

Abstract
Many eukaryotic proteins are regulated by modification with the ubiquitin‐like protein small ubiquitin‐like modifier (SUMO). This linkage is reversed by SUMO proteases, of which there are two in Saccharomyces cerevisiae, Ulp1 and Ulp2. SUMO‐protein conjugation regulates transcription, but the roles of SUMO proteases in transcription remain unclear. We report that Ulp2 is recruited to transcriptionally active genes to control local polysumoylation. Mutant ulp2 cells show impaired association of RNA polymerase II (RNAPII) with, and diminished expression of, constitutively active genes and the inducible CUP1 gene. Ulp2 loss sensitizes cells to 6‐azauracil, a hallmark of transcriptional elongation defects. We also describe a novel chromatin regulatory mechanism whereby histone‐H2B ubiquitylation stimulates histone sumoylation, which in turn appears to inhibit nucleosome association of the Ctk1 kinase. Ctk1 phosphorylates serine‐2 (S2) in the RNAPII C‐terminal domain (CTD) and promotes transcript elongation. Removal of both ubiquitin and SUMO from histones is needed to overcome the impediment to S2 phosphorylation. These results suggest sequential ubiquitin‐histone and SUMO‐histone modifications recruit Ulp2, which removes polySUMO chains and promotes RNAPII transcription elongation.
Keywords: CTD phosphorylation, histone, SUMO, ubiquitin, Ulp2
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Post-translational Modifications, Proteolysis & Proteomics
Introduction
The small ubiquitin‐like modifier (SUMO) protein is an evolutionarily conserved post‐translational modifier modulating proteins in diverse regulatory pathways (Flotho & Melchior, 2013). Mature SUMO is conjugated via its C‐terminal Gly‐Gly motif to substrate lysine side chains. Conjugation occurs through an enzyme cascade involving the E1 SUMO‐activating enzyme (Aos1‐Uba2), the E2 SUMO‐conjugating enzyme E2 (Ubc9), and one of a small number of E3 SUMO ligases (Hendriks & Vertegaal, 2016). Sumoylation is readily reversed by SUMO proteases; nine SUMO proteases have been described in humans and two, Ulp1 and Ulp2, in Saccharomyces cerevisiae (Hickey et al, 2012). While there are numerous reports of functions and substrates of Ulp1, most cellular roles of Ulp2 remain unclear. Ulp2 has particularly high activity toward polySUMO chains (Bylebyl et al, 2003), and it preferentially localizes to the nucleus (Li & Hochstrasser, 2000). Its loss results in a pleiotropic mutant phenotype including defects in cell growth; sensitivity to heat, DNA damage, or aberrant spindle formation; multi‐chromosome aneuploidy; and upregulated expression of ribosomal proteins (Li & Hochstrasser, 2000; Ryu et al, 2016).
SUMO modification serves both positive and negative roles in transcription in yeast as well as mammals (Chymkowitch et al, 2015b). For example, SUMO conjugation to many gene‐specific transcription factors, including C/EBP, c‐Jun, ELK‐1, and the TFIID subunit TAF5, suppresses the transcription of their target genes (Muller et al, 2000; Kim et al, 2002; Yang et al, 2003; Boyer‐Guittaut et al, 2005). Another demonstrated mechanism for SUMO‐dependent repression is sumoylation of the transcriptional coactivator p300; this modification promotes p300 binding to histone deacetylase complexes (HDACs), which locally deactivate chromatin (Girdwood et al, 2003).
In some cases, however, transcription factor sumoylation stimulates transcription (Lyst & Stancheva, 2007; Guo & Sharrocks, 2009). For example, sumoylation of ZNF76, a repressor targeting the TATA‐binding protein (TBP), inhibits ZNF76 repression activity (Zheng & Yang, 2004). Intriguingly, the histone methyltransferase SETDB1 is differentially modulated by sumoylation of distinct cofactors (Lyst et al, 2006; Ivanov et al, 2007). Such conjugation can either promote gene expression (Lyst et al, 2006) or repress it (Ivanov et al, 2007).
In S. cerevisiae, SUMO is usually localized at constitutively transcribed genes and promotes their transcription as well as the activation of inducible genes (Rosonina et al, 2010). In a genome‐wide study, SUMO was found to be enriched at genes encoding tRNA and ribosomal protein (RP) genes, and sumoylation of the Rap1 transcriptional regulator promotes recruitment of the basal transcription machinery to these genes (Chymkowitch et al, 2015a. Interestingly, SUMO plays an important role not only in transcriptional activation by the Gcn4 transcription factor but also in subsequent transcriptional deactivation through a combination of SUMO‐dependent clearance of Gcn4 and stabilization of Tup1 corepressor binding at promoters (Ng et al, 2015). The yeast SUMO proteases also can modulate transcription. Ulp1 localized at nuclear pore complexes accelerates GAL1 de‐repression kinetics (Texari et al, 2013), while association of Ulp2 with RP and snoRNA genes limits accumulation of SUMO at these loci (Ryu et al, 2018).
The amino‐ and carboxy‐terminal tails of histones are major targets for multiple post‐translational modifications including acetylation, phosphorylation, methylation, and ubiquitylation. Additionally, many reports suggest that chromatin structure or gene expression is regulated by crosstalk among different histone modifications (Berger, 2002). A good example of such crosstalk is a trans‐tail modification pathway in which histone H2B monoubiquitylation is linked to histone H3 methylation in promoting gene transcription and telomeric silencing (Henry & Berger, 2002; Sun & Allis, 2002). All four core histones and the histone H2A variant H2A.Z are also all subject to SUMO modification in S. cerevisiae (Nathan et al, 2006; Kalocsay et al, 2009), whereas only sumoylated histones H3 and H4 have been identified in mammalian cells to date (Shiio & Eisenman, 2003; Hendriks et al, 2014). A small number of SUMO‐modification sites have been identified in yeast histones, but there appear to be many such sites within each histone (Nathan et al, 2006). In human cells, histone H4 sumoylation recruits the histone deacetylase HDAC1 and heterochromatin protein 1 (HP1), thereby attenuating transcription (Shiio & Eisenman, 2003). SUMO‐conjugated histones may reduce transcriptional activity by opposing activating histone marks such as acetylation or ubiquitylation (Nathan et al, 2006). However, whether histone sumoylation contributes more generally to transcription regulation is unclear.
The C‐terminal domain (CTD) of the largest subunit of RNA polymerase II (RNAPII), Rpo21/Rpb1, is composed of heptapeptide repeats (YSPTSPS) that are subject to extensive post‐translational modifications, including phosphorylation, glycosylation, proline isomerization, acetylation, methylation, and ubiquitylation (Egloff et al, 2012). The best studied of these modifications is phosphorylation of CTD serine 5 (S5‐P) and serine 2 (S2‐P), which occurs primarily at the initiation and elongation stages, respectively, of the transcription cycle. S5 phosphorylation is catalyzed by the TFIIH‐associated kinase Kin28 and promotes recruitment of mRNA‐capping enzyme and the nuclear cap‐binding complex to nascent transcripts. Subsequently, the cyclin‐dependent kinase Ctk1 (pTEFb or CDK9 in metazoans) phosphorylates S2, which couples the later stages of transcriptional elongation to mRNA 3′‐end processing (Ahn et al, 2004). In addition, the Bur1 kinase phosphorylates S2 at the 5′‐end of genes and stimulates Ctk1 activity (Murray et al, 2001).
These CTD phosphorylations are also associated with cotranscriptional histone modifications and chromatin remodeling (Srivastava & Ahn, 2015). CTD modifications control the recruitment of regulatory factors to chromatin, and reciprocally, histone modifications modulate CTD‐cofactor associations. For instance, S5‐P promotes cotranscriptional recruitment, via the PAF elongation complex, of the Rad6 E2 enzyme responsible for H2B ubiquitylation and the H3K4 methyltransferase complex (SET1/COMPASS; Ng et al, 2003; Wood et al, 2003; Xiao et al, 2005). In another example, deletion of CTK1 prevents H3K36 trimethylation by inhibiting the recruitment of the Set2 H3K36 methyltransferase (Wyce et al, 2007).
Here, we report that Ulp2 is preferentially recruited to actively transcribed genes, and its loss impedes association of RNAPII, resulting in decreased gene expression. Whereas the N‐terminal domain of Ulp2 is sufficient to promote Ulp2 chromatin localization, its C‐terminal domain is important for efficient transcriptional elongation and Ulp2 interaction with nucleosomes. A genetic screen revealed a synthetic lethal interaction between mutations in ULP2 and the RAD6‐ and BRE1‐encoded H2B ubiquitylation enzymes. Notably, histone sumoylation and chromatin localization of Ulp2 both depend on H2B ubiquitylation, and this Ulp2 localization is required for subsequent histone desumoylation. Persistent polySUMO conjugation to H2B or loss of ULP2 inhibits recruitment of the Ctk1 kinase to constitutively active genes, limiting CTD‐S2 phosphorylation. A similar block to Ctk1 recruitment correlates with the persistent H2B ubiquitylation observed in cells lacking the Ubp8 ubiquitin protease.
Taken together, these data suggest that Ulp2 acts as a general transcription factor to control RNAPII recruitment and SUMO levels, thereby promoting gene transcription. Furthermore, our results indicate a new crosstalk pathway in which ubiquitin and SUMO are sequentially conjugated to histones during the transcription cycle. These transient histone modifications are coupled with Ctk1‐mediated RNAPII S2‐P modification, facilitating later transcription elongation steps. The Ulp2 SUMO protease therefore appears to promote transcription by coordinating histone‐SUMO and RNAPII CTD‐S2‐P modifications.
Results
Ulp2 localizes to actively transcribed genes
Although there have been attempts to explore the relationship between Ulp2 and transcription, these studies included no evidence for direct involvement of the SUMO protease in gene expression. To investigate this further, we first grouped previously published RNA‐seq data comparing wild‐type (WT) and ulp2Δ strains (Ryu et al, 2018) into three categories: high, medium, or low expression levels (Fig 1A and B). Because expression of RPs is a well‐known target of the SUMO pathway (Chymkowitch et al, 2015a; Ryu et al, 2018), we excluded from our re‐analysis the 130 RP genes with significantly changed values in ulp2Δ strains. We found that loss of ULP2 impaired transcription of 91.1% of highly expressed genes (Fig 1B). Also, 69.9% of genes with moderate expression were significantly down‐regulated in ulp2Δ cells. Expression levels of three representative genes—PMA1, ADH1, and PYK1—that have relatively high constitutive transcription rates were measured by quantitative RT–PCR (qRT–PCR) in cells lacking ULP2 (Fig 1C). We confirmed down‐regulation of their expression in the ulp2Δ strain. Therefore, these results suggest that loss of Ulp2 preferentially diminishes transcription of relatively highly expressed genes.
Figure 1. Ulp2 is recruited to actively transcribed genes.
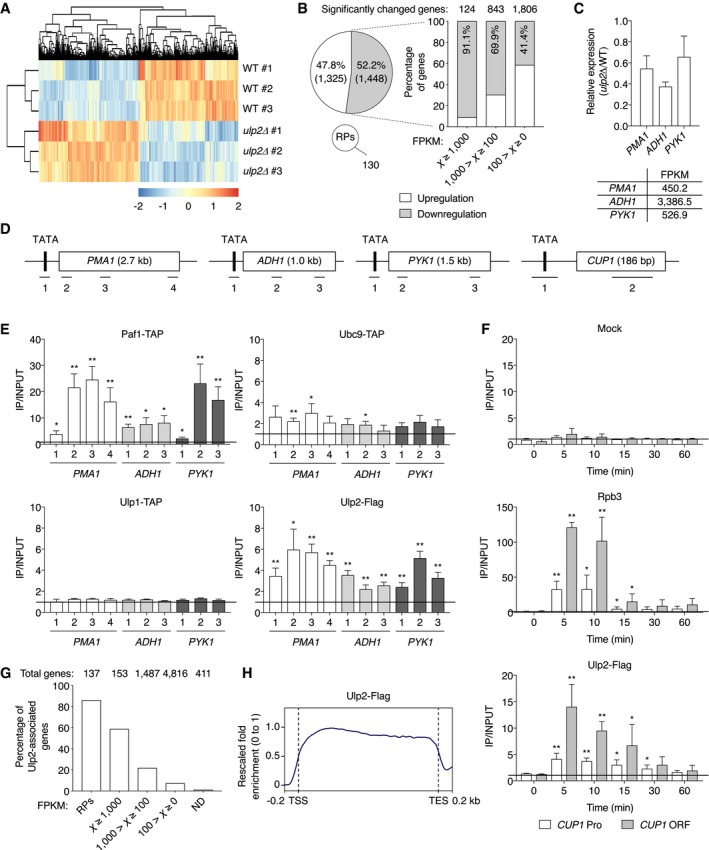
- Transcriptome re‐analysis of ulp2Δ strains compared with WT (MHY1379) from previous RNA‐seq experiments (Ryu et al, 2018). Two‐dimensional agglomerative hierarchical clustering shows 2,903 significantly up‐ or down‐regulated genes (P < 0.05) in triplicate RNA samples. Red and blue indicate up‐ and down‐regulation of genes, respectively, and their intensity represents the relative gene expression changes.
- Summary of transcriptome data in (A). White and gray colors indicate up‐ and down‐regulated genes, respectively, in ulp2Δ cells. The pie graph shows the percentages of significantly changed genes, except for 130 ribosomal protein genes (RPs), and are classified by FPKM (Fragments Per Kilobase of Million reads mapped) values in the bar graph.
- qRT–PCR analysis of the highly transcribed PMA1, ADH1, and PYK1 genes in ulp2Δ cells. Expression was measured relative to WT cells, and data were normalized to SPT15 expression. Error bars indicate the standard deviations (SDs) from three independent RNA preparations. FPKMs of each gene in WT are shown in the bottom graph.
- Schematic diagram of PMA1, ADH1, PYK1, and CUP1 genes. The TATA/promoter (Pro) and open reading frame (ORF) are represented by black and white boxes, respectively. Bars with numbers below the genes show the relative positions of the PCR products used in the ChIP and qRT–PCR analyses and are used for identification in all later figures.
- ChIP analysis using IgG‐Sepharose or anti‐Flag‐agarose beads in strains expressing TAP‐tagged Paf1, Ubc9, or Ulp1 or Flag‐tagged Ulp2. An untagged strain (MHY500) was used as a negative control for immunoprecipitation of Ulp2‐Flag (Fig EV1A). The qPCR signals of the indicated genes were quantitated and normalized to an internal background control and the input DNA. The primer pairs used are indicated in (D). Quantification (described in Materials and Methods) presented as fold over background; a value of 1 indicates no signal detected above background signal at a nontranscribed locus, as marked with the horizontal line. Error bars indicate SDs calculated from three independent chromatin preparations.
