

Abstract
Background
The aim of the current work was to determine thermodynamical properties of 5-(nitrophenyl)-2-furaldehyde oximes and 3-[5-(nitrolphenyl)-2-furyl]acrylic acids.
Results
The temperature dependences of saturated vapor pressures of 5-(nitrophenyl)-2-furaldehyde oximes and 3-[5-(nitrolphenyl)-2-furyl]acrylic acids were determined by the Knudsen effusion method. The results are presented by the Clapeyron–Clausius equation in linear form, and via this form, the standard enthalpies of sublimation of compounds were calculated at 298.15 K. The standard molar formation enthalpies of compounds in crystalline state at 298.15 K were determined indirectly from the corresponding standard molar combustion enthalpy, obtained using combustion bomb calorimetry. The non-nearest neighbour interactions (strain) in molecule were defined. The ideal-gas enthalpies of investigated compounds formation and the data available from the literature were used for calculation of group-additivity parameters and the correction terms useful in the application of the Benson correlation.
Conclusion
Determining the thermodynamic properties for these compounds will contribute to solving practical problems pertaining to optimization processes of their synthesis, purification and application. It will also provide a more thorough insight regarding the theoretical knowledge of their nature and are necessary for the application of the Benson group-contribution correlation for calculation of (g)calc.
Electronic supplementary material
The online version of this article (10.1186/s13065-019-0619-2) contains supplementary material, which is available to authorized users.
Keywords: Arylfuran derivatives, Vapor pressure, Combustion enthalpy, Formation enthalpy, Sublimation enthalpy, Isomerization, Group-additivity correlation
Introduction
The rapid growth of pharmaceutical and chemical industries using nitrogen-containing heterocyclic compounds requires a continuous diversification of these products. New synthesized compounds with complex structure have no description of their thermodynamic properties. Recently, numerous reactions of synthesis of nitrogen-containing compounds with a phenyl furan fragment, which exhibit various types of biological activity, have been investigated. This allows them to be widely used in various fields of medical chemistry [1–10].
The furfural oximes are used as inhibitors of soil nitrification [1], as intermediates in the synthesis of anti-TB [11] and antifungal [12] drugs, and also as starting materials for the synthesis of disubstituted derivatives of furan [13]. Phenyl derivatives of furfural oxime show antispasmodic [14], vasodilator [15], cardiotropic [8] and antiviral [9] properties.
3-(2-Furyl)acrylic acids are widely used in the synthesis of polymeric materials for the production of polymeric glass, light stabilizers and luminophores [10], as well as for the synthesis of compounds with antimicrobial properties [16].
Previously, we have published several studies on thermodynamic properties of 5-(nitrophenyl)-2-furaldehydes and ethyl esters of cyan acrylic acids [17–19].
This paper follows this course and concerns the investigation of 5-(nitrophenyl)-2-furaldehyde oximes and 3-[5-(nitrolphenyl)-2-furyl] acrylic acids.
The analysis of the properties of positional isomers of disubstituted benzene derivatives shows that a change in the functional group position in an aromatic ring can substantially change the applied properties of compounds, whereas the change in their thermodynamic properties is often unknown. Therefore, the aim of this work is to investigate enthalpy properties of 5-(nitrophenyl)-2-furaldehyde oximes and 3-[5-(nitrolphenyl)-2-furyl]acrylic acids differ in the position of the nitro group.
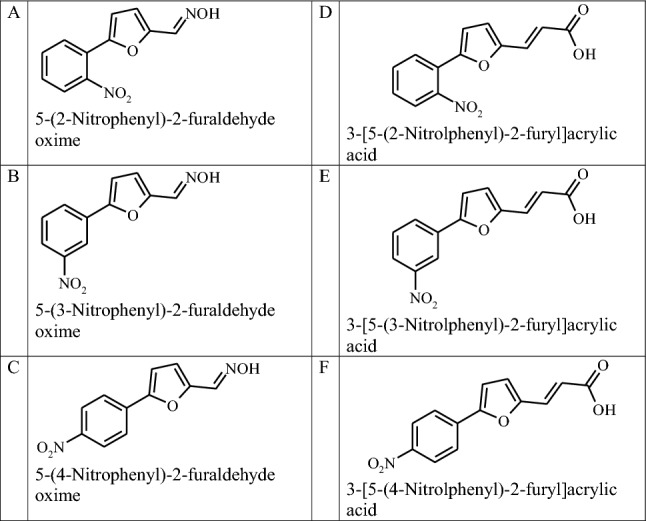
Investigated 5-(2-nitrophenyl)-2-furaldehyde oxime (A), 5-(3-nitrophenyl)-2-furaldehyde oxime (B), 5-(4-nitrophenyl)-2-furaldehyde oxime (C), 3-[5-(2-nitrolphenyl)-2-furyl]acrylic acid (D), 3-[5-(3-nitrolphenyl)-2-furyl] acrylic acid (E) and 3-[5-(4-nitrolphenyl)-2-furyl] acrylic acid (F) (Table 1) are crystalline substances under normal conditions.
Table 1.
Names and structural formulas of investigated compounds
Thermodynamic properties allow finding the most energetically favourable ways of synthesis and application of compounds with the maximum economic benefit.
Joint analysis of thermodynamic properties of 5-(nitrophenyl)-2-furaldehyde oximes and 3-[5-(nitrolphenyl)-2-furyl] acrylic acids will reveal many theoretically important patterns of mutual influence of atoms in a molecule and enable to calculate the formation enthalpies of free radicals, energy relations, tension, cyclization, determine the group contributions to the additive schemes.
Results and discussion
Effusion measurements
Primary effusion measurement results, including the saturated vapor pressure P of the researched compounds, are shown in Table 2. The measurement results were processed by the least squares method and presented as a linear equation:
Table 2.
