

 Benzoporphyrins with varying non-noble metal centers can reduce carbon dioxide to carbon monoxide with faradaic efficiencies changing between 33 and 48%.
Benzoporphyrins with varying non-noble metal centers can reduce carbon dioxide to carbon monoxide with faradaic efficiencies changing between 33 and 48%.
Abstract
We report the synthesis and electrochemical properties of freebase tetraphenyltetrabenzoporphyrin and its complexes of Zn(ii), Co(ii), Ni(ii), Cu(ii) and Sn(iv) towards electrochemical reduction of carbon dioxide (CO2). Based on cyclic voltammetry, it is shown that central metals significantly affect the electrocatalytic performance in the reduction of CO2 in terms of reduction potential and catalytic current enhancement. At an applied potential of –1.90 V vs. an Ag/AgCl quasi reference electrode for 20 h, the electrocatalytic reduction of CO2 realized by Zn(ii)- and Cu(ii)-tetraphenyltetrabenzoporphyrins to carbon monoxide resulted in faradaic efficiencies of around 48% and 33%, respectively.
Introduction
Rapid global economic and population growth has led to an increase in fuel consumption in industrial, transportation, commercial and residential sectors. As a major source of energy, vast quantities of fossil fuels are burnt, creating environmental problems due to subsequent creation of greenhouse gases and pollutants. A greenhouse relevant product from fuel combustion processes is carbon dioxide (CO2). Therefore, assiduous scientific attention is directed towards identification of optimal methods to reduce the generation of CO2 and, at the same time, to convert CO2 into other useful compounds. One way to address this issue might be the cyclic use of carbon in human society, which stabilizes the CO2 content in the atmosphere.
The electrochemical reduction processes of CO2 using catalysts have attracted much attention because they require lower overpotential, compared to the direct reduction of CO2, and provide higher product selectivity.1–5 As economically friendly substitutes for precious-metal catalysts, organometallic electrocatalysts have become popular.6–13 Among such materials, porphyrin derivatives have been investigated continuously as potential electrochemical catalysts for the reduction of CO2.14,15 The great advantages of porphyrin compounds regarding this application originate from their high stability and tunability of their electrochemical properties by changing metal centers and substituents at the macrocyclic peripheral positions. Such structural modifications do not only enable improvement in the electrocatalytic performance of the systems, but also allow one to investigate the possible catalytic mechanisms.16–19
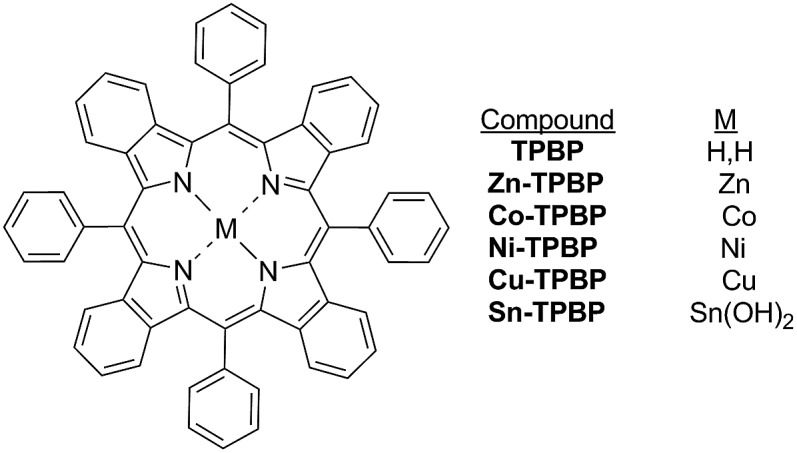
Tetrabenzoporphyrins are one of the very intriguing porphyrin derivatives that exhibit unique photophysical and electrochemical behaviors caused by extension of a π-conjugated structure at the β-positions of the porphyrin core and highly distorted macrocycle core due to meso-substitutions.20–24 Our previous work described systematic investigation of the photophysical and electrochemical properties of meso-substituted porphyrin and benzoporphyrin derivatives as ternary components for highly efficient bulk-heterojunction solar cells.22 Our observations on the electrochemical properties of the meso-substituted benzoporphyrin derivatives obtained from the previous study have led to extended investigation towards their catalytic activities for the electrochemical reduction of CO2 in this work. To the best of our knowledge, until now there are only a few studies on the electrochemical properties and catalysis of benzoporphyrin derivatives for CO2 reduction. Ramirez et al. described the electrochemical and photoelectrochemical reduction of CO2 using a Co(ii)-tetrabenzoporphyrin-modified electrode.25,26 To gain insights into the electrochemical properties of benzoporphyrins towards the electrochemical reduction of CO2, a series of metal-free and metallated tetraphenyltetrabenzoporphyrins as shown in Chart 1 were synthesized, characterized and investigated for their electrocatalytic activities for the reduction of CO2. Complexes of interest for this study had non-precious-metal centers like Zn(ii)–, Co(ii)–, Ni(ii)–, Cu(ii)– and Sn(iv)–, which were previously studied in porphyrin analogs for the electrochemical reduction of CO2.27–31 The overview presented in this work on the effect of the central metals on the electrochemical behavior and electrocatalytic activities of these benzoporphyrins for the reduction of CO2 will become a useful guideline for further development of several precious-metal-free oligopyrrole-based electrocatalytic systems.
