

Abstract
Titin is a giant protein with multiple functions in cardiac and skeletal muscles. Rat cardiac titin undergoes developmental isoform transition from the neonatal 3.7 MDa N2BA isoform to primarily the adult 2.97 MDa N2B isoform. An autosomal dominant mutation dramatically altered this transformation. Titins from eight skeletal muscles: Tibialis Anterior (TA), Longissimus Dorsi (LD) and Gastrocnemius (GA), Extensor Digitorum Longus (ED), Soleus (SO), Psoas (PS), Extensor Oblique (EO), and Diaphram (DI) were characterized in wild type and in homozygous mutant (Hm) rats with a titin splicing defect. Results showed that the developmental reduction in titin size is eliminated in the mutant rat so that the titins in all investigated skeletal muscles remain large in the adult. The alternative splicing of titin mRNA was found repressed by this mutation, a result consistent with the large titin isoform in the mutant. The developmental pattern of titin mRNA alternative splicing differs between heart and skeletal muscles. The retention of intron 49 reveals a possible mechanism for the absence of the N2B unique region in the expressed titin protein of skeletal muscle.
Keywords: TITIN ISOFORM TRANSITION, EXON SKIPPING, N2B, MIDDLE Ig AND PEVK
Titin [Wang et al., 1979], also named connectin [Maruyama, 1976], is the largest and third most abundant protein in mammalian striated muscle [Tskhovrebova and Trinick, 2003]. The titin molecule is an elastic filament approximately 1 μm long and 3–4 nm wide [Maruyama et al., 1984; Trinick et al., 1984; Wang et al., 1984] that spans half of the sarcomere with the amino terminus located in Z-line and the carboxyl terminus in the M-line [Fürst et al., 1988; Labeit and Kolmerer, 1995; Obermann et al., 1997; Gregorio et al., 1998]. The segment of titin in the A-band region is inextensible; the elasticity of skeletal muscle titin mainly comes from two elastic regions located in the I-band: polymeric Ig regions (middle Ig) and PEVK domain [Labeit and Kolmerer, 1995]. A unique N2B region found in cardiac muscle can also be lengthened with stretch, and is marginally more stable than the PEVK [Helmes et al., 1999; Linke et al., 1999; Trombitás et al., 1999; Watanabe et al., 2002]. The giant size and the specialized structure enable titin to play a mechanical role in maintaining the structure of the sarcomere, and titin accounts for most of the passive tension of striated muscles in the physiological extension range [Horowits and Podolsky, 1987; Funatsu et al., 1990; Wang et al., 1991; Granzier and Irving, 1995]. Besides its mechanical effects, titin also has many other physiological roles. Titin is necessary for sarcomere assembly by acting as a platform for myofibrillar construction [Labeit et al., 1992; Ohtsuka et al., 1997; Sorimachi et al., 1997]. Titin is involved in the protein turnover signaling pathway by interacting with MURF1 and MURF2 with its carboxyl kinase domain [Centner et al., 2001; McElhinny et al., 2002; Pizon et al., 2002], and the protein also control the balance between muscle growth and degradation by interacting with myostatin and calpain-3 [Nicholas et al., 2002].
Titin is expressed from a single gene, but due to alternative splicing of the titin mRNA, its protein isoforms have diverse sizes in different muscles during muscle development [Labeit and Kolmerer, 1995; Freiburg et al., 2000; Bang et al., 2001; Greaser et al., 2005; Prado et al., 2005; Ottenheijm et al., 2009]. The alternative splicing of titin mRNA primarily occurs in the regions corresponding to I-band middle Ig and PEVK regions [Labeit and Kolmerer, 1995; Bang et al., 2001; Greaser et al., 2002]. Our lab previously reported the developmental changes in rat cardiac titin isoforms and an autosomal mutation that eliminated these changes [Greaser et al., 2005, 2008]. Here we present results from eight skeletal titin isoforms in wild type and mutant rats and the alternative splicing of titin mRNA underlying these titin isoforms.
MATERIALS AND METHODS
SAMPLE PREPARATION
Skeletal muscle Tibialis Anterior (TA), Longissimus Dorsi (LD), Gastrocnemius (GA) Extensor Digitorum Longus (ED), Soleus (SO), Psoas (PS), Extensor Oblique (EO), Diaphragm (DI), and cardiac muscle Left ventricle (LV) were taken at 180 days from three wild type (Wt) and three homozygous mutant (Hm) rats for adult comparisons. The TA, LD, and GA samples were also taken at 1, 20, and 60 days after birth from both Wt and Hm rats for analyzing the developmental titin isoform transition (three animas per genotype and time point). Protein samples were prepared with urea-thiourea SDS buffer as described previously [Warren et al., 2003]. RNA samples were prepared from 180 days WtLD, Hm LD,WtTA, andHm TA for analyzing the alternative splicing of skeletal titin mRNA, 180 day Wt LV and Hm LV for the analysis of cardiac titin mRNA. RNA from neonatal Wt TA (5 days) and Wt LV (1 day) were used to compare with adult Wt TA and Wt LV for developmental study of titin alternative splicing (one animal per genotype and time point). RNAs are prepared using TRIzol reagent (Invitrogen) following the manufacture’s protocol. All animals are from a hybrid strain composed of 50% Brown Norway–25% Fisher 344–25% Sprague Dawley [Greaser et al., 2008].
GEL ELECTROPHORESIS AND MOLECULAR WEIGHT ESTIMATION
Titin protein isoforms were analyzed by a vertical sodium dodecyl sulfate agarose gel electrophoresis system [Warren et al., 2003]. Silver stained agarose gels were scanned and analyzed by NIH Image version 1.62. Human soleus titin isoform: N2A (3.7 MDa) and adult cardiac titin isoform: N2B (2.97 MDa) were used as size standards to estimate the molecular weights of titin in skeletal muscle [Warren et al., 2003].
