

Abstract
In this study, a new series of N-alkyl-3,6-dibromocarbazole and N-alkyl-5-bromoindole derivatives have been synthesized and evaluated in vitro as anti-cancer and anti-migration agents. Cytotoxic and anti-migratory effects of these compounds were evaluated in MCF-7 and MDA-MB-231 breast cancer cell lines and an insight on the structure-activity relationship was developed. Preliminary investigations of their anti-cancer activity demonstrated that several compounds have moderate antiproliferative effects on cancer cell lines with GI50 values in the range of 4.7–32.2 µM. Moreover, carbazole derivatives 10, 14, 15, 23, and 24 inhibit migration activity of metastatic cell line MDA-MB-231 in the range of 18–20%. The effect of compounds 10, 14, and 15 in extension of invadopodia and filopodia was evaluated by fluorescence microscopy and results demonstrated a reduction in actin-based cell extensions by compounds 10 and 15.
Keywords: 3,6-dibromocarbazole; 5-bromoindole; carbazole; actin; breast cancer; migration
1. Introduction
In women, breast cancer is the leading cause of death, mainly due to metastasis [1]. If breast cancer is detected and treated prior to metastasis, the patient has a higher probability of being cured of their disease. Cancer cell invasion involves cell migration through the extracellular matrix (ECM) and the accompanying degradation of the ECM [2]. Several proteins play a key role in this process, by the extension of structures known as invadopodia. Invadopodia are actin-rich protrusive structures with associated matrix degradation activity and are believed to be important for tumor cells to penetrate the basement membrane of epithelia and blood vessels [3]. In cell migration, the reorganization of the actin cytoskeleton produces the force necessary for cell migration [4]. The Rho GTPases, Rac, and Cdc42 are key molecular switches activated by a myriad of cell surface receptors to promote breast cancer cell migration/invasion, proliferation, and survival [5]. Unlike Ras, Rac and Cdc42 are not mutated in breast cancer, but activated via the deregulation of expression and/or activity of their upstream regulators, guanine nucleotide exchange factors (GEFs) [6]. The WASP family proteins are key regulators of the actin cytoskeleton and cell migration through induction of membrane protrusions at the leading edge [7]. In cancer cells, when N-WASP interacts and activates the Arp2/3 complex, it catalyzes actin polymerization and assembly into filopodia and invadopodia [7,8]. To initiate this process, the Rho GTPase Cdc42, in its GTP-activated form, binds and activates N-WASP by inducing a conformational change that liberates the autoinhibited structure, thereby interacting with Arp2/3 complex, and regulating the protrusive formation in membrane structures promoting extracellular matrix (ECM) degradation [8]. Therefore, inhibition of these processes decreases cell motility and invasion, and may greatly improve the potential therapeutic applications of such inhibitors against cancer metastasis.
Natural and synthetic carbazole derivatives comprise a wide variety of biologically active agents with diverse pharmacological activities, including antitumoral, antioxidant, anti-inflammatory, antibacterial, anticonvulsant, antipsychotic, antidiabetic, and larvicidal properties [9,10,11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28]. Carbazoles are tricyclic aromatic compounds with a benzene ring fused to the 2,3-positions of an indole ring [29]. The antitumor properties of carbazole derivatives have been correlated to their polycyclic, planar aromatic structure, and large π-conjugated backbone that noncovalently bind with DNA base pairs, hydrophobic pockets, and forms electrostatic interactions to intercalate into DNA [9,30]. In particular, among a wide variety of carbazoles, a series of N-alkyl-3,6-disubstituted carbazole derivatives has been discovered and evaluated for their potential as neuroprotective agents [31], antimalarial [32], antitumoral [33,34], anti-apoptotic [35], and antibacterial activities [36]. Selected examples of bioactive N-alkyl-3,6-disubstituted carbazole derivatives are represented in Figure 1.
Figure 1.

Structure of representative N-alkyl-3,6-dihalogencarbazole derivatives.
The compound P7C3 (Figure 1) was discovered from a library of 200,000 drug-like molecules, and showed proneurogenic and neuroprotective properties, stabilized mitochondrial membrane potential, and inhibits neuronal apoptosis [37]. Several derivatives of P7C3 have been synthesized with modifications at the linker chain. Replacement of the hydroxyl group with a fluorine atom, and an additional methylene group between the hydroxyl group and the aniline, increases activity [37]. The aromatic ring was replaced with heteroaromatic groups, but activity was found to be less effective. The commercially available TDR30137 (Figure 1) was discovered and characterized as an inhibitor of P. falciparum K1 (Pf-K1) in human red blood cells with an IC50 of 57 nM [32]. However, TDR30137 was not active in in vivo studies with the Plasmodium berghei mouse model. In structure-activity relationship (SAR) studies, the importance of 3,6-halogen substitution, hydroxyl group, and the tertiary amine correlated with improved activity on Pf-K1 strains [32]. A carbazole derivative named Wiskostatin (Figure 1) was identified to bind within a pocket in the GBD regulatory module that maintains N-WASP in an inactive, autoinhibited conformation [38]. In a pyrene-actin polymerization assay, using purified proteins, it was demonstrated that Wiskostatin inhibited full-length N-WASP activation of the Arp2/3 complex at IC50 = 10 μM [38]. The specific binding site of Wiskostatin was determined to be within the GBD of the autoinhibited conformation of N-WASP after performing the experiment with activated Cdc42-GTP. Unfortunately, a recent report described that Wiskostatin inhibited other cellular functions that are not believed to be N-WASP dependent [39]. These studies revealed that Wiskostatin caused an irreversible decrease in cellular ATP levels, and that it does not function as a selective inhibitor of N-WASP dependent functions in intact cells, and caused an overall change in the energy status of cells; thus, inhibiting normal transport processes [39]. Herein, we designed and synthesized a new series of N-alkyl-3,6-dibromocarbazole and 5-bromoindole derivatives, tested for their antiproliferative and antimigratory activities in MCF-7 and MDA-MB-231 breast cancer cell lines, and analyzed the effect of the most active migration inhibitor on actin dynamics and actin cytoskeleton rearrangement.
2. Results and Discussion
The aim of this study is to design and synthesize a new series of N-alkyl-3,6-dibromocarbazole and N-alkyl-5-bromoindole derivatives and analyze their cytotoxic effect and potential to inhibit actin cytoskeleton rearrangement and cancer cell migration. The structural elements of Wiskostatin and derivatives identified as pharmacophoric unit are a 3,6-dihalogen carbazole and a dialkylamino-2-propanol chain. Our strategy was to design and synthesize a new series of compounds with a 3,6-dibromocarbazole or 5-bromoindole ring connected, via a three-carbon atom aliphatic chain, to an amide group. The influence of different N-alkyl or aromatic substituents at the amide group was examined.
We screened all compounds to determine their cytotoxic effect against MCF-7 and MDA-MB-231 breast cancer cells using the Sulforhodamine B (SRB) assay [40] (Figure 2). In addition, anti-migratory activity was determined using the wound healing assay (scratch method) [41] on the metastatic MDA-MB-231 cancer cells. In this assay, the relative migration of MDA-MB-231 breast cancer cells in the presence of carbazole or indole derivatives at a concentration of 10 µM (or at concentrations that do not affect cell viability) was compared to the migration in the presence of vehicle (0.02% DMSO). Representative photomicrographs of the migration inhibition of compounds 10, 14, 15, and Wiskostatin are represented in Figure 3. Results show that in the vehicle-treated control experiment, wound healing is progressing considerably, and after 24 h, the wound is completely healed. When cells are incubated with compounds 10, 14, and 15 after 24 h, the wound healing is inhibited. However, Wiskostatin did not elicit an inhibitory effect on wound healing when incubated with MDA-MB-231 cells after 24 h at a concentration of 2 µM. Actin and Arp2/3 regulation by active WASP induce de novo actin polymerization and assembly to generate the F-actin structures filopodia and invadopodia used for cell migration [42]. Therefore, to investigate the effect of compounds 10, 14, 15, and Wiskostatin on actin dynamics, we performed immunofluorescence microscopy to detect polymerized actin on MDA-MB-231 cancer cells (Figure 4). The structure and biological activities of new compounds are summarized in Table 1 and Table 2.
Figure 2.

Log-dose response curve for compounds 7, 8, 10, 18, 21, and Wiskostatin on MCF-7 and MDA-MB-231 breast cancer cell lines. Each data point represents the mean of three replicates and error bars represent ± SD. Each GI50 value was calculated based on sigmoidal curve fitting to the respective data set.
Figure 3.

Inhibitory effect of compounds 10, 14, 15, and Wiskostatin on MDA-MB-231 cells migration detected by wound-healing assay. MDA-MB-231 cells were treated with vehicle or with compounds 10, 14, 15, and Wiskostatin. The photomicrographs were obtained at 0 and 24 h. Percent relative migration values are the average of three independent experiments. Dotted lines show the area occupied by the initial scraping for 0 h, and the wound edge for 24 h.
Figure 4.

Effect of Wiskostatin and compounds 10, 14, and 15 on actin cytoskeleton of metastatic cancer cell MDA-MB-231. MDA-MB-231 metastatic breast cancer cells were treated with vehicle or Wiskostatin (2 µM) and compounds 10, 14, or 15 at 10 µM for 24 h to determine changes in actin cytoskeletal structures. Cells were fixed, permeabilized, and stained with rhodamine phalloidin to visualize F-actin. Arrows, lamellipodia; arrowheads, filopodia.
Table 1.
Cell growth inhibition and anti-migration activity of 3,6-dibromocarbazole derivatives.
| Comp. |
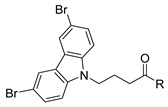 R = |
GI50 (µM)a | Migration (%)b,c | |
|---|---|---|---|---|
| MCF-7 | MDA-MB-231 | |||
| 4 |
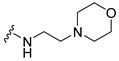
|
>50 | >50 | 99 ± 6.03 |
| 5 |
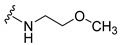
|
>50 | >50 | 99 ± 5.98 |
| 6 |
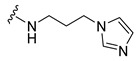
|
16.8 | 16 | 94 ± 3.93 |
| 7 |
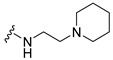
|
6.8 | 10 | 99 ± 0.03 (at 2 μM) |
| 8 |
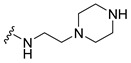
|
8 | 4.7 | 97 ± 4.90 (at 1 μM) |
| 9 |
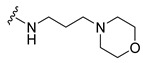
|
13.4 | 15.4 | 99 ± 1.58 (at 3.1 μM) |
| 10 |
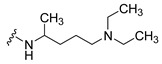
|
8.1 | 10.5 | 87 ± 4.65 (at 2.1 μM) |
| 11 |
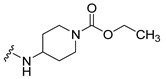
|
>50 | >50 | 99 ± 0.02 |
| 12 |
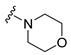
|
>50 | >50 | 97 ± 6.41 |
| 13 |
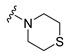
|
>50 | 25 | 99 ± 0.03 |
| 14 |
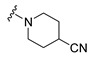
|
11.8 | 16.7 | 81 ± 8.96 |
| 15 |
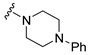
|
>50 | >50 | 80 ± 5.87 |
| 16 |
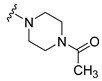
|
18.2 | 23 | 82 ± 7.52 |
| 17 |
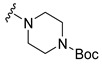
|
17.5 | >50 | 99 ± 2.09 |
| 18 |
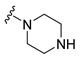
|
7.5 | 6.7 | 99 ± 0.02 |
| 19 |
|
12.4 | >50 | 99 ± 0.97 |
| 20 |
|
9.1 | 13.4 | 99 ± 0.05 (at 2.7 μM) |
| 21 |
|
6.5 | 8 | 99 ± 0.48 (at 1.6 μM) |
| 22 |
|
>50 | >50 | 97 ± 4.19 |
| 23 |
|
>50 | 19 | 90 ± 6.60 |
| 24 |
|
32.2 | >50 | 82 ± 5.19 |
| 25 |
|
>50 | >50 | 96 ± 4.69 |
| 26 |
|
>50 | >50 | 99 ± 6.52 |
| 27 |
|
>50 | >50 | 99 ± 0.05 |
| Wiskostatin | 9.7 | 8.3 | 95 ± 6.79 (at 2 μM) |
|
a GI50 = compound concentration required to inhibit MDA-MB-231 proliferation by 50% after 48 h treatment. Values are expressed as the mean of triplicate experiments, and standard deviation (SD) is <10%. b After 24 h, MDA-MB-231 cellular migration was determined by measuring the distance traveled from the edge of the scratch toward the center of the scratch, relative to control. c Percent relative migration values at 10 µM (or at concentrations that do not affect cell viability). Results are presented as means ± SD of three independent experiments.
