

Abstract
Background
Holobionts comprising nitrogen-fixing diazotrophs and phytoplankton or zooplankton are ubiquitous in the pelagic sea. However, neither the community structure of plankton-associated diazotrophs (PADs) nor their nitrogenase transcriptional activity are well-understood. In this study, we used nifH gene Illumina sequencing and quantitative PCR to characterize the community composition and nifH expression profile of PADs with > 100 μm size fraction in the euphotic zone of the northern South China Sea.
Results
The results of DNA- and RNA-derived nifH gene revealed a higher alpha-diversity in the active than in the total community. Moreover, the compositional resemblance among different sites was less for active than for total communities of PADs. We characterized the 20 most abundant OTUs by ranking the sum of sequence reads across 9 sampling stations for individual OTUs in both nifH DNA and RNA libraries, and then assessed their phylogenetic relatedness. Eight of the 20 abundant OTUs were phylogenetically affiliated with Trichodesmium and occurred in approximately equal proportion in both the DNA and RNA libraries. The analysis of nifH gene expression level showed uneven attribute of the abundance and nitrogenase activities by the remaining 12 OTUs. Taxa belonging to cluster III and Betaproteobacteria were present at moderate abundance but exhibited negligible nitrogenase transcription activity. Whereas, the abundances of Richelia, Deltaproteobacteria and Gammaproteobacteria were low but the contribution of these groups to nitrogenase transcription was disproportionately high.
Conclusions
The substantial variation in community structure among active dizatrophic fractions compared to the total communities suggests that the former are better indicators of biological response to environmental changes. Altogether, our study highlights the importance of rare PADs groups in nitrogen fixation in plankton holobionts, evidenced by their high level of nitrogenase transcription.
Electronic supplementary material
The online version of this article (10.1186/s12866-019-1565-9) contains supplementary material, which is available to authorized users.
Keywords: Symbiosis, Nitrogen fixation, Presence-absence, Transcriptional activity, Dormancy, South China Sea
Background
Nitrogen fixation by marine diazotrophs compensates for the loss of nitrogen through sinking and denitrification [1] and supports primary productivity in marine ecosystems [2–4]. The dominant N2 fixers in the world’s oceans are diazotrophic cyanobacteria, which are composed of three major groups: filamentous non-heterocyst-forming cyanobacteria, heterocystous cyanobacteria, and unicellular cyanobacteria [5]. Trichodesmium, a genus of filamentous non-heterocystous cyanobacteria, is thought to be the major contributor to marine nitrogen fixation globally [6–8]. As members of heterocystous cyanobacteria, the Richelia co-exist with diatoms and are commonly present in the open oceans [9, 10]. A recent investigation of a unicellular cyanobacteria group (UCYN-A) pointed out the need to re-evaluate the underestimated contribution of unicellular cyanobacteria to marine nitrogen fixation [11]. In recent years, the nifH gene, a gene encoding a subunit of nitrogenase enzyme, has been frequently used to explore the diversity of diazotroph [12]. In addition to autotrophic diazotrophic cyanobacteria, the nifH gene maker have identified highly diversified non-cyanobacterial heterotrophic diazotrophs in a wide oligotrophic aquatic environments [12, 13].
Holobionts comprising nitrogen-fixing diazotrophs and phytoplankton or zooplankton are ubiquitous in the pelagic sea [14, 15]. Trichodesmium in the holobionts form large colonies (millimeter size), and provide a micro-niche for diverse assemblages of microorganisms including autotrophic and heterotrophic diazotrophs. Despite that Trichodesmium is non-symbiotic diazotrophs, its colony-forming lifestyle sustains the growth of diverse symbiotic diazotrophic species [16, 17]. Various symbiotic strategies between plankton-associated diazotrophs (PADs) and their hosts facilitate marine biologic nitrogen fixation in the holobionts. Symbionts of diazotrophic cyanobacteria are exemplified by the genus Richelia, displaying symbiotic association with eukaryotic algae Rhizosolenia and other diatoms [9, 18–21]. The symbiotic strategies of autotrophic cyanobacterial diazotrophs, including symbiotic unicellular cyanobacteria [22], have been extensively documented. Nonetheless, the symbiotic phenomenon is not unique to autotrophic cyanobacterial diazotrophs and also exists in heterotrophic nitrogen-fixing bacteria [23–25]. This was demonstrated in a 15N-incorporation experiment that showed N2 fixation in the copepod gut by heterotrophic symbiotic diazotrophs, and the vital role of that symbiosis in marine nitrogen cycles [26].
The diversity and distribution of PADs have been extensively studied by means of DNA-derived nifH clone libraries [13, 24–26] and high-throughput sequencing [27]. However, due to the high degree of dormancy of environmental microbes [28, 29], the contribution of nitrogen fixation and community dynamics may be incorrectly estimated if based solely on data obtained from nifH DNA analysis [11, 30]. Instead, the use of both DNA- and RNA-derived assessments of microbial communities allows the additional identification of the rare but metabolically active microbes present in natural environments [31–33]. In the case of PADs, despite their key role in converting nitrogen in marine ecosystems, their nitrogenase transcription activity is poorly understood. Moreover, differences in the abundance and activity of different PADs groups have yet to be investigated.
Non-cyanobacteria predominated the bulk diazotrophic communities of the northern South China Sea in previous field studies [34, 35] whereas the community structure of the PADs remains largely unexplored. Thus, in the present study, we extended previous work and used high-throughput nifH gene amplicon sequencing to characterize the total and active communities of PADs inhabiting the northern South China Sea. In addition, by analyzing nifH gene expression, we were able to identify differences in nitrogenase transcription activity by the different phylogenetic groups of PADs depending on their symbiotic strategies.
Results
Alpha- and beta-diversity
We retrieved a total of 762,968 valid nifH sequences from the 18 DNA- and RNA-derived libraries. After quality filtering, the valid sequences (310–380 bp) were clustered into 731 OTUs at the 94% sequence similarity level. From a total of 731 OTUs, 153 OTUs were present in all nine samples of the DNA library, 109 OTUs in all nine samples of the RNA library, and 104 OTUs in both the DNA and the RNA libraries. For simplicity, we refer to nifH DNA-sequence composition as ‘total community’, while nifH RNA-sequence composition as ‘active community’ throughout the manuscript.
To evaluate the diversity of the diazotrophic assemblages in the total and active PADs communities, the Shannon index, Simpson index, Pielou evenness, and Chao 1 index, as alpha-diversity parameters, were determined in each sample based on the number of nifH-sequence-derived OTUs. Generally, each alpha-diversity parameter was significantly higher in the active than in the total communities (ANOVA, P < 0.05; Fig. 1), despite some variation in each diversity index among the stations (Additional file 1: Table S2).
Fig. 1.

Alpha-diversity of the total and active plankton-associated diazotrophs (PADs) communities. Shannon index (a), Simpson index (b), Pielou evenness (c) and Chao 1 index (d) of the total and active PADs community based on nifH-derived DNA and RNA libraries. The dots indicate the diversity index value of each sample
The beta-diversity of the total and active PADs communities was investigated as well, with the differences in between-group community composition visualized in a NMDS plot (Fig. 2a). The total PADs communities were taxonomically distinct according to their sampling origin (average Bray-Curtis distance, 0.06 ± 0.02). However, regardless of their origin, the total PADs communities clustered together in the NMDS scatter plot whereas the active PADs communities were more dispersed (average Bray-Curtis distance, 0.22 ± 0.11; Fig. 2a). To better understand the mean between-community differences in dependence of presence-absence or metabolic activity, a beta-dispersion analysis was performed to quantify the between-group variation within the total and active PADs communities. Between-community variation was significantly lower in the total PADs communities than in the active fractions (ANOVA, P < 0.001; Fig. 2b), suggesting that the differences in the diazotrophic communities across all stations with respect to the presence-absence of the nifH gene were smaller than the differences in nifH expression.