Results of effusion measurements of investigated substances
| T, K | τ, s | m·103, g | P, Pa | m·103, g | P, Pa | m·103, g | P, Pa |
|---|---|---|---|---|---|---|---|
| Membrane № 1 | Membrane № 2 | Membrane № 3 | |||||
| 5-(2-Nitrophenyl)-2-furaldehyde oxime | |||||||
| 382.6 | 7350 | 1.50 | 0.097 | 1.60 | 0.100 | 1.50 | 0.097 |
| 386.1 | 7285 | 2.45 | 0.161 | 2.60 | 0.165 | 2.40 | 0.158 |
| 391.4 | 3655 | 1.90 | 0.250 | 2.00 | 0.255 | 2.00 | 0.264 |
| 393.3 | 3643 | 2.65 | 0.350 | 2.50 | 0.320 | 2.50 | 0.332 |
| 396.4 | 3643 | 4.05 | 0.538 | 4.15 | 0.534 | 3.90 | 0.520 |
| 402.1 | 3660 | 6.05 | 0.805 | 6.55 | 0.845 | 6.30 | 0.842 |
| 405.3 | 3640 | 10.2 | 1.37 | 10.4 | 1.35 | 9.70 | 1.31 |
| 5-(3-Nitrophenyl)-2-furaldehyde oxime | |||||||
| 390.0 | 10,827 | 2.50 | 0.111 | 2.60 | 0.112 | 2.60 | 0.116 |
| 394.1 | 10,828 | 3.90 | 0.174 | 4.00 | 0.173 | 3.90 | 0.174 |
| 395.7 | 10,822 | 4.70 | 0.210 | 4.90 | 0.212 | 4.80 | 0.215 |
| 399.4 | 10,821 | 6.70 | 0.301 | 6.80 | 0.296 | 6.70 | 0.302 |
| 402.1 | 10,825 | 8.90 | 0.401 | 9.00 | 0.392 | 8.70 | 0.393 |
| 406.0 | 7240 | 8.20 | 0.555 | 8.40 | 0.550 | 8.10 | 0.550 |
| 410.2 | 10,830 | 19.6 | 0.891 | 21.0 | 0.924 | 20.0 | 0.912 |
| 414.0 | 10,838 | 27.5 | 1.25 | 28.5 | 1.26 | 27.7 | 1.27 |
| 419.8 | 7222 | 30.2 | 2.08 | 29.9 | 2.00 | 29.5 | 2.04 |
| 5-(4-Nitrophenyl)-2-furaldehyde oxime | |||||||
| 414.8 | 3619 | 1.75 | 0.239 | 1.80 | 0.238 | 1.75 | 0.240 |
| 417.6 | 3619 | 2.40 | 0.329 | 2.50 | 0.332 | 2.40 | 0.330 |
| 419.6 | 3620 | 2.95 | 0.406 | 3.05 | 0.406 | 2.95 | 0.407 |
| 421.9 | 3696 | 3.75 | 0.506 | 3.85 | 0.504 | 3.75 | 0.508 |
| 423.1 | 3622 | 4.20 | 0.579 | 4.25 | 0.568 | 4.15 | 0.575 |
| 427.6 | 3622 | 6.60 | 0.915 | 6.80 | 0.914 | 6.55 | 0.912 |
| 429.1 | 3626 | 7.70 | 1.07 | 7.90 | 1.06 | 7.60 | 1.06 |
| 430.2 | 3600 | 8.35 | 1.17 | 8.55 | 1.16 | 8.30 | 1.17 |
| 432.1 | 3618 | 10.2 | 1.42 | 10.7 | 1.45 | 10.3 | 1.44 |
| 3-[5-(2-Nitrolphenyl)-2-furyl]acrylic acid | |||||||
| 422.8 | 14,418 | 4.30 | 0.141 | 4.40 | 0.140 | 4.40 | 0.145 |
| 426.2 | 7218 | 3.30 | 0.217 | 3.50 | 0.223 | 3.30 | 0.218 |
| 426.6 | 7219 | 3.55 | 0.234 | 3.60 | 0.229 | 3.50 | 0.231 |
| 428.9 | 3620 | 2.30 | 0.302 | 2.40 | 0.306 | 2.30 | 0.304 |
| 429.7 | 7218 | 5.30 | 0.350 | 5.40 | 0.345 | 5.30 | 0.351 |
| 430.8 | 7218 | 6.20 | 0.410 | 6.30 | 0.404 | 6.00 | 0.398 |
| 432.2 | 7220 | 7.20 | 0.477 | 7.60 | 0.487 | 7.10 | 0.472 |
| 433.9 | 3619 | 4.60 | 0.609 | 4.70 | 0.603 | 4.50 | 0.598 |
| 434.7 | 7226 | 10.0 | 0.663 | 10.2 | 0.656 | 9.70 | 0.646 |
| 437.2 | 7223 | 13.5 | 0.899 | 13.9 | 0.896 | 13.7 | 0.915 |
| 3-[5-(3-Nitrolphenyl)-2-furyl]acrylic acid | |||||||
| 438.6 | 10,820 | 1.55 | 0.069 | 1.55 | 0.067 | 1.50 | 0.067 |
| 442.3 | 14,420 | 3.20 | 0.107 | 3.20 | 0.104 | 3.25 | 0.109 |
| 444.7 | 14,428 | 4.15 | 0.139 | 4.35 | 0.142 | 4.20 | 0.142 |
| 447.8 | 14,418 | 6.10 | 0.206 | 5.80 | 0.190 | 5.90 | 0.200 |
| 448.4 | 14,422 | 6.50 | 0.219 | 6.10 | 0.199 | 6.00 | 0.203 |
| 449.3 | 14,416 | 7.00 | 0.237 | 6.90 | 0.226 | 6.70 | 0.227 |
| 451.1 | 10,836 | 6.40 | 0.288 | 6.60 | 0.288 | 6.55 | 0.296 |
| 454.7 | 14,418 | 11.6 | 0.394 | 12.2 | 0.402 | 11.0 | 0.376 |
| 456.5 | 14,417 | 14.0 | 0.477 | 13.9 | 0.459 | 13.1 | 0.448 |
| 3-[5-(4-Nitrolphenyl)-2-furyl]acrylic acid | |||||||
| 441.4 | 21,624 | 0.65 | 0.015 | 0.65 | 0.014 | 0.65 | 0.015 |
| 446.2 | 14,435 | 0.80 | 0.027 | 0.80 | 0.026 | 0.80 | 0.027 |
| 448.6 | 14,400 | 1.10 | 0.037 | 1.20 | 0.039 | 1.15 | 0.039 |
| 452.9 | 14,427 | 1.80 | 0.061 | 1.90 | 0.062 | 1.70 | 0.058 |
| 454.2 | 14,424 | 1.95 | 0.066 | 1.95 | 0.064 | 1.95 | 0.067 |
| 455.1 | 14,422 | 2.35 | 0.080 | 2.35 | 0.077 | 2.30 | 0.079 |
| 457.8 | 14,423 | 3.10 | 0.106 | 3.10 | 0.102 | 2.90 | 0.099 |
| 460.2 | 14,422 | 3.95 | 0.135 | 4.00 | 0.132 | 3.85 | 0.132 |
| 463.7 | 14,422 | 6.00 | 0.206 | 6.30 | 0.209 | 6.10 | 0.210 |
lnP (Pa) = A + B/T with correlation coefficient ρ, by means of which the standard molar enthalpies = B·R were calculated at average temperatures of measurement interval (Table 3). The error of all experimentally determined thermodynamic values was calculated with the Student’s confidence coefficient of 95%.
Table 3.
Coefficients of a linear equation: ln P (Pa) = A + B/T. standard sublimation enthalpie of investigated substances
| Compound | Tm, K | A | − B, K | ρ | , kJ mol−1 |
|---|---|---|---|---|---|
| A | 394.0 | 43.2 ± 1.8 | 17,394 ± 726 | 0.9934 | 144.6 ± 6.0 |
| B | 404.9 | 38.83 ± 0.61 | 15,990 ± 248 | 0.9988 | 132.9 ± 2.4 |
| C | 423.5 | 42.9 ± 0.36 | 18,367 ± 152 | 0.9996 | 152.7 ± 1.3 |
| D | 430.0 | 54.38 ± 0.50 | 23,793 ± 216 | 0.9992 | 197.8 ± 2.0 |
| E | 447.5 | 46.3 ± 1.5 | 21,475 ± 673 | 0.9951 | 178.5 ± 5.6 |
| F | 452.5 | 50.2 ± 1.3 | 24,011 ± 579 | 0.9971 | 199.6 ± 4.8 |
Standard enthalpies of sublimation can be adjusted to 298.15 K by the equations:
| 1 |
The changes of standard phase transitions heat capacity values at indicated temperature ranges for the probed compounds are unknown. Therefore, Eq. (2) was used to calculate the enthalpies of sublimation at 298.15 K [20].
| 2 |
Heat capacities in solid state were calculated by the additive method [20] and were equal: (298.15 K) = 260.4 J mol−1 K−1; (298.15 K) = 300.7 J mol−1 K−1. Standard enthalpies of sublimation at the mean experiment temperatures are shown in Table 3.