- Occupancy of Rpb3 and Ulp2 at CUP1 gene was determined by ChIP in a strain expressing Flag‐tagged Ulp2 using anti‐Rpb3 antibody and anti‐Flag agarose beads as shown in (E). “Mock” indicates use of protein G beads without added antibody. For CUP1 induction, cells were harvested at the indicated time points after adding CuSO4. “Pro” and “ORF” represent the positions of PCR fragments 1 and 2 from the CUP1 gene as described in (D). Black bar indicates a value of 1, the background signal. Error bars, SD from four independent experiments.
- A percentage graph of association of Ulp2‐Flag with genes classified by FPKM in the file Dataset EV1. RPs and ND indicate ribosomal protein genes and genes not detected genes in our earlier RNA‐seq experiments, respectively. The numbers above the graph indicate the total number of yeast genes in each expression category.
- Average plot of Ulp2‐Flag occupancy at transcribed genes. The values of the y‐axis indicate rescaled fold enrichment, setting the maximum occupancy to 1, and the minimum occupancy to 0, and the x‐axis indicates normalized distance from transcription start sites (TSS) and transcription end sites (TES). The dotted lines indicate the TSS and TES. The ChIP‐seq data were obtained from duplicate experiments.
To determine whether the Ulp2 enzyme is recruited to such genes, chromatin immunoprecipitation (ChIP) analysis was carried out on PMA1, ADH1, and PYK1 (Fig 1D and E). We used Paf1, a subunit of the PAF complex, as a positive control because it is known to associate strongly with gene coding regions (Kim et al, 2004). ChIP results with the SUMO‐conjugating enzyme Ubc9 indicated weak association with both promoter and open reading frame (ORF) sequences in these genes, while the Ulp1 SUMO protease was not significantly enriched at these sites. Interestingly, Flag‐tagged Ulp2 occupied the length of these three test genes, with substantially higher association within the ORF in PMA1 and PYK1. These results suggest SUMO‐conjugating (Ubc9) and SUMO‐deconjugating (Ulp2) enzymes contribute directly to both transcription initiation and elongation.
The CUP1 gene was then examined to determine whether Ulp2 is recruited to genes in a transcription‐dependent manner. CUP1 encodes the copper‐binding metallothionein protein (Hottiger et al, 1994). Its transcription is induced 10‐ to 20‐fold within 5 min of addition of 1 mM copper, and then it down‐regulates its own expression, resulting in a return to basal transcript levels by ~30 min of sustained copper exposure (Hamer et al, 1985; Pena et al, 1998). Unlike mock immunoprecipitation with only protein G beads (Fig 1F, top), we observed that occupancy by Rpb3, a subunit of RNAPII, at promoter and ORF regions of CUP1 increased by ~30‐fold and ~120‐fold, respectively, within 5 min of exposure to copper and subsequently dropped to uninduced levels by 15–30 min (Fig 1F, middle). Ulp2 also showed a strong and rapid increase in association with CUP1, increasing ~15‐fold at the ORF region within 5 min of copper addition, and then decreased in parallel with the drop in RNAPII association (Fig 1F, bottom).
To map Ulp2 binding sites on a genome‐wide scale, we carried out ChIP‐seq experiments (Fig 1G) and compared genome‐wide association of Ulp2 with the RNA‐seq data in Fig 1B. Consistent with our previous report (Ryu et al, 2018), we found that Ulp2 was enriched at most ribosomal protein gene loci. Furthermore, we observed a greater percentage of genes that were highly expressed associated with Ulp2, consistent with a potential role for Ulp2 in transcription. When Ulp2 binding sites derived from the ChIP‐seq data were mapped relative to transcribed regions throughout the genome (from transcription start site to transcription end site), we saw a clear enrichment in these regions (Fig 1H), consistent with the results in Figs 1E and EV1B. Although Ulp2 has previously been shown to associate specifically with rDNA, RP, and snoRNA genes (Liang et al, 2017; Ryu et al, 2018), these observations provide the first direct evidence for the transcription‐linked association of Ulp2 with actively transcribed genes in vivo.
Figure EV1. Genome‐wide analysis of Ulp2‐Flag binding sites in chromatin.
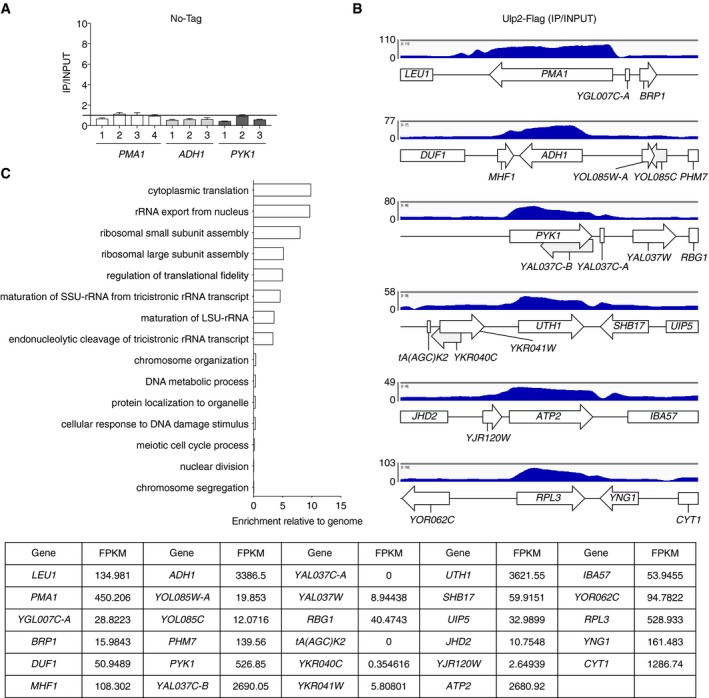
- ChIP analysis using anti‐Flag agarose in an untagged strain (MHY500), as described in Fig 1E. Black bar indicates value of 1, which is the background signal. Error bars indicate SDs calculated from three independent experiments. See Dataset EV2 for qPCR raw data.
- Representative data from ChIP‐seq analysis of Ulp2‐Flag. The y‐axis shows fold enrichment normalized to the input DNA. Arrows and boxes with gene names indicate locations of ORFs. FPKMs of each gene in WT are shown in the bottom graph.
- Gene Ontology (GO) enrichment analysis of the genes displayed in Fig 1G. Bar diagrams indicate the fold enrichment of categories of biological process to the genome using GO data from PANTHER. The genes used in GO biological process are listed in the file Dataset EV1.
Ulp2 is involved in transcription elongation
Our RNA‐seq, ChIP, and ChIP‐seq analyses in Fig 1 demonstrated that Ulp2‐chromatin association correlates with transcriptional activation. To explore further how Ulp2 might participate in transcription, we first tested the sensitivity of ulp2Δ cells to 6‐azauracil (6‐AU), a general indicator for involvement in transcription elongation (Conaway et al, 2000). Cells lacking RAD6 were used as a positive control for 6‐AU sensitivity (Xiao et al, 2005). On the control SD‐Ura plate, the ubc9ts and ulp1ts mutants showed modest growth defects at 30°C, whereas cell growth was more strongly impaired in the ulp2Δ strain (Fig 2A, upper panels), consistent with previous reports (Li & Hochstrasser, 2000). Notably, we observed an extremely strong sensitivity to 6‐AU in ubc9ts but not ulp1ts cells. Because the ubc9 mutant reduces RNAPII occupancy and association of SUMO with constitutive genes (Rosonina et al, 2010), these defects may sensitize cells to 6‐AU. By contrast, our ChIP results in Fig 1E and previous microscopic examination (Li & Hochstrasser, 2003) did not reveal any evidence of chromatin binding by Ulp1 in vivo, and ulp1ts cells grew well on 6‐AU plates (Fig 2A), suggesting Ulp1 is not directly involved in the transcription elongation process.
Figure 2. Ulp2 promotes transcription elongation and RNAPII localization.
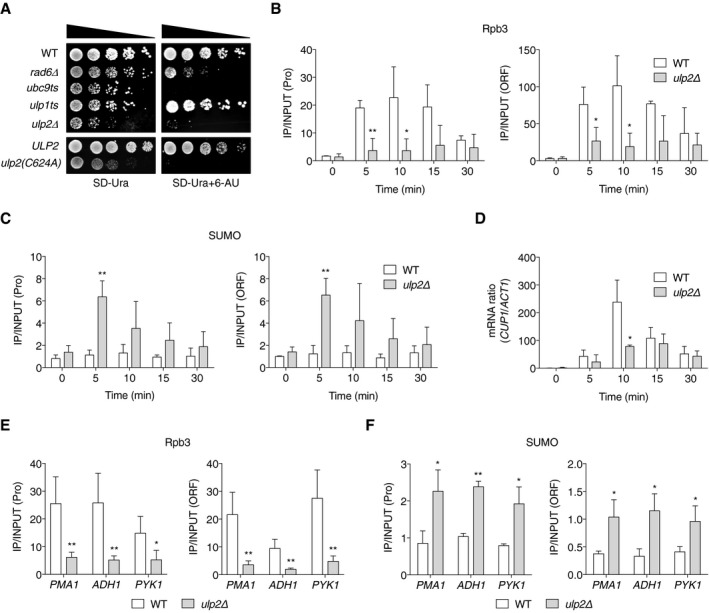
-
ASensitivity of the indicated mutants to 6‐AU. All strains carried a URA3 plasmid, pRS316, and were spotted on SD‐Ura with or without 6‐AU (100 μg/ml); plates were incubated for 2‐4 days at 30°C. ULP2 and ulp2(C624A) represent ulp2Δ::HIS3 cells containing either pRS314‐ULP2‐FLAG or pRS314‐ulp2(C624A)‐FLAG.
-
B, CChIP analyses using anti‐Rpb3 (B) and anti‐SUMO (C) antibodies in ulp2Δ cells as in Fig 1F. Error bars indicate the SD from three independent experiments.
-
DqRT–PCR analysis of CUP1 mRNA levels in ulp2Δ cells. Data were normalized to ACT1 mRNA levels. CUP1 gene induction was performed as in Fig 1F. Error bars represent the SD from three RNA samples.
-
E, FChIP assays using anti‐Rpb3 (E) and anti‐SUMO (F) antibodies in ulp2Δ cells as done in Fig 1E. “Pro” denotes the #1 PCR product of PMA1, ADH1, and PYK1 genes described in Fig 1D, while “ORF” indicates the #3 products of PMA1 and #2 of ADH1 and PYK1; these are used in all ensuing figures except where specified. Error bars indicate the SD from four (E) and three (F) independent assays.
Cells lacking ULP2 also showed increased sensitivity to 6‐AU. Because both SUMO‐conjugating and SUMO‐deconjugating enzymes had clear defects in cell growth on 6‐AU plates, our data suggest that properly maintained levels of SUMO conjugation at actively transcribed genes are required for efficient transcription elongation. To determine whether Ulp2 catalytic activity is required for this, the same 6‐AU plate assay was employed in ulp2Δ cells carrying plasmids encoding either WT ULP2 or the inactive ulp2‐C624A mutant, in which the catalytic Cys residue is replaced with Ala (Li & Hochstrasser, 2000; Fig 2A, bottom panels). As with ulp2Δ cells, ulp2‐C624A cells could barely grow on 6‐AU, suggesting that the SUMO protease activity of Ulp2 is crucial for regulating transcriptional elongation.
Ulp2 controls RNAPII and SUMO occupancy during transcription
Since our data indicated that Ulp2‐chromatin binding correlates closely with transcription levels, we monitored recruitment of RNAPII and SUMO during CUP1 gene activation in ulp2Δ cells (Fig 2B and C). A dramatic increase of Rpb3 recruitment is observed in WT cells at 5–10 min following CuSO4 addition. By contrast, Rpb3 binding to the promoter and ORF of CUP1 was greatly reduced by loss of ULP2 (Fig 2B). Concomitantly, the deletion of ULP2 led to a rapid and abnormally high accumulation of SUMO at both the promoter and ORF within 5 min of induction (Fig 2C). SUMO levels gradually dropped with continuous exposure to copper. Since a change in SUMO levels at CUP1 was observed in ulp2Δ but not WT cells, local sumoylation must normally be tightly regulated by Ulp2 protease during transcription. In line with the Rpb3 ChIP results, levels of CUP1 mRNA increased strikingly at 10 min following the addition of copper to WT cells, but this was strongly attenuated in ulp2Δ cells (Fig 2D). Similarly, Rpb3 occupancy was reduced and, conversely, SUMO levels were enhanced in ulp2Δ at the constitutive test genes PMA1, ADH1, and PYK1 (Fig 2E and F). Taken together, the results indicate that localization of RNAPII and SUMO is tightly regulated by Ulp2 on transcribed genes with significant impact on transcription.
The C‐terminal domain of Ulp2 is important for transcription elongation
We previously found that Ulp2 has a poorly conserved N‐terminal domain (NTD) adjacent to the catalytic domain, but the NTD is nonetheless necessary and sufficient for nuclear localization and is required for most Ulp2 functions (Kroetz et al, 2009). By contrast, deletion of the C‐terminal domain (CTD) has more modest effects on cell growth, and the C‐terminally truncated protein still localizes to the nucleus. To determine the potential contribution of the NTD and CTD of Ulp2 to transcription elongation, we examined the 6‐AU sensitivity of the ULP2 strain and mutants expressing Ulp2ΔN, Ulp2ΔC, and the NTD of Ulp2, all from the native chromosomal locus (Fig 3A). Expression of the Ulp2 derivatives was undiminished relative to full‐length Ulp2 (Fig 3B). The ulp2ΔN strain and cells expressing just the NTD showed severe growth defects on SD‐Ura, and they were almost inviable on the plate with 6‐AU, suggesting that both nuclear localization and the catalytic domain of Ulp2 are likely required to overcome the stress caused by exposure to this drug. Remarkably, whereas ulp2ΔC cells grew only slightly slower than congenic WT cells on SD‐Ura, they displayed very strong sensitivity to 6‐AU. The CTD of Ulp2 was known to be required for efficient depolymerization of large polySUMO conjugates (Kroetz et al, 2009) and for nucleolar substrate targeting (de Albuquerque et al, 2018); these new data reveal a potential function of the CTD in transcription elongation.
Figure 3. Ulp2 C‐terminal domain is required for efficient association with nucleosomes.
