

Abstract
Background
Constitutional loss of function (LOF) single nucleotide polymorphisms (SNPs) in pattern recognition receptors FPR1, TLR3, and TLR4 have previously been reported to predict oxaliplatin benefit in colorectal cancer. Confirmation of this association could substantially improve patient stratification.
Methods
We performed a retrospective biomarker analysis of the Short Course in Oncology Therapy (SCOT) and COIN/COIN-B trials. Participant status for LOF variants in FPR1 (rs867228), TLR3 (rs3775291), and TLR4 (rs4986790/rs4986791) was determined by genotyping array or genotype imputation. Associations between LOF variants and disease-free survival (DFS) and overall survival (OS) were analyzed by Cox regression, adjusted for confounders, using additive, dominant, and recessive genetic models. All statistical tests were two-sided.
Results
Our validation study populations included 2929 and 1948 patients in the SCOT and COIN/COIN-B cohorts, respectively, of whom 2728 and 1672 patients had functional status of all three SNPs determined. We found no evidence of an association between any SNP and DFS in the SCOT cohort, or with OS in either cohort, irrespective of the type of model used. This included models for which an association was previously reported for rs867228 (recessive model, multivariable-adjusted hazard ratio [HR] for DFS in SCOT = 1.19, 95% confidence interval [CI] = 0.99 to 1.45, P = .07; HR for OS in COIN/COIN-B = 0.92, 95% CI = 0.63 to 1.34, P = .66), and rs4986790 (dominant model, multivariable-adjusted HR for DFS in SCOT = 0.86, 95% CI = 0.65 to 1.13, P = .27; HR for OS in COIN/COIN-B = 1.08, 95% CI = 0.90 to 1.31, P = .40).
Conclusion
In this prespecified analysis of two large clinical trials, we found no evidence that constitutional LOF SNPs in FPR1, TLR3, or TLR4 are associated with differential benefit from oxaliplatin. Our results suggest these SNPs are unlikely to be clinically useful biomarkers.
The antitumor immune response is an important determinant of clinical outcome in colorectal cancer (CRC). To date, attention has primarily focused on the role of the adaptive immune system, and particularly the T-cell response, the increasing intensity of which correlates with reduced recurrence in early-stage CRC (1,2). Although the influence of the innate immune system to clinical outcome is less well understood, several studies have suggested that this may also exert a meaningful antitumor effect through the recognition of endogenous ligands presented by dying cells (3–7). This effect has been reported to be especially relevant in the context of cell death induced by anthracyclines and oxaliplatin (3–5), an analog of cisplatin used commonly in the systemic therapy of CRC (8). Pattern recognition receptors present endogenous ligands to macrophages and as such are essential components of the innate immune response (9). Constitutional variants in several genes encoding these proteins have been shown to alter the innate immune response to systemic infection (10). Recently, polymorphisms that result in putative loss of function (LOF) alterations in pattern recognition receptor genes have also been reported to influence benefit from anthracycline and oxaliplatin chemotherapy (4–7). These variants, which affect FPR1 [rs867228: c.1037A>C, p.Glu346Ala, where Ala is the LOF allele (11)], TLR3 [rs3775291: c.1234C>T, p.Leu412Phe, where Phe is the LOF allele (12)], and TLR4 [rs4986790: c.896A>G, p. Asp299Gly, where Gly is the LOF allele (4,7)] in strong linkage disequilibrium with rs4986791: c.1196C>T, p.Thr399Ile, are proposed to act by attenuating the immune response against the immunogenic cell death caused by these agents (4–7). These associations were reflected in statistically significant differences in both progression-free and overall survival (OS) between patients bearing LOF and functional alleles in these genes when treated with these agents (hazard ratios [HRs] for LOF allele of 1.37–2.13; summarized in Supplementary Table 1, available online) (4–7,13,14). If validated, these variants could be used as biomarkers to target these toxic therapies to those most likely to benefit from them, resulting in less harm to patients and cost savings for health-care providers. Because anthracyclines and oxaliplatin are the mainstays of systemic treatment against two common cancers (breast and colorectal, respectively) (15,16) and because these LOF polymorphisms are relatively common (prevalence of 5% to 80% in populations of European descent), confirmation of this association could affect many thousands of patients each year in Europe and the United States alone. The purpose of this validation study was to confirm this association in the context of oxaliplatin treatment for CRC by analysis of two well-defined, prospectively treated cohorts from the Short Course in Oncology Therapy (SCOT) and COIN/COIN-B trials (17,18), encompassing both early-stage and advanced disease.
Methods
Clinical Trials
Details of the SCOT (ISRCTN59757862), COIN (ISRCTN27286448), and COIN-B (ISRCTN38375681) trials have been published previously (17–20). Briefly, the SCOT trial compared the efficacy of 12 weeks of oxaliplatin-based adjuvant chemotherapy with the previous standard of care of 24 weeks of treatment in high-risk, stage II (defined as one or more of: pT4 primary tumor, tumor obstruction, fewer than 10 lymph nodes harvested, grade 3 histology, perineural invasion, or extramural venous or lymphatic vascular invasion), or stage III colon or rectal cancer. The trial randomized 6088 patients between March 2008 and November 2013, of whom 6065 consented for their data to be used for the intention to treat analyses. At its primary analysis, the attenuated course of chemotherapy was confirmed to be noninferior to the standard of care (HR = 1.01, 95% CI = 0.91 to 1.11, test for noninferiority P = .012) (17). As part of the study, participants at selected centers were invited to participate in a translational substudy, the TransSCOT study. Tissue and blood samples were collected from these patients and constitutional DNA was extracted for translational studies. Following informed consent, 3109 patients provided samples for analysis. The COIN trial examined both the efficacy of the anti-EGFR monoclonal antibody cetuximab added to oxaliplatin-based chemotherapy and the impact of interrupting treatment in patients with stable or responding metastatic CRC after 12 to 16 weeks of systemic therapy (18). The trial recruited 2445 patients between March 2005 and May 2008. At its primary analysis, no statistically significant difference was observed between the chemotherapy-only and the chemotherapy plus cetuximab groups (20), and the comparison between intermittent and continuous chemotherapy failed to confirm noninferiority of interrupting treatment (18). The COIN-B study compared intermittent chemotherapy with either intermittent or continuous cetuximab in 226 patients with metastatic CRC (19). Among 169 patients with KRAS wild-type disease, analysis suggested greater activity of continuous cetuximab, though this difference was not statistically significant. As part of ancillary translational studies, 2244 study participants in COIN and COIN-B donated blood samples for DNA extraction and analysis. Given their similar patient populations and treatments (21), the COIN and COIN-B biomarker cohorts were combined for all analyses.