Calorimetric measurements
Combustion energy ∆cU(cpd) of the investigated substances was calculated by the equation:
| 3 |
where m(cpd)—compound weight determined using gas analysis; ΔUΣ = W·∆T—total heat released in the experiment, W—energy equivalent of the calorimetric system, ∆T—real increase in temperature. Calculations were performed taking into account corrections for the combustion of cotton thread ΔUfuse, terylene container ΔUter, soot to carbon dioxide ΔUcarbon and also for formation in a bomb nitric acid solution , using the following data for heats of combustion (J g−1): terylene 22,944.2 [21]; cotton thread 16704.2 [21]; soot to carbon dioxide − 32,763 [22]; formation of nitric acid 59 kJ mol−1 [22].
The results of determination of the compounds combustion energies ∆cU(cpd) are listed in Table 4, which apart from the above notation, contains values of combustion completeness regarding CO2 (the ratio of experimentally determined to calculated weights of CO2 mexp/mcal).
Table 4.
Results of combustion energy determination for investigated compounds
| m(cpd), g | ΔT, V | ΔUfuse, J | ΔUHNO3, J | ΔUcarbon, J | ΔUter, J | − ∆cU(cpd), J g−1 | m exp /m cal |
|---|---|---|---|---|---|---|---|
| 5-(2-Nitrophenyl)-2-furaldehyde oxime | |||||||
| 0.13947 | 0.25347 | 106.7 | 7.7 | 22.3 | 412.4 | 23,464 | 0.9996 |
| 0.18548 | 0.33140 | 119.5 | 11.8 | 27.6 | 473.7 | 23,510 | 1.0002 |
| 0.16006 | 0.28787 | 106.2 | 4.1 | 20 | 434.6 | 23,520 | 0.9985 |
| 0.20085 | 0.35193 | 124.4 | 8.9 | 39.5 | 427.2 | 23,515 | 0.9961 |
| 0.17080 | 0.30603 | 118.1 | 8.9 | 23.6 | 441.8 | 23,507 | 0.9998 |
| 0.17019 | 0.30680 | 111.2 | 9.4 | 38.2 | 488 | 23,510 | 0.9987 |
| 0.19456 | 0.34651 | 121.3 | 11.2 | 43.5 | 505.5 | 23,483 | 0.9959 |
| − ∆cU(cpd)average = 23,501 ± 17 J g−1 | |||||||
| 5-(3-Nitrophenyl)-2-furaldehyde oxime | |||||||
| 0.15358 | 0.31158 | 82.0 | 10.0 | 28.5 | 982.4 | 23,421 | 0.9968 |
| 0.26843 | 0.46052 | 94.5 | 14.8 | 28.2 | 499.8 | 23,400 | 0.9952 |
| 0.14936 | 0.27347 | 82.4 | 7.1 | 48.4 | 534.1 | 23,431 | 0.9998 |
| 0.14361 | 0.26148 | 121.2 | 7.1 | 25.1 | 424.5 | 23,456 | 1.0000 |
| 0.15737 | 0.28468 | 120.7 | 7.1 | 27.4 | 453.2 | 23,438 | 0.9987 |
| 0.18331 | 0.32799 | 99.5 | 8.9 | 28.2 | 510.2 | 23,400 | 0.9990 |
| 0.13068 | 0.24331 | 99.4 | 7.7 | 25.3 | 483.8 | 23,416 | 0.9991 |
| − ∆cU(cpd)average = 23,429 ± 16 J g−1 | |||||||
| 5-(4-Nitrophenyl)-2-furaldehyde oxime | |||||||
| 0.20572 | 0.36079 | 106.5 | 14.2 | 27.7 | 486.0 | 23,319 | 1.0000 |
| 0.12488 | 0.23304 | 104.1 | 4.7 | 23.9 | 473.6 | 23,335 | 0.9989 |
| 0.17515 | 0.34180 | 117.1 | 8.3 | 32.3 | 902.4 | 23,395 | 0.9999 |
| 0.21358 | 0.40512 | 120.3 | 13.0 | 27.2 | 946.7 | 23,335 | 0.9992 |
| 0.18755 | 0.36144 | 136.0 | 9.4 | 21.3 | 884.7 | 23,338 | 0.9999 |
| 0.24203 | 0.44750 | 127.9 | 13.6 | 31.7 | 913.2 | 23,324 | 0.9987 |
| 0.16330 | 0.32067 | 120.1 | 8.9 | 23.6 | 858.2 | 23,360 | 0.9965 |
| − ∆cU(cpd)average = 23,344 ± 22 J g−1 | |||||||
| 3-[5-(2-Nitrolphenyl)-2-furyl]acrylic acid | |||||||
| 0.31440 | 0.49265 | 82.0 | 3.5 | 29.9 | – | 23,172 | 0.9968 |
| 0.37839 | 0.59087 | 72.3 | 4.7 | 28.4 | – | 23,140 | 0.9992 |
| 0.33991 | 0.53119 | 85.7 | 8.9 | 53.3 | – | 23,165 | 0.9973 |
| 0.36358 | 0.56796 | 78.7 | 9.4 | 30.8 | – | 23,120 | 0.9965 |
| 0.41155 | 0.64234 | 74.8 | 9.4 | 30.8 | – | 23,127 | 0.9978 |
| 0.13428 | 0.21518 | 129.3 | 4.7 | 34.1 | – | 23,134 | 0.9995 |
| 0.12360 | 0.19809 | 113.3 | 4.1 | 28.2 | – | 23,160 | 0.9997 |
| − ∆cU(cpd)average = 23,145 ± 20 J g−1 | |||||||
| 3-[5-(3-Nitrolphenyl)-2-furyl]acrylic acid | |||||||
| 0.21935 | 0.3439 | 97.3 | 7.1 | 46.9 | – | 23,100 | 0.9985 |
| 0.23066 | 0.36164 | 103.8 | 8.9 | 24.9 | – | 23,062 | 0.9962 |
| 0.19197 | 0.30458 | 110.9 | 7.7 | 21.3 | – | 23,135 | 1.0000 |
| 0.27004 | 0.42269 | 80.2 | 13.0 | 24.3 | – | 23,069 | 0.9998 |
| 0.24827 | 0.39045 | 97.7 | 10.6 | 22.8 | – | 23,090 | 0.9986 |
| 0.24804 | 0.38918 | 84.7 | 12.4 | 28.5 | – | 23,103 | 0.9991 |
| 0.24000 | 0.37684 | 91.5 | 10.0 | 24.4 | – | 23,076 | 0.9979 |
| − ∆cU(cpd)average = 23,091 ± 22 J g−1 | |||||||
| 3-[5-(4-Nitrolphenyl)-2-furyl]acrylic acid | |||||||
| 0.16072 | 0.25432 | 106.7 | 5.9 | 22.8 | – | 23,020 | 0.9999 |
| 0.17773 | 0.27984 | 101.4 | 7.7 | 25.1 | – | 22,989 | 0.9999 |
| 0.23568 | 0.37014 | 115.7 | 10.0 | 27.2 | – | 22,985 | 0.9996 |
| 0.16126 | 0.25492 | 101.1 | 5.9 | 23.3 | – | 23,036 | 0.9998 |
| 0.19640 | 0.30968 | 99.9 | 8.3 | 23.0 | – | 23,062 | 0.9997 |
| 0.16949 | 0.26797 | 112.3 | 6.5 | 23.5 | – | 22,997 | 0.9999 |
| 0.16084 | 0.25462 | 102.3 | 4.7 | 20.3 | – | 23,050 | 0.9995 |
| − ∆cU(cpd)average = 23,020 ± 26 J g−1 | |||||||
The high rate of consistency of carbon dioxide content in substances (0.9952 to 1.0002) calculated by the formula (the results of its experimental determination are shown by the Rossini method) can also serve as an indirect confirmation of the sufficient purity of the compounds.