ALTERNATIVE SPLICING ANALYSIS
RT-PCR was used to investigate the alternative splicing of titin mRNAs from various tissues, primers (listed in Supplemental Table I) were designed in the range from exon 40 to exon 226 based on the rat titin sequence (accession number AC098426). PCR was performed in 20 μl of final volume containing 2 μl 10× PCR buffer, 1.5 mM MgCl2, 0.2 μM dNTP, 0.2 μM of forward and reverse primers, 1 μl cDNA and 2 U of Platinum Taq DNA polymerase (Invitrogen). Temperature parameters for PCR were: 94°C 1 min, 8 touchdown cycles at 92°C 10 s, 68°C for 30 s (−1°C per cycle), 68°C 1 min/kb, followed by 30 cycles at 92°C 10 s, 60°C for 30 s, 68°C 1 min/kb and 68°C 10 min for extension. PCR products were analyzed on 1% agarose gels. Bands of interest were directly sequenced or cloned into the pGEM-T Easy Vector System II (Promega) and then sequenced. Sequences were identified by comparing the human (AJ277892) and rat genomic titin sequence (AC098426) using “Blast two sequences” method (http://blast.ncbi.nlm.nih.gov/bl2seq/wblast2.cgi).
RESULTS
SKELETAL MUSCLE TITIN IN WILD TYPE AND MUTANT RATS
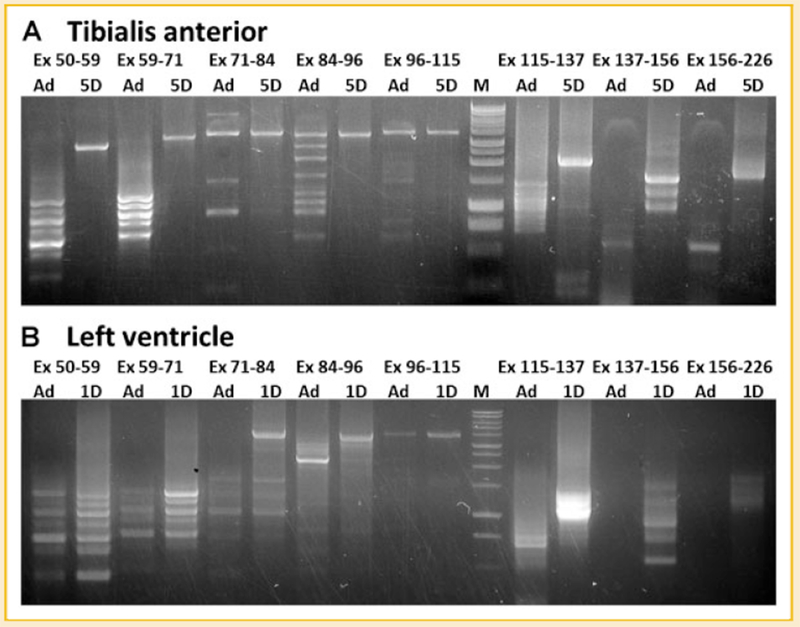
The survey on titin isoforms in adult TA, LD, GA, EDL, SO, PS, EO, and DI demonstrated that titin size between skeletal muscles varies in the wild type but remains constant in the mutant (Fig. 1A,B; Table I). Developmental titin isoform transitions in wild type TA, LD, and GA all showed same trend: progressing from larger sizes to small ones, only the extent varies (Fig. 1C–E). Titin in TA undergoes the largest developmental size reduction. In 1 day TA, the major titin has an approximate size of 3.70 MDa. At 20 days the 3.70 MDa titin has disappeared and been replaced with a 3.54 MDa isoform. At 60 days, the two major titin isoforms in TA are 3.45 and 3.31 MDa, respectively with the proportion of the two isoforms being almost equal. At 180 days, the two major isoforms are essentially the same sizes (3.43 and 3.29 MDa), but the quantity of the smaller one now dominates (Fig. 1C). In LD, the size of the titin isoform at 1 day of age was 3.70 MDa, and it was only slightly changed to 3.65 MDa in the adult (Fig. 1D). In GA, the titin isoform undergoes progressive changes from 3.70 MDa at 1 day to 3.49 MDa at 180 days (Fig. 1E). Developmental size reduction of titin isoform was completely eliminated in the mutant; the size of titin in Hm TA, Hm LD, and Hm GA remains at 3.75 MDa for all developmental stages.
Fig. 1.
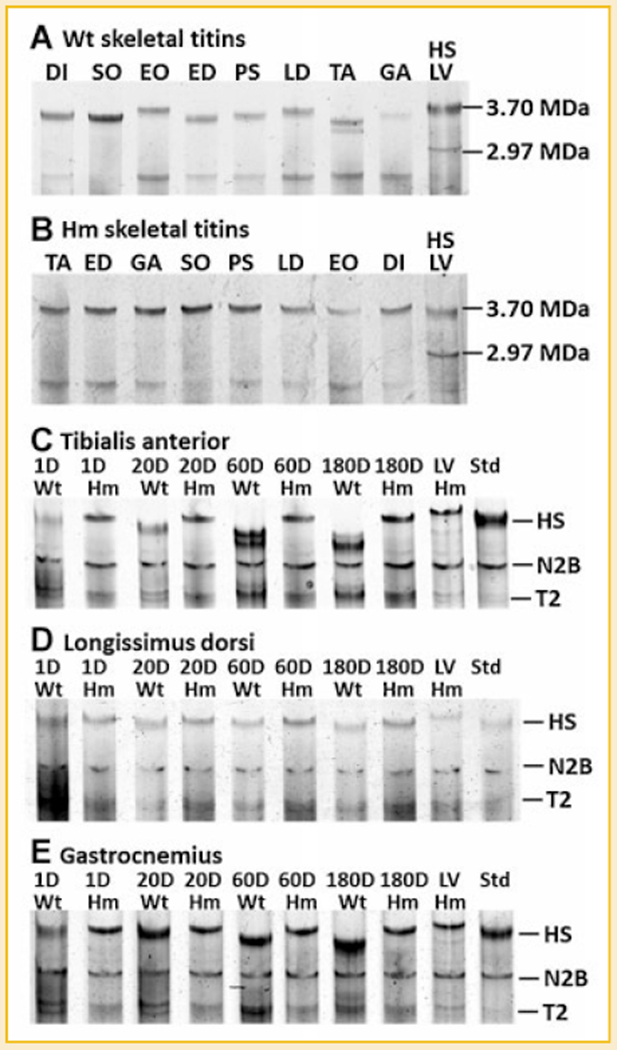
Titin isoforms in different skeletal muscles and the developmental transition of titin isoform in wild type and homozygote animals. A: The size of titin varies in different skeletal muscles of wild type rat; (B) The size of titin remains the same in all mutant skeletal muscles. A mixture of human soleus (HS, titin size of 3.7 MDa) and rat left ventricle (LV, titin size of 2.97 MDa) is included as a size ruler; (C–E) Titin isoforms in TA, LD, and GA was investigated at four time points: 1 day (D), 20, 60, and 180 days for both genotypes. Results show that titin in Wt TA and Wt GA changed dramatically from 1 to 180 days where as the Wt LD titin isoforms barely changed during this period. Titin size in mutant skeletal muscles remained constant during development. Wild type LV was mixed with each sample as a migration distance marker. HS, human Soleus; N2B, the major adult ventricle titin isoform; T2, titin proteolytic fragment.