Chart 1. Chemical structure of the benzoporphyrins.
Results and discussion
Synthesis
Synthesis of the benzoporphyrins started from previously reported demetallation of Cu-TPBP32 by sulfuric acid at room temperature for 30 min, resulting in TPBP in 81% yield (Scheme 1).33 In its 1H-NMR spectrum, a broad singlet signal of two inner protons at δ –1.17 ppm confirmed the formation of TPBP. To obtain the desired metallated benzoporphyrins, TPBP was reacted with Zn(OAc)2·2H2O,34 Co(OAc)2·4H2O35 and SnCl2·2H2O36 as described in the previous studies, leading to Zn-TPBP, Co-TPBP and Sn-TPBP, respectively, in 80–97% yield. Complete metallation of TPBP in these reactions was monitored by the absence of the broad singlet signal of the benzoporphyrinic inner protons at δ –1.17 ppm in the 1H-NMR spectra and disappearance of the characteristic emission peak of TPBP at 787 nm. HR-ESI-MS confirmed the formation of both Zn-TPBP and Co-TPBP by showing their molecular ion peaks at m/z 876.2231 and 871.2273, respectively. Due to difficulties in chromatographic purification, Sn-TPBP could not be completely separated from other byproducts and was obtained in >90% purity based on 1H-NMR spectroscopy. However, its formation could be confirmed by MALDI-TOF MS showing its molecular ion peak at m/z 966.292. Ni-TPBP was obtained from a known procedure described in detail by Finikova et al.32
Scheme 1. Synthesis of Zn-TPBP, Co-TPBP and Sn-TPBP.

Photophysical properties
As mentioned above, variation of the metal center of the benzoporhyrin derivatives does not only significantly affect the electrochemical behavior, but also the photophysical properties of the molecules. Therefore, absorption and emission of TPBP, Zn-TPBP, Co-TPBP, Ni-TPBP, Cu-TPBP and Sn-TPBP were investigated in this study to provide additional information that should be useful for further applications of these materials in optoelectronics and photoelectrocatalysis. By using UV-Vis spectrophotometry, normalized absorption spectra of all compounds in toluene were obtained as shown in Fig. 1. All compounds exhibited characteristic absorption patterns of the metallated benzoporphyrins having intense Soret bands in the range of 446–466 nm with absorption coefficients (ε) of 1.6 × 105–5.3 × 105 M–1 cm–1, and the Q-bands in the range of 592–698 nm, as summarized in Table 1. Upon excitation at their absorption maxima, TPBP, Zn-TPBP and Sn-TPBP showed emission peaks in the range of 658–787 nm as shown in Fig. 2 and Table 1, while Ni-TPBP, Cu-TPBP and Co-TPBP gave no significant emission.
Fig. 1. Normalized absorption spectra of (a) TPBP (black solid line), Zn-TPBP (red dashed line) and Co-TPBP (blue dotted line), and (b) Ni-TPBP (black solid line), and Cu-TPBP (red dashed line).

