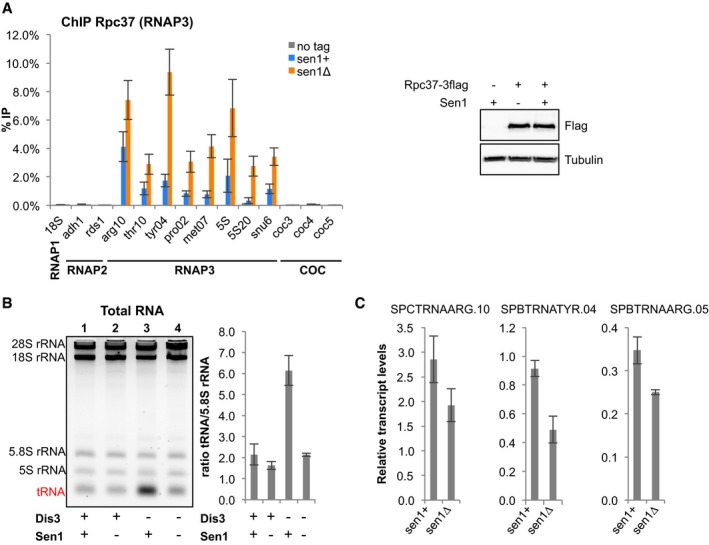
- (left) ChIP‐qPCR analysis of Rpc37 in the presence or absence of Sen1 at the indicated loci in a population of cycling cells (mean ± SD from four biological replicates). (right) Western blot analysis of Rpc37 protein levels in the presence or absence of Sen1. Tubulin was used as a loading control.
- (left) Total RNA from the indicated strains was separated on a 2.8% agarose gel. (right) Quantification of overall tRNA levels (mean ± SD from three biological replicates).
- Strand‐specific RT–qPCR was used to quantify the indicated RNAP3 transcripts. Transcript levels were normalized to act1 (mean ± SD from three biological replicates).
The accumulation of RNAP3 on its target genes in the absence of Sen1 is independent of R‐loops
Fission yeast Sen1 is able to unwind RNA‐DNA hybrids in vitro (Kim et al, 1999), and the budding yeast and human homologues of Sen1 are believed to antagonize R‐loop formation in vivo (Mischo et al, 2011; Skourti‐Stathaki et al, 2011). Interestingly, R‐loops were shown to interfere with transcription elongation, at least when they form close to the TSS (Belotserkovskii et al, 2017), and we have shown previously that RNase H‐sensitive R‐loops form at tRNA genes in fission yeast (Legros et al, 2014; Hartono et al, 2018). To test whether the stabilization of R‐loops at tRNA genes could underlie the accumulation of RNAP3 in the absence of Sen1, we expressed RNase H1 from Escherichia coli (RnhA) under the control of the strong nmt1 promoter in fission yeast cells. We showed previously that this strategy was sufficient to remove R‐loops at tRNA genes (Legros et al, 2014; Hartono et al, 2018). Using R‐ChIP to monitor R‐loop formation at tRNA genes (Legros et al, 2014), we confirmed that RnhA expression was sufficient to completely remove R‐loops in the absence of Sen1 (Fig 3A). However, this treatment did not alter the accumulation of RNAP3 (Fig 3B), indicating that R‐loops do not contribute to the accumulation of RNAP3 in the sen1∆ mutant. Conversely, stabilization of R‐loops at tRNA genes by the deletion of both endogenous RNase H1 and RNase H2 (rnh1∆rnh201∆) (Legros et al, 2014) did not result in the accumulation of RNAP3 (Fig 3B). These results therefore establish that the stabilization of R‐loops does not account for the accumulation of RNAP3 in Sen1‐deficient cells.
Figure 3. Sen1 regulates RNAP3 recruitment in an R‐loop‐independent manner.
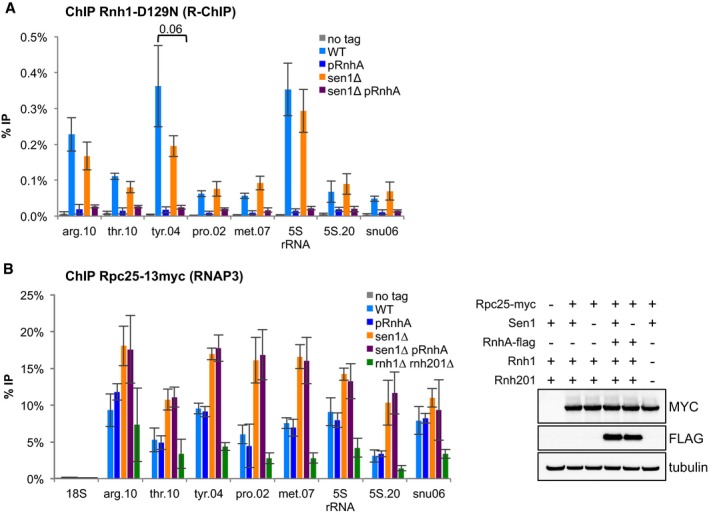
- R‐ChIP using a catalytically inactive RNase H1 (Rnh1‐D129N) was used to quantify R‐loop formation at RNAP3‐transcribed genes (mean ± SD from four biological replicates).
- (left) ChIP‐qPCR of the 13myc‐tagged RNAP3 subunit Rpc25 in the indicated strains at the indicated loci (mean ± SD of four biological replicates). (right) Western blot analysis of Myc‐tagged Rpc25 and Flag‐tagged RnhA protein levels in the indicated strains. Tubulin was used as a loading control.
Sen1 is required for effective RNAP3 transcription termination
We next analysed the effect of a Sen1 deficiency on the genome‐wide distribution of RNAP3 by comparing ChIP‐seq profiles of Rpc1 and Rpc2 in sen1+ and sen1Δ strains. Strikingly, in the absence of Sen1, the distribution of both Rpc1 and Rpc2 displayed increased density downstream of most tRNA and 5S rRNA genes (as exemplified on Fig 4A and B), as well as at srp7 (Fig 4C), consistent with read‐through transcription by RNAP3. Evidence of delayed transcription termination in the sen1Δ mutant was also noted at the snu6 gene, albeit at more modest levels (Appendix Fig S4). Importantly, averaging Rpc1 and Rpc2 ChIP‐seq signals over all isolated tRNA and 5S rRNA genes confirmed that the distribution pattern of RNAP3 is globally extended at the 3′ end in the absence of Sen1 (Fig 4D). ChIP followed by qPCR analysis at several candidate loci confirmed that the domain occupied by RNAP3 was wider in the absence of Sen1 and that RNAP3 accumulated downstream of its natural transcription termination sites (Fig 4E). Notably, this accumulation downstream of the transcription termination site was not altered upon RnhA expression, confirming that it did not result from the stabilization of R‐loops (Fig EV2). Together, these results reveal that Sen1 is required for RNAP3 termination at the genome‐wide level in an R‐loop‐independent manner.
Figure 4. Sen1 is required for efficient RNAP3 transcription termination.