Table 2.
Cell growth inhibition and anti-migration activity of 5-bromoindole derivatives.
| Comp. |
 R = |
GI50 (µM)a | Migration (%)b,c | |
|---|---|---|---|---|
| MCF-7 | MDA-MB-231 | |||
| 31 |
|
>50 | >50 | 99 ± 0.05 |
| 32 |
|
>50 | >50 | 99 ± 7.78 |
| 33 |
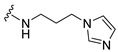
|
>50 | >50 | 99 ± 0.02 |
| 34 |
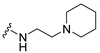
|
18.4 | >50 | 99 ± 6.60 |
a GI50 = compound concentration required to inhibit MDA-MB-231 proliferation by 50% after 48 h treatment. Values are expressed as the mean of triplicate experiments, and standard deviation (SD) is <10%. b After 24 h, MDA-MB-231 cellular migration was determined by measuring the distance traveled from the edge of the scratch toward the center of the scratch, relative to control. c Percent relative migration values at 10 µM. Results are presented as means ± SD of three independent experiments.
2.1. N-Alkyl-3,6-Dibromocarbazole Derivatives
The synthetic method to construct the 3,6-dibromocarbazole derivatives library is described in Scheme 1 (see the Supplementary Materials for representative 1H and 13C NMR spectral data). The N-alkyl-3,6-dibromocarbazole derivatives were generated in a three-step synthesis using 3,6-dibromocarbazole 1 as origin of the carbazole derivatives core. Compound 1 was reacted with ethyl 4-bromobutyrate to introduce the aliphatic side chain by nucleophilic substitution (Scheme 1), followed by hydrolysis to afford the corresponding 3,6-diromocarbazole-4-butyric acid 2. For the generation of 3,6-dibromocarbazole-4-butyramide derivatives 3, compound 2 was therefore used as starting material, which reacted with different amines via an amide coupling reaction using N-(3-Dimethylaminopropyl)-N’-ethylcarbonate (EDAC) with Hydroxybenzotriazole (HOBt) as an additive dissolved in methylene chloride (CH2Cl2).
Scheme 1.

General synthetic procedure of 3,6-dibromocarbazole-4-butyramide derivatives 3. Reagents and conditions: (a) (i) Ethyl 4-bromobutyrate, K2CO3, DMF, 80 °C, 2h; (ii) KOH, DMF/water, 80 °C, 2-6 h, 86%; (b) HOBt, EDAC, CH2Cl2, Et3N, rt, amine: R-NH2 or HNR1R2, 2-8 h.
The in vitro anti-proliferative and anti-migratory activities of compounds 4–27, and Wiskostatin are represented in Table 1. From the twenty-four compound derivatives of 3,6-dibromocarbazole, it can be observed that in the MCF-7 (ER+) cancer cell line, compounds 6–10, 14, 16–21, and 32 showed good to moderate antiproliferative activity with a GI50 in the range of 6.8–32.2 µM. In the MDA-MB-231 cell line, compounds 6–10, 14, 16, 18, 20–21, and 23 inhibited cell proliferation with a GI50 in the range of 4.7–23 µM. The remaining compounds in that series had a GI50 above 50 µM in both breast cancer cell lines. Compound 8 with a 2-piperazinyl ethyl butyramide chain showed very good anticancer activity against both cancer cell lines MCF-7 and MDA-MB-231 with GI50 values of 8 and 4.7 µM, respectively. Also, compound 18 with a piperazinyl amide showed significant anticancer activity against both cancer cell lines with GI50 values of 7.5 and 6.7 µM, respectively. Similarly, compound 21, another piperazinyl amide derivative, showed very good in vitro anticancer activity against both cancer cell lines with GI50 values of 6.5 and 8 µM, respectively. Shortening the aliphatic chain between the N-atom of the amide group and the morpholine from C3 (9), over C2 (4), to C0 (12) led to a complete loss of antiproliferative activity on both cancer cell lines. On the other hand, introduction of an aromatic ring or aromatic heterocycle (22–27, Table 1) in the amide group led to lack of antiproliferative activity with GI50 above 50 µM on both cancer cell lines. Thus, in general, compounds with a piperazinyl butyramide group attached to the N-atom of the carbazole appear to be more cytotoxic than compounds with other butyramide group in this series of compounds. In addition, three compounds—8, 18, and 21—were found to be more cytotoxic against both cancer cell lines MCF-7 and MDA-MB-231 than Wiskostatin, which in this assay showed GI50 values of 9.7 and 8.3 µM, respectively (Table 1, Figure 2).
To further assess the anti-migratory activity of carbazole derivatives in vitro, we examined its inhibitory effects on the migration of the metastatic breast cancer cell line MDA-MB-231 using the wound-healing assay at concentrations that do not affect cell viability. We chose the MDA-MB-231 breast cancer cells over MCF-7 due to its enhanced metastatic and migratory properties, with concomitant Rac and Cdc42 expression, compared to the non-metastatic and poorly migrating MCF-7 cells. The relative migration of treated cells with 3,6-dibromocarbazole derivatives compared with control (MDA-MB-231 cells) are summarized in Table 1.
Among the twenty-four compounds tested for their anti-migratory effect, five compounds (10, 14, 15, 23, 24) inhibited migration in the range of 10–20%. While compounds 7, 8, 18, and 21 were among the most cytotoxic compounds tested against MDA-MB-231 and MCF-7 cancer cell lines, they did not show significant anti-migratory effect. In contrast, compounds 14–16 and 24 inhibited migration in the range of 18–20%, compared to Wiskostatin that inhibit 5% of migration at 2 µM. Compound 14, a carbazole derivative with a piperidine-4-carbonitrile amide group that showed moderate anti-proliferative activity on both MCF-7 and MDA-MB-231 cell lines, inhibit 19% of migration. Both compounds 15 and 16, with phenyl- and acetyl-piperazine amide group, respectively, showed comparable anti-migratory activity with 20% and 18%, respectively. The anti-proliferative effect of 16 on both cancer cell lines was moderate, compared to 15, which lacks cytotoxic activity. On the other hand, the 3-aminoquinoline amide 24, showed anti-migratory activity of 18% with GI50 above 50 µM on MDA-MB-231 cell line. When comparing Wiskostatin and carbazole derivatives from Table 1, compound 10 exhibited anti-proliferative activity with GI50 values comparable with Wiskostatin on both cancer cell lines. However, compound 10 showed higher anti-migratory potency with 13% at 2.1 µM, compared with 5% anti-migratory effect of Wiskostatin at 2 µM on MDA-MB-231 breast cancer cells. Therefore, compound 10 exhibits similar cytotoxicity with improved anti-migratory potential compared to Wiskostatin.
The Rho GTPases are believed to stimulate plasma membrane protrusion by inducing actin filament nucleation and polymerization on or close to membranes [43]. In particular, the Rho GTPase protein Cdc42, through activation of N-WASP/Arp2/3 pathway, is an important mediator of actin polymerization and filopodium extension [44,45]. Therefore, compounds that interfere with this process might be potentially useful molecular probes for the study of cell migration and invasion. To determine changes in actin cytoskeletal structures, we treated MDA-MB-231 breast cancer cells with vehicle or compounds 10, 14, 15, and Wiskostatin at 10 µM for 24 h (Figure 4).
To identify F-actin based cell surface extensions, cells were stained with rhodamine phalloidin to localize F-actin. The results showed that untreated cells (control) demonstrated a strong formation of lamellipodia extensions, membrane ruffles, and stress fibers, with few filopodia. Wiskostatin and compounds 14 and 15 moderately reduced lamellipodia extensions when compared with control cells at concentrations that do not affect cell viability. In addition, compound 10, at concentrations that inhibit 13% cell migration (2 µM), exhibited a marked reduction in lamellipodia formation when compared to vehicle, and in particular with Wiskostatin, which inhibits cell migration in 5% at 2 µM (Table 1). Compound 15 demonstrates a marked reduction in polymerized actin indicating inhibition of Arp2/3 mediated actin nucleation. Specific inhibition of filopodia, as would be predicted by WASP inhibition, could not be determined since the vehicle control cells exhibited more invadopodia and lamellipodia than filopodia. In general, these results suggest that new N-alkyl-3,6-dibromocarbazoles and compounds derivatives of Wiskostatin can be explored as new probes to study actin dynamics in cancer cells, or to further develop new anti-cancer and anti-metastatic drugs.
2.2. N-alkyl-5-Bromoindole Derivatives
To further explore the N-alkyl-5-bromoindole butyramide derivative series, we synthesized several compounds in which the carbazole core was replaced by a 5-bromoindole ring (Scheme 2). The strategy is to analyze the effect of using a smaller ring system as a core connected to the C3 linker side chain and the amide group. The synthesis of N-alkyl-5-bromoindole derivatives is described in Scheme 2. The 5-bromoindole 28 was reacted with ethyl 4-bromobutyrate, followed by hydrolysis to yield the corresponding 5-bromoindole-4-butyric acid 29. Since N-alkylation in indoles is more difficult than in carbazoles, the reaction rate to obtain 29 was improved by using Cs2CO3 as a base over K2CO3, where Cs2CO3 solubility is ten times higher in organic solvents than K2CO3. For the generation of 5-bromoindole-4-butyramide derivative 30, carboxylic acid 29 was therefore used as starting material, which reacted with different amines via an amide coupling reaction using reaction conditions similar as described in Scheme 1 (see the Supplementary Materials for representative 1H and 13C NMR spectral data).
Scheme 2.

General synthetic procedure of 5-bromoindole-4-butyramide derivative 30. Reagents and conditions: (a) (i) Ethyl 4-bromobutyrate, Cs2CO3, DMF, 100 °C, 16 h; (ii) KOH, DMF/water, 80 °C, 2-6 h, 80%; (b) HOBt, EDAC, CH2Cl2, Et3N, rt, amine: R-NH2 or HNR1R2, 2-8 h.