TABLE I.
Titin Size in the Wild Type and Homozygous Mutant
| Dia | Soleus | EO | EDL | Psoas | LD | TA | GA | LV | |
|---|---|---|---|---|---|---|---|---|---|
| Wt (KDa) | 3569 | 3521 | 3664 | 3503 | 3567 | 3646 | 3440/3296 | 3574 | 2970 |
| Hm (KDa) | 3752 | 3831 |
The titin size in wild type skeletal muscles varies but all were smaller than neonatal skeletal muscle (3.7 MDa). Titin size in all mutant skeletal muscles remains same (3.75 MDa).
MUTANT SKELETAL MUSCLE TITIN IS SMALLER THAN MUTANT CARDIAC TITIN
The previous study showed that the developmental titin isoform transitions did not occur in cardiac titin from mutant homozygotes [Greaser et al., 2008]. The current data demonstrated that developmental titin isoform transitions were absent in skeletal muscles from mutant homozygotes as well. We then compared the size of mutant cardiac titin with mutant skeletal titin. Mixtures of proteins from the two tissues in the same lane revealed that mutant cardiac titin is slightly larger than that of mutant skeletal muscle. The size of mutant cardiac titin was 3.83 MDa (Fig. 2).
Fig. 2.
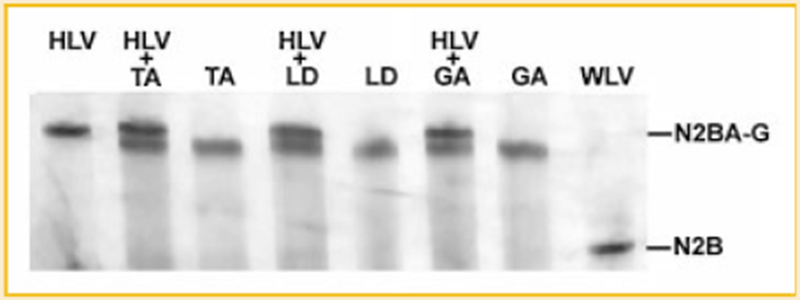
Size comparison of mutant cardiac titin and mutant skeletal titin. Hm skeletal titins from TA, LD, and GA are used to compare with Hm cardiac titin, showing that Hm skeletal titin is smaller than Hm cardiac titin. HLV, Hm left ventricle; TA, (from Hm); LD, (from Hm); GA, (from Hm); WLV, wild type left ventricle.
cDNA ANALYSIS OF MIDDLE Ig AND PEVK REGIONS IN SKELETAL MUSCLES
Titin in Wt LD is relative large with a similar molecular weight to titin in Hm skeletal muscles. Since the alternative splicing in titin mRNA corresponding to the middle Ig and PEVK region (from exon 50 to exon 226) contributes to the size difference between titin isoforms [Supplemental Fig. S1, Freiburg et al., 2000], titin mRNA in Wt LD should have similar length with the mutant for this alternatively spliced region. We then compared the splicing of middle Ig and PEVK region in Wt LD with that in Hm LD and Hm TA.
The splicing pattern in these three tissues was found to be very similar: constitutive splicing from exon 50 to 156 and alternative splicing from exon 156 to 226 (Fig. 3A,C). The prevalent constitutive splicing in middle Ig and PEVK regions of titin mRNA in Wt LD, Hm TA, and Hm LD is consistent with the large titin size in these tissues. In contrast, Wt TA has the smallest titin size in all the tested skeletal muscles, and its titin mRNA undergoes extensive alternative splicing. Almost the entire middle Ig and PEVK regions (from exon 50 to 226) are alternatively spliced; constitutive splicing can only be detected mixed with alternative splicing from exon 71 to 115 (Fig. 3B,C). The exon composition of titin mRNAs in different tissues is summarized in Figure 4.
Fig. 3.
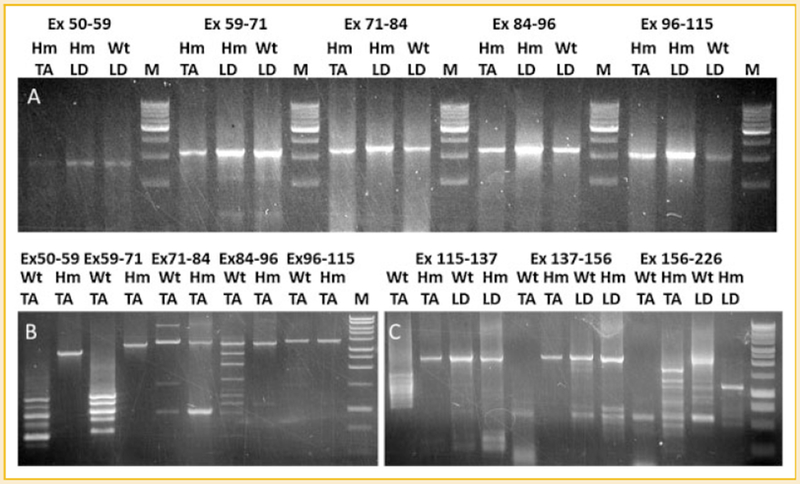
Splicing analysis of titin mRNA in wild type and mutant skeletal muscles. cDNAs from Wt TA, Hm TA, Wt LD, and Hm LD are amplified by primer sets: 50F-59R, 59F-71R, 71F-84R, 84F-96R 96F-115R, 115F-137R, 137F-156R, and 156F-226R for the corresponding exons (Ex, exon; F, forward primer; R, reverse primer). A: only constitutive splicing can be found in the middle Ig region (exon 50-115) of Hm TA, Wt LD, and Hm LD. B: alternative splicing of middle Ig region can be found in Wt TA but only constitutive splicing in Hm TA. C: Only alternative splicing can be detected in the PEVK region (exon 115-226) of Wt TA but constitutive splicing exists in wt LD, Hm LD, and Hm TA from exon 115 to 156; the region from exon 156 to 226 is alternatively spliced in all tested tissues.