DNA Extraction, Genotyping, and Imputation
DNA was extracted from EDTA-venous bloods using standard methods. After exclusion of samples that failed DNA extraction (n = 28) and those for which trial IDs were missing or duplicates (n = 14), 3067 DNA samples from the SCOT cohort were genotyped using the Global Screening Array (Illumina, San Diego, CA). Genotyping quality control entailed removal of any sample or single nucleotide polymorphism (SNP) with more than 2% missing data, any sample with an outlying heterozygosity rate, any sample with discordant reported sex and genotype imputed sex, and any SNP violating Hardy-Weinberg equilibrium at P less than 1 × 10–10 (n = 66 samples removed; n = 32 850 SNPs removed). Identity by descent analysis was conducted in PLINK 1.9 (22) and population stratification was examined using EIGENSTRAT (23). Related individuals (n = 8) were removed (IBD > 0.185) along with those with non-European ancestry (n = 54, as assessed by merging SCOT with HapMap release 23a and removing outliers based on eigenvector 1). Genotypes for 2939 remaining individuals were phased using SHAPEIT (24) and imputed using IMPUTE2 (25) and the UK10K + 1000 genomes merged reference panel. Of the SNPs analyzed in this study, rs3775291, rs4986790, and rs4986791 were directly genotyped. The fourth, rs867228, was imputed with an info score of 0.95. For this imputed SNP, genotype probabilities were converted to genotypes using gtool (http://www.well.ox.ac.uk/∼cfreeman/software/gwas/gtool.html) with a minimum probability threshold of .9 set for specifying per sample genotypes.
Cases from the COIN and COIN-B studies were genotyped using Affymetrix Axiom Arrays according to the manufacturer’s recommendations (Affymetrix, Santa Clara, CA) at the King Faisal Specialist Hospital and Research Center, Saudi Arabia (under IRB approval 2110033). We excluded individuals from analysis if they failed one or more of the following thresholds: overall successfully genotyped SNPs less than 95% (n = 122), discordant sex information (n = 8), classed as out of bounds by Affymetrix (n = 30), duplication or cryptic relatedness (identity by descent >0.185, n = 4), and evidence of non-white European ancestry by principal components analysis-based analysis in comparison with HapMap samples (n = 130). Imputation was performed using 1000 Genomes Project Pilot data as a reference panel (26). Genetic linkage of SNPs was determined by calculation of D’ and R2 using PLINK 1.9 (22).
Statistical Analyses
Comparison between groups was made using unpaired Student t test for continuous variables (eg, age) and either χ2 or Fisher exact test for categorical variables (eg, mutation present vs absent, responder vs nonresponder). Biomarker analyses in this study were performed and are reported in accordance with the REMARK guidelines (27). All analyses were prespecified and are detailed in Supplementary Table 2 (available online). Survival endpoints included disease-free survival (DFS, defined as time from study randomization to CRC recurrence or death from any cause in SCOT only) and OS (defined as time from randomization to death from any cause in both cohorts). Progression-free survival was not used as an endpoint in the COIN/COIN-B trials in view of the difficulty in defining its duration in the context of intermittent chemotherapy, which was tested in both studies. Survival curves for SNP genotypes were plotted using the Kaplan-Meier method and analyzed by the log-rank test. Survival endpoints were also analyzed by univariate and multivariable Cox proportional hazards models, under additive, recessive, and dominant genetic models (eg, for rs867228, which has alleles A and C—of which C is the LOF allele—the additive model implies CC [2] vs CA [1] vs AA [0], modelled as a continuous variable; the recessive model implies CC [1] vs both CA and AA [0]; and the dominant model implies both CC and CA [1] vs AA). Proportionality of hazards was confirmed by inspection of scaled Schoenfeld residuals. For the multivariable analyses, adjustment was made for baseline demographic variables (age, sex), clinicopathological and molecular covariables of known prognostic value where available, and treatment type and schedule depending on the cohort. In the SCOT analyses, these comprised age, sex, disease site (colon vs rectum), primary tumor stage (pT1–2 vs pT3 vs pT4), nodal status (N0 vs N1 vs N2), treatment regimen (FOLFOX or CAPOX), and treatment duration (24 vs 12 weeks). In the COIN/COIN-B analyses, these comprised age, sex, disease site (colon vs rectum), World Health Organization (WHO) performance status (0 or 1 vs 2), primary tumor resection (unresected vs resected), tumor KRAS, NRAS, and BRAF mutation status (mutated vs wild type), patient white blood cell count (<10 000 cells per μL vs ≥10 000 cells per μL), cetuximab treatment (yes vs no), chemotherapy regimen (FOLFOX vs CAPOX), and chemotherapy schedule (intermittent vs continuous). In both cases, covariables were prespecified and no selection procedure (eg, backwards elimination) was performed. Models included all cases for which data were available and excluded those with missing data. P values for individual predictors in Cox models were calculated by the Wald test. Statistical analyses were performed in R version 3.4.4 (CRAN Corporation) and STATA version 13 (StataCorp, College Station, TX). All statistical tests were two-sided. Statistical significance was accepted at P less than .05. No correction for multiple testing was applied.