The in vitro anti-proliferative and anti-migratory activities of compounds 31–34 are represented in Table 2. In this assay, the replacement of the rigid carbazole ring core by an indole resulted in loss of cytotoxic activity on both MCF-7 and MDA-MB-231 cell lines. For example, from the four representative 5-bromoindole derivatives synthesized, it can be observed that only compound 34 showed moderate cytotoxic effect on MCF-7 cells with GI50 of 18.4 µM, while GI50 value on MDA-MB-231 breast cancer cells was above 50 µM. Furthermore, the remaining compounds in that series had GI50s above 50 µM in both breast cancer cell lines. Additionally, after 24 h treatment at 10 µM using the wound-healing assay in the MDA-MB-231 cell line, no migration inhibition could be observed. Hence, the absence of any activity of 5-bromoindole derivative series, together with the fact that several carbazole derivatives show promising activity, a structure-activity relationship (SAR) can be established. For example, it appears that the presence of a carbazole ring core, the C3 linker region, and amide group improve both anti-proliferative and anti-migratory activity.
3. Materials and Methods
3.1. General Methods
All experiments were carried out in pre-dried glassware (≥1 h, 80–90 °C) under a nitrogen atmosphere. Nuclear magnetic resonance (NMR) spectra were obtained using a 400 MHz Bruker Avance UltraShield™ spectrometer. 1H (400 MHz) and 13C (100 MHz) NMR were recorded in CDCl3 or DMSO-d6, unless otherwise used, and the chemical shift was expressed in parts per million (ppm) relative to CDCl3 (δ 7.26 for 1H and δ 77.0 for 13C) or DMSO-d6 (δ 2.50 for 1H and δ 39.5 for 13C) as the internal standard. 1H NMR data is reported as position (δ), relative integral, multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; dt, doublet of triplets; dd, doublet of doublets; dq, doublet of quartets; m, multiplet; br, broad peak), coupling constant (J) in hertz (Hz), and the assignment of the atom. The shift in ppm for multiplets correspond to the centermost value of the entire splitting pattern. 13C NMR data are reported as position (δ) and assignment of the atom. Microwave reactions were conducted in a CEM Discovery Microwave for Drug Discovery, SP- 1445. High resolution electrospray ionization mass spectrometry (ESI-HRMS) data were obtained on a Thermo Scientific™ Q Exactive™ Hybrid Quadrupole-Orbitrap Mass Spectrometer with high performance liquid chromatography (HPLC) Agilent 1200 utilizing a Zorbax SB-C18 column (2.1 mm × 50 mm, 1.8µm) at 40 °C, and a mobile phase of acetonitrile containing 5% Milli Q water at a flow rate of 0.35 mL/min (run time: 6.5 min), and 1 µL injection volume.
3.2. Synthesis Methods
Progress of the reaction was monitored via TLC analysis using general purpose silica gel on glass 5 × 20 cm with UV indicator, 250 μm, 60 Å medium pore diameter, UV indicator, and visualized by UV fluorescent Spectroline E Series Ultraviolet lamps, in most cases followed by staining with I2. The compounds were purified via column chromatography over silica gel (70–230 mesh, 60 Å) with the appropriate size column (24/40, 12 in. × 0.5 in.) or (24/40, 12 in. × 0.72 in.). Wiskostatin compound was obtained from MilliporeSigma.
3.2.1. General Procedure for the Synthesis of 4-(3,6-Dibromo-Carbazol-9-yl)-Butyric Acid (2)
A 50 mL three-neck round-bottom flask, equipped with a reflux condenser, was charged with 3,6-dibromocarbazole 1 (0.325 g, 1.0 mmol), K2CO3 (0.1382 g, 1 mmol), and ethyl 4-bromobutyrate 2 (0.4436 mL, 3.1 mmol), dissolved in DMF (5 mL). After 15 min of stirring at room temperature, the reaction mixture was refluxed at 80 °C for 2 h. After completion of the reaction (analyzed by TLC), water (1 mL) and KOH (1.0 mmol) was added and the reaction mixture, refluxed at 80 °C for 2 h. After the reaction was completed (analyzed by TLC), the mixture was allowed to reach room temperature. The mixture was washed with water (20 mL) and the product was extracted using dichloromethane (3 × 10 mL). The organic layer was washed with brine and dried with Na2SO4, and filtered and concentrated under reduced pressure. The crude oil was purified via column chromatography over silica gel and 50% ethyl acetate in hexane, and the product obtained as a white solid for the precursor 4-(3,6-Dibromo-carbazol-9-yl)-butyric acid 2 (0.3535 g, 0.86 mmol, 86%). TLC analysis in ethyl acetate-hexane (1:1), Rf = 0.19. 1H NMR (400 MHz, CDCl3) δ 1.98 (2H, m), 2.27 (2H, t, J = 7.2 Hz), 4.41 (2H, t, J = 7.2 Hz), 7.30 (2H, d, J = 8.9 Hz), 7.56 (2H, dd, J = 2.0, 8.8 Hz), 8.14 (1H, d, J = 2.0 Hz); 13C NMR (100 MHz, CDCl3) 24.2, 31.1, 42.2, 111.8, 112.0, 123.4, 123.9, 139.5, 174.3. HR-FTMS (ESI) m/z calcd. for C16H13Br2NO2, [M + H]+ 411.9365, found 411.9365.
3.2.2. Synthesis of 3,6-Dibromocarbazole-4-butyramide Derivatives (4–27)
3.2.3. General Procedure for the Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-N-(2-Morpholin-4-Ylethyl)Butyramide (4), and for Compounds 5–27
A 50 mL three-neck round-bottom flask was charged with 4-(3,6-Dibromo-carbazol-9-yl)-butyric acid 2 (0.4111 g, 1.0 mmol), HOBT (0.2027 g, 1.5 mmol), and EDAC (0.2876 g, 1.5 mmol). The mixture was dissolved in CH2Cl2 (10 mL), stirred for 30 min, and 2-(4-morpholinyl)ethanamine 2 (0.133 g, 1.0 mmol) was added. After 15 min, Et3N (0.43 mL, 3.0 mmol) was added and the mixture was stirred at room temperature for 16 h. After completion of the reaction (analyzed by TLC), water was added (30 mL) and the product was extracted using dichloromethane (3 × 10 mL). The organic layer was washed with brine and dried with Na2SO4, and filtered and concentrated under reduced pressure. The crude oil was purified via column chromatography over silica gel and 10% methanol in dichloromethane, and the product obtained as a white solid (0.382 g, 0.73 mmol, 73%). TLC analysis in CH2Cl2-MeOH (9:1), Rf = 0.26. 1H NMR (400 MHz, CDCl3) δ 2.0 (2H, m), 2.22 (2H, t, J = 6.8 Hz), 2.35 (2H, t, J = 4 Hz), 2.50 (4H, t, J = 1.6 Hz), 3.19 (2H, m), 3.56 (4H, t, J = 4.4 Hz), 4.39 (2H, t, J = 6.8 Hz), 7.60 (2H, d, J = 1.6 Hz), 8.0 (2H, bs), 8.46 (2H, bs); 13C NMR (100 MHz, CDCl3) 24.0, 31.2, 34.3, 41.8, 53.15, 53.2, 54.9, 57.2, 66.1, 111.3, 111.5, 122.9, 123.4, 128.8, 139.0, 161.0. HR-FTMS (ESI) m/z calcd. for C22H25Br2N3O2, [M + H]+ 524.0366, found 524.0367.
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-N-(2-methoxyethyl)butyramide (5)
The crude oil was purified via column chromatography over silica gel and 10% methanol in dichloromethane, and the product obtained as a white solid (0.096 g, 0.21 mmol, 63%). TLC analysis in CH2Cl2-MeOH (9:1), Rf = 0.56. 1H NMR (400 MHz, CDCl3) δ 1.63 (2H, bs), 2.15 (2H, t, J = 6.0 Hz), 2.21 (2H, m), 3.63 (3H, s), 3.46 (2H, d, J = 2.0 Hz), 4.39 (2H, t, J = 6.4 Hz), 7.35 (2H, d, J = 8.8 Hz), 7.57 (2H, dd, J = 1.6, 8.8 Hz), 8.16 (2H, d, J = 1.6 Hz); 13C NMR (100 MHz, CDCl3) 24.1, 32.4, 39.2, 42.3, 58.7, 71.0, 110.5, 112.1, 123.2, 123.4, 129.1, 139.2, 171.6. HR-FTMS (ESI) m/z calcd. for C19H20Br2N2O2, [M + H]+ 468.9944, found 468.9947.
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-N-(3-imidazol-1-yl-propyl)butyramide (6)
The crude oil was purified via column chromatography over silica gel and 10% methanol in dichloromethane, and the product obtained as a white solid (0.078 g, 0.15 mmol, 49%). TLC analysis in CH2Cl2-MeOH (9:1), Rf = 0.22. 1H NMR (400 MHz, CDCl3) δ 1.96 (2H, t, J = 6.8 Hz), 2.07 (2H, t, J = 6.4 Hz), 2.20 (2H, m), 3.23 (2H, q, J = 6.4 Hz), 3.98 (2H, t, J = 6.8 Hz), 4.38 (2H, t, J = 6.8 Hz), 6.93 (1H, bs), 7.05 (1H, Bs), 7.33 (2H, d, J = 8.4 Hz), 7.51 (1H, bs), 7.56 (2H, dd, J = 2.0, 8.8 Hz), 8.15 (2H, d, J = 1.6 Hz); 13C NMR (100 MHz, CDCl3) 24.0, 31.0, 32.3, 36.9, 42.2, 44.8, 110.5, 112.2, 112.3, 123.3, 123.4, 129.1, 129.3, 129.4, 139.3, 171.9. HR-FTMS (ESI) m/z calcd. for C22H22Br2N4O, [M + H]+ 519.0213, found 519.0211.
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-N-(2-piperidin-1-ylethyl)butyramide (7)
The crude oil was purified via column chromatography over silica gel and 10% methanol in dichloromethane, and the product obtained as a white solid (0.1033 g, 0.2 mmol, 59%). TLC analysis in CH2Cl2-MeOH (9:1), Rf = 0.47. 1H NMR (400 MHz, CDCl3) δ 1.43 (2H, q, J = 4. 9 Hz), 1.52 (4H, m), 2.11 (4H, dt, J = 3.3, 11.7 Hz), 2.34 (3H, bs), 2.38 (3H, t, J = 6.0 Hz), 3.31 (2H, q, J = 5.5 Hz), 4.29 (2H, t, J = 6.4 Hz), 6.05 (1H, bs), 7.28 (2H, d, J = 8.7 Hz), 7.5 (2H, dd, J = 1.9, 8.7 Hz), 8.06 (2H, d, J = 1.9 Hz); 13C NMR (100 MHz, CDCl3) 24.0, 24.2, 32.2, 35.9, 42.2, 54.2, 57.0, 110.5, 112.0, 123.1, 123.2, 128.9, 139.2, 171.4. HR-FTMS (ESI) m/z calcd. for C23H27Br2N3O, [M + H]+ 522.0573, found 522.0572.
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-N-(2-piperazin-1-ylethyl)butyramide (8)
The crude oil was purified via column chromatography over silica gel and 10% methanol in dichloromethane, and the product obtained as a white solid (0.1425 g, 0.27 mmol, 71%). TLC analysis in CH2Cl2-MeOH (9:1), Rf = 0.23. 1H NMR (400 MHz, CDCl3) δ 1.9 (1H, bs), 2.11 (2H, t, J = 6.4 Hz), 2.20 (2H, m), 2.38 (3H, bs), 2.42 (4H, t, J = 5.9 Hz), 2.84 (4H, t, J = 4.7 Hz), 3.32 (2H, q, J = 5.4 Hz), 4.38 (2H, t, J = 6.7 Hz), 7.33 (2H, d, J = 8.7 Hz), 7.54 (2H, dd, J = 1.9, 10.6 Hz), 8.14 (2H, s); 13C NMR (100 MHz, CDCl3) 24.0, 32.2, 35.6, 42.4, 45.9, 54.1, 57.0, 110.6, 112.1, 123.2, 123.5, 139.4, 171.4. HR-FTMS (ESI) m/z calcd. for C23H27Br2N3O, [M + H]+ 523.0526, found 523.0526.