Table 1. Absorption and emission spectral data of the benzoporphyrins.
| Compound | λ abs/nm (ε × 105/M–1 cm–1) | λ em/nm |
| TPBP | 465 (5.3), 588 a , 626 a , 640 a , 698 a | 720, 787 |
| Zn-TPBP | 461 (2.8), 607 a , 652 a | 658, 724 |
| Co-TPBP | 446 (1.7), 595 a , 640 (0.8) | — b |
| Ni-TPBP | 449 (2.2), 592 a , 644 (0.9) | — b |
| Cu-TPBP | 449 (1.6), 463 (1.9), 601 (0.1), 648 (0.8) | — b |
| Sn-TPBP | 430 (0.3), 466 (4.1), 612 a , 660 (0.9) | 665, 745 |
aDue to low absorption, the ε value could not be determined.
bNo emission peak was observed.
Fig. 2. Normalized emission spectra of TPBP (black solid line), Zn-TPBP (red dashed line) and Sn-TPBP (blue dotted line).

Electrochemical characterization and catalytic activity
All benzoporphyrins were studied by means of cyclic voltammetry to evaluate their catalytic activities towards electrochemical CO2 reduction. The cyclic voltammograms were recorded in the potential range of 0.0 V to –2.0 V from a 0.1 M TBAPF6 solution in DMF containing 1.0 mM benzoporphyrin with a scan rate of 50 mV s–1 under the N2- and CO2-saturated conditions at ambient temperature and pressure. Fig. 3a shows that under the N2-saturated conditions, the first and the second reduction steps of TPBP are characterized by reversible reduction peaks at –1.22 V and –1.52 V, respectively. However, when the solution of TPBP was saturated with CO2, the first irreversible reduction was also observed at –1.22 V with a two-fold increase in peak current from 0.04 mA to 0.08 mA, while its second irreversible reduction peak appeared at –1.84 V with a negative shift by 0.32 V and a peak current enhancement from 0.05 mA to 0.13 mA. The great difference in the electrochemical behavior of TPBP observed in this case, compared with that under the N2-saturated conditions, may be attributed to possible binding between CO2 and inner pyrrolenine nitrogen atoms of the freebase benzoporphyrin macrocycle. The binding CO2 might form an ionic salt that is stabilized by the TBA cation which may present itself as a peak at more negative potentials. Moreover, new anodic signals were observed in the potential range of –0.40 V to –0.80 V, indicating the generation of unknown products from the irreversible reduction process(es) or the release of captured carbon dioxide. As for the electrochemical reduction of Zn-TPBP (Fig. 3b), a fully reversible first reduction (around –1.6 V) and as suggested by the scan-rate dependency of the peak current, a quasi-reversible second reduction peak (around –1.8 V) can be observed under N2-saturated conditions. A slight current increase for the first reduction peak and a two-fold current increase (from 0.05 mA to 0.10 mA) for the second one were monitored under CO2-saturated conditions signaling the reduction of CO2. Unknown oxidation signals were found under both N2- and CO2-saturated conditions in the potential range of –0.30 V to –0.70 V, which may be attributed to the side products formed during the reduction process. This observation of the increase in current and the change in the shape of the second ligand-based peak suggested that the doubly-reduced Zn-TPBP is the responsible molecule for the reduction of CO2.
Fig. 3. Cyclic voltammograms of a 0.1 M TBAPF6 solution in DMF containing 1.0 mM (a) TPBP, (b) Zn-TPBP, (c) Co-TPBP, (d) Ni-TPBP, (e) Cu-TPBP and (f) Sn-TPBP under the N2-(black solid line) and CO2-saturated (red dashed line) conditions recorded at a scan rate of 50 mV s–1 in the potential range of 0.00 to –2.00 V.