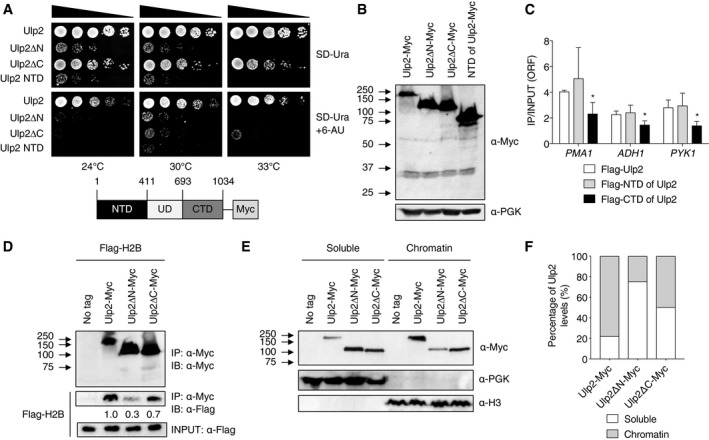
- Sensitivity to 6‐AU of the indicated cells expressing chromosomally integrated ULP2‐Myc derivatives. Assays were performed as in Fig 2A. The schematic diagram below shows full‐length Ulp2 with a 9Myc epitope tag and the segments defining the N‐ and C‐terminal domains and the catalytic ULP domain (UD).
- Immunoblot assay of the Ulp2‐Myc derivatives expressed in the strains used in (A).
- ChIP analysis of Flag‐Ulp2 derivatives performed with the same PCR probes used in Fig 2E. W303a cells carrying pRS424‐GAL1 plasmids that expressed N‐terminally Flag‐tagged Ulp2 derivatives—specifically, full‐length, NTD (1‐403), and CTD (667–1,034)—were grown to mid‐log phase in SD‐Trp medium containing 2% galactose. Error bars indicate the SD from three independent experiments.
- Co‐IP assay for interaction between Ulp2‐Myc derivatives and Flag‐H2B in strains from (A) transformed with a pRS314 plasmid expressing Flag‐tagged H2B. Immunoprecipitated (IP) proteins from anti‐Myc agarose beads were analyzed by immunoblotting (IB) using anti‐Myc or anti‐Flag antibodies. The upper and middle panels show anti‐Myc blotting and anti‐Flag blotting, respectively. The levels of bound H2B proteins were measured relative to INPUT H2B levels shown in the lower panel.
- Chromatin association assays using strains from (A). After fractionation into chromatin (pellet) and soluble (supernatant) components, fractions were analyzed by immunoblotting using the indicated antibodies.
- Quantitation of chromatin association of Ulp2‐Myc derivatives in (E). The levels of Ulp2 protein in each fraction were measured by ImageJ, and then, each signal was divided by the total values for each Ulp2 derivative.
To determine whether the Ulp2 CTD or NTD binds chromatin, we compared binding of Flag‐tagged full‐length, NTD and CTD versions of Ulp2 to constitutively active genes (Fig 3C). The NTD alone bound comparably to the full‐length Ulp2 protein at these sites, revealing a previously unknown function for the NTD. The CTD by itself showed little if any binding at any of the loci. We also analyzed the association between Ulp2 derivatives and nucleosomes in strains with Myc‐tagged ULP2, ulp2ΔN, and ulp2ΔC alleles and carrying a plasmid expressing Flag‐tagged histone H2B; interactions were tested by co‐immunoprecipitation (co‐IP) analysis (Fig 3D). Relative to WT ULP2, both the ulp2ΔN and ulp2ΔC strains exhibited reduced Ulp2 interaction with H2B; loss of the CTD had a milder effect than did loss of the NTD. As an alternative to the above co‐IP analysis, we carried out chromatin association assays in strains expressing Myc‐tagged Ulp2, ulp2ΔN, and ulp2ΔC proteins (Fig 3E and F). Separation of soluble proteins from those that bind chromatin was verified by immunoblotting using anti‐PGK (soluble) and anti‐H3 (chromatin), respectively. About 80% of full‐length Ulp2 partitioned into the chromatin fraction. Loss of either the Ulp2 CTD or NTD substantially reduced Ulp2 localization to the chromatin fraction, paralleling the histone H2B co‐IP data. These results suggest that the Ulp2 CTD contributes to, but is not essential for, Ulp2‐chromatin binding; its function in transcription may be related to the SUMO‐interacting motif (SIM) in this domain or one of two other conserved motifs in the CTD (Kroetz et al, 2009; de Albuquerque et al, 2018).
Polysumoylated histones are substrates of Ulp2
The co‐IP data in Fig 3D showed Ulp2 protein interacts physically with histone H2B in vivo, congruent with a previous report that sumoylated histones H3 and H4 are substrates of Ulp2 (Nathan et al, 2006). We therefore tested the level of sumoylated histone H2B in WT and ulp2Δ strains (Fig 4A, lanes 4 and 6). Compared to WT cells, ulp2Δ contains slightly lower levels of monosumoylated H2B (*) but much higher levels of presumptive polysumoylated H2B, which accumulated at the top of the resolving gel and in the stacking gel (***). This result suggests that Ulp2 is responsible for processing polySUMO chains attached to histones. Nathan and colleagues previously observed only monosumoylated histones accumulating in ulp2Δ cells compared to WT (Nathan et al, 2006). This difference from our results is likely due to different purification methods. We immunoprecipitated cell lysates prepared by denaturing TCA precipitation, thereby preserving most sumoylated species. In the previous study, polysumoylated histones in the ulp2Δ cell samples might have been processed into monosumoylated species by the Ulp1 SUMO protease during chromatin fractionation steps.
Figure 4. Histone ubiquitylation and sumoylation and Ulp2 function.
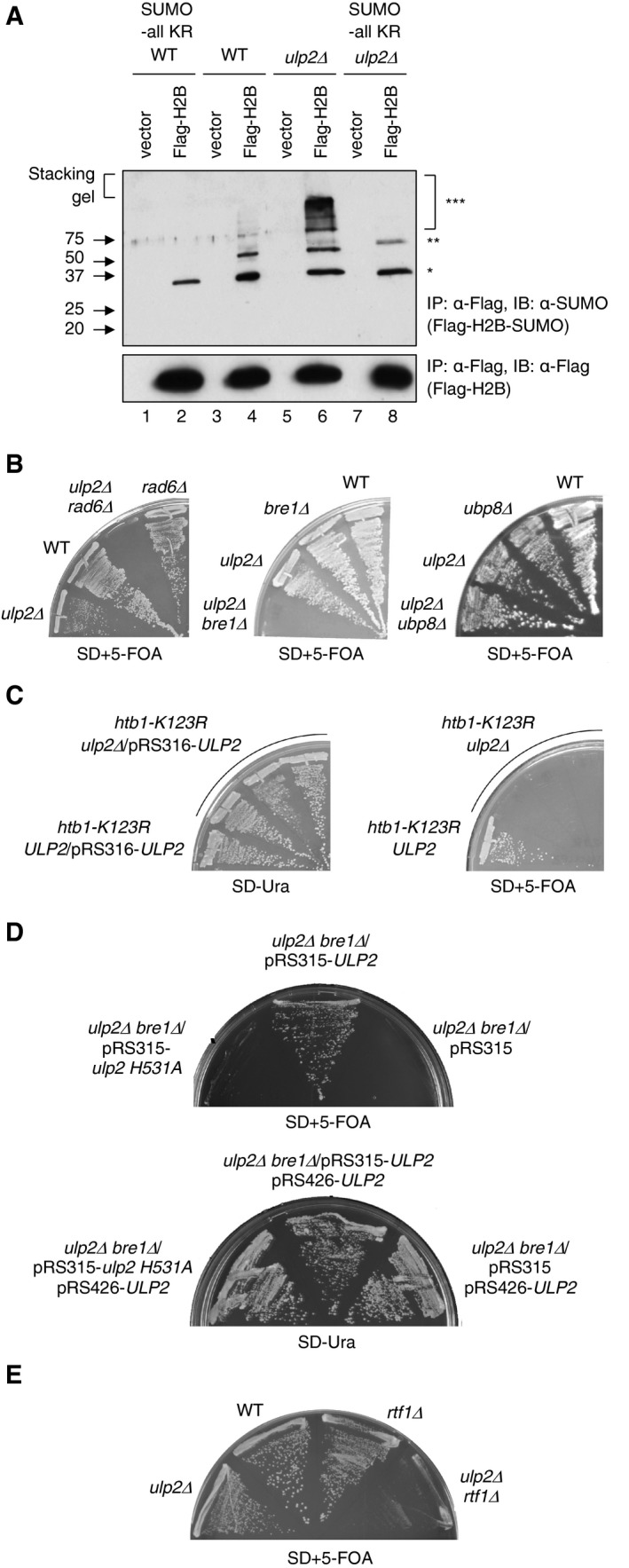
- Immunoblot analysis of immunoprecipitated Flag‐tagged histone H2B with anti‐Flag (bottom panel) or anti‐SUMO (upper panel) antibodies in WT, ulp2Δ, SUMO‐all KR, or ulp2Δ SUMO‐all KR strains carrying empty vector or pRS426‐FLAG‐HTB1. One, two, or three asterisks represent mono‐, di‐, and polysumoylated histones, respectively. Note that the apparent diSUMO‐H2B species (**) in ulp2Δ SUMO‐all KR cells migrates more slowly than H2B linked to a SUMO dimer, suggesting that this species likely represents a low level of H2B with SUMO attached to two different histone lysines (lane 8; also see weaker band in lane 6).
- Yeast strains of the indicated genotypes and carrying a YCplac33‐ULP2 cover plasmid were streaked on SD+5‐FOA to evict YCplac33‐ULP2; cells were grown at 30°C for 3 days.
- Mutant htb1‐K123R ulp2Δ and htb1‐K123R ULP2 strains with a pRS316‐ULP2 cover plasmid were grown on the indicated plates at 30°C for 2–3 days. Three double mutant isolates were tested in parallel.
- Mutant ulp2Δ bre1Δ strains carrying a pRS426‐ULP2 (URA3) cover plasmid and the indicated plasmids were grown on SD‐Ura and SD+5‐FOA at 30°C for 2–3 days.
- Yeast strains of the indicated genotypes and carrying a YCplac33‐ULP2 cover plasmid were streaked on SD+5‐FOA to evict YCplac33‐ULP2; cells were grown at 30°C for 3 days.
Source data are available online for this figure.
To test whether the high‐molecular‐weight (HMW) SUMO‐conjugated species were H2B proteins with multiple monoSUMO additions or polySUMO chains, we tested the sumoylation of histone H2B in ulp2Δ cells expressing SUMO‐allKR, in which all the SUMO lysines were substituted with arginine, which prevents SUMO polymerization (Fig 4A, lanes 2 and 8). Strikingly, HMW sumoylated H2B was undetectable in both WT and ulp2Δ cells bearing the SUMO‐allKR allele, strongly suggesting that these HMW species are polySUMO chains attached to H2B. It is unlikely that the SUMO‐allKR is simply unable to conjugate to any protein since the monosumoylated protein is still readily detected, consistent with earlier work (Bylebyl et al, 2003).
Mutations in ULP2 and RAD6 or BRE1 are synthetically lethal
In a previous genomic synthetic interaction screen with ulp2Δ, we characterized esc1Δ as a ulp2Δ suppressor; Esc1 is a nuclear envelope protein important for Ulp1 localization to nuclear pore complexes (Lewis et al, 2007). From the same screen, we also had identified deletions of two genes, RAD6 and BRE1, that had the apparent opposite effect, a synthetic lethal interaction with ulp2Δ. Rad6 and Bre1 are the E2 and E3 enzymes, respectively, responsible for histone H2B monoubiquitylation. To confirm our preliminary observations, tetrad segregants from crosses between ulp2Δ [pRS316‐ULP2] and rad6Δ or bre1Δ strains were streaked on plates containing 5‐fluoroorotic acid (5‐FOA) to evict the covering ULP2 plasmid (Fig 4B). Disruption of RAD6 or BRE1 was indeed lethal when combined with ulp2Δ. Consistent with the hypothesis that histone H2B was the critical ubiquitylation target responsible for this genetic interaction, the htb1‐K123R mutation, which specifically eliminates H2B ubiquitylation, was also lethal in ulp2Δ cells (Fig 4C). By contrast, combining a deletion of the gene encoding the histone H2B‐ubiquitin protease Ubp8 did not alter the growth defect of cells lacking ULP2 (Fig 4B, right panel).
Loss of Ulp2 catalytic activity was sufficient for the synthetic interaction with bre1Δ (Fig 4D). Specifically, cells with the ulp2‐H531A catalytic mutation, which express the inactive enzyme at levels similar to wild type (Schwartz et al, 2007), were synthetically lethal with bre1Δ. Furthermore, disruption of RTF1, encoding a component of the RNAPII‐associated chromatin remodeling PAF complex, which promotes H2B ubiquitylation (Van Oss et al, 2016), was also lethal in combination with ulp2Δ (Fig 4E). These data strongly imply that histone H2B ubiquitylation and Ulp2 have overlapping functions in the regulation of transcription.
Histone H2B ubiquitylation promotes histone sumoylation
Based on the results in Fig 4, we tested whether histone H2B ubiquitylation might alter histone‐SUMO conjugate levels or vice versa. First, we examined levels of SUMO‐conjugated H2B in rad6Δ, bre1Δ, and ubp8Δ mutants (Fig 5A, upper panel). Levels of H2B with one (*) or two (**) attached SUMO molecules, which are readily detected in WT cells, were strongly reduced in the ubiquitin E2 and E3 mutants (lanes 2 and 3), which eliminate the ubiquitin‐H2B conjugate (lower panel, arrowhead). Histone H4 sumoylation was also examined and yielded results similar to those seen with H2B (Fig EV2A). When we examined global levels of SUMO conjugates in the same mutants (Fig 5B), loss of RAD6 or BRE1, but not UBP8, led to diminished levels only of relatively low‐molecular‐weight SUMO conjugates (below ~80 kDa). These are likely to represent, at least in part, histone‐SUMO conjugates.
Figure 5. Histone H2B ubiquitylation promotes histone sumoylation.
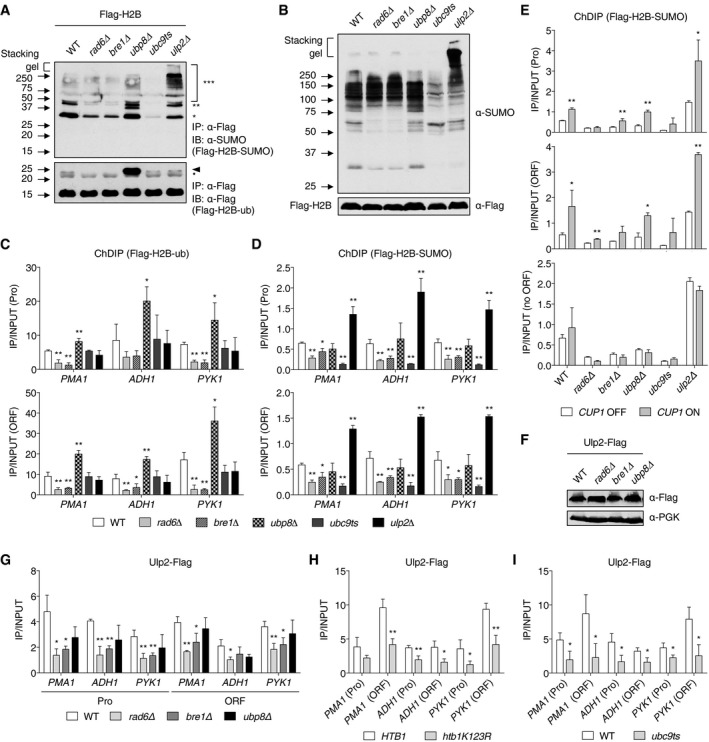
-
AImmunoblot analysis of immunoprecipitated Flag‐tagged histone H2B using anti‐Flag or anti‐SUMO antibodies in the indicated mutants, as described in Fig 4A. The upper and lower panels show Flag‐H2B‐SUMO and Flag‐H2B‐ub, respectively. One, two, or three asterisks indicate mono‐, di‐, and polysumoylated histones (upper panel), respectively, and an arrowhead and bullet point represent mono‐ubiquitylated H2B, and a non‐specific band, respectively.