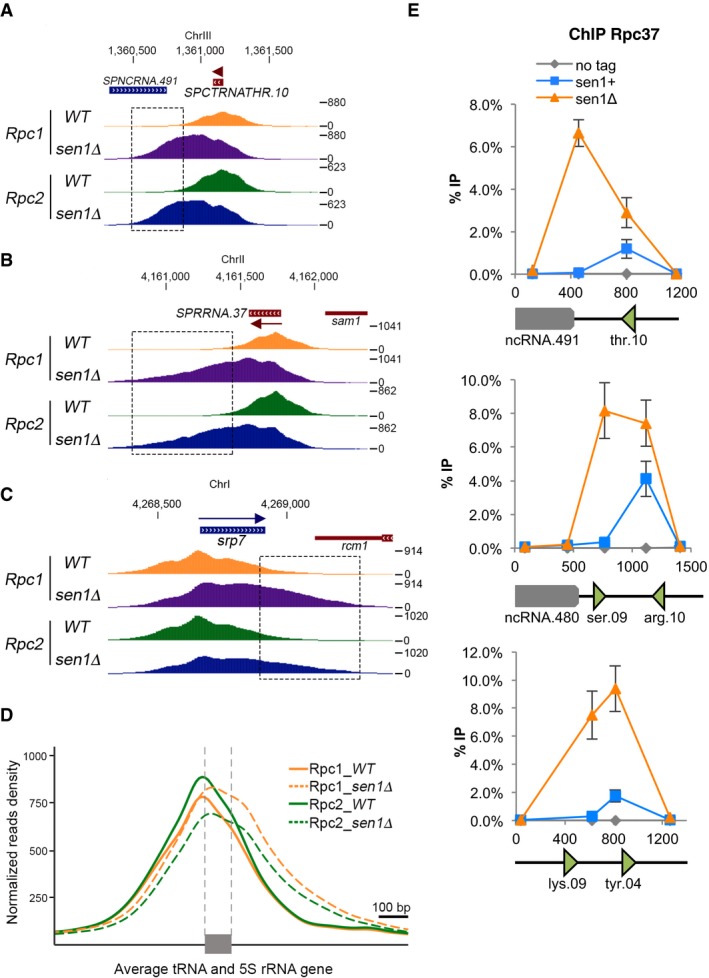
-
A–CSnapshots of ChIP‐seq signals of the RNAP3 subunits Rpc1 and Rpc2 in the presence or absence of Sen1 across a representative (A) tRNA gene, (B) 5S rRNA gene and (C) srp7. Boxed regions highlight the increased density of reads in the downstream region of genes in the absence of Sen1.
-
DAverage ChIP‐seq profile of Rpc1 and Rpc2 across all isolated tRNA and 5S rRNA genes in the presence and absence of Sen1.
-
EScanning of Rpc37‐3flag occupancy at three different tDNA loci in the absence of Sen1 by ChIP‐qPCR (mean ± SD from six biological replicates).
Figure EV2. Loss of R‐loops upon expression of RnhA does not impact the distribution of RNAP3 in the absence of Sen1.
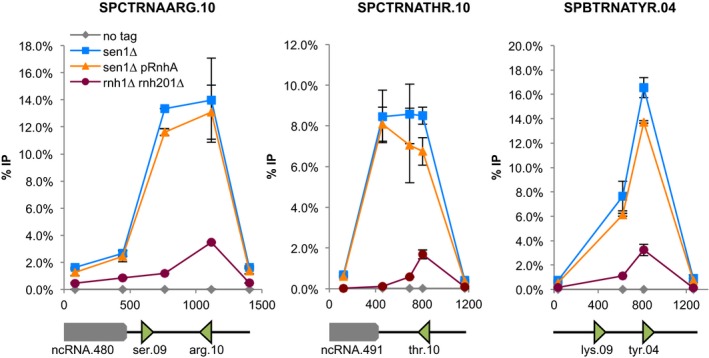
ChIP‐qPCR analysis of the RNAP3 subunit Rpc25 in the indicated genotypes and at the indicated loci in a population of cycling cells (mean ± SD from two biological replicates).
Read‐through tRNA transcripts accumulate in the absence of Sen1
We used several independent assays to demonstrate that the transcription termination defects associated with lack of Sen1 resulted in the production of 3′‐extended transcripts. First, we used a genetic assay that translates a transcription termination defect at the synthetic tRNA DRT5T construct into a change of colour of yeast colonies from red to white (Iben et al, 2011; Fig 5A, see scheme of the construct at the top). Briefly, a transcription termination defect allows the synthesis of a suppressor tRNA that suppresses the accumulation of a red pigment caused by the ade6‐704 mutation, resulting in white colonies in limiting adenine conditions. As a positive control for this assay, we mutated the valine residue at position 189 in the Rpc37 subunit of RNAP3 into an aspartate residue (rpc37‐V189D), as overexpression of this mutant was shown to interfere with transcription termination in a dominant‐negative manner (Rijal & Maraia, 2013). Here, we mutated the endogenous rpc37 gene and established that the rpc37‐V189D mutant is viable but displays transcription termination defects (Fig 5A). Similarly, in the absence of Sen1 but not in the absence of its close homologue Dbl8, colonies turned white in the presence of the DRT5T construct, indicating that Sen1 but not Dbl8 is required for robust transcription termination at DRT5T (Fig 5A). Consistent with the idea that Sen1 contributes to transcription termination of RNAP3‐transcribed genes, we found that Sen1 becomes essential for cell viability when the termination is impaired by the rpc37‐V189D mutant (Fig 5B).
Figure 5. Lack of Sen1 produces extended read‐through tRNA transcripts.
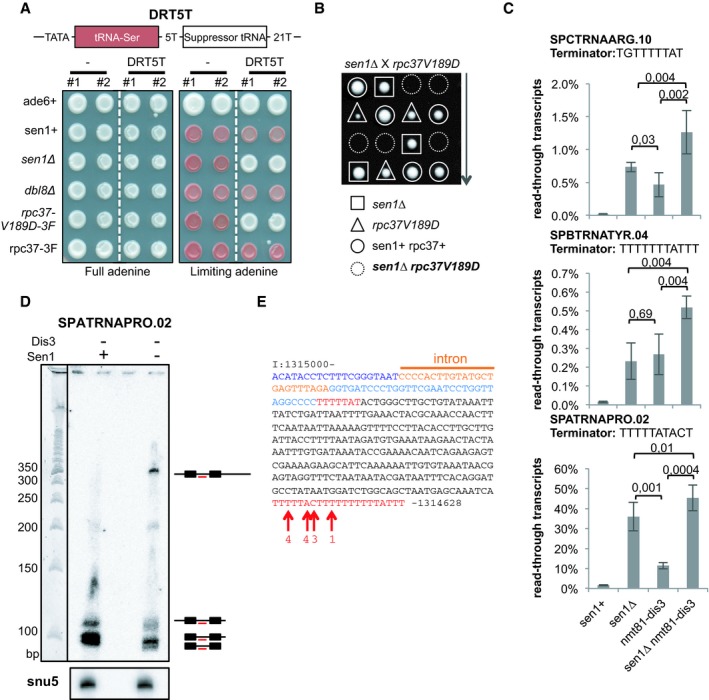
- Cells of the indicated genotypes that carried or not the DRT5T dimeric tRNA construct (schematized on top) were grown either in the presence of the optimum concentration of adenine (left) or in the presence of a limiting concentration of adenine (right). Two independent clones of the same genotype (#1 and #2) were used. See text for details. 3F refers to the 3Flag epitope tag at the C‐terminus of Rpc37.
- Tetrad dissection was used to show that the double‐mutant sen1∆ rpc37‐V189D is dead.
- Strand‐specific RT–qPCR was used to quantify the levels of read‐through transcripts (see Materials and Methods). The mean ± SD from four biological replicates is represented here. P‐values were obtained using the Wilcoxon–Mann–Whitney statistical test.
- Northern blot analysis of the tRNA SPATRNAPRO.02 using an intron‐specific probe (TCTAAACTCAGCATACAAGTGGGG). U5 snRNA was used as a loading control.
- Sequence of the ˜350‐nt‐long read‐through transcript at SPATRNAPRO.02. Residues in blue represent the sequence of the mature tRNA. Residues in red represent potential terminator sequences. Red arrows show the 3′ end nucleotide of the read‐through transcripts. The numbers indicate the number of times the sequenced transcripts terminated at the indicated position.