The cyclic voltammogram of Co-TPBP in the N2-saturated solution revealed a reversible reduction peak at –0.75 V stemming from the metal-centered reduction of the molecule. This was followed by two quasi-reversible peaks at –1.29 V and –1.55 V which may be attributed to the multi-step ligand-centered reduction of the meso-substituted benzoporphyrin macrocycle (Fig. 3c). Under the CO2-saturated conditions, Co-TPBP showed a very similar reduction pattern with a slight current increase. Compared with planar porphyrin and phthalocyanine derivatives reported as potential catalysts.37 The difference in the catalytic behavior of Co-TPBP observed herein should stem from the unique saddle-shaped macrocycle distortion of the metallated benzoporphyrins previously studied by several groups.20–24
The cyclic voltammogram of Ni-TPBP under the N2-saturated conditions showed the first reversible reduction peak at –1.10 V which is ligand-centred and the second quasi-reversible one at –1.90 V that originates from metal-centered reduction.38 (Fig. 3d). Under the CO2-saturated conditions, similar electrochemical features were observed with a 3.6-fold current enhancement of the second reduction from 0.05 mA to 0.18 mA and a positive shift of its onset potential (Eonset) by 0.20 V, suggesting the catalytic activity of Ni-TPBP towards the electrochemical reduction of CO2. In a similar manner, the N2-saturated solution of Cu-TPBP exhibited two major reversible reduction peaks at –1.27 V and –1.90 V which are attributed to the formation of a dianion radical and dianion of the macrocycle, respectively as shown in Fig. 3e.38,39 In the presence of CO2, a considerable current increase from 0.05 mA to 0.13 mA was observed at –1.85 V with a positive shift of Eonset by 0.18 V, suggesting the catalytic activity of Cu-TPBP for the electrochemical reduction of CO2. Sn-TPBP showed rather complex electrochemical behavior with two major reduction peaks at –0.92 V and –1.36 V, followed by at least five anodic peaks under the N2-saturated conditions, suggesting irreversible structural changes of the molecules upon the reduction processes (Fig. 3f). In the presence of CO2, its cyclic voltammogram also exhibited two main reduction peaks at a similar peak potential with even more complex anodic signals and a negligible current enhancement. This was attributed to possible interaction of the axial hydroxyl ligands of Sn-TPBP and CO2 that might lead to unexpected formation of side products upon reduction.
To further investigate the electrocatalytic performance of each benzoporphyrin towards the electrochemical reduction of CO2, all compounds were subjected to constant-potential electrolysis at an applied potential of –1.90 V for 20 h. The results from GC headspace analysis showed that the CO2-saturated electrolyte solutions containing TPBP, Co-TPBP, Ni-TPBP and Sn-TPBP did not yield any detectable reduction product. In the case of TPBP, it is likely that the above-mentioned binding between CO2 and the benzoporphyrin core was relatively strong and therefore suppressed the favorable reduction process of CO2 molecules. The low current enhancement observed for the above-described cyclic voltammograms of Co-TPBP and Sn-TPBP under the CO2 atmosphere could imply their low catalytic efficiencies for the electrochemical reduction of CO2. As for Ni-TPBP, although the current enhancement was found to be quite high in the cyclic voltammetry study, the reduction process did not lead to any detectable amount of CO or any other gaseous products. It is possible that the reduction process proceeded via other mechanisms and further optimization of the reduction conditions, such as addition of proton sources, pH adjustment and use of a co-catalyst, may be required. When Zn-TPBP and Cu-TPBP were used as catalysts, 8.67 μmol and 12 μmol of CO were detected, corresponding to a faradaic efficiency of 33% and 48% with turnover numbers of 1.7 and 6.6, respectively.
Conclusions
Six tetraphenyltetrabenzoporphyrins, including the freebase derivative and the metal-chelated ones having Zn, Co, Ni, Cu and Sn metal centers, were successfully synthesized and characterized. According to UV-visible and fluorescence spectrophotometry, and cyclic voltammetry, the introduction and variation of the metal centers obviously affected the photophysical properties, electrochemical behaviors and catalytic activities for the electrochemical reduction of CO2 to CO. The results from cyclic voltammetry showed that, compared with the N2-saturated conditions, TPBP, Ni-TPBP, Zn-TPBP and Cu-TPBP exhibited a significant current enhancement, while Co-TPBP and Sn-TPBP gave only a slight increase in the peak current when CO2 was introduced into the solution. The constant-potential electrolysis revealed the formation of CO as the main reduction product with faradaic efficiencies of 33% and 48% and turnover numbers of 1.7 and 6.6 for Zn-TPBP and Cu-TPBP, respectively. This study suggests that, among the metallobenzoporphyrins of interest, Zn- and Cu-chelated analogs exhibited superior electrocatalytic activities towards the electrochemical reduction of CO2 to CO. Here, we show the catalytic potential of the metallated benzoporphyrins as alternative CO2 reduction catalysts using cheaper and more abundant metal centers like Zn and Cu, compared to Re and Pd. Optimization of the reduction conditions, together with structural modification of benzoporphyrin ligands will be further studied and described elsewhere.
