

Summary
In mammals, ∼100 deubiquitinases act on ∼20,000 intracellular ubiquitination sites. Deubiquitinases are commonly regarded as constitutively active, with limited regulatory and targeting capacity. The BRCA1-A and BRISC complexes serve in DNA double-strand break repair and immune signaling and contain the lysine-63 linkage-specific BRCC36 subunit that is functionalized by scaffold subunits ABRAXAS and ABRO1, respectively. The molecular basis underlying BRCA1-A and BRISC function is currently unknown. Here we show that in the BRCA1-A complex structure, ABRAXAS integrates the DNA repair protein RAP80 and provides a high-affinity binding site that sequesters the tumor suppressor BRCA1 away from the break site. In the BRISC structure, ABRO1 binds SHMT2α, a metabolic enzyme enabling cancer growth in hypoxic environments, which we find prevents BRCC36 from binding and cleaving ubiquitin chains. Our work explains modularity in the BRCC36 DUB family, with different adaptor subunits conferring diversified targeting and regulatory functions.
Keywords: BRCA1-A, BRISC, ubiquitin signaling, deubiquitination, DNA repair, regulation, SHMT2
Graphical Abstract
Highlights
-
•
Detailed structural studies of the BRCA1-A and BRISC-SHMT2 protein complex
-
•
Complex-specific subunits functionalize BRCC36, regulating its activity and targeting
-
•
BRCA1-A sequesters BRCA1 with nanomolar affinity
-
•
Cancer-associated metabolic enzyme SHMT2 inhibits BRISC deubiquitination activity
Structural studies of the BRCA1-A and BRISC complexes reveal a common enzymatic core that is functionalized by complex-specific proteins. BRCA1-A localizes to DNA double-strand breaks, sequesters BRCA1, and suppresses DNA resection. BRISC regulates protein degradation by the endosomal-lysosomal pathway through deubiquitination and is inhibited by the metabolic enzyme SHMT2.
Introduction
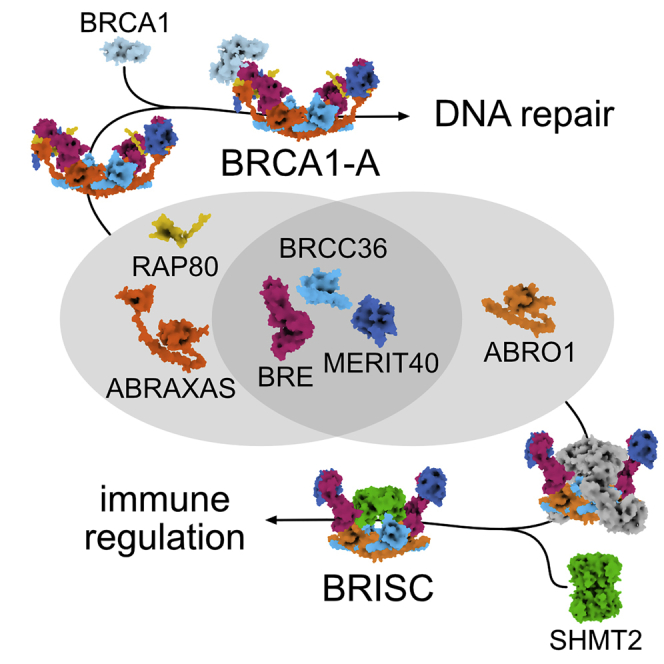
The deubiquitinase (DUB) BRCC36 is the catalytic subunit for two multi-protein complexes: BRCA1-A, which safeguards genome integrity by regulating DNA repair pathway choice, and BRISC, which serves cellular stress response and immune signaling functions (Wu et al., 2012, Yin et al., 2012, Zheng et al., 2013). Both BRCC36-containing complexes are specific for lysine-63-linked ubiquitin (K63-Ub) chains (Cooper et al., 2009, Zeqiraj et al., 2015). BRCA1-A and BRISC also contain proteins MERIT40 (Feng et al., 2009, Shao et al., 2009, Wang et al., 2009) and BRE (Feng et al., 2009, Hu et al., 2011a) as core components. The key compositional differences between the two complexes are the two scaffolding partners of BRCC36 (Figure S1A): in BRCA1-A, BRCC36 is supported by ABRAXAS (Wang et al., 2007), whereas in BRISC, it is paired with ABRO1 (Hu et al., 2011a, Wang et al., 2007). ABRAXAS and ABRO1 are evolutionarily related as they share ∼30% identity in humans and contain Mpr1, Pad1 N-terminal (MPN) domains like BRCC36 (Figures S1B and S1C).
BRCA1 and BRCA2 facilitate DNA double-strand break (DSB) repair by homologous recombination (HR), whereas BRCA1-A opposes HR by suppressing resection (Coleman and Greenberg, 2011, Hu et al., 2011b). The BRCA1-A assembly constitutively includes the repair protein RAP80 (Kim et al., 2007, Sobhian et al., 2007, Wang et al., 2007), a protein that recognizes K63-Ub chromatin domains generated by ɣ-H2AX-dependent DNA damage signaling through RNF4/RNF8/RNF168 (Guzzo et al., 2012, Typas et al., 2015, Uckelmann and Sixma, 2017).
Familial mutations in the BRCA1-A proteins ABRAXAS and RAP80 predispose carriers to early-onset breast cancer, analogous to mutations in BRCA1 and BRCA2 (Nikkilä et al., 2009, Solyom et al., 2012). BRCA1-A requires the tandem ubiquitin (UIM2)- and SUMO-interacting motifs (SIM) in RAP80 and the BRCC36 DUB to function in DNA repair (Guzzo et al., 2012, Hu et al., 2012, Lombardi et al., 2017, Ng et al., 2016). BRCA1-A recruits BRCA1 by binding its BRCT domains upon phosphorylation of a motif near the C terminus of ABRAXAS (Wang et al., 2007). BRCA1 binding to BRCA1-A sequesters the HR activator BRCA1 ∼2–10 kb distal from DNA break sites (Goldstein and Kastan, 2015, Kakarougkas et al., 2013), which is posited to limit HR (Goldstein and Kastan, 2015). It is currently unclear how BRCA1-A is functionalized and targeted by RAP80 and ABRAXAS and how BRCA1 is inhibited when bound to the complex.
While BRCA1-A is predominantly confined to the nucleus, BRISC localizes to the nucleus and the cytoplasm, where its wide range of reported substrates includes ATF4, THAP5, NLRP3, and the cytosolic domain of IFNAR1, an interferon receptor (Ambivero et al., 2012, Cilenti et al., 2011, Py et al., 2013, Zheng et al., 2013). BRISC has been noted to co-purify with serine hydroxy methyltransferase 2 (SHMT2), a metabolic enzyme that converts serine to glycine and a tetrahydrofolate-bound one-carbon unit, and occurs in cytosolic (SHMT2α) and mitochondrial (mSHMT2) forms (Giardina et al., 2015, Sowa et al., 2009, Zheng et al., 2013). SHMT2 is implicated in regulating two medically important aspects of BRISC function, interferon signaling, and viral protein degradation (Xu et al., 2018, Zheng et al., 2013). Since overexpression of SHMT2 plays a pivotal role in the metabolic adaptation of cancer cells to hypoxia (Kim et al., 2015, Lee et al., 2014, Sowa et al., 2009, Zheng et al., 2013), we aimed to understand the functional interplay between BRISC and SHMT2.
While some DUBs have specificity for cleaving distinct ubiquitin-chain linkages, they are not typically regarded as modular, multimeric enzymes and are not assumed to exhibit specificity for individual proteins or pathways. The available data for the BRISC and BRCA1-A complexes, however, suggested that a shared DUB core might assume different roles in DNA repair and immune signaling. To examine the mechanistic circuitry in these multimeric DUB assemblies, we set out to study the molecular basis of BRCA1-A and BRISC regulation and targeting.
Results
Common Architectural Cores of the BRCA1-A and BRISC Assemblies
We determined the structure of the 10-protein mouse BRCA1-A complex (overall molecular weight of 338 kDa; highly similar to human BRCA1-A), comprising two copies each of BRCC36, ABRAXAS, BRE, MERIT40, and RAP80 (residues 274–334) by X-ray crystallography using low-resolution native Zn-SAD phasing methods and carried out final refinement at 3.75 Å resolution (Figures 1A and 1B; Figure S1D; Table 1). In addition, we characterized the structure of the 10-protein human BRISC-SHMT2α complex (overall molecular weight 445 kDa) by cryoelectron microscopy (cryo-EM) at an overall resolution of 3.86 Å (Figures 1B and 1C; Figures S1D, S2A–S2E; Table 2). Analogous to BRCA1-A, the BRISC structure comprises two copies each of proteins BRCC36, ABRO1, BRE, MERIT40, and SHMT2α. We used the A264T mutant of SHMT2α (A285T in mSHMT2) for all structural studies and biochemical characterization, which was originally used to solve the crystal structure of the dimeric apo-SHMT2 (PDB: 6DK3). SHMT2 residue numbers in the text refer to SHMT2α.
Figure 1.

Structures of the BRCA1-A and BRISC-SHMT2α Complexes
(A) A cartoon representation of the BRCA1-A complex crystal structure.
(B) Schematic representation of the domain boundaries of BRCA1-A and BRISC-SHMT2α, showing parts present in the structure (domain cartoons and solid line), parts that are present in the construct but disordered in the structure (dashed line), and parts not present in the construct (grayed out domain cartoons and solid gray line).
(C) A cartoon representation of the BRISC-SHMT2α complex cryo-EM structure.
Table 1.
BRCA1-A Crystallographic Data Collection and Refinement Statistics
| Native Zn-SAD | PDB: 6GVWa | |
|---|---|---|
| Data Collection | ||
| Space group | P212121 | P212121 |
| Cell dimensions | ||
| a, b, c (Å) | 97.07, 122.71, 431.94 | 97.10, 112.64, 431.33 |
| Resolution (Å) | 118–4.30 (4.374–4.300)b | 30–3.75 (3.814–3.750) |
| Rmeas | 0.217 (>3.000) | 0.175 (>3.000) |
| Rpim | 0.024 (1.814) | 0.025 (2.173) |
| CC1/2 | 0.999 (0.305) | 0.952 (0.476) |
| Mean I/σI | 18.3 (0.5) | 11.6 (0.4) |
| Multiplicity | 79.6 (79.1) | 51.9 (52.8) |
| Completeness (%) | ||
| Spherical | 100 (100) | 100 (100) |
| Ellipsoidal | – | 82.6 (33.2)c |
| Refinement | ||
| Resolution (Å) | – | 30–3.75 |
| No. reflections | – | 42,193 |
| Rwork/Rfree (%) | – | 22.72/25.91 |
| No. atoms | ||
| Protein | – | 20,138 |
| Zn2+ ions | – | 2 |
| Water | – | 2 |
| B factors | ||
| Protein | – | 229 |
| Zn2+ ions | – | 193 |
| Water | – | 163 |
| RMSDs | ||
| Bond lengths (Å) | – | 0.006 |
| Bond angles (°) | – | 1.147 |
Data from 7 crystals were combined
Values in parentheses are for highest-resolution shell
After anisotropic truncation with STARANISO as used for refinement
Table 2.
Cryo-EM Data Collection, Refinement, and Validation Statistics
| BRISC-SHMT2 (EMDB: EMDB-0132; PDB: 6H3C) | |
|---|---|
| Data Collection and Processing | |
| Magnification | 58,140 |
| Voltage (kV) | 300 |
| Electron exposure (e–/Å2) | 45 |
| Defocus range (μm) | −0.5 to −5 |
| Pixel size (Å) | 0.86 |
| Symmetry imposed | C2 |
| Initial particle images (no.) | 332,598 |
| Final particle images (no.) | 35,595 |
| Map resolution (Å) | 3.86 |
| FSC threshold | 0.143 |
| Map resolution range (Å) | 3.86–15 |
| Refinement | |
| Initial model used (PDB code) | BRCA1-A (PDB: 6GVW), SHMT2 (PDB: 5V7I) |
| Model resolution (Å) | 3.86 |
| FSC threshold | 0.143 |
| Map sharpening B factor (Å2) | N/A |
| Model composition | |
| Non-hydrogen atoms | 25,134 |
| Protein residues | 3,154 |
| Water | 2 |
| B factors (Å2) | |
| Protein | 90.25 |
| Water | 64.48 |
| RMSDs | |
| Bond lengths (Å) | 0.020 |
| Bond angles (°) | 1.93 |
| Validation | |
| MolProbity score | 1.32 |
| Clashscore | 1.88 |
| Poor rotamers (%) | 0.07 |
| Ramachandran plot | |
| Favored (%) | 94.56 |
| Allowed (%) | 4.8 |
| Disallowed (%) | 0.64 |
In BRCA1-A, two heteropentamers composed of BRCC36, ABRAXAS, BRE, and MERIT40 form an arc-shaped complex with approximate dimensions 213 × 117 × 87 Å3 (Figure 1A). The two half-arcs contact each other through BRCC36 and its scaffold partner ABRAXAS. While BRCC36 and ABRAXAS both contain MPN domains, only that of BRCC36 is catalytically active (Figure S3A). In our crystals, the BRCC36 catalytic center is captured in an active configuration, with a single catalytic Zn2+ ion tetrahedrally coordinated by His122, His124, Asp135, and a water molecule, oriented by Glu33 (Zeqiraj et al., 2015) (Figure S3A). We find that BRCC36 in BRCA1-A and BRISC is activated by assembly due to interaction between Glu30 of BRCC36 and Asn170 in ABRAXAS and Asn164 in ABRO1, respectively, which structures the activation loop and positions the catalytic Glu33 (Figure S3B). This mechanism is homologous to what has been observed in the ancestral BRCC36-KIAA0157 complex (Zeqiraj et al., 2015) but is distinct from that seen in the COP9 signalosome (CSN) and the proteasome lid, where the position of the scaffold MPN domain differs (Figure S3B) (Lingaraju et al., 2014, Pathare et al., 2014, Worden et al., 2014, Worden et al., 2017).
In the BRCA1-A complex structure, the ABRAXAS MPN domain plays a key structural role connecting the catalytically active BRCC36 subunit to the distal part of the arc consisting of BRE, MERIT40, and RAP80 (Figure 1A). BRE, in turn, forms the central bridge between the ABRAXAS-BRCC36 MPN domain dimer and the Von-Willebrand factor A (VWA) domain protein MERIT40 (Figures 1A and 2A). BRE contains N- and C-terminal ubiquitin E2 variant (UEV) domains (Figure 2A). We identify a new, third domain in BRE, a previously unidentified central RING-finger containing, WD-repeat-containing proteins and yeast DEAD (DEXD)-like helicase domain (RWD) located between the UEV domains (Figures 1B and 2A; Figure S3C). The BRE RWD and C-terminal UEV (UEV-C) domains are connected by a continuous helix, similar to the arrangement of the UEV and RWD domain observed in human DNA repair protein FANCL (Hodson et al., 2011) (Figure S3D). The BRE N-terminal UEV domain (UEV-N) binds the ABRAXAS MPN domain, and the BRE UEV-C holds MERIT40 at the extremity of the arc (Figure 2A).
Figure 2.

Integration of RAP80 and DUB Specificity of BRCA1-A
(A) The position of the UEV-N, RWD, and UEV-C domains of BRE and the VWA domain of MERIT40 (shown as cartoon) within the BRCA1-A complex (shown as surface). RAP80 is embedded into the complex through contacts with BRE, MERIT40, and ABRAXAS.