-
BImmunoblot assay of sumoylated proteins in extracts prepared from the strains in (A). Anti‐Flag blotting for Flag‐H2B was used to verify the similar loading.
-
CChromatin double immunoprecipitation (ChDIP) of Flag‐H2B‐ub in the indicated strains expressing Flag‐H2B. The first ChIP was performed with anti‐Flag agarose, and the eluted samples were immunoprecipitated with an antibody against ubiquitin. PCR signals from the indicated genes were normalized to an internal control and the input DNA. The error bars indicate the SD from three independent chromatin samples.
-
DChDIP of Flag‐H2B‐SUMO in the indicated strains expressing both Flag‐H2B and HA‐SUMO. Chromatin samples were sequentially immunoprecipitated with anti‐HA agarose followed by anti‐Flag agarose. PCR signals were normalized to the input DNA. The error bars indicate the SD from three independent experiments.
-
EChDIP of Flag‐H2B‐SUMO in the indicated mutant strains during CUP1 gene induction, analyzed as in (D). The CUP1 induction was performed as in Fig 1F. CUP1 OFF and ON indicate uninduced and induced (5 min) conditions, respectively. The error bars represent the SD from three independent ChDIP assays.
-
FImmunoblot assay of Flag‐tagged Ulp2 in the indicated strains. Anti‐PGK blotting shows similar loading.
-
G–IChIP assays in the indicated strains expressing Flag‐tagged Ulp2 as in Fig 1E. The error bars indicate the SD from three experiments.
Figure EV2. The analysis of histone sumoylation in mutants.
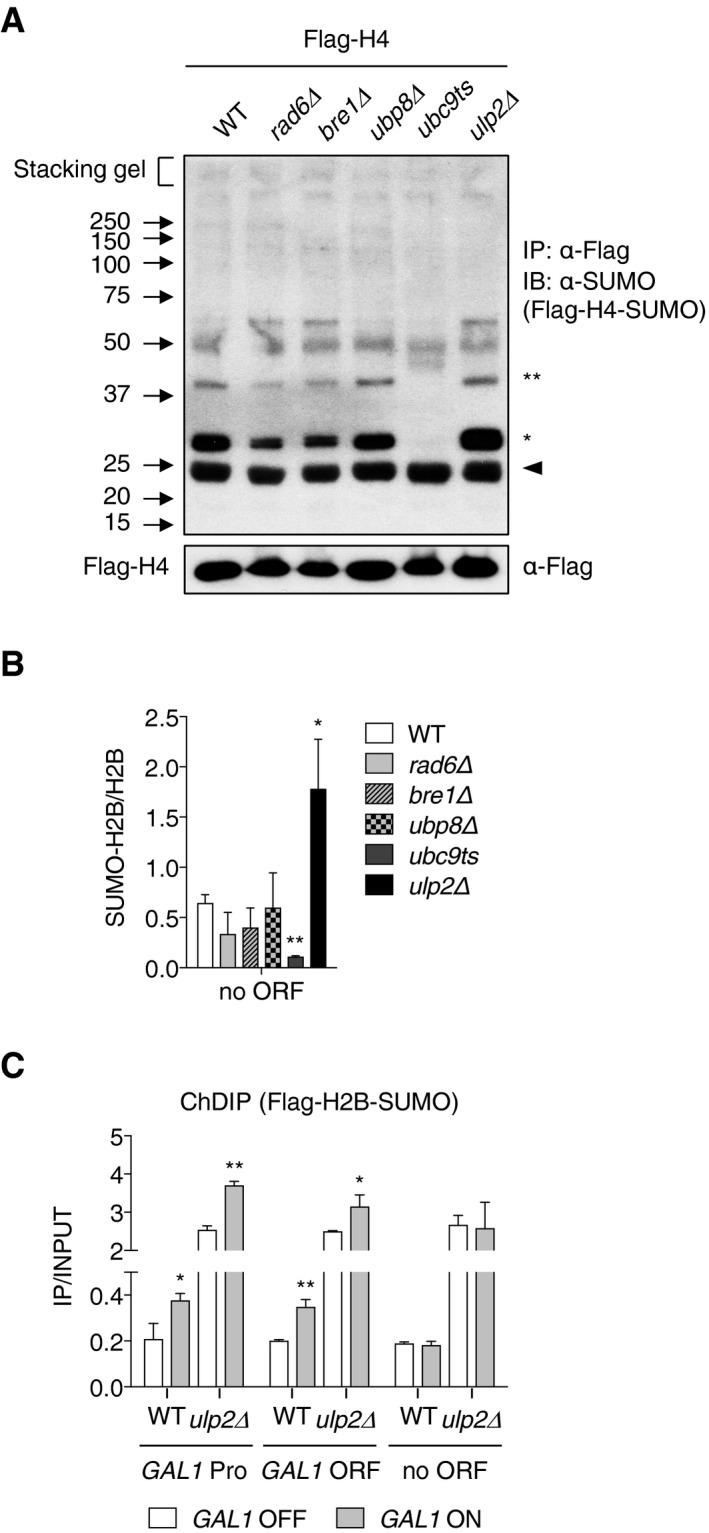
- Immunoblot analysis of immunoprecipitated Flag‐tagged histone H4 using anti‐SUMO antibodies in the indicated mutants, as described in Fig 4A. The upper and lower panels show Flag‐H4‐SUMO and Flag‐H4 (loading control), respectively. One and two asterisks indicate mono‐ and di‐sumoylated histones (upper panel), respectively, and the arrowhead represents a non‐specific band.
- ChDIP of Flag‐H2B‐SUMO in the indicated strains at a nontranscribed site (“no ORF”) as in Fig 6D. Error bars indicate the SD from three independent assays.
- ChDIP of Flag‐H2B‐SUMO in WT and ulp2Δ strains during GAL1 gene induction, described as in Fig 5D. For GAL1 induction, cells grown in SD‐Trp medium with 2% glucose were shifted to SD‐Trp medium containing 2% raffinose. After 2 h, the medium was replaced with SD‐Trp medium containing 2% galactose and then incubated for 1 h. GAL1 OFF and ON indicate uninduced and induced conditions, respectively. The error bars represent the SD from three ChDIP assays.
To verify that the bulk in vivo histone ubiquitylation and sumoylation changes in Fig 5A reflected alterations within chromatin, we performed chromatin double immunoprecipitation (ChDIP) experiments to investigate these modifications of histone H2B at specific loci (Fig 5C and D). Genome‐wide studies indicate that H2B ubiquitylation is principally enriched in the ORF regions of almost all active genes and correlates with transcriptional activity (Shieh et al, 2011; Bonnet et al, 2014). We tested ubiquitylation of Flag‐tagged histone H2B by sequential ChIP using anti‐Flag antibody‐conjugated beads in the first round and an antibody against ubiquitin in the second round, as previously used (Chandrasekharan et al, 2009). As expected, we observed slightly higher levels of ubiquitin‐H2B in ORF regions of the highly transcribed test genes compared to promoters in WT cells. Levels of H2B ubiquitylation were significantly decreased at these sites in both rad6Δ and bre1Δ strains, and the opposite was true in cells lacking UBP8 (Fig 5C). Mutations in SUMO pathway components UBC9 or ULP2 appeared to have no effect on H2B ubiquitylation at these genes.
Because we failed to find a SUMO‐specific antibody suitable for ChDIP, we generated an htb1‐1 htb2‐1 smt3Δ triple mutant (which lacked the chromosomal genes for histone H2B and SUMO) that carried low‐copy plasmids expressing Flag‐tagged H2B and HA‐tagged SUMO. Sumoylated H2B levels were evaluated by ChDIP at the three test genes using anti‐Flag beads and then anti‐HA beads (Fig 5D). Whereas levels of sumoylated H2B were reduced in ubc9ts cells, ULP2 loss increased the ratio of sumoylated H2B forms to total H2B at both promoters and ORFs by twofold to fourfold. Remarkably, the occupancy of sumoylated H2B at the tested loci was also significantly decreased in the rad6Δ and bre1Δ strains.
Taken together, our results indicate that the promoters and ORFs of actively transcribed genes are general sites of histone sumoylation, and furthermore, that H2B ubiquitylation stimulates histone sumoylation, but not the reverse, suggesting a novel sequential histone modification pathway associated with transcription.
Histone sumoylation is linked with transcriptional activation
Despite these links to transcription, we also observed changes in H2B sumoylation at untranscribed intergenic regions in response to mutations in Ulp2 and Ubc9 (Fig EV2B). These ChDIP data and previous reports (Nathan et al, 2006) demonstrating that sumoylated histones were not restricted to transcribed genes make the role of this modification in transcription uncertain. To determine whether H2B sumoylation levels correlate with changes in transcriptional activity, we evaluated changes in SUMO‐H2B occupancy at promoter and ORF sites of CUP1 during copper‐mediated activation of its transcription and compared it to a transcriptionally silent site (“no ORF”) (Fig 5E). In WT cells, but not ubc9ts cells, sumoylated H2B levels increased significantly in the activated chromatin of both the CUP1 promoter and ORF, compared with the uninduced condition. Sumoylated H2B levels did not change significantly in the “no ORF” genomic region during CUP1 induction, suggesting that relative histone sumoylation levels are stimulated during local transcription activation. We also confirmed significantly increased occupancy of SUMO‐H2B at the induced GAL1 promoter and ORF, but not at an untranscribed region (Fig EV2C). As we had observed at constitutively expressed genes (Fig 5D), much higher amounts of SUMO‐H2B were detected at the induced CUP1 and GAL1 loci in cells lacking ULP2 (Figs 5E and EV2C). Together, these results provide evidence that SUMO conjugation to histones is linked to gene expression status.
Histone H2B ubiquitylation is required for Ulp2 localization at active genes
These results prompted us to investigate the correlation between histone H2B ubiquitylation and Ulp2 SUMO protease localization. First, we examined the association of Ulp2 with actively transcribed genes in cells lacking RAD6, BRE1, or UBP8 (Fig 5G). Ulp2 recruitment to the promoters and ORFs of the three test genes, PMA1, ADH1, and PYK1, was significantly decreased by loss of RAD6 but not UBP8. Loss of BRE1 led to ChIP results similar to the rad6Δ data. Deletion of RAD6 or BRE1 did not affect overall Ulp2 protein levels (Fig 5F), arguing instead that rad6Δ and bre1Δ suppressed Ulp2‐chromatin association.
To test directly whether it was the loss of histone H2B ubiquitylation that reduced Ulp2 binding to chromatin, we used ChIP analysis to compare Ulp2 binding between cells expressing WT HTB1 or htb1‐K123R; the latter strain lacks the H2B lysine ubiquitylated by Rad6‐Bre1 (Fig 5H). The results were very similar to those with rad6Δ and bre1Δ in Fig 5G. These data indicate that H2B ubiquitylation drives Ulp2 recruitment to constitutively expressed genes.
Because our results above suggested histones are sequentially modified by ubiquitin and SUMO during transcription, we speculated that the diminished association of Ulp2 with the tested genes, which resulted from the block to H2B ubiquitylation, was due more proximately to reduced histone sumoylation. Therefore, we performed ChIP assays (Fig 5I) to examine the association of Ulp2 with these same genes in a ubc9ts mutant, which largely eliminates SUMO conjugation to H2B (Fig 5A). As predicted, association of Ulp2 with active genes was significantly diminished by mutation of UBC9. These data suggest that sumoylation of histones and/or other chromatin factors is essential for Ulp2 recruitment to active chromatin.
Ulp2 promotes recruitment of Ctk1 kinase to both SAGA‐ and TFIID‐dominated genes
Histone H2B ubiquitylation plays an important role in transcription, and its level is dynamically regulated by ubiquitin hydrolases Ubp8 and Ubp10 (Henry et al, 2003; Daniel et al, 2004; Emre et al, 2005). During transcriptional activation, Ubp8 removes ubiquitin from H2B to stimulate recruitment of the CTD‐S2 kinase Ctk1 to nucleosomes (Wyce et al, 2007). Given the results in the preceding section, we tested for effects of Ulp2 in H2B ubiquitylation‐dependent transcription pathways. To begin, we performed ChIP analysis of S2‐phosphorylated RNAPII levels using PCR probes to the 3′ ORF regions of constitutively active genes (Fig 6A). Consistent with a previous study that reported CTD‐S2 hypophosphorylation at the active GAL1 gene in ubp8Δ cells (Wyce et al, 2007), we detected a significantly reduced fraction of the CTD in its S2 phosphorylated (S2‐P) state at the tested genomic regions in cells lacking UBP8. More importantly, the fractional CTD S2‐P level was also significantly reduced by loss of ULP2. A strong functional connection between Ulp2 and CTD‐S2‐P is supported by a synthetic lethal interaction between ulp2Δ and ctk1Δ (Fig 6B). These results lead us to suggest that Ulp2 controls CTD S2‐P levels during transcript elongation by RNAPII.
Figure 6. Ctk1 recruitment and CTD S2‐P of RNAPII are diminished in ulp2Δ cells.