Ethical Approval
Informed consent for the collection and analysis of samples was provided by study participants at the time of study recruitment under trial-specific ethical approval. Molecular analysis of samples from the SCOT cohort was performed under North West – Liverpool Central Research Committee approval (17/NW/0252). Molecular analysis of COIN/COIN-B samples was performed under REC approval (04/MRE06/60).
Results
Patient Characteristics and SNP Genotyping
The CONSORT diagram demonstrating the flow of patients eligible for this biomarker study is shown in Figure 1. Demographic and clinicopathological characteristics of the 2929 SCOT cases with samples informative for this analysis were broadly similar to those of the SCOT trial population as a whole, although they differed statistically significantly, albeit modestly, from the nonbiomarker population in age, disease site, disease stage, and nodal status (Supplementary Table 2, available online). Characteristics of 2244 patients in the COIN/COIN-B biomarker subgroup were similar to the combined COIN/COIN-B trial population (not shown). Details of baseline demographic, clinicopathological, and molecular variables, and SNP genotypes in cases from both biomarker cohorts are provided in Table 1. Of 2929 patients in the SCOT cohort, 2728 (93.1%), 2924 (99.9%), and 2929 (100%) underwent successful genotyping or imputation and were informative for analysis of rs867228, rs3775291, and rs4986790/rs4986791 respectively. The slightly lower number of cases informative for rs867228 reflects the exclusion of those in which the genotype could not be imputed with high confidence. The corresponding numbers in the COIN/COIN-B cohort of 1948 patients were 1672 (85.6%), 1948 (100%), and 1948 (100%) respectively. The allelic frequencies of all SNPs in both cohorts were concordant with the reported population frequency in ExAC (28), EVS (29), and UK10K (30). As expected, rs4986790 and rs4986791 were in strong linkage disequilibrium in both the SCOT (D’ = 0.99 and r2 = 0.93) and COIN/COIN-B (D’ = 0.99 and r2 = 0.89) cohorts. Because analyses of these two SNPs individually yielded essentially identical results (Supplementary Figure 1, available online), we largely limited subsequent investigations to rs4986790.
Figure 1.

CONSORT diagram showing flow of patients analyzed in the study. ITT = intention to treat.
Table 1.
Baseline characteristics of SCOT and combined COIN/COIN-B cohorts
| Variable | SCOT | COIN and COIN-B | P |
|---|---|---|---|
| No. (%) | No. (%) | ||
| Total | 2929 (100) | 1948 (100) | — |
| Median age, y (range) | 65 (23–84) | 53 (18–87) | <.001* |
| Sex | |||
| Male | 1795 (61.3) | 1270 (65.2) | <.001† |
| Female | 1134 (38.7) | 678 (34.8) | |
| Unknown | 0 (0.0) | 0 (0.0) | |
| Disease stage | |||
| II | 585 (20.0) | 0 (0.0) | — |
| III | 2344 (80.0) | 0 (0.0) | |
| IV | 0 (0.0) | 1948 (100.0) | |
| Unknown | 0 (0.0) | 0 (0.0) | |
| Primary tumor stage | |||
| pT1 | 94 (3.2) | NA | — |
| pT2 | 285 (9.7) | NA | |
| pT3 | 1694 (57.8) | NA | |
| pT4 | 856 (29.2) | NA | |
| Unknown | 0 (0.0) | NA | |
| Nodal stage | |||
| N0 | 585 (20.0) | NA | — |
| N1 | 1695 (57.9) | NA | |
| N2 | 649 (22.2) | NA | |
| Unknown | 0 (0.0) | NA | |
| Primary tumor location | |||
| Colon | 2346 (80.1) | 1325 (67.9) | <.001† |
| Rectum | 583 (19.9) | 621 (31.9) | |
| Unknown | 0 (0.0) | 2 (0.2) | |
| Primary tumor resected | |||
| No | 0 (0.0) | 821 (42.1) | — |
| Yes | 2929 (100.0) | 1127 (57.9) | |
| Unknown | 0 (0.0) | 0 (0.0) | |
| Peritoneal metastases | |||
| No | NA | 1519 (78.0) | — |
| Yes | NA | 259 (13.3) | |
| Unknown | NA | 170 (8.7) | |
| KRAS mutation status | |||
| Wild-type | ND | 989 (50.8) | — |
| Mutant | ND | 636 (32.6) | |
| Unknown | ND | 323 (16.6) | |
| NRAS mutation status | |||
| Wild type | ND | 1506 (77.3) | — |
| Mutant | ND | 69 (3.6) | |
| Unknown | ND | 373 (19.1) | |
| BRAF mutation status | |||
| Wild type | ND | 1438 (73.8) | — |
| Mutant | ND | 143 (7.3) | |
| Unknown | ND | 367 (18.9) | |
| FPR1 rs867228 genotype | |||
| AA | 116 (4.0) | 49 (2.5) | .003† |
| AC | 813 (27.8) | 444 (22.8) | |
| CC | 1799 (61.4) | 1179 (60.5) | |
| Unknown | 201 (6.9) | 276 (14.2) | |
| TLR3 rs3775291 genotype | |||
| CC | 1486 (50.7) | 934 (47.9) | .005† |
| CT | 1207 (41.2) | 810 (41.6) | |
| TT | 231 (7.9) | 204 (10.5) | |
| Unknown | 5 (0.2) | 0 (0.0) | |
| TLR4 rs4986790 genotype | |||
| AA | 2581 (88.1) | 1744 (89.5) | .11† |
| AG | 333 (11.4) | 200 (10.3) | |
| GG | 15 (0.5) | 4 (0.2) | |
| Unknown | 0 (0.0) | 0 (0.0) | |
| TLR4 rs4986791 genotype | |||
| CC | 2568 (90.7) | 1726 (88.6) | .12† |
| CT | 344 (11.7) | 218 (11.2) | |
| TT | 17 (0.6) | 4 (0.2) | |
| Unknown | 0 (0.0) | 0 (0.0) |
*Determined by two-sided unpaired Student t test. NA = not applicable; ND = not determined; pT = pathological tumor (T) stage; SCOT = Short Course in Oncology Therapy.
