

Abstract
Background
To investigate the mechanism underlying the chondroprotective effect of estrogen in AMP-activated protein kinase (AMPK) deficiency mice.
Methods
Female cartilage-specific AMPKα double knockout (AMPKα cDKO) mice were generated and subjected to ovariectomy (OVX). The model of osteoarthritis (OA) was induced by destabilization of medial meniscus (DMM). Histopathological changes were evaluated by using OARSI scoring systems. Autophagy changes were analyzed by immunofluorescence staining. Human chondrocytes were subjected to mechanical stress to mimic OA development. and incubated in presence of or absence of 17β-estradiol or/and compound C (AMPK inhibitor) or/and U0126 (ERK inhibitor). The expression levels of ERK1/2 phosphorylation, p70S6K phosphorylation and light chain 3 (LC3) were detected by Western blot.
Results
Compared with in OVX-sham AMPKα cDKO and OVX-sham WT mice, DMM-induced OA is more severe, and significantly low level of LC3 was observed in articular cartilage in OVX AMPK cDKO mice. Both mechanical stress and compound C were shown to induce an increase in phosphorylation of p70S6K, respectively. 17β-estradiol stimulation led to a reduction in the basal level of p70S6K phosphorylation as well as in the compound C or mechanical stress-induced level of p70S6K phosphorylation. 17β-estradiol stimulation not only led to an increase in LC3 conversion but also overrode the inhibitory effect of compound C on LC3 conversion. The effects of 17β-estradiol were abrogated by blocking ERK signaling pathway.
Conclusions
Our findings suggest that estrogen can protect articular cartilage from damage during OA development by promoting chondrocyte autophagy via ERK-mammalian target of rapamycin (mTOR) signaling, and give new insight into the mechanism of the chondroprotective effect of estrogen.
Keywords: Estrogen, osteoarthritis (OA), chondrocytes, AMP-activated protein kinase (AMPK), mammalian target of rapamycin (mTOR)
Introduction
Osteoarthritis (OA) is one of the most common diseases of the joint, who characterized by extracellular matrix (ECM) degradation and loss of cartilage cellularity (1). Multiple factors have been implicated in the development of OA, such as mechanical stress and aging (2,3). Unfortunately there is no effective disease-modifying dug available for OA on clinic to date.
AMP-activated protein kinase (AMPK) is serine/threonine protein kinase and exists as a heterotrimer composed of one catalytic subunit (α1, α2), and two regulatory subunits β (β1, β2) and γ (γ1, γ2, γ3) (4,5). AMPK has emerged as a pivotal regulator of signals that control cellular energy metabolism through the regulation of diverse biochemical pathways, and its dysfunction has been linked to numerous human diseases (6).
Several lines of evidence suggested that compromised AMPK activity in chondrocytes has been associated with the development of OA. In human knee OA chondrocytes, AMPK activity was reduced. The surgical destabilization of medial meniscus (DMM) model is widely used for simulating spontaneous models of OA (7). The reduction of AMPKα phosphorylation was found in chondrocytes in DMM-induced OA mouse model and bovine after mechanical injury (8,9). AMPK activators suppressed Interleukin-1β-induced pro-catabolic response in human chondrocytes (9). Meanwhile, AMPK activators alleviated cartilage degeneration and led to an anti-apoptotic effect on articular cartilage in rat OA model (10). We demonstrated that accelerated OA progression in AMPK deficiency mice at 8 weeks post-DMM surgery (11). Recently, it was demonstrated that AMPK stimulated autophagy by inhibiting mammalian target of rapamycin complex (mTORC) (12). Autophagy is an essential cellular homeostasis mechanism and compromised autophagy has been shown to be associated with the development of OA (13-15). Cartilage-specific deletion of mTOR upregulated chondrocytes autophagy and alleviated cartilage damage from DMM-induced OA in mouse model (16). Intra-articular or intraperitoneal (IP) injection of rapamycin, a pharmacological inhibitor of mTOR, reduced the severity of experimental OA by autophagy activation (17).