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-N-(3-morpholin-4-ylpropyl)butyramide (9)
The crude oil was purified via column chromatography over silica gel and 10% methanol in dichloromethane, and the product obtained as a white solid (0.1496 g, 0.28 mmol, 69%). TLC analysis in CH2Cl2-MeOH (9:1), Rf = 0.59. 1H NMR (400 MHz, CDCl3) δ 1.56 (2H, m, J = 6.0 Hz), 1.95 (2H, t, J = 6.8 Hz), 2.14 (4H, m), 2.23 (2H, bs), 2.36 (2H, t, J = 6.4 Hz), 3.25 (2H, q, J = 5.6 Hz), 3.42 (1H, bs), 4.31 (2H, t, J = 6.8 Hz), 7.31 (2H, d, J = 8.8 Hz), 7.54 (2H, dd, J = 2.0, 8.8 Hz), 8.12 (2H, d, J = 1.6 Hz); 13C NMR (100 MHz, CDCl3) 24.0, 24.4, 32.3, 39.6, 42.1, 53.5, 58.0, 66.8, 110.6, 112.1, 123.2, 123.4, 129.1, 139.3, 171.1. HR-FTMS (ESI) m/z calcd. for C23H27Br2N3O2, [M + H]+ 538.0522, found 538.0521.
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-N-(4-diethylamino-1-methylbutyl)butyramide (10)
The crude oil was purified via column chromatography over silica gel and 10% methanol in dichloromethane, and the product obtained as a white solid (0.0063 g, 0.040 mmol, 9.1%). TLC analysis in CH2Cl2-MeOH (9:1), Rf = 0.21. 1H NMR (400 MHz, CDCl3) δ 0.89 (2H, m), 1.19 (6H, t, J = 7.6 Hz), 1.64 (2H, m), 2.20 (3H, bs), 2.25 (2H, t, J = 6.8 Hz), 2.62 (2H, t, J = 6.4 Hz), 3.29 (4H, m), 3.60 (2H, t, J = 7.20 Hz), 4.37 (2H, t, J = 7.20 Hz), 7.33 (2H, d, J = 8.8 Hz), 7.55 (2H, dd, J = 2.0, 8.8 Hz), 8.13 (2H, d, J = 2.0 Hz), 9.28 (1H, bs); 13C NMR (100 MHz, CDCl3) 8.5, 21.2, 21.3, 24.4, 29.7, 32.7, 33.6, 42.6, 44.6, 47.0, 52.3, 110.7, 112.0, 123.2, 123.5, 129.1, 139.4, 172.0. HR-FTMS (ESI) m/z calcd. for C25H33Br2N3O2, [M + H]+ 552.1043, found 552.1044.
Synthesis of 4-[4-(3,6-Dibromocarbazol-9-yl)-butyrylamino]piperidine-1-carboxylic acid ethyl ester (11)
The crude oil was purified via column chromatography over silica gel and 10% methanol in dichloromethane, and the product obtained as a white solid (0.1254 g, 0.22 mmol, 85.3%). TLC analysis in CH2Cl2-MeOH (9:1), Rf = 0.63. 1H NMR (400 MHz, CDCl3) δ 1.25 (3H, t, J = 7.2 Hz), 1.88 (2H, dd, J = 2.8, 12.4 Hz), 2.08 (2H, t, J = 6.4 Hz), 2.19 (2H, m, J = 6.8 Hz), 2.88 (2H, t, J = 12.0 Hz), 3.92 (1H, m, J = 3.6 Hz), 4.12 (4H, q, J = 6.8 Hz), 4.37 (2H, t, J = 6.8 Hz), 5.16 (1H, d, J = 7.6 Hz), 7.32 (2H, d, J = 8.8 Hz), 7.55 (2H, dd, J = 2.0, 8.8 Hz), 8.14 (2H, d, J = 1.6 Hz); 13C NMR (100 MHz, CDCl3) 14.7, 24.0, 32.0, 32.4, 42.2, 42.7, 46.8, 61.4, 110.5, 112.2, 123.3, 123.5, 129.2, 139.3, 155.4, 170.1. HR-FTMS (ESI) m/z calcd. for C24H27Br2N3O3, [M + H]+ 566.0471, found 566.0470.
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-1-morpholin-4-ylbutan-1-one (12)
The crude oil was purified via column chromatography over silica gel and 10% methanol in dichloromethane, and the product obtained as a white solid (0.0768 g, 0.16 mmol, 52%). TLC analysis in CH2Cl2-MeOH (9:1), Rf = 0.94. 1H NMR (400 MHz, CDCl3) δ 2.15 (4H, t, J = 6.0 Hz), 2.21 (2H, m, J = 6.8 Hz), 3.16 (2H, t, J = 4.8 Hz), 3.50 (2H, t, J = 4.4 Hz), 3.64 (4H, dt, J = 2.8, 6.4 Hz), 7.31 (2H, d, J = 8.8 Hz), 7.55 (2H, dd, J = 1.6, 8.4 Hz), 8.14 (2H, d, J = 1.6 Hz); 13C NMR (100 MHz, CDCl3) 23.6, 28.6, 41.5, 41.9, 45.1, 65.9, 66.0, 111.3, 111.5, 122.9, 123.41, 128.8, 139.0, 170.0. HR-FTMS (ESI) m/z calcd. for C20H20Br2N2O2, [M + H]+ 480.9944, found 480.9945.
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-1-thiomorpholin-4-ylbutan-1-one (13)
The crude oil was purified via column chromatography over silica gel and 10% methanol in dichloromethane, and the product obtained as a white solid (0.0712 g, 0.41 mmol, 35%). TLC analysis in CH2Cl2-MeOH (9:1), Rf = 0.60. 1H NMR (400 MHz, CDCl3) δ 2.11 (2H, t, J = 6.3 Hz), 2.21 (2H, m), 2.34 (2H, t, J = 5.0 Hz), 2.62 (2H, t, J = 5.2 Hz), 3.46 (2H, t, J = 5.0 Hz), 3.89 (2H, t, J = 5.0 Hz), 4.41 (2H, t, J = 6.7 Hz), 7.32 (2H, d, J = 8.6 Hz), 7.56 (2H, dd, J = 1.9, 8.7 Hz), 8.15 (2H, d, J = 1.7 Hz); 13C NMR (100 MHz, CDCl3) 23.5, 27.3, 27.4, 29.0, 42.1, 44.3, 47.8, 110.6, 112.2, 123.3, 123.5, 129.1, 139.4, 169.9. HR-FTMS (ESI) m/z calcd. for C20H20Br2N3OS, [M + H]+ 496.9715, found 496.9713.
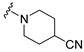
Synthesis of 1-[4-(3,6-Dibromocarbazol-9-yl)butyryl]piperidine-4-carbonitrile (14)
The crude oil was purified via column chromatography over silica gel and 10% methanol in dichloromethane, and the product obtained as a white solid (0.0775 g, 0.16 mmol, 44%). TLC analysis in CH2Cl2-MeOH (9:1), Rf = 0.97. 1H NMR (400 MHz, CDCl3) δ 1.63 (2H, t, J = 5.6 Hz), 1.85 (1H, m), 2.15 (2H, m), 2.84 (2H, m), 3.14 (2H, dt, J = 5.2, 13.6 Hz), 3.35 (2H, dt, J = 6.0, 14.4 Hz), 3.63 (2H, dq, J = 4.4, 13.6 Hz), 3.76 (2H, dq, J = 3.2, 13.6 Hz), 4.41 (2H, m), 7.31 (2H, d, J = 8.4 Hz), 7.54 (2H, dd, J = 1.6, 8.4 Hz), 8.14 (2H, d, J = 1.6 Hz); 13C NMR (100 MHz, CDCl3) 23.4, 26.2, 28.1, 28.6, 28.7, 39.5, 42.1, 43.0, 110.6, 112.2, 120.5, 123.3, 123.5, 129.1, 139.4, 169.8. HR-FTMS (ESI) m/z calcd. for C22H21Br2N3O, [M + H]+ 504.0104, found 504.0104.
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-1-(4-phenylpiperazin-1-yl)butan-1-one (15)
The crude oil was purified via column chromatography over silica gel and 10% methanol in dichloromethane, and the product obtained as a white solid (0.2658 g, 0.27 mmol, 69%). TLC analysis in CH2Cl2-MeOH (9:1), Rf = 0.53. 1H NMR (400 MHz, CDCl3) δ 1.55 (2H, bs), 2.22 (2H, m), 2.97 (2H, t, J = 5.3 Hz), 3.15 (2H, t, J = 4.9 Hz), 3.34 (2H, t, J = 4.5 Hz), 3.30 (2H, t, J = 4.7 Hz), 4.43 (2H, t, J = 3.4 Hz), 6.92 (2H, t, J = 7.9 Hz), 7.29 (3H, t, J = 7.9 Hz), 7.34 (2H, d, J = 8.7 Hz), 7.54 (2H, dd, J = 1.8, 8.6 Hz), 8.15 (2H, d, J = 1.7 Hz); 13C NMR (100 MHz, CDCl3) 23.6, 28.9, 41.6, 42.2, 45.1, 49.4, 49.5, 110.6, 112.1, 116.8, 120.7, 123.3, 123.5, 129.2, 129.3, 139.4, 150.9, 170.0. HR-FTMS (ESI) m/z calcd. for C26H25Br2N3O, [M + H]+ 556.0417, found 556.0417.
Synthesis of 1-(4-Acetylpiperazin-1-yl)-4-(3,6-dibromocarbazol-9-yl)butan-1-one (16)
The crude oil was purified via column chromatography over silica gel and 10% methanol in dichloromethane, and the product obtained as a white solid (0.0775 g, 0.15 mmol, 62%). TLC analysis in CH2Cl2-MeOH (9:1), Rf = 0.67. 1H NMR (400 MHz, CDCl3) δ 1.57 (3H, s), 2.08 (2H, bs), 2.12 (2H, bs), 2.21 (2H, bs), 3.19 (2H, t, J = 5.3 Hz), 3.45 (2H, t, J = 6.2), 3.64 (2H, t, J = 5.0 Hz), 4.42 (2H, t, J = 6.6 Hz), 7.32 (2H, dd, J = 4.2, 8.7 Hz), 7.54 (2H, dd, J = 1.9, 8.7 Hz), 8.14 (2H, d, J = 1.8 Hz); 13C NMR (100 MHz, CDCl3) 21.4, 28.8, 31.0, 41.5, 42.0, 42.2, 44.8, 45.9, 110.5, 112.2, 123.4, 129.1, 139.3, 169.3, 170.4. HR-FTMS (ESI) m/z calcd. for C22H23Br2N3O2, [M + H]+ 522.0209, found 522.0209.
