

Abstract
During MHC‐I‐restricted antigen processing, peptides generated by cytosolic proteasomes are translocated by the transporter associated with antigen processing (TAP) into the endoplasmic reticulum, where they bind to newly synthesized MHC‐I molecules. Dendritic cells and other cell types can also generate MHC‐I complexes with peptides derived from internalized proteins, a process called cross‐presentation. Here, we show that active proteasomes within cross‐presenting cell phagosomes can generate these peptides. Active proteasomes are detectable within endocytic compartments in mouse bone marrow‐derived dendritic cells. In TAP‐deficient mouse dendritic cells, cross‐presentation is enhanced by the introduction of human β2‐microglobulin, which increases surface expression of MHC‐I and suggests a role for recycling MHC‐I molecules. In addition, surface MHC‐I can be reduced by proteasome inhibition and stabilized by MHC‐I‐restricted peptides. This is consistent with constitutive proteasome‐dependent but TAP‐independent peptide loading in the endocytic pathway. Rab‐GTPase mutants that restrain phagosome maturation increase proteasome recruitment and enhance TAP‐independent cross‐presentation. Thus, phagosomal/endosomal binding of peptides locally generated by proteasomes allows cross‐presentation to generate MHC‐I‐peptide complexes identical to those produced by conventional antigen processing.
Keywords: antigen cross‐presentation, MHC‐I, proteasomes, Rab‐GTPase
Subject Categories: Immunology, Membrane & Intracellular Transport
Introduction
Cytotoxic T lymphocytes (CTL) that eliminate virus‐infected cells or tumors do so by recognizing MHC‐I molecules associated with short peptides derived from viral or tumor‐specific protein antigens. Initiation of a CTL response requires priming of naïve CD8‐positive T lymphocytes by professional antigen presenting cells, normally dendritic cells (DCs), by a mechanism called cross‐priming or cross‐presentation (Grotzke et al, 2017). This involves endocytosis of antigenic proteins or phagocytosis of virally infected cells or tumor cells followed by antigen proteolysis and binding of resulting peptides by MHC‐I molecules. This is very different from the way MHC‐I‐peptide complexes are generated by the virally infected cells or tumor cells themselves. Here, the protein antigens are synthesized conventionally on ribosomes and peptides derived from them are generated in the cytosol by proteasomal degradation. The peptides are then translocated by TAP into the endoplasmic reticulum (ER) where those with the appropriate sequence and length bind to MHC‐I molecules transiently associated with TAP via tapasin within the peptide loading complex (PLC). Trimming in the ER by dedicated aminopeptidases (ERAP‐1 and ‐2 in humans, ERAAP in mice) may also be required for the generation of antigenic peptides of a suitable length. Ultimately, the MHC‐I‐peptide complexes are expressed on the plasma membrane (Blum et al, 2013).
For an effective cytotoxic response, CTLs primed by cross‐presenting DCs must recognize endogenous MHC‐peptide complexes displayed by the infected cells or tumor. How the cross‐presentation and endogenous processing pathways result in the same MHC‐I complexes is poorly understood, but the predominant explanation is that exogenous antigens internalized by the DCs are translocated across endosomal or phagosomal membranes into the cytosol (Kovacsovics‐Bankowski & Rock, 1995; Delamarre et al, 2003; Lu et al, 2018). Although the precise mechanism of translocation remains a matter of debate, subsequent events, including proteasomal degradation and peptide translocation by TAP, would then be similar to those involved in conventional processing of cytosolic antigens. A major difference is that TAP‐mediated peptide translocation may occur in phagosomes or endosomes containing membrane recruited from the ER (Gagnon et al, 2002). MHC‐I peptide binding could then occur within these compartments, either to TAP‐associated MHC‐I molecules also recruited from the ER (Ackerman et al, 2003, 2006; Guermonprez et al, 2003; Houde et al, 2003) or to recycling MHC‐I molecules derived from the plasma membrane (Nair‐Gupta et al, 2014).
An alternative mechanism, commonly called the vacuolar pathway, postulates that the peptides are generated by endocytic or phagosomal proteases before binding to recycling MHC‐I molecules (Song & Harding, 1996). However, this mechanism implies that the production of identical MHC‐I‐binding peptides by vacuolar enzymes in cross‐presenting DCs and cytosolic proteasomes in the target cell is a matter of chance (Grotzke et al, 2017). Here, we present an alternative to this model for the vacuolar pathway. We propose that active proteasomes are imported into phagosomes/endolysosomes and degrade internalized antigens within them, generating the same spectrum of peptides that are produced by cytosolic proteasomes in conventional MHC‐I antigen processing. This eliminates the role of coincidence in the production of the same MHC‐I‐peptide complexes by cross‐priming DCs and the target cells ultimately recognized by mature CD8‐positive CTL.
Results
Human β2‐microglobulin rescues loss of surface MHC‐I and antigen cross‐presentation in BMDCs derived from TAP1−/− mice
Multiple groups have shown that surface MHC‐I expression is reduced on bone marrow‐derived dendritic cells (BMDC) derived from TAP1−/− mice, and that these cells fail to cross‐present exogenous antigens (Van Kaer et al, 1992; Kovacsovics‐Bankowski & Rock, 1995; Singh & Cresswell, 2010). Two potential explanations exist for the loss of cross‐presentation. In the first, peptides generated by cytosolic proteolysis are not translocated across the ER or phagosomal membrane because TAP is absent. In the second, peptides are generated in vacuolar compartments but fewer MHC‐I‐β2m dimers are available for binding because their assembly and transport from the ER is reduced.
To distinguish these possibilities, we expressed human β2‐microglobulin (hβ2m) in TAP1−/− BMDCs. The introduction of hβ2m is known to increase surface expression of H2‐Kb and Db molecules on TAP1‐negative mouse RMA‐S cells, and Kb is well expressed on the surface when introduced into TAP‐negative human T2 cells (Anderson et al, 1993). We predicted that the expression of hβ2m in TAP1−/− BMDCs would enhance surface Kb expression, and that cross‐presentation would be restored if TAP is only necessary to increase the MHC‐I available for vacuolar loading. As expected, TAP1−/− DCs exhibited low cell surface Kb, measured by flow cytometry, that was partially restored by the introduction of hβ2m (Fig 1A). Cross‐presentation by BMDCs of phagocytosed OVA associated with latex beads was assessed by measuring the stimulation of IL‐2 release by the OVA‐specific Kb‐restricted hybridoma B3Z. Virtually, no cross‐presentation was observed in TAP1−/− BMDCs transduced with a control vector (Fig 1B) but it was partially restored by a vector expressing hβ2m (Fig 1B). This is consistent with the hypothesis that the lack of cross‐presentation by TAP1−/− BMDCs is at least partially a consequence of a reduction of available MHC‐I‐β2m dimers compared to wild‐type cells.
Figure 1. Expression of hβ2m partially rescues surface Kb expression and antigen cross‐presentation in TAP1−/− BMDC, but does not rescue endogenous antigen presentation.
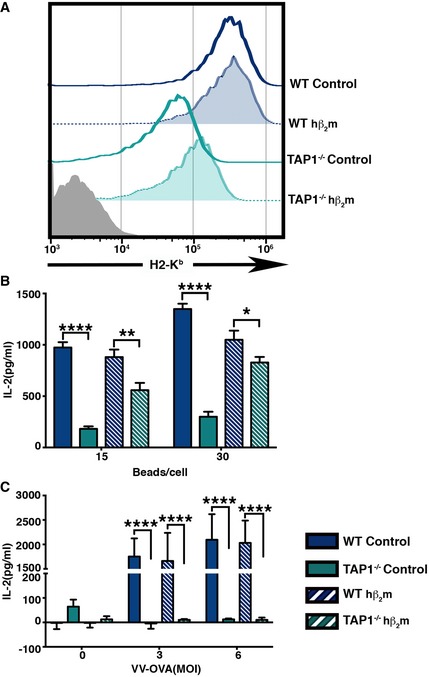
- Cell surface H2‐Kb levels of wild type and TAP1−/− BMDC transduced with control vector or a vector expressing hβ2m were analyzed.
- Cross‐presentation of OVA by wild type and TAP1−/− BMDC expressing hβ2m was compared to control BMDC, by measuring IL‐2 production by a Kb‐SIINFEKL‐specific T‐cell hybridoma (B3Z) cultured with paraformaldehyde‐fixed BMDC that were incubated with varying numbers of OVA‐coated latex beads for 6 h, prior to fixation.
- The effect of hβ2m on the presentation of peptides derived from endogenous antigen was assessed by measuring IL‐2 levels in the culture supernatant of the B3Z hybridoma incubated with fixed VV‐OVA‐infected WT and TAP1−/− BMDC transduced with control vector or hβ2m.
As a control, we asked whether the expression of hβ2m impacted conventional antigen presentation. To test this, TAP1−/− BMDC with or without hβ2m were infected with vaccinia virus‐encoding OVA (VV‐OVA; Ackerman et al, 2006) and their ability to mediate direct presentation of OVA was assessed. In contrast to cross‐presentation (Fig 1B), endogenous antigen presentation by TAP1−/− BMDC was not rescued by the introduction of hβ2m (Fig 1C), consistent with the known requirement of TAP for delivery of OVA‐derived peptides for binding to assembling Kb‐β2m dimers (Androlewicz et al, 1993; Procko & Gaudet, 2009).
Rab‐GTPase mutants that restrict phagosomal degradation enhance TAP‐independent cross‐presentation
Cross‐presentation via the vacuolar pathway is thought to depend on the generation of the appropriate peptides by phagosomal or lysosomal proteases (Song & Harding, 1996; Campbell et al, 2000; Shen et al, 2004), while cross‐presentation by the cytosolic pathway is believed to be enhanced by a reduction in lysosomal proteolysis that allows intact proteins or large fragments of them to be translocated into the cytosol for proteasomal degradation. Vacuolar proteolysis is restrained in cross‐presenting DCs by an increase in lysosomal pH relative to non‐cross‐presenting cells, such as macrophages (Savina et al, 2006; Samie & Cresswell, 2015). If the TAP‐independent cross‐presentation observed in Fig 1 is a consequence of conventional phagosomal proteolysis, one would predict that introducing Rab‐GTPase mutants that restrain the maturation of phagosomes to phagolysosomes would inhibit it. We therefore independently expressed three different Rab‐GTPase mutants, Rab5ACA, Rab22ACA, and Rab7ADN, each of which has been shown to impair phagosomal maturation and degradation in distinct cell types (Duclos et al, 2000; Harrison et al, 2003; Roberts et al, 2006), together with hβ2m, in TAP1−/− bone marrow‐derived cells to assess their impact on cross‐presentation after differentiation into DCs. Constitutive expression of Rab5ACA and Rab22ACA affected BMDC differentiation so they were placed under the control of a doxycycline‐inducible (Tet‐ON)‐based promoter (Appendix Fig S1A). Rab7ADN did not affect differentiation and was expressed under a constitutive promoter (Appendix Fig S1B). The expression of the Rab mutants does not alter the cell surface MHC‐I levels or the differentiation of BMDCs (Appendix Fig S1C–E).