Epidemiological studies suggest that the prevalence of OA significantly increases in women around the age of 50 compared with OA in men, which coincides with the beginning of menopause. Some studies have shown that estrogen replacement therapy (ERT) significantly reduced the risk of radiographic OA, suggesting a protective effect of ERT on OA (18,19). In vivo, OVX female mice developed significantly severe OA than control females, whereas the severity of OA was back to the level of control females when supplemented with exogenous 17β-estradiol (20,21). In vitro, 17β-estradiol upregulates the expression of Collagen 2 and protects chondrocytes from mechanical injury-induced cell death (22). Although great efforts have been made, the mechanism underlying the protective effect of estrogen still remains largely unknown.
Our previous study revealed that AMPKα mutant females did not exhibit more severe OA compared with WT littermates, indicating that estrogen may exert a compensation effect for AMPK dysfunction (11). The objective of this study was to investigate the potential mechanism underlying the protective effect of estrogen on articular cartilage in a mouse model of AMPK deficiency. Here, our findings suggest that estrogen may inhibit mTOR signaling pathway via activating ERK, which will promote chondrocytes autophagy to protect AMPK mutant mice from OA.
Methods
Generation of inducible cartilage-specific AMPKα double knockout mice
Inducible cartilage-specific AMPKα double knockout (AMPKα cDKO) mice were generated by crossing C57BL/6-AMPKα1flox/flox α2flox/flox mice, which were kindly provided by Professor Shuai Chen (Nanjing University, Nanjing, China), with C57BL/6-Col2a1-CreERT2 mice, provided by Minghao Zheng (University of Western Australia, Perth, Australia). AMPKα1flox/flox, AMPKα2flox/flox and Cre transgene were genotyped by PCR. The eight weeks old cartilage-specific AMPKα cDKO mice and WT control were injected by IP with tamoxifen (1 mg/10 g body weight, Sigma) for 5 succession days (11,23). All animal experimental operations were approved by Institutional Animal Care and Use Committee (IACUC) of the Model Animal Research Center of Nanjing University (Animal Care and Use Protocol Permit Number: JQ05).
Murine model of OVX and OA
Ten-week-old AMPKα female cDKO and corresponding WT littermates were subjected to OVX or OVX-sham as described previously (20). Two weeks later, OA was surgically induced in 12-week-old OVX-sham WT, OVX-sham AMPKα cDKO and OVX AMPKα cDKO mice by DMM surgery in the right knee, as previously described (24). Briefly, AMPK cKO and WT control females received IP anesthesia with 100 mg/kg ketamine and 5 mg/kg xylazine. To give DMM, a micro-surgical scissors is used to transect of the medial meniscotibial ligament (MMTL), which anchors the medial meniscus to the tibial plateau. The contralateral left knee joint was sham-operated with medial capsulotomy only. Histological analysis was performed at 8 weeks post-DMM surgery.
Histological analysis of mouse knee joints
Mouse joint at 8 weeks post-OA surgery were fixed in 4% paraformaldehyde at 4 °C for 24 h and embedded in paraffin after decalcification for 7 days in 0.5 M EDTA (pH 7.4). Serial frontal sections (5 µm thick) were cut and stained with Safranin O/Fast Green staining. Cartilage deterioration was evaluated using the Osteoarthritis Research Society International (OARSI) scoring method (24). All slides stained were evaluated by two independent investigators in a blinded fashion.
Immunofluorescence
We performed immunohistochemical staining to detect the expression of light chain 3 (LC3) in the articular cartilage. Rabbit polyclonal antibodies to LC3 (Cell Signaling, Beverly, MA, USA), were used at a 1:200 dilution and incubated overnight at 4 °C. Alexa Fluor 488-conjugated donkey anti-rabbit IgG (Cell Signaling) was used at 1:200 dilution as the secondary antibody against the primary antibodies at room temperature for 1 hour. Nuclei were counterstained with DAPI (Invitrogen). After staining, we evaluated the expression levels in the articular cartilage using a Leica TCS SP5 laser confocal microscope.