(B) BRCA1-A complex processes (Ub)4 faster than (Ub)3 and (Ub)2. K63-linked ubiquitin substrate (150 ng/lane) was incubated with 5 nM BRCA1-A on ice and analyzed by silver-stained SDS-PAGE. Under these conditions, (Ub)4 is cleaved within 5 min, whereas (Ub)2 and (Ub)3 are not cleaved within 7 h.
(C) Tetra-ubiquitin chains are pre-oriented on BRCA1-A complex during cleavage, as evident from comparison of the degradation of K63-linked (Ub)4 substrate labeled with TAMRA at the proximal and distal ubiquitin, respectively. Degradation products of 1 μM substrate incubated with 10 nM BRCA1-A at room temperature were resolved on an SDS-PAGE; TAMRA was subsequently visualized.
In the BRCA1-A complex, the two heteropentamers are intimately linked by a bundle of four ⍺helices comprising the C-terminal tails of BRCC36 and ABRAXAS (Figure 1A; Figure S3E). An ABRAXAS linker segment and an α helix crossover from one site of the half-arc to the other effectively interlock the two pentamers (Figure S3E). The same arrangement is found in BRISC (Figure S3E). This crossover arrangement is not observed in the ancestral BRCC36/KIAA0157 subcomplex (Figure S3E), where dissociation of the two pentamers has been suggested as a putative mechanism to regulate activity (Zeqiraj et al., 2015). Given the observed pseudo-knotted arrangement, regulation by dissociation is unlikely in the case of mammalian BRCA1-A, and instead, we find that accessory subunits, such as RAP80, have appeared in the evolutionary lineage to confer targeting and regulatory capabilities.
The BRCA1-A/BRISC Architecture Offers a Scaffold for Ubiquitin Binding
In light of the multiple ubiquitin-binding modules in BRE, we tested BRCA1-A with K63-linked chains of different length ranging from di-ubiquitin (Ub)2 and tri-ubiquitin (Ub)3 to tetra-ubiquitin (Ub)4. BRE contains UEV/RWD binding domains, with a K63-linked ubiquitin-binding site previously reported for the BRE-MERIT40 complex (Kyrieleis et al., 2016). We saw preferential cleavage of (Ub)4, while no cleavage of (Ub)2 and (Ub)3 chains was observed under these conditions (Figure 2B). BRISC similarly exhibited preferential cleavage for longer chains (Figure S3F). Cleavage of (Ub)2, however, was seen when reactions were run at higher temperatures and enzyme concentrations (Figure S3G). (Ub)4 units span ∼90 Å and may thus connect the BRCC36 active site to BRE and potentially MERIT40. In light of the structure, we tested whether the VWA domain of the distal MERIT40 itself also potentially contributed to ubiquitin binding. NMR binding studies using 15N-labeled ubiquitin found mono-ubiquitin binding to BRE-MERIT40 complex with estimated micromolar affinity (Figure S3H). Additionally, the isolated MERIT40 subunit bound ubiquitin with estimated high micromolar affinity (Figure S3H), comparable to established ubiquitin-binding domains (Hicke et al., 2005). The arc-shaped BRCA1-A/BRISC architecture thus offers a ubiquitin-binding scaffold potentially ranging from the BRCC36 active site located at the middle of the arc up to the MERIT40 subunits located at the tip.
We then tested whether this arrangement pre-orientates the (Ub)4 chains for cleavage or whether the ubiquitin-binding modules predominately serve to generate high local concentrations. For this, we introduced fluorescent labels on either the proximal or the distal end of (Ub)4 chains (Figure 2C). Incubating the proximally labeled substrate with BRCA1-A, we mostly observed (Ub)2 and (Ub)3 cleavage products, while the distally labeled sample resulted in a different cutting pattern, accumulating predominantly Ub cleavage products (Figure 2C). These findings are consistent with alignment of the (Ub)4 chain with the distal ubiquitin, pointing toward BRCC36, being cleaved first, in a process likely supported by the multiple ubiquitin-binding domains on BRE and MERIT40.
BRCA1-A RAP80 Integration
BRCA1-A activity in DNA repair critically depends on the presence of RAP80. Our crystal structure now demonstrates how the RAP80 subunit functionalizes the complex. We find that the RAP80 protein is solubilized and stabilized by the presence of BRCA1-A and stably maintained in all purification steps. Our BRCA1-A structure reveals that RAP80 is deeply integrated into the BRCA1-A assembly by interactions with three subunits: ABRAXAS, BRE, and MERIT40 (Figures 1A and 2A). The N-terminal portion (Gly272-Trp278) of the RAP80 ABRAXAS-interacting region (AIR) engages MERIT40, the trailing ⍺ helix (Asn290-Leu314) binds across the BRE UEV-C domain, with the RAP80 C terminus forming a β strand (Val315-Phe330) sandwiched by the RWD and UEV-N domains of BRE. The ABRAXAS C terminus binds the ⍺ helix of RAP80 AIR through an ⍺-helical domain (Ile277-Phe287), and the RAP80 AIR β sheet stacks in-between β sheets from MERIT40 and ABRAXAS (Figure 2A). The structural elements of RAP80 AIR are highly conserved (Figure S4A).
While the region in RAP80 that mediates BRCA1-A binding is short, it is essential for BRCA1-A function (Figures S4B–S4D). It also gives rise to a relatively large interface between RAP80 and BRE (1,951 Å2 buried surface area) and MERIT40 (822 Å2). It is the interaction with ABRAXAS (1,146 Å2), however, that drives specific incorporation of RAP80 into BRCA1-A: while BRISC alone integrates co-expressed RAP80 substoichiometrically (Figure S4E), a hybrid BRISC complex that contains a fusion scaffold protein, in which the C-terminal tail of ABRO1 (261–415) is exchanged for that of ABRAXAS (269–407), tightly integrates RAP80 (Figure 3A). The BRCA1-A-specific ABRAXAS subunit drives RAP80 integration, while the BRE/MERIT40 subunits present in both BRISC/BRCA1-A complexes play an important accessory role. Since BRISC and RAP80 are targeted to different cellular compartments, association between BRISC and RAP80 is not observed in vivo despite the presence of BRE and MERIT40 (Feng et al., 2010).
Figure 3.

RAP80 Controls BRE Conformation and Prevents Dimerization
(A) A fusion scaffold protein containing residues 1–260 of mouse ABRO1 fused to residues 269–407 of mouse ABRAXAS disrupts dimerization (native PAGE) and integrates RAP80 stoichiometrically into the complex (SDS-PAGE).
(B) The structure of BRISC dimer. A pseudoatomic model of BRISC was generated by rigid-body fitting of the atomic BRISC model derived from the cryo-EM structure determination of BRISC-SHMT2α complex into the density map of BRISC dimer.
(C) Integration of RAP80 into BRCA1-A results in a substantial conformation change of BRE as evidenced by a comparison of the structures of BRCA1-A and BRISC.
Our structure of BRCA1-A explains the observation that loss of BRE, or MERIT40, significantly impairs BRCA1-A function, resulting in DNA repair defects and loss of cancer suppression similar to what was seen for loss of RAP80 (Feng et al., 2009, Hu et al., 2011a, Patterson-Fortin et al., 2010, Rebbeck et al., 2011, Shao et al., 2009). The high degree of structural interdependence between RAP80, BRE, and MERIT40 means that the physical link between RAP80 and ABRAXAS, which is essential for DNA repair, can no longer be efficiently retained in BRCA1-A once BRE (accounts for ∼50% of the RAP80 interface) or MERIT40 (accounts for ∼20%) is lost.
RAP80 binding further affects the BRCA1-A quaternary structure. A previous ∼20 Å low-resolution structure of truncated BRCA1-A determined in the absence of RAP80 and using chemical crosslinking reported the dimerization of two full BRCA1-A arcs (Kyrieleis et al., 2016). We find that BRISC in the absence of SHMT2 exists in an equilibrium between single arc and dimer, which can be shifted toward the dimer by chemical crosslinking, analogous to truncated BRCA1-A without RAP80 (Figures 3A and 3B; Figures S4F and S4G). We observe a single arc of RAP80-bound BRCA1-A in our crystal structure (Figure 1A) and in negative-stain EM studies, irrespective of the use of chemical crosslinking (Figure S4H). The presence of RAP80 binding results in a structural change in BRE and MERIT40, which in turn prevents dimerization of the two arcs (Figure 3C; Figure S4I). RAP80 is essential for BRCA1-A complex assembly and stability in vivo (Bian et al., 2012), and its cellular concentration exceeds that of ABRAXAS (Mok and Henderson, 2012). This, together with the observed intricate structural integration of RAP80 into BRCA1-A, suggests that RAP80 is a constitutive member of the BRCA1-A complex and that the single arc observed in our structure likely depicts the physiological assembly.
SUMO-Dependent Targeting of BRCA1-A
The RAP80 N-terminal region contains SIM (residues 41–43) and K63-linkage- specific tandem UIM domains (residues 80–99 and 105–124) that bind mixed ubiquitin-SUMO chains with high affinity (Guzzo et al., 2012, Hu et al., 2012). These domains recruit BRCA1-A to sites of DNA damage (Guzzo et al., 2012, Hu et al., 2012) and are connected to BRCA1-A via a flexible linker (Figure S4J).
We thus tested whether the RAP80 SIM/UIM units serve in recruitment only or whether they also contribute to preferential cleavage of mixed SUMO ubiquitin chains. For this, we monitored degradation of (Ub)2 and SUMO/di-ubiquitin (SUMO-K63-diUb) chains by BRCA1-A in gel-based cleavage assays (Figure S5A). We found a small preference in cleavage of SUMO-K63-Ub2 chains in the presence of full-length RAP80, but not with BRCA1-A variants that lack the RAP80 SIM-UIM2 (Figure S5B). In light of the preferential cleavages of (Ub)4 over (Ub)2 chains, we then switched to longer SUMO-K63 chains with, on average, more than 6 ubiquitin units. In the context of these longer chains, the RAP80 SIM/UIM module did not confer a robust effect on cleavage (Figure S5C). This result suggests that the role of the RAP80 SIM/UIM module lies predominantly in recruiting the complex to sites of SUMO and ubiquitin modifications.
BRCA1 Sequestration by BRCA1-A Complex
A key BRCA1-A function is to regulate BRCA1 activity in DSB repair. BRCA1-A and BRCA1 form a complex that sequesters BRCA1 distant from the site of damage, an important regulatory step in HR (Goldstein and Kastan, 2015, Kakarougkas et al., 2013). This is dependent on the presence of RAP80, and a correctly assembled BRCA1-A complex, but does not require BRCC36 DUB activity (Ng et al., 2016). The ABRAXAS C terminus phosphorylated at Ser406 binds the BRCA1-BRCT 1 (residues 1,642–1,736) and BRCT 2 (residues 1,756–1,855) domains (BRCA1-BRCT) with an apparent Kd of ∼1.2 μM, an affinity comparable to that of other BRCA1-BRCT interaction partners (Badgujar et al., 2013, Wu et al., 2016). Given comparable affinities with other BRCA1-BRCT binders, it was unclear how BRCA1-A, an inhibitor of resection, can potentially sequester BRCA1 away from other binding partners such as BACH1 or CtIP that typically activate HR (Badgujar et al., 2013, Clapperton et al., 2004, Shiozaki et al., 2004, Varma et al., 2005, Wu et al., 2016).
We set out to quantify BRCA1 binding to BRCA1-A in the context of the fully assembled complex. Activation of the DSB repair checkpoint has been suggested to proceed via the double-phosphorylated ABRAXAS variant p-Ser404/406 in conjunction with induced dimerization of the BRCA1-BRCT (Wu et al., 2016). We generated the entire BRCA1-A complex with site-specifically phosphorylated ABRAXAS by ligating phosphorylated C-terminal peptides by sortase-mediated transpeptidation (Figures S5D and S5E). In microscale thermophoresis (MST) experiments, measuring binding of labeled BRCA1-BRCT domains to the entire BRCA1-A complex bearing ABRAXAS p-Ser404/406, we observed substantially higher affinity (Kd ∼80 ± 7 nM) (Figure 4A) than what is typically reported for BRCA1-BRCT domain binding. The doubly phosphorylated ABRAXAS p-Ser404/406 integrated in the BRCA1-A complex thus provides a high-affinity docking cradle for BRCA1, exceeding the affinity of other known BRCA1 interactors by more than 10-fold (Badgujar et al., 2013, Liu and Ladias, 2013, Ray et al., 2006, Shiozaki et al., 2004, Varma et al., 2005).
Figure 4.

BRCA1-A Forms a Defined High-Affinity Complex with BRCA1
(A) In an MST assay measuring binding of labeled BRCA1-BRCT to full-length BRCA1-A complex including phosphorylated p-Ser404/406 ABRAXAS C terminus, BRCA1-BRCT is bound with nanomolar affinity. The assay measures change of relative fluorescence during heating; values are Fnorm = Fhot / Fcold. Error bars represent mean ± SD of n = 4 replicates. Back titration with unlabeled BRCA1-BRCT confirms nanomolar affinity. Error bars represent mean ± SD of n = 4 replicates.
(B) Crosslinking network of the BRCA1-A-BRCA1 complex. Proteins are shown schematically as bars. Crosslinks are shown as black lines. The crosslink between K360 of BRE and K1750 of BRCA1 is conditional on the presence of p-Ser404/406 phosphorylation on ABRAXAS C terminus.
(C) Model of the BRCA1-A-BRCA1 high-affinity complex. One protomer of ABRAXAS, BRE, and RAP80 is shown as cartoon, while the remainder of the BRCA1-A complex is shown as gray surface. A BRCA1-BRCT dimer (blue, cartoon) is depicted in a position that localizes K1750 of BRCA1 proximal to K360 of BRE. The unstructured C-terminal regions of ABRAXAS are depicted schematically as orange and gray lines. A di-ubiquitin (green, surface) is shown modeled into the active site.
The increased interaction between BRCA1 and the BRCA1-A complex likely arises from increased avidity due to the dimeric structure of the BRCA1-A arc but also potentially suggests further contacts extending beyond the BRCA1-BRCT ABRAXAS phosphopeptide interface. We compared the interaction between BRCA1-BRCT and non-modified or p-Ser404/406 phosphorylated BRCA1-A in a crosslinking mass spectrometry experiment and observed a specific crosslink between BRCA1-A subunit BRE (Lys360) and BRCT (Lys1750) that was dependent on the presence of p-Ser404/406 modifications in ABRAXAS (Figure 4B). The unstructured linker in ABRAXAS that separates the RAP80-binding domain from the phosphorylated BRCA1-binding motif allows the BRCA1-BRCT dimer to reach BRE (UEV-C) at either side of the arc (Figure 4C).
We have not observed higher-order assemblies of multiple BRCA1-A arcs stabilized by BRCA1-BRCT in negative-stain EM (data not shown) or mobility shift assays (Figure S5E) that had previously been proposed (Kyrieleis et al., 2016). We also did not detect a difference in activity when assaying the phosphorylated BRCA1-A complex in the presence or absence of the BRCA1-BRCT (Figure S5F).
Our data instead support a model where ABRAXAS integrates RAP80 specifically into BRCA1-A, thereby preferentially targeting the BRCC36 DUB to DNA repair foci with both SUMO and K63-Ub chains. Following checkpoint activation, the phosphorylated ABRAXAS subunit in BRCA1-A provides the high-affinity binding site for BRCA1 sequestration and inhibition away from the break site.