†Determined by two-sided χ2 test or Fisher exact test in the case of rs4986791 (in cases of SNP genotypes, values are calculated from cases in which SNP status was determined).
The effect sizes (hazard ratios) of each SNP detectable in multivariable analyses using recessive and dominant genetic models, based on a power (1-β) of 0.8 and a two-sided α of 0.05, are shown for both cohorts in Supplementary Table 4 (available online). For comparison with previous reports, our power to detect an association of identical effect size using the same (recessive) model to that previously reported for the FPR1 rs867228 SNP was 1.0 and 0.995 for DFS and OS, respectively, in the SCOT cohort and 1.0 for OS in the COIN/COIN-B cohort. Our power to detect an association of the same effect size as that previously reported for the TLR4 rs4986790 SNP using the same (dominant) model was 0.65 and 0.31 for DFS and OS, respectively, in the SCOT cohort and 0.96 for OS in the COIN/COIN-B cohort.
Pattern Recognition SNPs and Clinical Outcome in the SCOT Cohort
Biomarker analyses were performed with data used for the primary analysis of the SCOT trial, at which point the 2929 patients in the biomarker cohort had a median follow-up of 36.8 months, and 538 DFS events and 186 deaths had occurred (Table 2). Comparing survival curves by the log-rank test, univariate and multivariable Cox models demonstrated no statistically significant association of any SNP irrespective of genetic model imposed (Figure 2, Table 2, details of covariables in multivariable models provided in Supplementary Table 5, available online). This included models for which an association was previously reported for rs867228 (5) (recessive model, multivariable-adjusted HR for DFS = 1.19, 95% CI = 0.99 to 1.45, P = .07) and rs4986790 (4) (dominant model, multivariable-adjusted HR for DFS = 0.86, 95% CI = 0.65 to 1.13, P = .27) (Table 2, Supplementary Table 5, available online).
Table 2.
Univariate and multivariable analyses of DFS and OS in SCOT cohort by LOF SNP*
| Polymorphism/genetic model | No. | DFS events | OS events | Univariate analysis |
Multivariable analysis |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DFS |
OS |
DFS |
OS |
|||||||||
| HR (95% CI) | P† | HR (95% CI) | P† | HR (95% CI) | P† | HR (95% CI) | P† | |||||
| rs867228 (FPR1 c.1037A>C) | 2728 | 487 | 167 | |||||||||
| Additive | — | — | — | 1.13 (0.96 to 1.32) | .15 | 1.09 (0.83 to 1.44) | .53 | 1.16 (0.98 to 1.37) | .08 | 1.10 (0.84 to 1.47) | .48 | |
| Recessive | — | — | — | 1.15 (0.95 to 1.40) | .15 | 1.07 (0.77 to 1.49) | .67 | 1.19 (0.99 to 1.45) | .07 | 1.10 (0.79 to 1.53) | .56 | |
| Dominant | — | — | — | 1.17 (0.73 to 1.88) | .50 | 1.40 (0.57 to 3.41) | .46 | 1.16 (0.73 to 1.87) | .53 | 1.32 (0.54 to 3.22) | .54 | |
| rs3775291 (TLR3 c.1234C>T) | 2924 | 536 | 186 | |||||||||
| Additive | — | — | — | 1.05 (0.92 to 1.19) | .52 | 1.15 (0.93 to 1.44) | .29 | 1.02 (0.90 to 1.17) | .68 | 1.13 (0.91 to 1.41) | .27 | |
| Recessive | — | — | — | 1.24 (0.92 to 1.66) | .15 | 1.46 (0.92 to 2.32) | .11 | 1.14 (0.85 to 1.52) | .38 | 1.32 (0.83 to 2.10) | .24 | |
| Dominant | — | — | — | 1.01 (0.85 to 1.19) | .95 | 1.12 (0.84 to 1.49) | .44 | 1.00 (0.85 to 1.19) | .97 | 1.12 (0.84 to 1.49) | .44 | |
| rs4986790 (TLR4 c.896A>G) | 2929 | 538 | 186 | |||||||||
| Additive | — | — | — | 0.92 (0.71 to 1.19) | .52 | 0.89 (0.57 to 1.39) | .62 | 0.89 (0.69 to 1.16) | .39 | 0.87 (0.56 to 1.36) | .54 | |
| Recessive | — | — | — | 1.49 (0.55 to 4.00) | .42 | 1.93 (0.48 to 7.76) | .36 | 1.58 (0.59 to 4.25) | .36 | 1.82 (0.44 to 7.40) | .40 | |
| Dominant | — | — | — | 0.89 (0.67 to 1.17) | .40 | 0.83 (0.51 to 1.35) | .45 | 0.86 (0.65 to 1.13) | .27 | 0.81 (0.50 to 1.32) | .39 | |
*Both univariate and multivariable analyses use all informative cases. Hazard ratios show risk associated with reported LOF allele (underscored) for each SNP as follows: rs867228: FPR1 c.1037A>C p.Glu346Ala; rs3775291: TLR3 c.1234C>T, p.Leu412Phe; rs4986790: TLR4 c.896A>G, p. Asp299Gly. Corresponding associations from rs4986791 (TLR4 c.1196C>T, p.Thr399Ile), which is tightly linked to rs4986790, were essentially identical to those obtained from analysis of rs4986790 and are not shown. Multivariable-adjusted HRs were adjusted for age, sex, disease site (colon vs rectum), primary tumor stage (pT1–2 vs pT3 vs pT4), nodal status (N0 vs N1 vs N2), treatment regimen (FOLFOX or CAPOX), and treatment duration (24 vs 12 weeks). Prognostic associations of covariables are shown in Supplementary Table 5 (available online). CI = confidence interval; DFS = disease-free survival; HR = hazard ratio; LOF = loss of function; OS = overall survival; pT = pathological tumor (T) stage; SCOT = Short Course in Oncology Therapy.