Synthesis of 4-[4-(3,6-Dibromocarbazol-9-yl)butyryl]piperazine-1-carboxylic acid tert-butyl ester (17)
The crude oil was purified via column chromatography over silica gel and hexane-ethyl acetate (1:1) as the mobile phase, and the product obtained as a white solid (0.1977 g, 0.4 mmol, 80%). TLC analysis in hexane-ethyl acetate (1:1), Rf = 0.35. 1H NMR (400 MHz, CDCl3) δ 1.47 (9H, s), 2.20 (4H, m), 3.16 (2H, bs), 3.26 (2H, bs), 3.40 (2H, t, J = 5.2 Hz), 3.60 (2H, t, J = 4.4 Hz), 4.41 (2H, t, J = 6.4 Hz), 7.32 (2H, d, J = 8.4 Hz), 7.54 (2H, dd, J = 1.6, 8.4 Hz), 8.14 (2H, d, J = 2.0 Hz); 13C NMR (100 MHz, CDCl3) 23.5, 28.4, 29.0, 41.4, 42.2, 45.0, 80.4, 110.5, 112.2, 123.3, 123.5, 129.1, 139.4, 154.5, 170.2. HR-FTMS (ESI) m/z calcd. for C25H29Br2N3O3, [M + Na]+ 602.0447, found 602.0446.
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-1-piperazin-1-ylbutan-1-one (18)
The crude oil was purified via column chromatography over silica gel and 10% methanol in dichloromethane, and the product obtained as a white solid (0.0383 g, 0.080 mmol, 80%). TLC analysis in CH2Cl2-MeOH (9:1), Rf = 0.38. 1H NMR (400 MHz, CDCl3) δ 1.25 (1H, s), 2.18 (4H, m), 2.68 (2H, t, J = 4.8 Hz), 2.84 (2H, t, J = 5.2 Hz), 3.16 (2H, t, J = 4.8 Hz), 3.61 (2H, t, J = 5.2 Hz), 4.41 (2H, t, J = 6.4 Hz), 7.32 (2H, d, J = 8.4 Hz), 7.54 (2H, dd, J = 2.0, 8.8 Hz), 8.14 (2H, d, J = 1.6 Hz); 13C NMR (100 MHz, CDCl3) 22.6, 23.6, 42.2, 42.7, 45.8, 46.0, 46.3, 110.6, 112.1, 123.3, 123.5, 129.1, 139.4, 170.0. HR-FTMS (ESI) m/z calcd. for C20H21Br2N3O, [M + H]+ 480.0104, found 480.0101.
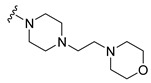
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-1-[4-(2-morpholin-4-ylethyl)piperazin-1-yl]butan-1-one (19)
The crude oil was purified via column chromatography over silica gel and 10% methanol in dichloromethane, and the product obtained as a white solid (0.2036 g, 0.34 mmol, 76.4%). TLC analysis in CH2Cl2-MeOH (9:1), Rf = 0.52. 1H NMR (400 MHz, CDCl3) δ 2.12 (2H, t, J = 6.0 Hz), 2.18 (2H, t, J = 7.2 Hz), 2.24 (2H, m), 2.44 (2H, t, J = 5.2 Hz), 2.48 (4H, t, J = 4.4 Hz), 3.17 (2H, t, J = 4.8 Hz), 3.63 (2H, t, J = 5.2 Hz), 3.70 (4H, t, J = 4.8 Hz), 4.41 (2H, t, J = 6.4 Hz), 7.28 (2H, d, J = 6.0 Hz), 7.37 (2H, dd, J = 6.4 Hz, 8.8 Hz), 8.15 (2H, d, J = 2.1 Hz); 13C NMR (100 MHz, CDCl3) 23.5, 28.7, 41.6, 42.1, 45.0, 53.2, 53.3, 54.1, 55.4, 56.3, 66.9, 110.7, 112.1, 123.3, 123.4, 129.2, 139.4, 169.8. HR-FTMS (ESI) m/z calcd. for C26H32Br2N4O2, [M + H]+ 593.0944, found 593.0945.
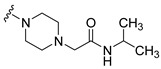
Synthesis of 2-{4-[4-(3,6-Dibromocarbazol-9-yl)-butyryl]piperazin-1-yl}-N-isopropylacetamide (20)
The crude oil was purified via column chromatography over silica gel and 10% methanol in dichloromethane, and the product obtained as a white solid (0.2273 g, 0.39 mmol, 87.3%). TLC analysis in CH2Cl2-MeOH (9:1), Rf = 0.68. 1H NMR (400 MHz, CDCl3) δ 1.17 (6H, d, J = 6.6 Hz), 2.10 (2H, t, J = 6.3 Hz), 2.21 (2H, m), 2.26 (2H, t, J = 4.7 Hz), 2.47 (2H, t, J = 4.5 Hz), 3.15 (2H, t, J = 4.5 Hz), 3.64 (2H, t, J = 4.5 Hz), 4.10 (1H, m), 4.43 (2H, t, J = 6.6 Hz), 6.73 (1H, bs), 7.31 (2H, d, J = 8.7 Hz), 7.54 (2H, dd, J = 1.9, 8.7 Hz), 8.14 (2H, d, J = 1.9 Hz); 13C NMR (100 MHz, CDCl3) 22.8, 23.4, 28.7, 40.8, 41.6, 42.0, 45.0, 53.0, 53.1, 61.5, 110.64, 112.1, 123.3, 123.5, 129.2, 139.4, 168.4, 170.0. HR-FTMS (ESI) m/z calcd. for C25H30Br2N4O2, [M + H]+ 579.0788, found 579.0790.
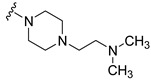
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-1-[4-(2-dimethylaminoethyl)piperazin-1-yl]butan-1-one (21)
The crude oil was purified via column chromatography over silica gel and 10% methanol in dichloromethane, and the product obtained as a white solid (0.0228 g, 0.04 mmol, 7%). TLC analysis in CH2Cl2-MeOH (9:1), Rf = 0.50. 1H NMR (400 MHz, CDCl3) δ 1.25 (6H, s), 2.12 (2H, t, J = 6.0 Hz), 2.19 (2H, m), 2.24 (4H, t, J = 5.6 Hz), 2.43 (4H, t, J = 4.8 Hz), 3.19 (2H, t, J = 4.4 Hz), 3.64 (2H, t, J = 4.4 Hz), 4.40 (2H, t, J = 6.4 Hz), 7.31 (2H, d, J = 8.8 Hz), 7.53 (2H, dd, J = 1.6, 8.4 Hz,), 8.21 (2H, d, J = 2.0 Hz); 13C NMR (100 MHz, CDCl3) 23.5, 28.7, 41.5, 42.1, 45.0, 45.6, 53.1, 53.3, 56.1, 56.5, 110.6, 112.1, 123.2, 123.4, 129.2, 139.4, 170.0. HR-FTMS (ESI) m/z calcd. for C24H30Br2N4O, [M + H]+ 551.0839, found 551.0842.
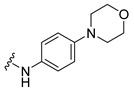
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-N-(4-morpholin-4-ylphenyl)butyramide (22)
The crude oil was purified via column chromatography over silica gel and 10% methanol in dichloromethane, and the product obtained as a white solid (0.0689 g, 0.12 mmol, 40%). TLC analysis in CH2Cl2-MeOH (9:1), Rf = 0.80. 1H NMR (400 MHz, DMSO-d6) δ 2.05 (2H, t, J = 6.8 Hz), 2.30 (2H, m), 3.02 (4H, t, J = 4.8 Hz), 3.71 (4H, t, J = 4.4 Hz), 4.44 (2H, t, J = 6.8 Hz), 6.86 (2H, d, J = 8.8 Hz), 7.41 (2H, d, J = 8.8 Hz), 7.61 (2H, dd, J = 0.8, 8.8 Hz), 7.64 (2H, d, J = 8.4 Hz), 8.48 (2H, s), 9.62 (1H, s); 13C NMR (100 MHz, DMSO-d6) 24.1, 32.7, 42.0, 48.9, 66.1, 111.3, 111.6, 115.4, 120.3, 122.9, 123.4,128.8, 131.5, 139.0, 147.0, 169.7. HR-FTMS (ESI) m/z calcd. for C26H25Br2N3O2, [M + H]+ 572.0366, found 572.0365.
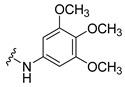
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-N-(3,4,5-trimethoxyphenyl)butyramide (23)
The crude oil was purified via column chromatography over silica gel and 10% methanol in dichloromethane, and the product obtained as a white solid (0.051 g, 0.10 mmol, 30%). TLC analysis in CH2Cl2-MeOH (9:1), Rf = 0.17. 1H NMR (400 MHz, DMSO-d6) δ 2.08 (2H, m), 2.28 (2H, t, J = 7.2 Hz), 3.60 (3H, s), 3.71 (3H, s), 3.72 (3H, s), 4.46 (2H, t, J = 6.8 Hz), 6.90 (2H, s), 7.60 (2H, dd, J = 1.6, 8.4 Hz), 7.64 (2H, d, J = 8.8 Hz), 8.47 (2H, d, J = 1.6 Hz), 9.71 (1H, s); 13C NMR (100 MHz, DMSO-d6) 6.96, 24.0, 32.0, 42.0, 55.6, 60.0, 96.9, 111.3, 111.6, 122.9, 123.4, 128.8, 133.3, 135.2, 139.1, 152.6, 170.1. HR-FTMS (ESI) m/z calcd. for C25H24Br2N2O4, [M + H]+ 577.0155, found 577.0142.
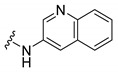
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-N-quinolin-3-ylbutyramide (24)
The crude oil was purified via column chromatography over silica gel and 10% methanol in dichloromethane, and the product obtained as a white solid (0.0275 g, 0.052 mmol, 15%). TLC analysis in hexane-ethyl acetate (1:1), Rf = 0.20. 1H NMR (400 MHz, DMSO-d6) δ 2.13 (2H, m), 2.44 (2H, t, J = 6.8 Hz), 4.50 (2H, t, J = 6.5 Hz), 7.41 (1H, t, J = 7.4 Hz), 7.54 (1H, t, J = 8.0 Hz), 7.62 (1H, t, J = 7.9 Hz), 7.67 (1H, d, J = 8.6 Hz), 7.71 (1H, d, J = 8.3 Hz), 7.92 (2H, t, J = 8.4 Hz), 7.98 (2H, d, J = 8.4 Hz), 8.47 (2H, s), 8.64 (1H, s), 8.82 (1H, s), 10.3 (1H, s); 13C NMR (100 MHz, DMSO-d6) 23.9. 32.9, 42.0, 109.6, 111.4, 111.7, 119.2, 122.0, 123.0, 123.5, 124.5, 127.0, 127.4, 127.6, 127.7, 127.8, 128.5, 128.9, 132.8, 139.1, 144.1, 144.5, 171.3. HR-FTMS (ESI) m/z calcd. for C25H19Br2N3O, [M + H]+ 537.9947, found 537.9946.
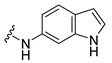
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-N-(1H-indol-6-yl)butyramide (25)
The crude oil was purified via column chromatography over silica gel and 10% methanol in dichloromethane, and the product obtained as a white solid (0.1152 g, 0.22 mmol, 38%). TLC analysis in hexane-ethyl acetate (1:1), Rf = 0.50. 1H NMR (400 MHz, DMSO-d6) δ 2.08 (2H, m), 2.35 (2H, t, J = 7.1 Hz), 4.47 (2H, t, J = 7.0 Hz), 6.33 (1H, s), 6.97, (1H, d, J = 9.1 Hz), 7.24 (1H, s), 7.40 (1H, d, J = 8.4 Hz), 7.61 (2H, d, J = 7.2 Hz), 7.66 (2H, d, J = 8.7 Hz), 7.95 (1H, s), 8.49 (2H, s), 9.75 (1H, s); 13C NMR (100 MHz, DMSO-d6) 24.2, 32.9, 42.1, 100.9, 102.2, 111.3, 111.6, 112.3, 119.7, 123.0, 123.5, 123.7, 124.8, 128.9, 133.3, 135.9, 139.1. HR-FTMS (ESI) m/z calcd. for C24H19Br2N3O, [M + H]+ 525.9947, found 9946.