Chondrocytes culture and exposure to mechanical stress
All human chondrocytes experiments were approved by the Human Ethics Committee of Drum Tower Hospital, School of Medicine, Nanjing University (No. 2009022). Human chondrocytes isolation was as described previously (25). Briefly, chondrocytes from donors were digested with 0.1% type II collagenase (Gibco) for 12–16 hours at 37 °C. Chondrocytes were seeded on a 10-cm dish maintained in phenol red-free Dulbecco’s modified Eagle’s medium and Ham’s F-12 (DMEM/F12) containing 10% fetal bovine serum (Gibco) and 1% penicillin/streptomycin (HyClone). The fetal bovine serum was pretreated with dextran-coated charcoal to remove estrogen according to the previous study (26). At passage 1, chondrocytes were seeded on collagen1-coated six-well Bioflex plates (Flexcell International, Hillsborough, NC, USA) at a density of 1×105 cells per wells. Cyclic tensile strain (CTS) experiments were performed using the FX-5000TM Tension system at 10% elongation (0.5 Hz) for 24 hours (27).
Western blotting
Cells were lysed in RIPA lysis buffer containing protease and phosphatase inhibitors and the protein content of the lysates was determined by a bicinchoninic acid protein assay reagent (Pierce), with bovine serum albumin (BSA) as the standard. Equal equivalents of protein were separated on 8%, 10%, or 12% SDS-PAGE gels. Proteins were transferred onto 0.45 or 0.22 µm polyvinylidene fluoride membranes (Millipore), blocked in 5% BSA for 1 h, then incubated overnight at 4 °C in the presence of the respective primary antibodies: anti-phospho-P70S6K (9204; 1:1,000 dilution; Cell Signaling Technology), anti-LC3A/B (12741; 1:1,000 dilution; Cell Signaling Technology), anti-phospho-ERK (4370; 1:1,000 dilution; Cell Signaling Technology), anti-GAPDH (5174; 1:1,000 dilution; Cell Signaling Technology). Membranes were then washed three times using Tris-buffered saline containing 0.1% Tween-20 and probed with horseradish peroxidase-conjugated secondary immunoglobulin G mouse anti-rabbit (5127; 1:2,000; Cell Signaling Technology) for 1 h at room temperature. Membranes were scanned, and western blotting results were quantified using an Odyssey Infrared Imaging System (Li-Cor).
Statistical analysis
All the data were expressed as the mean ± SD. Unpaired two-tailed Student t-test, one-way analysis of variance (ANOVA) was applied for multiple comparisons between independent groups. P values less than 0.05 were considered statistically significant. SPSS version 11.5 (SPSS, Inc.) was used for statistical analysis.
Results
Aggravated DMM-induced OA in AMPK mutant mice following OVX
Inducible AMPKα cDKO mouse exhibited no structural defects at meniscus identified in sham-operated knees till 12 weeks post-birth. As our group described previously, male AMPKα cDKO mice exhibited exacerbated OA after DMM induction compared with WT littermates. However, female OVX-sham AMPKα cDKO mice and OVX-sham WT littermates only displayed a slight fibrillation and proteoglycans loss in articular cartilage at 8 weeks post-DMM and no significant differences on OARSI scores were detected (Figure 1A,B; one-way ANOVA; femur: 8 W, P=0.9070; tibia: 8 W, P=0.7379). However, as compared with OVX-sham AMPKα cDKO females, OVX cDKO females exhibited a significant increase in loss of proteoglycan and articular chondrocytes cellularity at 8 weeks post-DMM. Almost complete loss of the articular cartilage and exposed subchondral bone were observed in the medial tibial plateau. Two blind observers further confirmed a significant increase in OARSI scores at both medial tibial plateau and medial femoral condyle in OVX AMPKα cDKO females compared with OVX-sham AMPKα cDKO females (Figure 1B; one-way ANOVA; femur: 8 W, P<0.001; tibia: 8 W, P<0.001). However, DMM sham-operated knees from OVX-sham AMPKα cDKO, OVX-sham WT and OVX AMPKα cDKO exhibited no obvious cartilage damage (Figure 1C).