The absence of significant systematic errors while measuring at the calorimetry installation was confirmed by the coincidence of our results of combustion enthalpies (kJ/mol) of secondary etalons (salicylic acid) and biphenyl − 3026.6 ± 3.1 and − 6246.9 ± 7.3 with recommended ones: − 3025.0 ± 5.0 and − 6250 ± 20 respectively [23].
The standard combustion enthalpies (298.15K) of compounds were calculated taking into account the correction for the volume expansion work ∆nRT and the Washburn correction [24].
The calculation of formation enthalpies in condensed phase by Eq. (4) was based on the following key values of (kJ mol−1): − 285.830 ± 0.042 (H2O; l) and − 393.514 ± 0.046 (CO2; g) [25].
| 4 |
The gaseous state standard formation enthalpies of the investigated compounds were determined by the summation of the corresponding solid state formation enthalpies and their sublimation enthalpies according to the Eq. (5).
| 5 |
Standard combustion and formation (,) enthalpies of the investigated compounds are listed in Table 5.
Table 5.
Combustion and formation enthalpies of investigated compounds at 298.15 K (kJ/mol)
| Compound | δ | |||||
|---|---|---|---|---|---|---|
| exp | calc | |||||
| A | 5458.8 ± 4.0 | 13.1 ± 4.0 | 148.4 ± 7.5 | 135.3 ± 8.5 | 108.3 | 27.0 |
| B | 5442.0 ± 3.6 | 29.9 ± 3.6 | 137.2 ± 3.9 | 107.3 ± 5.3 | − 0.5 | |
| C | 5422.2 ± 5.2 | 49.7 ± 5.2 | 157.7 ± 2.7 | 108.0 ± 5.9 | − 0.3 | |
| D | 6003.0 ± 5.3 | 398.7 ± 5.3 | 203.9 ± 2.3 | − 194.8 ± 5.8 | − 256.7 | 61.9 |
| E | 5989.0 ± 5.7 | 412.9 ± 5.7 | 185.4 ± 6.5 | − 227.5 ± 8.6 | 29.2 | |
| F | 5970.6 ± 6.8 | 431.3 ± 6.8 | 206.7 ± 5.6 | − 224.6 ± 8.8 | 32.1 | |
The standard gaseous state formation enthalpies of compounds (g)calc are also calculated according to Benson additive scheme [26]. According to the scheme, the formation enthalpy of a compound in the gaseous state is the sum of the contributions of individual groups. Each group consists of a central atom and atoms of its first environment. Structural formulas of substances with indication of the serial number of increment, along with the values of group contributions (g)calc which were used for calculations of the formation enthalpies are shown in the Tables 6 and 7. Comparison of the experimentally obtained formation enthalpies and those calculated according to the Benson additive scheme allows us to determine the contributions of unknown groups to the additive scheme. In case of experimental and calculated values coincidence, it is possible to calculate these values for other representatives of the investigated compounds. In case of differences, one can find the interaction between non-nearest atoms in a molecule, predict the peculiarities of the compound structure and correct these interactions in Benson additive scheme.
Table 6.
Calculation of the formation enthalpies of the studied substances using the addictive Benson scheme for 5-nitrophenyl-2-furaldehyde oxime isomers
| Substance | № | Increment | ΔfH298.15, kJ mol−1 |
|---|---|---|---|
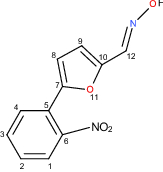
|
1–4 | Cb–(Cb)2(H) | 13.8 |
| 5 | Cb–(Cb)2(Cd) | 23.8 | |
| 6 | Cb–(Cb)2(NO2) | − 0.5 | |
| 7 | Cd–(Cd)(Cb)(O) | 59.7 | |
| 8,9 | Cd–(Cd)2(H) | 28.4 | |
| 10 | Cd–(Cd)2(O) | 43.4 | |
| 11 | O–(Cd)2 | − 137.2 | |
| 12 | Cd–(Cd)(H)(N–OH)* | 33.0 | |
| Furan cycle | − 25.9 | ||
ΔfH298,15 = 4·ΔfH298,15(Cb–(Cb)2(H)) + ΔfH298,15(Cb−(Cb)2(Cd)) + ΔfH298,15(Cb–(Cb)2(NO2)) + ΔfH298,15(Cd–(Cd)(Cb)(O)) + 2·ΔfH298,15(Cd–(Cd)2(H)) + ΔfH298,15(Cd–(Cd)2(O)) + ΔfH298,15(O–(Cd)2) + ΔfH298,15(Cd–(Cd)(H)(N–OH)) + ΔfH298,15(Furan cycle) = 4·13.8 + 23.8 − 0.5 + 59.7 + 2·28.4 + 43.4 − 137.2 + 33.0 − 25.9 = 108.3 kJ/mol
Table 7.