To verify that the Rab mutants affect phagosomal degradation, we adapted a previously described assay (Savina et al, 2010). BMDCs expressing the Rab mutants were incubated with latex beads covalently conjugated with Alexa 647‐labeled OVA. Following a 1‐h pulse to allow phagocytosis, the cells were extracted with detergent (1% Triton X‐100 plus 0.1% SDS) at different time points. Phagosomal degradation of the OVA was assessed using flow cytometry to measure the reduction in Alexa 647 fluorescence of the released beads. Consistent with previous reports, the rate of phagosomal degradation was reduced compared to control cells in BMDC expressing Rab5ACA, Rab22ACA, or Rab7ACA (Fig EV1A and B).
Figure EV1. Expression of Rab5ACA, Rab22ACA, and Rab7ADN restricts the degradation of phagocytosed antigen.
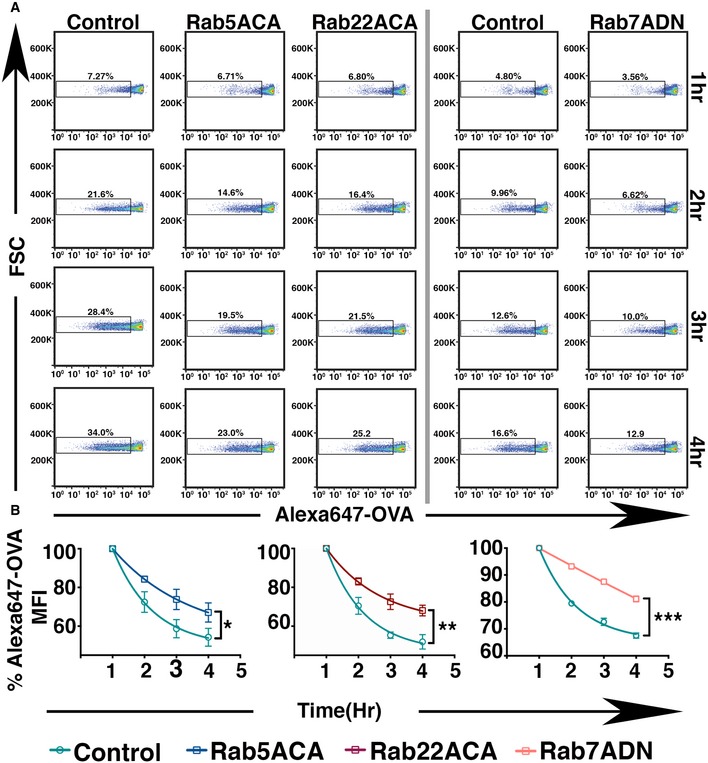
- Phagocytosed latex beads covalently conjugated with Alexa 647‐OVA were extracted from BMDC expressing GFP or GFP‐Rab5ACA or GFP‐Rab22ACA or Rab7ADN at 1‐h intervals, and analyzed for the loss of Alexa 647 fluorescence by flow cytometry.
- The mean Alexa 647 fluorescence on the beads normalized to the fluorescence at the 1‐h time point is plotted (n = 3).
To investigate the role of phagosomal degradation in TAP‐independent cross‐presentation, we co‐expressed the individual Rab mutants with hβ2m in BMDCs derived from wild‐type and TAP1−/− mice and analyzed cell surface Kb expression and cross‐presentation efficiency. Kb levels on TAP1−/− BMDCs co‐expressing hβ2m plus the Rab mutants were not significantly different from TAP1−/− BMDC expressing hβ2m alone (Fig 2A–C). However, simultaneous expression of the Rab mutants and hβ2m further enhanced the cross‐presentation efficiency of both TAP1−/− BMDC and wild‐type BMDC (Fig 2D–F). The expression of hβ2m was confirmed by flow cytometry (Appendix Fig S2).
Figure 2. Expression of Rab5ACA, Rab22ACA, and Rab7ADN, respectively, increases hβ2m‐enhanced TAP‐independent cross‐presentation.
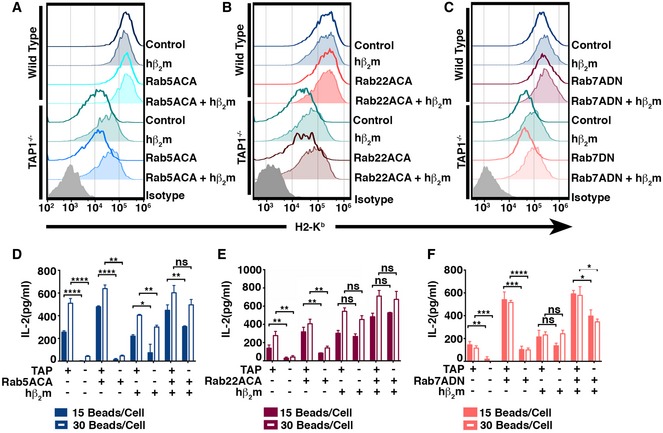
-
A–CSurface H2‐Kb levels of wild‐type and TAP1−/− BMDC co‐expressing Rab mutants and hβ2m were analyzed by flow cytometry.
-
D–FCross‐presentation of OVA by wild‐type and TAP1−/− BMDC co‐expressing Rab mutants and hβ2m was analyzed, and IL‐2 release is shown (D–F).
The above data suggest that reduced phagosomal maturation combined with the availability of a stable pool of cell surface Kb molecules may be the key requirements for efficient cross‐presentation in the absence of a functional TAP transporter. TAP‐dependent cross‐presentation is thought to require limited endocytic proteolysis followed by antigen translocation into the cytosol and subsequent proteasomal degradation. However, the above experiments show that TAP‐independent cross‐presentation is enhanced in the presence of Rab‐GTPase mutants that restrict lysosomal degradation. Clearly, a protease must be required to produce the epitope, raising the possibility that proteasomes may remain a critical protease even in TAP‐independent antigen cross‐presentation.
TAP‐independent cell surface stability of MHC‐I and antigen cross‐presentation are proteasome‐dependent
Restricting peptide delivery to the ER, for example, by inhibiting TAP, results in a reduction in MHC‐I cell surface expression. We hypothesized that if the residual surface MHC‐I observed in TAP1−/− BMDCs depends on peptides generated by proteasomes, then a proteasome inhibitor would also reduce cell surface expression. We tested this prediction by measuring surface Kb levels on TAP1−/− BMDCs in response to varying doses of epoxomicin and found that after 6 h treatment there was a concentration‐dependent reduction of Kb expression (Fig 3A and B). To confirm that this was a result of loss of peptide generation, we repeated the experiment in the presence of exogenously added SIINFEKL peptide and found that this rescued the loss of Kb surface expression (Fig 3A and B). We repeated this analysis using TAP1−/− BMDC transduced with hβ2m and again observed that surface Kb expression was reduced by proteasome inhibition and that this was reversed by adding SIINFEKL peptide (Fig 3C and D). These findings suggest that peptide generation by proteasomes is required for maintaining the stability of surface MHC‐I molecules in TAP1−/− BMDCs.
Figure 3. Proteasomal activity is required for stable surface Kb expression in TAP1−/− dendritic cells and antigen cross‐presentation.
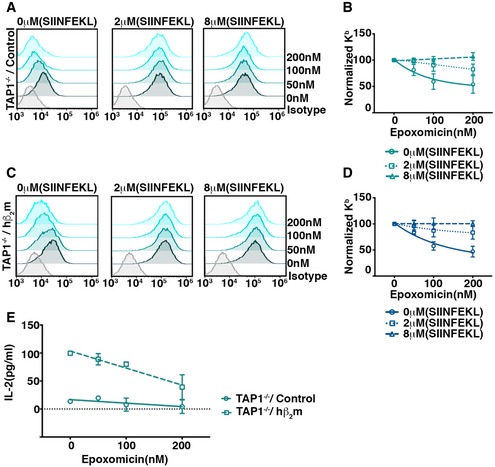
-
A–DTAP1−/− BMDC transduced with vector control (A) or hβ2m (C) were incubated with varying concentrations of epoxomicin for 6 h in the absence or presence of exogenously added Kb‐binding SIINFEKL peptide and analyzed for surface expression of Kb by flow cytometry. Representative plots are shown (A, C). The impact of epoxomicin on cell surface Kb was analyzed by plotting Kb surface expression on cells incubated with epoxomicin, normalized to that on cells incubated without epoxomicin (n = 3) (B, D).
-
EThe effect of epoxomicin on cross‐presentation of OVA‐coated beads by TAP1−/− BMDC expressing hβ2m was determined by incubating the BMDC with OVA‐coated latex beads in the presence of varying doses of epoxomicin and 10 μM gB peptide. After 6 h, the cells were fixed and incubated with B3Z cells and IL‐2 production was measured.
Conceivably, the reduction in Kb cell surface expression by the inhibition of proteasomes could play a role in the inhibition of cross‐presentation in TAP1−/− BMDCs independently of a reduction in the generation of the SIINFEKL epitope derived from OVA. To dissect the potential roles of the two parameters, we used the peptide stabilization strategy to uncouple proteasome‐dependent epitope generation from down‐regulation of cell surface MHC‐I. We used a different Kb‐binding peptide, an epitope derived from HSV‐glycoprotein B (gB; Singh & Cresswell, 2010), to prevent the down‐regulation of Kb in response to epoxomicin (Fig EV2). We then measured the impact of varying doses of epoxomicin on OVA cross‐presentation by TAP1−/− BMDC expressing or not expressing hβ2m in the presence of the HSV‐gB peptide. Down‐regulation of Kb was indeed inhibited, but the increased cross‐presentation induced by hβ2m remained sensitive to proteasome inhibition (Figs 3E and EV2).
Figure EV2. Rescue of epoxomicin‐mediated down‐regulation of surface Kb by HSV‐gB peptide.