Fig. 2.

Beta-diversity of the total and active PADs communities. a Similarity among total and active PADs communities as determined by non-metric multidimensional scaling ordination. b Beta-dispersion illustrating the difference in the between-community variation of the nifH DNA and nifH RNA libraries. The significance of the between-community variation within total and active PADs was statistically tested by ANOVA
The composition and environmental relationships of diazotrophs
To visualize the phylogenetic diversity of the diazotrophs, a phylogenetic tree was constructed using the nifH sequences of the 20 most abundant OTUs (comprising 90.3% of the total nifH DNA and RNA sequences) and reference sequences (Fig. 3). The retrieved nifH sequences belonged to a wide range of diazotrophic groups, including Trichodesmium, heterocystous cyanobacteria, unicellular cyanobacteria, Gammaproteobacteria, Deltaproteobacteria, Betaproteobacteria and diazotrophs of cluster III (Fig. 3).
Fig. 3.
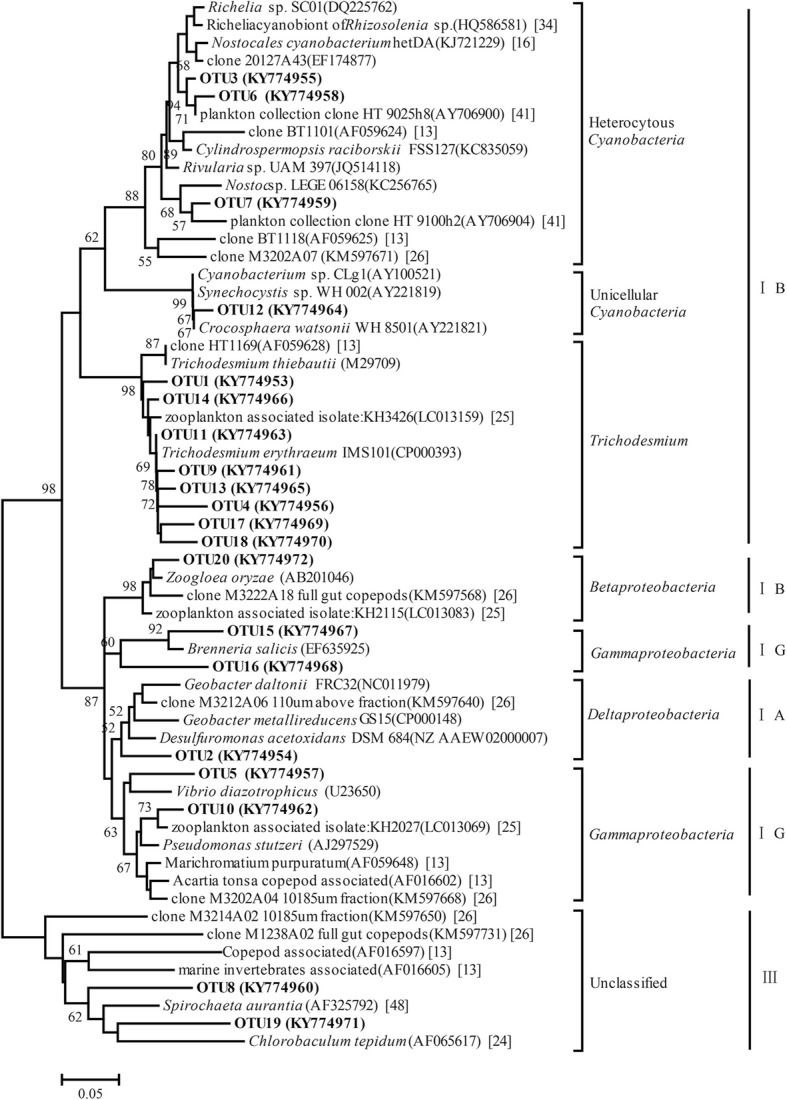
Unrooted neighbor-joining phylogenetic tree based on the nifH amino acid sequences of the 20 most abundant OTUs and reference sequences. The bootstrap values (> 50%) of relevant nodes are shown based on 1000 replicates. Sequences from this study are shown in bold. Scale bars: 5% of the estimated sequence divergence. The number in square brackets refers to the studies in which the reference sequences were previously reported
Trichodesmium predominated in both the total and the active fractions of the PADs communities across all stations, representing, on average, 98% and 87% of the nifH DNA and nifH RNA sequences, respectively (Fig. 4). Eight of the 20 most abundant OTUs were affiliated with Trichodesmium, thus indicating a divergent phylogenesis of the Trichodesmium nitrogenase gene (Fig. 3). Specifically, the relative abundance of Trichodesmium was relatively stable at all stations, ranging from 95.5 to 99.7% within the individual DNA libraries (Fig. 4). However, the variation in the relative abundance of this genus was more pronounced in the RNA libraries, ranging from 65.7% at station F to 99.4% at station B (Fig. 4).
Fig. 4.
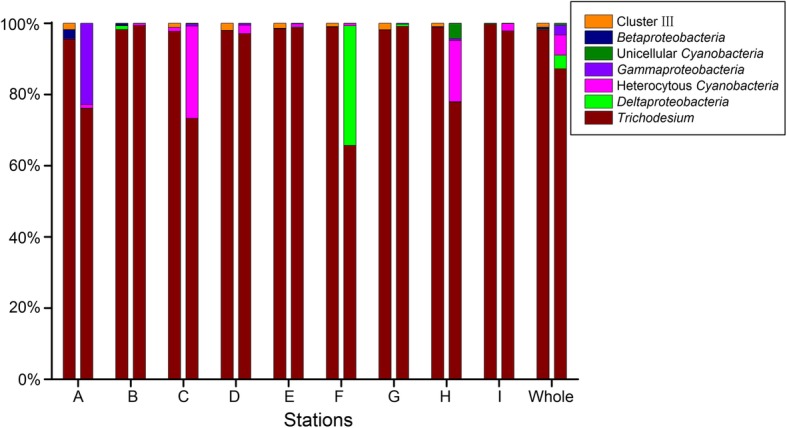
Composition of nifH taxa based on DNA- and RNA-derived libraries. For all stations (A–I), the left bars represent the DNA-derived libraries and the right bars the RNA-derived libraries; “whole” refers to the average relative abundance of all samples
Some diazotrophs were enriched in the RNA libraries compared to their relative abundance in the DNA libraries, while for others the opposite was the case (Fig. 4). For example, Richelia-like heterocystous cyanobacteria were absent or of lower abundance (0.2%) in the total PADs communities but reached moderate abundances in eight of the nine stations (Fig. 4). Plankton-associated Deltaproteobacteria were over-represented in the active PADs community of station F, and a similar pattern was observed for Gammaproteobacteria at station A (Fig. 4). Unicellular cyanobacteria, whose nifH sequences clustered together with those of Crocosphaera watsonii WH 8501 (Fig. 3), accounted for only 0.01% of the nifH DNA sequences, but their contribution to the nifH RNA sequences was higher (0.5%). Conversely, taxa affiliated with cluster III and Betaproteobacteria accounted for an average of 1.11% and 0.33%, respectively, of the abundance based on the DNA libraries across stations, but their abundances were negligible in the RNA libraries (Fig. 4).