Figure 1.

Accelerated DMM-induced OA in OVX AMPKα cDKO mice. Ten-week-old C57Bl/6J mice were subjected to OVX or OVX-sham operation and then received DMM surgery or sham operation two weeks post-OVX. The left knee was not subjected to surgery and was used as a control. Each group included 8 mice, with 24 mice totally. (A) Representative photographs of articular cartilage destruction in OVX-sham WT (n=8), OVX-sham AMPKα cDKO (n=8) and OVX AMPKα cDKO (n=8) mice 8 weeks post-DMM. Knee joints were analyzed by staining with Safranin O. Scale bars =100 µm. (B) Histological scores containing three DMM-operated groups (n=24) showed a significant increase in osteoarthritis scores after ovariectomy. (C) Histological scores for the DMM-sham experiments. Values are mean ± SD. ***, P<0.001. NS, not significant; DMM, destabilization of medial meniscus; OA, osteoarthritis; OVX, ovariectomy.
Reduced autophagy activity in articular cartilage in OVX AMPKα cDKO mice
To investigate whether accelerated OA progression in OVX AMPKα cDKO females was coupled with the autophagy process, we examined the levels of autophagic activity marker by immunofluorescence staining (28). As shown in Figure 2, the autophagosome marker LC3-positive puncta, which represents the presence of autolysosomes, were apparently decreased in female OVX AMPK cDKO mice compared with OVX-sham AMPK mutant and OVX-sham WT females, while no significant difference was found with expression levels of LC3 between OVX-sham AMPK mutant and OVX-sham WT females.
Figure 2.

Decreased autophagy activity in articular cartilage in OVX AMPKα cDKO mice. Immunofluorescence image of WT, AMPKα cDKO and OVX AMPKα cDKO mice stained with LC3 (green)/DAPI (blue). Scale bars =20 µm. OVX, ovariectomy; LC3, light chain 3.
Estrogen inhibited mTOR activity in mechanical stress induced OA model in vitro
To further investigate the potential mechanism underlying the protective effect of estrogen on chondrocytes, we performed in vitro experiments. It is demonstrated that compromised mTOR activity resulted in increased autophagy activity and protection from DMM-induced OA (16,17,28). In the second part of our study, we performed Western Blot analysis to examine the effect of 17β-estradiol on mTOR activity. Our study applied CTS with 10% elongation for 24 h to human chondrocytes to mimic the OA model in vitro as described previously (Figure 3A) (27). We detected mTOR activity by determining phosphorylation level of p70S6K.
Figure 3.

17β-estradiol stimulation inhibited mTOR activity and enhanced chondrocytes autophagy via ERK-mTOR signaling in mechanical stress induced OA model in vitro. Human chondrocytes cultured in serum-free medium were pretreated with 5 mM Compound or 10 uM U0126 and incubated with or without exogenous 10 nM 17β-estradiol. Human chondrocytes were then exposed to mechanical stress at 10% elongation (0.5 Hz) for 24 hours. (A) Schematic diagram of the FX-5000TM Tension system. (B,C) Western blotting analysis of phosphorylation level of p70S6K under mechanical stress or unstrained conditions. Comparisons among human chondrocytes treated with compound C or 17β-estradiol are shown in the histograms for phospho-p70S6K normalized with GAPDH. (D,E) Western blotting analysis of expression of autophagy marker of LC3. Comparisons among human chondrocytes incubated in presence of or absence of 17β-estradiol or/and compound C or/and U0126 are shown in the histograms for LC3-II normalized with LC3-I. (F,G,H) Western blotting analysis of phosphorylation level of ERK1/2 and p70S6K. Comparisons among human chondrocytes incubated in presence of or absence of 17β-estradiol or/and U0126 are shown in the histograms for phospho-ERK1/2 or phospho-p70S6K normalized with GAPDH. Results are presented as the mean ± SD. *, P<0.05; **, P<0.01; ***, P<0.001. NS, not significant.