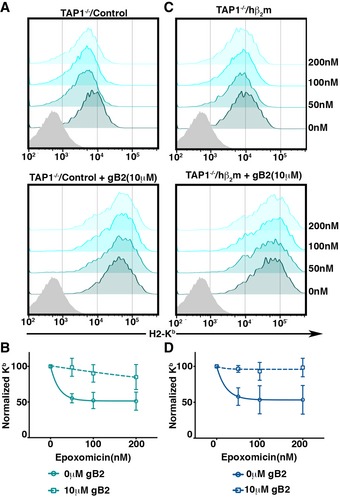
-
A–DCell surface Kb expression of TAP1−/− BMDC transduced with control vector (A) or hβ2m expressing vector (C), incubated with varying doses of epoxomicin in the presence and absence of 10 μM gB peptide, was analyzed by flow cytometry. (B, D) Mean (±SEM) surface Kb levels normalized to untreated cells were plotted (n = 2).
Reconstitution of TAP‐independent antigen cross‐presentation in non‐immune cells
The proteasome‐dependent but TAP‐independent antigen cross‐presentation pathway we observed could be a specialized mechanism evolved in mouse BMDC or result from loss of TAP as a PLC structural component rather than a peptide delivery function. To examine this question, we reconstituted the pathway in a non‐immune human cell line and combined this with an alternative means of inhibiting TAP function. We previously showed that the non‐hematopoietic 293T cell line can efficiently mediate cross‐presentation of OVA immune complexes when supplied with human FcRγIIb and Kb (Giodini et al, 2009). We expressed two different viral TAP inhibitors in these 293T‐FcR‐Kb cells. One, US6, is encoded by human cytomegalovirus (HCMV), and the second, ICP47, is encoded by herpes simplex virus‐1 (HSV‐1). US6 and ICP47 inhibit human TAP function via distinct mechanisms, on the luminal and cytosolic sides of the ER, respectively (Lehner et al, 1997; Neumann et al, 1997). TAP inhibition was confirmed by measuring the resulting decrease in surface H2‐Kb levels by flow cytometry (Fig EV3A). The impact of the viral inhibitors on cross‐presentation was assessed by comparing 293T‐FcR‐Kb cells expressing the viral genes to cells expressing LacZ as a control. The cross‐presentation efficiency of the cells expressing the US6 or ICP47 was reduced by approximately 80% compared to cells expressing LacZ (Figs 4A and B, and EV3C), even though surface Kb levels remained substantial, suggesting that the availability of stable pool of cell surface Kb may not be sufficient to drive maximum TAP‐independence. We hypothesized that reduced phagosomal degradation in the cells expressing the viral TAP inhibitors could also be required, and used the Rab mutants to inhibit phagosomal maturation as described above for the BMDCs. Indeed, co‐expression of US6 or ICP47 with any of the Rab mutants (Fig EV3A and B) that delayed phagosomal maturation reversed the ability of the viral TAP inhibitors to inhibit cross‐presentation (Figs 4A and B, and EV3C); 293T‐FcR‐Kb cells expressing US6 or ICP47 plus the respective Rab mutants cross‐presented OVA at an efficiency close to 80% of the control cells expressing LacZ plus the Rab mutants (Fig 4A and B). The residual 20% reduction in cross‐presentation may be attributable to the reduction of surface Kb molecules caused by the viral proteins (Fig EV3A).
Figure EV3. Expression of Rab mutants in 293T‐FcR‐Kb and impact of co‐expression of US6 or ICP47 with Rab mutants of the cell surface H2‐Kb and cross‐presentation.
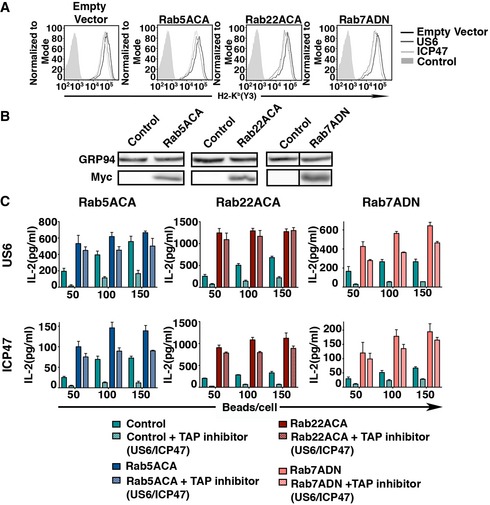
- H2‐Kb on the cell surface of 293T‐FcR‐Kb co‐expressing US6 or ICP47 along with Rab mutants was measured by flow cytometry.
- Transient expression of Rab5ACA, Rab22ACA, and Rab7ADN in 293T‐FcR‐Kb was evaluated by immunoblotting with anti‐Myc.
- Representative plots of at the least three independent experiments of 293T‐FcR‐Kb cells co‐expressing US6 or ICP47 with LacZ or Rab mutants were incubated with opsonized OVA‐coated latex beads for 12 h. The effect of US6 or ICP47 along with the Rab mutants on cross‐presentation was assessed by paraformaldehyde fixation and measuring IL‐2 production by added B3Z cells. The assay was set up in triplicates, and the mean (±SD) of the triplicates is plotted.
Source data are available online for this figure.
Figure 4. Reconstitution of TAP‐independent cross‐presentation in 293T‐FcR‐Kb cells by expressing Rab5ACA, Rab22ACA, and Rab7ADN .
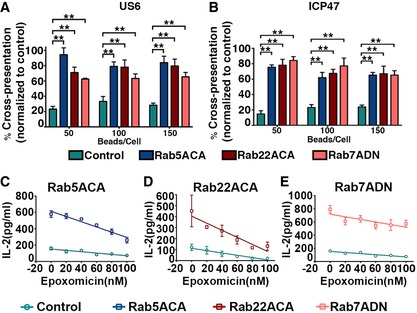
-
A, B293T‐FcR‐Kb cells co‐expressing empty vector or Rab mutants (Rab5ACA, Rab22ACA, and Rab7ADN) individually, with either LacZ as a control or US6 (A) or ICP47 (B), were analyzed for cross‐presentation of OVA using B3Z cells. The effects of US6 and ICP47 on cross‐presentation were analyzed by plotting the mean percentage of IL‐2 release by cells co‐expressing US6 (A) or ICP47 (B) with control vector or Rab mutants compared to cells co‐expressing LacZ with control vector or Rab mutants, respectively.
-
C–E293T‐FcR‐Kb cells expressing Rab5ACA (C), Rab22ACA (D), and Rab7ADN (E) were incubated with opsonized OVA‐coated latex beads in the presence of varying doses of epoxomicin. After 6 h, the cells were fixed and incubated with B3Z cells, and IL‐2 production was measured.
Overall, the data above suggest that a major fraction of cross‐presentation by 293T‐FcR‐Kb cells expressing the Rab mutants is TAP‐independent, and, as for the TAP1−/− BMDCs, we further found that this enhancement of cross‐presentation remained sensitive to proteasome inhibition (Fig 4C–E). The requirement for both reduced phagosomal degradation and a stable pool of cell surface Kb molecules for proteasome‐dependent cross‐presentation cannot be explained by current models.
Proteasomes are imported into the endolysosomal/phagosomal lumen
We postulated that the entry of active proteasomes that generate the SIINFEKL epitope into the endolysosomal lumen could explain the proteasome dependence we observe in cells lacking TAP activity. To approach this question, we first used immunoelectron microscopy to examine endolysosomal compartments in BMDCs for the presence of proteasomes. Endolysosomal membranes were labeled with anti‐LAMP1 antibody (5 nm gold particles, white arrowhead), and proteasome distribution was first analyzed using a rabbit antibody specific for the immunoproteasome subunit LMP2 (Appendix Fig S3A), detected using 15 nm gold particles (black arrowhead). In addition to cytosolic labeling, we also observed LMP2 subunits localized to LAMP1‐positive vacuoles in BMDC (Fig 5A and B). As a specificity control, we compared the labeling of INF‐γ‐treated MEF cells derived from wild‐type and LMP2 knockout mice. The labeling of LMP2 knockout MEF cells was much lower than wild‐type MEF cells (Fig 5A and B, Appendix Fig S3B), establishing a background level of labeling by the LMP2 antibody. To further verify proteasome localization within the endolysosomal compartment, we performed immunoelectron microscopy using LAMP1 antibody together with antibodies raised against three different subunits of constitutive proteasomes (15 nm gold particles, black arrowhead), namely α5 and β5, which are components of the outer and inner rings of the 20S particle, respectively, and S2, located in the 19S cap of the 26S particle. In addition to cytosolic labeling, all three components were located in membrane‐bound compartments labeled with LAMP1 (Fig 5C and D, Appendix Fig S3C). Quantitative analysis revealed that approximately 15% of the proteasomes, defined by labeling with antibodies against three distinct proteasome subunits, were within the LAMP1‐positive membrane compartment (Fig 5E).
Figure 5. Proteasomes access the endolysosomal and phagosomal lumen.
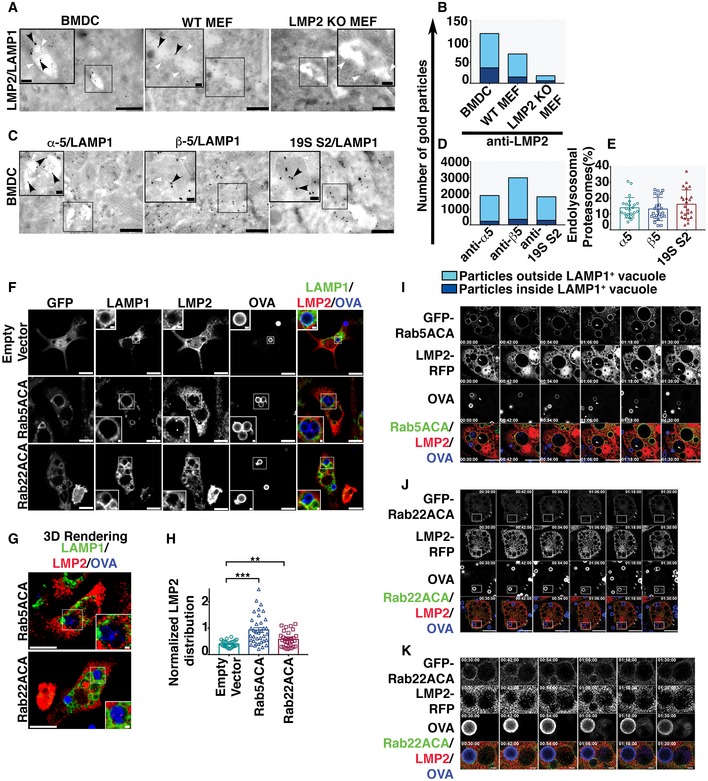
-
AEM micrographs of double immuno‐gold labeling using antibodies against immunoproteasome subunit LMP2 and the endolysosomal membrane marker LAMP1 in BMDC, WT MEF, and LMP2 KO MEF. Large gold particles (15 nm, black arrowhead) label LMP2 and small gold particles label LAMP1 (5 nm, white arrowhead; Scale bars = 500 nm). The insets are the magnification of region of interest containing LAMP1‐positive vacuole (Scale bars = 100 nm), marked by the black rectangle.