†P values were calculated by two-sided Wald test.
Figure 2.

FPR1, TLR3, and TLR4 loss of function (LOF) single nucleotide polymorphisms (SNPs), disease-free survival (DFS), and overall survival (OS) in Short Course in Oncology Therapy (SCOT) cohort. Kaplan Meier curves showing DFS for patients in SCOT cohort by pattern recognition receptor SNPs rs867228 (FPR1 c.1037A>C p.Glu346Ala) (A), rs3775291 (TLR3 c.1234C>T p.Leu412Phe) (B), and rs4986790 (TLR4 c.896A>G, p.Asp299Gly) (C) (LOF allele/amino acid underscored in each case). Corresponding results for OS are shown in D–F. Analyses of the rs4987691 (TLR4 c.1196C>T, p. Thr399Ile) polymorphism, which is strongly linked with rs4986790, were essentially identical to C and F and are provided as Supplementary Figure 1 (available online). Shaded areas represent 95% confidence intervals. P values indicate comparison of all groups by the two-sided log-rank test.
A previous study reported that the association of the FPR1 LOF polymorphism rs867228 was only evident in patients with functional TLR3 or TLR4, consistent with their participation in the same pathway (5). We therefore examined this in the SCOT biomarker cohort after stratifying by TLR3 (rs3775291) and TLR4 (rs4986790) status. These analyses did not confirm the previously reported, statistically significant association with DFS in the context of either functional TLR3 background (multivariable-adjusted HR for additive model = 1.02, 95% CI = 0.82 to 1.27, P = .85; recessive model HR = 1.01, 95% CI = 0.78 to 1.31, P = .91; dominant model HR = 1.09, 95% CI = 0.59 to 2.00, P = .78) or functional TLR4 background (additive model HR = 1.17, 95% CI = 0.99 to 1.40, P = .07; recessive model HR = 1.20, 95% CI = 0.98 to 1.48, P = .08; dominant model HR = 1.31, 95% CI = 0.79 to 1.20, P = .30). Similarly, no statistically significant association of rs867228 with DFS was observed in cases with functional polymorphisms at both of these loci (multivariable-adjusted HR for additive model = 0.97, 95% CI = 0.77 to 1.21, P = .76; recessive model HR = 0.92, 95% CI = 0.70 to 1.22, P = .58; dominant model HR = 1.14, 95% CI = 0.60 to 2.16, P = .68) (Supplementary Figure 2, available online).
Pattern Recognition SNPs, Clinical Outcome, and Oxaliplatin Response in the COIN/COIN-B Cohort
Corresponding analyses were performed on the COIN/COIN-B cohort in which the median follow-up of the 1948 patients was 23.2 months, by which time 1453 deaths had occurred. Similar to the SCOT analyses, there was no statistically significant association of either SNP with OS by either log-rank test or univariate or multivariable Cox regression, regardless of model (Figure 3 , Table 3, details of covariables in multivariable models provided in Supplementary Table 6, available online). Again, this included the recessive model for rs867228 (5) (multivariable-adjusted HR for OS = 0.92, 95%CI = 0.63 to 1.34, P = .66), and the dominant model for rs4986790 (4) (multivariable-adjusted HR for OS = 1.08, 95% CI = 0.90 to 1.31, P = .40) (Table 3, Supplementary Table 6, available online). Likewise, prespecified subgroup analyses stratified by TLR3 and TLR4 status revealed no evidence of an association between FPR1 status and OS in the context of functional TLR3 (multivariable-adjusted HR for additive model = 0.93, 95% CI = 0.78 to 1.10, P = .37; recessive model HR = 0.91, 95% CI = 0.56 to 1.48, P = .71; dominant model HR = 0.91, 95% CI = 0.74 to 1.12, P = .36), or functional TLR4 (additive model HR = 1.03, 95% CI = 0.90 to 1.17, P = .66; recessive model HR = 1.00, 95% CI = 0.68 to 1.49, P = .99; dominant model HR = 1.04, 95% CI = 0.89 to 1.21, P = .62). Similar to the results from the SCOT cohort, no statistically significant association was observed in cases with functional polymorphisms at both loci (multivariable-adjusted HR for additive model = 0.99, 95% CI = 0.82 to 1.19, P = .87; recessive model = 1.22, 95% CI = 0.73 to 2.04, P = .43; dominant model HR = 0.95, 95% CI = 0.76 to 1.18, P = .63) (Supplementary Figure 3, available online).
Figure 3.