We then explored the relationship between community composition and the corresponding environmental variables. Since the physical and chemical parameters of the water column vary with water depth and sampling site (see Additional file 1: Table S3 and Figure S1), separate RDA analyses were performed for surface and averaged (across depths) environmental parameters. Total and active PADs communities significantly correlated with sea surface pH, water column average pH, concentrations of average nitrate plus nitrite, and the average ammonium concentration (Additional file 1: Table S4). Several environmental factors significantly influenced the abundance and activity of Trichodesmium. Both total and active Trichodesmium highly correlated with sea surface pH (P = 0.019), water column average pH (P = 0.021), and marginally correlated with average nitrate plus nitrite concentrations (P = 0.100) (Additional file 1: Figure S3 and Table S4). However, there was no significant correlation between total and/or active heterocystous cyanobacteria and any of the environmental variables (Additional file 1: Figure S3).
Proportions of abundant OTUs
To assess whether the 20 most abundant OTUs accounted for a disproportionate amount of nifH gene activity, we compared the relative abundances (frequencies) of each OTU in the DNA and RNA libraries (Fig. 5). The relative abundance of OTU1 was > 50% in both libraries in all samples, while the remaining 19 OTUs were less abundant (Fig. 5). The frequencies of all eight OTUs affiliated with Trichodesmium were approximately equal in the DNA and RNA libraries, with disproportionate frequencies determined for the remaining 12 OTUs. For example, OTUs affiliated with members of heterocystous cyanobacteria, unicellular cyanobacteria, Gammaproteobacteria, and Deltaproteobacteria were of relatively high but varying frequencies in the nifH RNA libraries (Fig. 5) whereas the OTUs of members of the Betaproteobacteria and diazotrophs of cluster III were of negligible frequency in the RNA but not the DNA libraries (Fig. 5).
Fig. 5.
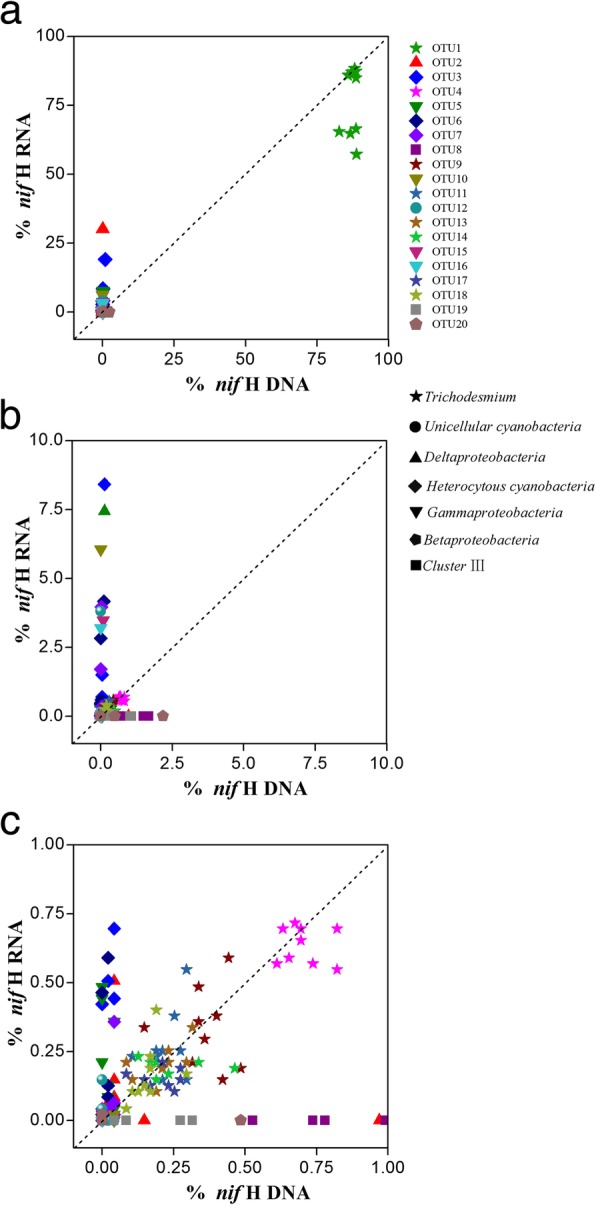
Relationship between nifH RNA and nifH DNA frequencies of the dominant OTUs. The panels show all the 20 most abundant OTUs (a) as well as abundant OTUs accounting for < 10% (b) and < 1% (c) of the abundance in both the DNA and RNA libraries. The dotted line indicates a 1:1 ratio. A deviation from the 1:1 line is expected for OTUs with a notably higher proportion in either the DNA or the RNA library, resulting in a distribution of the dots towards the x or y axis
NifH gene expression level
The nifH gene expression level of the bulk PADs communities ranged from 0.0042 to 0.52, with an average nifH RNA/DNA ratio of 0.17 (Fig. 6). To further quantify nifH gene expression by the different PADs groups, we calculated the nifH RNA/DNA ratio of the main groups by combining the bulk community nifH RNA/DNA ratio with the frequencies of the dominant groups. Heterocystous cyanobacteria were the most active group, based on nifH RNA/DNA ratios ranging from 0.39 to 13.82 (average nifH RNA/DNA ratio 3.51). Trichodesmium predominated in the total and active PADs communities (Fig. 4), but nifH gene expression by this genus (average nifH RNA/DNA ratio 0.15) was somewhat lower than that of the bulk community (0.17). Unicellular cyanobacteria, Deltaproteobacteria and Gammaproteobacteria were present at low abundances (Fig. 4) but exhibited high-level nifH expression (Fig. 6), whereas nifH mRNA expression by diazotrophs of cluster III and Betaproteobacteria was nearly undetectable. Due to high between-sample variations, which might be caused by environmental differences, there was no statistically significant difference in the nifH RNA/DNA ratio of the analyzed PADs groups.
Fig. 6.

The nifH mRNA expression level of the main groups. The expression ratio is based on the quantitative PCR results of each sample combined with the average taxonomic frequencies in the nifH DNA and RNA libraries. The dots represent the nifH RNA/DNA ratio of each sample. For some samples, the ratio is not provided because no sequence was found in the DNA libraries
Discussion
The aim of this study was to elucidate the community structure and nifH mRNA expression of diazotrophs associated with plankton with a particle size > 100 μm in northern South China Sea. Our results showed that the active PADs communities were taxonomically distinct from the total communities. Trichodesmium predominated among the PADs of the euphotic zone, with approximately equal abundances in the DNA and RNA libraries. Differences in the nifH gene expression levels of the different phylogenetic groups of diazotrophs may be related to the life forms and symbiotic strategies of these organisms.