To confirm the production of read‐through transcripts at endogenous tRNA genes in the absence of Sen1, we used strand‐specific RT–qPCR. Using this approach, we detected read‐through transcripts in the absence of Sen1 at several tRNA genes (Fig EV3). Importantly, read‐through transcripts were not detected in the absence of Dbl8, confirming that Sen1 plays a non‐redundant role in RNAP3 transcription (Fig EV3). To rule out that those read‐through transcripts only resulted from the defective degradation of naturally occurring longer transcripts (Turowski et al, 2016), we quantified these read‐through transcripts in cells deficient for the RNA exosome subunit Dis3. If Sen1 was only involved in the RNA exosome‐dependent degradation of naturally occurring read‐through transcripts, the amount of read‐through transcripts found in RNA exosome mutants should not change upon deletion of Sen1. As shown in Fig 5C, we found that the amount of read‐through transcripts detected in RNA exosome mutants increased significantly in the absence of Sen1, suggesting that the accumulation of read‐through tRNAs in the absence of Sen1 is independent of RNA exosome activity. Using Northern blots, we detected a predominant ~350‐nt‐long extended transcript at the intron‐containing SPATRNAPRO.02 (tRNAPRO CGG) in the absence of Sen1 (Fig 5D). Sequencing of the 3′ end of this transcript confirmed that it included a 3′ extension and showed that it terminated at a strong distal terminator sequence (TTTTTACTTTTTTTTTTATTT) located 274 bp downstream of the primary terminator (Fig 5E). This is consistent with our ChIP‐seq data and confirms that RNAP3 continues transcribing downstream of the primary terminator sequence in the absence of Sen1. Finally, we showed that the accumulation of read‐through transcripts in the absence of Sen1 was unchanged after RnhA expression, reinforcing the idea that read‐through transcription in the absence of Sen1 was not a consequence of R‐loop stabilization (Appendix Fig S5). Taken together, our data indicate that Sen1 prevents the synthesis of long, aberrant read‐through tRNAs by promoting efficient termination of RNAP3.
Figure EV3. Lack of Sen1 but not lack of its close homologue Dbl8 results in read‐through transcripts at tRNA genes.
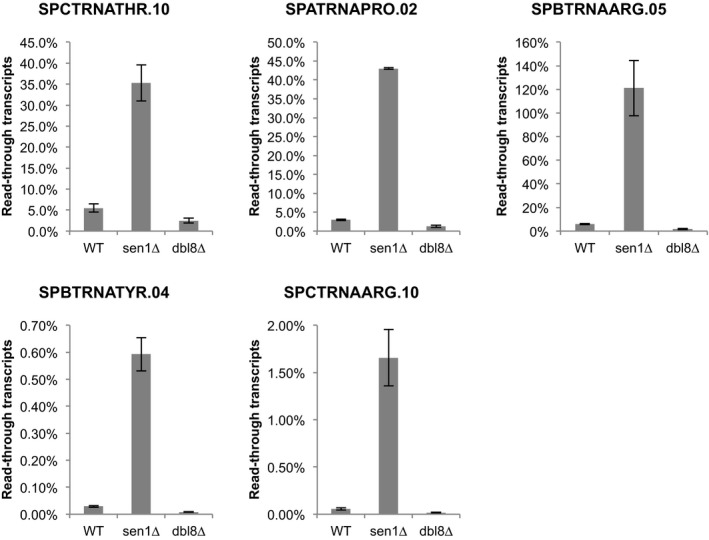
Strand‐specific RT–qPCR was used to quantify the levels of read‐through transcripts (see Materials and Methods). The mean ± SD from two biological replicates is represented here.
A strong terminator sequence compensates for lack of Sen1
It was shown previously that a stretch of 21 thymine residues is sufficient to provide robust transcription termination even in the presence of a termination‐defective RNAP3 (Iben et al, 2011; Rijal & Maraia, 2013) and our analysis of read‐through transcripts at SPATRNAPRO.02 (Fig 5C and D) was consistent with the idea that a strong terminator sequence could overcome the requirement for Sen1 for RNAP3 transcription termination. We therefore tested whether strengthening the primary terminator sequence could rescue the defects observed in the absence of Sen1. To do this, we replaced the endogenous terminator sequence of SPCTRNAARG.10 by a stretch of 23 thymine residues (Fig 6A). Strikingly, the presence of such a strong terminator sequence was sufficient to suppress both the accumulation of RNAP3 downstream of the terminator (Fig 6B, compare orange to green lines) and the accumulation of read‐through transcripts in the absence of Sen1 (Fig 6C). Note, however, that the presence of this super‐terminator at SPCTRNAARG.10 did not reduce the production of read‐through transcripts at the neighbouring SPCTRNASER.09 (Fig 6C). Similarly, the introduction of a terminator composed of 20 consecutive thymine residues at SPCTRNATHR.10 was sufficient to suppress the accumulation of RNAP3 downstream of the terminator (Fig EV4). Altogether, these observations demonstrate that the RNAP3 molecules that accumulate downstream of the terminator sequence in the absence of Sen1 correspond to RNAP3 molecules that overrode the terminator sequence and conclusively establish that Sen1 is required for robust transcription termination of RNAP3‐transcribed genes.
Figure 6. A strong terminator sequence compensates for lack of Sen1.
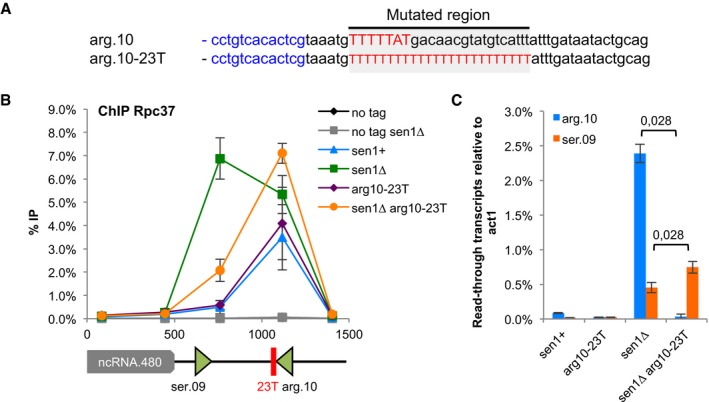
- Sequence of the engineered strong terminator (arg10‐23T) at the SPCTRNAARG.10 gene.
- ChIP‐qPCR analysis of Rpc37 around SPCTRNAARG.10 gene in the strong terminator mutant (mean ± SD from four biological replicates).
- Strand‐specific RT–qPCR was used to quantify read‐through transcripts at SPCTRNAARG.10 (arg.10) and SPCTRNASER.09 (ser.09) in the strong terminator mutant (mean ± SD from four biological replicates; P‐value obtained using the Wilcoxon–Mann–Whitney statistical test).
Figure EV4. A super‐terminator at SPCTRNATHR.10 suppresses the accumulation of RNAP3 in 3′ when Sen1 is missing.
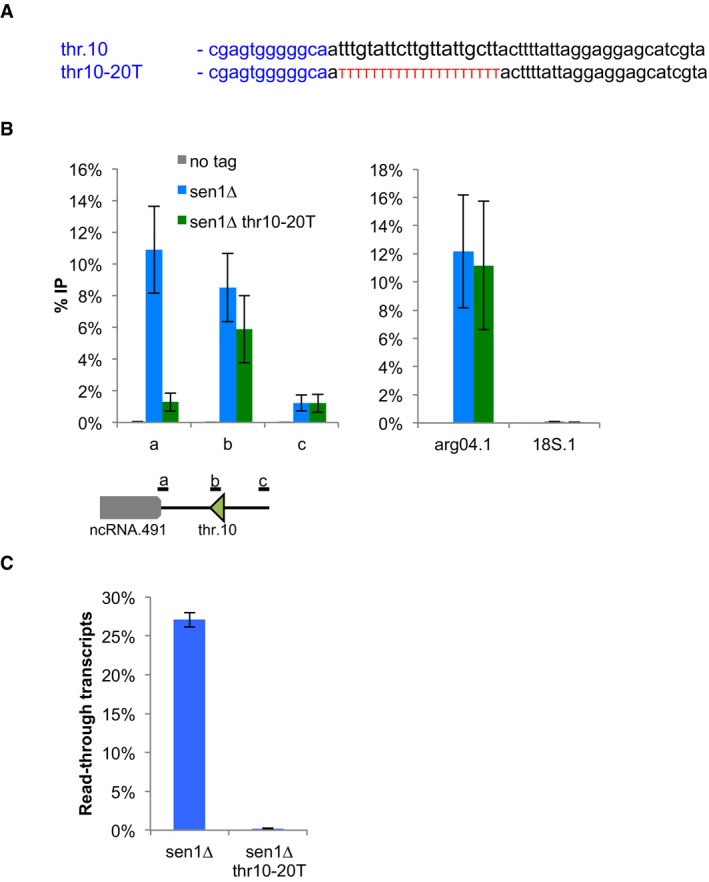
- Sequences of the engineered strong super‐terminator (thr10‐20T) at the SPCTRNATHR.10 gene.