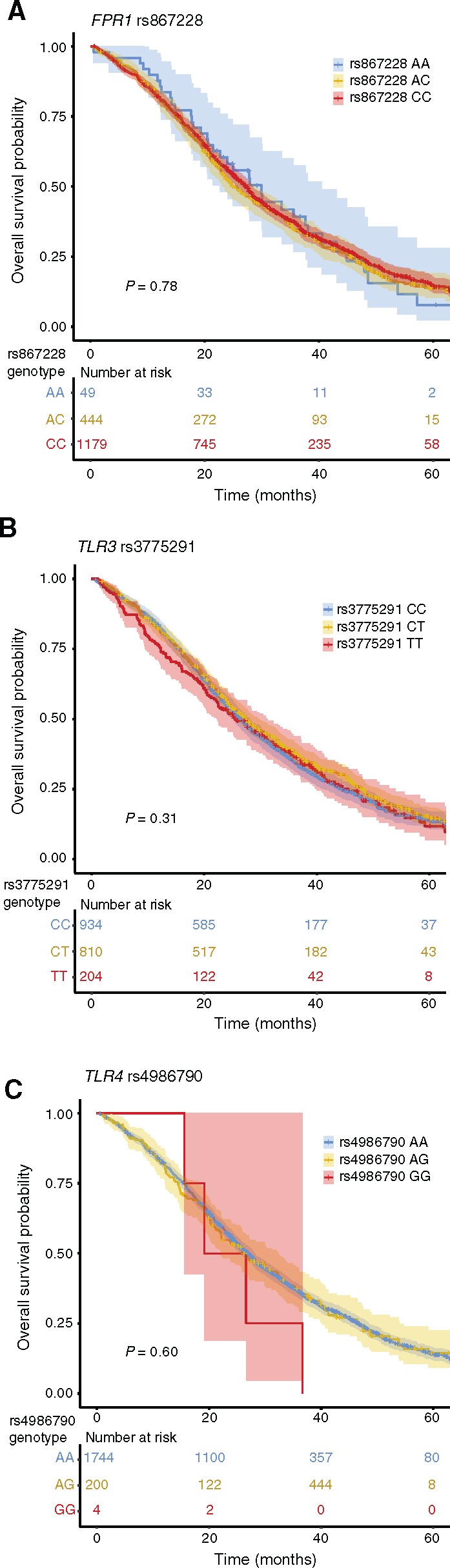
FPR1, TLR3, and TLR4 loss of function (LOF) single nucleotide polymorphisms (SNPs) and overall survival (OS) in COIN/COIN-B cohort. Kaplan Meier curves showing OS for patients in combined COIN/COIN-B cohort by pattern recognition receptor SNPs rs867228 (FPR1 c.1037A>C p.Glu346Ala) (A), rs3775291 (TLR3 c.1234C>T p.Leu412Phe) (B), and rs4986790 (TLR4 c.896A>G, p.Asp299Gly) (C) (LOF allele/amino acid underscored in each case). Analyses of the rs4987691 (TLR4 c.1196C>T, p. Thr399Ile) polymorphism, which is strongly linked with rs4986790, were essentially identical to C and are not shown. Shaded areas represent 95% confidence intervals. P values indicate comparison of all groups by the two-sided log-rank test.
Table 3.
Univariate and multivariable analyses of OS in combined COIN/COIN-B cohort by LOF SNP*
| Polymorphism/genetic model | Univariate analysis |
Multivariable analysis |
||||||
|---|---|---|---|---|---|---|---|---|
| No. | OS events | HR (95% CI) | P† | No. | OS events | HR (95% CI) | P† | |
| rs867228 (FPR1 c.1037A>C) | 1672 | 1241 | — | — | 1336 | 970 | — | — |
| Additive | — | — | 1.03 (0.93 to 1.14) | .60 | — | — | 0.99 (0.88 to 1.12) | .91 |
| Recessive | — | — | 0.98 (0.71 to 1.37) | .93 | — | — | 0.92 (0.63 to 1.34) | .66 |
| Dominant | — | — | 1.04 (0.92 to 1.18) | .52 | — | — | 1.05 (0.90 to 1.23) | .53 |
| rs3775291 (TLR3 c.1234C>T) | 1948 | 1453 | — | — | 1563 | 1150 | — | — |
| Additive | — | — | 0.98 (0.91 to 1.06) | .67 | — | — | 0.97 (0.89 to 1.06) | .56 |
| Recessive | — | — | 1.07 (0.90 to 1.26) | .45 | — | — | 1.08 (0.90 to 1.31) | .41 |
| Dominant | — | — | 0.94 (0.86 to 1.05) | .31 | — | — | 0.93 (0.83 to 1.04) | .20 |
| rs4986790 (TLR4 c.896A>G) | 1948 | 1453 | — | — | 1563 | 1150 | — | — |
| Additive | — | — | 1.03 (0.88 to 1.21) | .71 | — | — | 1.10 (0.91 to 1.33) | .31 |
| Recessive | — | — | 1.65 (0.61 to 4.40) | .31 | — | — | 2.91 (0.93 to 9.12) | .07 |
| Dominant | — | — | 1.02 (0.86 to 1.20) | .81 | — | — | 1.08 (0.90 to 1.31) | .40 |
*Both univariate and multivariable analyses use all informative cases (ie, cases lacking covariable data were excluded from multivariable models). Hazard ratios show risk associated with reported LOF allele (underscored) for each SNP as follows: rs867228: FPR1 c.1037A>C p.Glu346Ala; rs3775291: TLR3 c.1234C>T, p.Leu412Phe; rs4986790: TLR4 c.896A>G, p. Asp299Gly. Corresponding associations from rs4986791 (TLR4 c.1196C>T, p.Thr399Ile), which is tightly linked to rs4986790, were essentially identical to those obtained from analysis of rs4986790 and are not shown. Multivariable-adjusted HRs are adjusted for age, sex, disease site (colon vs rectum), World Health Organization (WHO) performance status (0 or 1 vs 2), primary tumor resection (unresected vs resected), tumor KRAS, NRAS, and BRAF mutation status (mutated vs wild type), patient white blood cell count (<10 000 cells/μL vs ≥10 000 cells/μL), cetuximab treatment (yes vs no), chemotherapy regimen (FOLFOX vs CAPOX), and chemotherapy schedule (intermittent vs continuous). Prognostic associations of covariables are shown in Supplementary Table 6 (available online). CI = confidence interval; HR = hazard ratio; LOS = loss of function; OS = overall survival; pT = pathological tumor (T) stage; SNP = single nucleotide polymorphism.
†P values were calculated by two-sided Wald test.
An additional analysis according to radiological response to oxaliplatin-based chemotherapy after 12 weeks of therapy [complete or partial response vs stable or progressive disease by RECIST 1.0 (31)] revealed no difference in the proportions of functional and LOF alleles between responders and nonresponders for rs867228 (P = .90, χ2 test), rs3775291 (P = .68, χ2 test), or rs4986790 (P = .64, Fisher exact test).