Active communities are more diverse and divergent than total communities
In determinations of the microbial communities in a particular environment, higher community diversity in DNA- than in RNA-derived libraries is typical and has been demonstrated in several 16S rRNA-based studies [33, 36–39]. Here we directly tracked nifH-defined communities, because nifH gene may provide better insight into the diversity and composition of the communities that are capable of nitrogen fixation in an environment than those extrapolated from 16S rRNA data. However, our nifH sequence analysis revealed a higher alpha-diversity in the active (based on RNA) than in the total (based on DNA) communities of PADs (Fig. 1). The evenness of the total PADs communities was also lower than that of the metabolically active communities, as indicated by the very small number of highly dominant taxa. The nifH gene-based OTU abundances from the DNA analysis do not always predict which diazotrophic OTU is actively expressing nifH assessed in the RNA analysis. The former one is presumably considered a proxy for the dominant diazotrophs in a given sample, whereas the latter offers a representation of metabolically active diazotrophs with regard to nitrogen fixation processes. A high alpha diversity in a given community could be a result of high evenness. As such, a higher evenness obtained from nifH RNA-based OTU analysis accounts for closer number among individuals that were actively expressing nifH, relative to the communities comprising a few highly dominant diazotrophs detected in nifH DNA based approach. This is supported by the prevalence of Trichodesium in all DNA-derived communities and the decline in its abundance in the RNA-derived communities, whereas some taxa that were rare in the total communities were of moderate abundance in the active communities (Fig. 4). Thus, although the low-abundance groups represented a small proportion of the total PADs community biomass, they contributed a disproportionately high amount of the activity, presumably due to active transcription of nitrogenase mRNA under suitable growth conditions.
Previous studies have confirmed the dissimilarities between total and active diazotrophic communities [40, 41], but the variability within communities of total and active diazotrophshas not been quantitatively investigated. Given that changes occur faster in RNA than in DNA [42], we hypothesized a more pronounced variability in the active diazotrophic communities (RNA) across stations than in the total communities (DNA). Indeed, variations in the community structures of PADs among sampling sites were observed and were independent of the presence or activity of the cells. However, in accordance with our hypothesis, this variation was significantly higher in the active fraction of the diazotrophic communities than in the total communities (Fig. 2b). Moreover, the differences between the active communities at different sites were mainly caused by the replacement of taxa, rather than changes in the abundance of common taxa (Fig. 4), suggesting that different selective pressures determine the turnover in membership of metabolically active diazotrophs in the studied ecosystems. Accordingly, active diazotrophic assemblages may better reflect the response of PADs to environmental alterations.
A varying level of metabolic activity among rare taxa
Given that some taxa tend be to conditionally rare [43], investigations of membership turnover in rare but active populations of natural communities may shed light on microbial diversity and succession. In our study, rare but active PADs groups included heterocystous cyanobacteria, unicellular cyanobacteria, Deltaproteobacteria and Gammaproteobacteria (Figs. 4 and 6). Members of the heterocystous cyanobacterial genus Richelia were the most transcriptionally active group. The heterocystous cyanobacteria Richelia has been extensively reported as a diatom symbiote, including the investigations in the South China Sea [34, 35], and was identified to actively transfer nitrogen to their diatom hosts [15]. The association of diazotrophic unicellular cyanobacteria with phytoplankton has been the subject of recent studies [11, 27, 44], including a report describing a high nitrogen fixation rate and rapid nitrogen transfer from symbiotic unicellular cyanobacteria to their hosts, evidenced by N-isotope-based nano-SIMS technology [11]. The highly active transcription of nitrogenase mRNA by intracellular symbiotic Richelia and unicellular cyanobacteria detected in our study is in agreement with previous studies reporting high nitrogen fixation rates by those diazotrophs [11, 15]. Intracellular symbiotic strategies and nutrient exchange mechanisms may facilitate a high efficiency of nitrogen fixation by Richelia as well as unicellular cyanobacteria.
High nitrogenase mRNA transcription level was also detected in Deltaproteobacteria and Gammaproteobacteria, previously reported to be dominant diazotrophic groups [35, 45]. In our study, although Deltaproteobacteria was present at low abundance, its level of nifH mRNA transcription activity was high. This group was detected in copepods in previous studies [24, 26] whereas in the case of diazotrophic Gammaproteobacteria little is known about its symbiotic relationships. Fluorescence in situ hybridization studies of diazotrophic Gammaproteobacteria may lead to the identification of its host and thus provide insights into the ecological importance of this PADs group.
The inactive diazotrophic groups
Nitrogenase is a multi-component enzyme with several different forms, although the nifH gene encodes a highly conserved subunit [30]. However, the nifH gene also shares a similarity with nifH-like genes in non-diazotrophic organisms, such that many nifH sequences retrieved from environmental clones are in fact pseudogenes [46, 47]. In this study, nifH sequences belonging to cluster III were the second most abundant group in the nifH DNA libraries, but few transcripts were detected in the nifH RNA libraries for all stations (Figs. 4 and 5). Considering that the cluster III nifH sequences obtained in this study were not transcribed, whether they were pseudogenes or indeed belonged to inactive groups was unclear. The reference sequences that clustered closely with the sequences of cluster III (Fig. 3) originated from zooplankton and the termite gut [24, 48]. In addition, the nitrogenase activity of PADs was shown to be much higher in starved copepods than in full-gut copepods [26]. It was therefore hypothesized that, the presence of sufficient bio-available nitrogen in the zooplankton gut down-regulates nitrogenase gene expression [26]. Similarly, the cluster III diazotrophs found in our study might be those inactive members associated with zooplankton, which would imply that zooplankton-associated heterotrophic diazotrophs are suppliers of extra nitrogen for their hosts, especially under conditions of starvation.
Trichodesmium dominate the PADs
The results of microscopy-based cell counting showed a relatively lower Trichodesmium abundance (1.29 × 103–8.22 × 103 trichomes m− 2 in the euphotic waters, Additional file 1: Figure S2) in the South China Sea than previously reported [34, 35, 49]. The differences may have been due to the use of different sampling methods, as the net-tow (100-μm mesh) collection of plankton in our study would have excluded free-living or smaller aggregates of Trichodesmium cells, both of which are captured in previous studies using smaller pore-size filters (0.2-, 10- or 20-μm) [34, 35, 49]. Nevertheless, the sampling method used in our study was chosen to ensure collection of the PADs fraction while excluding free-living diazotrophs, as that different patterns for PADs and whole diazotrophic communities were expected.
Trichodesmium is one of the dominant diazotrophs in tropical and subtropical oceans [5, 6, 50]. In the South China Sea, heterotrophic Proteobacteria was shown to dominate the diazotrophic communities [34, 35]. However, consistent with the results of Farnelid and colleagues, who reported the predominance and enrichment of cyanobacteria, including Trichodesmium, in diazotrophic communities associated with sinking particles [27], we found that Trichodesmium accounted for nearly all of the PADs (~ 98%) in our samples. It seems that autotrophic cyanobacterial diazotrophs such as Trichodesmium tend to dominate in large particle-size fraction of the diazotrophic community inhabiting the euphotic-zone, while heterotrophic diazotrophs tend to be free-living. Alternatively, the disproportional enrichment of Trichodesmium in the > 100 μm size fraction could be resulted from colonization of their cells. Thus, our collection method revealed a difference in the community structure of bulk diazotrophs vs. PADs associated with large particles.