Fig. 4.
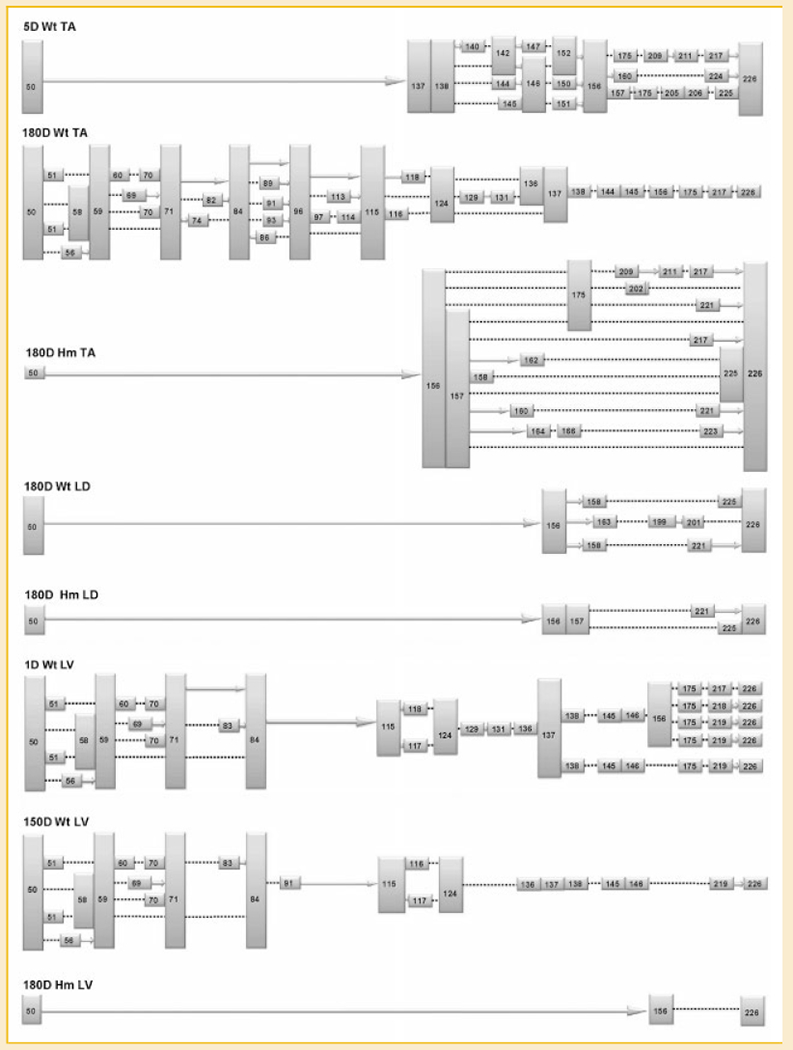
Summary of the exon profile of titin mRNA corresponding to middle Ig and PEVK region in different tissues. The colored boxes are exons (with their numbers), the solid arrows indicate consecutive exons. The dotted lines denote direct attachment between the adjacent numbered exons, the information between exon 156 to 226 in 180D Hm LV is missing.
cDNA ANALYSIS OF MIDDLE IG AND PEVK REGION IN CARDIAC MUSCLES
Because Hm cardiac titin is larger than Hm skeletal titin, we examined if the splicing of middlg Ig and PEVK region in Hm cardiac titin mRNA is different from that in the Hm skeletal. Results showed that the Hm cardiac titin mRNA is constitutively spliced from exon 50 to 156, which is the same as the Hm skeletal but Hm cardiac cDNA can barely be amplified between exons 156 to 226 (Fig. 5). Alternative splicing dominates in the entire region from exon 50 to 137 in Wt cardiac; constitutive splicing mixed with alternative splicing occurs between exons 96 to 115. After exon 137, no PCR products can be amplified from wild type cardiac (Fig. 5). The failure to amplify exon 137–156 and exon 156–226 suggests the absence of exon 156 in Wt cardiac titin mRNA, which is verified by sequencing the PCR product obtained with primer pair 137F-226R (see Fig. 4).
Fig. 5.
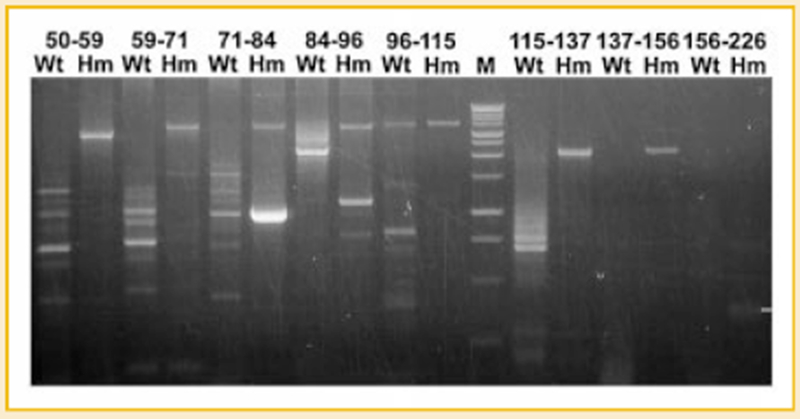
Splicing analysis of titin mRNA in wild type and mutant cardiac muscles. Numbers correspond to the exons for the primer pairs. Alternative splicing dominates in wild type cardiac titin mRNA but only constitutive splicing was detected in the mutant. The bright band found in 71-84 Hm with size smaller than constitutively spliced product does not contain titin sequence.