-
BThe distribution of LMP2‐positive signals (n = 22 images) assessed by plotting the number of gold particles labeling LMP2 within the LAMP1‐positive membrane compartment versus those outside the LAMP1‐positive organelles.
-
CImmuno‐gold labeling with antibodies against constitutive proteasome subunits, α5, β5, and 19 S2 (15 nm, black arrowhead), co‐labeled with an anti‐LAMP1 antibody (5 nm, white arrowhead) in BMDC (Scale bars = 500 nm). Regions of interest containing LAMP1‐positive vacuoles are marked by the black rectangles and magnified in the insets (Scale bars = 100 nm).
-
D, EImmuno‐electron microscopy images (n = 23) were analyzed by plotting the total number of gold particles labeling proteasome subunits inside and outside the LAMP1‐positive membrane compartments (D). These data are also presented as the percentage of the total gold particles that are within the LAMP1‐positive organelle (E).
-
F, GBMDC transduced with genes encoding GFP, GFP‐Rab5ACA, and GFP‐Rab22ACA under the control of an inducible promoter were fixed and stained for LMP2 and LAMP1 and analyzed by confocal microscopy (F, G) 24 h after doxycycline induction and 4 h after Alexa 647‐OVA‐coated bead uptake. A single optical section (F) and the 3D‐rendering (G) of 10 optical sections of a representative cell are shown (Scale Bar 10 μm). Region of interest containing LAMP1‐positive vacuole containing beads coated with Alexa 647‐conjugated OVA, marked by white rectangles, is magnified as inset (Scale bars = 1 μm).
-
HThe same cells were analyzed for LMP2 fluorescence intensity within the phagosomal lumen as defined by a region of interest positive for Alexa 647‐OVA enclosed within a limiting membrane positive for LAMP1. The fluorescence intensity of LMP2 within the phagosomal lumen normalized to Alexa 647‐OVA was calculated, and data compiled from independent experiments (n = 3) are plotted (H). For each transduced cell type, images of at the least 27 cells acquired across three independent experiments were analyzed and each cell is represented as a data point.
-
I–KBMDC co‐expressing LMP2‐RFP and GFP‐Rab5ACA (I) or GFP‐Rab22ACA (J, K) were live imaged 16 h after doxycycline induction and 30 min after uptake of Alexa 647‐coated latex beads (Bar 10 μm). Images were acquired every 3 min, and images taken at 12‐min interval are presented (I–K). The arrowheads in panel (I and J) mark the phagosome or vacuoles containing LMP2, respectively. The boxed region in panel (J) marks a phagosome that has been enlarged in panel (K) (Bar 1 μm).
Efforts to perform immunoelectron microscopy on phagosomes containing latex beads were inconclusive, so in an attempt to confirm that proteasomes enter the phagosomal lumen we performed confocal microscopy on BMDCs that had phagocytosed latex beads coated with Alexa 647‐conjugated fluorescent OVA. Phagosomal membranes were delineated with an antibody to the LAMP1, and proteasomes were detected using an antibody to the immunoproteasome subunit LMP2. We observed some LMP2 staining associated with the phagosomes (Fig 5F), but because of the limitations of optical microscopy, we could not determine whether the proteasomes were within the lumen or simply proximal to it. However, as expected based on previous findings in HeLa and BHK cells (Simonsen et al, 1998; Kauppi et al, 2002; Vieira et al, 2003), the expression of Rab5ACA and Rab22ACA resulted in enlargement of the endolysosomes and phagosomes in BMDC (Fig 5F and G), and LMP2‐positive structures were readily observed within these compartments (Fig 5F and G). To quantitate the luminal proteasomal content, we normalized the LMP2 fluorescence intensity to the Alexa 647‐OVA content associated with the LAMP1‐positive structures and found significantly higher proteasomal content in cells expressing Rab5ACA and Rab22ACA (Fig 5H). These results were confirmed with an antibody to the immunoproteasome subunit, LMP7 (Fig EV4A–C). The punctate staining of the lumenal proteasomes suggests that, at least initially, they may be within small membrane‐bound vesicles within the endolysosomal compartment, potentially recruited from the cytosol (Figs 5F and G, and EV4A and B).
Figure EV4. Immunofluorescence analysis of localization of immunoproteasome subunit, LMP7, with respect to phagosomal lumen.
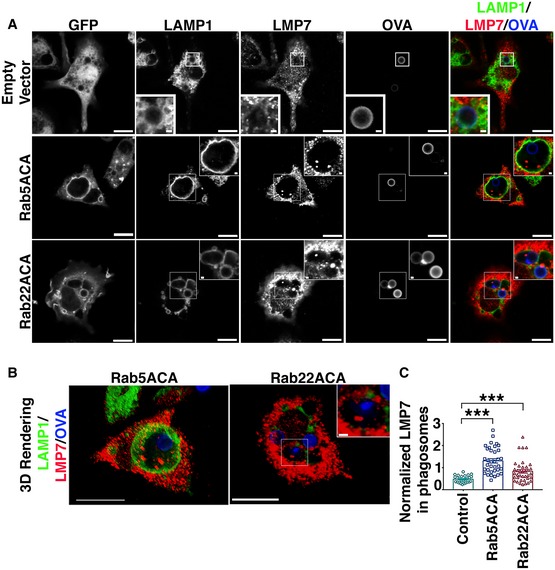
-
A, BBMDCs expressing GFP, GFP‐Rab5ACA, or GFP‐Rab22ACA were stained for LMP7 and LAMP1, and analyzed by confocal microscopy (A, B), 4 h after uptake of Alexa 647‐OVA‐coated bead. A single optical section (A) and a 3D‐rendering (B) of 10 optical sections of a representative cell are shown (Bar 10 μm). White rectangle boxes marking the regions of interest containing LAMP1‐positive vacuole containing beads coated with Alexa 647‐conjugated OVA are magnified as inset (Scale bars = 1 μm).
-
CThe same cells were analyzed for LMP7 fluorescence intensity within the phagosomal lumen as defined by a region of interest positive for Alexa 647‐OVA enclosed within a limiting membrane positive for LAMP1. The fluorescence intensity of LMP7 within the phagosomal lumen normalized to Alexa 647‐OVA was calculated, and the data from 3 (n = 3) independent experiments were compiled and plotted (C). At the least 28 transduced BMDC, cells were analyzed for each transduction and each cell is represented as a data point.
To investigate the phenomenon kinetically, we performed live time‐lapse imaging of BMDCs after uptake of Alexa 647 OVA‐coated latex beads, with frames 3 min apart. To detect the proteasomes, the cells were co‐transduced with LMP2 tagged with red fluorescent protein (RFP) together with GFP‐tagged Rab5ACA (Fig 5I, Movie EV1) or GFP‐tagged Rab22ACA (Fig 5J and K, Movies EV2 and EV3). The time‐lapse images revealed interesting differences between BDMCs expressing the two Rab mutants. Beads with associated OVA were present in Rab5ACA‐positive phagosomes that over time became positive for punctate LMP2‐RFP (Fig 5I, Movie EV1 arrowhead), while bead‐associated fluorescent OVA in Rab22ACA‐positive phagosomes appeared to be released into a proximal but separate Rab22ACA‐negative vacuolar compartment that was positive for LMP2‐RFP (Fig 5J and K; Movies EV2 and EV3). However, in both cases vacuoles containing OVA also contained the proteasomal marker, consistent with the hypothesis that proteasomal degradation within the phagocytic pathway generates the Kb‐restricted OVA epitope.
The difference between the effects of the two Rab mutants could be because they differentially affect the rate of endosome–phagosome fusion. However, Rab22A has been hypothesized to play a role in sorting cargo from early phagosomes/endosomes into a pathway leading to degradation rather than one leading to recycling (Weigert et al, 2004; Cebrian et al, 2016). Thus, the exit of OVA from the Rab22ACA‐positive phagosomes could be a result of a sorting process mediated by Rab22A. Nevertheless, these live imaging experiments confirm that proteasomes gain access to the phagosomal lumen. They also indicate that the LMP2‐positive structures within the phagosomes disappear over time. The punctate structures initially visible may represent small vesicles that rupture after arrival in the phagosome, diffusing the fluorescent signal so that it is no longer visible.
Phagosomal content undergoes ubiquitination and degradation by proteasomes
For phagosomal proteasomes to generate antigenic peptides in situ, they would have to be active. We first examined this question by using flotation to purify phagosomes from Rab mutant‐expressing 293T‐FcR‐Kb cells that had internalized opsonized latex beads (Desjardins et al, 1994). The phagosomes were treated with detergent to remove membrane, which was efficient as assessed by Western blot analysis of the pelleted beads and supernatant using an anti‐LAMP1 antibody (Appendix Fig S4A). Importantly, proteasomes remained associated with the beads after detergent treatment, determined by Western blot analysis with an antibody to the β5 subunit (Appendix Fig S4A). The beads were then assayed for proteasome activity using a fluorogenic proteasome substrate (LLVY‐AMC) and an ATP regenerating system. Fluorescence was measured at hourly intervals, and specificity was confirmed by including epoxomicin in a parallel control incubation. Beads purified from 293T‐FcR‐Kb cells expressing Rab mutants had 2‐ to 3‐fold higher epoxomicin‐inhibitable activity than those from cells expressing a control vector (Fig 6A, Appendix Table S1). The results support the hypothesis that active proteasomes within the early phagosomes are active.
Figure 6. Phagosomal proteasomes are active.
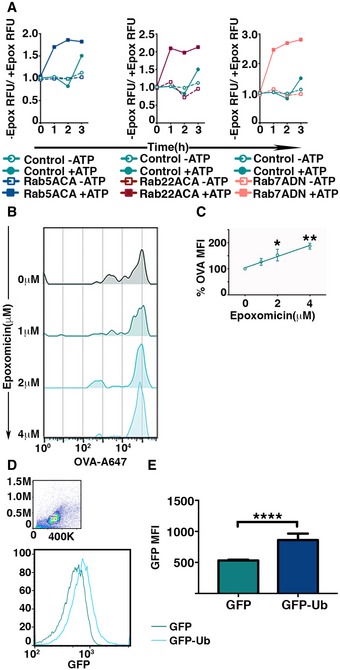
-
AActivity of proteasomes within the phagosomes was determined by measuring the fluorescence intensity of fluorogenic proteasome substrate incubated with latex beads from detergent‐treated phagosomes treated or untreated with epoxomicin. The fluorescence intensities were measured at 1‐h intervals, and values plotted represent the ratio of the signals in the absence versus the presence of epoxomicin (A).