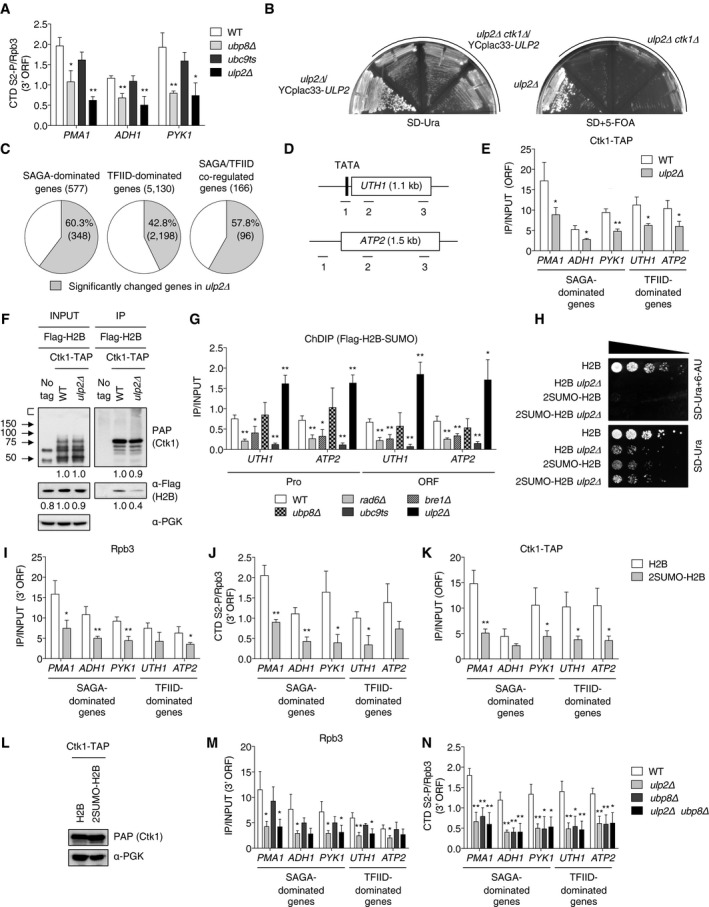
-
AThe ratio of CTD S2‐P relative to Rpb3 at 3′ ORF regions was analyzed by ChIP using anti‐S2‐P and anti‐Rpb3 antibodies. The error bars represent the SD from triplicate experiments.
-
BA ulp2Δ single mutant and three isolates of ulp2Δ ctk1Δ with YCplac33‐ULP2 were grown on SD‐Ura or SD+5‐FOA plates at 30°C for 2–3 days.
-
CPie graphs showing percentages of genes in ulp2Δ cells (gray color) with significantly changed expression compared with WT (MHY1379). RNA‐seq data were subdivided into groups of genes regulated by SAGA, TFIID, or both factors based on a previous report (Huisinga & Pugh, 2004). Gene numbers are shown in parentheses.
-
DSchematic diagram of TFIID‐dominated genes UTH1 and ATP2. The TATA/promoter region and ORF are represented by black and white boxes, respectively. Lines and numbers below the genes show that the relative positions of the ChIP PCR products used in all later panels.
-
EChIP analyses of Ctk1‐TAP in the indicated strains. The ORF of TFIID‐dominated genes indicates position 2 of PCR fragments from the UTH1 and ATP2 genes shown in (D). Error bars represent the SD from triplicate experiments.
-
FCo‐IP analysis of Ctk1 and histone H2B interaction. WT and ulp2Δ strains expressing the indicated tagged proteins were precipitated with IgG‐Sepharose. IP and INPUT proteins were subjected to immunoblotting with PAP (Ctk1‐TAP) and anti‐Flag (Flag‐H2B) antibodies. Protein signals were quantified relative to PGK and then normalized to the WT value.
- G
-
HSensitivity to 6‐AU of indicated htb1‐1 htb2‐1 (null alleles for both chromosomal H2B genes) strains carrying pRS314 plasmids that express Flag‐H2B or 2SUMO‐H2B. Assays were performed as in Fig 2A.
-
I, JChIP analyses of the indicated strains used in (H). Occupancy of Rpb3 (I) and the ratio of CTD S2‐P relative to Rpb3 (J) at 3′ ORF regions of the indicated genes were individually analyzed by ChIP, as described in Fig 1E and (A). Error bars show SD from three independent analyses.
-
KChIP analyses of Ctk1‐TAP in the indicated strains. The ORF of TFIID‐dominated genes indicates position 2 of PCR fragments from the UTH1 and ATP2 genes shown in (D). Error bars represent the SD from triplicate experiments.
-
LImmunoblot analysis of TAP‐tagged Ctk1 proteins in extracts prepared from the strains in (K). Anti‐PGK blotting shows similar loading.
- M, N
The H2B ubiquitin hydrolase Ubp8 is part of the DUB (deubiquitylating) module (Henry et al, 2003) in the general transcriptional coactivator complex called SAGA; SAGA shares several subunits with another general factor, TFIID (Grant et al, 1998). A genome‐wide expression study previously suggested that TFIID and SAGA contribute to the expression of ~90% and ~10% of all yeast genes, respectively (Huisinga & Pugh, 2004). In our transcriptome analysis of WT and ulp2Δ cells, loss of Ulp2 led to significantly altered expression of 60%, 43%, and 58% of genes whose expression is dominated by SAGA, TFIID, or both factors, respectively (Fig 6C). This suggests Ulp2 has broad effects on RNAPII transcription that go beyond this classification of gene types.
The previously analyzed test genes (PMA1, ADH1, and PYK1) were all SAGA‐dominated, so we decided to also examine TFIID‐dominated genes, UTH1 and ATP2 (Fig 6D). As predicted from our RNA‐seq data, ChIP analysis of ulp2Δ cells revealed significantly diminished Ctk1 recruitment to both SAGA‐ and TFIID‐dominated (Fig 6E). In support of the ChIP data, we found by co‐IP analysis in cells coexpressing Ctk1‐TAP and Flag‐H2B that Flag‐H2B was consistently co‐precipitated with Ctk1‐TAP, but the amount of coprecipitating Flag‐H2B was reduced by ~60% in ulp2Δ compared to WT cells (Fig 6F). We conclude that Ulp2 promotes nucleosomal binding of Ctk1 and limits SUMO‐histone accumulation at both SAGA‐dependent and SAGA‐independent genes.
Histone sumoylation impedes CTD‐S2 phosphorylation
We documented histone sumoylation not only at SAGA‐dominated genes (Fig 5D) but also at TFIID‐dominated genes (Fig 6G). At both locus types, SUMO‐H2B was suppressed by mutation of RAD6, BRE1, or UBC9, and strongly enhanced in cells lacking ULP2.
As a test for whether the inhibition of CTD‐S2 phosphorylation in ulp2Δ cells was due to accumulation of polySUMO‐conjugated histones, we generated yeast strains bearing plasmids expressing 2SUMO‐fused H2B, in which two SUMO molecules were translationally linked in tandem to the N‐terminus of H2B. The two SUMOs lacked the Gly‐Gly motif required for SUMO protease processing, which could mimic the failure to deconjugate sumoylated histones in ulp2Δ cells. Indeed, 2SUMO‐H2B slowed yeast growth to a degree comparable to the ULP2 deletion (Fig 6H). No additional growth defect was observed when 2SUMO‐fused H2B was expressed in ulp2Δ cells expressing 2SUMO‐fused H2B, and WT cells expressing 2SUMO‐fused H2B showed strong sensitivity to 6‐AU. These results are consistent with the accumulation of polySUMO‐conjugated histones being a major cause of the growth defects and 6‐AU sensitivity in ulp2 mutants.
When we measured ORF occupancy of RNAPII subunit Rpb3 or the fraction of the RNAPII CTD with S2‐P at these sites in cells stably expressing 2SUMO‐fused H2B, we observed diminished Rpb3 occupancy and low levels of CTD S2‐P at all tested SAGA‐dominated genes and at least one tested TFIID‐dominated gene (Fig 6I and J). Also, a defect in the recruitment of Ctk1 was observed in cells expressing 2SUMO‐fused H2B without changes in Ctk1 protein levels (Fig 6K and L). 2SUMO‐fused H2B thus appeared to phenocopy the ulp2Δ mutant (Figs 2E and 6A and E). Taken together, the results suggest that persistent histone sumoylation limits recruitment of RNAPII and the Ctk1 kinase to genes regulated principally by either SAGA or TFIID.
Ulp2 and Ubp8 function in the same pathway to control CTD S2‐P
Recent genome‐wide localization studies revealed that the SAGA complex is present on most active genes and the DUB activity of SAGA is detected on almost all yeast genes (Bonnet et al, 2014; Baptista et al, 2017). Furthermore, upon mutation or depletion of SAGA subunits, recruitment of RNAPII even to “TFIID‐dominated” genes was significantly impaired, as was synthesis of new mRNA (Bonnet et al, 2014; Baptista et al, 2017). This suggests SAGA is a more general cofactor for RNAPII‐mediated transcription than previously thought (Huisinga & Pugh, 2004). We addressed the potential association of Ubp8, which is a part of the SAGA complex, as well as that of Ulp2, with so‐called TFIID‐dominated genes (UTH1 and ATP2) using ChIP assays (Fig EV3). As was found with the SAGA‐dominated PMA1 gene, recruitment of TAP‐tagged Ubp8 was observed at all the analyzed regions. Binding of Ulp2 enzyme also was seen at these chromatin sites. Our results therefore concur with the recent reports indicating the SAGA complex is likely involved in transcription of both SAGA‐ and TFIID‐dominated genes.
Figure EV3. Ubp8 (SAGA) is also associated with TFIID‐dominated genes.
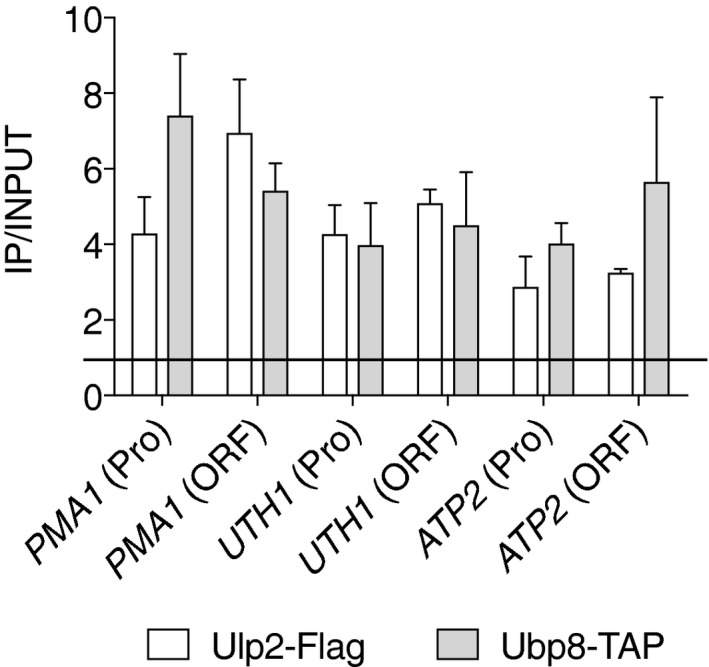
ChIP analysis using anti‐Flag agarose or IgG‐Sepharose beads in strains with the indicated tags, analyzed as in Fig 1E. The qPCR signals in the promoter (Pro) or ORF of PMA1, UTH1, and ATP2 were quantitated and normalized to an internal background control and input DNA. Horizonal line indicates value of 1, the background signal. Error bars indicate SDs calculated from four ChIP assays. See Dataset EV2 for qPCR raw data.
To determine whether Ulp2 and Ubp8 function in the same Ctk1‐regulated transcription pathway, we compared levels of Rpb3 and CTD S2‐P at the test‐gene ORFs in WT, ulp2Δ, ubp8Δ, and ulp2Δ ubp8Δ double mutant cells (Fig 6M and N). Rpb3 association with all genes was significantly impaired by loss of ULP2 but not UBP8. The latter finding matches a previous report that Rpb3 recruitment is unaffected by ubp8Δ (Wyce et al, 2007), suggesting a step other than RNAPII association is impaired. The same ORFs displayed low CTD S2‐P levels in both single mutants (Fig 6N). Importantly, we found no additional defects in the ulp2Δ ubp8Δ double mutant, suggesting Ulp2 and Ubp8 regulate CTD‐S2 phosphorylation as part of the same pathway, consistent with growth data (Fig 4B).
Discussion
The results presented here have revealed two previously unknown regulatory steps in transcription elongation in yeast. First, the Ulp2 SUMO protease associates preferentially with active RNAPII‐regulated genes, reducing local (poly)SUMO conjugation and promoting association of RNAPII. Second, sequential histone ubiquitylation and sumoylation are coupled to RNAPII CTD‐S2 phosphorylation. These two sets of events are integrated by the Ulp2 protease. Deconjugation of SUMO from histones by Ulp2 is associated with Ctk1‐mediated S2 phosphorylation of RNAPII, which helps drive transcription elongation.
Considering the results obtained in this study with previous reports, we propose a model of Ulp2‐mediated transcriptional elongation (Fig 7). At the promoter during the transition from transcriptional initiation to elongation, the CTD S5‐P form of RNAPII interacts with the histone H2B ubiquitylation enzymes Rad6‐Bre1 and the Set1/COMPASS histone methylation complex through the PAF complex (Wood et al, 2003) (Fig 7A). This interaction is essential for H2B ubiquitylation and H3K4 methylation (Krogan et al, 2002; Ng et al, 2003; Xiao et al, 2005). Subsequently, the Ubc9 E2 SUMO‐conjugating enzyme (and presumably a SUMO E3 ligase) is loaded onto the RNAPII transcription machinery or nearby, where it catalyzes histone polysumoylation (Fig 7B). Ubc9 directly interacts with Rad6 (Hoege et al, 2002), but its role in transcription has been unknown. The presence of both histone ubiquitylation and sumoylation blocks the recruitment of Ctk1 kinase to nucleosome, preventing further phosphorylation of CTD S2 on RNAPII (Wyce et al, 2007; and our data). The Ulp2 protease is recruited to the transcription machinery through its binding to polySUMO‐modified chromatin proteins (Fig 7C). Histones modified by polySUMO and ubiquitin are deconjugated by Ulp2 and Ubp8 (within the SAGA DUB module), respectively. Upon removal of SUMO and ubiquitin, the Ctk1 kinase can associate with RNAPII and phosphorylate CTD‐S2, promoting transcript elongation along with other elongation factors.
Figure 7. Proposed model for Ulp2 function in transcription elongation.