- ChIP‐qPCR analysis of Rpc37 around SPCTRNATHR.10 gene in the strong terminator mutant (mean ± SD from four biological replicates). The RNAP3‐transcribed SPBTRNAARG.04 (arg.04) and the RNAP1‐transcribed rDNA (18S) were used as specificity controls.
- RT–qPCR demonstrates the loss of read‐through transcripts in the strong terminator mutant (mean ± SD from 3 biological replicates).
Discussion
RNAP3 transcribes the abundant structural tRNA and 5S rRNA transcripts. In contrast to RNAP2, whose transcription termination relies on the coordinated assembly of dedicated protein complexes at the 3′ of genes, termination of RNAP3 is generally thought to rely on the autonomous destabilization of the elongation complex upon reaching dedicated terminator DNA sequences. Here, we challenge this view by showing that the highly conserved DNA/RNA helicase Sen1 associates physically with RNAP3, is enriched primarily at RNAP3‐transcribed genes and prevents both the accumulation of RNAP3 downstream of the primary terminator and the production of RNA exosome‐sensitive read‐through transcripts. Our results also indicate that very strong terminator sequences can overcome the need for Sen1 in RNAP3 transcription termination, suggesting that Sen1 acts in complement to the intrinsic RNAP3 transcription termination mechanisms. We conclude that Sen1 is a cofactor of RNAP3 required for robust termination of RNAP3 transcription in fission yeast. Importantly, we found no evidence that Dbl8, the other Senataxin homologue in fission yeast, impacts RNAP3 transcription, suggesting that Sen1 and Dbl8 have evolved separate functions.
Senataxin homologues are conserved regulators of transcription termination
The function of Senataxin homologues in the termination of transcription at specific RNAP2‐transcribed genes is well established in budding yeast and in mammals (Ursic et al, 1997; Steinmetz et al, 2001, 2006; Skourti‐Stathaki et al, 2011; Porrua & Libri, 2013; Zhao et al, 2016) but was previously ruled out for the fission yeast homologues Sen1 and Dbl8 (Lemay et al, 2016; Larochelle et al, 2018). Here, we show that the function of Sen1 in transcription termination is conserved in fission yeast, albeit for another RNA polymerase. This suggests that a contribution to transcription termination is an ancestral function of Senataxin homologues and it is tempting to speculate that their mode of action in promoting transcription termination is conserved for RNAP3.
In budding yeast, Sen1 was shown to interact with all three RNA polymerases (Yüce & West, 2013), opening the possibility that it might contribute to transcription termination of all three RNA polymerases. Consistent with this, it was proposed that Sen1 contributes also to RNAP1 transcription termination in budding yeast (Kawauchi et al, 2008). Although the contribution of Dbl8 to RNAP1 transcription remains to be determined, we showed here that Dbl8 associates with subunits of the RNAP1 complex (Fig EV1), suggesting that the two Senataxin homologues in fission yeast are active at different classes of genes. As a physical interaction between mammalian Senataxin and RNAP3 was not detected previously (Suraweera et al, 2009; Yüce & West, 2013; Miller et al, 2015), it remains unclear whether Senataxin or another DNA/RNA helicase could also contribute to RNAP3 transcription termination in mammalian cells.
Fission yeast Sen1 promotes robust RNAP3 termination in vivo
In vitro studies have led to the conclusion that RNAP3 could terminate transcription autonomously, without the need for accessory factors (reviewed in Arimbasseri et al 2013). In contrast to this view, our data clearly show that Sen1 is required for efficient termination of RNAP3 transcription in vivo. This suggests that in vitro transcription assays, although extremely informative, do not fully recapitulate RNAP3 transcription. In vitro RNAP3 transcription termination assays do not yet recapitulate the chromatin environment and often, although not always, rely on the artificial assembly of an elongating complex (EC) without the need for TFIIIB or TFIIIC (Arimbasseri & Maraia, 2015). It is possible that the role of Sen1 in transcription termination becomes dispensable in these conditions. This could also indicate that the way RNAP3 is loaded onto the DNA template has an impact on the way transcription terminates, as suggested previously (Wang & Roeder, 1998). Alternatively, it is possible that transcription elongation rates are higher in vivo than in vitro and demand a more efficient Sen1‐dependent transcription termination mechanism, in line with what has been shown for Sen1‐mediated RNAP2 termination in budding yeast (Hazelbaker et al, 2013).
Lack of Sen1 impacts RNAP3 tRNA levels
Our ChIP‐qPCR experiments indicate that RNAP3 levels increased at RNAP3‐transcribed genes in the absence of Sen1. Interestingly, this accumulation was not accompanied by an increase in the amount of mature tRNA. On the contrary, we measured a threefold reduction in the amount of tRNA in the absence of Sen1 when Dis3‐dependent tRNA degradation was impaired (Fig 2B), suggesting that nascent tRNA production is reduced. Interestingly, Dis3 depletion did not alter overall tRNA abundance in the absence of Sen1 (Fig 2B, compare lanes 2–4). This could suggest that Dis3 is less active towards tRNA when Sen1 is missing, thereby reducing tRNA degradation and partially compensating for the reduction in tRNA production.
It is important to note that the conclusion that tRNA production is reduced in the absence of Sen1 differs significantly from what we concluded in a previous study (Legros et al, 2014). The reason for this is merely technical. We previously measured the abundance of tRNA species using RT–qPCR primed with random hexamers, whereas gene‐specific oligonucleotides were used in the current study. Figure EV5 shows that tRNAs are poorly reverse‐transcribed using random hexamers, probably because they are small and highly structured (Zheng et al, 2015). Conversely, the long read‐through transcripts produced in the absence of Sen1 are more efficiently reverse‐transcribed, giving the false impression that tRNA levels were more abundant (Fig EV5).
Figure EV5. RT with random hexamers predominantly retro‐transcribed the long read‐through tRNA transcripts produced in the absence of Sen1.