Discussion
Previous studies have suggested that LOF polymorphisms in the pattern recognition receptors FPR1 (rs867228), TLR3 (rs3775291), and TLR4 (rs4986790/rs4986791) decrease the presentation of ligand to the innate immune system by dying cells (3–7). This, in turn, is proposed to reduce the efficacy of anthracycline and oxaliplatin chemotherapy, the activities of which depend in part on the induction of immunogenic cell death (3–5,7). In this study of nearly 5000 patients with CRC treated with oxaliplatin, we failed to confirm any of these associations. The 95% confidence intervals for the association of each SNP with DFS and OS in the SCOT cohort and OS in the COIN/COIN-B cohort all included the estimate of no effect. Although our data by no means exclude an immunomodulatory effect of these SNPs, they suggest that they are very unlikely to be clinically useful as predictive biomarkers for oxaliplatin benefit in CRC. The discordance between our results and those from previous studies may be explained by the increased risk of false-positive associations in the smaller cohorts they used, and in the case of rs867228, an apparent misclassification of the functional and LOF alleles in the survival analyses (the functional FPR1 allele c.1037A, p.346Glu appeared to be incorrectly classified as LOF in all analyses in the study by Vacchelli et al.) (5). Our results underscore the importance of validation of encouraging findings from modestly sized studies in large, meticulously curated trial cohorts, even where preclinical data provide a plausible mechanism for an association.
Strengths of our study include its large size, defined clinical trial cohorts, standardized therapy, comprehensively annotated clinicopathological variables, and, in the case of the COIN/COIN-B cohort, molecular variables and mature outcome data. Consequently, our analyses were powered to detect even a modest association of most SNPs with clinical outcome and had a power of greater than 0.95 to detect an association of similar strength to that previously reported for the rs867228 and rs4986790 LOF variants (4,6). Limitations include the lack of molecular profiling in the SCOT trial, which meant that we were unable to test for an association of the SNPs with clinical outcome in specific tumor subgroups such as those with enhanced immunogenicity due to defective DNA mismatch repair or POLE exonuclease domain mutation.
In summary, in this study of two large clinical trial cohorts, we find no evidence that LOF SNPs in the pattern recognition receptors FPR1, TLR3, and TLR4 are associated with differential benefit from oxaliplatin in CRC. Future studies may better define the complex relationship between cytotoxic therapeutic-induced cell death, pattern recognition SNPs, and the innate immune system.
Funding
This work was supported by The Oxford NIHR Comprehensive Biomedical Research Centre, Cancer Research UK (C6199/A10417 and C399/A2291), the European Union Seventh Framework Programme (FP7/207– 2013) grant 258236 collaborative project SYSCOL, European Research Council project EVOCAN, and core funding to the Wellcome Trust Centre for Human Genetics from the Wellcome Trust (090532/Z/09/Z). The SCOT trial was supported by the Medical Research Council (transferred to NETSCC—Efficacy and Mechanism Evaluation; grant reference G0601705), the Swedish Cancer Society, and Cancer Research UK Core Clinical Trials Unit Funding (funding reference C6716/A9894). The TRIAL sponsor was NHS Greater Glasgow & Clyde and University of Glasgow (Eudract reference 2007–003957–10; ISRCTN number 23516549). COIN and COIN-B were coordinated by the Medical Research Council Clinical Trials Unit and conducted with the support of the National Institutes of Health Research Cancer Research Network. COIN and COIN-B translational studies were supported by the Bobby Moore Fund from Cancer Research UK, Tenovus, the Kidani Trust, Cancer Research Wales, and the National Institute for Social Care and Health Research Cancer Genetics Biomedical Research Unit.
SB is funded by an MRC Clinical Research Training Fellowship. CP is funded by a University of Birmingham Fellowship. NAA, BFM, and SMW were funded and supported by KFSHRC. RSH is supported by Cancer Research UK. DNC is funded by a Health Foundation/Academy of Medical Sciences Clinician Scientist Fellowship.
The cost of open access publication was provided by core funding to the Wellcome Centre for Human Genetics from the Wellcome Trust (203141/Z/16/Z).
Notes
Division of Cancer and Genetics, School of Medicine, Cardiff University, Cardiff, UK (VG, MGS, JPC); Wellcome Centre for Human Genetics, University of Oxford, Oxford, UK (SB, EJ, LG, DNC); Cancer Genetics and Evolution Laboratory, Institute of Cancer and Genomic Sciences, University of Birmingham, Edgbaston, Birmingham, UK (SB, EJ, IT); Gastrointestinal Cancer Genetics Laboratory, Institute of Cancer and Genomic Sciences, University of Birmingham, Edgbaston, Birmingham, UK (CP); Southampton University Hospital NHS Foundation Trust, Southampton, UK (TI, TSM); Department of Oncology, Old Road Campus Research Building, University of Oxford, Oxford, UK (RK, EJ, HW); Christie Hospital NHS Foundation Trust, Manchester, UK (MPS); Cancer Research UK Clinical Trials Unit, Institute of Cancer Sciences, University of Glasgow, Glasgow, UK (JP, AH, JM); MRC Clinical Trials Unit at UCL, London, UK (RK); Institute of Psychological Medicine and Clinical Neurosciences, School of Medicine, Cardiff University, Cardiff, UK (VE-P); Department of Genetics, King Faisal Specialist Hospital and Research Center, Riyadh, Saudi Arabia (NAA-T, BFM, SMW); Division of Genetics and Epidemiology, The Institute of Cancer Research, London, UK (RSH); NIHR Oxford Comprehensive Biomedical Research Centre, Oxford University Hospitals NHS Foundation Trust, Oxford, UK (DNC).
The authors have no disclosures. The funders had no role in the design of the study; the collection, analysis, and interpretation of the data; the writing of the manuscript; and the decision to submit the manuscript for publication. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR, the Department of Health, or the Wellcome Trust. We are grateful to the participants in the SCOT, COIN, and COIN-B trials who consented for the donation of samples for research, and to the investigators and pathologists who recruited patients and collected samples.
Author contributions: Study design: CP, JPC, IT, DNC; data collection: SB, CP, EJ, TI, RK, MPS, JP, AH, JM, MGS, EJ, HW, LG, TSM, RK, VE-P, NA-T, BFM, SMW, RSH; data analysis: VG, CP, JPC, IT, DNC; manuscript writing: DNC; manuscript approval: all authors.