Experimental section
Materials and methods
All chemicals were of analytical grade, purchased from commercial suppliers and used as received without further purification. 1H-NMR and 13C-NMR spectra were obtained in deuterated chloroform (CDCl3) at 400 megahertz (MHz) for 1H nuclei and 100 MHz for 13C nuclei. Chemical shifts (δ) are reported in parts per million (ppm) relative to the residual CHCl3 peak (7.26 ppm for 1H-NMR and 77.0 ppm for 13C-NMR spectroscopy). Mass spectra were obtained using high-resolution electron spray ionization mass spectrometry (HR-ESI-MS) and matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS) with dithranol as a matrix. Ultraviolet-visible (UV-vis) and fluorescence spectrophotometry were performed in toluene at room temperature. Molar extinction coefficients (ε) are expressed in M–1 cm–1.
Non-commercial compounds
meso-tetraphenyltetrabenzoporphyrinatonickel(ii) (Ni-TPBP)32 and meso-tetraphenyltetrabenzoporphyrinatocopper(ii) (Cu-TPBP)32 were prepared by a published procedure.
Synthesis and characterization of benzoporphyrin derivatives
meso-Tetraphenyltetrabenzoporphyrin (TPBP)
Following a previously published procedure,33Cu-TPBP32 (0.052 g, 0.060 mmol) was reacted with concentrated sulfuric acid (10 mL) at room temperature for 30 min. The resulting reaction mixture was poured into a water/ice mixture and then extracted with dichloromethane. The combined organic phase was dried over anhydrous Na2SO4 and concentrated to dryness. The crude mixture was purified by column chromatography (silica gel, CH2Cl2/hexanes (2 : 1)) to afford TPBP as a green solid (0.039 g, 81%). 1H-NMR: δH –1.17 (s, 2H), 7.34–7.43 (m, 8H), 7.82–8.02 (m, 16H), 8.37 (d, J = 7.6 Hz, 4H), 8.56 (d, J = 7.2 Hz, 8H); 13C-NMR: δC 114.5, 115.8, 124.3, 124.7, 125.9, 128.6, 129.0, 129.3, 129.5, 130.1, 131.5, 134.7, 136.2, 139.9, 141.5, 142.1; MALDI-TOF-MS m/z (%): found 814.554 (100) [M+]; calcd 814.971 (M = C60H38N4); HR-ESI-MS m/z: [M + H]+ calcd for M = C60H38N4, 815.3175; found 815.3174; λabs(ε × 105) 465(5.3), 588, 626, 640, 698 nm; λem (λex = 465 nm) 720, 787 nm.
meso-Tetraphenyltetrabenzoporphyrinatozinc (Zn-TPBP)
Following a previously published procedure,34 a solution of TPBP (0.131 g, 0.161 mmol) in chloroform (117 mL) was reacted with a solution of Zn(OAc)2·2H2O (0.177 g, 0.805 mmol) in methanol (13 mL) at room temperature for 12 h. After that, the resulting mixture was washed with water, and the organic layer was separated and dried over anhydrous Na2SO4. After removal of the solvent, the crude mixture was purified by column chromatography (silica gel, CH2Cl2/hexanes (2 : 1)) to afford Zn-TPBP as a greenish blue solid (0.136 g, 97%). 1H-NMR: δH 7.16 (dd, J = 6.0, 2.8 Hz, 8H), 7.28 (dd, J = 6.0, 2.8 Hz, 8H) 7.86 (t, J = 7.2 Hz, 8H), 7.93 (t, J = 7.2 Hz 4H), 8.30 (d, J = 7.2 Hz, 8H); 13C-NMR: δC 117.3, 124.3, 124.4, 125.5, 128.8, 129.0, 129.1, 132.7, 134.1, 134.2, 138.6, 143.2, 143.4; MALDI-TOF-MS m/z (%): found 875.913 (100) [M+], calcd 875.365 (M = C60H36N4Zn); HR-ESI-MS m/z: [M+] calcd for M = C60H36N4Zn, 876.2231; found 876.2231; λabs(ε × 105) 461(2.8), 607, 652 nm; λem (λex = 461 nm) 658, 724 nm.