We also found that Trichodesmium accounted for the majority (87%) of the nitrogenase mRNA transcripts of PADs (Fig. 4). However, an analysis of the nifH RNA/DNA ratio showed that the nifH gene expression level of Trichodesmium was lower than that of rare but active groups (e.g., heterocystous cyanobacteria) (Fig. 6). In a previous isotope-based investigation, more than half of the Trichodesmium cells were incapable of fixing nitrogen [11]. Therefore, changes in the abundance of diazotrophic groups, determined using nifH DNA sequences, may not reflect changes in the nitrogen fixation activity of the respective members, whose ability to fix nitrogen varies. However, newly developed isotope techniques, especially NanoSIMS, allow the nitrogen fixation rate of specific groups to be investigated at the single cell level [11, 51].
Conclusions
In this study, the total and active communities of PADs in the northern South China Sea were inventoried, and the nifH gene expression level by different PADs groups were estimated. Active communities of PADs were significantly more divergent than the total communities. Overall, however, the PADs communities were taxonomically diverse, with different phylogenetic groups differing in their nifH mRNA transcription activity. Despite the low abundance of heterocystous cyanobacteria, unicellular cyanobacteria, Deltaproteobacteria, and Gammaproteobacteria, the nifH transcriptional activity of these groups was high, whereas Trichodesmiumwas highly abundant but its transcriptional activity was moderate, and Betaproteobacteria and diazotrophs of cluster III were of high abundance but of low nifH transcriptional activity. Our results extend those of previous studies by specifically characterizing the distribution, abundance, and transcriptional activity of PADs in the northern South China Sea.
Nonetheless, much remains to be learned about the diversity and structure of PADs communities, including the symbiotic relationships between the various species of PADs and their hosts. Determinations of the amount of nitrogen fixation contributed by each PADs group and its impact on both host metabolism and the functioning of marine ecosystems are needed as well. Manual sorting of plankton prior to amplification and sequencing is a feasible way to assess the specificity of the symbiotic relationships between PADs and host plankton species [25, 26]. The sequence information obtained in our study will aid in designing nucleotide probes for the in-situ visualization of specific diazotrophs in their plankton hosts. Furthermore, our findings highlight the importance of low-abundance PADs community members in understanding nitrogen fixation in marine ecosystems, but also the need for more detailed insights into the symbiotic strategies and contribution to nitrogen fixation of diazotrophic communities.
Methods
Study sites and sample collection
Seawater used for this study was collected on R/V Shiyan3 cruise WPOS (20 October–10 November 2014) to the northern South China Sea. Plankton samples were obtained at nine sites using a vertical haul with a 100-μm-mesh, 0.4-m-diameter plankton net (Fig. 7; see Additional file 1: Table S1 for more information on the sampling locations). The samples were rinsed with 0.22-μm-filtered seawater and the final volume was adjusted to 1 L. The collected samples were thoroughly mixed and a 250-ml aliquot was fixed with 0.1% Lugol’s iodine solution for Trichodesmium counting by microscopy [52]. The remaining volume of each sample was incubated in DNA/RNA Sample Protector Buffer (Takara BioInc. Japan), and stored at − 20 °C until nucleic acid extraction. Additionally, seawater samples were collected using a CTD system (Sea-Bird Scientific Inc. WA, USA) fitted with temperature, conductivity, pH and dissolved oxygen probes. The seawater was filtered through a 0.22-μm PEGF membrane (Millipore, Darmstadt, Germany) and then stored at − 20 °C for nutrient concentration measurements [53].
Fig. 7.

Location of the sampling stations. The map was generated using ODV software (Schlitzer, R., Ocean Data View, http://odv.awi.de, 2015)
Nucleic acid extraction, cDNA synthesis and Illunima sequencing
By centrifuging at 5000×g for 5 min, the supernatant of DNA/RNA protector buffer was removed from each sample prior to nucleic acid extraction. DNA and RNA were simultaneously extracted using a total DNA/RNA isolation kit (Omega Bio-tek, Inc. GA, USA) and their concentrations were measured using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific Inc. DE, USA). Genomic DNA was removed from the RNA extracts by DNase digestion (Takara BioInc. Japan) prior to cDNA synthesis using a PrimeScript RT reagent kit (Takara BioInc. Japan). The 20-μl reverse transcription reaction contained 2 μl (< 2 μg) of RNA. A negative control without reverse transcriptase was included to check for genomic DNA contamination of the cDNA.
The nifH gene of the DNA and cDNA were amplified using the primer pair PolF and PolR [54] combined with barcode sequences. Amplification was performed in reaction mixtures consisted of 25 μl Ex Taq (2×) (Takara BioInc. Japan), 1 μl of primer PolF (10 μM), 1 μl of primer PolR (10 μM) and 1 μl of DNA/cDNA in a 50-μl final volume. The thermal profile was as follows: initial denaturation at 94 °C for 5 min, followed by 35 cycles of denaturation at 94 °C for 30 s, annealing at 54 °C for 45 s and extension at 72 °C for 45 s, and final elongation at 72 °C for 10 min. Libraries were constructed from the purified PCR products of each sample. The 2 × 250-bp paired-ends sequencing was conducted on a HiSeq 2500 platform, Illumina (Genewiz, Suzhou, China).
Phylogenetic analysis
Quality trimming was done using Btrim with a quality score threshold to 20 [55]. Forward and reverse reads were merged using FLASH [56]; the overlaps of the merged reads more than 30 bp and mismatch less than 5% were kept, otherwise filtered out. Sequences with a length < 300 base pairs and ambiguous bases > 0 were removed. The forward and reverse primers were trimmed before further analysis. Operational taxonomic units (OTUs) were classified using UCLUST, based on 94% sequence similarity, as described previously [57]. Chimeric OTUs were removed using UCHIME in de novo mode [58]. Frame-shift errors in the sequences were checked and corrected using RDP FrameBot [59]. All libraries were subsampled with 100 iterations to 4746 sequence reads (smallest library size) to standardize uneven sequencing depths, resulting in a normalized OTUs table.
The validated nifH sequences were translated into amino acid sequences for phylogenetic analysis. Among the OTUs in the nifH DNA and RNA libraries, whose normalized reads in all samples were aggregated, the 20 most abundant OTUs across samples were assigned by ranking the aggregated reads. Representative sequences of the 20 most abundant OTUs (accounting for 90.3% of the total sequences) were checked against the closest related sequences in the GenBank database and in an ARB nifH database [60]. We selected the reference sequences based on the nucleotide sequence similarity and the score obtained from the BLAST: 1) sequences from the top 3–5 matching hits and 2) one sequence derived from pure culture isolates that scored highest. Based on these criteria, a total of 39 sequences were retrieved, where 26 sequences were derived from environmental clones, and the other 13 sequences from pure culture isolates. The nifH protein sequences of the 20 most abundant OTUs and 39 reference sequences were used to construct the phylogenetic tree. A neighbor-joining phylogenetic tree was constructed using MEGA software (v6.02) [61]. Bootstrap confidence values of the nodes were obtained by 1000 iterations. Substitution model of Poisson, uniform rates among sites and homogeneous pattern among lineages were used for MEGA. The taxonomic affiliation of the 20 abundant OTUs was assigned based on their phylogenetic relatedness to the reference sequences on the tree. Representative nifH gene sequences were deposited in the GenBank database under accession numbers KY774953–KY774972.