Calculation of the formation enthalpies of the studied substances using the addictive Benson schemefor 3-[5-nitrolphenyl-2-furyl]acrylic acid isomers
| Substance | № | Increment | ΔfH298.15, kJ mol−1 |
|---|---|---|---|
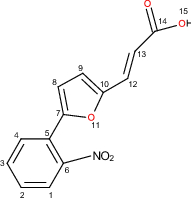
|
1–4 | Cb–(Cb)2(H) | 13.8 |
| 5 | Cb–(Cb)2(Cd) | 23.8 | |
| 6 | Cb–(Cb)2(NO2) | − 0.5 | |
| 7 | Cd–(Cd)(Cb)(O) | 59.7 | |
| 8,9,12 | Cd–(Cd)2(H) | 28.4 | |
| 10 | Cd–(Cd)2(O) | 43.4 | |
| 11 | O–(Cd)2 | − 137.2 | |
| 13 | Cd–(Cd)(H)(CO) | 32.1 | |
| 14 | CO–(Cd)(O) | − 140.2 | |
| 15 | O–(CO)(H) | − 252.3 | |
| Furan cycle | − 25.9 | ||
ΔfH298,15 = 4·ΔfH298,15(Cb–(Cb)2(H)) + ΔfH298,15(Cb–(Cb)2(Cd)) + ΔfH298,15(Cb–(Cb)2(NO2)) + ΔfH298,15(Cd–(Cd)(Cb)(O)) +3·ΔfH298,15(Cd–(Cd)2(H)) + ΔfH298,15(Cd–(Cd)2(O)) + ΔfH298,15(O–(Cd)2) + ΔfH298,15(Cd–(Cd)(H)(CO)) + ΔfH298,15(CO–(Cd)(O)) +ΔfH298,15(O–(CO)(H)) + ΔfH298,15(Furan cycle) = 4·13.8 + 23.8 − 0.5 + 59.7 + 3·28.4 + 43.4 − 137.2 + 32.1 − 140.2 − 252.3 − 25.9 = 256.7 kJ/mol
The investigated compounds are rather complex, therefore, some group contributions, necessary for the calculations of the formation enthalpies, are absent. Such contributions are marked with asterisks in Tables 6 and 7. They were calculated using reliable gaseous state formation enthalpies of the compounds. The contributions of the groups Cd–(Cd)2(O), C–(Cb)2(NO2) and Cd–(Cd)(O)(Cb) which are absent in the additive scheme were defined from the gaseous state formation enthalpy of vinylfuran [27], nitrobenzene [27] and ethyl-2-cyano-3-(furan-2-yl)-prop-2-enoate [18]. In addition, we had to use the contribution of the whole fragment (Cd)–CH=N–OH for calculations. This contribution is the sum of contributions of Cd–(Cd)(N)(H), N–(Cd)(OH), and OH–(N). According to the results published in [24], the contribution of Ni–(OH) group is approximately − 20.9 kJ/mol, but in the following works [28, 29] this group is absent at all. Contributions of the groups Cd–(Cd)(N)(H) and N–(Cd)(OH) in the Benson scheme are also absent. Therefore, the contribution of the fragment (Cd)–CH=N–OH was calculated from the formation enthalpy of furfural oxime, which we have defined in [30]. It is the optimal choice because the oximes A, B, C can be considered as its derivatives.
The values of the gaseous state standard formation enthalpies of the investigated compounds which were obtained experimentally expΔfH298.15(g) and calculated theoretically calcΔfH298.15(g), as well as the difference between them δ are given in Table 5. The resulting difference cannot be explained by the errors of experiments or calculations.
In Ref. [31] interactions between atoms which were by at least two other atoms were called non-nearest neighbour interactions. The result of such interactions caused in general a strain of a molecule. Authors define the strain of a molecule, as the difference between the experimental standard enthalpy of formation (g)exp and the calculated sum of the strain-free increments (g)calc. A strain of molecules between benzene and furan rings in tert-butylbenzene (9 kJ/mol) is determined [31]. It can exist in the compounds investigated by us. The strain of adjacent nitro and tert-butyl groups (− 25 kJ/mol) is defined in [31].
Moreover, there is an alternation of single and double conjugated bonds in the molecular structure of all studied compounds. It is known for such systems that the entire molecule is usually situated in the same plane due to the presence of common π-cloud [32]. To verify the assertion, we simulated the most energy-efficient spatial structures of the investigated molecules using the HyperChem software (PM3 geometry optimization method). Other computational calculations of different thermodynamical properties for various nitrogen-containing organical compounds can be found in [33–36].The results confirmed that for the oximes A, B, C the flat configuration of the molecule is the most energy-efficient. However, for the acids D, E, F the minimum internal energy of the molecules is observed when the chain of atoms –CH=CH–C(O)–OH is in a plane almost perpendicular to the furan cycle plane. An example of a geometric model for 3-(furan-2-yl) acrylic acid is shown in Fig. 1. Thus, the strain caused by the furan cycle rotation relative to the chain in the acids D, E and F may exist.
Fig. 1.

Geometric model of 3-(furan-2-yl)-acrylic acid molecule with minimal internal energy. a Frontal projection, b side view
According to the above-mentioned data, we predicted the presence of three different strains in the investigated compounds. The first one is provided by the rotation of the side chain plane relative to the furan cycle in the derivatives of furanacrylic acids (X), the second one corresponds to the interaction of the furan cycle and nitro group in the ortho-position (Y), and the third one is the interaction of the benzene and furan cycles (Z). To calculate interaction corrections in Benson additive scheme a redefined system of six linear equations with two unknowns (X) and (Y) was composed of the corresponding experimentally obtained formation enthalpies and strain-free increments. The correction (Z) of 9 kJ/mol was taken from [31]. The same correction was used to determine the contribution of the group Cd–(Cd)(O)(CB) from the formation enthalpy of 3-substituted 2-cyano-acrylic acid ethyl ester [18]. As a result of the system solution, the correction terms (kJ/mol) were defined: X = − 30.7, Y = − 29.5 It is clear that the corrections were determined for a small set of substances, and will be defined more exactly as additional experimental material is gained.
Experimental
Materials
Furyl-2-oxime (A–C) derivatives were synthesized according to the following procedure. A mixture of appropriated furyl-2-carbaldehyde (0.023 mol), hydroxylamine hydrochloride (0.03 mol) and fused sodium acetate 2 g in 20 mL of ethanol was boiled for 4 h, 30 mL of water was added to the mixture under stirring after cooling. The resulting precipitate was filtered off and recrystallized from ethanol.
Synthesis of 3-[5-(2-nitrolphenyl)-2-furyl] acrylic acids (D–F) was carried out according to the following procedure. 2–3 Drops of piperidine were added to a mixture consisting of appropriated furyl-2-carbaldehyde (0.01 mol) and malonic acid (0.01 mol) in 10 mL of pyridine. The reaction mixture was heated for 2–3 h in a boiling water bath, then cooled, diluted with water (20 mL) and acidified with diluted (1:1) hydrochloric acid. The solution was filtered off, washed with water and dried. The acids were recrystallized from ethanol or mixture of ethanol–DMF solvents. We used the samples obtained after 3- and 4-fold recrystallization.
The identification of substances was confirmed by NMR1H spectroscopy data. NMR 1H spectra were recorded on Varian 600 (600 MHz) spectrometers in DMSO-d6 or acetone-d6. Chemical shifts (δ, ppm) were determined in regards to the signal of DMSO (2.50 ppm). Spectral data for the investigated substances are shown below:
(A)—1H NMR (600 MHz, Acetone-d6), δ: 6.96 (d, J = 3.5 Hz, 1H, furan). 7.41 (d, J = 3.5 Hz, 1H, furan), 7.47 (s, 1H, CH), 7.64 (t, J = 8.4 Hz, 1H, C6H4), 7.78 (t, J = 8.1, Hz, 1H, C6H4), 7.88 (d, J = 8.1 Hz, 1H, C6H4), 7.91 (d, J = 7.8 Hz, 1H, C6H4), 11.20 (s, 1H, NOH).