meso-Tetraphenyltetrabenzoporphyrinatocobalt(Co-TPBP)
Following a previously published procedure,35 a solution of TPBP (0.048 g, 0.059 mmol) in chloroform (45 mL) was reacted with a solution of Co(OAc)2·4H2O (0.073 g, 0.30 mmol) in methanol (5 mL) at room temperature for 4 h. After the mixture was washed with water, the organic phase was separated and dried over anhydrous Na2SO4. The solvent was removed and the resulting crude product was purified by column chromatography (silica gel, CH2Cl2/MeOH (99 : 1)) to afford Co-TPBP as a dark green solid (0.047 g, 89%). MALDI-TOF-MS m/z (%): found 870.594 (100) [M+], calcd 871.888 (M = C60H36N4Co); HR-ESI-MS m/z: [M+] calcd for M = C60H36N4Co, 871.2272; found 871.2273; λabs(ε × 105) 446(1.7), 595, 640(0.8) nm. Upon excitation at 446 nm, no emission peak was observed.
meso-Tetraphenyltetrabenzoporphyrinatotin (Sn-TPBP)
Following a previously published procedure,36 a solution of TPBP (0.102 g, 0.125 mmol) and SnCl2·2H2O (0.141 g, 0.625 mmol) in dimethylformamide (DMF, 5 mL) was treated with pyridine (0.05 mL) and refluxed for 4 h. A blue green precipitate was formed and collected by filtration. After that, the resulting crude product was purified by column chromatography (silica gel, CH2Cl2/MeOH (99 : 1)) to afford Sn-TPBP as a deep green solid (0.047 g, 90%). Due to incomplete purification of column chromatography, the achieved product was obtained with >90% purity, based on 1H-NMR spectroscopy. 1H-NMR: δH 7.23 (dd, J = 6.4, 3.2 Hz, 8H), 7.43 (dd, J = 6.4, 3.2 Hz, 8H) 7.90 (t, J = 7.6 Hz, 8H), 8.00 (t, J = 7.6 Hz 4H), 8.34 (d, J = 7.6 Hz, 8H); 13C-NMR: δC 116.5, 125.6, 127.2, 127.6, 129.7, 129.8, 133.9, 137.4, 137.6, 137.8, 141.3, 141.6, 141.7, 141.8, 142.0; MALDI-TOF-MS m/z (%): found 966.292 (100) [M+], calcd 965.679 (M = C60H38N4O2Sn); λabs(ε × 105) 430(0.3), 466(4.1), 612, 660(0.9) nm; λem (λex = 466 nm) 665, 745 nm.
Electrochemical studies
Background current
A background cyclic voltammogram was obtained in an anhydrous solution (10 mL) of 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) in DMF. A three-electrode one-compartment cell was used throughout the experiments. A glassy carbon electrode served as a working electrode while a Pt plate was used as a counter electrode. A silver wire coated with silver chloride (Ag/AgCl) was used as a quasi-reference electrode (QRE). The Ag/AgCl QRE was prepared using a procedure described elsewhere40 and externally calibrated with a ferrocene/ferrocenium redox couple using a potential of 0.72 V vs. a normal hydrogen electrode (NHE) as a reference value.41 The cyclic voltammograms were recorded at potentials ranging from 0.00 V to –2.00 V vs. a Ag/AgCl QRE at a scan rate of 50 mV s–1. The potential values were therefore reported with respect to the Ag/AgCl QRE. The solution was purged with nitrogen (N2) or CO2 for 20 min before each measurement with a flow rate of 0.2 L min–1. As a control experiment, constant-potential electrolysis of the electrolyte solution under CO2-saturated conditions in the absence of the catalyst for 20 h did not yield any CO2 reduction product.
Electrochemical reduction of CO2
The catalytic activity of the benzoporphyrins towards electrochemical reduction of CO2 was investigated by means of cyclic voltammetry and the constant-potential electrolysis using the above-mentioned electrochemical setup at potentials ranging from 0.00 to –2.00 V. Each cyclic voltammogram was collected from a 0.1 M TBAPF6 solution in anhydrous DMF containing 1.0 mM benzoporphyrin (10 mL) at a scan rate of 50 mV s–1 under N2- or CO2-saturated conditions. The solution was purged with N2 or CO2 for 20 min before each measurement with a flow rate of 0.2 L min–1. The constant-potential electrolysis of each benzoporphyrin was performed at ambient temperature using the above-mentioned electrochemical setup and at an applied potential of –1.90 V. After 20 h, a 2 mL gas sample from headspace gas (total volume was 10 mL) was taken from a reaction vial and analyzed by gas chromatography (GC) equipped with a thermal conductivity detector (TCD). Curves for CO obtained from the experiments were integrated to get the peak area, which was then used to calculate the amount of CO in the headspace using a pre-measured calibration curve.