BRISC-SHMT2α Architecture
BRISC and BRCA1-A are functionalized by the ABRO1 and ABRAXAS subunits, respectively, that work together with dedicated adaptor proteins. The BRISC core (BRCC36-ABRO1 MPN dimer, UEV-N and RWD domain of BRE) has the same 2-fold symmetrical arc shape and is structurally similar to BRCA1-A (RMSD 2.8 Å). The conformation of BRE UEV-C and MERIT40, however, differs markedly, with MERIT40 in BRISC rotated by ∼56° in respect to its position in the BRCA1-A crystal structure. We found the structural change to be induced by RAP80 binding to BRCA1-A (Figures 1A, 1C, 3A, and 3C). Whereas RAP80 is constitutively integrated into BRCA1-A, SHMT2 has an established BRISC-independent role in one-carbon metabolism (Giardina et al., 2015, Kim et al., 2015). SHMT2 is expressed in a mitochondrial and a cytosolic form (SHMT2α) (Anderson and Stover, 2009). SHMT2α is the form encountered by BRISC in vivo, because BRISC does not enter mitochondria. BRISC in isolation is present in a two-arc assembly (Figures 3A and 3B; Figures S4F, S4G, and S4I) not suitable for high-resolution structure determination due to structural heterogeneity. SHMT2α binding converts BRISC into a single-arc arrangement (Figure 1C). The BRISC-SHMT2 complex is recruited to the K63-modified IFNAR1 receptor and has been implicated in IFNAR1 deubiquitination (Zheng et al., 2013). Furthermore, BRISC-SHMT2 controls viral Tat protein degradation in HIV-1-infected cells (Xu et al., 2018). The BRISC-SHMT2α structure now illustrates how ABRO1 allows SHMT2α binding to BRISC and how SHMT2α functionalizes the complex beyond what is expected from an adaptor.
The BRISC-bound SHMT2α is catalytically inactive (Zheng et al., 2013), which is explained by our structure. Active SHMT2α when bound to its pyridoxal-5′-phosphate (PLP) cofactor is a tetramer (Giardina et al., 2015). In the absence of its cofactor, mammalian SHMT2 forms a structurally “open” apo-SHMT2 dimer that is enzymatically inactive (Giardina et al., 2015). SHMT2α bound to the center of the BRISC arc is present as a dimer, and each protomer forms an extended 1,161 Å2 interface involving subunits ABRO1, BRCC36, and BRE in BRISC (Figure 1C; Figure S5G). In complex with BRISC, the SHMT2α dimer adopts the open conformation, the structural hallmarks of the inactive PLP cofactor-free enzyme (Figures S5H and S5I).
Two key hydrophobic SHMT2α residues, Leu190 and Leu194, pack against ABRO1 residues Ile133, Ser134, and Thr135 (Figure 5A). An extended loop in SHMT2α (residues Gly274-Tyr288) contacts BRE residues Lys162-Ala167, and Phe195 of SHMT2α interacts with Trp130 of BRCC36 (Figures S5J and S5K). Mutation of the ABRO1-interacting SHMT2α residues Leu190 and Leu194 to lysine reduced BRISC binding in bio-layer interferometry by more than 10-fold (SHMT2α binding to BRISC: apo Kd ∼46 nM, PLP-preincubated Kd ∼39 nM; SHMT2α Leu190Lys, Leu194Lys: Kd ∼530 nM) (Figure S6A). Analogous to what we saw for RAP80/ABRAXAS in BRAC1-A, it is the BRISC-specific ABRO1 subunit, with help of the remainder of the complex, that is required for SHMT2α integration.
Figure 5.

Metabolic Enzyme SHMT2α Binds Specifically to BRISC, Inactivating It
(A) The cryo-EM structure of BRISC-SHMT2α reveals why SHMT2α binds BRISC, but not BRCA1-A. While the ABRO1 isoleucine (Ile133) proximal to SHMT2α is conserved in ABRAXAS, it points away from SHMT2α due to insertion of a proline (Pro137) into ABRAXAS.
(B) SHMT2 clashes with the position of the proximal ubiquitin of a ubiquitin dimer bound to the active site. Modeling di-ubiquitin (red, surface) into the BRCC36 active site reveals a substantial clash with the position of SHMT2α (green) in the BRISC-SHMT2α map (semitransparent surface).
(C) SHMT2α is a potent inhibitor of BRISC (2 nM), but not of BRCA1-A (5 nM), as shown by SHMT2α-mediated inactivation of BRISC in a DUB-activity assay using IQF K63-linked di-Ubiquitin. A BRISC-RAP80 fusion construct (2 nM) containing the MPN domain of ABRO1 combined with the C terminus of ABRAXAS integrates RAP80 like BRCA1-A but is inhibited by SHMT2α like BRISC. Error bars represent mean ± SD of n = 3 replicates.
SHMT2α Functions as a BRISC Inhibitor
A model of K63-linked di-ubiquitin chains on BRCC36 using AMSH-LP as a template (Sato et al., 2008) finds SHMT2α potentially blocking substrate access to the active site (Figure 5B). We therefore tested whether BRISC is inhibited when in complex with SHMT2α. For this, we monitored the cleavage of K63-linked (Ub)2 substrates by BRISC in the presence of SHMT2α. In fluorescent di-ubiquitin cleavage assays, SHMT2α inhibited K63-linked (Ub)2 cleavage by BRISC in a concentration-dependent manner, with an estimated apparent Ki ∼7 ± 0.3 nM, which is comparable to the binding constant between SHMT2α and BRISC determined by bio-layer interferometry (Kd ∼39 nM) (Figures S6A and S6B). Mutating SHMT2α residues Leu190 and Leu194 required for ABRO1 binding to Lys190 and Lys194 abolished BRISC inhibition by SHMT2α (Figure S6C). In addition to a possible role of SHMT2α in targeting BRISC to membranes, SHMT2α serves as a soluble DUB inhibitor when engaged. We find that the inhibitory effect of SHMT2α on BRISC is not affected by concentrations of PLP, serine, glycine, or tetrahydrofolate tested (Figures S6D–S6G).
We then tested whether SHMT2α inhibition was specific to BRISC and examined which epitopes of ABRO1 are involved. Unlike BRISC, BRCA1-A was not inhibited by SHMT2α, but a chimeric BRCA1-A complex bearing the MPN domain of ABRO1 (1-260) instead of ABRAXAS (1-269) was inhibited by SHMT2α (Figure 5C). Although in BRISC the SHMT2α-binding interface consists of BRE, ABRO1, and BRCC36, it is primarily the BRISC-specific subunit ABRO1, this time through its MPN domain, that confers specific SHMT2α binding. While ABRAXAS contains the equivalent of the SHMT2α-binding ABRO1 Ile133 residue (Ile139), an adjacent deletion of one residue flips the isoleucine side chain away from the interface, providing the structural basis of specificity (Figure 5A). SHMT2α thus functions as a specific, high-affinity protein inhibitor of the BRISC MPN DUB complex by restricting access to the active site, and this property is specifically conferred by ABRO1. Since BRCA1-A does not bind SHMT2α, BRCA1-A activity in DNA repair signaling is unaffected by nuclear SHMT2α.
SHMT2α as a Cell-Type-Dependent Regulator of BRISC Activity
The observed high-affinity binding and potent mutual inhibition between BRISC and SHMT2α in vitro led us to examine whether the concentration of BRISC-SHMT2α and its subcellular distribution would permit regulation in vivo. Pre-processed SHMT2 and a mature form cleaved after residue Ser29 predominantly localized to the mitochondria in mouse liver cells (Anderson and Stover, 2009). SHMT2α, however, is found in comparable amounts in the nucleus, cytosol, and mitochondria (Anderson and Stover, 2009). All SHMT2 isoforms are expected to inhibit BRISC based on our structure. To determine BRISC and total SHMT2 subcellular localization, we carried out immunofluorescence experiments using a SHMT2 antibody for an epitope common to all three forms of SHMT2 (Figure S6H). We found endogenous SHMT2 in HEK293 cells present in the cytosol (62.1% ± 11.7%), nucleus (30.3% ± 10.0%), and mitochondria (7.6% ± 2.2%). The BRISC-specific subunit ABRO1 was found in the cytosol (66.2% ± 8.8%) and in the nucleus (32.6% ± 8.2%) but was not detected in mitochondria (Figure 6A). Using quantitative mass spectrometry, concentrations on the order of ∼1 μM SHMT2 and ∼20–100 nM BRISC were measured in HeLa cells (Hein et al., 2015). To determine the expected extent of BRISC inhibition by SHMT2, we carried out in vitro internally quenched fluorescent (IQF) di-ubiquitin cleavage assays. At concentrations of BRISC and SHMT2α found in HeLa cells, BRISC DUB activity is expected to be inhibited by SHMT2 up to 50% (Figure 6B). In cancer cell lines surveyed by quantitative mass spectrometry, endogenous SHMT2 is present in concentrations exceeding 1 μM (Hein et al., 2015, Wilhelm et al., 2014), which corresponds to BRISC inhibition of more than 80% in our assays (Figure 6C). Across a variety of cell lines and tissues, SHMT2 and BRISC therefore are present at concentrations that permit BRISC inhibition, allowing SHMT2 to potentially regulate BRISC-mediated K63 deubiquitination activity.
Figure 6.

BRISC and SHMT2 Share the Same Cellular Compartments In Vivo and Vary in Concentration across Cell Lines and Tissues
(A) Endogenous SHMT2 and ABRO1 colocalize to nucleus and cytosol in quantitative immunofluorescence experiments. Error bars represent mean ± SD. SHMT2: n = 13 replicates. ABRO1: n = 9 replicates. Every image contained 2–10 individual cells.
(B) BRISC DUB activity at substrate and SHMT2 concentrations encountered inside healthy and diseased human cells ranges from fully active to completely inhibited. BRISC DUB activity corresponding to the concentrations of SHMT2α and ABRO1 in HeLa cells is shown in red.
(C) Concentrations of ABRO1 and endogenous SHMT2 in healthy human tissues (gray squares) and cancer cell lines (black circles) suggest that BRISC is attenuated by no more than 50% in healthy human cells but mostly inhibited in cancer cell lines. Concentrations of SHMT2 and ABRO1 corresponding to HeLa cells are shown in red.
Discussion
Our results demonstrate how a near identical deubiquitinase core is functionalized for radically different cellular purposes through the ABRO1 and ABRAXAS subunits and how these subunits confer differential BRCC36 targeting and regulation.
The BRCA1-A complex is an important safeguard for genome stability, which is frequently mutated in inherited breast cancers. BRCA1-A serves three roles in DSB repair: (1) recruitment to foci by stable RAP80 integration (Kim et al., 2007, Sobhian et al., 2007, van Wijk et al., 2012, Wang et al., 2007), (2) delimiting the K63-Ub boundary around breaks (Ng et al., 2016), and (3) inhibiting resection by sequestering BRCA1 and limiting its availability for the BRCA1/2-PALB2-RAD51 complex (Goldstein and Kastan, 2015, Typas et al., 2015).
Our work demonstrates that ABRAXAS functionalizes BRCA1-A for all three tasks. We show that RAP80 is an integral, structural part of the BRCA1-A complex as it interacts not only with ABRAXAS, but has substantial interaction surfaces with two additional subunits, MERIT40 and BRE. Loss of MERIT40, or BRE, compromises BRCA1-A integrity and genome safeguarding, which is explained by the structure. Loss of the RAP80 SIM-UIM2 domains gives rise to substantial deficiencies in BRCA1-A targeting to breaks and concomitant repair defects (Guzzo et al., 2012). The BRCA1-A architecture with the RAP80 SIM-UIM2 domains bestows preferential recruitment to mixed SUMO/K63-Ub2 chains. ABRAXAS recruits RAP80, and thereby localizes BRCA1-A to mixed chains, and likely interlinks K63-ubiquitin and SUMO signaling at repair sites and delimits the K63-ubiquitin boundary around breaks.
BRCA1-A plays an important role engaging and stabilizing BRCA1 at 2–10 kb distance from the breaks and inactivating it (Goldstein and Kastan, 2015, Typas et al., 2015). Binding of the BRCA1-BRCT domains to the ABRAXAS phospho-peptide is mutually incompatible with binding phosphorylated CtIP or BACH1 (CtIP: Kd∼3.7 μM; BACH1: Kd∼0.9 μM) (Shiozaki et al., 2004, Varma et al., 2005, Wu et al., 2016). The BRCA1-A arc architecture, with phosphorylation sites on ABRAXAS close to its C terminus, sequesters BRCA1 in a Kd ∼60–80 nM complex, which is significantly tighter than the affinities reported for other BRCA1-BRCT interactions. Cradled distal from break sites within the high-affinity BRCA1-A scaffold contacting BRE and ABRAXAS, BRCA1 is unable to engage other activators of resection providing a structural rationale for the observed BRCA1 inhibition.
The BRCA1-A architecture provides the circuitry for BRCA1 sequestration and directed deubiquitination through distinct structured modules. A mutation in BRCA1-BRCT (e.g., Lys1702Met), which impairs ABRAXAS binding, gives rise to a hyper-resection phenotype equivalent to the loss of RAP80/BRCA1-A (Dever et al., 2011), demonstrating that both modules are essential for BRCA1-A function. BRCA1 sequestration and targeting to SUMO-ubiquitin marks critically depend on ABRAXAS yet at the same time involve other core subunits present in both BRISC and BRCA1-A.
The BRISC complex largely operates outside the nucleus, regulating K63-ubiquitin levels in immune signaling. Whether BRISC activity can be regulated was unclear previously. We find that in BRISC, it is also the subcomplex-specific subunit ABRO1 that functionalizes the complex, enabling SHMT2α binding. Cellular proteases are typically produced as inactive proenzymes (Khan and James, 1998), complexed to soluble proteinaceous inhibitors (Farady and Craik, 2010), or embedded in macromolecular complexes that are autoinhibited in the absence of a substrate (Lingaraju et al., 2014, Pathare et al., 2014, Worden et al., 2014, Worden et al., 2017). Overall, the MPN DUB family is a notable exception, with no soluble protein inhibitors described so far (Leznicki and Kulathu, 2017, Sahtoe et al., 2015, Vander Linden et al., 2015). While assembled BRISC and BRCA1-A complexes assume an active, non-autoinhibited, default state, BRISC binding to the soluble metabolic enzyme SHMT2α results in mutual inhibition of both SHMT2α and BRISC activity.
Endogenous co-purification (Sowa et al., 2009, Zheng et al., 2013) and subcellular co-localization of BRISC and SHMT2 establish that a reciprocally inhibited BRISC-SHMT2α complex forms in target cells. The interaction between SHMT2α and BRISC, and the ensuing inhibition, could be regulated, for example, through posttranslational modifications at the interface. How SHMT2α inhibition of BRISC can facilitate the reported BRISC-dependent deubiquitination of IFNAR1 presently remains unclear (Zheng et al., 2013). Combining the results of in vitro enzymatic studies and the reported variations in SHMT2α and ABRO1 levels across different human cell lines and tissues, we expect that the relative BRISC activity is impacted by SHMT2 levels in some tissues (e.g., in lymph nodes, colon, and liver) and is largely unaffected in others (e.g., in brain and reproductive organs) (Uhlén et al., 2015, Uhlén et al., 2017, Wilhelm et al., 2014). We predict inhibition to apply predominantly to the BRISC/DUB and not to the metabolic enzyme SHMT2, because SHMT2 is present in large (>10-fold) excess over BRISC in many tissues (Figure S6I). We note that cancer cells frequently increase reliance on one-carbon metabolism by overexpression of SHMT2 by >10-fold to adapt to the hypoxic conditions inside tumors (Amelio et al., 2014, Kim et al., 2015, Wilhelm et al., 2014) (Figure S7). Future work is needed to assess how SHMT2 expression affects K63-Ub homeostasis across different tissues in health and disease.