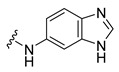
Synthesis of N-(3H-Benzoimidazol-5-yl)-4-(3,6-dibromocarbazol-9-yl)butyramide (26)
The crude oil was purified via column chromatography over silica gel and 10% methanol in dichloromethane, and the product obtained as a white solid (0.100 g, 0.20 mmol, 44%). TLC analysis in hexane-ethyl acetate (1:1), Rf = 0.70. 1H NMR (400 MHz, DMSO-d6) δ 2.16 (2H, m, J = 7.2 Hz), 3.18 (2H, t, J = 7.6 Hz), 4.52 (2H, t, J = 7.2 Hz), 6.66 (1H, dd, J = 2.0, 8.8 Hz), 7.60 (1H, dd, J = 2.0, 8.8 Hz), 7.68 (1H, d, J = 8.8 Hz), 8.05 (1H, s), 8.47 (1H, dd, J = 1.6, 8.0 Hz); 13C NMR (100 MHz, DMSO-d6) 23.6, 32.0, 42.3, 97.2, 111.8, 112.0, 114.3, 117.2, 122.3, 123.5, 124.0, 129.4, 139.5, 141.0, 141.2, 151.7, 173.0. HR-FTMS (ESI) m/z calcd. for C23H18Br2N4O, [M + H]+ 526.9900, found 526.9904.
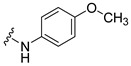
Synthesis of 4-(3,6-Dibromocarbazol-9-yl)-N-(4-methoxyphenyl)butyramide (27)
The crude oil was purified via column chromatography over silica gel and 10% methanol in dichloromethane, and the product obtained as a white solid (0.1830 g, 0.35 mmol, 94%). TLC analysis in hexane-ethyl acetate (1:1), Rf = 0.64. 1H NMR (400 MHz, DMSO-d6) δ 2.05 (2H, m), 2.30 (2H, t, J = 7.2 Hz), 3.70 (3H, s), 4.45 (2H, t, J = 7.2 Hz), 6.84 (2H, d, J = 9.2 Hz), 7.44 (2H, d, J = 9.2 Hz), 7.60 (2H, dd, J = 2.0, 8.8 Hz), 7.65 (2H, d, J = 8.8 Hz), 8.48 (2H, d, J = 2.0 Hz), 9.68 (1H, s); 13C NMR (100 MHz, DMSO-d6) 24.1, 32.7, 42.0, 55.1, 111.3, 111.6, 113.8, 120.7, 123.0, 123.5, 128.7, 132.3, 139.1, 155.1, 169.9. HR-FTMS (ESI) m/z calcd. for C23H20Br2N2O2, [M + H]+ 516.9944, found 516.9941.
3.2.4. Synthesis of N-Alkyl-5-Bromoindole Derivatives (31–34)
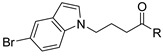
3.2.5. General Procedure for the Synthesis of 4-(5-Bromoindol-1-yl)Butyric Acid (29)
A 50 mL three-neck round-bottom flask, equipped with a reflux condenser, was charged with 5-bromoindole 28 (0.1960 g, 1.0 mmol), Cs2CO3 (0.9770 g, 3 mmol) and ethyl 4-bromobutyrate (0.43 mL, 3.0 mmol), dissolved in DMF (3 mL). After 15 min of stirring at room temperature, the reaction mixture was refluxed at 100 °C for 16 h. After completion of the reaction (analyzed by TLC), water (1 mL) and KOH (1.0 mmol) were added and the reaction mixture refluxed at 80 °C for 2 h. After the reaction was completed (analyzed by TLC), the mixture was allowed to reach room temperature and 1N HCl solution was added until reach neutral pH. The mixture was washed with water (20 mL) and the product was extracted using dichloromethane (3 × 10 mL). The organic layer was washed with brine and dried with Na2SO4, and filtered and concentrated under reduced pressure. The crude oil was purified via column chromatography over silica gel and 10% CH2Cl2 in MeOH, and the product obtained as a white solid for the precursor 4-(5-Bromoindol-1-yl)Butyric acid 29 (0.2260 g, 0.801 mmol, 80%). TLC analysis in CH2Cl2-MeOH (9:1), Rf = 0.25. 1H NMR (400 MHz, CDCl3) δ 2.16 (2H, m), 2.34 (2H, t, J = 7.2 Hz), 4.20 (2H, t, J = 6.80 Hz), 6.44 (1H, d, J = 2.4 Hz), 7.08 (1H, 2, J = 3.2 Hz), 7.22 (1H, d, J = 6.4 Hz), 7.29 (1H, dd, J = 2.0, 8.8 Hz), 7.74 (1H, d, J = 1.6 Hz); 13C NMR (100 MHz, CDCl3) 25.1, 30.5, 45.3, 101.1, 110.7, 112.8, 123.5, 124.5, 128.9, 130.3, 134.6, 177.4. HR-FTMS (ESI) m/z calcd. for C12H12BrNO2, [M + H]+ 284.0104, found 284.0102.
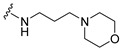
3.2.6. General Procedure for the Synthesis of 4-(5-Bromoindol-1-yl)-N-(3-Morpholin-4-ylpropyl)Butyramide (31), and for Compounds 32–34
A 50 mL three-neck round-bottom flask was charged with 4-(5-Bromoindol-1-yl)Butyric acid 29 (0.098 g, 0.35 mmol), HOBT (0.2027 g, 1.5 mmol), and EDAC (0.2876 g, 1.5 mmol). The mixture was dissolved in DMF (5 mL), stirred for 30 min, and 3-morpholinopropylamine (0.0512 mL, 0.35 mmol) was added. After 15 min, Et3N (0.43 mL, 3.0 mmol) was added and the mixture stirred at room temperature for 16 h. After the reaction was completed (analyzed by TLC), water was added (30 mL) and the product was extracted using dichloromethane (3 × 10 mL). The organic layer was washed with brine and dried with Na2SO4, and filtered and concentrated under reduced pressure. The crude oil was purified via column chromatography over silica gel and 10% methanol in dichloromethane, and the product obtained as a white solid (0.0748 g, 0.18 mmol, 53%). TLC analysis in hexane-ethyl acetate (1:1), Rf = 0.36. 1H NMR (400 MHz, CDCl3) δ 1.62 (2H, m), 2.01 (2H, t, J = 6.8 Hz), 2.21 (2H, m), 2.35 (4H, bs), 2.42 (2H, t, J = 6.4 Hz), 3.32 (2H, q, J = 5.6 Hz), 3.47 (4H, bs), 4.39 (2H, t, J = 6.8 Hz), 7.0 (1H, bs), 7.26 (1H, d, J = 1.2 Hz), 7.32 (1H, d, J = 8.8 Hz), 7.55 (1H, dd, J = 2.0, 8.8 Hz), 8.14 (1H, d, J = 1.6 Hz); 13C NMR (100 MHz, CDCl3) 24.5, 25.8, 32.6, 39.6, 45.2, 53.6, 58.0, 67.0, 101.1, 110.9, 112.6, 123.4, 124.3, 128.8, 130.2, 134.9, 171.2. HR-FTMS (ESI) m/z calcd. for C19H26BrN3O2, [M + H]+ 410.1261, found 410.1257.
Synthesis of 1-[4-(5-Bromoindol-1-yl)butyryl]piperidine-4-carbonitrile (32)
The crude oil was purified via column chromatography over silica gel and 10% methanol in dichloromethane, and the product obtained as a white solid (0.0356 g, 0.10 mmol, 24.4%). TLC analysis in CH2Cl2-MeOH (9:1), Rf = 0.76. 1H NMR (400 MHz, DMSO-d6) δ 1.58 (4H, m), 1.81 (2H, bs), 1.94 (2H, t, J = 7.2 Hz), 2.26 (2H, t, J = 7.2 Hz), 3.07 (1H, m), 3.50 (2H, dt, J = 4.8, 15.2 Hz), 3.78 (2H, dt, J = 4.8, 13.6Hz), 4.18 (2H, t, J = 6.8Hz), 6.42 (1H, d, J = 3.2 Hz), 7.24 (1H, dd, J = 2.0, 8.4 Hz), 7.41 (1H, d, J = 3.2 Hz), 7.47 (1H, d, J = 8.8 Hz), 7.73 (1H, d, J = 2.0 Hz); 13C NMR (100 MHz, DMSO-d6) 22.7, 25.3, 28.2, 29.4, 29.7, 39.5, 43.1, 45.3, 101.0, 111.0, 112.7, 120.6, 123.5, 124.4, 128.9, 130.2, 134.9, 169.9. HR-FTMS (ESI) m/z calcd. for C18H20BrN3O, [M + H]+ 376.0842, found 376.0837.
Synthesis of 4-(5-Bromoindol-1-yl)-N-(3-imidazol-1-yl-propyl)butyramide (33)
The crude oil was purified via column chromatography over silica gel and 10% methanol in dichloromethane, and the product obtained as a white solid (0.1255 g, 0.32 mmol, 57%). TLC analysis in CH2Cl2-MeOH (9:1), Rf = 0.44. 1H NMR (400 MHz, CDCl3) δ 1.81 (2H, m, J = 6.8 Hz), 1.98 (2H, m, J = 6.8 Hz), 2.06 (2H, t, J = 6.4 Hz), 3.01 (2H, q, J = 5.6 Hz), 3.95 (2H, t, J = 6.8 Hz), 4.17 (2H, t, J = 7.2 Hz), 6.42 (1H, d, J = 2.8 Hz), 6.95 (1H, bs), 7.23 (2H, dd, J = 1.6, 8.4 Hz), 7.40 (1H, d, J = 2.8 Hz), 7.45 (1H, d, J = 8.8 Hz), 7.72 (1H, d, J = 2.0 Hz), 7.90 (1H, t, J = 5.2 Hz); 13C NMR (100 MHz, CDCl3) 25.7, 31.0, 32.2, 36.5, 44.2, 45.6, 101.0, 111.0, 112.5, 119.1, 123.3, 124.4, 128.7, 128.8, 130.2, 134.7, 136.9, 172.4. HR-FTMS (ESI) m/z calcd. for C18H21BrN4O, [M + H]+ 391.0951, found 391.0956.
Synthesis of 4-(5-Bromoindol-1-yl)-N-(2-piperidin-1-ylethyl)butyramide (34)
The crude oil was purified via column chromatography over silica gel and 10% methanol in dichloromethane, and the product obtained as a white solid (0.1967 g, 0.50 mmol, 50%). TLC analysis in CH2Cl2-MeOH (9:1), Rf = 0.47. 1H NMR (400 MHz, DMSO-d6) δ 1.35 (2H, m, J = 5.2 Hz), 1.47 (4H, m, J = 5.6 Hz), 1.96 (2H, m, J = 6.4 Hz), 2.01 (2H, t, J = 6.0 Hz), 2.30 (2H, t, J = 6.8 Hz), 2.35 (2H, bs), 3.13 (2H, q, J = 6.4 Hz), 4.18 (2H, t, J = 6.4 Hz) 7.35 (1H, dd, J = 2.0, 8.8 Hz), 7.54 (2H, d, J = 2.4 Hz), 7.56 (1H, s), 7.68 (1H, s), 7.71 (1H, t, J = 5.6 Hz); 13C NMR (100 MHz, DMSO-d6) 24.1, 25.5, 25.7, 32.4, 35.7, 45.8, 54.3, 57.2, 100.8, 109.4, 111.0, 118.5, 120.3, 128.9, 129.0, 134.7, 171.7. HR-FTMS (ESI) m/z calcd. for C25H30Br2N4O2, [M + H]+ 394.1312, found 394.1313.