Both mechanical stress and compound C were shown to induce an increase phosphorylation of p70S6K, and no synergistic effect was detected between mechanical stress and compound C. 17β-estradiol (10 nM) stimulation led to a reduction not only in the basal level of p70S6K phosphorylation, but also in the compound C or mechanical stress-induced level of p70S6K phosphorylation (Figure 3B,C; one-way ANOVA; P<0.001). These data supported the notion that estrogen operated through an AMPK-independent mechanism to inhibit mTOR signaling.
Estrogen enhanced chondrocytes autophagy via ERK-mTOR signaling
To investigate whether chondrocytes autophagy was modulated by estrogen, human chondrocytes were exposed to mechanical stress for 24 h in presence of or absence of 17β-estradiol or/and compound C or/and U0126 to monitor the autophagy induction by determining the conversion from LC3I to LC3II. As shown in Figure 3D,E, compound C stimulation resulted in a significant reduction in LC3 conversion. 17β-estradiol stimulation not only led to an increase in LC3 conversion but also over-ride the inhibitory effect of compound C on LC3 conversion.
17β-estradiol could modulate cell stress and autophagy in osteoblast through extracellular signal-regulated kinase-1/2 (ERK1/2) signaling pathway (29). To further investigate whether 17β-estradiol acted in a similar mechanism, we examined the phosphorylation levels of ERK1/2, and p70S6K by Western blotting. As shown in Figure 3F, 17β-estradiol stimulation not only resulted in an apparent increase of phosphorylation of ERK1/2, but also led to a reduction of phosphorylation of p70S6K. U0126 was shown to decrease the phosphorylation of ERK1/2 but increase the phosphorylation of p70S6K. However, increased ratios of LC3-II/LC3-I induced by 17β-estradiol was completely suppressed by co-treatment with U0126 (Figure 3F,G,H).
Discussion
Over the past decades, AMPK has emerged as a key mediator that regulates cellular energy metabolism in response to stress, cytokines and nutrients, which has been linked with many physiological and pathological processes (4). OA is the most common chronic and degenerative disease of the joint, which affects millions of people worldwide (2,3). Compromised AMPK activity in chondrocytes has been linked to the development of OA. It has been reported that AMPKα activity in OA patients and OA animal models was greatly reduced (8,9). Our previous study provided in vivo evidence that AMPK deficiency is correlated with OA development (11). Interestingly, our data also showed that AMPKα cDKO females did not exhibit more severe cartilage deterioration in DMM-induced OA compared with their WT control, whereas AMPK mutant females developed severe OA after OVX compared with OVX-sham AMPKα cDKO mice and OVX-sham WT. In the present study, to the best of our knowledge, it is the first time to demonstrate that estrogen can protect articular cartilage from damage during OA development by promoting chondrocytes autophagy via ERK-mTOR signaling.
OA is a slow progressing disease resulting in articular cartilage deterioration and DMM is a widely used OA model in animals to mimic the development of OA in human, which is characterized by articular cartilage destruction, little or no synovitis and osteophyte formation (7). In the present study, we provide comprehensive in vivo and in vitro evidence that estrogen may compensate for AMPK dysfunction during OA development.