cDNA ANALYSIS OF N2B UNIQUE REGION IN SKELETAL AND CARDIAC MUSCLE
Titin N2B unique region is absent in skeletal muscles because the exon 49 which encodes N2B unique is skipped from skeletal titin mRNA by alternative splicing [Freiburg et al., 2000]. Since the alternative splicing in our mutant rats are significantly repressed, we wanted to check if exon 49 was expressed in mutant skeletal muscles. Primers were designed on exon 40 and 50 to amplify the entire region with cDNA from neonatal Wt TA, adult Wt TA, Hm TA, Wt LD, Hm LD, Wt LV, and Hm LV. Results showed that exon 49 can only be found from cardiac titin mRNA for both wild type and mutant (Fig. 6A). In Hm TA and Hm LD, even the mutation can largely repress the exon skipping in the region from exon 50 to 225, but it cannot repress the skipping of exon 49, which indicates the alternative splicing of exon 49 was regulated by a mechanism independent of the mutation we characterized. Intriguingly, together with the skipping of exon 49, we found partial and complete retention of intron 49 in skeletal titin mRNA; the retention of intron 49 cannot be found in the cardiac titin mRNA (Fig. 6B). It should be noted that both intron retention and partial intron retention result in stop codons and block skeletal titin mRNA translation.
Fig. 6.
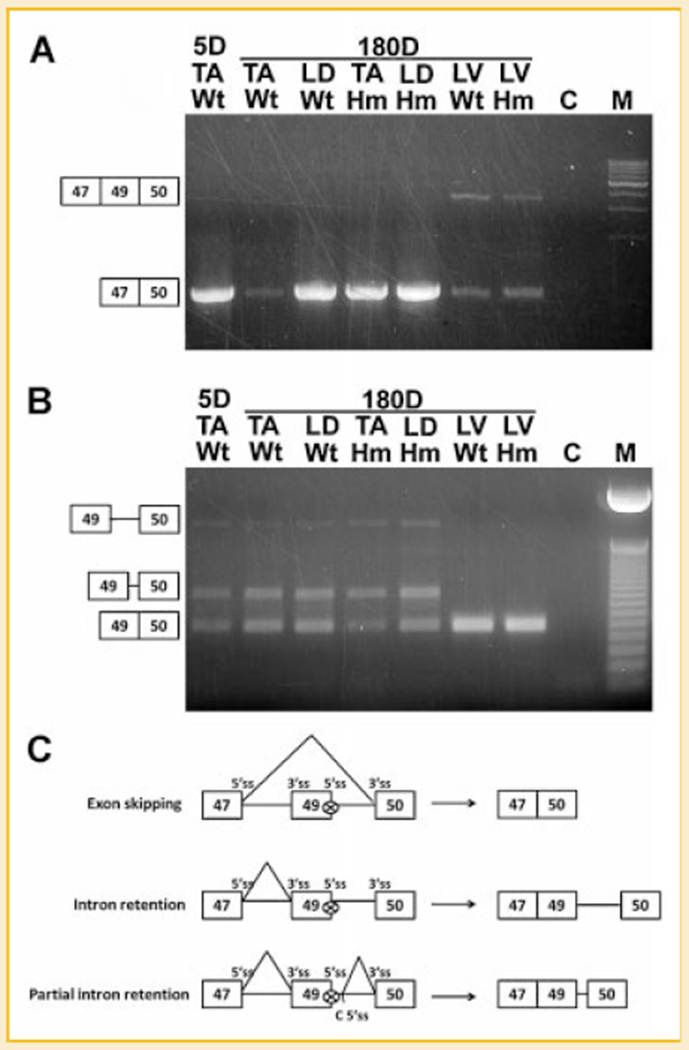
The skipping of exon 49 and retention of intron 49. A: Exon 49 is absent from skeletal muscles but present in both wild type and mutant left ventricle (LV). Exons are illustrated by boxes. Exon 48 has not been found in rat titin mRNA; the constitutive splicing pattern is exon 47-49-50 in cardiac muscles and alternative splicing of exon 47 to 50 in skeletal muscles. B: Complete intron 49 retention and partial intron 49 retention occurs in titin mRNA of skeletal muscles but not in cardiac muscles. Solid lines connecting the exons indicate complete and partial intron retention (from the sequenced products). C: Schematic diagram of exon 49 skipping and intron 49 retention, 5′ splice site (ss) and 3′ ss of each exon are illustrated, “C 5′ ss” in partial intron 49 retention refers to cryptic 5′ ss.
DEVELOPMENTAL CHANGES OF TITIN ALTERNATIVE SPLICING IN WT TA AND WT LV
Since cardiac titin undergoes even more developmental change in size [from 3.7 to 2.9 MDa – Warren et al., 2004] than TA titin in wild type rats, the cDNA from neonatal TA (5 day), adult TA (180 day), neonatal LV (1 day), and adult LV (180 day) were analyzed to investigate the developmental changes in titin alternative splicing. Results showed that constitutive splicing dominated the middle Ig region of TA titin mRNA at 5 days of age (Fig. 7A); in contrast the middle Ig region of cardiac titin mRNAs are already alternatively spliced by 1 day of age (Fig. 7B). The comparison between TA and LV reveals the different timing of titin alternative splicing between these two tissues. The early alternative splicing of cardiac titin mRNA explains why small titin isoform: N2B (2.9 MDa) can be found in 1 day LV [Greaser et al., 2008] and why the titin isoform in neonatal LV is smaller than that in neonatal TA. The observation that different tissues share the same alternative splicing pattern was unexpected; splicing in the region from exon 50 to 71 was exactly the same in 1D LV, 180D LV, and 180D TA although the splicing patterns in other regions differed in these three tissues.
Fig. 7.
Developmental changes of alternative splicing of titin mRNA corresponding to middle Ig and PEVK regions. A: splicing of titin mRNA in adult TA and 5 day neonatal TA. B: Splicing of titin mRNA in adult LV and 1 day neonatal LV. Results show that titin mRNA in the adult undergoes more extensive alternative splicing than the neonatal. Titin mRNA in neonatal, LV is alternatively spliced earlier than that in TA.