Quantification of nifH mRNA expression level
The primers PolF and PolR [54] were used to quantify the ratio of nifH mRNA/nifH DNA. The amplification was performed in 20-μl reaction mixtures containing 10 μl of SYBR Premix Ex TaqII (2×) (Takara BioInc. Japan), 0.8 μl of primer PolF (10 μM), 0.8 μl of primer PolR (10 μM) and 1 μl of DNA/cDNA sample. Quantitative PCR was carried out using the Light Cycler 480 System (Roche Molecular Systems, Inc. CA, USA) and the following PCR conditions: 2 min at 95 °C, 45 cycles of 94 °C for 30 s, 54 °C for 45 s, and 72 °C for 45 s. All reactions were performed in triplicate. The specificity of the amplification was confirmed by melting-curve analysis and electrophoresis in a 2% agarose gel.
The ratio of nifH mRNA and nifH DNA in the bulk communities was calculated according to Eq. 1:
| 1 |
where R is the resulting ratio of nifH mRNA/nifH DNA, E is the amplification efficiency, and ΔCt the cycle threshold difference between nifH mRNA and nifH DNA. The amplification efficiency was calculated using established standards, as described in [62]. The nifH RNA/DNA ratio of the main PADs groups was further calculated by multiplying the nifH RNA/DNA ratio obtained from the bulk community by the respective frequencies of the dominant groups (Eq. 2):
| 2 |
where f is the frequency of each group in either the RNA or the DNA library.
Statistical analysis
Diversity indexes were calculated based on 100 resampled OTUs tables. The Chao1 index, Shannon index (H), Simpson index (D), and Pielou evenness (J) were calculated to determine the alpha-diversity of the nifH-derived communities. Student’s t-test was used to identify the significance of the difference in the diversity indexes between the RNA and DNA libraries. A Monte Carlo permutation test was performed to evaluate the correlation between environmental attributes and nifH sequences. Differences in community composition among the nine sampling stations and between the total and active PADs communities were identified using non-metric multidimensional scaling (NMDS) with a Bray-Curtis dissimilarity distance. An analysis of beta-dispersion [63] was used to quantify between-community variations in the total and the active PADs communities, and a redundancy analysis (RDA) to explore the relationship between diazotrophic groups and environmental parameters. Statistical analyses in this study were performed using the R vegan package (v3.5.1) and an R-based pipeline, as previously described [62].
Additional file
Figure S1. Water quality profiles at each station as determined by the CTD sampler. Figure S2. Abundance of Trichodesmium in at the nine stations. Cell numbers were determined by microscopic counting. Figure S3. Redundancy analysis (RDA) ordination plot showing the relationships between environmental variables and taxa in the DNA and RNA libraries. Table S1. Geolocation of the stations, sampling depth, and sample volume. Table S2. Alpha-diversity indexes of the total and active communities in each sample. Table S3. Nutrient concentrations at the sampling stations. Table S4. Monte Carlo permutation test of the effects of environmental variables on the nifH-derived communities. Table S5. The CT values obtained from the technical triplicate of quantitative PCR. (PDF 899 kb)
Acknowledgments
We thank Dr. Jiexin Xu for CTD data processing, and all members of the R/V Shiyan3 for their work on the cruises. We thank two anonymous reviewers for their constructive comments on the earlier version of the manuscript.
Authors’ contributions
YYZ, JDD, YHT and QSY conceived the research. QSY, WGZ, YZZ and JL collected sample and performed the experiments. QSY and DDS performed data analysis. DDS, MA and QSY wrote the manuscript. QSY and DDS edited the manuscript. All authors approved the manuscript.
Funding
This research was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA13020300 and XDA11020202), the National Natural Science Foundation of China (41676107, 41276114, and 41676163), and the Science and Technology Planning Project of Guangdong Province, China (2017B030314052). The postdoctoral fellowship of DDS was funded by the Leibniz Institute for Baltic Sea Research (IOW) in Germany.
Availability of data and materials
The datasets used and/or analyzed for the present study are available from the corresponding author on reasonable request. The nucleotide sequences have been deposited in GenBank nucleotide database of NCBI under accession numbers (KY774953–KY774972).
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Dan-Dan Shen, Email: dand.shen@gmail.com.
Yan-Ying Zhang, Email: zyy@scsio.ac.cn.
References
- 1.Zehr JP, Kudela RM. Nitrogen cycle of the open ocean: from genes to ecosystems. Annu Rev Mar Sci. 2011;3:197–225. doi: 10.1146/annurev-marine-120709-142819. [DOI] [PubMed] [Google Scholar]
- 2.Zehr JP, Ward BB. Nitrogen cycling in the ocean: new perspectives on processes and paradigms. Appl Environ Microb. 2002;68(3):1015–1024. doi: 10.1128/AEM.68.3.1015-1024.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Karl DM, Letelier RM. Nitrogen fixation-enhanced carbon sequestration in low nitrate, low chlorophyll seascapes. Mar Ecol Prog Ser. 2008;364:257–268. [Google Scholar]
- 4.Karl DM, Church MJ, Dore JE, Letelier RM, Mahaffey C. Predictable and efficient carbon sequestration in the North Pacific Ocean supported by symbiotic nitrogen fixation. P Natl Acad Sci USA. 2012;109(6):1842–1849. doi: 10.1073/pnas.1120312109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zehr JP. Nitrogen fixation by marine cyanobacteria. Trends Microbiol. 2011;19(4):162–173. doi: 10.1016/j.tim.2010.12.004. [DOI] [PubMed] [Google Scholar]
- 6.Capone DG, Zehr JP, Paerl HW, Bergman B, Carpenter EJ. Trichodesmium, a globally significant marine cyanobacterium. Science. 1997;276(5316):1221–1229. [Google Scholar]
- 7.Carpenter EJ, Romans K. Major role of the cyanobacterium Trichodesmium in nutrient cycling in the North-Atlantic Ocean. Science. 1991;254(5036):1356–1358. doi: 10.1126/science.254.5036.1356. [DOI] [PubMed] [Google Scholar]
- 8.Carpenter EJ, Price CC. Marine oscillatoria (Trichodesmium) - explanation for aerobic nitrogen-fixation without heterocysts. Science. 1976;191(4233):1278–1280. doi: 10.1126/science.1257749. [DOI] [PubMed] [Google Scholar]
- 9.Carpenter EJ, Montoya JP, Burns J, Mulholland MR, Subramaniam A, et al. Extensive bloom of a N-2-fixing diatom/cyanobacterial association in the tropical Atlantic Ocean. Mar Ecol Prog Ser. 1999;185:273–283. [Google Scholar]
- 10.Villareal TA, Brown CG, Brzezinski MA, Krause JW, Wilson C. Summer diatom blooms in the north pacific subtropical gyre: 2008-2009. PLoS One. 2012;7(4):15. doi: 10.1371/journal.pone.0033109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martinez-Perez C, Mohr W, Loescher CR, Dekaezemacker J, Littmann S, et al. The small unicellular diazotrophic symbiont, UCYN-A, is a key player in the marine nitrogen cycle. Nat Microbiol. 2016;1(11):16163. doi: 10.1038/nmicrobiol.2016.163. [DOI] [PubMed] [Google Scholar]