-
B, CIntraluminal proteasome‐dependent degradation of phagocytosed antigen was measured by flow cytometric analysis of phagocytosed latex beads covalently conjugated with Alexa 647‐OVA. Latex beads were extracted by mechanical disruption of BMDC and incubated with ATP, lysosomal protease inhibitor cocktail, and varying dose of epoxomicin at 37°C for 16 h. After incubation, latex beads were treated with detergent and analyzed by flow cytometry (B). The mean of Alexa 647 fluorescence associated with the latex beads incubated with epoxomicin was normalized to fluorescence of untreated beads from independent experiments were plotted (n = 4) (C).
-
D, EUbiquitination of phagosomal cargo within the phagosomes was assessed by measuring the acquisition of GFP fluorescence by phagocytosed latex beads conjugated with OVA post‐detergent extraction from BMDC expressing GFP or GFP‐Ub (D). The mean of fluorescence intensities of GFP on the beads from independent experiments was plotted (E).
As an additional approach, we developed a flow cytometry‐based phagosomal degradation assay, which is a modification of the phagosomal degradation assay we used to measure the impact of Rab mutants on phagosomal degradation (Fig EV1). BMDCs were allowed to internalize latex beads with covalently bound Alexa 647‐OVA for 3 h. After internalization, a crude phagosome preparation was incubated for 16 h at 37°C in the presence of a lysosomal protease inhibitor cocktail but with an ATP regenerating system to allow proteasomal degradation. Various concentrations of epoxomicin were added. The degradation of OVA was measured by flow cytometry of the beads after removal of the phagosomal membrane with detergent. Prior to detergent treatment, the phagosomes were incubated with a rabbit anti‐OVA antiserum to coat any latex beads that had failed to internalize or that were in phagosomes lacking intact membranes. After detergent treatment, the beads were incubated with an Alexa 488‐conjugated secondary antibody to rabbit IgG, to allow exclusion of non‐phagocytosed beads upon subsequent flow cytometric analysis (Appendix Fig S4B). Flow cytometry clearly showed that degradation of bead‐bound OVA occurred and that it was inhibited by epoxomicin (Fig 6B and C), confirming that active proteasomes were present within the lumen of the purified phagosomes.
Finally, a key requirement for cytosolic proteasome degradation is ubiquitination of the substrate. To ask whether the proteasomal degradation in phagosomes could involve ubiquitination, we expressed GFP (control) or GFP‐tagged ubiquitin (GFP‐Ub) (Dantuma et al, 2006) in BMDCs and allowed them to internalize beads covalently conjugated with OVA. After 4 h at 37°C, the cells were detergent extracted and analyzed for the ubiquitination of phagosomal cargo by measuring GFP fluorescence on the beads. Those isolated from BMDCs expressing GFP‐Ub acquired a significantly higher fluorescence signal than the control (Fig 6D and E). This indicates that phagosomal cargo in BMDCs is subject to ubiquitination, further supporting the concept that an active ubiquitin/proteasome system in phagosomes is involved in cross‐presentation.
Discussion
We demonstrate here that active proteasomes can enter phagosomal/endosomal compartments and drive TAP‐independent peptide accumulation within them. We hypothesize that this mediates cross‐presentation that is particularly evident when phagosomal maturation is inhibited using gain‐of‐function Rab mutants that limit this process and therefore proteolysis by lysosomal proteases. In contrast with the conventional vacuolar pathway proposed to rely on lysosomal proteases, epitopes produced by this novel pathway would match those generated by cytosolic processing in virus‐infected cells or tumor cells. We hypothesize that compartmentalization of proteasomes within phagosomes drives a high local concentration of peptides that facilitates efficient MHC‐I loading, evidenced by the reduction of MHC‐I surface expression when proteasomes are inhibited and its reversal when exogenous peptide is added.
Earlier studies have described TAP‐independent and proteasome‐dependent MHC‐I‐restricted antigen processing. We recently reported that the presentation of an epitope derived from endogenous PMEL protein in melanoma cells depends on proteasomal degradation but is independent of TAP function (Vigneron et al, 2018). Our studies revealed this phenotype in cross‐presentation by expressing hβ2m in TAP1−/− BMDCs to increase surface Kb expression and availability. A prior study used the alternate strategy of incubating TAP‐deficient cells at lower temperature to achieve the same ends and rendered cross‐presentation TAP‐independent and proteasome‐dependent (Merzougui et al, 2011). Interestingly, it has also been shown that MHC‐II‐restricted epitopes derived from influenza virus can be generated by the proteasome and presented by a TAP‐independent mechanism (Miller et al, 2015). This observation could also be explained by the presence of active proteasomes within the phagosomal/endolysosomal lumen. It has been speculated that an alternate peptide transporter might be driving these pathways (Lawand et al, 2016), and our data certainly do not eliminate the possibility of such a transporter working in tandem with luminal proteasomes. TAP‐Like (TAP‐L), a TAP homologue localized to lysosomal membranes, has been suggested as a candidate, although it was recently shown that BMDC derived from TAP‐L knockout mice do not exhibit impaired cross‐presentation (Lawand et al, 2018).
Our current understanding of proteasome function within cells is based on the presence of active proteasomes in the cytosol, nucleus, or the cytosolic side of membrane compartments (Wojcik & DeMartino, 2003). The majority of proteasome substrates identified have been cytosolic, nuclear, or membrane proteins. Recent cryo‐electron microscopy analysis has shown that proteasomes localize to the cytosol and cytosolic protein aggregates in neuronal cells (Asano et al, 2015; Guo et al, 2018). However, studies in yeast and plant cells have shown that endocytic compartments contain proteasomes, although no evidence was presented that they were active (Marshall et al, 2015; Cohen‐Kaplan et al, 2016). Work by Houde et al (2003) used a protease protection approach with purified DC phagosomes and showed that most of the proteasomes were degraded and therefore associated with the cytosolic face. However, a fraction of the proteasomes was not degraded and may correspond to those present in the lumen that we describe. No data on activity are available in this case, but here we used two different approaches to demonstrate proteasomal activity in the lumen of phagosomes. In the first, we assayed for ATP‐dependent, epoxomicin‐sensitive cleavage of a fluorescent proteasome substrate by phagosomes isolated from 293T‐FcR‐Kb cells. This required the expression of Rab mutants, which presumably increases proteasomal content. In the second, we demonstrated post‐purification epoxomicin‐sensitive degradation of fluorescent OVA covalently linked to latex beads that were isolated from phagosomes. In addition, we showed that OVA‐conjugated beads in the lumen of phagosomes can recruit ubiquitin. In the latter two sets of experiments, no Rab mutants were required to observe the effects. Overall, the data strongly support the hypothesis that active proteasomes within phagosomes can mediate cross‐presentation.
TAP‐independent cross‐presentation was evident in human 293T‐FcR‐Kb cells, but functional activity by TAP‐deficient BMDCs was only readily observed when the level of surface MHC‐I molecules was boosted by the introduction of human β2m. This suggests that the critical antigen presenting molecules may be mature, recycling MHC‐I molecules derived from the cell surface rather than directly delivered from the ER. This is in line with previous suggestions that trafficking of cell surface MHC‐I through sorting endosomes and recycling endosomes facilitates cross‐presentation (Lizee et al, 2003; Nair‐Gupta et al, 2014). In our experiments, we showed that enhancing cell surface expression of H2‐Kb molecules on hβ2m‐positive TAP1−/− BMDCs by adding an irrelevant Kb‐binding peptide resulted in increased cross‐presentation of OVA, consistent with peptide exchange occurring during recycling.
Previous studies have reported that phagosomes within cross‐presenting DCs maintain a close to neutral pH and a weak proteolytic environment (Savina et al, 2006; Samie & Cresswell, 2015). This is thought to preserve protein antigens, or large fragments of them, allowing their translocation into the cytosol (Kovacsovics‐Bankowski & Rock, 1995). These conditions are also likely to provide a suitable environment for the maintenance of proteasomal activity. Such sequestration would therefore be expected to have a significant impact on cross‐presentation in cells that demonstrate slower kinetics of phagosomal/endosomal maturation, consistent with the increased cross‐presentation we observe upon expression of the Rab mutants. Notably, ER‐derived aminopeptidases that can trim peptides following TAP‐mediated translocation into the ER and generate MHC‐I‐binding peptides from N‐terminally extended precursors can be recruited to phagosomes (Ackerman et al, 2003). Co‐localization of these enzymes in the phagosome would be expected to further facilitate cross‐presentation of functionally relevant epitopes. Whether the peptide editor tapasin, or its homologue TAPBPR (Neerincx & Boyle, 2017), or the endosome‐resident aminopeptidase, IRAP(Saveanu et al, 2009), play a role are questions that merits analysis.
A remaining important question is how delivery of proteasomes into endocytic organelles is mediated. The immunoelectron microscopic analysis of BMDCs, as well as the disappearance over time of proteasome‐containing puncta in BMDCs expressing the Rab mutants, suggests that entry of small vesicles containing proteasomes that are derived from the cytosol may be involved. A number of cellular pathways involved in the delivery of cytosolic proteins into the endolysosomal compartment have been described. The more notable ones are macroautophagy, chaperone‐mediated autophagy, and budding of intraluminal vesicles (Crotzer & Blum, 2008). Macroautophagy or budding of intraluminal vesicles could deliver large protein complexes such as proteasomes, which would be consistent with our immunoelectron microscopy data. However, the molecular machinery involved in these processes also impacts other cellular functions, making simple interpretation of inhibition, knockout, or knockdown experiments difficult. For example, components involved in macroautophagy play a role in maintaining cell surface MHC‐I expression (Loi et al, 2016). The ESCRT components have been identified as key proteins that drive intraluminal vesicle budding but they also play a role in endolysosomal membrane repair (Skowyra et al, 2018), which also could potentially impact cross‐presentation. Experiments to examine the possible involvement of one or more of these potential mechanisms in cross‐presentation that may avoid these complicating issues are ongoing.
It is unlikely that the pathway we describe here is the exclusive mechanism that mediates cross‐presentation. There is considerable evidence that internalized antigenic proteins can be translocated into the cytosol from endocytic organelles, facilitating the cytosolic pathway of cross‐presentation (Cebrian et al, 2011; Lu et al, 2018), and for a role for lysosomal proteases such as cathepsin S (Shen et al, 2004). A combination of these processes, perhaps mediated by different subsets of DCs, would be expected to expand the repertoire of peptides available for cross‐presentation. However, only the translocation‐dependent pathway and the process we have outlined here would routinely produce peptides that match those generated by conventional antigen processing. Future studies should focus on the relative roles of these pathways in the initiation of CD8‐positive T‐cell responses to a variety of pathogens and tumors.