3.3. Biological Evaluation
3.3.1. Cell Culture
MCF-7 and MDA-MB-231 cells were cultured in Minimum Essential Medium Eagle (MEME) 10% FBS supplemented with Earle’s Balanced Salt Solution (EBSS), Non-essential Amino Acids (NEAA), Sodium Pyruvate, Pen/Strep, and L-glutamine 37 °C in 5% CO2.
3.3.2. Sulforhodamine B (SRB) Assay
A stock solution of compounds was prepared at 50 mM in 100% DMSO. For cell preparation, a flask of 75 cm2 or 25 cm2 was used for 2.6 × 105 cells/mL or 1.44 × 105 cells/mL, respectively, with an 80–90% confluence. Cells were washed with PBS and trypsinized. The concentration of cells was determined using a 1:2 dilution with Trypan Blue and a hemocytometer. After cell count, the concentration was adjusted to have a 7.0–10.0 × 104 cells/mL. Approximately 100 μL of cells suspension, compounds, control positive and control negative were added in triplicates to a 96 well plate. The positive control used was doxorubicin, and the negative control was DMSO 0.1%. All compounds at 50, 25, 12.5, 6.3, and 1.6 μM were incubated with cells at 37 °C for 48 hrs. For fixation, cold TCA 50% was used and incubated at 4 °C for 1 hr. Wells were washed and dried prior to tincture with 100 μL of SRB 0.4%. To remove excess SRB, acetic acid was used. For analysis, TRIS-Base Solution (pH = 10.5) was used and shaken prior to reading using an ELISA reader at 540 nm and the software SoftMax Pro 4.8. For each compound, 50% growth inhibition (GI50) was calculated from sigmoidal dose-response curves (variable-slope) that were generated with data obtained from experiments carried out in triplicates (GI50 values were generated with GraphPad Prism V. 6.02, GraphPad Software, Inc.).
3.3.3. Wound Healing Assay (Scratch Method) Using MDA-MB-231 Cancer Cells
Prior to assays, cells were grown until 80–90% confluence was observed. We used a 75 cm2 flask for 2.6 × 105 cells/mL in 10 mL, and for a 25 cm2 flask 1.44 × 105 cells/mL in 5 mL. The cells were washed with PBS to remove all traces of FBS. We added trypsin at 2 mL for a 25 cm2 flask or 4 mL for a 75 cm2 flask, and incubated 5–10 min at 37 °C. At the end of the incubation time, cells were re-suspended and counted with hemocytometer using 1:2 dilutions with Trypan Blue. Subsequently, cell viability was calculated. In a 12 multiwell plate. Cells were seeded at 1.5–2.2 × 105 cells/mL in 1 mL and incubated for 24 h. Cells were then rinsed with PBS and incubated in starving media (0.5% FBS) overnight. All controls and drugs were tested in triplicate. The vehicle control for each drug was prepared according to the drug’s DMSO concentration. Drugs were diluted and the final concentration at each well was 10 µM (or GI50/5 on MDA-MB-231 cells). The wound was made using a sterile pipette tip of 200 μL. Cells were then rinsed very gently with media without FBS and media with vehicle controls or drugs were added. After a 24 h incubation, the gap distance was evaluated using the software Lumera Infinity Analyze 6.4.0. Pictures were taken at 0, 8, 12, and 24 h using a 10X objective in an Inverted Laboratory Microscope Leica DM IL LED, and an Infinity1-3 3.1 Megapixel USB 2.0 camera CMOS. The percentage of migration was calculated using the following formula:
100 − [ (X₀⁄Ẍ₀)]*100 for time 0 h measurements
100 − [ (X₂₄⁄Ẍ₀)]*100 for time 24 h measurements
3.3.4. Actin Polymerization Assay
MDA-MB-231 cells were treated with vehicle or compound derivatives at 10 μM (or at concentrations that do not affect cell viability) for 24 h at 37 °C in 5% CO2. Cells were fixed in 3.7% of formaldehyde, permeabilized using 0.2% Triton, and stained with Rhodamine Phalloidin to visualize F-actin. Fluorescence micrographs were acquired at 60x in an Eclipse E400 fluorescence microscope using a DS-Qi2 monochrome digital camera from Nikon for fluorescence imaging.
4. Conclusions
In this study, a new series of N-alkyl-3,6-dibromocarbazole and N-alkyl-5-bromoindole derivatives were designed, synthesized, and biologically evaluated for their anti-cancer and anti-migration effect on MCF-7 and MDA-MB-231 cancer cells. In addition, the effect of compounds on the actin cytoskeleton of MDA-MB-231 cancer cells was explored. From the results, we have established the Structure-Activity Relationships of novel derivatives of the lead compound Wiskostatin. We found that the carbazole moiety is needed for in vitro anti-cancer and anti-migratory potency. The replacement of the carbazole by an indole ring resulted in loss of activity. Moreover, the aliphatic or aromatic amide group was extended 3-carbon atoms away from the carbazole moiety. The most potent anti-proliferative compounds were those bearing a 3,6-dibromocarbazole group with GI50 values between 7–32 µM and 4.7–23 µM on MCF-7 and MDA-MB-231 cancer cells, respectively. The most promising results in terms of anti-migratory effect was exhibited by carbazole derivatives 10, 23, 14–16, and 24 with migration inhibition in the range of 10–20% on the highly metastatic breast cancer cells MDA-MB-231. Future studies will improve these derivatives and also test their effects at higher concentrations and shorter times of migration to reduce potential cytotoxic effects, which may confound inhibition of cell migration. The carbazole derivatives 10, 14, and 15 were further examined as actin polymerization inhibitors and results demonstrated reduction in actin polymerization and extension of F-actin based structures. Further studies of mechanism of actions focused on the effect of these novel compounds in the Cdc42/N-WASP/Arp2/3 pathway is needed to fully characterize and analyze the potential of this new series of compounds as in vitro and in vivo anti-metastatic drugs.
Acknowledgments
We thank Fernando González-Illán from the US FDA for assisting with the high-resolution electrospray ionization mass spectrometry data and analysis.
Supplementary Materials
The following are available online. 1H and 13C NMR spectral data for representative compounds. Figure S1: 1H NMR Spectral Data of 2; Figure S2: 13C NMR Spectral Data of 2; Figure S3: 1H NMR Spectral Data of 7; Figure S4: 13C NMR Spectral Data of 7; Figure S5: 1H NMR Spectral Data of 12; Figure S6: 13C NMR Spectral Data of 12; Figure S7: 1H NMR Spectral Data of 14; Figure S8: 13C NMR Spectral Data of 14; Figure S9: 1H NMR Spectral Data of 16; Figure S10: 1H NMR Spectral Data of 22; Figure S11: 13C NMR Spectral Data of 22; Figure S12: 1H NMR Spectral Data of 24; Figure S13: 13C NMR Spectral Data of 24; Figure S14: 1H NMR Spectral Data of 29; Figure S15: 13C NMR Spectral Data of 29; Figure S16: 1H NMR Spectral Data of 33; Figure S17: 13C NMR Spectral Data of 33.
Author Contributions
K.B., Z.R., and A.F. performed the experiments and data collection; J.B. performed data analysis of GCMS; S.D. developed a methodology for actin assay experiments; and E.H. supervised, administrated, and was in charge of funding acquisition of the project. All authors read and approved the final manuscript.
Funding
This research was funded by the National Institutes of Health (MBRS-SCORE NIH/NIGMS SC2GM116712).
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Not available.
References
- 1.Gupta G.P., Massagué J. Cancer Metastasis: Building a Framework. Cell. 2006;127:679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 2.Walker C., Mojares E., Hernández A.D.R. Role of Extracellular Matrix in Development and Cancer Progression. Int. J. Mol. Sci. 2018;19:3028. doi: 10.3390/ijms19103028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen W.T. Proteolytic activity of specialized surface protrusions formed at rosette contact sites of transformed cells. J. Exp. Zool. 1989;251:167–185. doi: 10.1002/jez.1402510206. [DOI] [PubMed] [Google Scholar]
- 4.Pollard T.D., Borisy G.G. Cellular Motility Driven by Assembly and Disassembly of Actin Filaments. Cell. 2003;113:549. doi: 10.1016/S0092-8674(03)00357-X. [DOI] [PubMed] [Google Scholar]
- 5.Wertheimer E., Gutierrez-Uzquiza A., Rosemblit C., Lopez-Haber C., Sosa M.S., Kazanietz M.G. Rac signaling in breast cancer: A tale of GEFs and GAPs. Cell. Signal. 2012;24:353–362. doi: 10.1016/j.cellsig.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Muise A.M., Walters T., Xu W., Shen-Tu G., Guo C.H., Fattouh R., Lam G.Y., Wolters V.M., Bennitz J., Van Limbergen J., et al. Single nucleotide polymorphisms that increase expression of the guanosine triphosphatase RAC1 are associated with ulcerative colitis. Gastroenterology. 2011;141:633–641. doi: 10.1053/j.gastro.2011.04.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Worthylake R.A., Lemoine S., Watson J.M., Burridge K. RhoA is required for monocyte tail retraction during transendothelial migration. J. Cell Biol. 2001;154:147–160. doi: 10.1083/jcb.200103048. (b) Takenawa, T.; Miki, H. WASP and WAVE family proteins: key molecules for rapid rearrangement of cortical actin filaments and cell movement. J. Cell Sci.2001, 114, 1801–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rohatgi R., Ma L., Miki H., Lopez M., Kirchhausen T., Takenawa T., Kirschner M.W. The Interaction between N-WASP and the Arp2/3 Complex Links Cdc42-Dependent Signals to Actin Assembly. Cell. 1999;97:221–231. doi: 10.1016/S0092-8674(00)80732-1. [DOI] [PubMed] [Google Scholar]