- 12.Zehr JP, Jenkins BD, Short SM, Steward GF. Nitrogenase gene diversity and microbial community structure: a cross-system comparison. Environ Microbiol. 2003;5(7):539–554. doi: 10.1046/j.1462-2920.2003.00451.x. [DOI] [PubMed] [Google Scholar]
- 13.Zehr JP, Mellon MT, Zani S. New nitrogen-fixing microorganisms detected in oligotrophic oceans by amplification of Nitrogenase (nifH) genes. Appl Environ Microb. 1998;64(9):3444–3450. doi: 10.1128/aem.64.9.3444-3450.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bonnet S, Biegala IC, Dutrieux P, Slemons LO, Capone DG. Nitrogen fixation in the western equatorial Pacific: rates, diazotrophic cyanobacterial size class distribution, and biogeochemical significance. Global Biogeochem Cy. 2009;23(3):1874–1892. [Google Scholar]
- 15.Foster RA, Kuypers MM, Vagner T, Paerl RW, Musat N, et al. Nitrogen fixation and transfer in open ocean diatom-cyanobacterial symbioses. ISME J. 2011;5(9):1484–1493. doi: 10.1038/ismej.2011.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mague TH, Weare NM, Holmhans O. Nitrogen-fixation in north pacific ocean. Mar Biol. 1974;24(2):109–119. [Google Scholar]
- 17.Gomez F, Furuya K, Takeda S. Distribution of the cyanobacterium Richelia intracellularis as an epiphyte of the diatom Chaetoceros compressus in the western Pacific Ocean. J Plankton Res. 2005;27(4):323–330. [Google Scholar]
- 18.Momper LM, Reese BK, Carvalho G, Lee P, Webb EA. A novel cohabitation between two diazotrophic cyanobacteria in the oligotrophic ocean. ISME J. 2015;9(4):882–893. doi: 10.1038/ismej.2014.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frischkorn KR, Rouco M, Van Mooy BAS, Dyhrman ST. Epibionts dominate metabolic functional potential of Trichodesmium colonies from the oligotrophic ocean. ISME J. 2017;11(9):2090–2101. doi: 10.1038/ismej.2017.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Venrick EL. Distribution and significance of richelia-intracellularis schmidt in north pacific central gyre. Limnol Oceanogr. 1974;19(3):437–445. [Google Scholar]
- 21.Rai AN, Soderback E, Bergman B. Cyanobacterium-plant symbioses. New Phytol. 2000;147(3):449–481. doi: 10.1046/j.1469-8137.2000.00720.x. [DOI] [PubMed] [Google Scholar]
- 22.Bothe H, Tripp HJ, Zehr JP. Unicellular cyanobacteria with a new mode of life: the lack of photosynthetic oxygen evolution allows nitrogen fixation to proceed. Arch Microbiol. 2010;192(10):783–790. doi: 10.1007/s00203-010-0621-5. [DOI] [PubMed] [Google Scholar]
- 23.Proctor LM. Nitrogen-fixing, photosynthetic, anaerobic bacteria associated with pelagic copepods. Aquat Microb Ecol. 1997;12(2):105–113. [Google Scholar]
- 24.Braun ST, Proctor LM, Zani S, Mellon MT, Zehr JP. Molecular evidence for zooplankton-associated nitrogen-fixing anaerobes based on amplification of the nifH gene. FEMS Microbiol Ecol. 1999;28(3):273–279. [Google Scholar]
- 25.Azimuddin KM, Hirai J, Suzuki S, Haider MN, Tachibana A, et al. Possible association of diazotrophs with marine zooplankton in the Pacific Ocean. MicrobiologyOpen. 2016;5(6):1016–1026. doi: 10.1002/mbo3.385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Scavotto RE, Dziallas C, Bentzon-Tilia M, Riemann L, Moisander PH. Nitrogen-fixing bacteria associated with copepods in coastal waters of the North Atlantic Ocean. Environ Microbiol. 2015;17(10):3754–3765. doi: 10.1111/1462-2920.12777. [DOI] [PubMed] [Google Scholar]
- 27.Farnelid Hanna, Turk-Kubo Kendra, Ploug Helle, Ossolinski Justin E., Collins James R., Van Mooy Benjamin A. S., Zehr Jonathan P. Diverse diazotrophs are present on sinking particles in the North Pacific Subtropical Gyre. The ISME Journal. 2018;13(1):170–182. doi: 10.1038/s41396-018-0259-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones SE, Lennon JT. Dormancy contributes to the maintenance of microbial diversity. P Natl Acad Sci USA. 2010;107(13):5881–5886. doi: 10.1073/pnas.0912765107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lennon JT, Jones SE. Microbial seed banks: the ecological and evolutionary implications of dormancy. Nat Rev Microbiol. 2011;9(2):119–130. doi: 10.1038/nrmicro2504. [DOI] [PubMed] [Google Scholar]
- 30.Zehr JP, Turner PJ. Nitrogen fixation: Nitrogenase genes and gene expression. Method Microbiol. 2001;30:271–286. [Google Scholar]
- 31.Zhang Y, Zhao Z, Dai M, Jiao N, Herndl GJ. Drivers shaping the diversity and biogeography of total and active bacterial communities in the South China Sea. Mol Ecol. 2014;23(9):2260–2274. doi: 10.1111/mec.12739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wilhelm L, Besemer K, Fasching C, Urich T, Singer GA, et al. Rare but active taxa contribute to community dynamics of benthic biofilms in glacier-fed streams. Environ Microbiol. 2014;16(8):2514–2524. doi: 10.1111/1462-2920.12392. [DOI] [PubMed] [Google Scholar]
- 33.Klein AM, Bohannan BJ, Jaffe DA, Levin DA, Green JL. Molecular evidence for metabolically active bacteria in the atmosphere. Front Microbiol. 2016;7:772. doi: 10.3389/fmicb.2016.00772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang Y, Zhao Z, Sun J, Jiao N. Diversity and distribution of diazotrophic communities in the South China Sea deep basin with mesoscale cyclonic eddy perturbations. FEMS Microbiol Ecol. 2011;78(3):417–427. doi: 10.1111/j.1574-6941.2011.01174.x. [DOI] [PubMed] [Google Scholar]
- 35.Kong L, Jing H, Kataoka T, Sun J, Liu H. Phylogenetic diversity and spatio-temporal distribution of nitrogenase genes (nifH) in the northern South China Sea. Aquat Microb Ecol. 2011;65(1):15–27. [Google Scholar]
- 36.Romanowicz Karl J., Freedman Zachary B., Upchurch Rima A., Argiroff William A., Zak Donald R. Active microorganisms in forest soils differ from the total community yet are shaped by the same environmental factors: the influence of pH and soil moisture. FEMS Microbiology Ecology. 2016;92(10):fiw149. doi: 10.1093/femsec/fiw149. [DOI] [PubMed] [Google Scholar]
- 37.Freedman ZB, Romanowicz KJ, Upchurch RA, Zak DR. Differential responses of total and active soil microbial communities to long-term experimental N deposition. Soil Biol Biochem. 2015;90:275–282. [Google Scholar]
- 38.Hunt DE, Lin Y, Church MJ, Karl DM, Tringe SG, et al. Relationship between abundance and specific activity of bacterioplankton in open ocean surface waters. Appl Environ Microb. 2012;79(1):177–184. doi: 10.1128/AEM.02155-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang J, Bao JT, Li XR, Liu YB. Molecular ecology of nifH genes and transcripts along a chronosequence in revegetated areas of the tengger desert. Microb Ecol. 2016;71(1):150–163. doi: 10.1007/s00248-015-0657-9. [DOI] [PubMed] [Google Scholar]