N2B SPLICE ROUTE, N2B LIKE SPLICE ROUTE AND OTHER SPECIAL SPLICING EVENTS
It has been hypothesized that the cardiac N2B titin isoform results from direct ligation of exon 50 to 219 (N2B splice route) in rabbit, human, pig, and rat [Freiburg et al., 2000; Greaser et al., 2002]. Here we wanted to check if this splice route can be inhibited in our mutant rats. First the cDNA from Wt LV and Hm LV were analyzed, and the reported N2B splice route was detected in Wt LV but not in the mutant. The splice route obtained from Hm LV has four more exons skipped than the wild type such that exon 50 is directly ligated to exon 223 instead of exon 219 (Fig. 8). Splicing similar to the N2B splice route was detected also in Wt TA, Hm TA, Wt LD, and Hm LD (Fig. 8). These “N2B like” splice routes were independent of the mutation. Interestingly, the extensive alternative splicing routes detected in various tissues always showed ligation of the first few exons to the last few ones in the defined region, no exons in the middle part of the skipped region can be included. Another interesting finding was that some alternative splicings of titin mRNA do not occur at the exon-intron boundary; instead exons were truncated at certain intra-exon positions. These cryptic splice sites were detected in exons 51, 95, 96, 225, and 226, with multiple cryptic sites in exons 51 and 95 (Supplemental Figs. S2 and S3).
Fig. 8.

N2B and N2B like splice routes in various tissues. Boxes with numbers correspond to exons. The dotted lines denote direct attachment between the numbered exons.
DISCUSSION
CHANGES IN TITIN PROTEIN ISOFORMS DURING DEVELOPMENT
Previous work has reported that the titin protein size in the left ventricle declined during development [Lahmers et al., 2004; Opitz et al., 2004; Warren et al., 2004]. In contrast the apparent cardiac titin size in mutant rats did not change through ages up to one and a half years [Greaser et al., 2008]. The current study was designed to determine whether the same large to small titin size transformation occurred in skeletal muscles of wild type and mutant rats. Results showed that titin protein size reduction with age also occurs in wild type skeletal muscles, but the extent and developmental progression were not the same between muscles. Thus, although TA and GA titins became smaller, the LD remained essentially the same large size through 180 days of age (Table I). A single major band was expressed during the early postnatal period in the skeletal muscles examined. In contrast cardiac titin development showed discrete sized N2BA isoforms that are gradually replaced with smaller ones and with multiple bands being visible between ages 1 and 20 days [Greaser et al., 2008]. Another difference between the tissues was the time course of development; cardiac changes in titin size were completed by 20 days after birth while titins continued to become smaller in the TA and GA between 20 and 60 days of age, but showed little change with development in the LD. The gradual developmental change of titin size in skeletal muscles confirms results with mouse and rabbit skeletal muscles found previously [Ottenheijm et al., 2009]. The mutant skeletal muscles showed no developmental change in titin size in any of the muscles examined.
WHY CONSTITUTIVE SPLICING BEFORE EXON 156 BUT ALTERNATIVE SPLICING AFTER
Our results clearly showed that titin mRNAs in TA and LV, where titin isoforms are small in the wild type but large in the mutant, were alternatively spliced in the wild type but constitutively spliced in the mutant from exon 50 to 156. However, for the region from exon 156 to 226, constitutive splicing was not detected in the mutant; instead no product was amplified in the Hm LV but many alternative spliced products were observed in Hm TA and Hm LD. A possible explanation might be that the region from exon 156 to 226 is too large in the mutant to be amplified by RT-PCR: 70 exons total and the 6.5 kb in length (human). The primers used to amplify exon 156–226 were originally designed according to human cardiac titin mRNA: AJ277892, which yields an alternatively spliced titin mRNA 2.5 kb in length containing only 26 exons. However, consideration needs to be taken that the alternative splicing of titin mRNA is largely inhibited in our mutant rats, it is possible that the alternative splicing in the region from exon 156 to 226 is inhibited also. If this happened, the length of the resulting titin mRNA might be out of the reasonable range for RT-PCR. The full length of titin mRNA from exon 156 to 226 is unknown in rat because the exons in this region have not been characterized. For the same reason, alternative splicing from exon 156 to 226 may be inhibited in mutant skeletal muscles; detection of some alternative splicings in Hm TA and Hm LD might only because they are easier to be amplified by RT-PCR. The alternatively spliced titin mRNA in Hm skeletal muscles should not account for major proportion, otherwise the titin sizes in different mutant skeletal muscles should vary, which is inconsistent with the fact that titins in all mutant skeletal muscles have exactly same size.
WHY CARDIAC TITIN IS LARGER THAN SKELETAL TITIN IN THE MUTANT
A significant size difference (3.75 MDa skeletal and 3.83 MDa cardiac) was found when mutant skeletal and mutant cardiac titins were compared (Fig. 2). Based on our analysis, if the middle Ig and PEVK region of titin mRNA is constitutively spliced in both Hm skeletal and Hm cardiac muscles, there should be another region on titin mRNA responsible for the size difference between Hm skeletal and Hm cardiac titin. The N2B unique region encoded by exon 49 is a candidate, because it only exists in cardiac titin. The size of the N2B unique region is approximately 95 kDa; it can reasonably be assumed that the approximately 80 kDa size difference between Hm cardiac and Hm skeletal titins results mainly from inclusion or exclusion of exon 49.