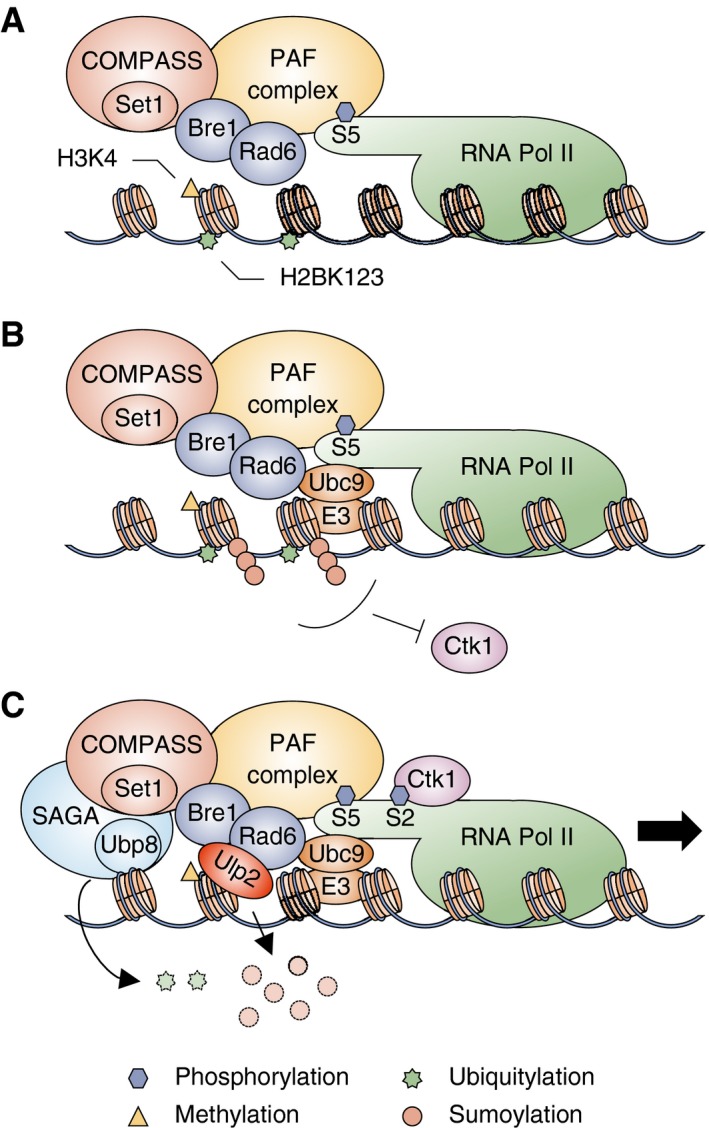
- At the early stages of transcription, the PAF complex and CTD S5‐P form of RNAPII are required for monoubiquitylation of histone H2B on lysine 123 by Rad6‐Bre1. H2B ubiquitylation drives promoter association of the COMPASS/Set1 methyltransferase, which methylates histone H3 on lysine 4.
- Histones are then conjugated to polySUMO chains by Ubc9 and E3 SUMO ligases. Both H2B ubiquitylation and histone sumoylation inhibit nucleosomal binding of Ctk1 kinase.
- Removal of ubiquitin and polySUMO from histones by Ubp8 within SAGA and by Ulp2, respectively, promotes Ctk1 association to phosphorylate S2 within the CTD and subsequently facilitate RNAPII elongation.
Our results reveal that Ulp2 protease is required for full transcriptional activation at both constitutively active and transiently inducible loci. Ulp2 is associated with highly transcribed genes, and its loss impairs maintenance of elongating RNAPII transcription complexes at these sites. Prior work indicated specific localization of Ulp2 at rDNA, RP, and snoRNA loci (Ryu et al, 2016, 2018; Liang et al, 2017). It has been unclear, however, how the enzyme is recruited to chromatin. No known chromatin‐ or DNA‐binding domains are present in the Ulp2 protein sequence. Our results indicate that elimination of histone H2B ubiquitylation inhibits SUMO conjugation to histones, and loss of SUMO‐histone conjugation impedes Ulp2 association with constitutively active genes (Fig 5D and G–I).
These data are consistent with a serial histone modification model for gene activity in which histone H2B ubiquitylation promotes transcription in part through its ability to stimulate histone (poly)sumoylation. This, in turn, is required for Ulp2 localization and ultimately RNAPII CTD‐S2 phosphorylation and transcriptional elongation, although we cannot exclude the possibility that SUMO conjugation to other chromatin‐bound factors is directly required for Ulp2 binding. Notably, histone H2B ubiquitylation also promotes histone H3 methylation at K4 and K79, thereby stimulating local transcription (Briggs et al, 2002; Dover et al, 2002; Sun & Allis, 2002); another histone methylation mark, H3K36 di‐methylation, is reduced by persistent H2B ubiquitylation in ubp8Δ cells (Henry et al, 2003). How histone methylation and sumoylation are jointly regulated by H2B ubiquitylation is currently under investigation (H‐Y Ryu and M Hochstrasser, unpublished data).
Prior to this study, only monosumoylated histones were observed in S. cerevisiae (Nathan et al, 2006). However, our findings show that bulk histones H2B and H4 have not only monoSUMO—but also polySUMO‐conjugated forms in vivo, whose relative levels were specifically regulated by the Ulp2 protease. To analyze sumoylated H2B levels at specific genes, we generated a strain coexpressing Flag‐tagged H2B and HA‐tagged SUMO for ChDIP analysis. However, the ChDIP technique cannot distinguish monoSUMO and polySUMO modifications of H2B. Therefore, at present we do not know for certain the types of SUMO‐histone conjugates present at specific chromatin sites. We also do not know whether SUMO and ubiquitin moieties are simultaneously conjugated to the same H2B polypeptide within the nucleosome or even to the same nucleosome. In addition, SUMO polypeptides can form branched chains and even be subjected to modifications such as ubiquitylation, phosphorylation, and acetylation, potentially producing complex signaling codes (Hendriks & Vertegaal, 2016).
In previous research investigating crosstalk between histone sumoylation and other histone modifications, sumoylation was reported to antagonize acetylation or ubiquitylation events that might occur at the same lysine residues (Shiio & Eisenman, 2003; Nathan et al, 2006). Our findings extend these earlier studies by revealing a new type of crosstalk, in which SUMO does not compete with these transcription‐promoting histone modifications but rather, is part of a dependent histone conjugation and deconjugation sequence during transcription in which both ubiquitin and SUMO play positive roles. This new wrinkle in the “histone code” is closely linked to the RNAPII “CTD code”, in which combinatorial CTD modifications of RNAPII orchestrate the sequential recruitment of co‐activators, co‐repressors, or other factors during transcription (Srivastava & Ahn, 2015).
We have shown here that Ulp2 is involved in the transcription of both SAGA‐ and TFIID‐dominated genes. On the other hand, our genome‐wide study of Ulp2‐chromatin association shows that Ulp2 is not localized to all genes (for example, Fig EV1B), nor is transcription of all genes altered in ulp2Δ cells. Histone sumoylation in metazoans has important roles in transcription through its interactions with other epigenetic marks, such as trimethylation of H3K9 and H4K20, LSD1‐CoREST complex‐mediated demethylation of H3K4, and histone deacetylation by HDAC1 in human cells (Shiio & Eisenman, 2003; Metzler‐Guillemain et al, 2008; Dhall et al, 2017); however, genome‐wide analyses of histone‐specific sumoylation have not yet been performed. Future genome‐wide chromatin studies in yeast that catalog fully those genes subject to histone sumoylation and Ulp2‐dependent transcriptional regulation will be informative. Such results should provide insights into the broader roles of the histone sumoylation network in transcription and chromatin organization in higher eukaryotes as well.
Materials and Methods
Yeast strains
Yeast strains used in this study are listed in Table EV1. Standard techniques were used for strain construction. To generate the MHY10241 strain, the C‐terminal tagging cassette from pFA6a‐6xGly‐3xFlag::kanMX4 (Funakoshi & Hochstrasser, 2009) was amplified by PCR and inserted into the RAD6 locus; for MHY10243, MHY10525, and MHY10526 strains, a tagging cassette was amplified from the CTK1‐TAP locus in the tagged yeast strain from Open Biosystems. Insertion cassettes were separately transformed into MHY500 (WT) or ulp2Δ::kanMX4 cells bearing a YCplac33‐ULP2 plasmid. MHY4120 was made by substitution of the codons for lysine 123 in HTB1 and HTB2 to arginine codons in a W303α strain (MHY1396). The mutations were introduced into primers used to amplify tagging cassettes from pFA6a‐3HA‐TRP1 and pFA6a‐13Myc‐kanMX, respectively (Longtine et al, 1998), and the PCR products were transformed into W303α. To make MHY4121 and MHY4122, MHY4120 was crossed with congenic ulp2Δ::HIS3 cells containing pRS316‐ULP2, and the histone substitution mutant and the triple mutant strain with the ULP2 plasmid were identified by tetrad analysis.
Diploid cells from a cross between GBY12 (MATa smt3‐allKR::TRP1 ulp2Δ::URA3) (Bylebyl et al, 2003) and the congenic JD53 (MATα) strain (Dohmen et al, 1995) were sporulated, and tetrads were dissected to generate MHY7212‐7215. To make MHY10244, the pRS314‐FLAG‐HTB1 CEN/TRP1 plasmid was transformed into MHY4027 and to make MHY10519, pRS314‐FLAG‐HHF1 and pRS317‐HHT1 plasmids were transformed into MHY4259, and then, the URA3‐marked YCp50‐HTB1 plasmid and YCp50‐copyII (HHT2‐HHF2) plasmids, respectively, were evicted by two consecutive streakings on SD plates containing 5‐fluoroorotic acid (5‐FOA, 1 g/l). To create MHY10250, endogenous SMT3 in MHY10244 was replaced by a HIS3 disruption cassette from MHY1538 (Hannich et al, 2005) after transformation with pRS425‐GPD‐HA‐SMT3. To generate MHY10256 and MHY10257, the pRS314‐HTB1 or pRS314‐htb1K123R plasmid was first transformed into MHY4027 followed by replacement of ULP2 with kanMX4; the resident YCp50‐HTB1 plasmid was evicted on 5‐FOA medium, and pRS316‐ULP2‐FLAG was then transformed into the cells. The ubc9ts mutants were made by replacing the chromosomal UBC9 gene with NAT or TRP1 markers amplified from yDS374 (Schwartz et al, 2007) or MHY3997 (Seufert et al, 1995) yeast DNA, respectively, after having transformed pRS416‐GPD‐ChFP‐ubc9ts into the cells. The rad6, bre1, ubp8, or ctk1 deletion strains were generated by replacing each ORF with kanMX4 modules constructed by PCR amplification from the corresponding strains obtained from Euroscarf. The kanMX disruption cassettes for ULP2, CCR4, and CLN3 were amplified using genomic DNA from MHY5033, MHY9388, and MHY9386 (Ryu et al, 2016), respectively, and were introduced into appropriate strains. All strains were verified by PCR and/or immunoblot analysis. The ulp2Δ strains MHY5816, MHY10249, and MHY10255 were freshly prepared from frozen stocks, and chromosome copy number and cell growth were tested (Fig EV4).
Figure EV4. Test of aneuploidy and cellular growth in ulp2Δ derivatives.
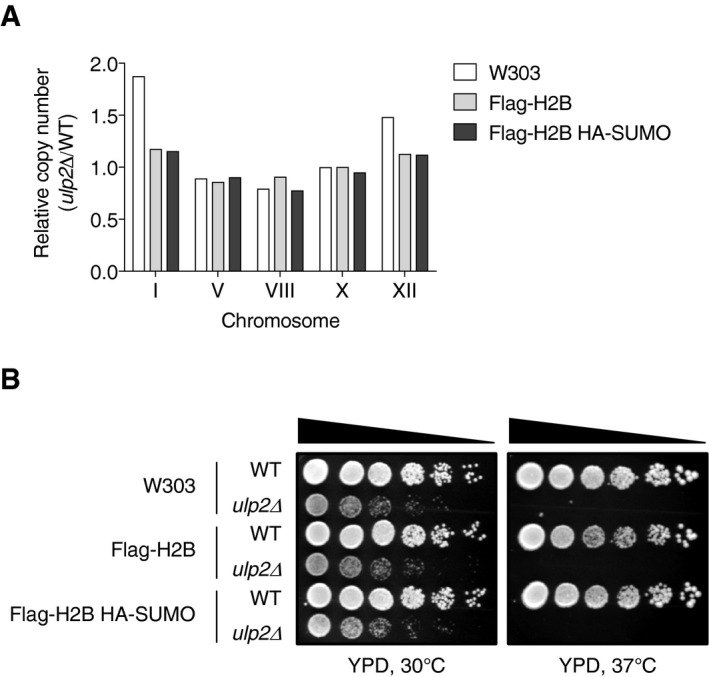
- qPCR ploidy assays of ulp2Δ strains (W303 background) expressing no tagged protein (MHY5816), Flag‐H2B (MHY10249), or Flag‐H2B and HA‐SUMO (MHY10255). Copy number of the five tested chromosomes was normalized to the euploid WT strains. See Dataset EV2 for qPCR raw data.
- Growth assay of ulp2Δ strains shown in (A). After spotting cells in fivefold serial dilutions, the YPD plates were incubated for 2 and 4 days at 30 and 37°C, respectively.
Yeast growth conditions
Cells were grown at 30°C in YPD (rich) or SD (minimal) medium with suitable supplements (Ryu et al, 2016). For plate growth assays, liquid cultures in exponential growth were normalized to an OD600 of 0.1 and subjected to fivefold serial dilutions. Cells were spotted onto SD medium with suitable supplements or 6‐azauracil (6‐AU, 100 μg/ml), and the plates were incubated at 30°C for 2–4 days. For induction of CUP1, CuSO4 was added to a final concentration of 1 mM to cells in mid‐exponential phase in SD medium, and then the cells were harvested at various times. For GAL1 induction, cells were grown in SD‐Trp medium with 2% glucose to mid‐log phase and then shifted to SD‐Trp medium containing 2% raffinose. After 2 h, the 2% raffinose medium was replaced with SD‐Trp medium containing 2% galactose and incubated for 1 hour. Between each shift, cells were washed twice with sterile water.
Plasmids
Plasmids used in this study are listed in Table EV2. ULP2 tagged with a sequence for the triplicated Flag epitope, including 500 base pairs (bp) upstream of and downstream of the ORF, was PCR‐amplified from MHY7863 (Ryu et al, 2016) and cloned into pRS314 and pRS316. The PCR‐amplified HHT1 gene with 500 bp upstream and 200 bp downstream of the ORF was cloned into pRS317. pRS314‐FLAG‐HHF1 was generated by subcloning the ApaI/BamHI fragment containing FLAG‐HHF1 from pRS424‐FLAG‐HHT1 into the ApaI/BamHI site of pRS314. To make pRS425‐GPD‐HA‐SMT3, HA‐SMT3 were amplified by PCR from the plasmid YATAG200‐HA‐SMT3 (Li & Hochstrasser, 1999) and then cloned into p425‐GPD (Mumberg et al, 1995). To create pRS314‐ulp2(C624A)‐FLAG and pRS315‐ulp2(H531A), QuikChange mutagenesis was used to mutate the ULP2 codon for the catalytic Cys 624 residue to Ala and His 531 to Ala, respectively. All constructs were confirmed by DNA sequencing.
Reverse transcription–quantitative polymerase chain reaction (RT–qPCR)
Total RNA was isolated as described previously (Ryu et al, 2016). One OD600 equivalent of cells grown to mid‐exponential phase was used for RNA preparation using the RNeasy kit (Qiagen) and the DNA‐free kit (Ambion). One μg of RNA was used in reverse transcription reactions using the iScript cDNA synthesis kit (Bio‐Rad). The oligonucleotide sequences used for qPCR are listed in Table EV3. Diluted cDNA (1:200) was used in qPCR reactions using the iQ SYBR Green Supermix kit (Bio‐Rad) on LightCycler 480 II (Roche). Gene expression level was normalized with ACT1 or SPT15 controls. Each qPCR reaction was performed in technical triplicate. Relative expression levels were calculated using the comparative C T method (ΔΔC T) (Schmittgen & Livak, 2008).