Conflicts of interest
There are not conflicts to declare.
Acknowledgments
This research was partially supported by the Ratchadapiseksomphot Endowment Fund under Outstanding Research Performance Program, Chulalongkorn University (Sci-Super III-003), and Graduate School of Chulalongkorn University (The 90th Anniversary of Chulalongkorn University Fund, Ratchadaphiseksomphot Endowment Fund). Staff mobility was carried out with funding from the Grant for Join Funding, Ratchadapiseksomphot Endowment Fund. Electrochemical characterization of the materials and product analysis were done with funding from the Austrian Science Foundation (FWF) within the framework of the Wittgenstein Prize of Prof. Sariciftci (Solare Energie Umwandlung Z222-N19). E. P. is thankful for the financial support from the FWF project P29645-N36.
References
- Michele A., Carbon Dioxide as Chemical Feedstock, Wiley-VCH Verlag GmbH&Co., 2010. [Google Scholar]
- Collin J. P., Sauvage J. P. Coord. Chem. Rev. 1989;93:245–268. [Google Scholar]
- Khoshro H., Zare H. R., Gorji A., Namazian M., Jafari A. A., Vafazadeh R. J. Electroanal. Chem. 2014;732:117–121. [Google Scholar]
- Ghobadi K., Zare H. R., Khoshro H., Gorji A., Jafari A. A. C. R. Chim. 2018;21:14–18. [Google Scholar]
- Khoshro H., Zare H. R., Jafari A. A., Gorji A. Electrochem. Commun. 2015;51:69–71. [Google Scholar]
- Agnew D. W., Sampson M. D., Moore C. E., Rheingold A. L., Kubiak C. P., Figueroa J. S. Inorg. Chem. 2016;55:12400–12408. doi: 10.1021/acs.inorgchem.6b02299. [DOI] [PubMed] [Google Scholar]
- Cometto C., Kuriki R., Chen L., Maeda K., Lau T. C., Ishitani O., Robert M. J. Am. Chem. Soc. 2018;140:7437–7440. doi: 10.1021/jacs.8b04007. [DOI] [PubMed] [Google Scholar]
- MacHan C. W., Kubiak C. P. Dalton Trans. 2016;45:17179–17186. doi: 10.1039/c6dt03243e. [DOI] [PubMed] [Google Scholar]
- Robert M., Rao H., Bonin J. ChemSusChem. 2017;10:4447–4450. doi: 10.1002/cssc.201701467. [DOI] [PubMed] [Google Scholar]
- Rao H., Bonin J., Robert M. Chem. Commun. 2017;53:2830–2833. doi: 10.1039/c6cc09967j. [DOI] [PubMed] [Google Scholar]
- Reineke M. H., Porter T. M., Ostericher A. L., Kubiak C. P. Organometallics. 2018;37:448–453. [Google Scholar]
- Zhanaidarova A., Steger H., Reineke M. H., Kubiak C. P. Dalton Trans. 2017;46:12413–12416. doi: 10.1039/c7dt02620j. [DOI] [PubMed] [Google Scholar]
- Takeda H., Cometto C., Ishitani O., Robert M. ACS Catal. 2017;7:70–88. [Google Scholar]
- Morris A. J., Meyer G. J., Fujita E. Acc. Chem. Res. 2009;42:1983–1994. doi: 10.1021/ar9001679. [DOI] [PubMed] [Google Scholar]
- Kumar S., Wani M. Y., Arranja C. T., E Silva J. D. A., Avula B., Sobral A. J. F. N. J. Mater. Chem. A. 2015;3:19615–19637. [Google Scholar]
- Anderson H. L. Chem. Commun. 1999:2323–2330. [Google Scholar]
- Krebs F. C., Spanggaard H. Sol. Energy Mater. Sol. Cells. 2005;88:363–375. [Google Scholar]
- Lindsey J. S. Acc. Chem. Res. 2010;43:300–311. doi: 10.1021/ar900212t. [DOI] [PubMed] [Google Scholar]