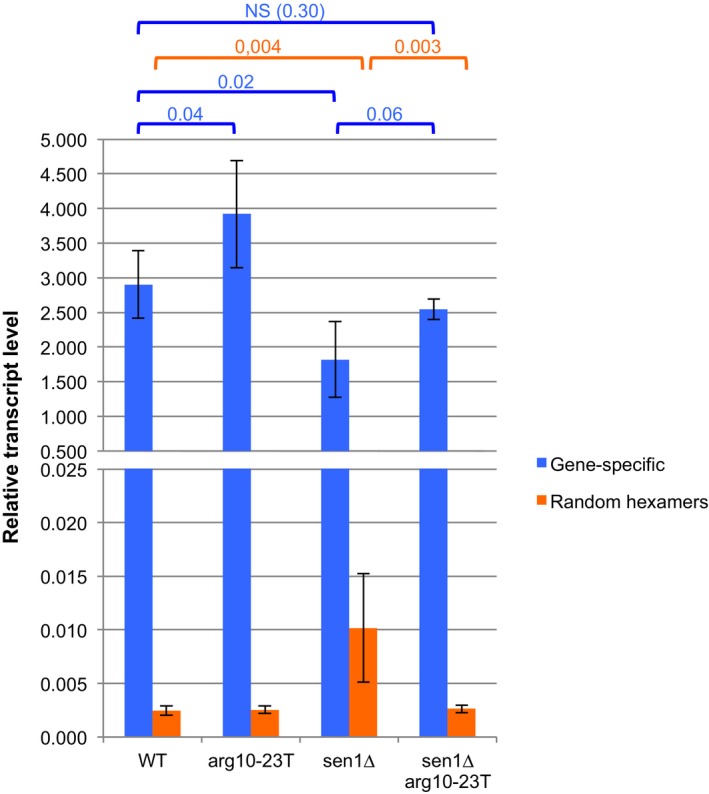
Total RNAs were extracted from the indicated strains, and RT–qPCR was performed using either random hexamers or a primer specific for SPCTRNAARG.10. The gene‐specific primer gave much stronger signals than random hexamers. Random hexamers retro‐transcribed predominantly the long read‐through transcripts produced in the absence of Sen1, as shown by the fact that relative RNA levels dropped significantly when read‐through transcription was prevented by the presence of a strong terminator (arg10‐23T) (mean ± SD from 6 biological replicates; P‐values from a Wilcoxon–Mann–Whitney test).
There are two non‐mutually exclusive models for interpreting the reduced tRNA accumulation in the absence of Sen1. It was previously suggested that efficient transcription termination facilitates the rapid recycling of RNAP3, thereby maintaining high levels of nascent transcription (Dieci & Sentenac, 1996). Conversely, abnormal transcription termination downstream of the canonical terminator in the absence of Sen1 might prevent efficient RNAP3 recycling by reducing the physical proximity between the terminating RNAP3 and the transcription factors required for initiation (Turowski & Tollervey, 2016). In this scenario, the reduced tRNA accumulation in sen1∆ cells would result directly from the transcription termination defects. Alternatively, lack of Sen1 might independently affect transcription elongation and termination. Interestingly, the introduction of a super‐terminator sequence at SPACTRNAARG.10 suppressed the transcription termination defects in sen1Δ cells but failed to restore RNAP3 levels back to normal on this tRNA gene (Fig 6B). Although we cannot completely rule out at this stage that some of the RNAP3 molecules that we detected at SPACTRNAARG.10 correspond to RNAP3 molecules that overrode the terminator sequence at the neighbouring SPCTRNASER.09, these observations might also suggest that the accumulation of RNAP3 on tRNA genes is at least partly independent from the transcription termination defects observed in Sen1‐deficient cells.
Could a Mfd‐like release and catch‐up mechanism explain the different roles of Sen1?
The precise mode of action of fission yeast Sen1 in RNAP3 transcription termination remains to be determined. It was recently proposed that the Mfd translocase in Escherichia coli could both nudge forward weakly paused RNAP and induce the dissociation of stalled RNAP using a release and catch‐up mechanism (Le et al, 2018). It is tempting to speculate that such a release and catch‐up mechanism could underlie the roles of fission yeast Sen1 in RNAP3 transcription: through translocation, Sen1 could nudge forward RNAP3 molecules that are weakly paused at gene‐internal pause sites such as TFIIIC‐binding sites (Turowski et al, 2016) and therefore facilitate transcription elongation, or release RNAP3 molecules that are stalled at primary terminator sequences to promote transcription termination. Importantly, the release and catch‐up mechanism was also proposed to underlie the role of Mfd in both transcription‐coupled repair and transcription–replication conflict resolution (Le et al, 2018) and budding yeast Sen1 has also been implicated in both transcription‐coupled repair (Li et al, 2016) and transcription–replication conflict resolution (Mischo et al, 2011; Alzu et al, 2012; Brambati et al, 2018), strengthening the analogy with Mfd. However, the molecular mechanisms involved in such a release and catch‐up mechanism might differ between Sen1 and Mfd, because Mfd was shown to translocate autonomously on double‐stranded DNA, whereas both budding yeast and fission yeast Sen1 were shown to translocate on both single‐stranded DNA and RNA, albeit at greater rate on DNA (Kim et al, 1999; Martin‐Tumasz & Brow, 2015; Han et al, 2017). In addition, translocation on the nascent RNA was sufficient to explain the ability of budding yeast Sen1 to dissociate RNAP2 elongation complexes in vitro (Han et al, 2017).
A unifying way of interpreting the different roles of Sen1 could be to propose that it is targeted to chromatin through its interaction with RNA polymerases and that it subsequently patrols chromatin locally to facilitate the resolution of R‐loops and/or stalled elongation complexes through a release and catch‐up mechanism. The identity of the RNA polymerase involved would change in different organisms or for different paralogs in the same organism. Such a scenario might also explain that RNAP2 accumulates over tRNA genes in an hypomorphic mutant of budding yeast Sen1 (Steinmetz et al, 2006). We propose that the ancestral function of Senataxin‐like helicases is to dissociate stalled transcription elongation complexes from chromatin.
Materials and Methods
Fission yeast strains and culture
The list of all the strains used in this study is given in Appendix Table S2. Standard genetic crosses were employed to construct all strains. sen1‐3flag and rpc37‐3flag were generated using a standard PCR procedure. Cells were grown at 30°C in complete YES+adenine medium or in synthetic PMG medium as indicated. The expression of the dis3 gene driven by the nmt81 promoter was repressed by the addition of 60 μM of thiamine to the PMG medium. To induce the expression of E. coli RnhA from by the nmt1 promoter, cells were grown in PMG minimal medium lacking thiamine for a minimum of 18 h.
Mutagenesis of Rpc37
To generate rpc37‐V189D‐3Flag, the 3′ end of rpc37 was first amplified by PCR and cloned into pCR‐Blunt II‐TOPO using the Zero Blunt II‐TOPO PCR Cloning Kit (Invitrogen Life Technologies). Site‐directed PCR mutagenesis was then carried out to mutate the codon corresponding to valine 189 (GTC into GAC) using the QuikChange II Site‐Directed Mutagenesis Kit (Agilent Technologies). Overlapping PCR was used to add a C‐terminal 3Flag epitope and a marker conferring resistance to Nourseothricin, and the resulting PCR fragment was transformed into fission yeast using routine protocols. Proper integration of the mutation at the endogenous locus was verified by PCR and sequencing.
Mutagenesis of SPCTRNAARG.10
The TATA‐less and super‐terminator mutants of SPCTRNAARG.10 (arg10 TATA‐less and arg10‐23T) were first synthesized (GeneCust Europe). The mutagenized SPCTRNAARG.10 gene was then transformed into fission yeast, and its correct integration was selected by counter‐selecting on FOA the loss of the ura4 gene previously integrated at SPCTRNAARG.10. The correct integration of the mutation was confirmed by sequencing.
Chromatin Immunoprecipitation
1.5 × 108 cells were cross‐linked with 1% formaldehyde (Sigma) at 18°C for 30 min. After three washes with cold PBS, the cells were frozen in liquid nitrogen. Frozen cells were then lysed in cold lysis buffer (HEPES‐KOH 50 mM [pH 7.5], NaCl 140 mM, EDTA 1 mM, Triton 1%, Na‐deoxycholate 0.1%, Phenylmethanesulphonyl fluoride (PMSF) 1 mM) with glass beads using a Precellys 24 mill (Bertin Technology). To fragment the chromatin, the lysates were sonicated at 4°C using a Covaris S220 or Diagenode Bioruptor sonicator. Immunoprecipitation was done overnight at 4°C using Protein A‐coupled or Protein G‐coupled Dynabeads previously incubated with anti‐GFP A11122 antibody (Invitrogen), anti‐Flag antibody (M2 Sigma) and anti‐Myc 9E10 (Sigma). The immunoprecipitated complexes were washed for 5′ successively with: Wash I buffer (20 mM Tris pH 8, 150 mM NaCl, 2 mM EDTA, 1% Triton X‐100, 0.1% SDS), Wash II buffer (20 mM Tris pH 8, 500 mM NaCl, 2 mM EDTA, 1% Triton X‐100, 0.1% SDS) and Wash III buffer (20 mM Tris pH 8, 1 mM EDTA, 0.5% Na‐deoxycholate, 1% Igepal, 250 mM LiCl). After two additional washes in Tris‐EDTA pH 8, the beads were resuspended in 10% Chelex resin (Bio‐Rad) and incubated at 98°C for 10′. After addition of 2 μl of 10 mg/ml proteinase K, the mixture was incubated at 43°C for 1 h, then at 98°C for another 10′. After centrifugation, the supernatant was collected and analysed by qPCR in a thermocycler Rotor‐Gene (Qiagen) using the primers listed in Appendix Table S3.