Materials and Methods
Mice
C57BL/6 and TAP1−/− breeding pairs were purchased from the Jackson Laboratories. The mice were housed and bred at Yale Animal Resource center according to institutional guidelines.
Plasmids
Human β2‐microglobulin was cloned into BamHI and NotI site of pMX‐IRES‐Puro and pRetroX‐Tight‐Pur. PCR fragment encoding GFP was cloned into BamHI and NotI site of pRetroX‐Tight‐Pur to create pRetroX‐Tight‐Pur‐GFP plasmid, and in doing so, the original BamHI site was disrupted and a new BamHI site was created at the 3′ end of GFP encoding region. The constitutively active Rab5AQ79L and Rab22AQ64L mutants were cloned into the newly created BamHI and NotI site of pRetroX‐Tight‐Pur‐GFP in frame with the GFP coding sequence. Myc‐tagged dominant‐negative Rab7T22N was cloned into NotI site of pMX‐IRES‐Puro. The Myc‐tagged constitutively active human Rab5AQ79L and Rab22AQ64L mutants were cloned into BamHI and NotI site of pMX‐IRES‐Thy1.1 (Seo & Cresswell, 2013), and dominant‐negative mouse Rab7T22N mutant was cloned into NotI site pMX‐IRES‐Thy1.1. Retroviral constructs expressing US6 and ICP47 have been previously described (Panter et al, 2012). LMP2 coding region in frame with RFP coding region was cloned into the BamHI and EcoRI site of pRetroX‐Tight‐Pur. PCR fragment encoding GFP‐ubiquitin was amplified from GFP‐Ub gifted from Nico Dantuma (Addgene plasmid #11928) and cloned in pRetroX‐Tight‐Pur.
Antibodies and reagents
Polyclonal antibodies against GFP (S3) and monoclonal Myc‐Tag antibody (9E10) were used for detecting the expression of tagged Rab mutants. Monoclonal antibodies against H2‐Kb (Y3) (Anderson & Cresswell, 1994) and human β2‐microglobulin antibody (BBM.1) (Abcam) were used for flow cytometry analysis. Anti‐LMP2 and anti‐LMP7 antibodies were generously provided by Dr. George N DeMartino, University of Texas Southwest Medical Center (Fabunmi et al, 2001). A monoclonal LAMP1 antibody (1D4B) was obtained from eBioscience. Antibodies to LMP2 purchased from Proteintech (14859‐AP) was used for immunoelectron microscopy. Antibody to the β5 subunit of proteasomes was purchased from Bethyl Laboratories (Cat No.‐A303‐847A). Antibodies raised against α‐5 and 19S S2 were obtained from Abcam (ab11437 and ab3315) Epoxomicin was purchased from Enzo Life Sciences. SIINFEKL and gB peptides were purchased from GenScript.
Cell culture
BMDCs were cultured from C57BL/6 and TAP1−/− mice. Bone marrow was harvested from the femurs and the tibia of 6‐ to 8‐week‐old mice, and the cells were differentiated for 5–7 days in RPMI 1640 containing 10% heat‐inactivated fetal calf serum (Gemini Bio‐Products), 100 units/ml penicillin (GIBCO), 100 μg/ml streptomycin (GIBCO), 25 mM HEPES (GIBCO), 1% nonessential amino acids (GIBCO), 1 mM l‐glutamine (GIBCO), 50 μM β‐mercaptoethanol (GIBCO), and conditioned media from a GM‐CSF producing cell line, J558. The B3Z hybridoma specific for OVA257–264 was a gift from Dr. Nilabh Shastri (University of California, Berkeley) and grown in RPMI 1640 medium supplemented with 10% heat‐inactivated fetal calf serum (Gemini Bio‐Products), 100 units/ml penicillin (GIBCO), 100 μg/ml streptomycin (GIBCO), 25 mM HEPES (GIBCO), (GIBCO), 1 mM l‐glutamine (GIBCO), 50 μM β‐mercaptoethanol (GIBCO), and 1 mM pyruvate. 293T‐FcR‐Kb cells were grown in IMDM with 10% heat‐inactivated fetal calf serum (Gemini Bio‐Products), 1× glutaMax (GIBCO), 2 mg/ml G418, and 2 μg/ml puromycin. MEF cells derived from WT and LMP2 knockout mice were processed for immunoelectron microscopy after being treated with 10 nM of INF‐γ for 72 h in a RPMI 1640 medium supplemented with 10% heat‐inactivated fetal calf serum (Gemini Bio‐Products), 100 units/ml penicillin (GIBCO), 100 μg/ml streptomycin (GIBCO), 25 mM HEPES (GIBCO), (GIBCO), 1 mM l‐glutamine (GIBCO), 50 μM β‐mercaptoethanol (GIBCO), and 1 mM pyruvate.
Transfection and retroviral transduction
293T‐FcR‐Kb cells were transfected with Rab mutants cloned into pMX‐IRES‐Thy1.1 using Lipofectamine 2000 (Invitrogen). Transfection was carried out according to the manufacturer's guidelines. In brief, 293T‐FcR‐Kb cells were transfected with 25 μg of the plasmid with 60 μl of Lipofectamine 2000 for 6 h. After overnight culture at 37°C, transfected cells were isolated by magnetic‐activated cell sorting using biotinylated anti‐Thy1.1 antibody (BioLegends) and streptavidin magnetic beads (Miltenyi Biotec) according to the manufacturer's manual.
BMDC were transduced with retroviral plasmids expressing Rab mutants as described previously (Samie & Cresswell, 2015). BMDC from wild‐type or TAP1−/− mice were transduced with pMX‐IRES‐Puro hβ2m to assess the impact of human β2m on cell surface Kb, and cross‐presentation BMDC from wild‐type mice were co‐transduced with pRetroX‐Tet‐ON advanced with pRetroX‐Tight‐Pur‐GFP Rab5AQ79L or pRetroX‐Tight‐Pur‐GFP‐Rab22AQ64L to assess the role of Rab mutants on cross‐presentation and confocal microscopy. BMDC derived from wild‐type mice were transduced with pMX‐IRES‐Puro Rab7AT22N to measure the effect of Rab7AT22N on cross‐presentation. BMDC from wild‐type or TAP1−/− mice were co‐transduced with pRetroX‐Tet‐ON advanced and pRetroX‐Tight‐Pur‐hβ2m along with pRetroX‐Tight‐Pur‐GFP‐Rab5AQ79L or pRetroX‐Tight‐Pur‐GFP‐Rab22AQ64L, in order to assess the role of TAP function on cross‐presentation. BMDC from wild‐type or TAP1−/− mice were co‐transduced with pMX‐IRES‐Puro Rab7AT22N and pMX‐IRES‐Puro hβ2m. For live cell imaging, wild‐type BMDC were co‐transduced with pRetroX‐Tet‐ON advanced and pRetroX‐Tight‐Pur‐LMP2‐RFP along with pRetroX‐Tight‐Pur‐GFP‐Rab5AQ79L or pRetroX‐Tight‐Pur‐GFP‐Rab22AQ64L. BMDCs transduced with inducible vectors were cultured in the presence of 1 μg/ml of doxycycline for 16 h prior to the experiment. For ubiquitination assay, BMDC were co‐transduced with pRetroX‐Tet‐ON advanced and pRetroX‐Tight GFP‐Ub.
In vitro cross‐presentation assay
Transduced and doxycycline‐induced BMDCs were seeded into wells of a 96‐well plate in triplicate, at a density of 1 × 105 cells per well. The cells were incubated with latex beads with non‐covalently bound OVA for 6 h at 37°C and fixed by incubating the cells in 1% paraformaldehyde for 5 min. Following the fixation, the cells were washed with 200 mM glycine and B3Z medium twice to quench the paraformaldehyde. B3Z cells were incubated with the fixed BMDCs for 16 h, and IL‐2 secreted by B3Z cells in the culture supernatant was measured by ELISA as the readout for cross‐presentation.
Sorted 293T‐FcR‐Kb cells expressing Myc‐tagged Rab mutants were seeded in triplicate into wells of 96‐well plates at a density of 1 × 105 cells per well. Cells were incubated for 12 h with opsonized latex beads (Polysciences) non‐covalently bound to OVA (Worthington Biochemical Corporation). Following the incubation with latex beads, the cells were fixed with 1% paraformaldehyde for 5 min and 200 mM glycine was added to stop fixation. After washing the cells with B3Z culture medium, 200 μl of B3Z cells at a cell density of 3.5 × 105 cells/ml was added to each well and cultured with fixed cells for 16 h. The concentration of released IL‐2 was measured using an IL‐2 ELISA Kit (BD OptEIA Mouse IL‐2 ELISA Kit).
Endogenous antigen presentation assay
Endogenous antigen presentation assay was performed as per the protocol described earlier (Ackerman et al, 2006). In brief, BMDC transduced with control vector or human β2m were infected with varying MOI of recombinant vaccinia virus‐expressing OVA (VV‐OVA). Six hours postinfection, cells were fixed with 1% paraformaldehyde and cultured with B3Z. Antigen presentation efficiency was evaluated by measuring IL‐2 concentration in the B3Z culture media.
Flow cytometry
Cell surface staining of H2‐Kb was done by incubating BMDC or 293T‐FcR‐Kb with 1–200 dilution of Alexa 647‐conjugated Y3 monoclonal antibody for 30 min at 4°C. BMDC were stained for cell surface CD11c using PE‐conjugated CD11c antibody (BD Biosciences). The expression of cell surface hβ2m was assessed by incubating BMDC with 1–100 dilution of unconjugated monoclonal antibody BBM.1 (Abcam) for 30 min at 4°C and washed with PBS containing 1% fetal calf serum. Cell surface‐bound unconjugated BBM.1 antibody was detected by incubating with 1–1,000 dilution of Alexa 647‐conjugated goat anti‐mouse IgG (Invitrogen) for 20 min at 4°C. Cell surface staining data were acquired on an Accuri flow cytometer system (BD Biosciences), and data were analyzed with FlowJo software.
Immunofluorescence staining, confocal microscopy, image quantification, and live cell imaging
Transduced BMDC were cultured with latex beads non‐covalently bound to Alexa 647‐conjugated OVA at a density of 5–10 beads per cell on coverslips in a 24‐well plate and incubated at 37°C for 4 h. The cells were fixed in 4% paraformaldehyde for 30 min and permeabilized in 0.5% saponin. Cells were stained with anti‐LMP2 or anti‐LMP7 antibody and co‐stained with an anti‐LAMP1 antibody. Z‐section images of cells were acquired using a Leica SP8 confocal microscope using a 63× oil immersion objective.