- Kadish K. M., Progress in Inorganic Chemistry, Wiley-VCH Verlag GmbH&Co., 1986, vol. 34. [Google Scholar]
- Chen Y., Wang L. Polyhedron. 1993;12:1353–1360. [Google Scholar]
- Kadish K. M., Finikova O. S., Espinosa E., Gros C. P., De Stefano G., Cheprakov A. V., Beletskaya I. P., Guilard R. J. Porphyrins Phthalocyanines. 2004;08:1062–1066. [Google Scholar]
- Keawsongsaeng W., Gasiorowski J., Denk P., Oppelt K., Apaydin D. H., Rojanathanes R., Hingerl K., Scharber M., Sariciftci N. S., Thamyongkit P. Adv. Energy Mater. 2016;6:1–11. [Google Scholar]
- Zanotti G., Angelini N., Mattioli G., Paoletti A. M., Pennesi G., Rossi G., Caschera D., De Marco L., Gigli G. RSC Adv. 2016;6:5123–5133. doi: 10.1039/c8ra01651h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solonenko D., Gasiorowski J., Apaydin D., Oppelt K., Nuss M., Keawsongsaeng W., Salvan G., Hingerl K., Serdar Sariciftci N., Zahn D. R. T., Thamyongkit P. J. Phys. Chem. C. 2017;121:24397–24407. [Google Scholar]
- Ramírez G., Lucero M., Riquelme A., Villagrán M., Costamagna J., Trollund E., Aguirre M. J. J. Coord. Chem. 2004;57:249–255. [Google Scholar]
- Ramírez G., Ferraudi G., Chen Y. Y., Trollund E., Villagra D. Inorg. Chim. Acta. 2009;362:5–10. [Google Scholar]
- Ogura K., Yoshida I. J. Mol. Catal. 1988;47:51–57. [Google Scholar]
- Zheng G., Stradiotto M., Li L. J. Electroanal. Chem. 1998;453:79–88. [Google Scholar]
- Behar D., Dhanasekaran T., Neta P., Hosten C. M., Ejeh D., Hambright P., Fujita E. J. Phys. Chem. A. 1998;102:2870–2877. [Google Scholar]
- Sonoyama N., Kirii M., Sakata T. Electrochem. Commun. 1999;1:213–216. [Google Scholar]
- Maeda C., Miyazaki Y., Ema T. Catal. Sci. Technol. 2014;4:1482–1497. [Google Scholar]
- Finikova O. S., V Cheprakov A., Beletskaya I. P., Carroll P. J., Vinogradov S. A. J. Org. Chem. 2004;69:522–535. doi: 10.1021/jo0350054. [DOI] [PubMed] [Google Scholar]
- Vogler A., Kunkely H., Rethwisch B. Inorg. Chim. Acta. 1980;46:101–105. [Google Scholar]
- Strohmeier M., Orendt A. M., Facelli J. C., Solum M. S., Pugmire R. J., Parry R. W., Grant D. M. J. Am. Chem. Soc. 1997;7863:7114–7120. [Google Scholar]
- Lysenko A. B., Thamyongkit P., Schmidt I., Diers J. R., Bocian D. F., Lindsey J. S. J. Porphyrins Phthalocyanines. 2006;10:22–32. [Google Scholar]
- Wang S., Tabata I., Hisada K., Hori T. Dyes Pigm. 2002;55:27–33. [Google Scholar]
- Manbeck G. F., Fujita E. J. Porphyrins Phthalocyanines. 2015;19:45–64. [Google Scholar]
- Fang Y., Senge M. O., Van Caemelbecke E., Smith K. M., Medforth C. J., Zhang M., Kadish K. M. Inorg. Chem. 2014;53:10772–10778. doi: 10.1021/ic502162p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolzenberg A. M., Schussel L. J. Inorg. Chem. 1991;30:3205–3213. [Google Scholar]
- Hassel A. W., Fushimi K., Seo M. Electrochem. Commun. 1999;1:180–183. [Google Scholar]
- Zoski C. G., Handbook of Electrochemistry, Elsevier B.V., 1st edn, 2007. [Google Scholar]