INTRON 49 RETENTION REVEALS POSSIBLE MECHANISM FOR EXON 49 SKIPPING IN SKELETAL TITIN mRNA
Expression of the N2B unique region is restricted to cardiac muscle in human and rabbit [Freiburg et al., 2000]. Our data shows that the N2B unique region is also apparently absent in skeletal muscles of rats (Fig. 6A). Using primers from within exon 49 to amplify exon 49-to-exon 50 segments showed intron 49 retention in skeletal titin mRNA, but not in cardiac (Fig. 6B). The intron 49 retention potentially provides clues for the mechanism of exon 49 skipping. A possible mechanism might be the inactivation of the 5′ splice site (ss) of exon 49 in skeletal titin pre-mRNA. During splicing, exons are ligated together and the introns are spliced out of the pre-mRNA. To accomplish this, the boundary of exons in pre-mRNA needs to be defined at the 5′ ss by U1 snRNP and at the 3′ ss by U2AFs and U2 snRNP in a process is called exon definition [Berget, 1995; Zorio and Blumenthal, 1999]. Failure in exon definition will lead to skipping of that exon. In this case, based on the retention of intron 49 in some skeletal titin mRNAs, we know the splicing between 5′ ss of exon 49 and 3′ ss of exon 50 is somehow inhibited (Fig. 6C, middle), so one or both of these two splice sites may be inactive for splicing. Based on the direct ligation of 5′ ss of exon 47 and 3′ ss of exon 50 in skeletal titin mRNA (Fig. 6C, top), we know the 3′ ss of exon 50 must be active, so it is very possible that the 5′ ss of exon 49 is inactivated in skeletal titin pre-mRNA. This hypothesis can also be supported by partial intron 49 retention found in some other skeletal titin mRNAs, in which the 3′ ss of exon 50 spliced at a cryptic 5′ ss within intron 49 instead of the authentic 5′ ss of exon 49 (Fig. 6C, bottom), indicating the 5′ ss of exon 49 is not available for splicing in these skeletal titin mRNAs. No intron 49 was found in cardiac titin mRNA (Fig. 6B), indicating the exon 49 is well defined in cardiac titin pre-mRNA, which is consistent with the constitutive splicing on exon 49 in cardiac titin mRNA.
PROBLEMS
Sequencing products from multiple primer sets will not give a full picture of the intact cDNA. There is no way, for example, to determine whether a segment obtained from primers for exons 50 to 59 is connected to another product from exons 59 to 71. Thus the construction of a valid full-length cDNA is impossible, even for the restricted regions explored here. However, the cDNA sequences and PCR product sizes found are consistent with the expressed protein sizes in the wild type and mutant muscles. Newer methods like RNA-Seq may provide more information, but would be unlikely to provide more insights on the extremely complex and varied pattern of splicing. It remains unclear whether the varied splicing pathways have functional importance even if the resulting overall size of the protein products may be very similar.
The results and conclusions from the current work regarding the PEVK exons (from exon 115 to 226) expressed in wild type hearts were somewhat different from those obtained previously [Greaser et al., 2008]. In the earlier work, the full PEVK region was amplified using primers from exons 115 and 225. None of the previous cloned sequences contained exons 137 or 156. Most of the earlier clones also had alternative splicing in the 115–137 region. The earlier failure to obtain constitutively spliced products from the 115 to 156 region in Hm LV may be due to the anticipated larger PCR product (>3,000 bp) if all these exons were amplified, a size that might be difficult to obtain, particularly when competing with amplification of shorter products.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the College of Agricultural & Life Sciences, University of Wisconsin-Madison and by grants from the National Institutes of Health (HL77196) and Hatch 1556.
Grant sponsor: National Institutes of Health; Grant number: HL77196; Grant sponsor: Hatch; Grant number: 1556.
Footnotes
Additional Supporting Information may be found in the online version of this article.
REFERENCES
- Bang ML, Centner T, Fornoff F, Geach AJ, Gotthardt M, McNabb M, Witt CC, Labeit D, Gregorio CC, Granzier H, Labeit S. 2001. The complete gene sequence of titin, expression of an unusual approximately 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ Res 89:1065–1072. [DOI] [PubMed] [Google Scholar]
- Berget SM. 1995. Exon recognition in vertebrate splicing. J Biol Chem 270: 2411–2414. [DOI] [PubMed] [Google Scholar]
- Centner T, Yano J, Kimura E, McElhinny AS, Pelin K, Witt CC, Bang ML, Trombitas K, Granzier H, Gregorio CC, Sorimachi H, Labeit S. 2001. Identification of muscle specific ring finger proteins as potential regulators of the titin kinase domain. J Mol Biol 306:717–726. [DOI] [PubMed] [Google Scholar]
- Freiburg A, Trombitas K, Hell W, Cazorla O, Fougerousse F, Centner T, Kolmerer B, Witt C, Beckmann JS, Gregorio CC, Granzier H, Labeit S. 2000. Series of exon-skipping events in the elastic spring region of titin as the structural basis for myofibrillar elastic diversity. Circ Res 86:1114–1121. [DOI] [PubMed] [Google Scholar]
- Funatsu T, Higuchi H, Ishiwata S. 1990. Elastic filaments in skeletal muscle revealed by selective removal of thin filaments with plasma gelsolin. J Cell Biol 110:53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fürst DO, Osborn M, Nave R, Weber K. 1988. The organization of titin filaments in the half-sarcomere revealed by monoclonal antibodies in immunoelectron microscopy: A map of ten nonrepetitive epitopes starting at the Z line extends close to the M line. J Cell Biol 106:1563–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granzier HL, Irving TC. 1995. Passive tension in cardiac muscle: Contribution of collagen, titin, microtubules, and intermediate filaments. Biophys J 68: 1027–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaser ML, Berri M, Warren CM, Mozdziak PE. 2002. Species variations in cDNA sequence and exon splicing patterns in the extensible I-band region of cardiac titin: Relation to passive tension. J Muscle Res Cell Motil 23:473–482. [DOI] [PubMed] [Google Scholar]
- Greaser ML, Krzesinski PR, Warren CM, Kirkpatrick B, Campbell KS, Moss RL. 2005. Developmental changes in rat cardiac titin/connectin: Transitions in normal animals and in mutants with a delayed pattern of isoform transition. J Muscle Res Cell Motil 26:325–332. [DOI] [PubMed] [Google Scholar]