Chromatin immunoprecipitation (ChIP)
ChIP experiments were performed as described previously (Ryu & Ahn, 2014; Ryu et al, 2016). TAP‐tagged and Flag‐tagged proteins were precipitated with IgG‐Sepharose beads (GE Healthcare, 17‐0969‐01) and anti‐Flag agarose beads (Sigma, A2220), respectively. The antibodies anti‐Rpb3 (BioLegend, 665004) or anti‐SUMO (Rockland, 200‐401‐428) were bound to protein G‐Sepharose (GE Healthcare, 17‐0618‐01) to precipitate chromatin. For immunoprecipitation of chromatin samples with the H5 antibody against CTD S2‐P (BioLegend, 920204), H5‐bound samples were reacted with anti‐mouse IgM antibodies coupled to agarose beads (Sigma, A4540) as described in Ahn et al (2004). H5‐precipitates were washed twice with FA lysis buffer containing 150 mM NaCl (50 mM HEPES‐KOH [pH 7.5], 1 mM EDTA, 0.1% sodium deoxycholate, 0.1% SDS, 150 mM NaCl), once with H5 wash buffer (2 mM Tris–HCl [pH 8.0], 0.02 mM EDTA, 50 mM LiCl, 0.1% Nonidet P‐40, 0.1% sodium deoxycholate), and once with TE (10 mM Tris–HCl [pH 8.0], 1 mM EDTA). The oligonucleotide sequences used for ChIP qPCR are listed in Table EV3. qPCR assays were carried out using diluted template DNA (1:8 dilution for IP DNA and 1:1,000 dilution for input DNA), and signals were normalized to the internal control (a fragment amplified from an untranscribed region on ChrIV, residues 1516109‐1516234; SGD) and input DNA. RNAPII CTD S2‐P levels were measured relative to total RNAPII levels (Rpb3‐precipitated). To analyze CTD S2‐P or SUMO, PhosSTOP (Roche) or N‐ethylmaleimide (NEM, Sigma), respectively, were added to the cell lysis buffer containing a protease inhibitor cocktail (Roche, 11697498001) with 1mM phenylmethylsulfonyl fluoride (PMSF, AmericanBio).
ChIP‐sequencing (seq) analysis
The libraries were constructed by protocols of the Yale Center for Genomic Analysis (https://medicine.yale.edu/keck/ycga/sequencing/Illumina/8_21598_284_12501_v1.pdf). Samples were sequenced using 100‐bp paired‐end sequencing on an Illumina HiSeq NovaSeq according to Illumina protocols. The 10 bp dual index was read during additional sequencing reads that automatically followed the completion of read 1. Signal intensities were converted to individual base calls during a run using the system's Real Time Analysis (RTA) software. Reads were trimmed using Trim Galore (v0.5.0) and then aligned to the yeast reference genome (sacCer3) using BWA (v0.7.17; preprint: Li, 2013). Peaks were called using MACS2 (v2.1.1; Feng et al, 2012) and annotated using HOMER (v4.10.4) (Heinz et al, 2010). The normalized bedGraph files were generated using MACS2 (“‐B –SPMR”) and then converted to bigwig files using the bedGraphToBigWig program downloaded from the website of UCSC Genome Browser. The genome‐wide profile was generated using the “computeMatrix scale‐regions” and plotProfile tools in the deepTools package (v3.1.3) (Ramirez et al, 2014).
Chromatin double immunoprecipitation (ChDIP)
ChDIP assays were carried out as described previously (Henry et al, 2003). After overnight immunoprecipitation with anti‐Flag agarose beads (Sigma), elution was performed with triple Flag peptide (Sigma). 5% of the elute was used as INPUT, and the remaining 95% was incubated, after clarifying centrifugation, with protein G‐Sepharose (GE Healthcare) bound with anti‐ubiquitin antibody (BioMol, PW8810) to analyze H2B monoubiquitylation and anti‐HA‐conjugated agarose (Thermo Scientific, 26182) to measure histone sumoylation (in HA‐SMT3 strains). The oligonucleotide sequences used in ChDIP qPCR are listed in Table EV3. DNA templates were diluted individually 1:10 for IP and 1:50 for INPUT for further qPCR assays. The ratio of IP to INPUT was normalized by the internal control for quantitation of H2B ubiquitylation, but not sumoylation. NEM (Sigma) was added to buffers at a final concentration of 20 mM for analysis of histone sumoylation.
Immunoblotting
Preparation of yeast whole‐cell extracts and immunoblotting were performed as described previously (Kroetz et al, 2009). Levels of Myc‐, Flag‐, or TAP‐tagged proteins; SUMO conjugates; PGK; or H3 were analyzed by immunoblotting with 1:2,000 dilution of 9E10 anti‐Myc (Covance, MMS‐150R), anti‐Flag (Sigma, F3165), peroxidase‐anti‐peroxidase complex (PAP; Sigma, P1291), anti‐SUMO (Li & Hochstrasser, 1999), anti‐PGK (Molecular Probes, 459250), or anti‐H3 (Abcam, ab1791) antibodies, respectively.
Co‐immunoprecipitation
Co‐immunoprecipitation experiments were performed essentially as described (Sa‐Moura et al, 2013) with some modifications. Frozen cell pellets were ground to a fine powder in a mortar with liquid nitrogen. The cell powder was resuspended in an equal volume of buffer (50 mM HEPES [pH 7.5], 100 mM NaCl, 10% glycerol, 0.1% Triton X‐100) with 1 mM PMSF (AmericanBio), 150 mM aprotinin (Roche), 10 μM leupeptin (Roche), and 15 μM pepstatin A (Sigma). Cell debris was removed by sequential centrifugation at 29,000 g for 30 min at 4°C and at 165,000 g for 1 h at 4°C. The supernatant was collected, and the protein concentration was determined using the Bradford protein assay (Bio‐Rad). Ten mg of protein was incubated with prewashed anti‐Myc agarose beads (Covance, AFC‐150P) or IgG‐Sepharose beads (GE Healthcare) for 4 h at 4°C. Following five washes with the same buffer (sans protease inhibitors), 50 μl of SDS sample buffer was added to the washed beads for elution at 100°C. Finally, eluates (IP) and INPUT were analyzed by SDS–PAGE and immunoblotting as described above.
Chromatin association assay
Chromatin association assays were carried out as described previously (Gilbert et al, 2014). Forty OD600 equivalents of cells grown to mid‐exponential phase were harvested, washed with water and SB buffer (20 mM Tris [pH 7.4], 1 M sorbitol), and then frozen at ‐80 °C until ready for isolation. Cells were resuspended in 1 ml of PSB buffer (20 mM Tris [pH 7.4], 2 mM EDTA, 100 mM NaCl, and 10 mM β‐ME), followed by addition of 1 ml of SB buffer. Cells were then spheroplasted with Zymolyase (MP Biomedicals, Inc) for 30 min at room temperature. Spheroplasts were spun down at 5,000 × g for 15 min and washed twice with LB (20 mM PIPES [pH 6.8], 0.4 M sorbitol, 150 mM potassium acetate, 2 mM magnesium acetate). Triton X‐100 was added to 0.1 ml of LB (final concentration of 1%). Cells were lysed for 15 min on ice. Chromatin was isolated by spinning down lysates at 10,000 × g for 15 min. The supernatant was collected and saved as the “soluble” fraction. The chromatin was washed once more with LB and then resuspended into an equal volume to that of the “soluble” fraction. Volume equivalents were resolved by SDS–PAGE and immunoblotting as described above. All buffers contained a protease inhibitor cocktail (Roche) plus 20 mM NEM (Sigma).
In vivo histone ubiquitylation and sumoylation assays
The relative levels of ubiquitylated or sumoylated histone H2B or H4 were analyzed as described previously (Robzyk et al, 2000) with minor modifications. TCA‐extracted histones from 50 OD600 equivalents of cells were resuspended in 0.6 ml of SDS sample buffer with 50 μl of unbuffered 2 M Tris, and then heated to 100°C for 10 min. After centrifugation, 0.4 ml of supernatants was diluted with 0.8 ml of IP buffer (50 mM Tris [pH 7.4], 150 mM NaCl, 0.5% NP‐40), immunoprecipitated with anti‐Flag agarose beads (Sigma) for 4 hr at 4°C and then finally eluted into SDS sample buffer by heating to 100°C for 5 min. Both IP and INPUT samples were analyzed by SDS–PAGE and immunoblotting as described above. IP buffer for analysis of histone sumoylation contained 20 mM NEM (Sigma).
Synthetic lethal assay
The synthetic lethal screen with ulp2Δ cells was performed previously (Lewis et al, 2007). To confirm that the identified gene deletions were indeed synthetically lethal with ulp2Δ, kanMX4‐based gene replacements for each gene were made in diploid cells that were heterozygous for ulp2Δ::HIS3 and carried the YCplac33‐ULP2 plasmid. After tetrad dissection of sporulated cultures, double mutant haploid cells carrying YCplac33‐ULP2 were streaked on 5‐FOA plates and grown at 30°C for 3 days. Similarly, double mutant of ulp2Δ with htb1‐K123R (MHY4122) or ctk1Δ (MHY10242) that also carried a URA3‐marked ULP2 plasmid was streaked on 5‐FOA plates to test for synthetic growth defects.
qPCR ploidy assay
Chromosome copy number was determined by qPCR ploidy assays as described previously (Ryu et al, 2016). Genomic DNA was extracted from 1 OD600 equivalent of cells using a standard phenol/chloroform extraction method and then diluted 1:8 for qPCR assays. The oligonucleotide sequences used for qPCR are listed in Table EV3. Triplicate qPCR reactions were performed using the iQ SYBR Green Supermix kit in a Roche LightCycler 480 instrument, and the qPCR results were analyzed by the modified Ct method (Schmittgen & Livak, 2008). Ct values for each chromosome were normalized to the median Ct value for each strain, and ΔCt was represented as a ratio relative to WT control.
Author contributions
H‐YR contributed to the experimental design, performed most of the experiments, and drafted the manuscript. DS and NRW‐E performed the in vivo histone sumoylation assays and synthetic lethal assays in Fig 4, respectively. DZ and FLG analyzed ChIP‐seq and previous RNA‐seq data, respectively. MH designed the study, analyzed the data, and wrote the final version of the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Dataset EV1
Dataset EV2
Source Data for Expanded View
Review Process File
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
This study was supported NIH grant GM053756 to M.H. We thank Jianhui Li for comments on the manuscript.
The EMBO Journal (2019) 38: e102003
Data availability
ChIP‐seq data were deposited on GEO with the record number GSE130623 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE130623). qPCR raw data are provided as Dataset EV2.