The image slices were analyzed to quantify LMP2 and LMP7 fluorescence intensity within the phagosomal lumen as defined by Alexa 647‐positive OVA enclosed within LAMP1‐positive limiting membrane. Using the freehand selection function of ImageJ (NIH), the region of interest marking the phagosomal lumen was identified. Average fluorescence intensity of the LMP2 or LMP7‐positive pixel within this region of interest was quantified using the “measure” function of ImageJ. Average fluorescence intensity of pixels positive for Alexa 647 OVA within the region of interest was also measured. The final data were plotted as a ratio of average fluorescence intensity of LMP2 or LMP7 to the average fluorescence intensity of Alexa 647 OVA.
Live cell images were acquired using a Leica SP8 laser scanning confocal microscope with a temperature, humidity, and CO2 controlled chamber. Transduced BMDC seeded onto glass bottom plate were cultured with latex beads with non‐covalently bound Alexa 647‐conjugated OVA at a density of 5–10 beads per cell. Following incubation for 30 min at 37°C for the cells to adhere, the plates were transferred to the live cell‐imaging chamber. Stacked images of the cells were acquired every 3 min. Using ImageJ (NIH), individual Z‐sections were stitched to generate contiguous movies.
OVA degradation assay
Alexa 647‐conjugated OVA was covalently coupled to 3 μm latex bead with carboxylate reactive group. The coupling was done in the presence of EDAC in a coupling buffer (50 mM MES pH 5.0).
BMDC expressing control vector or Rab mutants were pulsed with the latex beads covalently conjugated with Alexa 647 OVA for 1 h at 37°C, at a dilution of 10 beads per cell. Non‐internalized beads were removed by pelleting the cell on top of a FBS cushion. Cells were plated onto 96‐well tissue culture plate at a density of 2 × 105 cells per well. At different time point, detergent‐containing buffer was added to the wells (TBS, 1% Triton X‐100, 0.1% SDS). The cells were extracted in the buffer for 1 h at 4°C, and the latex beads released postextraction were analyzed by flow cytometry.
Immunoelectron microscopy and analysis
BMDC were fixed in 4% paraformaldehyde, 0.2% glutaraldehyde in PBS for 30 min followed by further fixation in 4% PFA for 1 h. After rinsed in PBS, the cells were re‐suspended in 10% gelatin and placed in 2.3 M sucrose overnight at 4°C before transferred to aluminum pins and frozen rapidly in liquid nitrogen. The frozen blocks were cut with a Leica EM FC6 Ultramicrotome, and 60‐nm‐thick sections were collected on carbon/formvar‐coated grids. The immunolabeling was performed using the Tokuyasu method (Tokuyasu, 1973). Briefly, after treated with 0.1 M ammonium chloride to quench free aldehyde groups, sections were blocked in 1% fish skin gelatin in PBS. The grids were then incubated with primary rabbit antibodies against proteasome α5, β5, 19S S2 at a dilution of 1:200, LMP2 at a dilution of 1:300 and mouse anti‐Lamp1 at 1:100. Sequential labeling was performed with secondary antibody followed by 15 and 5 nm Protein A Gold (Utrecht Medical Center). All grids were rinsed in PBS, fixed in 1% glutaraldehyde for 5 min, and transferred to a uranyl acetate/methylcellulose drop before being collected and dried. Grids were examined and imaged in a FEI Tecnai BioTwin TEM at 80 kV accelerating voltage. Images were recorded using Morada CCD and iTEM (Olympus) software.
For quantitation, all 22–23 images were captured at a magnification 43,000×. LMP2 gold particles were defined by setting the threshold in ImageJ. The numbers of gold particles were counted by using the “Analyze Particle” function of ImageJ.
Proteasome activity assay
293T‐FcR‐Kb cells transfect with Rab mutants were cultured with 3 μm latex beads coated with opsonized OVA for 12 h at a density of 50 beads per cell. Phagosomes were isolated by a previously described protocol (Desjardins et al, 1994) and incubated for 15 min at 4°C in 0.5% Triton X‐100 in PBS, and the isolated latex beads were washed twice with proteasome assay buffer (25 mM HEPES pH 7.5, 0.5 mM EDTA, 0.05% NP‐40, 0.001% SDS). The latex beads were pelleted and re‐suspended in proteasome assay buffer along with the fluorogenic proteasome substrate (Suc‐LLVY‐AMC) and an ATP regeneration buffer (0.5 mM ATP, 0.5 mM UTP, 50 μM GTP, 5 mM creatine phosphate, 25 μg/ml creatine phosphokinase, 0.05 mM EGTA, and 0.5 mM MgCl2). Parallel reactions were set up in the presence of 10 μM epoxomicin and in the absence of ATP, respectively. The reaction mix was incubated at 37°C, and fluorescence signals were measured every hour for 3 h.
Proteasome‐mediated OVA degradation and OVA ubiquitination assay
BMDC were incubated with latex beads covalently conjugated to Alexa 647 OVA at a dilution of 10 beads per cell for 3 h. Post bead uptake cells were layered onto 2 ml FBS and pelleted by centrifugation at 150 g for 5 min. Cells were re‐suspended in a hypotonic homogenization buffer (250 mM sucrose, 3 mM imidazole, pH 7.0) and incubated on ice for 5 min. The cells suspension was passed through 26 g needle four times to released the phagosomes. Crude phagosomes were obtained by collecting the supernatant of 500 g for 5‐min spin of the cell lysate. Phagosomes were re‐suspended in an assay buffer (50 mM HEPES pH‐ 7.4, 78 mM KCl, 4 mM MgCl2, 8.37 mM CaCl2, 10 mM EGTA) and incubated with protease inhibitor cocktail (Sigma‐Aldrich, P1869), ATP regeneration system and varying doses of epoxomicin for 16 h at 37°C. Following the incubation, phagosomes were incubated with rabbit anti‐OVA antibody at 4°C for 30 min, following which phagosomes were detergent extracted to solubilize the membrane and remove the degradation products. Detergent‐extracted beads were incubated with anti‐rabbit Alexa 488 for 30 min at 4°C. The beads were analyzed by flow cytometry using Accuri, BD. The beads that were negative for anti‐OVA staining were analyzed.
BMDC expressing GFP or GFP‐Ub were pulsed with latex beads covalently conjugated with OVA for 1 h at 37°C, at a dilution of 10 beads per cell. The non‐internalized beads were removed layering the cells on FBS and pelleting the cells at 150 g for 5 min. Cells were plated onto 96‐well tissue culture plate at a density of 2 × 105 per well and incubated at 37°C for 4 h. Cells were detergent extracted (1% Triton X‐100, 0.1% SDS) for 1 h at 4°C. The latex beads released after detergent extraction were analyzed by flow cytometry.
Author contributions
DS and PC designed the research and experiments. DS did the experiments. MG and XL performed Immunoelectron Microscopy.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Source Data for Expanded View and Appendix
Review Process File
Acknowledgements
We thank Dr. George DeMartino for antisera to proteasome subunits, Dr. Jacques Neefjes for the tagged‐LMP2 template plasmid, and Dr. Luc van Kaer for the TAP1‐negative MEFs. We also thank Drs. Ralf M. Leonhardt and Claudia S. Wagner for generating the initial cDNA clones of Rab5A, Rab22A, and Rab7A, and their mutants. The study was supported by NIH Grant R01‐AI097206 (to PC) and a Postdoctoral Fellowship from the Cancer Research Institute (to D.S.).
The EMBO Journal (2019) 38: e99266
See also: M Desjardins (August 2019)
References
- Ackerman AL, Kyritsis C, Tampe R, Cresswell P (2003) Early phagosomes in dendritic cells form a cellular compartment sufficient for cross presentation of exogenous antigens. Proc Natl Acad Sci USA 100: 12889–12894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackerman AL, Giodini A, Cresswell P (2006) A role for the endoplasmic reticulum protein retrotranslocation machinery during crosspresentation by dendritic cells. Immunity 25: 607–617 [DOI] [PubMed] [Google Scholar]
- Anderson KS, Alexander J, Wei M, Cresswell P (1993) Intracellular transport of class I MHC molecules in antigen processing mutant cell lines. J Immunol 151: 3407–3419 [PubMed] [Google Scholar]
- Anderson KS, Cresswell P (1994) A role for calnexin (IP90) in the assembly of class II MHC molecules. EMBO J 13: 675–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Androlewicz MJ, Anderson KS, Cresswell P (1993) Evidence that transporters associated with antigen processing translocate a major histocompatibility complex class I‐binding peptide into the endoplasmic reticulum in an ATP‐dependent manner. Proc Natl Acad Sci USA 90: 9130–9134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano S, Fukuda Y, Beck F, Aufderheide A, Forster F, Danev R, Baumeister W (2015) Proteasomes. A molecular census of 26S proteasomes in intact neurons. Science 347: 439–442 [DOI] [PubMed] [Google Scholar]
- Blum JS, Wearsch PA, Cresswell P (2013) Pathways of antigen processing. Annu Rev Immunol 31: 443–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DJ, Serwold T, Shastri N (2000) Bacterial proteins can be processed by macrophages in a transporter associated with antigen processing‐independent, cysteine protease‐dependent manner for presentation by MHC class I molecules. J Immunol 164: 168–175 [DOI] [PubMed] [Google Scholar]
- Cebrian I, Visentin G, Blanchard N, Jouve M, Bobard A, Moita C, Enninga J, Moita LF, Amigorena S, Savina A (2011) Sec22b regulates phagosomal maturation and antigen crosspresentation by dendritic cells. Cell 147: 1355–1368 [DOI] [PubMed] [Google Scholar]
- Cebrian I, Croce C, Guerrero NA, Blanchard N, Mayorga LS (2016) Rab22a controls MHC‐I intracellular trafficking and antigen cross‐presentation by dendritic cells. EMBO Rep 17: 1753–1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen‐Kaplan V, Livneh I, Avni N, Fabre B, Ziv T, Kwon YT, Ciechanover A (2016) p62‐ and ubiquitin‐dependent stress‐induced autophagy of the mammalian 26S proteasome. Proc Natl Acad Sci USA 113: E7490–E7499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotzer VL, Blum JS (2008) Cytosol to lysosome transport of intracellular antigens during immune surveillance. Traffic 9: 10–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantuma NP, Groothuis TA, Salomons FA, Neefjes J (2006) A dynamic ubiquitin equilibrium couples proteasomal activity to chromatin remodeling. J Cell Biol 173: 19–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delamarre L, Holcombe H, Mellman I (2003) Presentation of exogenous antigens on major histocompatibility complex (MHC) class I and MHC class II molecules is differentially regulated during dendritic cell maturation. J Exp Med 198: 111–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desjardins M, Celis JE, van Meer G, Dieplinger H, Jahraus A, Griffiths G, Huber LA (1994) Molecular characterization of phagosomes. J Biol Chem 269: 32194–32200 [PubMed] [Google Scholar]