(B)—1H NMR (600 MHz, DMSO-d6), δ: 7.32 (d, J = 3.3 Hz, 1H, furan), 7.43 (d, J = 3.3 Hz, 1H, furan), 7.68 (s, 1H, CH), 7.74 (t, J = 8.0 Hz, 1H, C6H4), 8.19 (d, J = 7.7 Hz, 1H, C6H4), 8.24 (d, J = 7.9 Hz, 1H, C6H4), 8.53 (s, 1H, C6H4), 8.05 (s, 1H, NOH).
(C)—1H NMR (600 MHz, DMSO-d6), δ: 7.36 (d, J = 3.6 Hz, 0.1H, furan), 7.45 (d, J = 3.6 Hz, 1H, furan), 7.67 (s, 1H, CH), 8.00 (d, J = 8.9 Hz, 2H, C6H4), 8.18 (d, J = 8.9 Hz, 2H, C6H4), 12.10 (s, 1H, COOH).
(D)—1H NMR (600 MHz, DMSO-d6), δ: 6.17 (d, J = 15.8 Hz, 1H, CH=), 7.10 (d, J = 3.6 Hz, 1H, furan), 7.17 (d, J = 3.6 Hz, 1H, furan), 7.43 (d, J = 15.8 Hz, 1H, CH=), 7.65 (t, J = 7.8 Hz, 1H, C6H4), 7.79 (t, J = 7.8 Hz, 1H, C6H4), 7.96 (d, J = 7.8 Hz, 1H, C6H4), 8.00 (d, J = 7.8 Hz, 1H, C6H4), 12.57 (s, 1H, COOH).
(E)—1H NMR (600 MHz, DMSO-d6), δ: 7.05 (d, J = 3.6 Hz, 1H, 3-H furan), 7.05 (d, J = 15.8 Hz, 1H, CH=), 7.34 (d, J = 3.6 Hz, 1H, 4-H furan), 7.51 (d, J = 15.8 Hz, 1H, CH=), 7.63 (t, J = 7.9 Hz, 1H, C6H4), 7.78 (d, J = 7.7 Hz, 1H, C6H4), 7.90 (d, J = 8.0 Hz, 1H, C6H4), 7.95 (s, J = 7.8 Hz, 1H, C6H4), 12.06 (s, 1H, COOH).
(F)—1H NMR (600 MHz, DMSO-d6), δ: 6.49 (d, J = 15.8 Hz, 1H, CH=), 7.16 (d, J = 3.6 Hz, 1H, furan), 7.46–7.51 (m, 2H, CH= + furan), 8.13 (d, J = 8.9 Hz, 2H, C6H4), 8.33 (d, J = 8.9 Hz, 2H, C6H4), 12.55 (s, 1H, COOH).
The compounds purity was confirmed by a high-performance liquid chromatography using an Agilent 1100 HPLC instrument with a diode matrix and a mass-selective detector on Zorbax SB-C18 column, 4.6 mm × 15 mm; eluent was acetonitrile–water with 0.1% TFA (95:5) under normal conditions. No admixtures in the samples were detected.
Effusion measurements
Taking into account low volatility of the analyzed substances, the temperature dependences of the saturated vapour pressures were determined by the integral Knudsen effusion method. The design of the apparatus has been adopted from [37]. Construction of the chamber, membranes and experimental procedure was conducted using the recommendations [38].
Three membranes with a diameter of holes № 1—2.050, № 2—2.100 and № 3—2.055 mm were used for effusional research presented in this paper. The membranes are made of nickel foil with a thickness of 0.09 mm.
The vacuum of 0.1 Pa was achieved for 25 ± 15 s. The weight of the effunded substance m was determined using analytical scales VLR-20 (± 5·10−6 g) as the difference of the effusion camera weight before and after the experiment. The measurement accuracy of the temperature (T) and effusion time (τ) was ± 0.5 K and ± 1 s, respectively. Effective time (estimated time of effusion in the steady state, in which the weight loss of the effunded substance is equal to that in the transient regime), determined in separate experiments with benzoic acid, equals to 25 ± 5 s and added to the total time of the experiment.
The vapor pressure in the effusion cell Pk was calculated by equation [39]:
| 6 |
where τ is the time of effusion through a hole in the membrane with area S; T—temperature, R—universal gas constant, M—molecular weight of the substance, α—condensation coefficient.
Investigated nitrophenyl-furyl derivatives are molecular crystals that sublime without a change in their geometry and molecule weight, which allowed us to admit α to be equal to 1 [40].
Clausing coefficient—K, which stands for the membrane’s resistance to molecular flow of vapor for the hole in the membrane, with ratio of length (l) to radius (r) from 0 to 1.5, was determined by the empirical Kennard formula K = 1/[1 + 0.5(l/r)] [41].
The vapor pressure was calculated using correction factor according to the recommendations [42]. For three membranes used in the present work, the factors are equal to 5.12, 5.21 and 5.14 respectively.
Prior to this, the reliability of the effusion installation was checked by benchmark benzoic acid brand K − 1 (the major component content—99.995% mol) by a series of forty experiments.
The dependences of saturated vapor pressure on temperature have the forms:
| 7 |
ΔsubH340,2 = 88.0 ± 3.8 kJ/mol; ρ = 0.9967.
| 8 |
ΔsubH340,2 = 87.4 ± 4.8 kJ/mol; ρ = 0.9944.
| 9 |
ΔsubH340,2 = 87.5 ± 2.9 kJ/mol; ρ = 0.9990.
The resulting dependence of saturated vapour pressure on temperature for three membranes has the form:
| 10 |
ΔsubH340,2 = 87.6 ± 1.7 kJ/mol; ρ = 0.9976.
The average value of the standard enthalpy of sublimation in the temperature range of (332.5–348.0) K was = 87.6 ± 1.7 kJ/mol. In order to adjust the standard enthalpy of sublimation to 298 K, from Eq. (1), the standard heat capacity of benzoic acid at 298.15 J/(mol K) in the solid Cps° = 146.76 ± 0.32 [43] and gaseous Cpg° = 103.47 [43] state were utilized. Good coincidence of benzoic acid’s sublimation enthalpy adjusted to 298.15 K (kJ/mol) according to the Eq. (1) 91.1 ± 1.8 with the recommended values 89.7 ± 1.0 [43], and 89.0 ± 4.0 [27] shows the absence of significant systematic errors in the effusion installation.
Calorimetric measurements
The combustion enthalpies of the substances were determined by upgraded calorimeter V-08MA with the isothermal shell.
The temperature in the thermostat was maintained ± 0.03 K. The energy equivalent of the calorimetric systems W was estimated by combustion of the reference benzoic acid grade K − 1 (the major component content—99.995% mol, the heat of combustion, taking into account the Jessup factor—26,434.4 J g−1) in a series of 12 experiments. The value of W was 14,901 ± 11 J V−1.