In response to stress like energy depletion, AMPK is activated and inhibits mTORC1 activity at the level of phosphorylates TSC2, leading to activation of autophagy (30). mTOR, the downstream target of AMPK, is a fundamental regulator of cell growth, survival, and metabolism (31,32). The inhibitory function of mTOR complex 1 (mTORC1) in autophagy is well established (33). Recent studies have demonstrated that cartilage-specific knockout of mTOR or injection of rapamycin would promote autophagy in articular cartilage and alleviate DMM-induced cartilage degradation (16,17). In this study, we showed that cartilage-specific deletion of AMPK did not exhibit exacerbated OA in female mice compared with WT littermates, whereas AMPK mutant females exhibited severe cartilage deterioration after OVX compared with OVX-sham AMPK cDKO females. Moreover, we reported that no significant difference was found with expression levels of LC3 between OVX-sham AMPK mutant and OVX-sham WT females, whereas a significant reduction in LC3 expression was shown in AMPK mutant mice after OVX, indicating that estrogen may stimulate chondrocytes autophagy to protect articular cartilage from DMM-induced OA in mouse model of AMPK deficiency.
There is accumulating evidence that estrogen played a protective role in the development of OA. Ma reported that OVX had a detrimental effect in the progression of DMM-induced OA while implanting 17β-estradiol pellet subcutaneously after OVX restored the severity of OA to the level of OVX-sham females, indicating that estrogen decreased the severity of OA in female mice (19). In vitro, physiological concentrations of 17β-estradiol not only alleviated the injury-related cell death and GAG release significantly in articular cartilage explants, but also enhanced the antioxidative effect of chondrocytes (22,34). Collectively, our in vivo results suggest that estrogen may have a protective effect on articular cartilage while the exact mechanism remains to be elucidated.
To further understand mechanism underlying the protective effect of estrogen on chondrocytes, we performed in vitro experiments. A recent study reported that estrogen could stimulate autophagy by inhibiting mTOR activity in the murine osteoblast-like MC3T3-E1 cells (29). In our study, we showed that 17β-estradiol inhibited mTOR activity and promoted human chondrocytes autophagy. We further revealed that 17β-estradiol-mediated inhibition of mTOR activity and elevation of autophagy activity were reversed by ERK inhibitor not AMPK inhibitor, respectively, suggesting the effect of 17β-estradiol is, at least partly, in an AMPK-independent manner. Thus, our findings indicate that chondroprotective effect of estrogen is involved in elevation of autophagy activity via inhibiting mTOR activity in an ERK signaling dependent-manner (Figure 4).
Figure 4.
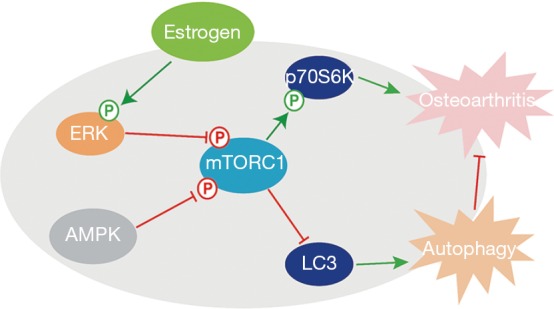
A schematic model of chondrocytes autophagy regulated by estrogen.
In summary, our results suggest that estrogen prevents articular cartilage destruction in a mouse model of AMPK deficiency by promoting chondrocyte autophagy via ERK-mTOR signalling and our findings also give new insight into the mechanism of the protective effect of estrogen on cartilage.
Acknowledgments
Funding: This work was supported by National Natural Science Foundation of China (No. 81472116), the Projects of International Cooperation and Exchanges NSFC (No. 81420108021), Key Program of NSFC (No. 81730067), Excellent Young Scholars NSFC (No. 81622033), Jiangsu Provincial Key Medical Center Foundation, Jiangsu Provincial Medical Outstanding Talent Foundation, Jiangsu Provincial Medical Youth Talent Foundation and Jiangsu Provincial Key Medical Talent Foundation.
Ethical Statement: The authors are accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All animal experimental operations were approved by Institutional Animal Care and Use Committee (IACUC) of the Model Animal Research Center of Nanjing University (Animal Care and Use Protocol Permit Number: JQ05). All human chondrocytes experiments were approved by the Human Ethics Committee of Drum Tower Hospital, School of Medicine, Nanjing University (No. 2009022).