More than 20,000 ubiquitination sites in mammalian cells are deubiquitinated by a pool of ∼100 DUBs (Leznicki and Kulathu, 2017, Mevissen and Komander, 2017). We find the BRCC36-containing BRCA1-A and BRISC complexes to be modular DUBs, with ABRAXAS and ABRO1 specifically integrating accessory factors RAP80, BRCA1, or SHMT2, respectively, and conferring targeting and regulation (Figure 7). Specific binding of these accessory factors is driven through interactions by the subcomplex-specific ABRO1/ABRAXAS subunits, combined with contacts by subunits shared between BRISC and BRCA1-A. Interestingly, it is the ABRO1 and ABRAXAS subunits that drive SHMT2 and RAP80/BRCA1 integration, respectively, but their integration involves additional contributions from the common core subunits. The BRISC/BRCA1-A structures and their functional dissection presented here reveal modularity in the MPN DUB family reminiscent to what has been described for ubiquitin ligases of the cullin-RING family (Petroski and Deshaies, 2005). We expect similar targeting and regulatory principles to apply to most multimeric DUB assemblies.
Figure 7.

Assembly and Regulation of BRCA1-A and BRISC
Schematic summary of BRCA1-A and BRISC function.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-BRCA1 | Santa Cruz Biotechnology | Cat# sc-6954; RRID: AB_626761 |
| Rabbit anti-MDC1 | Abcam | Cat# ab11171; RRID: AB_297810 |
| Rabbit anti-RAP80 | Bethyl Laboratories | Cat# A300-764; RRID: AB_2779443 |
| Mouse anti-GFP | Roche | Cat# 11814460001; RRID: AB_390913 |
| Mouse anti-tubulin | Sigma-Aldrich | Cat# T6199; RRID: AB_477583 |
| Rabbit anti-SHMT2 | Sigma-Aldrich | Cat# SAB1100388; RRID: AB_10610173 |
| Rabbit anti-ABRO1 | Abcam | Cat# ab74333; RRID: AB_1280902 |
| Alexa Fluor 488-conjugated donkey anti-rabbit antibody | ThermoFisher | Cat# R37118; RRID: AB_2536183 |
| Bacterial and Virus Strains | ||
| Escherichia coli BL21-CodonPlus (DE3)-RIL | Agilent Technologies | Cat# 230245 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| SigmaFast protease inhibitor cocktail | Sigma-Aldrich | Cat# S8820 |
| Strep-Tactin resin | iba | Cat# 2-1201-025 |
| TEV protease | This paper | N/A |
| Human and mouse BRCA1-A complexes | This paper | N/A |
| Human and mouse BRISC complexes | This paper | N/A |
| K63-linked di-ubiquitin (K63-diUb) | Emma Branigan, Ronald T. Hay | N/A |
| SUMO-2-K63-(Ub)2 | Emma Branigan, Ronald T. Hay | N/A |
| K63-(Ub)2 | Boston Biochem | Cat# UC-300B |
| K63-(Ub)3 | Boston Biochem | Cat# UC-315B |
| K63-(Ub)4 | Boston Biochem | Cat# UC-310B |
| peptide p-Ser404 (GGGFGEYpSRSPTF) | Biomatik | Custom synthesis |
| Peptide p-Ser406 (GGGFGEYSRpSPTF | Biomatik | Custom synthesis |
| peptides p-Ser404/406 (GGGFGEYpSRpSPTF) | Biomatik | Custom synthesis |
| BirA enzyme | This paper | N/A |
| MitoTracker Red CMXRos | Thermo Fisher Scientific | Cat# M7512 |
| Alexa Fluor 633-conjugated wheat germ agglutinin | Thermo Fisher Scientific | Cat# W21404 |
| K63-(Ub)4, TAMRA label on proximal ubiquitin | LifeSensors | Cat# SI6304T |
| K63-(Ub)4, TAMRA label on distal ubiquitin | Boston Biochem | Custom synthesis |
| Critical Commercial Assays | ||
| IQF DiUb K63 assay | Life Sensors | Cat# DU0102, DU6301 |
| Deposited Data | ||
| Original image files | This paper; Mendeley Data | https://dx.doi.org/10.17632/8ntswhp8d5.1 |
| Structure of BRCA1-A complex | This paper | PDB: 6GVW |
| Map of BRISC-SHMT2 complex | This paper | EMDB: EMDB-0132 |
| Structure of BRISC-SHMT2 complex | This paper | PDB: 6H3C |
| PLP-bound SHMT2 structure | Giardina et al., 2015 | PDB: 5V7I |
| Apo-SHMT2 | Unpublished data | PDB: 6DK3 |
| COP9 signalosome | Lingaraju et al., 2014 | PDB: 4D10 |
| APC2-UBCH10 complex | Brown et al., 2015 | PDB: 4YII |
| Crystal Structure of the SpRpn10 VWA domain | Riedinger et al., 2010 | PDB: 2X5N |
| Structure of CfBRCC36-CfKIAA0157 complex | Zeqiraj et al., 2015 | PDB: 5CW3 |
| Experimental Models: Cell Lines | ||
| Spodoptera frugiperda Sf9 cells | Thermo Fisher Scientific | Cat# 11496015 |
| Tni insect cells | Expression Systems | Cat# 94-002F |
| Human osteosarcoma (U2OS) cells | Dr. Durocher | N/A |
| U2OS Rap80-KO | Dr. Durocher | N/A |
| HEK293T | Dr. Peters | N/A |
| Recombinant DNA | ||
| pFastBac MmABRAXAS | This paper | N/A |
| pFastBac MmABRO1 | This paper | N/A |
| pFastBac MmBRCC36 | This paper | N/A |
| pFastBac MmBRE | This paper | N/A |
| pFastBac MmMERIT40 | This paper | N/A |
| pFastBac MmRAP80 | This paper | N/A |
| pFastBac MmRAP80 250-413 | This paper | N/A |
| pFastBac HsABRO1 | This paper | N/A |
| pFastBac HsBRCC36 | This paper | N/A |
| pFastBac HsBRE | This paper | N/A |
| pFastBac HsMERIT40 | This paper | N/A |
| pFastBac SHMT2 | This paper | N/A |
| pFastBac Strep-BirA-HsABRO1 | This paper | N/A |
| SHMT2α | SGC structural genomics consortium | 3OU5 |
| GFP-RAP80(WT) | Dr. A.M. Jetten | N/A |
| GFP-RAP80(ΔAIR) (Δ274-334) | This paper | N/A |
| pFasBac- mouse ABRO1 (1-260) - ABRAXAS (269-407) | This paper | N/A |
| Human BRCA1-BRCT | Wu et al., 2016 | N/A |
| pFastBac- ABRAXAS-sortase site -FLAG tag at the C terminus (ABRAXAS 1-394 - LPETGDYKDHDGDYKDHDIDYKDDDDK) | This paper | N/A |
| Software and Algorithms | ||
| Fiji | Schindelin et al., 2012 | https://fiji.sc |
| DYNAFIT | Kuzmič, 2009 | http://www.biokin.com/dynafit/ |
| Prism | https://www.graphpad.com | |
| XDS | Kabsch, 2010 | http://xds.mpimf-heidelberg.mpg.de |
| DIALS | Winter et al., 2018 | https://dials.diamond.ac.uk |
| POINTLESS | Evans, 2011 | http://www.ccp4.ac.uk |
| AIMLESS | Evans and Murshudov, 2013 | http://www.ccp4.ac.uk |
| STARANISO | Global Phasing | http://staraniso.globalphasing.org |
| SHELXD | Sheldrick, 2008 | http://shelx.uni-goettingen.de |
| PHASER | McCoy et al., 2007 | https://www.phaser.cimr.cam.ac.uk/ |
| PIRATE | Cowtan, 2000 | http://www.ccp4.ac.uk |
| PARROT | Cowtan, 2010 | http://www.ccp4.ac.uk |
| PHENIX | Adams et al., 2010 | https://www.phenix-online.org |
| COOT | Emsley et al., 2010 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| ISOLDE | Croll, 2018 | https://isolde.cimr.cam.ac.uk |
| REFMAC | Nicholls et al., 2012 | http://www.ccp4.ac.uk |
| AUTOBUSTER | Global Phasing | https://www.globalphasing.com |
| CNS | Schröder et al., 2010 | http://cns-online.org/v1.3/ |
| ROSETTA | Wang et al., 2016 | https://www.rosettacommons.org |
| PDB-REDO | Joosten et al., 2014 | https://pdb-redo.eu |
| MOLPROBITY | Chen et al., 2010 | http://molprobity.biochem.duke.edu |
| CryoFLARE | A.D.S., unpublished data | http://www.cryoflare.org |
| motioncor2 | Li et al., 2013 | https://msg.ucsf.edu/software |
| GCTF | Zhang, 2016 | https://www.mrc-lmb.cam.ac.uk/kzhang/ |
| Gautomatch | https://www.mrc-lmb.cam.ac.uk/kzhang/ | |
| RELION | Scheres, 2012 | https://www3.mrc-lmb.cam.ac.uk/relion |
| EMAN2 | Tang et al., 2007 | https://blake.bcm.edu/emanwiki/ |
| ResMap | Kucukelbir et al., 2014 | http://resmap.sourceforge.net |
Contact for Reagent and Resource Sharing
Requests for further information, resources, and reagents should be directed to and will be fulfilled by the lead contact Nicolas H. Thomä (nicolas.thoma@fmi.ch).
Experimental Model and Subject Details
Cell culture of human osteosarcoma (U2OS) cells
U2OS cells (female, a gift from Dr. Durocher) were cultured at 37°C in an atmosphere of 5% CO2 in DMEM supplemented with antibiotics, 10% fetal calf serum and glutaMAX (GIBCO). They were not otherwise authenticated.
Method Details
Protein expression and purification of BRCA1-A and BRISC
Expression constructs for the BRCA1-A and BRISC complex were synthesized (Genescript and Geneart, for Mus musculus and Homo sapiens, respectively) and cloned into pFastBac (ThermoFisher) vectors for insect cell expression using standard molecular biology techniques. Native codon usage was preserved with the exception of the removal of NotI and KpnI restriction sites. Truncated constructs were cloned from full-length constructs using standard molecular biology techniques. All constructs were verified by sequencing (Microsynth). Baculoviruses were generated in Spodoptera frugiperda Sf9 cells (ThermoFisher) using the Bac-to-Bac system (ThermoFisher). For recombinant protein expression of the BRCA1-A and BRISC complex or subcomplexes, Trichoplusia ni High Five cells (ExpressionSystems) were coinfected with baculoviruses encoding the desired proteins. BRCA1-A complex was expressed by co-infection of High Five cells with viruses encoding (full-length, engineered or truncated) ABRAXAS, BRCC36, BRE, MERIT40, and RAP80. BRISC complex was expressed by co-infection of High Five cells with viruses encoding (full-length, engineered or truncated) ABRO1, BRCC36, BRE, and MERIT40. The BRE-MERIT40 complex was expressed by co-infection of High Five cells with viruses encoding BRE and MERIT40. Cells were harvested by centrifugation 36 h after infection, resuspended in lysis buffer (50 mM HEPES pH 7.4, 200 mM NaCl, 0.1% (v/v) Triton X-100, 1 mM PMSF, 0.2 mM TCEP) supplemented with 1 × SigmaFast protease inhibitor cocktail (Sigma-Aldrich) and disrupted by sonication. Cell debris was removed by ultracentrifugation (45 min at 40,000g) and the supernatant was filtrated through Miracloth (EMD Millipore) and subsequently applied to Strep-Tactin resin (IBA Lifesciences). The affinity resin was washed (wash buffer: 50 mM HEPES pH 7.4, 200 mM NaCl, 0.2 mM TCEP) and bound protein was subsequently eluted (elution buffer: 50 mM HEPES pH 7.4, 200 mM NaCl, 0.2 mM TCEP, 2.5 mM D-desthiobiotin). The eluted complex was subjected to anion exchange purification on a Poros 50 HQ (ThermoFisher) column using a linear gradient (50 mM Tris-HCl pH 8.0, 0.2-1 M NaCl, 0.2 mM TCEP). Fractions containing protein were concentrated by ultrafiltration (Amicon Ultra-15, 30 kDa molecular weight cutoff). For crystallization, concentrated BRCA1-A was incubated at room temperature (RT) with 1% (w/w) tobacco etch virus (TEV) protease for 1 h to remove affinity tags. Samples were finally subjected to gel filtration on a Superose 6 column (ThermoFisher), where buffer was exchanged to crystallization buffer (20 mM HEPES pH 7.4, 150 mM NaCl, 0.2 mM TCEP) or storage buffer (50 mM HEPES pH 7.4, 200 mM NaCl, 0.2 mM TCEP).
BRCA1-A crystallization
BRCA1-A (10 mg/ml in crystallization buffer) was crystallized in Cryschem plates (Hampton Research) by vapor diffusion equilibration against 500 μl well solution. Crystallization trials were set up by mixing 1 μl BRCA1-A solution with 1 μl well solution (100 mM MES-KOH pH 5.6, 200 mM MgCl2, 8% (w/v) PEG6000) at room temperature. The plate was subsequently incubated at 4°C and rhombohedral plate-like crystals grew. They obtained their largest dimension after three days. Crystals were cryoprotected by gradual supplementation of the crystal growth drop with ethylene glycol to a final concentration of 25% (v/v), and flash-cooled in liquid nitrogen for X-ray diffraction analysis.
BRCA1-A crystal structure determination
BRCA1-A formed crystals belonging to the orthorhombic space group P212121 with unit cell parameters a = 97.1 Å, b = 112.6 Å, c = 431.3 Å, and contained a single copy of the complex, with a molecular weight of 333 kDa in the asymmetric unit and solvent content of ∼65%. These crystals typically diffracted X-rays anisotropically to 4.2 Å resolution. All diffraction data were collected at the Swiss Light Source (SLS) from crystals cooled to 100 K as described. In a native zinc single wavelength anomalous dispersion (SAD) phasing approach, high-multiplicity diffraction data were collected from a fixed position on a single crystal with a low-dose X-ray beam at the zinc K-edge absorption peak wavelength (1.2816 Å; fʹ = −9.23, f″ = 4.97) from SLS beamline X10SA with a Pilatus 6M detector (Dectris). Data were processed with XDS (Kabsch, 2010). Two sites corresponding to the BRCC36 active site Zn2+ ions were found using the anomalous intensities in the resolution range 118-7.5 Å with SHELXD (Sheldrick, 2008). SAD phases were calculated from the two sites after refinement in PHASER (McCoy et al., 2007) to 7.3 Å resolution, giving an overall figure-of-merit of 0.29. Density modification was carried out initially with PIRATE (Cowtan, 2000) from the CCP4 suite (Winn et al., 2011) at 7.3 Å resolution and thereafter with PARROT (Cowtan, 2010) or phenix.resolve (Adams et al., 2010). SAD phasing was reinitiated in PHASER using the density modified map as the partial structure (MR-SAD) and followed by a round of density modification to generate an improved electron density map. By iterating this procedure, the phases were progressively extended to 6 Å resolution, yielding substantially improved electron density. The atomic model was built interactively in COOT (Emsley et al., 2010). At first domain fragments were placed in the electron density from known structures for the ABRAXAS-BRCC36 MPN domain dimer (PDB: 4d10; Lingaraju et al., 2014), the BRE UEV-C domain (PDB: 4YII; Brown et al., 2015), and MERIT40 (PDB: 2X5N; Riedinger et al., 2010), and ideal ⍺-helices in other regions clearly defined by tubes of electron density. Dramatic improvement to the interpretability of the electron density was achieved through many cycles of poly-serine model building followed by consecutive refinement in CNS (Schröder et al., 2010) with non-crystallographic symmetry (NCS) and deformable elastic network restraints (ɣ = 0 and wDEN = 100), and REFMAC (Nicholls et al., 2012) with jelly-body restraints (σ = 0.01 for 100–500 cycles), followed by density modification with NCS averaging. Refinement was carried out using SAD phase restraints against the Zn-SAD data in resolution range 118-4.3 Å, or data to comparable resolution obtained from different crystals. Once the quality of the electron density indicated that completing the atomic model would be feasible, focus was shifted to assigning its amino acid sequence, and increasing consideration was given to the validation characteristics of the refined coordinates. Diffraction data for model completion and refinement were assembled by multi-crystal merging across seven crystals, improving overall data quality and the measurability of reflections in a cone around the a∗ (a) axis, which was the weakest direction of anisotropy. Diffraction data were obtained SLS beamline X06SA with an Eiger X 16M detector (Dectris) at a wavelength of 1 Å and processed using DIALS (Winter et al., 2018), with diffraction geometry parameters adjusted by joint refinement across multiple sweeps obtained from several crystals. Intensity data were combined with POINTLESS (Evans, 2011) and scaled and merged with AIMLESS (Evans and Murshudov, 2013). Structure factor amplitudes treated for anisotropy were calculated from the final intensities by STARANISO (Global Phasing Ltd.), applying a high-resolution cutoff of 1 I/σI. Amino acid sequence assignment was guided by the positions of methionine and cysteine residues determined by MR-SAD for the native sulfur atoms in combination with the mercury atoms found for a crystal soaked in p-chloromercuribenzoic acid. The model was rebuilt and extended with COOT in cycle with refinement against data extending to 3.75 Å resolution variously with autoBUSTER (Global Phasing Ltd.), phenix.refine, ROSETTA (Wang et al., 2016), and REFMAC. Development of the refinement strategy was guided by analysis with PDB-REDO (Joosten et al., 2014). Final refinement was performed with ISOLDE followed by REFMAC (Croll, 2018, Nicholls et al., 2012). Analysis of the final model with MOLPROBITY (Chen et al., 2010) indicates that 92.5% of the residues are in favorable regions of the Ramachandran plot, with 1.2% outliers. Detailed crystallographic data processing and refinement statistics are in Table 1.