- Duclos S, Diez R, Garin J, Papadopoulou B, Descoteaux A, Stenmark H, Desjardins M (2000) Rab5 regulates the kiss and run fusion between phagosomes and endosomes and the acquisition of phagosome leishmanicidal properties in RAW 264.7 macrophages. J Cell Sci 116(Pt 19): 3531–3541 [DOI] [PubMed] [Google Scholar]
- Fabunmi RP, Wigley WC, Thomas PJ, DeMartino GN (2001) Interferon gamma regulates accumulation of the proteasome activator PA28 and immunoproteasomes at nuclear PML bodies. J Cell Sci 114: 29–36 [DOI] [PubMed] [Google Scholar]
- Gagnon E, Duclos S, Rondeau C, Chevet E, Cameron PH, Steele‐Mortimer O, Paiement J, Bergeron JJ, Desjardins M (2002) Endoplasmic reticulum‐mediated phagocytosis is a mechanism of entry into macrophages. Cell 110: 119–131 [DOI] [PubMed] [Google Scholar]
- Giodini A, Rahner C, Cresswell P (2009) Receptor‐mediated phagocytosis elicits cross‐presentation in nonprofessional antigen‐presenting cells. Proc Natl Acad Sci USA 106: 3324–3329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grotzke JE, Sengupta D, Lu Q, Cresswell P (2017) The ongoing saga of the mechanism(s) of MHC class I‐restricted cross‐presentation. Curr Opin Immunol 46: 89–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guermonprez P, Saveanu L, Kleijmeer M, Davoust J, Van Endert P, Amigorena S (2003) ER‐phagosome fusion defines an MHC class I cross‐presentation compartment in dendritic cells. Nature 425: 397–402 [DOI] [PubMed] [Google Scholar]
- Guo Q, Lehmer C, Martinez‐Sanchez A, Rudack T, Beck F, Hartmann H, Perez‐Berlanga M, Frottin F, Hipp MS, Hartl FU et al (2018) In situ structure of neuronal C9orf72 Poly‐GA aggregates reveals proteasome recruitment. Cell 172: 696–705.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison RE, Bucci C, Vieira OV, Schroer TA, Grinstein S (2003) Phagosomes fuse with late endosomes and/or lysosomes by extension of membrane protrusions along microtubules: role of Rab7 and RILP. Mol Cell Biol 23: 6494–6506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houde M, Bertholet S, Gagnon E, Brunet S, Goyette G, Laplante A, Princiotta MF, Thibault P, Sacks D, Desjardins M (2003) Phagosomes are competent organelles for antigen cross‐presentation. Nature 425: 402–406 [DOI] [PubMed] [Google Scholar]
- Kauppi M, Simonsen A, Bremnes B, Vieira A, Callaghan J, Stenmark H, Olkkonen VM (2002) The small GTPase Rab22 interacts with EEA1 and controls endosomal membrane trafficking. J Cell Sci 115: 899–911 [DOI] [PubMed] [Google Scholar]
- Kovacsovics‐Bankowski M, Rock KL (1995) A phagosome‐to‐cytosol pathway for exogenous antigens presented on MHC class I molecules. Science 267: 243–246 [DOI] [PubMed] [Google Scholar]
- Lawand M, Abramova A, Manceau V, Springer S, van Endert P (2016) TAP‐dependent and ‐independent peptide import into dendritic cell phagosomes. J Immunol 197: 3454–3463 [DOI] [PubMed] [Google Scholar]
- Lawand M, Evnouchidou I, Baranek T, Montealegre S, Tao S, Drexler I, Saveanu L, Si‐Tahar M, van Endert P (2018) Impact of the TAP‐like transporter in antigen presentation and phagosome maturation. Mol Immunol 10.1016/j.molimm.2018.06.268 [DOI] [PubMed] [Google Scholar]
- Lehner PJ, Karttunen JT, Wilkinson GW, Cresswell P (1997) The human cytomegalovirus US6 glycoprotein inhibits transporter associated with antigen processing‐dependent peptide translocation. Proc Natl Acad Sci USA 94: 6904–6909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizee G, Basha G, Tiong J, Julien JP, Tian M, Biron KE, Jefferies WA (2003) Control of dendritic cell cross‐presentation by the major histocompatibility complex class I cytoplasmic domain. Nat Immunol 4: 1065–1073 [DOI] [PubMed] [Google Scholar]
- Loi M, Muller A, Steinbach K, Niven J, Barreira da Silva R, Paul P, Ligeon LA, Caruso A, Albrecht RA, Becker AC et al (2016) Macroautophagy proteins control MHC class I levels on dendritic cells and shape anti‐viral CD8(+) T cell responses. Cell Rep 15: 1076–1087 [DOI] [PubMed] [Google Scholar]
- Lu Q, Grotzke JE, Cresswell P (2018) A novel probe to assess cytosolic entry of exogenous proteins. Nat Commun 9: 3104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall RS, Li F, Gemperline DC, Book AJ, Vierstra RD (2015) Autophagic degradation of the 26S proteasome is mediated by the dual ATG8/ubiquitin receptor RPN10 in Arabidopsis . Mol Cell 58: 1053–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merzougui N, Kratzer R, Saveanu L, van Endert P (2011) A proteasome‐dependent, TAP‐independent pathway for cross‐presentation of phagocytosed antigen. EMBO Rep 12: 1257–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MA, Ganesan AP, Luckashenak N, Mendonca M, Eisenlohr LC (2015) Endogenous antigen processing drives the primary CD4+ T cell response to influenza. Nat Med 21: 1216–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair‐Gupta P, Baccarini A, Tung N, Seyffer F, Florey O, Huang Y, Banerjee M, Overholtzer M, Roche PA, Tampe R et al (2014) TLR signals induce phagosomal MHC‐I delivery from the endosomal recycling compartment to allow cross‐presentation. Cell 158: 506–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neerincx A, Boyle LH (2017) Properties of the tapasin homologue TAPBPR. Curr Opin Immunol 46: 97–102 [DOI] [PubMed] [Google Scholar]
- Neumann L, Kraas W, Uebel S, Jung G, Tampe R (1997) The active domain of the herpes simplex virus protein ICP47: a potent inhibitor of the transporter associated with antigen processing. J Mol Biol 272: 484–492 [DOI] [PubMed] [Google Scholar]
- Panter MS, Jain A, Leonhardt RM, Ha T, Cresswell P (2012) Dynamics of major histocompatibility complex class I association with the human peptide‐loading complex. J Biol Chem 287: 31172–31184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Procko E, Gaudet R (2009) Antigen processing and presentation: TAPping into ABC transporters. Curr Opin Immunol 21: 84–91 [DOI] [PubMed] [Google Scholar]
- Roberts EA, Chua J, Kyei GB, Deretic V (2006) Higher order Rab programming in phagolysosome biogenesis. J Cell Biol 174: 923–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samie M, Cresswell P (2015) The transcription factor TFEB acts as a molecular switch that regulates exogenous antigen‐presentation pathways. Nat Immunol 16: 729–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saveanu L, Carroll O, Weimershaus M, Guermonprez P, Firat E, Lindo V, Greer F, Davoust J, Kratzer R, Keller SR et al (2009) IRAP identifies an endosomal compartment required for MHC class I cross‐presentation. Science 325: 213–217 [DOI] [PubMed] [Google Scholar]
- Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, Lennon‐Dumenil AM, Seabra MC, Raposo G, Amigorena S (2006) NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell 126: 205–218 [DOI] [PubMed] [Google Scholar]
- Savina A, Vargas P, Guermonprez P, Lennon AM, Amigorena S (2010) Measuring pH, ROS production, maturation, and degradation in dendritic cell phagosomes using cytofluorometry‐based assays. Methods Mol Biol 595: 383–402 [DOI] [PubMed] [Google Scholar]
- Seo JY, Cresswell P (2013) Viperin regulates cellular lipid metabolism during human cytomegalovirus infection. PLoS Pathog 9: e1003497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Sigal LJ, Boes M, Rock KL (2004) Important role of cathepsin S in generating peptides for TAP‐independent MHC class I crosspresentation in vivo . Immunity 21: 155–165 [DOI] [PubMed] [Google Scholar]
- Simonsen A, Lippe R, Christoforidis S, Gaullier JM, Brech A, Callaghan J, Toh BH, Murphy C, Zerial M, Stenmark H (1998) EEA1 links PI(3)K function to Rab5 regulation of endosome fusion. Nature 394: 494–498 [DOI] [PubMed] [Google Scholar]
- Singh R, Cresswell P (2010) Defective cross‐presentation of viral antigens in GILT‐free mice. Science 328: 1394–1398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowyra ML, Schlesinger PH, Naismith TV, Hanson PI (2018) Triggered recruitment of ESCRT machinery promotes endolysosomal repair. Science 360: eaar5078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song R, Harding CV (1996) Roles of proteasomes, transporter for antigen presentation (TAP), and beta 2‐microglobulin in the processing of bacterial or particulate antigens via an alternate class I MHC processing pathway. J Immunol 156: 4182–4190 [PubMed] [Google Scholar]
- Tokuyasu KT (1973) A technique for ultracryotomy of cell suspensions and tissues. J Cell Biol 57: 551–565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Kaer L, Ashton‐Rickardt PG, Ploegh HL, Tonegawa S (1992) TAP1 mutant mice are deficient in antigen presentation, surface class I molecules, and CD4‐8+ T cells. Cell 71: 1205–1214 [DOI] [PubMed] [Google Scholar]
- Vieira OV, Bucci C, Harrison RE, Trimble WS, Lanzetti L, Gruenberg J, Schreiber AD, Stahl PD, Grinstein S (2003) Modulation of Rab5 and Rab7 recruitment to phagosomes by phosphatidylinositol 3‐kinase. Mol Cell Biol 23: 2501–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigneron N, Ferrari V, Van den Eynde BJ, Cresswell P, Leonhardt RM (2018) Cytosolic processing governs TAP‐independent presentation of a critical melanoma antigen. J Immunol 201: 1875–1888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigert R, Yeung AC, Li J, Donaldson JG (2004) Rab22a regulates the recycling of membrane proteins internalized independently of clathrin. Mol Biol Cell 15: 3758–3770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik C, DeMartino GN (2003) Intracellular localization of proteasomes. Int J Biochem Cell Biol 35: 579–589 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Movie EV1
Movie EV2
Movie EV3
Source Data for Expanded View and Appendix
Review Process File