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Takayama K, Ishida K, Matsushita T, et al. SIRT1 regulation of apoptosis of human chondrocytes. Arthritis Rheum 2009;60:2731-40. 10.1002/art.24864 [DOI] [PubMed] [Google Scholar]
- 2.Aigner T, Sachse A, Gebhard PM, et al. Osteoarthritis: pathobiology-targets and ways for therapeutic intervention. Adv Drug Deliv Rev 2006;58:128-49. 10.1016/j.addr.2006.01.020 [DOI] [PubMed] [Google Scholar]
- 3.Loeser RF, Goldring SR, Scanzello CR, et al. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheum 2012;64:1697-707. 10.1002/art.34453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Witczak CA, Sharoff CG, Goodyear LJ. AMP-activated protein kinase in skeletal muscle: from structure and localization to its role as a master regulator of cellular metabolism. Cell Mol Life Sci 2008;65:3737-55. 10.1007/s00018-008-8244-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Salminen A, Kaarniranta K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res Rev 2012;11:230-41. 10.1016/j.arr.2011.12.005 [DOI] [PubMed] [Google Scholar]
- 6.Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev 2009;89:1025-78. 10.1152/physrev.00011.2008 [DOI] [PubMed] [Google Scholar]
- 7.Glasson SS, Blanchet TJ, Morris EA. The surgical destabilization of the medial meniscus (DMM) model of osteoarthritis in the 129/SvEv mouse. Osteoarthritis Cartilage 2007;15:1061-9. 10.1016/j.joca.2007.03.006 [DOI] [PubMed] [Google Scholar]
- 8.Petursson F, Husa M, June R, et al. Linked decreases in liver kinase B1 and AMP-activated protein kinase activity modulate matrix catabolic responses to biomechanical injury in chondrocytes. Arthritis Res Ther 2013;15:R77. 10.1186/ar4254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Terkeltaub R, Yang B, Lotz M, et al. Chondrocyte AMP-activated protein kinase activity suppresses matrix degradation responses to proinflammatory cytokines interleukin-1β and tumor necrosis factor α. Arthritis Rheum 2011;63:1928-37. 10.1002/art.30333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou Y, Liu SQ, Yu L, et al. Berberine prevents nitric oxide-induced rat chondrocyte apoptosis and cartilage degeneration in a rat osteoarthritis model via AMPK and p38 MAPK signaling. Apoptosis 2015;20:1187-99. 10.1007/s10495-015-1152-y [DOI] [PubMed] [Google Scholar]
- 11.Zhou S, Lu W, Chen L, et al. AMPK deficiency in chondrocytes accelerated the progression of instability-induced and ageing-associated osteoarthritis in adult mice. Sci Rep 2017;7:43245. 10.1038/srep43245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gwinn DM, Shackelford DB, Egan DF, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 2008;30:214-26. 10.1016/j.molcel.2008.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caramés B, Taniguchi N, Otsuki S, et al. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis Rheum 2010;62:791-801. 10.1002/art.27305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 2011;13:132-41. 10.1038/ncb2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qin N, Wei L, Li W, et al. Local intra-articular injection of resveratrol delays cartilage degeneration in C57BL/6 mice by inducing autophagy via AMPK/mTOR pathway. J Pharmacol Sci 2017;134:166-74. 10.1016/j.jphs.2017.06.002 [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Vasheghani F, Li YH, et al. Cartilage-specific deletion of mTOR upregulates autophagy and protects mice from osteoarthritis. Ann Rheum Dis 2015;74:1432-40. 10.1136/annrheumdis-2013-204599 [DOI] [PubMed] [Google Scholar]