Protein expression and purification of SHMT2α
The expression construct for SHMT2 encoding the cytosolic form SHMT2α was obtained from the SGC structural genomics consortium and contains a mutation A264T, which is distal from the BRISC interaction surface. In comparison with the PLP-bound SHMT2 structure (PDB: 4PVF) there is no indication that the mutation affects the overall structure (PDB: 6DK3). SHMT2α was expressed at 15°C overnight in Escherichia coli BL21 (DE3) pRIL (Agilent Technologies) in auto-induction medium (Studier, 2005). Cells were resuspended in lysis buffer (50 mM HEPES pH 7.4, 200 mM NaCl, 0.1% (v/v) Triton X-100, 1 mM PMSF, 1 × SigmaFast protease inhibitor (Sigma-Aldrich), 0.2 mM TCEP) and lysed by sonication. Cell debris was subsequently removed by ultracentrifugation. The supernatant was subjected to Ni-NTA (Sigma-Aldrich) affinity chromatography. The protein was eluated using steps of 5, 20, 50, 100, 500 mM imidazole (buffer: 100 mM HEPES pH 7.4, 200 mM NaCl, 0.2 mM TCEP) and pooled protein fractions were concentrated by ultrafiltration (Amicon Ultra-15, 30 kDa molecular weight cutoff, Merck) and separated by gel filtration on a Superdex 200 column (ThermoFisher). SHMT2α was purified in its apo-form lacking pyridoxal 5ʹ-phosphate (PLP). To assemble SHMT2 tetramers with bound cofactor, PLP (Sigma-Aldrich) was added to SHMT2α prior to gel filtration. The protein fractions obtained showed the characteristic yellow color indicative of PLP binding.
Cryo-electron microscopy
For structure determination of BRISC-SHMT2 complex by cryo-electron microscopy equal volumes of human BRISC (12 mg/ml) and SHMT2 (9.3 mg/ml) were mixed and incubated at room temperature for 15 min. The complex was purified by gel filtration (Superose 6, GE Healthcare) and the peak fraction (0.44 mg/ml) was collected. Tween-20 was added to a final concentration of 0.001% (v/v). Gold foil grids UltrAuFoil 1.2/1.3 300 mesh (Quantifoil) were glow discharged in a Solarus plasma cleaner (Gatan) for 12 s in a H2/O2 environment prior to vitrification. A 4 μl sample was applied to the grid and a protocol consisting of 30 s pre-blot incubation, 2 s blotting and no post-blot incubation was utilized for vitrification using a Leica EM GP plunge freezer (Leica Microsystems) operated at 4°C and 80% humidity. Data were collected automatically with EPU (ThermoFisher) on a Cs corrected (CEOS) FEI Titan Krios (ThermoFisher) electron microscope at 300 kV. Zero-energy loss micrographs were recorded using a Gatan K2 summit direct electron detector (Gatan) in counting mode located after a Quantum-LS energy filter operated with a slit width of 20 eV. The acquisition was performed at a calibrated magnification of 58140x in EFTEM mode yielding a pixel size of 0.86 Å and a dose rate of 4.7 e−/(px·s). Exposures of 7 s were fractionated into 40 frames leading to a total dose of 45 e−/Å2. The defocus values of the dataset ranged from −0.5 to −5 μm.
Image processing
Parallel to acquisition with EPU the micrograph stacks were corrected for drift, the CTF was determined, and particles were auto picked using CryoFLARE (in house development; www.cryoflare.org) for automation of the process. The drift correction was performed with the program motioncor2 (Li et al., 2013). A sum of all 40 frames was generated with and without applying a dose weighting scheme. The CTF was fitted using GCTF (Zhang, 2016) on the non-dose-weighted sums and the particles were picked using Gautomatch (Dr. Kai Zhang, Cambridge) on the dose-weighted sums. A total of 1822 micrograph movies were acquired from which 332,598 particles were selected. The corrected and picked dataset was subsequently subjected to extraction with a box size of 350 px from the dose-weighted sums and resulting particles were 2D classified. Six rounds of 2D classification in RELION (Scheres, 2012) and selection of good classes were performed to remove ice contamination, junk particles, and particles touching each other due to micro-aggregation. The 2D classfication and selection process yielded a refined dataset of 44,381 particles. An initial 3D model was generated in RELION without imposing symmetry. The initial model was rotated in chimera to align the twofold symmetry axis of the complex with Z and afterward symmetrized in C2 using EMAN2 (Tang et al., 2007). The symmetrized map was used as a starting model for a 3D classification into 3 classes in RELION. The largest class containing 35,595 particles was selected for 3D refinement. The refined dataset was used to re-extract particles from dose-weighted sums where the first 3 and the last 20 frames were excluded. The re-extracted dataset was subjected to another round of 3D refinement yielding a map at 3.9 Å resolution. The refinements followed the gold-standard procedure and the resolution estimates are based on the Fourier shell correlation curve (FSC) at the 0.143 criterion (Rosenthal and Henderson, 2003) after post-processing. The final map was corrected for the modulation transfer function (MTF) of the K2 detector and sharpened by applying a negative B factor estimated automatically within RELION. Variations in the local resolution were estimated from the independent half-maps of the refinement using the program ResMap (Kucukelbir et al., 2014). Our BRCA1-A atomic model and SHMT2 (PDB: 5V7I, Ducker et al., 2017) were docked into the BRISC-SHMT2α cryo-EM map with COOT. Using tools from ROSETTA suite and PROMALS3D (Pei et al., 2008), the human BRISC amino acid sequence was assigned by threading, the protomer conformations were optimized, and refinement with iterative local rebuilding was carried out. The final model was obtained after minimal cycles of rebuilding with COOT and refinement with phenix.real_space_refine. Detailed cryo-EM data collection, refinement, and validation statistics are in Table 2.
Chain length specific degradation assay
To assess chain length dependence of human BRISC and BRCA1-A (truncated RAP80) DUB activity, 4 μg K63-linked (Ub)2, (Ub)3, or (Ub)4 (Boston Biochem) were incubated with 5 nM DUB complex in 210 μl volume (gel filtration buffer). Samples (10 μl) were taken at regular intervals, reactions were stopped by addition of SDS-PAGE loading buffer and subsequently samples were analyzed qualitatively by SDS-PAGE (AnyKD, Bio-Rad) and silver stain (Bio-Rad). Initial velocity of cleavage of (Ub)2 at variable BRCA1-A concentrations (0-500 nM) was measured using 200 nM internally quenched K63-linked (Ub)2 as substrate in assay buffer (50 mM HEPES pH 7.0, 100 mM NaCl, 0.5% (w/v) BSA, 0.03% (w/v) Brij-35, 0.2 mM TCEP). Relative TAMRA fluorescence was monitored in 10 s time increments (excitation at 540 nm, emission at 590 nm) and assays were carried out at 22°C in black flat-bottom high-base 386-well plates (Greiner Bio-One) using a Pherastar plate reader (BMG Labtech). Initial velocity in relative fluorescence units per second was determined by linear regression.
Nuclear magnetic resonance experiments
NMR experiments were conducted at 25°C on a Varian INOVA 600-MHz spectrometer equipped with 1H, 13C, 15N triple-resonance, z axis pulsed-field-gradient probe. All samples were prepared in a buffer 50 mM HEPES pH 7.4, 200 mM NaCl, 0.2 mM TCEP, and 5% D2O. All NMR data were processed with NMRPipe and NMRDraw (Delaglio et al., 1995). NMR data were obtained from standard 1D single-resonance experiments acquired on 100 μM uniformly 15N–labeled Ubiquitin purchased from BostonBiochem. 15N-HSQC titration of 50 μM 15N-Ubiquitin proteins was done by stepwise addition of MERIT40 truncated, MERIT40 full-length mixed 1:1 with BRE, or Merit40 truncated mixed 1:1 with BRE from high concentration stocks. 15N-HSQC titration data were analyzed with SPARKY (https://www.cgl.ucsf.edu/home/sparky/).
(Ub)4 pre-orientation assay
Human BRCA1-A (containing truncated RAP80) at a concentration of 10 nM was mixed with 1 μM (Ub)4 with a TAMRA label on the proximal (Life Sensors) and distal (Boston Biochem) Ubiquitin, respectively, in assay buffer (50 mM HEPES pH 7.0, 100 mM NaCl, 0.5% (w/v) BSA, 0.03% (w/v) Brij-35, 0.2 mM TCEP). Samples (10 μl) were taken at regular intervals and reactions were stopped by addition of SDS-PAGE loading buffer. Gels were analyzed by SDS-PAGE (AnyKD, Bio-Rad) and imaged with a Typhoon FLA 9500 imager (GE Healtchare Life Sciences).
Cell biology and fluorescence microscopy of DNA repair foci
U2OS parental and RAP80 knockout cells were used. The knockout of RAP80 was confirmed by western blot analysis using RAP80-specific antibody. The GFP-RAP80(WT) plasmid was a gift of Dr. A.M. Jetten (Yan et al., 2002). This plasmid was used to remove the AIR domain (residues 274-334) to generate GFP-RAP80(ΔAIR). Both parental and RAP80 knockout cells were seeded on 18 mm coverslips and transiently transfected with 25 ng GFP-NLS, 500 ng GFP-RAP80(WT) or 500 ng GFP-RAP80(ΔAIR) using lipofectamine 2000 according to the manufacturer’s instructions. At 24 h after transfection, cells were irradiated with 2 Gy using a YXlon X-ray generator (YXlon International, 200 KV, 10 mA, dose rate 2 Gy/min) and after 1 h cells were fixed with 4% formaldehyde in PBS for 15 min at room temperature. Cells were treated with 100 mM glycine in PBS for 10 min to block unreacted aldehyde groups. Cells were rinsed with phosphate-buffered saline and equilibrated in WB (PBS containing 0.5% BSA, and 0.05% Tween 20; Sigma-Aldrich). Antibody steps and washes were in WB. The primary antibodies (mouse BRCA1 Santa Cruz sc-6954; 1/100, and rabbit MDC1 Abcam ab11171; 1/1000) were incubated overnight at 4°C. Detection was done using goat anti-mouse or goat anti-rabbit Ig coupled to Alexa 555 or 647 (1/1000; Invitrogen Molecular probes). Samples were incubated with 0.1 μg/ml DAPI and mounted in Polymount. Images of fixed samples were acquired on a Zeiss AxioImager M2 widefield fluorescence microscope equipped with 63x PLAN APO (1.4 NA) oil-immersion objectives (Zeiss) and an HXP 120 metal-halide lamp used for excitation. Fluorescent probes were detected using the following filters: DAPI (excitation filter: 350/50 nm, dichroic mirror: 400 nm, emission filter: 460/50 nm), GFP (excitation filter: 470/40 nm, dichroic mirror: 495 nm, emission filter: 525/50 nm), Alexa 555 (excitation filter: 545/25 nm, dichroic mirror: 565 nm, emission filter: 605/70 nm), and Alexa 647 (excitation filter: 640/30 nm, dichroic mirror: 660 nm, emission filter: 690/50 nm). Images were recorded using ZEN 2012 software. The number, intensity, and size of BRCA1 foci was analyzed for all conditions in ImageJ using a custom-built macro that enabled automatic and objective analysis of the foci as described previously (Typas et al., 2015). Cell extracts were generated by boiling cell pellets in Laemmli buffer, separated by SDS-PAGE and transferred to Immobilon-FL PVDF membranes (Millipore). Membranes were probed with the following antibodies: rabbit anti-RAP80 (Bethyl A300-764, 1/1000), mouse anti-GFP (Roche 11814460001, 1/2000), and mouse anti-tubulin (Sigma T6199, 1/5000). Protein detection was done using the Odyssey infrared imaging scanning system (LI-COR Biosciences). Secondary antibodies were purchased from Biotium.
Co-expression in insect cells and pull-down
High Five cells were grown to 5⋅106 cells/ml and 5 ml were mixed with 5 ml of fresh medium supplemented with penicillin and streptomycin and infected by addition of 200 μl of P2 virus. Cells were harvested after 40 h, pelleted. and lysed by sonication in lysis buffer (50 mM HEPES pH 7.4, 200 mM NaCl, 0.1% Triton X-100, 0.2 mM TCEP). Lysate was cleared by centrifugation (13200 rpm at 30 min, 4°C). 25 μl Ni-NTA (Sigma-Aldrich) or 15 μl Strep-Macro-Prep (IBA AG) were added and sample was incubated on a shaking platform at 4°C for 30 min. After two washes with lysis buffer, the resin was resuspended in SDS-PAGE sample buffer and analyzed by SDS-PAGE.
Analysis of RAP80 incorporation and double arc formation
A fusion gene of mouse ABRO1 (residues 1-260) and ABRAXAS (residues 269-407) was synthesized by IDT and cloned into pFastBac baculovirus expression vector. Fusion complex was prepared as described for BRCA1-A complex, using fusion virus instead of ABRAXAS. Oligomerization was analyzed by SDS-PAGE (AnyKD gels, BioRad) and native PAGE electrophoresis (7.5% (w/v) acrylamide gels, BioRad) using 10 μg of complex for each lane. Gels were stained with Coomassie brilliant blue.