Western blotting
Protein extraction was performed using the TCA (trichloroacetic acid)–glass beads method. 108 cells were centrifuged for 3′ at 2,000 g, resuspended in 20% TCA and then lysed using glass beads in a Precellys 24 mill (Bertin Technology). After centrifugation (4′ at 13 krpm), the resulting pellets were resuspended in sample buffer (0.1 M Tris–HCl pH 9.5, 20% glycerol, 4% SDS (sodium dodecyl sulphate), 0.2% bromophenol blue, 715 mM β‐mercaptoethanol), incubated for 5′ at 100°C and centrifugated again at 13 krpm for 4′. The resulting supernatants were separated using SDS–PAGE on 7.5% polyacrylamide gels and transferred onto nitrocellulose using a semi‐dry transfer system. Anti‐Myc (A‐14 Santa Cruz Biotechnology) and anti‐Flag (M2 Sigma) antibodies were used for immunodetection of proteins and revealed using ECL‐based reagents. An anti‐tubulin antibody (TAT1), courtesy of Prof. Keith Gull (Oxford), was used as loading control.
Mass spectrometry analysis
The immunoprecipitation of Flag‐tagged Sen1 and Dbl8 and the analysis of their protein partners were performed as previously described (Legros et al, 2014). The complete list of the proteins identified is shown in Appendix Table S1.
RNA techniques
Total RNA was extracted from logarithmical growing cells (2 × 108) by the standard hot‐phenol method. The remaining traces of genomic DNA digest were digested with DNAse I (Ambion), and the integrity of RNAs was verified by electrophoresis on 0.8% agarose gels. Total RNA was reverse‐transcribed using SuperScript III (Invitrogen) according to the manufacturer's instructions using the strand‐specific primers listed in Appendix Table S3. To quantify read‐through transcripts at specific tRNA genes, two independent RT reactions were carried out in parallel: RT1 used a priming oligonucleotide placed downstream of the primary terminator, and RT2 used a priming oligonucleotide placed in the gene body upstream of the primary terminator. For both RT1 and RT2, an act1‐specific priming oligonucleotide was also used as internal control. The resulting cDNAs were quantified by quantitative PCR (qPCR) using a Rotor‐Gene machine (Qiagen) and primers specific for act1 and the gene body of the tRNA of interest. The proportion of read‐through transcripts was expressed using the ratio RT1/RT2 and expressed as a percentage (read‐through tRNA transcripts/total tRNA transcripts). At SPBTRNAARG.05, this ratio can exceed 100% in the absence of Sen1, presumably because RT1 is more efficient than RT2 and because most RNAs produced are read‐through RNAs. For Northern blots, 10 μg of total RNAs was separated on 10% polyacrylamide–8M urea gels and transferred onto a nylon membrane (GE Healthcare Amersham Hybond‐N+). The membrane was then UV‐cross‐linked and dried at 80°C for 30 minutes. After incubation with Church buffer for 30 min at 37°C, the membrane was hybridized overnight at 37°C with a 32P‐labelled DNA oligo antisense to the intron of SPATRNAPRO.02. The blot was then washed four times with 1X SSC + 0.1% SDS and scanned using a Phosphorimager Typhoon FLA 9500—GE Healthcare.
Mapping of the 3′ end of read‐through transcripts at SPATRNAPRO.02
20 μg of total RNA was separated on a 10% polyacrylamide–8M urea gel. The 200‐ to 600‐bp‐long RNAs were extracted from the gel and purified, before a pre‐adenylated RNA adaptor was ligated in 3′ as described previously (Heyer et al, 2015). This 3′ adaptor was used for retro‐transcription as described (Heyer et al, 2015), and SPATRNAPRO.02‐derived cDNAs were amplified by PCR using the primer Pro.02 qL1 (5′‐ACATACCTCTTTCGGGTAATCC‐3′). The PCR fragment obtained was cloned into pCR‐Blunt II‐TOPO using the Zero Blunt II‐TOPO PCR Cloning Kit (Invitrogen Life Technologies) and sequenced.
Transcription termination assay
Strains carrying the ade6‐704 mutation and the DRT5T dimeric construct were obtained from the Maraia laboratory. Standard genetic crosses were employed to introduce these reporter constructs in the strains of interest. At least two independent strains for each genotype were then plated on YES medium depleted or not of adenine for 3 days at 30°C.
Library preparation and Illumina sequencing
DNA libraries for ChIP‐seq experiments were prepared as described previously (Lemay et al, 2016) using the SPARK DNA Sample Prep Kit Illumina Platform (Enzymatics) according to the manufacturer's instructions.
ChIP‐seq processing
Briefly, the raw reads were trimmed using Trimmomatic version 0.32 (Bolger et al, 2014) with parameters ILLUMINACLIP:2:30:15 LEADING:30 TRAILING:30 MINLEN:23, and quality inspection was conducted using FastQC version 0.11.4 (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). The trimmed reads from all data sets were aligned using BWA version 0.7.12‐r1039 (Li & Durbin, 2010) with the algorithm mem and the parameter ‐split onto the sequence of the S. pombe ASM294v2. Note that no filtering on mapQ was performed in order to avoid discarding the signal at regions of the genome that are duplicated (BWA is randomly assigning the reads), but only primary alignments were kept and we generated mappability tracks for various read length to help the interpretation of particular regions. Signal density files in BedGraph format were then generated using BEDTools genomecov version 2.17 (Quinlan & Hall, 2010) with default parameters, then converted in uniform 10 nt bin WIG files for further normalization steps (inspired by the script bedgraph_to_wig.py; https://gist.github.com/svigneau/8846527).
ChIP‐seq data analysis
Each signal density file was scaled based on sample's sequencing depth, and then, the signal of the input data set was subtracted from its corresponding IP data set. The normalized WIG files were then encoded in bigWig format using the Kent utilities (Rhead et al, 2010). Visual inspection of the data was performed using an AssemblyHub on the UCSC Genome Browser (Casper et al, 2018).
The Versatile Aggregate Profiler (VAP) tool (Coulombe et al, 2014; Brunelle et al, 2015) version 1.1.0 was used to generate the average profile using the following parameters: annotation mode, absolute analysis method, 10 bp windows size, mean aggregate value and smoothing of 6, and missing data were considered as “0”. 131 isolated tRNA and 35 isolated 5S rRNA were used for the VAP analyses. We used the 3′ one reference point mode to generate the graph values and identified the average 5′ start using the 5′ one reference point mode (both graphs overlapped perfectly for all curves). Genome‐wide Pearson's correlation coefficients were calculated using the epiGeEC tool version 1.0 (Laperle et al, 2019).
To generate the % normalized hit distribution (Fig 1D), we used summarizeOverlaps from the R package GenomicAlignments (Lawrence et al, 2013) with the Union mode to count the aligned reads overlapping with each gene, both with the input and IP data. The counts were then normalized based on sample's sequencing depth. Then, the normalized input count was subtracted from the normalized IP count. Finally, the resulting counts were further normalized based on gene length. The % normalized hits for every type of gene were computed as the sum of the positive counts for a gene type divided by the sum of all positive count. All scripts used for data processing and statistical analysis were written in Python, Perl or R, and are available upon request.