- 17.Takayama K, Kawakami Y, Kobayashi M, et al. Local intra-articular injection of rapamycin delays articular cartilage degeneration in a murine model of osteoarthritis. Arthritis Res Ther 2014;16:482. 10.1186/s13075-014-0482-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Srikanth VK, Fryer JL, Zhai G, et al. A meta-analysis of sex differences prevalence, incidence and severity of osteoarthritis. Osteoarthritis Cartilage 2005;13:769-81. 10.1016/j.joca.2005.04.014 [DOI] [PubMed] [Google Scholar]
- 19.Ma HL, Blanchet TJ, Peluso D, et al. Osteoarthritis severity is sex dependent in a surgical mouse model. Osteoarthritis Cartilage 2007;15:695-700. 10.1016/j.joca.2006.11.005 [DOI] [PubMed] [Google Scholar]
- 20.Sniekers YH, Weinans H, Bierma-Zeinstra SM, et al. Animal models for osteoarthritis: the effect of ovariectomy and estrogen treatment – a systematic approach. Osteoarthritis Cartilage 2008;16:533-41. 10.1016/j.joca.2008.01.002 [DOI] [PubMed] [Google Scholar]
- 21.Maneix L, Servent A, Poree B, et al. Up-regulation of type II collagen gene by 17beta-estradiol in articular chondrocytes involves Sp1/3, Sox-9, and estrogen receptor alpha. J Mol Med (Berl) 2014;92:1179-200. 10.1007/s00109-014-1195-5 [DOI] [PubMed] [Google Scholar]
- 22.Imgenberg J, Rolauffs B, Grodzinsky AJ, et al. Estrogen reduces mechanical injury-related cell death and proteoglycan degradation in mature articular cartilage independent of the presence of the superficial zone tissue. Osteoarthritis Cartilage 2013;21:1738-45. 10.1016/j.joca.2013.07.007 [DOI] [PubMed] [Google Scholar]
- 23.Chen M, Lichtler AC, Sheu TJ, et al. Generation of a transgenic mouse model with chondrocyte-specific and tamoxifen-inducible expression of Cre recombinase. Genesis 2007;45:44-50. 10.1002/dvg.20261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glasson SS, Chambers MG, Van Den Berg WB, et al. The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage 2010;18 Suppl 3:S17-23. 10.1016/j.joca.2010.05.025 [DOI] [PubMed] [Google Scholar]
- 25.Liu Q, Zhang X, Dai L, et al. Long noncoding RNA related to cartilage injury promotes chondrocyte extracellular matrix degradation in osteoarthritis. Arthritis Rheumatol 2014;66:969-78. 10.1002/art.38309 [DOI] [PubMed] [Google Scholar]
- 26.Dang ZC, Lowik CW. Removal of serum factors by charcoal treatment promotes adipogenesis via a MAPK-dependent pathway. Mol Cell Biochem 2005;268:159-67. 10.1007/s11010-005-3857-7 [DOI] [PubMed] [Google Scholar]
- 27.Liu Q, Hu X, Zhang X, et al. Effects of mechanical stress on chondrocyte phenotype and chondrocyte extracellular matrix expression. Sci Rep 2016;6:37268. 10.1038/srep37268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caramés B, Hasegawa A, Taniguchi N, et al. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann Rheum Dis 2012;71:575-81. 10.1136/annrheumdis-2011-200557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yang YH, Chen K, Li B, et al. Estradiol inhibits osteoblast apoptosis via promotion of autophagy through the ER-ERK-mTOR pathway. Apoptosis 2013;18:1363-75. 10.1007/s10495-013-0867-x [DOI] [PubMed] [Google Scholar]
- 30.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003;115:577-90. 10.1016/S0092-8674(03)00929-2 [DOI] [PubMed] [Google Scholar]
- 31.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012;149:274-93. 10.1016/j.cell.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Laplante M, Sabatini DM. mTOR signaling at a glance. J Cell Sci 2009;122:3589-94. 10.1242/jcs.051011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jung CH, Ro SH, Cao J, et al. mTOR regulation of autophagy. FEBS Lett 2010;584:1287-95. 10.1016/j.febslet.2010.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Claassen H, Schluter M, Schunke M, et al. Influence of 17beta-estradiol and insulin on type II collagen and protein synthesis of articular chondrocytes. Bone 2006;39:310-7. 10.1016/j.bone.2006.02.067 [DOI] [PubMed] [Google Scholar]