Before combustion beginning the crystalline A, B and C samples were grinded in chalcedony mortar, screened, placed in Terylene ampoules and ignited in the platinum cup. 1 mL of distilled water was added before combustion. The initial pressure of the oxygen, previously purified from the combustible impurities, carbon dioxide and water, was equal to 3.0 MPa. The duration of the initial, main and end periods was—25, 40 and 30 counts, respectively. The initial temperature of the main period in all experiments was 298.15 K. The quantitative analysis of the combustion products for the presence of carbon oxide by the Rossini method [44] with the accuracy of ± 2·10−4 g and nitric acid content by titration of the liquid phase in a bomb with a 0.1 M solution of NaOH was carried out after every experiment. The quantities of the carbon dioxide, formed from the combustion of 1 g of Terylene and the cotton thread, were equal to 2.2872 g and 1.6284 g respectively [45]. The anticipated carbon monoxide to be formed during the combustion of products by using detector tubes within ± 5·10−6 g, was not encountered. The soot mass was determined by the weighting of the platinum cup before and after combustion with the accuracy of ± 5 × 10−6 g. The reliability of gas analyses was controlled by benzoic acid combustion.
Combustion of the investigated compounds is represented by reaction:
| 11 |
A more detailed description of the diffusion unit and combustion calorimeter, experimental procedure and calculations of primary results are presented in [17].
Conclusions
The determined thermodynamic properties of these compounds will contribute to solving practical problems pertaining to optimization processes of their synthesis, purification and application.
Temperature dependences of vapor pressure have their own practical value for calculation of the parameters for individual stages of the synthesis.
Determining of the thermodynamic properties for these compounds also provides a more thorough insight regarding the theoretical knowledge of their nature.
Using Hyper Chem software (PM3 geometry optimization method) the flat configuration of the phenyl-furan oxime derivatives molecules was established.
Phenyl-furan acid derivatives molecules have the minimum internal energy of the molecule, when the chain of atoms –CH=CH–C(O)–OH is in a plane almost perpendicular to the furan cycle plane.
The comparison of the formation enthalpies of the investigated substances in a gaseous state obtained experimentally with the ones calculated by the Benson scheme allowed to determine the energy of the interaction of a furan ring with a nitro group in the ortho position of the benzene ring (− 30 kJ/mol) as well as the energy of the chain –CH=CH–C(O)–OH rotation in the molecules of phenyl-furan acid derivatives (− 31 kJ/mol).
The new group-additivity parameters and the correction terms for substituted nitrophenyl-furyl derivatives of oximes and acids allowed to applicate the Benson group—contribution correlation to estimate (g)calc. of the compounds has not been investigated yet.
Additional file
Additional file 1: Appendix S1. Cartesian coordinates and computation results for the investigated compounds.
Acknowledgements
Not applicable.
Authors’ contributions
DV—guiding thermodynamic studies, analysis of the results; writing of the text. AM—performance of effusional and calorimetric measurements, processing of the results. IS—processing and analysis of thermodynamic measurements. YH—synthesis, purification and identification of substances. MO—synthesis and purification of substances. References. NV—performance of the NMR-spectroscopy. LG—statistical work with data. All authors read and approved the final manuscript.
Funding
Not applicable.
Availability of data and materials
All of the experimental data is available in the corresponding tables in this article (Additional file 1: Appendix S1).
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Volodymyr Dibrivnyi, Phone: +380505653184, Email: vdibriv@polynet.lviv.ua.
Andriy Marshalek, Email: yourowndarkness@gmail.com.
Iryna Sobechko, Email: phys.chem.lp@gmail.com.
Yuriy Horak, Email: horrak@gmail.com.
Mykola Obushak, Email: obushak@in.lviv.ua.
Nadiia Velychkivska, Email: velnadya@gmail.com.
Lubomyr Goshko, Email: avanttes@gmail.com.
References
- 1.Datta A, Walia S, Parmar B. Some furfural derivatives as nitrification inhibitors. J Agric Food Chem. 2001;49:4726–4731. doi: 10.1021/jf001318d. [DOI] [PubMed] [Google Scholar]
- 2.Horak YuI, Matiychuk VS, Obushak MD, Kutsyk RV, Lytvyn RZ, Kurovets LM. 2-(5-Aryl-2-furyl)quinolin-4-carboxylic acids and their antimicrobial activity. Ukr Bioorg Acta. 2008;1:49–50. [Google Scholar]
- 3.Lv W, Banerjee B, Molland KL, Seleem MN, Ghafoor A, Hamed MI, Wan B, Franzblau SG, Mesecar AD, Cushman M. Synthesis of 3-(3-aryl-pyrrolidin-1-yl)-5-aryl-1,2,4-triazines that have antibacterial activity and also inhibit inorganic pyrophosphatase. Bioorg Med Chem. 2014;22:406–418. doi: 10.1016/j.bmc.2013.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gavande NS, Vere-Carozza PV, Mishra AK, Vernon TL, Pawelczak KS, Turchi JJ. Design and structure-guided development of novel inhibitors of the xeroderma pigmentosum group A (XPA) protein–DNA interaction. J Med Chem. 2017;60:8055–8070. doi: 10.1021/acs.jmedchem.7b00780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hosoya T, Aoyama H, Ikemoto T, Kihara Y, Hiramatsu T, Endo M, Suzuki M. Dantrolene analogues revisited: general synthesis and specific functions capable of discriminating two kinds of Ca2+ release from sarcoplasmic reticulum of mouse sceletal muscle. Bioorg Med Chem. 2003;11:663–673. doi: 10.1016/S0968-0896(02)00600-4. [DOI] [PubMed] [Google Scholar]
- 6.Hansen SW, Erichsen MN, Fu B, Bjorn-Yoshimoto WE, Abrahamsen B, Hansen JC, Jensen AA, Bunch L. Identification of a new class of selective excitatory amino acid transporter subtype 1 (EAAT1) inhibitors followed by a structure–activity relationship study. J Med Chem. 2016;59:8757–8770. doi: 10.1021/acs.jmedchem.6b01058. [DOI] [PubMed] [Google Scholar]
- 7.Wang G, Wang X, Yu H, Wei S, Williams N, Holmes DL, Halfmann R, Naidoo J, Wang L, Li L, Chen S, Harran P, Lei X, Wang X. Small-molecule activation of the TRAIL receptor DR5 in human cancer cells. Nat Chem Biol. 2013;9:84–89. doi: 10.1038/nchembio.1153. [DOI] [PubMed] [Google Scholar]
- 8.Laforest J. Germany Patent 2922799. 1979