Negative stain electron microscopy
BRISC was prepared for electron microscopy using the GraFix method (Kastner et al., 2008). BRISC complex (10 mg/ml) was layered on top of a 10%–30% (w/v) glycerol gradient (50 mM HEPES pH 7.4, 150 mM NaCl, 0.2 mM TCEP) with an increasing concentration (0%–0.2% w/v) of glutaraldehyde (EMS) and subjected to ultracentrifugation (Beckman SW40Ti rotor, 32000 rpm, 19 h, 4°C). The fraction containing BRISC was directly applied to glow discharged Quantifoil grids (S7/2, Cu 400 mesh, Quantifoil Micro Tools GmbH), blotted, and stained with 2% (w/v) uranyl acetate. Data were collected using a Tecnai T12 electron microscope (FEI) at 100 kV, with a pixel size of 3.08 Å/pixel at the specimen level. Images were recorded with a TVIPS TemCam F416 with varying defocus (−0.5 to −1.5 μm) and processed with the EMAN2 image processing suite (Tang et al., 2007).
Di-ubiquitin preparation
K63-linked di-Ubiquitin (K63-(Ub)2) contains WT Ubiquitin linked by K63 to an N-terminally his-tagged Ubiquitin bearing a K63R mutation. To prepare K63-diUb, 300 μM his-tagged Ubiquitin K63R and 100 μM wild-type (WT) Ubiquitin were incubated with 0.1 μM E1, 2.5 μM Ubc13, 2.5 μM Ube2V2 and 0.55 μM RNF4∼RING at 20°C overnight in a buffer containing 3 mM ATP, 5 mM MgCl2, 50 mM Tris pH 7.5, 150 mM NaCl, 0.5 mM TCEP and 0.1% NP40. RNF4∼RING contains one full-length RNF4 linearly fused to a second RING domain of RNF4 (Plechanovová et al., 2011). The reaction mixture was purified by Ni-NTA chromatography. The Ni-NTA elution contained His-tagged (Ub)2 and His-tagged ubiquitin K63R, which were separated on a HiLoad Superdex 75 16/600 gel filtration column (ThermoFisher) in a buffer containing 50 mM Tris pH 7.5, 150 mM NaCl and 0.5 mM TCEP.
Preparation of SUMO/Ubiquitin mixed chains
SUMO-2-K63-(Ub)2 was prepared using SUMO-2-Ub as a substrate. SUMO-2-Ub contained WT ubiquitin linearly fused to a C-terminally His-tagged SUMO-2 (Bett et al., 2015). A second Ubiquitin bearing a K63R mutation was enzymatically linked to this substrate via K63 to generate SUMO-2-K63-(Ub)2. In the reaction, 300 μM ubiquitin K63R and 100 μM his-tagged SUMO-2-Ub were incubated with 0.1 μM E1, 2.5 μM Ubc13, 2.5 μM and 0.55 μM RNF4∼RING at 20°C overnight in a buffer containing 3 mM ATP, 5 mM MgCl2, 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5 mM TCEP and 0.1% (v/v) NP40. The reaction mixture was purified by Ni-NTA chromatography. The Ni-NTA elution contained his-tagged SUMO-2-K63-(Ub)2 and his-tagged SUMO-2-Ub, which were separated on a HiLoad Superdex 75 16/600 gel filtration column (ThermoFisher) in a buffer containing 50 mM Tris-HCl pH 7.5, 150 mM NaCl and 0.5 mM TCEP. Long K63-linked ubiquitin chains linked to tetra-SUMO were produced with the same method, using tetra-SUMO and WT Ubiquitin. Reaction products were purified by size exclusion chromatography with a Superdex 200 16/200 gel filtration column (ThermoFisher) and fractions containing chains larger than 100 kDa were pooled.
SUMO/Ubiquitin mixed chains degradation assay
Human BRCA1-A including full-length RAP80 (residues 1-719), or truncated RAP80 (residues 250-413) was prepared recombinantly from insect cells. DUB reactions were started by mixing 1:1 BRCA1-A with the substrate (100 nM BRCA1-A was mixed with 2 μM K63-diUb or SUMO-K63-diUb, and 20 nM BRCA1-A was mixed with 400 ng of Ubn∼ub∼4xSUMO-2). Samples were taken at the time points indicated and immediately mixed with SDS-PAGE sample buffer (62.5 mM Tris-HCl pH 6.8, 2% (w/v) SDS, 25% (v/v) glycerol, 0.05% (v/v) bromophenol blue, 5% (v/v) β-mercaptoethanol) in order to stop the DUB reaction. Samples were analyzed by SDS-PAGE (Any kD Mini-PROTEAN TGX Precast Protein Gel, Bio-Rad Laboratories) and subsequently visualized by staining with SYPRO Ruby (Sigma-Aldrich). Stained gels were imaged with a Typhoon FLA 9500 imager (GE Healthcare Life Sciences). Band densitometry was performed with Fiji (Schneider et al., 2012).
Protein expression and purification of BRCA1-BRCT
Qian Wu and Tom Blundell kindly provided the expression construct for the human BRCA1-BRCT domain (Wu et al., 2016). BRCA1-BRCT was expressed recombinantly in E. coli BL21-CodonPlus (DE3)-RIL cells (Agilent Technologies). Cell pellets were resuspended in lysis buffer containing 50 mM Tris-HCl pH 8.0, 200 mM NaCl, 0.1% Triton X-100, 1 mM PMSF, 0.2 mM TCEP and Roche EDTA-free protease inhibitor (1 tablet per 500 ml) and lysed by sonication. After removal of cell debris by ultracentrifugation (45 min at 40,000g), the supernatant was filtrated through Miracloth (EMD Millipore) and subsequently applied to Ni-NTA resin equilibrated with lysis buffer in a gravity flow column. The Ni-NTA resin was washed with wash buffer (50 mM Tris-HCl pH 8.0, 200 mM NaCl, 0.2 mM TCEP) and bound protein was eluted by step gradient using wash buffer supplemented with 10, 50, 200 and 500 mM imidazole, respectively. The eluate was concentrated by ultrafiltration (Amicon Ultra-15, 10 kDa molecular weight cutoff). Gel filtration with a Superdex 75 was used to purify the protein and exchange the buffer to 50 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.2 mM TCEP.
Sortase ligation of phosphorylated peptides to BRCA1-A
Phosphorylated versions of peptides from the C-terminal region of the BRCA1-A subunit ABRAXAS were covalently ligated to BRCA1-A complex using the sortase ligation technique (Guimaraes et al., 2013). A modified human ABRAXAS expression construct was designed adding a sortase recognition site followed by a FLAG tag at the C terminus (replacing the residues C-terminal of residue I394 with the sequence LPETGDYKDHDGDYKDHDIDYKDDDDK). The FLAG tag allowed removal of unlabeled protein by affinity chromatography and detection of successful labeling by SDS-PAGE (3.1 kDa decrease in protein mass). The modified human ABRAXAS expression construct was synthesized (Integrated DNA Technologies) and cloned into a pFastBac expression vector. BRCA1-A containing sortase-tagged ABRAXAS was expressed in insect cells and purified. Synthetic phosphorylated peptides p-Ser404 (GGGFGEYpSRSPTF), p-Ser406 (GGGFGEYSRpSPTF) and p-Ser404/406 (GGGFGEYpSRpSPTF) (Biomatik) were dissolved in peptide buffer (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 5 mM DTT) at 10 mg/ml and adjusted to pH 8.0. For sortase labeling, a final concentration of 8.5 μM BRCA1-A in 50 mM HEPES pH 7.4, 200 mM NaCl, 0.2 mM TCEP), 870 μM peptide in peptide buffer, and 20 μM sortase in 50 mM Tris-HCl, 200 mM NaCl, 10% (v/v) glycerol) were mixed to a final volume of 500 μl. Reactions were incubated at 22°C and monitored for FLAG tag removal by SDS-PAGE analysis; reactions completed within 6 h. Sortase and excess peptide were removed by gel filtration (Superose 6, ThermoFisher) in gel filtration buffer (50 mM HEPES pH 7.4, 150 mM NaCl, 0.2 mM TCEP). Binding of BRCT to phosphorylated BRCA1-A was assessed by band shift in Coomassie Brilliant Blue-stained native PAGE (7.5% Mini-PROTEAN TGX precast protein gel, Bio-Rad Laboratories).
Microscale thermophoresis binding assays
BRCA1-BRCT was labeled with the amine reactive RED-NHS labeling kit (NanoTemper Technologies). The labeled protein was purified by gel filtration over a Superdex 75 column (ThermoFisher) and diluted to 40 nM with assay buffer (50 mM HEPES pH 7.0, 100 mM NaCl, 0.5% BSA, 0.03% Brij-35, 0.2 mM TCEP). To determine the affinity of BRCA1-A p-Ser404/406 for BRCA1-BRCT, BRCA1-A p-Ser404/406 at concentrations between 0 nM and 853 nM was mixed with 20 nM BRCA1-BRCT and non-labeled BRCT in concentrations of 0 nM, 100 nM, 200 nM, 500 nM, 1000 nM was added to replicates. After 10 min incubation at RT, samples were loaded into Monolith NT.115 Premium capillaries (NanoTemper Technologies) and microscale thermophoresis was measured using a Monolith NT.115 (NanoTemper Technologies). For the back-titration, 0-46.6 μM unlabeled BRCA1-BRCT was titrated into 200 nM BRCA1-A p-Ser404/406 and 20 nM labeled BRCA1-BRCT. Affinities were determined by global fit using DYNAFIT (Kuzmič, 2009).
Crosslinking mass spectrometry
The approximate position of BRCA1-BRCT in the BRCA1-A-BRCA1-BRCT complex was determined by crosslinking mass spectrometry of BRCA1-A p-Ser404/406 with BRCA1-BRCT.50 μg of BRCA1-A p-Ser404/406 was mixed with 20 μg BRCA1-BRCT in a total volume of 117 μl. A 1 mg aliquot of the CID-cleavable DSSO crosslinking reagent (disuccinimidyl sulfoxide, MW = 388.35, Spacer Arm = 10.3 Å) was brought to room temperature and dissolved in 25 μl of anhydrous DMSO (100 mM stock). First the sample buffer was transferred into a 100 kDa Amicon Ultra spin filter reactor (0.5 ml), followed by protein stock solution. Finally, the crosslinking reagent was added and incubated at room temperature for 1 h. Excess of unreacted reagent was quenched by addition of 2.5 μl of 1M Tris (50 mM final conc.). After 50 min, filters were centrifuged for 2 min at 14000 g to remove crosslinking reagent and non-crosslinked protein. Filters were topped off with 400 μl (max 450 μl) of 8 M urea in 50 mM HEPES pH 8.5 for denaturing and washing, and then centrifuged for 2 min at 14000 g. This step was repeated twice. At the end of each spin, the sample was concentrated to 40-50 μl. The sample was reduced and alkylated by addition of 5.5 μl of reduction/alkylation solution (stock: 50 mM TCEP, 100 mM 2-chloroacetamide, final concentration 5 mM TCEP and 10 mM 2-chloroacetamide) and subsequent incubation for 30 min in the dark. The samples were centrifuged for 2 min at 14000 g and topped-off with 50 mM HEPES pH 8.5, 8M urea, centrifuged 2 min at 14000 g. This procedure was repeated twice for washing. The sample was digested by addition of 0.5 μg Lys-C (5 μl of 0.1 μg/μl stock, 1:100 enzyme to protein ratio) and incubation for 4 h at room temperature. The Lys-C digest was diluted 4-fold by addition of 240 μl of 50 μl HEPES pH 8.5 followed by addition of 0.5 μg of trypsin (2.5 μl of 0.2 μg/μl stock, 1:100 enzyme to protein ratio) and digestion overnight. Next, the sample was incubated for four h after addition of an additional aliquot of 0.5 μg of trypsin (1:100) and 35 μl of acetonitrile to a final concentration of 10%. The filter containing the trypsin digest was inverted and spun 1 min at 1000 g to collect the digest into a fresh Eppendorf tube. The digest was acidified with TFA to 1% final concentration, then sonicated, and centrifuged for 5 min at 20000 g. LC-MS analysis was performed using a 2 cm trapping column and a 50 cm analytical column coupled to an Orbitrap Fusion LUMOS mass spectrometer using a 90 h gradient and the MS2_MS3 method.
Bio-layer interferometry
Affinity between BRISC and SHMT2 was measured by bio-layer interferometry with BRISC immobilized on the sensor and SHMT2 in solution. BRISC was expressed with a Strep-BirA tag fused to the N terminus of ABRO1. The reaction mixture of 1 ml BirA-BRISC (4.6 mg/ml), 60 μl BirA enzyme (2.4 mg/ml), 20 μl 1 M MgCl2, 200 μl 2 mM biotin, 100 μl 400 mM ATP pH 7.5 was adjusted to a final volume of 1.5 ml by addition of 620 μl phosphate buffered saline (PBS), incubated at RT for 1 h, and subsequently at 4°C overnight. Biotinylated BRISC complex was purified by gel filtration (Superdex 200, ThermoFisher). Association and dissociation constants were measured in BLI buffer (50 mM HEPES pH 7.4, 100 mM NaCl, 0.2 mM TECP, 2mg/ml BSA) with a BLItz bio-layer interferometer using Streptavidin-coated sensors (FortéBio).
Internally quenched fluorophore (IQF) deubiquitination assays
Cleavage of K63-diUb was measured using an internally quenched (Ub)2 substrate, which carries a fluorophore (TAMRA) on one ubiquitin molecule and a quencher on the other. Following DUB cleavage of the K63 isopeptide bond, a quencher-bound ubiquitin is released and TAMRA fluorescence increases. BRISC activity was tested against six available fluorophore/quencher positions (DU0102, LifeSensors) and compared to cleavage of unlabeled (Ub)2 in a gel-based assay. Subsequent DUB activity assays were performed with linkage type 1 substrate (DU6301, LifeSensors). The deubiquitination reactions were initiated by mixing 5 μl of protein in storage buffer (50 mM HEPES pH 7.4, 200 mM NaCl, 0.2 mM TCEP) with 5 μl of substrate at 400 nM (200 nM final concentration) in assay buffer (50 mM HEPES pH 7.0, 100 mM NaCl, 0.5% BSA, 0.03% Brij-35, 0.2 mM TCEP). Fluorescence was monitored in 10 s time increments (excitation at 540 nm, emission at 590 nm) and molar reaction rates were determined by comparison to TAMRA-ubiquitin (SI270T, LifeSensors) controls. Assays were carried out at 22°C in black flat-bottom high-base 386-well plates (Greiner Bio-One) using a Pherastar plate reader (BMG Labtech). Reaction progress data were fitted globally with DYNAFIT (Kuzmič, 2009).
Quantitative fluorescence microscopy of ABRO1 and SHMT2
HEK293T cells were grown in DMEM supplemented with 10% (v/v) FBS on glass coverslips in six-well dishes for 48 h. For mitochondrial labeling, 100 nM MitoTracker Red CMXRos (ThermoFisher) was added to media and cells were incubated for 30 min at 37°C. Cells were washed once in PBS and fixed for 10 min at RT with 4% PFA in PBS. Cells were washed twice with PBS and plasma membranes were then labeled with 5 μg/ml Alexa Fluor 633-conjugated wheat germ agglutinin (ThermoFisher) for 10 min at RT. Cells were subsequently washed three times with PBS, permeabilized for 15 min at RT in PBS supplemented with 0.2% (v/v) Triton X-100, then blocked for 1 h in PBS supplemented with 10% (v/v) donkey serum and 2% (w/v) BSA. Samples were incubated overnight at 4°C in a humid chamber with primary antibodies diluted in blocking solution (Rabbit anti-SHMT2 (SAB1100388, Sigma-Aldrich, 1:200); Rabbit anti-ABRO1 (ab74333, abcam, 1:100). Samples were washed in PBS then incubated overnight at 4°C with an Alexa Fluor 488-conjugated donkey anti-rabbit antibody (Sigma-Aldrich, 1:500). Samples were washed with PBS and mounted with VectorShield with DAPI (VectorLabs) and sealed with nail polish. Slides were imaged with a W1 Spinning-Disk (Yokogawa) mounted on an Axio Imager Z2 microscope (Zeiss) and controlled with VISIVIEW (Visitron). Fluorescence quantification in the different cell compartments was carried out with FIJI (Schindelin et al., 2012) on 3D Z stacks as follows: signal was smoothed with a Gaussian filter in all channels; masks for the nuclei and the mitochondria were generated using the DAPI and MitoTracker channels respectively, applying auto-threshold with the Otsu or Moments methods after background subtraction (rolling ball). The same treatment was applied to the Alexa Fluor 488 channel and the resulting image was combined with the masks for nuclei and mitochondria to create a mask for the whole cell; these three masks were then used to quantify in each plane the amount of fluorescence in the Alexa Fluor 488 channel (SHMT2 or ABRO1 staining).