Author contributions
VV conceived the study with input from PB, JR and FB. JR, CT, EPR, AM and VV carried out all experiments except for the ChIP‐seq experiments. ML, FG and FB carried out and analysed the ChIP‐seq experiments. VV, JR, FB and PB discussed and interpreted the results. VV wrote the manuscript with input from FB and PB, and all authors revised it.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Review Process File
Acknowledgements
We are very grateful to Richard Maraia for sending reagents. We thank Thomas Diot for helping with the genome annotations of tRNA and their terminators in fission yeast. We thank the members of the LBMC Biocomputing Center for their advice, in particular Laurent Modolo and Hélène Polvèche. We are grateful to Domenico Libri and Odil Porrua for helpful discussions. This work was supported by a “Chaire d'Excellence” awarded to VV by the Agence Nationale pour la Recherche (ANR, Project TRACC, CHX11), by a “Projet Pluri‐Equipe” (PPE 2016–2018) awarded by la Ligue contre le Cancer, comité du Rhône to VV and by the PRCE project “GeneSilencingByCondensin” (ANR‐15‐CE12‐0002‐01) and a “Projet Fondation ARC” (PJA 20151203343) awarded to PB by the Agence Nationale pour la Recherche (ANR) and the Fondation ARC pour la recherche sur le cancer, respectively. Work in the Bachand laboratory was supported by funding from the Natural Sciences and Engineering Research Council of Canada (NSERC) to F.B. (RGPIN‐2017‐05482). F.B. holds a Canada Research Chair in Quality Control of Gene Expression.
The EMBO Journal (2019) 38: e101955
Data availability
The data sets produced in this study are available:
ChIP‐seq data: Gene Expression Omnibus GSE130709 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE130709).
References
- Alzu A, Bermejo R, Begnis M, Lucca C, Piccini D, Carotenuto W, Saponaro M, Brambati A, Cocito A, Foiani M et al (2012) Senataxin associates with replication forks to protect fork integrity across RNA‐polymerase‐II‐transcribed genes. Cell 151: 835–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews AM, McCartney HJ, Errington TM, D'Andrea AD, Macara IG (2018) A senataxin‐associated exonuclease SAN1 is required for resistance to DNA interstrand cross‐links. Nat Commun 9: 2592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimbasseri AG, Rijal K, Maraia RJ (2013) Transcription termination by the eukaryotic RNA polymerase III. Biochim Biophys Acta 1829: 318–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimbasseri AG, Maraia RJ (2015) Biochemical analysis of transcription termination by RNA polymerase III from yeast Saccharomyces cerevisiae . Methods Mol Biol 1276: 185–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belotserkovskii BP, Soo Shin JH, Hanawalt PC (2017) Strong transcription blockage mediated by R‐loop formation within a G‐rich homopurine‐homopyrimidine sequence localized in the vicinity of the promoter. Nucleic Acids Res 45: 6589–6599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambati A, Zardoni L, Achar YJ, Piccini D, Galanti L, Colosio A, Foiani M, Liberi G (2018) Dormant origins and fork protection mechanisms rescue sister forks arrested by transcription. Nucleic Acids Res 46: 1227–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunelle M, Coulombe C, Poitras C, Robert M‐A, Markovits AN, Robert F, Jacques P‐É (2015) Aggregate and heatmap representations of genome‐wide localization data using VAP, a versatile aggregate profiler. Methods Mol Biol 1334: 273–298 [DOI] [PubMed] [Google Scholar]
- Casper J, Zweig AS, Villarreal C, Tyner C, Speir ML, Rosenbloom KR, Raney BJ, Lee CM, Lee BT, Karolchik D et al (2018) The UCSC genome browser database: 2018 update. Nucleic Acids Res 46: D762–D769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y‐Z, Bennett CL, Huynh HM, Blair IP, Puls I, Irobi J, Dierick I, Abel A, Kennerson ML, Rabin BA et al (2004) DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet 74: 1128–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Chen J‐Y, Zhang X, Gu Y, Xiao R, Shao C, Tang P, Qian H, Luo D, Li H et al (2017) R‐ChIP using inactive RNase H reveals dynamic coupling of R‐loops with transcriptional pausing at gene promoters. Mol Cell 68: 745–757.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen S, Puget N, Lin Y‐L, Clouaire T, Aguirrebengoa M, Rocher V, Pasero P, Canitrot Y, Legube G (2018) Senataxin resolves RNA:DNA hybrids forming at DNA double‐strand breaks to prevent translocations. Nat Commun 9: 533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulombe C, Poitras C, Nordell‐Markovits A, Brunelle M, Lavoie M‐A, Robert F, Jacques P‐É (2014) VAP: a versatile aggregate profiler for efficient genome‐wide data representation and discovery. Nucleic Acids Res 42: W485–W493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieci G, Sentenac A (1996) Facilitated recycling pathway for RNA polymerase III. Cell 84: 245–252 [DOI] [PubMed] [Google Scholar]
- El Hage A, Webb S, Kerr A, Tollervey D (2014) Genome‐wide distribution of RNA‐DNA hybrids identifies RNase H targets in tRNA genes, retrotransposons and mitochondria. PLoS Genet 10: e1004716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groh M, Albulescu LO, Cristini A, Gromak N (2017) Senataxin: genome guardian at the interface of transcription and neurodegeneration. J Mol Biol 429: 3181–3195 [DOI] [PubMed] [Google Scholar]
- Gudipati RK, Xu Z, Lebreton A, Séraphin B, Steinmetz LM, Jacquier A, Libri D (2012) Extensive degradation of RNA precursors by the exosome in wild‐type cells. Mol Cell 48: 409–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada M, Huang Y, Lowe TM, Maraia RJ (2001) Widespread use of TATA elements in the core promoters for RNA polymerases III, II, and I in fission yeast. Mol Cell Biol 21: 6870–6881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Z, Libri D, Porrua O (2017) Biochemical characterization of the helicase Sen1 provides new insights into the mechanisms of non‐coding transcription termination. Nucleic Acids Res 45: 1355–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartono SR, Malapert A, Legros P, Bernard P, Chédin F, Vanoosthuyse V (2018) The affinity of the S9.6 antibody for double‐stranded RNAs impacts the accurate mapping of R‐loops in fission yeast. J Mol Biol 430: 272–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazelbaker DZ, Marquardt S, Wlotzka W, Buratowski S (2013) Kinetic competition between RNA Polymerase II and Sen1‐dependent transcription termination. Mol Cell 49: 55–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyer EE, Ozadam H, Ricci EP, Cenik C, Moore MJ (2015) An optimized kit‐free method for making strand‐specific deep sequencing libraries from RNA fragments. Nucleic Acids Res 43: e2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iben JR, Mazeika JK, Hasson S, Rijal K, Arimbasseri AG, Russo AN, Maraia RJ (2011) Point mutations in the Rpb9‐homologous domain of Rpc11 that impair transcription termination by RNA polymerase III. Nucleic Acids Res 39: 6100–6113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawauchi J, Mischo H, Braglia P, Rondon A, Proudfoot NJ (2008) Budding yeast RNA polymerases I and II employ parallel mechanisms of transcriptional termination. Genes Dev 22: 1082–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HD, Choe J, Seo YS (1999) The sen1(+) gene of Schizosaccharomyces pombe, a homologue of budding yeast SEN1, encodes an RNA and DNA helicase. Biochemistry 38: 14697–14710 [DOI] [PubMed] [Google Scholar]