Supplementary Material
References
- 1. Galon J, Costes A, Sanchez-Cabo F et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;3135795:1960–1964. [DOI] [PubMed] [Google Scholar]
- 2. Pages F, Berger A, Camus M et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med. 2005;353(25):2654–2666. [DOI] [PubMed] [Google Scholar]
- 3. Ghiringhelli F, Apetoh L, Tesniere A et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15(10):1170–1178. [DOI] [PubMed] [Google Scholar]
- 4. Tesniere A, Schlemmer F, Boige V et al. Immunogenic death of colon cancer cells treated with oxaliplatin. Oncogene. 2010;294:482–491. [DOI] [PubMed] [Google Scholar]
- 5. Vacchelli E, Ma Y, Baracco EE et al. Chemotherapy-induced antitumor immunity requires formyl peptide receptor 1. Science. 2015;3506263:972–978. [DOI] [PubMed] [Google Scholar]
- 6. Vacchelli E, Enot DP, Pietrocola F et al. Impact of pattern recognition receptors on the prognosis of breast cancer patients undergoing adjuvant chemotherapy. Cancer Res. 2016;7611:3122–3126. [DOI] [PubMed] [Google Scholar]
- 7. Apetoh L, Ghiringhelli F, Tesniere A et al. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13(9):1050–1059. [DOI] [PubMed] [Google Scholar]
- 8. Andre T, Boni C, Mounedji-Boudiaf L et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med. 2004;350(23):2343–2351. [DOI] [PubMed] [Google Scholar]
- 9. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;1406:805–820. [DOI] [PubMed] [Google Scholar]
- 10. Netea MG, van der Meer JW. Immunodeficiency and genetic defects of pattern-recognition receptors. N Engl J Med. 2011;3641:60–70. [DOI] [PubMed] [Google Scholar]
- 11. Seifert R, Wenzel-Seifert K. The human formyl peptide receptor as model system for constitutively active G-protein-coupled receptors. Life Sci. 2003;7318:2263–2280. [DOI] [PubMed] [Google Scholar]
- 12. Ranjith-Kumar CT, Miller W, Sun J et al. Effects of single nucleotide polymorphisms on Toll-like receptor 3 activity and expression in cultured cells. J Biol Chem. 2007;28224:17696–17705. [DOI] [PubMed] [Google Scholar]
- 13. Chen DN, Song CG, Yu KD et al. A prospective evaluation of the association between a single nucleotide polymorphism rs3775291 in Toll-Like Receptor 3 and breast cancer relapse. PLoS One. 2015;107:e0133184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Castro FA, Forsti A, Buch S et al. TLR-3 polymorphism is an independent prognostic marker for stage II colorectal cancer. Eur J Cancer. 2011;478:1203–1210. [DOI] [PubMed] [Google Scholar]
- 15. Senkus E, Kyriakides S, Ohno S et al. Primary breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2015;26(suppl 5):v8–v30. [DOI] [PubMed] [Google Scholar]
- 16. Labianca R, Nordlinger B, Beretta GD et al. Early colon cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2013;24(suppl 6):vi64–vi72. [DOI] [PubMed] [Google Scholar]
- 17. Iveson TJ, Kerr RS, Saunders MP et al. 3 versus 6 months of adjuvant oxaliplatin-fluoropyrimidine combination therapy for colorectal cancer (SCOT): an international, randomised, phase 3, non-inferiority trial. Lancet Oncol. 2018;194:562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Adams RA, Meade AM, Seymour MT et al. Intermittent versus continuous oxaliplatin and fluoropyrimidine combination chemotherapy for first-line treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. Lancet Oncol. 2011;127:642–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wasan H, Meade AM, Adams R et al. Intermittent chemotherapy plus either intermittent or continuous cetuximab for first-line treatment of patients with KRAS wild-type advanced colorectal cancer (COIN-B): a randomised phase 2 trial. Lancet Oncol. 2014;156:631–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maughan TS, Adams RA, Smith CG et al. Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. Lancet. 2011;3779783:2103–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Summers MG, Smith CG, Maughan TS et al. BRAF and NRAS locus-specific variants have different outcomes on survival to colorectal cancer. Clin Cancer Res. 2017;2311:2742–2749. [DOI] [PubMed] [Google Scholar]
- 22. Purcell S, Neale B, Todd-Brown K et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;813:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Price AL, Patterson NJ, Plenge RM et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;388:904–909. [DOI] [PubMed] [Google Scholar]
- 24. Delaneau O, Marchini J, Zagury JF. A linear complexity phasing method for thousands of genomes. Nat Methods. 2011;92:179–181. [DOI] [PubMed] [Google Scholar]
- 25. Marchini J, Howie B, Myers S et al. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet. 2007;397:906–913. [DOI] [PubMed] [Google Scholar]
- 26. Al-Tassan NA, Whiffin N, Hosking FJ et al. A new GWAS and meta-analysis with 1000Genomes imputation identifies novel risk variants for colorectal cancer. Sci Rep. 2015;5:10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McShane LM, Altman DG, Sauerbrei W et al. Reporting recommendations for tumor marker prognostic studies (REMARK). J Natl Cancer Inst. 2005;9716:1180–1184. [DOI] [PubMed] [Google Scholar]
- 28. Lek M, Karczewski KJ, Minikel EV et al. Analysis of protein-coding genetic variation in 60, 706 humans. Nature. 2016;5367616:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tennessen JA, Bigham AW, O’Connor TD et al. Evolution and functional impact of rare coding variation from deep sequencing of human exomes. Science. 2012;3376090:64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Walter K, Min JL, Huang J et al. The UK10K project identifies rare variants in health and disease. Nature. 2015;5267571:82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Therasse P, Arbuck SG, Eisenhauer EA et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada .J Natl Cancer Inst. 2000;923:205–216. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.