Quantification and Statistical Analysis
Assays were conducted at least in triplicate, with SD reported, with the exception of experiments that were conducted as dilution replicates and fitted globally. Sample size was not determined a priori.
Data and Software Availability
The accession number for the crystal structure of BRCA1-A reported in this paper is PDB: 6GVW. The accession number for the cryo-EM density map of BRISC-SHMT2α complex reported in this paper is EMDB: EMDB-0132. The accession number for the model of BRISC-SHMT2α complex reported in this paper is PDB: 6H3C. The link for access to raw imaging data reported in this paper is Mendeley Data: https://dx.doi.org/10.17632/8ntswhp8d5.1.
Acknowldegements
We gratefully acknowledge the support of the FMI core facilities: D. Hess, V. Iesmantavicius, and J. Seebacher (Proteomics and Protein Analysis Facility) for mass spectrometry; H.R. Hotz (Computational Biology Platform) for phylogenetics; A. Graff Meyer and C. Genoud (Facility for Advanced Imaging and Microscopy) for microscopy; J. Keusch (Protein Structure Facility) for crystallization facilities. For sharing material, we thank D. Durocher (RAP80 knockout U2OS cells), A. Jetten (GFP-RAP80 construct), and T. Blundell (BRCA1-BRCT construct). We acknowledge the Paul Scherrer Institut, Villigen, Switzerland for access to beamline X06SA (PXI) of the Swiss Light Source and thank M. Wang for assistance.
J.R. was funded by a Novartis Presidential Fellowship and C.G. by the Danish Cancer Society (grant no. R146-RP11394). This work was funded by the Novartis Research Foundation (M.E.G., A.H.F.M.P.), NWO-VENI (863.11.007) and NWO-VIDI (016.161.320) grants to M.S.L., a Consolidator grant from the European Research Council (ERC-CoG) to H.v.A., NIH grants R01 GM124148 and R01 GM127707 to S.M.R., the Lundbeck Foundation (N.M.), a Senior Investigator Award from the Wellcome Trust (098391/Z/12/7) to E.B. and R.T.H., and the Swiss National Science Foundation through Sinergia grant number CRSII3_160734/1 and by the European Research Council under the European Union’s Horizon 2020 Research and Innovation program grant agreement, grant number 666068, to N.H.T.
Author Contributions
Conceptualization, J.R. and N.H.T.; Methodology, J.R., R.D.B., A.D.S., S.C., M.S.L., H.v.A., A.F.M.I., A.H.M., C.G., U.H., and L.G.; Validation, J.R. and N.H.T.; Formal Analysis, J.R., R.D.B., A.D.S., S.C., A.A.-P., M.S.L., A.H.M., U.H., and L.G.; Investigation, J.R., R.D.B., A.D.S., S.C., M.E.G., W.A., A.A.-P., M.S.L., A.F.M.I., A.H.M., and C.G.; Resources, J.R., W.A., A.A.-P., A.F.M.I., E.B., J.D.A., C.G., T.B., and M.R.; Data Curation, J.R., R.D.B., A.D.S., and S.C.; Writing – Original Draft, J.R. and N.H.T.; Writing – Review and Editing, J.R., R.D.B., and N.H.T.; Visualization, J.R., R.D.B., A.D.S., S.C., M.E.G., A.A.-P., M.S.L., H.v.A. and A.H.M.; Supervision, N.H.T.; Project Administration, J.R. and N.H.T.; Funding Acquisition, A.H.F.M.P., S.M.R., N.M., M.S.L., H.v.A., R.T.H., and N.H.T.
Declaration of Interest
The authors declare no competing interests.
Published: June 25, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.molcel.2019.06.002.
Supporting Citations
The following references appear in the Supplemental Information: Chaneton et al., 2012, Putnam et al., 2008, Wishart et al., 2018.
Supplemental Information
References
- Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.-W., Kapral G.J., Grosse-Kunstleve R.W. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambivero C.T., Cilenti L., Zervos A.S. ATF4 interacts with Abro1/KIAA0157 scaffold protein and participates in a cytoprotective pathway. Biochim. Biophys. Acta. 2012;1823:2149–2156. doi: 10.1016/j.bbamcr.2012.08.020. [DOI] [PubMed] [Google Scholar]
- Amelio I., Cutruzzolá F., Antonov A., Agostini M., Melino G. Serine and glycine metabolism in cancer. Trends Biochem. Sci. 2014;39:191–198. doi: 10.1016/j.tibs.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson D.D., Stover P.J. SHMT1 and SHMT2 are functionally redundant in nuclear de novo thymidylate biosynthesis. PLoS ONE. 2009;4:e5839. doi: 10.1371/journal.pone.0005839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badgujar D.C., Sawant U., Yadav L., Hosur M.V., Varma A.K., Vikrant Preliminary crystallographic studies of BRCA1 BRCT-ABRAXAS complex. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2013;69:1401–1404. doi: 10.1107/S1744309113030649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bett J.S., Ritorto M.S., Ewan R., Jaffray E.G., Virdee S., Chin J.W., Knebel A., Kurz T., Trost M., Tatham M.H., Hay R.T. Ubiquitin C-terminal hydrolases cleave isopeptide- and peptide-linked ubiquitin from structured proteins but do not edit ubiquitin homopolymers. Biochem. J. 2015;466:489–498. doi: 10.1042/BJ20141349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian C., Wu R., Cho K., Yu X. Loss of BRCA1-A complex function in RAP80 null tumor cells. PLoS ONE. 2012;7:e40406. doi: 10.1371/journal.pone.0040406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown N.G., VanderLinden R., Watson E.R., Qiao R., Grace C.R.R., Yamaguchi M., Weissmann F., Frye J.J., Dube P., Ei Cho S. RING E3 mechanism for ubiquitin ligation to a disordered substrate visualized for human anaphase-promoting complex. Proc. Natl. Acad. Sci. USA. 2015;112:5272–5279. doi: 10.1073/pnas.1504161112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaneton B., Hillmann P., Zheng L., Martin A.C.L., Maddocks O.D.K., Chokkathukalam A., Coyle J.E., Jankevics A., Holding F.P., Vousden K.H. Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature. 2012;491:458–462. doi: 10.1038/nature11540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen V.B., Arendall W.B., 3rd, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J., Murray L.W., Richardson J.S., Richardson D.C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cilenti L., Balakrishnan M.P., Wang X.-L., Ambivero C., Sterlicchi M., del Monte F., Ma X.L., Zervos A.S. Regulation of Abro1/KIAA0157 during myocardial infarction and cell death reveals a novel cardioprotective mechanism for Lys63-specific deubiquitination. J. Mol. Cell. Cardiol. 2011;50:652–661. doi: 10.1016/j.yjmcc.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapperton J.A., Manke I.A., Lowery D.M., Ho T., Haire L.F., Yaffe M.B., Smerdon S.J. Structure and mechanism of BRCA1 BRCT domain recognition of phosphorylated BACH1 with implications for cancer. Nat. Struct. Mol. Biol. 2004;11:512–518. doi: 10.1038/nsmb775. [DOI] [PubMed] [Google Scholar]
- Coleman K.A., Greenberg R.A. The BRCA1-RAP80 complex regulates DNA repair mechanism utilization by restricting end resection. J. Biol. Chem. 2011;286:13669–13680. doi: 10.1074/jbc.M110.213728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper E.M., Cutcliffe C., Kristiansen T.Z., Pandey A., Pickart C.M., Cohen R.E. K63-specific deubiquitination by two JAMM/MPN+ complexes: BRISC-associated Brcc36 and proteasomal Poh1. EMBO J. 2009;28:621–631. doi: 10.1038/emboj.2009.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowtan K. General quadratic functions in real and reciprocal space and their application to likelihood phasing. Acta Crystallogr. D Biol. Crystallogr. 2000;56:1612–1621. doi: 10.1107/s0907444900013263. [DOI] [PubMed] [Google Scholar]
- Cowtan K. Recent developments in classical density modification. Acta Crystallogr. D Biol. Crystallogr. 2010;66:470–478. doi: 10.1107/S090744490903947X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croll T.I. ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D Struct. Biol. 2018;74:519–530. doi: 10.1107/S2059798318002425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F., Grzesiek S., Vuister G.W., Zhu G., Pfeifer J., Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Dever S.M., Golding S.E., Rosenberg E., Adams B.R., Idowu M.O., Quillin J.M., Valerie N., Xu B., Povirk L.F., Valerie K. Mutations in the BRCT binding site of BRCA1 result in hyper-recombination. Aging (Albany N.Y.) 2011;3:515–532. doi: 10.18632/aging.100325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducker G.S., Ghergurovich J.M., Mainolfi N., Suri V., Jeong S.K., Hsin-Jung Li S., Friedman A., Manfredi M.G., Gitai Z., Kim H., Rabinowitz J.D. Human SHMT inhibitors reveal defective glycine import as a targetable metabolic vulnerability of diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA. 2017;114:11404–11409. doi: 10.1073/pnas.1706617114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P.R. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr. D Biol. Crystallogr. 2011;67:282–292. doi: 10.1107/S090744491003982X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans P.R., Murshudov G.N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 2013;69:1204–1214. doi: 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farady C.J., Craik C.S. Mechanisms of macromolecular protease inhibitors. ChemBioChem. 2010;11:2341–2346. doi: 10.1002/cbic.201000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L., Huang J., Chen J. MERIT40 facilitates BRCA1 localization and DNA damage repair. Genes Dev. 2009;23:719–728. doi: 10.1101/gad.1770609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L., Wang J., Chen J. The Lys63-specific deubiquitinating enzyme BRCC36 is regulated by two scaffold proteins localizing in different subcellular compartments. J. Biol. Chem. 2010;285:30982–30988. doi: 10.1074/jbc.M110.135392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giardina G., Brunotti P., Fiascarelli A., Cicalini A., Costa M.G.S., Buckle A.M., di Salvo M.L., Giorgi A., Marani M., Paone A. How pyridoxal 5′-phosphate differentially regulates human cytosolic and mitochondrial serine hydroxymethyltransferase oligomeric state. FEBS J. 2015;282:1225–1241. doi: 10.1111/febs.13211. [DOI] [PubMed] [Google Scholar]
- Goldstein M., Kastan M.B. Repair versus checkpoint functions of BRCA1 are differentially regulated by site of chromatin binding. Cancer Res. 2015;75:2699–2707. doi: 10.1158/0008-5472.CAN-15-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimaraes C.P., Witte M.D., Theile C.S., Bozkurt G., Kundrat L., Blom A.E.M., Ploegh H.L. Site-specific C-terminal and internal loop labeling of proteins using sortase-mediated reactions. Nat. Protoc. 2013;8:1787–1799. doi: 10.1038/nprot.2013.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzzo C.M., Berndsen C.E., Zhu J., Gupta V., Datta A., Greenberg R.A., Wolberger C., Matunis M.J. RNF4-dependent hybrid SUMO-ubiquitin chains are signals for RAP80 and thereby mediate the recruitment of BRCA1 to sites of DNA damage. Sci. Signal. 2012;5:ra88. doi: 10.1126/scisignal.2003485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hein M.Y., Hubner N.C., Poser I., Cox J., Nagaraj N., Toyoda Y., Gak I.A., Weisswange I., Mansfeld J., Buchholz F. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell. 2015;163:712–723. doi: 10.1016/j.cell.2015.09.053. [DOI] [PubMed] [Google Scholar]
- Hicke L., Schubert H.L., Hill C.P. Ubiquitin-binding domains. Nat. Rev. Mol. Cell Biol. 2005;6:610–621. doi: 10.1038/nrm1701. [DOI] [PubMed] [Google Scholar]
- Hodson C., Cole A.R., Lewis L.P.C., Miles J.A., Purkiss A., Walden H. Structural analysis of human FANCL, the E3 ligase in the Fanconi anemia pathway. J. Biol. Chem. 2011;286:32628–32637. doi: 10.1074/jbc.M111.244632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X., Kim J.A., Castillo A., Huang M., Liu J., Wang B. NBA1/MERIT40 and BRE interaction is required for the integrity of two distinct deubiquitinating enzyme BRCC36-containing complexes. J. Biol. Chem. 2011;286:11734–11745. doi: 10.1074/jbc.M110.200857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y., Scully R., Sobhian B., Xie A., Shestakova E., Livingston D.M. RAP80-directed tuning of BRCA1 homologous recombination function at ionizing radiation-induced nuclear foci. Genes Dev. 2011;25:685–700. doi: 10.1101/gad.2011011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X., Paul A., Wang B. Rap80 protein recruitment to DNA double-strand breaks requires binding to both small ubiquitin-like modifier (SUMO) and ubiquitin conjugates. J. Biol. Chem. 2012;287:25510–25519. doi: 10.1074/jbc.M112.374116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joosten R.P., Long F., Murshudov G.N., Perrakis A. The PDB_REDO server for macromolecular structure model optimization. IUCrJ. 2014;1:213–220. doi: 10.1107/S2052252514009324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakarougkas A., Ismail A., Katsuki Y., Freire R., Shibata A., Jeggo P.A. Co-operation of BRCA1 and POH1 relieves the barriers posed by 53BP1 and RAP80 to resection. Nucleic Acids Res. 2013;41:10298–10311. doi: 10.1093/nar/gkt802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastner B., Fischer N., Golas M.M., Sander B., Dube P., Boehringer D., Hartmuth K., Deckert J., Hauer F., Wolf E. GraFix: sample preparation for single-particle electron cryomicroscopy. Nat. Methods. 2008;5:53–55. doi: 10.1038/nmeth1139. [DOI] [PubMed] [Google Scholar]
- Khan A.R., James M.N. Molecular mechanisms for the conversion of zymogens to active proteolytic enzymes. Protein Sci. 1998;7:815–836. doi: 10.1002/pro.5560070401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H., Chen J., Yu X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science. 2007;316:1202–1205. doi: 10.1126/science.1139621. [DOI] [PubMed] [Google Scholar]
- Kim D., Fiske B.P., Birsoy K., Freinkman E., Kami K., Possemato R.L., Chudnovsky Y., Pacold M.E., Chen W.W., Cantor J.R. SHMT2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature. 2015;520:363–367. doi: 10.1038/nature14363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kucukelbir A., Sigworth F.J., Tagare H.D. Quantifying the local resolution of cryo-EM density maps. Nat. Methods. 2014;11:63–65. doi: 10.1038/nmeth.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmič P. DynaFit--a software package for enzymology. Methods Enzymol. 2009;467:247–280. doi: 10.1016/S0076-6879(09)67010-5. [DOI] [PubMed] [Google Scholar]
- Kyrieleis O.J.P., McIntosh P.B., Webb S.R., Calder L.J., Lloyd J., Patel N.A., Martin S.R., Robinson C.V., Rosenthal P.B., Smerdon S.J. Three-dimensional architecture of the human BRCA1-A histone deubiquitinase core complex. Cell Rep. 2016;17:3099–3106. doi: 10.1016/j.celrep.2016.11.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G.Y., Haverty P.M., Li L., Kljavin N.M., Bourgon R., Lee J., Stern H., Modrusan Z., Seshagiri S., Zhang Z. Comparative oncogenomics identifies PSMB4 and SHMT2 as potential cancer driver genes. Cancer Res. 2014;74:3114–3126. doi: 10.1158/0008-5472.CAN-13-2683. [DOI] [PubMed] [Google Scholar]