- 9.Vlaar C.P., Castillo-Pichardo L., Medina J.I., Marrero-Serra C.M., Velez E., Ramos Z., Hernández E. Design, synthesis and biological evaluation of new carbazole derivatives as anti-cancer and anti-migratory agents. Biorganic. Med. Chem. 2018;26:884–890. doi: 10.1016/j.bmc.2018.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Montalvo-Ortiz B.L., Castillo-Pichardo L., Hernández E., Humphries-Bickley T., De La Mota-Peynado A., Cubano L.A., Vlaar C.P., Dharmawardhane S. Characterization of EHop-016, novel small molecule inhibitor of Rac GTPase. J. Biol. Chem. 2012;287:13228–13238. doi: 10.1074/jbc.M111.334524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Castillo-Pichardo L., Humphries-Bickley T., De La Parra C., Forestier-Roman I., Martinez-Ferrer M., Hernandez E., Vlaar C., Ferrer-Acosta Y., Washington A.V., Cubano L.A., et al. The Rac inhibitor EHop-016 inhibits mammary tumor growth and metastasis in a nude mouse model. Transl. Oncol. 2014;7:546–555. doi: 10.1016/j.tranon.2014.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dharmawardhane S., Hernandez E., Vlaar C. Development of EHop-016: a small molecule inhibitor of Rac. Enzymes. 2013;33:117–146. doi: 10.1016/B978-0-12-416749-0.00006-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Humphries-Bickley T., Castillo-Pichardo L., Corujo-Carro F., Duconge J., Hernandez-O’Farrill E., Vlaar C., Rodriguez-Orengo J.F., Cubano L., Dharmawardhane S. Pharmacokinetics of Rac inhibitor EHop-016 in mice by ultra-performance liquid chromatography tandem mass spectrometry. J. Chromatogr. B. 2015;981:19–26. doi: 10.1016/j.jchromb.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Głuszyńska A. Biological potential of carbazole derivatives. Eur. J. Med. Chem. 2015;94:405–426. doi: 10.1016/j.ejmech.2015.02.059. [DOI] [PubMed] [Google Scholar]
- 15.Guillonneau C., Pierré A., Charton Y., Guilbaud N., Kraus-Berthier L., Léonce S., Michel A., Bisagni E., Atassi G. Synthesis of 9-O-substituted derivatives of 9-hydroxy-5,6-dimethyl-6H-pyrido[4,3-b]carbazole-1-carboxylic acid (2-(Dimethylamino)-ethyl) amide and their 10-and 11-methyl analogues with improved antitumor activity. J. Med. Chem. 1999;42:2191–2203. doi: 10.1021/jm981093m. [DOI] [PubMed] [Google Scholar]
- 16.Saturnino C., Iacopetta D., Sinicropi M., Rosano C., Caruso A., Caporale A., Marra N., Marengo B., Pronzato M., Parisi O., et al. N-alkyl carbazole derivatives as new tools for Alzheimer’s disease: preliminary studies. Molecules. 2014;19:9307–9317. doi: 10.3390/molecules19079307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bandgar B.P., Adsul L.K., Chavan H.V., Jalde S.S., Shringare S.N., Shaikh R., Meshram R.J., Gacche R.N., Masand V. Synthesis, biological evaluation, and docking studies of 3-(substituted)-aryl-5-(9-methyl-3-carbazole)-1H-2-pyrazolines as potent anti-inflammatory and antioxidant agents. Bioorganic Med. Chem. Lett. 2012;22:5839–5844. doi: 10.1016/j.bmcl.2012.07.080. [DOI] [PubMed] [Google Scholar]
- 18.Biamonte M.A., Wanner J., Le Roch K.G. Recent advances in malaria drug discovery. Bioorganic Med. Chem. Lett. 2013;23:2829–2843. doi: 10.1016/j.bmcl.2013.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caruso A., Voisin-Chiret A.S., Lancelot J.-C., Sinicropi M.S., Garofalo A., Rault S. Efficient and Simple Synthesis of 6-Aryl-1,4-dimethyl-9H-carbazoles. Molecules. 2008;13:1312–1320. doi: 10.3390/molecules13061312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chakrabarty M., Ghosh N., Harigaya Y. A clay-mediated, regioselective synthesis of 2-(aryl/alkyl) amino-thiazolo [4, 5-c] carbazoles. Tetrahedron Lett. 2004;45:4955–4957. doi: 10.1016/j.tetlet.2004.04.129. [DOI] [Google Scholar]
- 21.Caruso A., Sinicropi M.S., Lancelot J.C., El-Kashef H., Saturnino C., Aubert G., Ballandonne C., Lesnard A., Cresteil T., Dallemagne P., et al. Synthesis and evaluation of cytotoxic activities of new guanidines derived from carbazoles. Bioorganic Med. Chem. Lett. 2014;24:467–472. doi: 10.1016/j.bmcl.2013.12.047. [DOI] [PubMed] [Google Scholar]
- 22.Iacopetta D., Rosano C., Puoci F., Parisi O.I., Saturnino C., Caruso A., Longo P., Ceramella J., Malzert-Fréon A., Dallemagne P., et al. Multifaceted properties of 1, 4-dimethylcarbazoles: Focus on trimethoxybenzamide and trimethoxyphenylurea derivatives as novel human topoisomerase II inhibitors. Eur. J. Pharm. Sci. 2017;96:263–272. doi: 10.1016/j.ejps.2016.09.039. [DOI] [PubMed] [Google Scholar]
- 23.Saturnino C., Palladino C., Napoli M., Sinicropi M.S., Botta A., Sala M., Carcereri de Prati A., Novellino E., Suzuki H. Synthesis and biological evaluation of new N-alkylcarbazole derivatives as STAT3 inhibitors: Preliminary study. Eur. J. Pharm. Sci. 2013;60:112–119. doi: 10.1016/j.ejmech.2012.11.004. [DOI] [PubMed] [Google Scholar]
- 24.Rizza P., Pellegrino M., Caruso A., Iacopetta D., Sinicropi M.S., Rault S., Lancelot J.C., El-Kashef H., Lesnard A., Rochais C., et al. 3-(Dipropylamino)-5-hydroxybenzofuro [2, 3-f] quinazolin-1 (2H)-one (DPA-HBFQ-1) plays an inhibitory role on breast cancer cell growth and progression. Eur. J. Pharm. Sci. 2016;107:275–287. doi: 10.1016/j.ejmech.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 25.Issa S., Walchshofer N., Kassab I., Termoss H., Chamat S., Geahchan A., Bouaziz Z. Synthesis and antiproliferative activity of oxazinocarbazole and N,N-bis(carbazolylmethyl)amine derivatives. Eur. J. Med. Chem. 2010;45:2567–2577. doi: 10.1016/j.ejmech.2010.02.045. [DOI] [PubMed] [Google Scholar]
- 26.Danish I.A., Prasad K.J.R. A one-pot synthesis of 1, 2, 4, 5-tetraazaspiro [5.5]-6, 7, 8, 9-tetrahydrocarbazol-3-thiones and their antibacterial activities. Indian J. Heterocycl. Chemstry. 2004;14:19–22. [Google Scholar]
- 27.Indumathi T., Fronczek F.R., Prasad K.R. Synthesis of 2-amino-8-chloro-4-phenyl-5, 11-dihydro-6H-pyrido[2,3-a]carbazole-3-carbonitrile: Structural and biological evaluation. J. Mol. Struct. 2012;1016:134–139. doi: 10.1016/j.molstruc.2012.01.032. [DOI] [Google Scholar]
- 28.Kantevari S., Yempala T., Surineni G., Sridhar B., Yogeeswari P., Sriram D. Synthesis and antitubercular evaluation of novel dibenzo[b,d]furan and 9-methyl-9H-carbazole derived hexahydro-2H-pyrano[3,2-c]quinolines via Povarov reaction. Eur. J. Med. Chem. 2011;46:4827–4833. doi: 10.1016/j.ejmech.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 29.Woon K.L., Ariffin A., Ho K.W., Chen S.-A. Effect of conjugation and aromaticity of 3,6 di-substituted carbazoles on triplet energy and the implication of triplet energy in multiple-cyclic aromatic compounds. RSC Adv. 2018;8:9850–9857. doi: 10.1039/C8RA00674A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bashir M., Bano A., Ijaz A.S., Chaudhary B.A. Recent Developments and Biological Activities of N-Substituted Carbazole Derivatives: A Review. Molecules. 2015;20:13496–13517. doi: 10.3390/molecules200813496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mac Millan K.S., Naidoo J., Liang J., Melito L., Williams L.S., Morlock L., Huntington P.J., Estill S.J., Longood J., Becker G., et al. Development of Proneurogenic, Neuroprotective Small Molecules. J. Am. Chem. Soc. 2011;133:1428–1437. doi: 10.1021/ja108211m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Molette J., Routier J., Abla N., Besson D., Bombrun A., Brun R., Burt H., Georgi K., Kaiser M., Nwaka S., et al. Identification and Optimization of an Aminoalcohol-Carbazole Series with Antimalarial Properties. ACS Med. Chem. Lett. 2013;4:1037–1041. doi: 10.1021/ml400015f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saturnino C., Caruso A., Iacopetta D., Rosano C., Caramella J., Muia N., Mariconda A., Grazia Bonomo M., Ponassi M., Rosace G., et al. Inhibition of Human Topoisomerase II by N,N,N-Trimethylethanammonium Iodide Alkylcarbazole Derivatives. Chem. Med. Chem. 2018;13:2635–2643. doi: 10.1002/cmdc.201800546. [DOI] [PubMed] [Google Scholar]
- 34.Caruso A., Iacopetta D., Pouci F., Cappello A.R., Saturnino C., Sinicropi M.S. Carbazole derivatives: A promising scenario for breast cancer treatment. Mini Rev. Med. Chem. 2016;16:630. doi: 10.2174/1389557515666150709111342. [DOI] [PubMed] [Google Scholar]
- 35.Bombrun A., Gerber P., Casi G., Terradillos O., Antonsson B., Halazy S. 3,6-dibromocarbazole piperazine derivatives of 2-propanol as first inhibitors of cytochrome c release via Bax channel modulation. J. Med. Chem. 2003;46:4365–4368. doi: 10.1021/jm034107j. [DOI] [PubMed] [Google Scholar]
- 36.Zhang Y., Tangadanchu V.K.R., Cheng Y., Yang R.G., Lin J.M., Zhou C.H. Potential Antimicrobial Isopropanol-Conjugated Carbazole Azoles as Dual Targeting Inhibitors of Enterococcus faecalis. ACS Med. Chem. Lett. 2018;9:244–249. doi: 10.1021/acsmedchemlett.7b00514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pieper A.A., Xie S., Capota E., Estill S.J., Zhong J., Long J.M., Becker G.L., Huntington P., Goldman S.E., Shen C.-H., et al. Discovery of a proneurogenic, neuroprotective chemical. Cell. 2010;142:39. doi: 10.1016/j.cell.2010.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peterson J.R., Bickford L.C., Morgan D., Kim A.S., Ouerfelli O., Kirschner M.W., Rosen M.K. Chemical inhibition of N-WASP by stabilization of a native autoinhibited conformation. Nat. Struct. Mol. Boil. 2004;11:747–755. doi: 10.1038/nsmb796. [DOI] [PubMed] [Google Scholar]
- 39.Guerriero C.J., Weisz O.A. N-WASP inhibitor wiskostatin non-selectively perturbs membrane transport by decreasing cellular ATP levels. Am. J. Physiol. Cell Physiol. 2007;292:C1562–C1566. doi: 10.1152/ajpcell.00426.2006. [DOI] [PubMed] [Google Scholar]
- 40.Orellana E.A., Kasinski A.L. Sulforhodamine B (SRB) Assay in Cell Culture to Investigate Cell Proliferation. Bio-protocol. 2016;6:e1984. doi: 10.21769/BioProtoc.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liang C.C., Park A.Y., Guan J.L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007;2:329–333. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 42.Olson M.F., Sahai E. The actin cytoskeleton in cancer cell motility. Clin. Exp. Metastasis. 2009;26:273. doi: 10.1007/s10585-008-9174-2. [DOI] [PubMed] [Google Scholar]
- 43.Ridley A.J. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Boil. 2006;16:522–529. doi: 10.1016/j.tcb.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 44.Takenawa T., Suetsugu S. The WASP–WAVE protein network: Connecting the membrane to the cytoskeleton. Nat. Rev. Mol. Cell Boil. 2007;8:37–48. doi: 10.1038/nrm2069. [DOI] [PubMed] [Google Scholar]
- 45.Frugtniet B., Jiang W.G., Martin T.A., Martin T. Role of the WASP and WAVE family proteins in breast cancer invasion and metastasis. Breast Cancer: Targets Ther. 2015;7:99–109. doi: 10.2147/BCTT.S59006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.