- 9.Brouwer WG. Canada Patent 2163175. 1996
- 10.Anthony VM. Russian Federation Patent 2039044. 1995
- 11.Meltzer RI, Lewis AD, King JA. Antitubercular substances. IV. Thioamides. J Am Chem Soc. 1955;77:4062–4066. doi: 10.1021/ja01620a029. [DOI] [Google Scholar]
- 12.Pandey OP, Sengupta SK, Chandra R. Efficacy of organophosphorus derivatives containing oximes against fungal pathogens of sugarcane. Met-Based Drugs. 2006;5:1515–1521. doi: 10.1155/MBD.2002.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ager DJ. The synthesis of 2,5-disubstituted furans. Tetrahedron Lett. 1983;24:5441–5444. doi: 10.1016/S0040-4039(00)94107-8. [DOI] [Google Scholar]
- 14.Bessin AP, Laforest J, Thuillier G. U.S. Patent 4207319. 1980
- 15.Thuillier G, Laforest J, Bessin AP. Germany Patent 2449205. 1975
- 16.Považanec F, Kováč J. Furan derivatives. CXXXV. Anhydrides of 3-(5-nitro-2-furyl)acrylic acid as starting materials for the synthesis of l-(5-nitro-2-furyl)-2-(1,3,4-oxadiazol-2-yl)-ethenes. Chem Zvesti. 1979;33:798–802. [Google Scholar]
- 17.Dibrivnyi V, Sobechko I, Puniak M, Horak Yu, Obushak M, Van-Chin-Syan Yu, Marshalek A, Velychkivska N. Thermodynamic properties of 5-(nitrophenyl)-furan-2-carbaldehyde isomers. Chem Cent J. 2015;9:67. doi: 10.1186/s13065-015-0144-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kos R, Sobechko I, Horak Y, Sergeev V, Dibrivnyi V. Thermodynamic characteristics of ethyl-2-cyano-3-(furan-2-yl)-prop-2-enoat derivatives. Modern Org Chem Res. 2017;2:74–80. doi: 10.22606/mocr.2017.22006. [DOI] [Google Scholar]
- 19.Kos RV, Sobechko IB, Horak YI, Sergeev VV, Goshko LV. Thermodynamic properties of isomeric ethyl esters of 2-cyano-3-[5-(2,3,4-nitrophenyl)-2-furan] acrylic acid. Issues Chem Chem Technol. 2017;2:15–20. [Google Scholar]
- 20.Chickos JS, Acree WE. Enthalpies of vaporization of organic and organometallic compounds 1880–2002. J Phys Chem Ref Data. 2003;32:519–878. doi: 10.1063/1.1529214. [DOI] [Google Scholar]
- 21.Dibrivnyi VN, Mel’nik GV, Van-Chin-Syan YY, Yuvchenko AP. The thermodynamic properties of four triphenylsilane acetylene peroxides. Russ J Phys Chem. 2006;80:330–334. doi: 10.1134/S0036024406030046. [DOI] [Google Scholar]
- 22.Hubbard WN, Scott DW, Waddington G. Experimental thermochemistry V1. In: Rossini FD, editor. Interscience. New York: Academic Press; 1956. p. 75. [Google Scholar]
- 23.Cox JD, Pilcher G. Thermochemistry of organic and organometallic compounds. New York: Academic Press; 1970. [Google Scholar]
- 24.Washburn EW. Standard states for bomb calorimetry. J Res Nat Bur Stand (US). 1933;10:525–546. doi: 10.6028/jres.010.037. [DOI] [Google Scholar]
- 25.Cox JD, Wagman DD, Medvedev VA. CODATA recommended key values for thermodynamics 1977. J Chem Thermodynam. 1978;10:903–906. doi: 10.1016/0021-9614(78)90050-2. [DOI] [Google Scholar]
- 26.Benson SW. Thermochemical kinetics. 2. New York: Wiley; 1976. [Google Scholar]
- 27.Chemistry Web-book. http://webbook.NIST.gov. Accessed 18 March 2018.
- 28.Eigenmann HK, Golden DM, Benson SW. Revised group additivity parameters for the enthalpies of formation of oxygen-containing organic compounds. J Phys Chem. 1973;77:1687–1691. doi: 10.1021/j100632a019. [DOI] [Google Scholar]
- 29.Benson SW. Bond energies. J Chem Educ. 1965;42:502–558. doi: 10.1021/ed042p502. [DOI] [Google Scholar]
- 30.Marshalek AS, Sobechko IB, Velychkivska NI, Horak YuI, Dibrivnyi VM. Thermodynamic properties of furfural oxime. Herald Lviv Natl Polytech Univ. 2016;841:26–31. [Google Scholar]
- 31.Verevkin SP. Thermochemisytry of nitro compounds. Experimental standtd enthalpies of formation and improved group—additivity values. Thermochim Acta. 1997;307:17–25. doi: 10.1016/S0040-6031(97)00359-6. [DOI] [Google Scholar]
- 32.Smith MB, March MJ. Advanced organic chemistry. Reactions, mechanisms, and structure. 6. Hoboken: Wiley; 2007. [Google Scholar]
- 33.Bikelyte G, Härtel M, Stierstorfer J, Klapötke TM, Pimerzin AA, Verevkin SP. Benchmark properties of 2-, 3- and 4-nitrotoluene: evaluation of thermochemical data with complementary experimental and computational methods. J Chem Thermodyn. 2017;111:271–278. doi: 10.1016/j.jct.2017.03.029. [DOI] [Google Scholar]
- 34.Verevkin SP, Emel’yanenko VN, Diky V, Dorofeeva OV. Enthalpies of formation of nitromethane and nitrobenzene: new experiments vs. quantum chemical calculations. J Chem Thermodyn. 2014;73:163–170. doi: 10.1016/j.jct.2013.12.013. [DOI] [Google Scholar]
- 35.Miranda MS, Duarte DJR, Liebman JF. What is the enthalpy of formation of pyrazine-2-carboxylic acid? J Chem Thermodyn. 2016;97:261–263. doi: 10.1016/j.jct.2016.02.004. [DOI] [Google Scholar]
- 36.Khan MAS, Dey A, Sikder JAAK. Calculation of enthalpies of formation and band gaps of polymeric binders. RSC Adv. 2014;4:32840–32846. doi: 10.1039/C4RA02847C. [DOI] [Google Scholar]
- 37.Ribeiro Da Silva MAV, Monte MJS. The construction, testing and use of a new Knudsen effusion apparatus. Thermochim Acta. 1990;171:169–183. doi: 10.1016/0040-6031(90)87017-7. [DOI] [Google Scholar]
- 38.Krasulin AP, Kozyro AA, Kabo GY. Saturated vapor pressure of urea at 329–403 K. Zhurnal Prikl Him. 1987;6:104–108. [Google Scholar]
- 39.Nesmeyanov AN. Vapour pressure of chemical elements. Moscow: USSR Academy of Sciences Publishing; 1961. [Google Scholar]
- 40.De Kruif GG (1984) Thermochemistry and its application to chemical, biochemical system. In: Proceedings of NATO Advanced Study Institute Thermochemistry. Today; D Reidel
- 41.Kennard EH. Kinetic theory of gases with an Introduction to statistical mechanics. New York: Academic Press; 1938. [Google Scholar]
- 42.Lebedev YuA, Miroshnichenko EA. Vapor formation thermochemistry of organic compounds. Moscow: Nauka; 1981. [Google Scholar]
- 43.Sabbah R, An Xu-wu, Chickos JS, Planas Leitão ML, Roux MV, Torres LA. Reference materials for calorimetry and differential thermal analysis. Thermochim Acta. 1999;331:93–204. doi: 10.1016/S0040-6031(99)00009-X. [DOI] [Google Scholar]
- 44.Rossini FD. J Res Nat Bur Standards. 1931;6:37–49. doi: 10.6028/jres.006.002. [DOI] [Google Scholar]
- 45.Gerasimov YaI, Akishin PA (1984) Chemical thermodynamics (Experimental research). (In Russian)
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Appendix S1. Cartesian coordinates and computation results for the investigated compounds.
Data Availability Statement
All of the experimental data is available in the corresponding tables in this article (Additional file 1: Appendix S1).