- 40.Short Steven M., Zehr Jonathan P. Methods in Enzymology. 2005. Quantitative Analysis of nifH Genes and Transcripts from Aquatic Environments; pp. 380–394. [DOI] [PubMed] [Google Scholar]
- 41.Church MJ, Short CM, Jenkins BD, Karl DM, Zehr JP. Temporal patterns of nitrogenase gene (nifH) expression in the oligotrophic North Pacific Ocean. Appl Environ Microb. 2005;71(9):5362–5370. doi: 10.1128/AEM.71.9.5362-5370.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.De Vrieze J, Regueiro L, Props R, Vilchez-Vargas R, Jauregui R, et al. Presence does not imply activity: DNA and RNA patterns differ in response to salt perturbation in anaerobic digestion. Biotechnol Biofuels. 2016;9:244. doi: 10.1186/s13068-016-0652-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shade A, Jones SE, Caporaso JG, Handelsman J, Knight R, et al. Conditionally rare taxa disproportionately contribute to temporal changes in microbial diversity. mBio. 2014;5(4):e01371–e01314. doi: 10.1128/mBio.01371-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Biegala IC, Raimbault P. High abundance of diazotrophic picocyanobacteria (<3 μm) in a Southwest Pacific coral lagoon. Aquat Microb Ecol. 2008;51:45–53. [Google Scholar]
- 45.Moisander PH, Serros T, Paerl RW, Beinart RA, Zehr JP. Gammaproteobacterial diazotrophs and nifH gene expression in surface waters of the South Pacific Ocean. ISME J. 2014;8(10):1962–1973. doi: 10.1038/ismej.2014.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mazard SL, Fuller NJ, Orcutt KM, Bridle O, Scanlan DJ. PCR analysis of the distribution of unicellular cyanobacterial diazotrophs in the Arabian Sea. Appl Environ Microb. 2004;70(12):7355–7364. doi: 10.1128/AEM.70.12.7355-7364.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mehta MP, Huber JA, Baross JA. Incidence of novel and potentially archaeal nitrogenase genes in the deep Northeast Pacific Ocean. Environ Microbiol. 2005;7(10):1525–1534. doi: 10.1111/j.1462-2920.2005.00836.x. [DOI] [PubMed] [Google Scholar]
- 48.Lilburn TC, Kim KS, Ostrom NE, Byzek KR, Leadbetter JR, et al. Nitrogen fixation by symbiotic and free-living spirochetes. Science. 2001;292(5526):2495–2498. doi: 10.1126/science.1060281. [DOI] [PubMed] [Google Scholar]
- 49.Wu J, Chung SW, Wen LS, Liu KK, Chen YL, et al. Dissolved inorganic phosphorus, dissolved iron, and Trichodesmiumin the oligotrophic South China Sea. Global Biogeochem Cy. 2003;17(1):1–10. [Google Scholar]
- 50.Villareal TA. Abundance and photosynthetic characteristics of Trichodesmium spp. along the Atlantic barrier reef at carrie bow cay, Belize. Mar Ecol. 1995;16(3):259–271. [Google Scholar]
- 51.Behrens S, Losekann T, Pett-Ridge J, Weber PK, Ng WO, et al. Linking microbial phylogeny to metabolic activity at the single-cell level by using enhanced element labeling-catalyzed reporter deposition fluorescence in situ hybridization (EL-FISH) and NanoSIMS. Appl Environ Microb. 2008;74(10):3143–3150. doi: 10.1128/AEM.00191-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guillou L, Nezan E, Cueff V, Erard-Le Denn E, Cambon-Bonavita MA, et al. Genetic diversity and molecular detection of three toxic dinoflagellate genera (Alexandrium, Dinophysis, and Karenia) from French coasts. Protist. 2002;153(3):223–238. doi: 10.1078/1434-4610-00100. [DOI] [PubMed] [Google Scholar]
- 53.Huang L, Tan Y, Song X, Huang X, Wang H, et al. The status of the ecological environment and a proposed protection strategy in Sanya Bay, Hainan Island. China Mar Pollut Bull. 2003;47(1–6):180–186. doi: 10.1016/S0025-326X(03)00070-5. [DOI] [PubMed] [Google Scholar]
- 54.Poly F, Monrozier LJ, Bally R. Improvement in the RFLP procedure for studying the diversity of nifH genes in communities of nitrogen fixers in soil. Res Microbiol. 2001;152(1):95–103. doi: 10.1016/s0923-2508(00)01172-4. [DOI] [PubMed] [Google Scholar]
- 55.Kong Y. Btrim: a fast, lightweight adapter and quality trimming program for next-generation sequencing technologies. Genomics. 2011;98(2):152–153. doi: 10.1016/j.ygeno.2011.05.009. [DOI] [PubMed] [Google Scholar]
- 56.Magoc T, Salzberg SL. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27(21):2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tu Q, Deng Y, Yan Q, Shen L, Lin L, et al. Biogeographic patterns of soil diazotrophic communities across six forests in the North America. Mol Ecol. 2016;25(12):2937–2948. doi: 10.1111/mec.13651. [DOI] [PubMed] [Google Scholar]
- 58.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27(16):2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang Q, Quensen JF, Fish JA, Lee TK, Sun Y, et al. Ecological patterns of nifH genes in four terrestrial climatic zones explored with targeted metagenomics using FrameBot, a new informatics tool. mBio. 2013;4(5):e00592–e00513. doi: 10.1128/mBio.00592-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heller P, Tripp HJ, Turk-Kubo K, Zehr JP. ARBitrator: a software pipeline for on-demand retrieval of auto-curated nifH sequences from GenBank. Bioinformatics. 2014;30(20):2883–2890. doi: 10.1093/bioinformatics/btu417. [DOI] [PubMed] [Google Scholar]
- 61.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol. 2013;30(12):2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Y, Yang Q, Ling J, Van Nostrand JD, Shi Z, et al. The shifts of diazotrophic communities in spring and summer associated with coral galaxea astreata, pavona decussata, and porites lutea. Front Microbiol. 2016;7:1870. doi: 10.3389/fmicb.2016.01870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Anderson MJ. Distance-based tests for homogeneity of multivariate dispersions. Biometrics. 2006;62(1):245–253. doi: 10.1111/j.1541-0420.2005.00440.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Water quality profiles at each station as determined by the CTD sampler. Figure S2. Abundance of Trichodesmium in at the nine stations. Cell numbers were determined by microscopic counting. Figure S3. Redundancy analysis (RDA) ordination plot showing the relationships between environmental variables and taxa in the DNA and RNA libraries. Table S1. Geolocation of the stations, sampling depth, and sample volume. Table S2. Alpha-diversity indexes of the total and active communities in each sample. Table S3. Nutrient concentrations at the sampling stations. Table S4. Monte Carlo permutation test of the effects of environmental variables on the nifH-derived communities. Table S5. The CT values obtained from the technical triplicate of quantitative PCR. (PDF 899 kb)
Data Availability Statement
The datasets used and/or analyzed for the present study are available from the corresponding author on reasonable request. The nucleotide sequences have been deposited in GenBank nucleotide database of NCBI under accession numbers (KY774953–KY774972).