- Leznicki P., Kulathu Y. Mechanisms of regulation and diversification of deubiquitylating enzyme function. J. Cell Sci. 2017;130:1997–2006. doi: 10.1242/jcs.201855. [DOI] [PubMed] [Google Scholar]
- Li X., Mooney P., Zheng S., Booth C.R., Braunfeld M.B., Gubbens S., Agard D.A., Cheng Y. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods. 2013;10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingaraju G.M., Bunker R.D., Cavadini S., Hess D., Hassiepen U., Renatus M., Fischer E.S., Thomä N.H. Crystal structure of the human COP9 signalosome. Nature. 2014;512:161–165. doi: 10.1038/nature13566. [DOI] [PubMed] [Google Scholar]
- Liu X., Ladias J.A.A. Structural basis for the BRCA1 BRCT interaction with the proteins ATRIP and BAAT1. Biochemistry. 2013;52:7618–7627. doi: 10.1021/bi400714v. [DOI] [PubMed] [Google Scholar]
- Lombardi P.M., Matunis M.J., Wolberger C. RAP80, ubiquitin and SUMO in the DNA damage response. J. Mol. Med. (Berl.) 2017;95:799–807. doi: 10.1007/s00109-017-1561-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J. Phaser crystallographic software. J. Appl. Cryst. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mevissen T.E.T., Komander D. Mechanisms of deubiquitinase specificity and regulation. Annu. Rev. Biochem. 2017;86:159–192. doi: 10.1146/annurev-biochem-061516-044916. [DOI] [PubMed] [Google Scholar]
- Mok M.T.S., Henderson B.R. The in vivo dynamic organization of BRCA1-A complex proteins at DNA damage-induced nuclear foci. Traffic. 2012;13:800–814. doi: 10.1111/j.1600-0854.2012.01355.x. [DOI] [PubMed] [Google Scholar]
- Ng H.-M., Wei L., Lan L., Huen M.S.Y. The Lys63-deubiquitylating enzyme BRCC36 limits DNA break processing and repair. J. Biol. Chem. 2016;291:16197–16207. doi: 10.1074/jbc.M116.731927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls R.A., Long F., Murshudov G.N. Low-resolution refinement tools in REFMAC5. Acta Crystallogr. D Biol. Crystallogr. 2012;68:404–417. doi: 10.1107/S090744491105606X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikkilä J., Coleman K.A., Morrissey D., Pylkäs K., Erkko H., Messick T.E., Karppinen S.-M., Amelina A., Winqvist R., Greenberg R.A. Familial breast cancer screening reveals an alteration in the RAP80 UIM domain that impairs DNA damage response function. Oncogene. 2009;28:1843–1852. doi: 10.1038/onc.2009.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathare G.R., Nagy I., Śledź P., Anderson D.J., Zhou H.-J., Pardon E., Steyaert J., Förster F., Bracher A., Baumeister W. Crystal structure of the proteasomal deubiquitylation module Rpn8-Rpn11. Proc. Natl. Acad. Sci. USA. 2014;111:2984–2989. doi: 10.1073/pnas.1400546111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson-Fortin J., Shao G., Bretscher H., Messick T.E., Greenberg R.A. Differential regulation of JAMM domain deubiquitinating enzyme activity within the RAP80 complex. J. Biol. Chem. 2010;285:30971–30981. doi: 10.1074/jbc.M110.135319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei J., Kim B.-H., Grishin N.V. PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Research. 2008;36:2295–2300. doi: 10.1093/nar/gkn072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petroski M.D., Deshaies R.J. Function and regulation of cullin-RING ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- Plechanovová A., Jaffray E.G., McMahon S.A., Johnson K.A., Navrátilová I., Naismith J.H., Hay R.T. Mechanism of ubiquitylation by dimeric RING ligase RNF4. Nat. Struct. Mol. Biol. 2011;18:1052–1059. doi: 10.1038/nsmb.2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putnam N.H., Butts T., Ferrier D.E.K., Furlong R.F., Hellsten U., Kawashima T., Robinson-Rechavi M., Shoguchi E., Terry A., Yu J.-K. The amphioxus genome and the evolution of the chordate karyotype. Nature. 2008;453:1064–1071. doi: 10.1038/nature06967. [DOI] [PubMed] [Google Scholar]
- Py B.F., Kim M.-S., Vakifahmetoglu-Norberg H., Yuan J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell. 2013;49:331–338. doi: 10.1016/j.molcel.2012.11.009. [DOI] [PubMed] [Google Scholar]
- Ray H., Moreau K., Dizin E., Callebaut I., Venezia N.D. ACCA phosphopeptide recognition by the BRCT repeats of BRCA1. J. Mol. Biol. 2006;359:973–982. doi: 10.1016/j.jmb.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Rebbeck T.R., Mitra N., Domchek S.M., Wan F., Friebel T.M., Tran T.V., Singer C.F., Tea M.K.M., Blum J.L., Tung N., Epidemiological Study of BRCA1 and BRCA2 Mutation Carriers (EMBRACE) Modification of BRCA1-associated breast and ovarian cancer risk by BRCA1-interacting genes. Cancer Res. 2011;71:5792–5805. doi: 10.1158/0008-5472.CAN-11-0773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedinger C., Boehringer J., Trempe J.F., Lowe E.D., Brown N.R., Gehring K., Noble M.E.M., Gordon C., Endicott J.A. Structure of Rpn10 and its interactions with polyubiquitin chains and the proteasome subunit Rpn12. J. Biol. Chem. 2010;285:33992–34003. doi: 10.1074/jbc.M110.134510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal P.B., Henderson R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol. 2003;333:721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- Sahtoe D.D., van Dijk W.J., El Oualid F., Ekkebus R., Ovaa H., Sixma T.K. Mechanism of UCH-L5 activation and inhibition by DEUBAD domains in RPN13 and INO80G. Mol. Cell. 2015;57:887–900. doi: 10.1016/j.molcel.2014.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato Y., Yoshikawa A., Yamagata A., Mimura H., Yamashita M., Ookata K., Nureki O., Iwai K., Komada M., Fukai S. Structural basis for specific cleavage of Lys 63-linked polyubiquitin chains. Nature. 2008;455:358–362. doi: 10.1038/nature07254. [DOI] [PubMed] [Google Scholar]
- Scheres S.H.W. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder G.F., Levitt M., Brunger A.T. Super-resolution biomolecular crystallography with low-resolution data. Nature. 2010;464:1218–1222. doi: 10.1038/nature08892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao G., Patterson-Fortin J., Messick T.E., Feng D., Shanbhag N., Wang Y., Greenberg R.A. MERIT40 controls BRCA1-Rap80 complex integrity and recruitment to DNA double-strand breaks. Genes Dev. 2009;23:740–754. doi: 10.1101/gad.1739609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick G.M. A short history of SHELX. Acta Crystallogr. A. 2008;64:112–122. doi: 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Shiozaki E.N., Gu L., Yan N., Shi Y. Structure of the BRCT repeats of BRCA1 bound to a BACH1 phosphopeptide: implications for signaling. Mol. Cell. 2004;14:405–412. doi: 10.1016/s1097-2765(04)00238-2. [DOI] [PubMed] [Google Scholar]
- Sobhian B., Shao G., Lilli D.R., Culhane A.C., Moreau L.A., Xia B., Livingston D.M., Greenberg R.A. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science. 2007;316:1198–1202. doi: 10.1126/science.1139516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solyom S., Aressy B., Pylkäs K., Patterson-Fortin J., Hartikainen J.M., Kallioniemi A., Kauppila S., Nikkilä J., Kosma V.-M., Mannermaa A. Breast cancer-associated Abraxas mutation disrupts nuclear localization and DNA damage response functions. Sci. Transl. Med. 2012;4:122ra23. doi: 10.1126/scitranslmed.3003223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowa M.E., Bennett E.J., Gygi S.P., Harper J.W. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier F.W. Protein production by auto-induction in high density shaking cultures. Protein Expr. Purif. 2005;41:207–234. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Tang G., Peng L., Baldwin P.R., Mann D.S., Jiang W., Rees I., Ludtke S.J. EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Typas D., Luijsterburg M.S., Wiegant W.W., Diakatou M., Helfricht A., Thijssen P.E., van den Broek B., Mullenders L.H., van Attikum H. The de-ubiquitylating enzymes USP26 and USP37 regulate homologous recombination by counteracting RAP80. Nucleic Acids Res. 2015;43:6919–6933. doi: 10.1093/nar/gkv613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uckelmann M., Sixma T.K. Histone ubiquitination in the DNA damage response. DNA Repair (Amst.) 2017;56:92–101. doi: 10.1016/j.dnarep.2017.06.011. [DOI] [PubMed] [Google Scholar]
- Uhlén M., Fagerberg L., Hallström B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Uhlén M., Zhang C., Lee S., Sjöstedt E., Fagerberg L., Bidkhori G., Benfeitas R., Arif M., Liu Z., Edfors F. A pathology atlas of the human cancer transcriptome. Science. 2017;357:357. doi: 10.1126/science.aan2507. [DOI] [PubMed] [Google Scholar]
- van Wijk S.J.L., Fiskin E., Putyrski M., Pampaloni F., Hou J., Wild P., Kensche T., Grecco H.E., Bastiaens P., Dikic I. Fluorescence-based sensors to monitor localization and functions of linear and K63-linked ubiquitin chains in cells. Mol. Cell. 2012;47:797–809. doi: 10.1016/j.molcel.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Linden R.T., Hemmis C.W., Schmitt B., Ndoja A., Whitby F.G., Robinson H., Cohen R.E., Yao T., Hill C.P. Structural basis for the activation and inhibition of the UCH37 deubiquitylase. Mol. Cell. 2015;57:901–911. doi: 10.1016/j.molcel.2015.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma A.K., Brown R.S., Birrane G., Ladias J.A.A. Structural basis for cell cycle checkpoint control by the BRCA1-CtIP complex. Biochemistry. 2005;44:10941–10946. doi: 10.1021/bi0509651. [DOI] [PubMed] [Google Scholar]
- Wang B., Matsuoka S., Ballif B.A., Zhang D., Smogorzewska A., Gygi S.P., Elledge S.J. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science. 2007;316:1194–1198. doi: 10.1126/science.1139476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B., Hurov K., Hofmann K., Elledge S.J. NBA1, a new player in the Brca1 A complex, is required for DNA damage resistance and checkpoint control. Genes Dev. 2009;23:729–739. doi: 10.1101/gad.1770309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R.Y.-R., Song Y., Barad B.A., Cheng Y., Fraser J.S., DiMaio F. Automated structure refinement of macromolecular assemblies from cryo-EM maps using Rosetta. eLife. 2016;5:352. doi: 10.7554/eLife.17219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm M., Schlegl J., Hahne H., Gholami A.M., Lieberenz M., Savitski M.M., Ziegler E., Butzmann L., Gessulat S., Marx H. Mass-spectrometry-based draft of the human proteome. Nature. 2014;509:582–587. doi: 10.1038/nature13319. [DOI] [PubMed] [Google Scholar]
- Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G.W., McCoy A. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G., Waterman D.G., Parkhurst J.M., Brewster A.S., Gildea R.J., Gerstel M., Fuentes-Montero L., Vollmar M., Michels-Clark T., Young I.D. DIALS: implementation and evaluation of a new integration package. Acta Crystallogr. D Struct. Biol. 2018;74:85–97. doi: 10.1107/S2059798317017235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D.S., Feunang Y.D., Marcu A., Guo A.C., Liang K., Vázquez-Fresno R., Sajed T., Johnson D., Li C., Karu N. HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res. 2018;46(D1):D608–D617. doi: 10.1093/nar/gkx1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worden E.J., Padovani C., Martin A. Structure of the Rpn11-Rpn8 dimer reveals mechanisms of substrate deubiquitination during proteasomal degradation. Nat. Struct. Mol. Biol. 2014;21:220–227. doi: 10.1038/nsmb.2771. [DOI] [PubMed] [Google Scholar]
- Worden E.J., Dong K.C., Martin A. An AAA motor-driven mechanical switch in Rpn11 controls deubiquitination at the 26S proteasome. Mol. Cell. 2017;67:799–811.e8. doi: 10.1016/j.molcel.2017.07.023. [DOI] [PubMed] [Google Scholar]
- Wu J., Liu C., Chen J., Yu X. RAP80 protein is important for genomic stability and is required for stabilizing BRCA1-A complex at DNA damage sites in vivo. J. Biol. Chem. 2012;287:22919–22926. doi: 10.1074/jbc.M112.351007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q., Paul A., Su D., Mehmood S., Foo T.K., Ochi T., Bunting E.L., Xia B., Robinson C.V., Wang B., Blundell T.L. Structure of BRCA1-BRCT/Abraxas complex reveals phosphorylation-dependent BRCT dimerization at DNA damage sites. Mol. Cell. 2016;61:434–448. doi: 10.1016/j.molcel.2015.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M., Moresco J.J., Chang M., Mukim A., Smith D., Diedrich J.K., Yates J.R., 3rd, Jones K.A. SHMT2 and the BRCC36/BRISC deubiquitinase regulate HIV-1 Tat K63-ubiquitylation and destruction by autophagy. PLoS Pathog. 2018;14 doi: 10.1371/journal.ppat.1007071. e1007071–e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z., Kim Y.-S., Jetten A.M. RAP80, a novel nuclear protein that interacts with the retinoid-related testis-associated receptor. J. Biol. Chem. 2002;277:32379–32388. doi: 10.1074/jbc.M203475200. [DOI] [PubMed] [Google Scholar]
- Yin Z., Menendez D., Resnick M.A., French J.E., Janardhan K.S., Jetten A.M. RAP80 is critical in maintaining genomic stability and suppressing tumor development. Cancer Res. 2012;72:5080–5090. doi: 10.1158/0008-5472.CAN-12-1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeqiraj E., Tian L., Piggott C.A., Pillon M.C., Duffy N.M., Ceccarelli D.F., Keszei A.F.A., Lorenzen K., Kurinov I., Orlicky S. Higher-order assembly of BRCC36-KIAA0157 is required for DUB activity and biological function. Mol. Cell. 2015;59:970–983. doi: 10.1016/j.molcel.2015.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K. Gctf: Real-time CTF determination and correction. J. Struct. Biol. 2016;193:1–12. doi: 10.1016/j.jsb.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H., Gupta V., Patterson-Fortin J., Bhattacharya S., Katlinski K., Wu J., Varghese B., Carbone C.J., Aressy B., Fuchs S.Y., Greenberg R.A. A BRISC-SHMT complex deubiquitinates IFNAR1 and regulates interferon responses. Cell Rep. 2013;5:180–193. doi: 10.1016/j.celrep.2013.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the crystal structure of BRCA1-A reported in this paper is PDB: 6GVW. The accession number for the cryo-EM density map of BRISC-SHMT2α complex reported in this paper is EMDB: EMDB-0132. The accession number for the model of BRISC-SHMT2α complex reported in this paper is PDB: 6H3C. The link for access to raw imaging data reported in this paper is Mendeley Data: https://dx.doi.org/10.17632/8ntswhp8d5.1.