References
- Ahn SH, Kim M, Buratowski S (2004) Phosphorylation of serine 2 within the RNA polymerase II C‐terminal domain couples transcription and 3′ end processing. Mol Cell 13: 67–76 [DOI] [PubMed] [Google Scholar]
- de Albuquerque CP, Suhandynata RT, Carlson CR, Yuan WT, Zhou H (2018) Binding to small ubiquitin‐like modifier and the nucleolar protein Csm1 regulates substrate specificity of the Ulp2 protease. J Biol Chem 293: 12105–12119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baptista T, Grunberg S, Minoungou N, Koster MJE, Timmers HTM, Hahn S, Devys D, Tora L (2017) SAGA is a general cofactor for RNA polymerase II transcription. Mol Cell 68: 130–143.e135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger SL (2002) Histone modifications in transcriptional regulation. Curr Opin Genet Dev 12: 142–148 [DOI] [PubMed] [Google Scholar]
- Bonnet J, Wang CY, Baptista T, Vincent SD, Hsiao WC, Stierle M, Kao CF, Tora L, Devys D (2014) The SAGA coactivator complex acts on the whole transcribed genome and is required for RNA polymerase II transcription. Genes Dev 28: 1999–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer‐Guittaut M, Birsoy K, Potel C, Elliott G, Jaffray E, Desterro JM, Hay RT, Oelgeschlager T (2005) SUMO‐1 modification of human transcription factor (TF) IID complex subunits: inhibition of TFIID promoter‐binding activity through SUMO‐1 modification of hsTAF5. J Biol Chem 280: 9937–9945 [DOI] [PubMed] [Google Scholar]
- Briggs SD, Xiao T, Sun Z‐W, Caldwell JA, Shabanowitz J, Hunt DF, Allis CD, Strahl BD (2002) Gene silencing: trans‐histone regulatory pathway in chromatin. Nature 418: 498 [DOI] [PubMed] [Google Scholar]
- Bylebyl GR, Belichenko I, Johnson ES (2003) The SUMO isopeptidase Ulp2 prevents accumulation of SUMO chains in yeast. J Biol Chem 278: 44113–44120 [DOI] [PubMed] [Google Scholar]
- Chandrasekharan MB, Huang F, Sun ZW (2009) Ubiquitination of histone H2B regulates chromatin dynamics by enhancing nucleosome stability. Proc Natl Acad Sci USA 106: 16686–16691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chymkowitch P, Nguea AP, Aanes H, Koehler CJ, Thiede B, Lorenz S, Meza‐Zepeda LA, Klungland A, Enserink JM (2015a) Sumoylation of Rap1 mediates the recruitment of TFIID to promote transcription of ribosomal protein genes. Genome Res 25: 897–906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chymkowitch P, Nguea PA, Enserink JM (2015b) SUMO‐regulated transcription: challenging the dogma. BioEssays 37: 1095–1105 [DOI] [PubMed] [Google Scholar]
- Conaway JW, Shilatifard A, Dvir A, Conaway RC (2000) Control of elongation by RNA polymerase II. Trends Biochem Sci 25: 375–380 [DOI] [PubMed] [Google Scholar]
- Daniel JA, Torok MS, Sun ZW, Schieltz D, Allis CD, Yates JR III, Grant PA (2004) Deubiquitination of histone H2B by a yeast acetyltransferase complex regulates transcription. J Biol Chem 279: 1867–1871 [DOI] [PubMed] [Google Scholar]
- Dhall A, Weller CE, Chu A, Shelton PMM, Chatterjee C (2017) Chemically sumoylated histone h4 stimulates intranucleosomal demethylation by the LSD1‐CoREST complex. ACS Chem Biol 12: 2275–2280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohmen RJ, Stappen R, McGrath JP, Forrova H, Kolarov J, Goffeau A, Varshavsky A (1995) An essential yeast gene encoding a homolog of ubiquitin‐activating enzyme. J Biol Chem 270: 18099–18109 [DOI] [PubMed] [Google Scholar]
- Dover J, Schneider J, Tawiah‐Boateng MA, Wood A, Dean K, Johnston M, Shilatifard A (2002) Methylation of histone H3 by COMPASS requires ubiquitination of histone H2B by Rad6. J Biol Chem 277: 28368–28371 [DOI] [PubMed] [Google Scholar]
- Egloff S, Dienstbier M, Murphy S (2012) Updating the RNA polymerase CTD code: adding gene‐specific layers. Trends Genet 28: 333–341 [DOI] [PubMed] [Google Scholar]
- Emre NC, Ingvarsdottir K, Wyce A, Wood A, Krogan NJ, Henry KW, Li K, Marmorstein R, Greenblatt JF, Shilatifard A et al (2005) Maintenance of low histone ubiquitylation by Ubp10 correlates with telomere‐proximal Sir2 association and gene silencing. Mol Cell 17: 585–594 [DOI] [PubMed] [Google Scholar]
- Feng J, Liu T, Qin B, Zhang Y, Liu XS (2012) Identifying ChIP‐seq enrichment using MACS. Nat Protoc 7: 1728–1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flotho A, Melchior F (2013) Sumoylation: a regulatory protein modification in health and disease. Annu Rev Biochem 82: 357–385 [DOI] [PubMed] [Google Scholar]
- Funakoshi M, Hochstrasser M (2009) Small epitope‐linker modules for PCR‐based C‐terminal tagging in Saccharomyces cerevisiae . Yeast 26: 185–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert TM, McDaniel SL, Byrum SD, Cades JA, Dancy BC, Wade H, Tackett AJ, Strahl BD, Taverna SD (2014) A PWWP domain‐containing protein targets the NuA3 acetyltransferase complex via histone H3 lysine 36 trimethylation to coordinate transcriptional elongation at coding regions. Mol Cell Proteomics 13: 2883–2895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girdwood D, Bumpass D, Vaughan OA, Thain A, Anderson LA, Snowden AW, Garcia‐Wilson E, Perkins ND, Hay RT (2003) P300 transcriptional repression is mediated by SUMO modification. Mol Cell 11: 1043–1054 [DOI] [PubMed] [Google Scholar]
- Grant PA, Schieltz D, Pray‐Grant MG, Steger DJ, Reese JC, Yates JR III, Workman JL (1998) A subset of TAF(II)s are integral components of the SAGA complex required for nucleosome acetylation and transcriptional stimulation. Cell 94: 45–53 [DOI] [PubMed] [Google Scholar]
- Guo B, Sharrocks AD (2009) Extracellular signal‐regulated kinase mitogen‐activated protein kinase signaling initiates a dynamic interplay between sumoylation and ubiquitination to regulate the activity of the transcriptional activator PEA3. Mol Cell Biol 29: 3204–3218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamer DH, Thiele DJ, Lemontt JE (1985) Function and autoregulation of yeast copperthionein. Science 228: 685–690 [DOI] [PubMed] [Google Scholar]
- Hannich JT, Lewis A, Kroetz MB, Li SJ, Heide H, Emili A, Hochstrasser M (2005) Defining the SUMO‐modified proteome by multiple approaches in Saccharomyces cerevisiae . J Biol Chem 280: 4102–4110 [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK (2010) Simple combinations of lineage‐determining transcription factors prime cis‐regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriks IA, D'Souza RC, Yang B, Verlaan‐de Vries M, Mann M, Vertegaal AC (2014) Uncovering global SUMOylation signaling networks in a site‐specific manner. Nat Struct Mol Biol 21: 927–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriks IA, Vertegaal AC (2016) A comprehensive compilation of SUMO proteomics. Nat Rev Mol Cell Biol 17: 581–595 [DOI] [PubMed] [Google Scholar]
- Henry KW, Berger SL (2002) Trans‐tail histone modifications: wedge or bridge? Nat Struct Biol 9: 565–566 [DOI] [PubMed] [Google Scholar]
- Henry KW, Wyce A, Lo WS, Duggan LJ, Emre NC, Kao CF, Pillus L, Shilatifard A, Osley MA, Berger SL (2003) Transcriptional activation via sequential histone H2B ubiquitylation and deubiquitylation, mediated by SAGA‐associated Ubp8. Genes Dev 17: 2648–2663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey CM, Wilson NR, Hochstrasser M (2012) Function and regulation of SUMO proteases. Nat Rev Mol Cell Biol 13: 755–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S (2002) RAD6‐dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419: 135–141 [DOI] [PubMed] [Google Scholar]
- Hottiger T, Furst P, Pohlig G, Heim J (1994) Physiological characterization of the yeast metallothionein (CUP1) promoter, and consequences of overexpressing its transcriptional activator, ACE1. Yeast 10: 283–296 [DOI] [PubMed] [Google Scholar]
- Huisinga KL, Pugh BF (2004) A genome‐wide housekeeping role for TFIID and a highly regulated stress‐related role for SAGA in Saccharomyces cerevisiae . Mol Cell 13: 573–585 [DOI] [PubMed] [Google Scholar]
- Ivanov AV, Peng H, Yurchenko V, Yap KL, Negorev DG, Schultz DC, Psulkowski E, Fredericks WJ, White DE, Maul GG et al (2007) PHD domain‐mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol Cell 28: 823–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalocsay M, Hiller NJ, Jentsch S (2009) Chromosome‐wide Rad51 spreading and SUMO‐H2A.Z‐dependent chromosome fixation in response to a persistent DNA double‐strand break. Mol Cell 33: 335–343 [DOI] [PubMed] [Google Scholar]
- Kim J, Cantwell CA, Johnson PF, Pfarr CM, Williams SC (2002) Transcriptional activity of CCAAT/enhancer‐binding proteins is controlled by a conserved inhibitory domain that is a target for sumoylation. J Biol Chem 277: 38037–38044 [DOI] [PubMed] [Google Scholar]
- Kim M, Ahn SH, Krogan NJ, Greenblatt JF, Buratowski S (2004) Transitions in RNA polymerase II elongation complexes at the 3′ ends of genes. EMBO J 23: 354–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroetz MB, Su D, Hochstrasser M (2009) Essential role of nuclear localization for yeast Ulp2 SUMO protease function. Mol Biol Cell 20: 2196–2206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan NJ, Dover J, Khorrami S, Greenblatt JF, Schneider J, Johnston M, Shilatifard A (2002) COMPASS, a histone H3 (Lysine 4) methyltransferase required for telomeric silencing of gene expression. J Biol Chem 277: 10753–10755 [DOI] [PubMed] [Google Scholar]
- Lewis A, Felberbaum R, Hochstrasser M (2007) A nuclear envelope protein linking nuclear pore basket assembly, SUMO protease regulation, and mRNA surveillance. J Cell Biol 178: 813–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SJ, Hochstrasser M (1999) A new protease required for cell‐cycle progression in yeast. Nature 398: 246–251 [DOI] [PubMed] [Google Scholar]
- Li SJ, Hochstrasser M (2000) The yeast ULP2 (SMT4) gene encodes a novel protease specific for the ubiquitin‐like Smt3 protein. Mol Cell Biol 20: 2367–2377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SJ, Hochstrasser M (2003) The Ulp1 SUMO isopeptidase: distinct domains required for viability, nuclear envelope localization, and substrate specificity. J Cell Biol 160: 1069–1081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA‐MEM. arXiv arXiv:13033997 [PREPRINT] [Google Scholar]
- Liang J, Singh N, Carlson CR, Albuquerque CP, Corbett KD, Zhou H (2017) Recruitment of a SUMO isopeptidase to rDNA stabilizes silencing complexes by opposing SUMO targeted ubiquitin ligase activity. Genes Dev 31: 802–815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR (1998) Additional modules for versatile and economical PCR‐based gene deletion and modification in Saccharomyces cerevisiae . Yeast 14: 953–961 [DOI] [PubMed] [Google Scholar]
- Lyst MJ, Nan X, Stancheva I (2006) Regulation of MBD1‐mediated transcriptional repression by SUMO and PIAS proteins. EMBO J 25: 5317–5328 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lyst MJ, Stancheva I (2007) A role for SUMO modification in transcriptional repression and activation. Biochem Soc Trans 35: 1389–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzler‐Guillemain C, Depetris D, Luciani JJ, Mignon‐Ravix C, Mitchell MJ, Mattei MG (2008) In human pachytene spermatocytes, SUMO protein is restricted to the constitutive heterochromatin. Chromosome Res 16: 761–782 [DOI] [PubMed] [Google Scholar]
- Muller S, Berger M, Lehembre F, Seeler JS, Haupt Y, Dejean A (2000) c‐Jun and p53 activity is modulated by SUMO‐1 modification. J Biol Chem 275: 13321–13329 [DOI] [PubMed] [Google Scholar]
- Mumberg D, Muller R, Funk M (1995) Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene 156: 119–122 [DOI] [PubMed] [Google Scholar]
- Murray S, Udupa R, Yao S, Hartzog G, Prelich G (2001) Phosphorylation of the RNA polymerase II carboxy‐terminal domain by the Bur1 cyclin‐dependent kinase. Mol Cell Biol 21: 4089–4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan D, Ingvarsdottir K, Sterner DE, Bylebyl GR, Dokmanovic M, Dorsey JA, Whelan KA, Krsmanovic M, Lane WS, Meluh PB et al (2006) Histone sumoylation is a negative regulator in Saccharomyces cerevisiae and shows dynamic interplay with positive‐acting histone modifications. Genes Dev 20: 966–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng HH, Robert F, Young RA, Struhl K (2003) Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol Cell 11: 709–719 [DOI] [PubMed] [Google Scholar]
- Ng CH, Akhter A, Yurko N, Burgener JM, Rosonina E, Manley JL (2015) Sumoylation controls the timing of Tup1‐mediated transcriptional deactivation. Nat Commun 6: 6610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena MM, Koch KA, Thiele DJ (1998) Dynamic regulation of copper uptake and detoxification genes in Saccharomyces cerevisiae . Mol Cell Biol 18: 2514–2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Dundar F, Diehl S, Gruning BA, Manke T (2014) deepTools: a flexible platform for exploring deep‐sequencing data. Nucleic Acids Res 42: W187–W191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robzyk K, Recht L, Osley MA (2000) Rad6‐dependent ubiquitination of histone H2B in yeast. Science 287: 501–504 [DOI] [PubMed] [Google Scholar]
- Rosonina E, Duncan SM, Manley JL (2010) SUMO functions in constitutive transcription and during activation of inducible genes in yeast. Genes Dev 24: 1242–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu HY, Ahn S (2014) Yeast histone H3 lysine 4 demethylase Jhd2 regulates mitotic rDNA condensation. BMC Biol 12: 75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu HY, Wilson NR, Mehta S, Hwang SS, Hochstrasser M (2016) Loss of the SUMO protease Ulp2 triggers a specific multichromosome aneuploidy. Genes Dev 30: 1881–1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu HY, Lopez‐Giraldez F, Knight J, Hwang SS, Renner C, Kreft SG, Hochstrasser M (2018) Distinct adaptive mechanisms drive recovery from aneuploidy caused by loss of the Ulp2 SUMO protease. Nat Commun 9: 5417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sa‐Moura B, Funakoshi M, Tomko RJ Jr, Dohmen RJ, Wu Z, Peng J, Hochstrasser M (2013) A conserved protein with AN1 zinc finger and ubiquitin‐like domains modulates Cdc48 (p97) function in the ubiquitin‐proteasome pathway. J Biol Chem 288: 33682–33696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ (2008) Analyzing real‐time PCR data by the comparative C(T) method. Nat Protoc 3: 1101–1108 [DOI] [PubMed] [Google Scholar]
- Schwartz DC, Felberbaum R, Hochstrasser M (2007) The Ulp2 SUMO protease is required for cell division following termination of the DNA damage checkpoint. Mol Cell Biol 27: 6948–6961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seufert W, Futcher B, Jentsch S (1995) Role of a ubiquitin‐conjugating enzyme in degradation of S‐ and M‐phase cyclins. Nature 373: 78–81 [DOI] [PubMed] [Google Scholar]
- Shieh GS, Pan C‐H, Wu J‐H, Sun Y‐J, Wang C‐C, Hsiao W‐C, Lin C‐Y, Tung L, Chang T‐H, Fleming AB (2011) H2B ubiquitylation is part of chromatin architecture that marks exon‐intron structure in budding yeast. BMC Genom 12: 627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiio Y, Eisenman RN (2003) Histone sumoylation is associated with transcriptional repression. Proc Natl Acad Sci USA 100: 13225–13230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava R, Ahn SH (2015) Modifications of RNA polymerase II CTD: connections to the histone code and cellular function. Biotechnol Adv 33: 856–872 [DOI] [PubMed] [Google Scholar]
- Sun Z‐W, Allis CD (2002) Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature 418: 104–108 [DOI] [PubMed] [Google Scholar]
- Texari L, Dieppois G, Vinciguerra P, Contreras MP, Groner A, Letourneau A, Stutz F (2013) The nuclear pore regulates GAL1 gene transcription by controlling the localization of the SUMO protease Ulp1. Mol Cell 51: 807–818 [DOI] [PubMed] [Google Scholar]
- Van Oss SB, Shirra MK, Bataille AR, Wier AD, Yen K, Vinayachandran V, Byeon IL, Cucinotta CE, Heroux A, Jeon J et al (2016) The histone modification domain of Paf1 complex subunit Rtf1 directly stimulates H2B ubiquitylation through an Interaction with Rad6. Mol Cell 64: 815–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood A, Schneider J, Dover J, Johnston M, Shilatifard A (2003) The Paf1 complex is essential for histone monoubiquitination by the Rad6‐Bre1 complex, which signals for histone methylation by COMPASS and Dot1p. J Biol Chem 278: 34739–34742 [DOI] [PubMed] [Google Scholar]
- Wyce A, Xiao T, Whelan KA, Kosman C, Walter W, Eick D, Hughes TR, Krogan NJ, Strahl BD, Berger SL (2007) H2B ubiquitylation acts as a barrier to Ctk1 nucleosomal recruitment prior to removal by Ubp8 within a SAGA‐related complex. Mol Cell 27: 275–288 [DOI] [PubMed] [Google Scholar]
- Xiao T, Kao CF, Krogan NJ, Sun ZW, Greenblatt JF, Osley MA, Strahl BD (2005) Histone H2B ubiquitylation is associated with elongating RNA polymerase II. Mol Cell Biol 25: 637–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, Jaffray E, Hay RT, Sharrocks AD (2003) Dynamic interplay of the SUMO and ERK pathways in regulating Elk‐1 transcriptional activity. Mol Cell 12: 63–74 [DOI] [PubMed] [Google Scholar]
- Zheng G, Yang YC (2004) ZNF76, a novel transcriptional repressor targeting TATA‐binding protein, is modulated by sumoylation. J Biol Chem 279: 42410–42421 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Dataset EV1
Dataset EV2
Source Data for Expanded View
Review Process File
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Data Availability Statement
ChIP‐seq data were deposited on GEO with the record number GSE130623 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE130623). qPCR raw data are provided as Dataset EV2.