- Laperle J, Hébert‐Deschamps S, Raby J, de Lima Morais DA, Barrette M, Bujold D, Bastin C, Robert M‐A, Nadeau J‐F, Harel M et al (2019) The epiGenomic Efficient Correlator (epiGeEC) tool allows fast comparison of user datasets with thousands of public epigenomic datasets. Bioinformatrics 35: 674–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larochelle M, Robert M‐A, Hébert J‐N, Liu X, Matteau D, Rodrigue S, Tian B, Jacques P‐É, Bachand F (2018) Common mechanism of transcription termination at coding and noncoding RNA genes in fission yeast. Nat Commun 9: 4364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pagès H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, Carey VJ (2013) Software for computing and annotating genomic ranges. PLoS Comput Biol 9: e1003118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le TT, Yang Y, Tan C, Suhanovsky MM, Fulbright RM, Inman JT, Li M, Lee J, Perelman S, Roberts JW et al (2018) Mfd dynamically regulates transcription via a release and catch‐up mechanism. Cell 172: 344–357.e15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legros P, Malapert A, Niinuma S, Bernard P, Vanoosthuyse V (2014) RNA processing factors Swd2.2 and Sen1 antagonize RNA Pol III‐dependent transcription and the localization of condensin at Pol III genes. PLoS Genet 10: e1004794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemay J‐F, Marguerat S, Larochelle M, Liu X, van Nues R, Hunyadkürti J, Hoque M, Tian B, Granneman S, Bähler J et al (2016) The Nrd1‐like protein Seb1 coordinates cotranscriptional 3′ end processing and polyadenylation site selection. Genes Dev 30: 1558–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R (2010) Fast and accurate long‐read alignment with Burrows‐Wheeler transform. Bioinformatics 26: 589–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Selvam K, Rahman SA, Li S (2016) Sen1, the yeast homolog of human senataxin, plays a more direct role than Rad26 in transcription coupled DNA repair. Nucleic Acids Res 44: 6794–6802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin‐Tumasz S, Brow DA (2015) Saccharomyces cerevisiae Sen1 helicase domain exhibits 5′‐ to 3′‐helicase activity with a preference for translocation on DNA rather than RNA. J Biol Chem 290: 22880–22889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MS, Rialdi A, Ho JSY, Tilove M, Martinez‐Gil L, Moshkina NP, Peralta Z, Noel J, Melegari C, Maestre AM et al (2015) Senataxin suppresses the antiviral transcriptional response and controls viral biogenesis. Nat Immunol 16: 485–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mischo HE, Gómez‐González B, Grzechnik P, Rondón AG, Wei W, Steinmetz L, Aguilera A, Proudfoot NJ (2011) Yeast Sen1 helicase protects the genome from transcription‐associated instability. Mol Cell 41: 21–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishra S, Maraia RJ (2019) RNA polymerase III subunits C37/53 modulate rU:dA hybrid 3′ end dynamics during transcription termination. Nucleic Acids Res 47: 310–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreira M‐C, Klur S, Watanabe M, Németh AH, Le Ber I, Moniz J‐C, Tranchant C, Aubourg P, Tazir M, Schöls L et al (2004) Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia‐ocular apraxia 2. Nat Genet 36: 225–227 [DOI] [PubMed] [Google Scholar]
- Noma K, Cam HP, Maraia RJ, Grewal SIS (2006) A role for TFIIIC transcription factor complex in genome organization. Cell 125: 859–872 [DOI] [PubMed] [Google Scholar]
- Orioli A, Pascali C, Quartararo J, Diebel KW, Praz V, Romascano D, Percudani R, van Dyk LF, Hernandez N, Teichmann M et al (2011) Widespread occurrence of non‐canonical transcription termination by human RNA polymerase III. Nucleic Acids Res 39: 5499–5512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrua O, Libri D (2013) A bacterial‐like mechanism for transcription termination by the Sen1p helicase in budding yeast. Nat Struct Mol Biol 20: 884–891 [DOI] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhead B, Karolchik D, Kuhn RM, Hinrichs AS, Zweig AS, Fujita PA, Diekhans M, Smith KE, Rosenbloom KR, Raney BJ et al (2010) The UCSC Genome Browser database: update 2010. Nucleic Acids Res 38: D613–D619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard P, Feng S, Manley JL (2013) A SUMO‐dependent interaction between Senataxin and the exosome, disrupted in the neurodegenerative disease AOA2, targets the exosome to sites of transcription‐induced DNA damage. Genes Dev 27: 2227–2232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rijal K, Maraia RJ (2013) RNA polymerase III mutants in TFIIFα‐like C37 that cause terminator readthrough with no decrease in transcription output. Nucleic Acids Res 41: 139–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C, Kudla G, Wlotzka W, Tuck A, Tollervey D (2012) Transcriptome‐wide analysis of exosome targets. Mol Cell 48: 422–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm L, Hernandez N (2002) Recruitment of RNA polymerase III to its target promoters. Genes Dev 16: 2593–2620 [DOI] [PubMed] [Google Scholar]
- Skourti‐Stathaki K, Proudfoot NJ, Gromak N (2011) Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2‐dependent termination. Mol Cell 42: 794–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmetz EJ, Conrad NK, Brow DA, Corden JL (2001) RNA‐binding protein Nrd1 directs poly(A)‐independent 3′‐end formation of RNA polymerase II transcripts. Nature 413: 327–331 [DOI] [PubMed] [Google Scholar]
- Steinmetz EJ, Warren CL, Kuehner JN, Panbehi B, Ansari AZ, Brow DA (2006) Genome‐wide distribution of yeast RNA polymerase II and its control by Sen1 helicase. Mol Cell 24: 735–746 [DOI] [PubMed] [Google Scholar]
- Suraweera A, Lim Y, Woods R, Birrell GW, Nasim T, Becherel OJ, Lavin MF (2009) Functional role for senataxin, defective in ataxia oculomotor apraxia type 2, in transcriptional regulation. Hum Mol Genet 18: 3384–3396 [DOI] [PubMed] [Google Scholar]
- Turowski TW, Tollervey D (2016) Transcription by RNA polymerase III: insights into mechanism and regulation. Biochem Soc Trans 44: 1367–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turowski TW, Leśniewska E, Delan‐Forino C, Sayou C, Boguta M, Tollervey D (2016) Global analysis of transcriptionally engaged yeast RNA polymerase III reveals extended tRNA transcripts. Genome Res 26: 933–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursic D, Himmel KL, Gurley KA, Webb F, Culbertson MR (1997) The yeast SEN1 gene is required for the processing of diverse RNA classes. Nucleic Acids Res 25: 4778–4785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Roeder RG (1998) DNA topoisomerase I and PC4 can interact with human TFIIIC to promote both accurate termination and transcription reinitiation by RNA polymerase III. Mol Cell 1: 749–757 [DOI] [PubMed] [Google Scholar]
- Xu W, Xu H, Li K, Fan Y, Liu Y, Yang X, Sun Q (2017) The R‐loop is a common chromatin feature of the Arabidopsis genome. Nat Plants 3: 704–714 [DOI] [PubMed] [Google Scholar]
- Yu Y, Ren J‐Y, Zhang J‐M, Suo F, Fang X‐F, Wu F, Du L‐L (2013) A proteome‐wide visual screen identifies fission yeast proteins localizing to DNA double‐strand breaks. DNA Repair 12: 433–443 [DOI] [PubMed] [Google Scholar]
- Yüce Ö, West SC (2013) Senataxin, defective in the neurodegenerative disorder ataxia with oculomotor apraxia 2, lies at the interface of transcription and the DNA damage response. Mol Cell Biol 33: 406–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao DY, Gish G, Braunschweig U, Li Y, Ni Z, Schmitges FW, Zhong G, Liu K, Li W, Moffat J et al (2016) SMN and symmetric arginine dimethylation of RNA polymerase II C‐terminal domain control termination. Nature 529: 48–53 [DOI] [PubMed] [Google Scholar]
- Zheng G, Qin Y, Clark WC, Dai Q, Yi C, He C, Lambowitz AM, Pan T (2015) Efficient and quantitative high‐throughput tRNA sequencing. Nat Methods 12: 835–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Review Process File
Data Availability Statement
The data sets produced in this study are available:
ChIP‐seq data: Gene Expression Omnibus GSE130709 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE130709).