- Greaser ML, Warren CM, Esbona K, Guo W, Duan Y, Parrish AM, Krzesinski PR, Norman HS, Dunning S, Fitzsimons DP, Moss RL. 2008. Mutation that dramatically alters rat titin isoform expression and cardiomyocyte passive tension. J Mol Cell Cardiol 44:983–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorio CC, Trombitas K, Centner T, Kolmerer B, Stier G, Kunke K, Suzuki K, Obermayr F, Herrmann B, Granzier H, Sorimachi H, Labeit S. 1998. The NH2 terminus of titin spans the Z-disc: Its interaction with a novel 19-kD ligand (T-cap) is required for sarcomeric integrity. J Cell Biol 143:1013–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmes M, Trombitas K, Centner T, Kellermayer M, Labeit S, Linke WA, Granzier H. 1999. Mechanically driven contour-length adjustment in rat cardiac titin’s unique N2B sequence: Titin is an adjustable spring. Circ Res 84:1339–1352. [DOI] [PubMed] [Google Scholar]
- Horowits R, Podolsky RJ. 1987. The positional stability of thick filaments in activated skeletal muscle depends on sarcomere length: Evidence for the role of titin filaments. J Cell Biol 105:2217–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labeit S, Gautel M, Lakey A, Trinick J. 1992. Towards a molecular understanding of titin. EMBO J 11:1711–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labeit S, Kolmerer B. 1995. Titins: Giant proteins in charge of muscle ultrastructure and elasticity. Science 270:293–296. [DOI] [PubMed] [Google Scholar]
- Lahmers S, Wu Y, Call DR, Labeit S, Granzier H. 2004. Developmental control of titin isoform expression and passive stiffness in fetal and neonatal myocardium. Circ Res 94:505–513. [DOI] [PubMed] [Google Scholar]
- Linke WA, Rudy DE, Centner T, Gautel M, Witt C, Labeit S, Gregorio CC. 1999. I-band titin in cardiac muscle is a three-element molecular spring and is critical for maintaining thin filament structure. J Cell Biol 146:631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama K 1976. Connectin, an elastic protein from myofibrils. J Biochem 80:405–407. [DOI] [PubMed] [Google Scholar]
- Maruyama K, Kimura S, Yoshidomi H, Sawada H, Kikuchi M. 1984. Molecular size and shape of beta-connectin, an elastic protein of striated muscle. J Biochem 95:1423–1433. [DOI] [PubMed] [Google Scholar]
- McElhinny AS, Kakinuma K, Sorimachi H, Labeit S, Gregorio CC. 2002. Muscle-specific RING finger-1 interacts with titin to regulate sarcomeric M-line and thick filament structure and may have nuclear functions via its interaction with glucocorticoid modulatory element binding protein-1. J Cell Biol 157:125–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas G, Thomas M, Langley B, Somers W, Patel K, Kemp CF, Sharma M, Kambadur R. 2002. Titin-cap associates with, and regulates secretion of myostatin. J Cell Physiol 193:120–131. [DOI] [PubMed] [Google Scholar]
- Obermann WM, Gautel M, Weber K, Furst DO. 1997. Molecular structure of the sarcomeric M band: Mapping of titin and myosin binding domains in myomesin and the identification of a potential regulatory phosphorylation site in myomesin. EMBO J 16:211–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohtsuka H, Yajima H, Maruyama K, Kimura S. 1997. The N-terminal Z repeat 5 of connectin/titin binds to the C-terminal region of alpha-actinin. Biochem Biophys Res Commun 235:1–3. [DOI] [PubMed] [Google Scholar]
- Opitz CA, Leake MC, Makarenko I, Benes V, Linke WA. 2004. Developmentally regulated switching of titin size alters myofibrillar stiffness in the perinatal heart. Circ Res 16(94):967–975. [DOI] [PubMed] [Google Scholar]
- Ottenheijm CA, Knottnerus AM, Buck D, Luo X, Greer K, Hoying A, Labeit S, Granzier H. 2009. Tuning passive mechanics through differential splicing of titin during skeletal muscle development. Biophys J 97:2277–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado LG, Makarenko I, Andresen C, Krüger M, Opitz CA, Linke WA. 2005. Isoform diversity of giant proteins in relation to passive and active contractile properties of rabbit skeletal muscles. J Gen Physiol 126:461–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizon V, Iakovenko A, Van Der Ven PF, Kelly R, Fatu C, Fürst DO, Karsenti E, Gautel M. 2002. Transient association of titin and myosin with microtubules in nascent myofibrils directed by the MURF2 RING-finger protein. J Cell Sci 115:4469–4482. [DOI] [PubMed] [Google Scholar]
- Sorimachi H, Freiburg A, Kolmerer B, Ishiura S, Stier G, Gregorio CC, Labeit D, Linke WA, Suzuki K, Labeit S. 1997. Tissue-specific expression and alpha-actinin binding properties of the Z-disc titin: Implications for the nature of vertebrate Z-discs. J Mol Biol 270:688–695. [DOI] [PubMed] [Google Scholar]
- Trinick J, Knight P, Whiting A. 1984. Purification and properties of native titin. J Mol Biol 180:331–356. [DOI] [PubMed] [Google Scholar]
- Trombitás K, Freiburg A, Centner T, Labeit S, Granzier H. 1999. Molecular dissection of N2B cardiac titin’s extensibility. Biophys J 77:3189–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tskhovrebova L, Trinick J. 2003. Titin: Properties and family relationships. Nat Rev Mol Cell Biol 4:679–689. [DOI] [PubMed] [Google Scholar]
- Wang K, McClure J, Tu A. 1979. Titin: Major myofibrillar components of striated muscle. Proc Natl Acad Sci USA 76:3698–3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Ramirez-Mitchell R, Palter D. 1984. Titin is an extraordinarily long, flexible, and slendermyofibrillar protein. Proc Natl Acad Sci USA 81:3685–3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, McCarter R, Wright J, Beverly J, Ramirez-Mitchell R. 1991. Regulation of skeletal muscle stiffness and elasticity by titin isoforms: A test of the segmental extension model of resting tension. Proc Natl Acad Sci USA 88:7101–7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren CM, Krzesinski PR, Greaser ML. 2003. Vertical agarose gel electrophoresis and electroblotting of high-molecular-weight proteins. Electrophoresis 24:1695–1702. [DOI] [PubMed] [Google Scholar]
- Warren CM, Krzesinski PR, Campbell KS, Moss RL, Greaser ML. 2004. Titin isoform changes in rat myocardium during development. Mech Dev 121: 1301–1312. [DOI] [PubMed] [Google Scholar]
- Watanabe K, Nair P, Labeit D, Kellermayer MS, Greaser M, Labeit S, Granzier H. 2002. Molecular mechanics of cardiac titin’s PEVK and N2B spring elements. J Biol Chem 277:11549–11558. [DOI] [PubMed] [Google Scholar]
- Zorio DA, Blumenthal T. 1999. Both subunits of U2AF recognize the 3’ splice site in Caenorhabditis elegans. Nature 402:835–838. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.