

Abstract
Addiction to conventional opioid pain analgesics is a major societal problem that is increasing at an alarming rate. New drugs to combat the effects of opioid abuse are desperately needed. Kappa-opioid agonists are efficacious in peripheral pain models but suffer from centrally-mediated effects. In this article, we discuss our efforts in developing peripheral kappa-based opioid receptor agonists that have the potential analgesic activity of opioids but do not manifest the negative side-effects of opioid use and abuse. Further, derivatives of the tetra-peptide D-Phe-D-Phe-D-Nle-D-Arg-NH2, such as CR665, exhibit high peripheral to central selectivity in analgesic models when administered intravenously (i.v.); however, they are inactive when administered orally. Application of our laboratory’s proprietary nonnatural amino acid technology to CR665 produced derivatives that exhibit peripheral analgesic activity when dosed orally but do not promote CNS-based effects. Lead compound JT09 activates the kappaopioid receptor with EC50s in the low nM range, while agonist selectivity for kappa over other peripheral opioid receptors was >33,400 fold. Results indicate that JT09 is approximately as efficacious as morphine in alleviating peripheral pain, while failing to produce undesired CNS-mediated activity. Additionally, JT09 did not promote other CNS-mediated effects associated with morphine (addiction, sedation, dysphoria, tolerance, addiction). Thus, we propose that JT09 has potential for development as a novel analgesic.
Perspective:
This article presents data supporting the analgesic properties of an orally available, peripherally-restricted, kappa-opioid agonist for peripheral pain. A potential out-patient pharmaceutical that acts as efficacious as morphine in alleviating peripheral pain, while failing to produce undesired CNS-mediated effects, could help reduce the current health care burden associated with prescription opioids.
Keywords: Analgesic, kappa, opioid, pain, peripheral
1. Introduction
One in five people globally suffer from chronic pain, which includes visceral, thermal, bone, neuropathic pain, and pain associated with cancer (Goldberg and McGee, 2011; Kumar, 2014; Nitu et al., 2003). In the United States, approximately 100 million adults are affected by chronic pain, costing our society an estimated 635 billion dollars annually (Gaskin, 2011; Hughes et al., 2013). Patients with chronic pain are often prescribed mu-opioid agonists (MOAs), which have potent analgesic activity but are associated with several untoward side-effects, most notably, abuse liability and respiratory depression. Approximately two million Americans suffer from a substance abuse disorder, with four out of five heroin users admitting to starting out with prescription opioid medications (2019). In 2017, approximately 47,000 Americans died from an accidental opioid overdose, with more than 130 people dying per day. Thus, the current opioid crisis poses a significant public health problem that could be mitigated by new therapeutic approaches to pain management.
Of the opioid analgesics, kappa-opioid receptor agonists (KOAs) exhibit the greatest efficacy in visceral pain models (American Society of Addiction Medicine, 2018; Kennedy, 1997; Kennedy et al., 1997). Peptide-derived KOAs have demonstrated a limited side-effect profile when compared to conventional opioid analgesics that target the mu-opioid receptor (American Society of Addiction Medicine, 2018; Stein et al., 2008). The analgesic properties of KOAs are mediated in the periphery by acting on kappa-opioid receptors (KORs); however, KOAs also act centrally, leading to untoward psychoactive side effects such as dysphoria, hallucinations, sedation, and psychosis (Riviere 2004). Thus, the development of a peripherally-restricted KOA that does not cross the blood brain barrier (BBB) could serve as a potential broad-spectrum analgesic for chronic pain. The peptidic KOA CR665 [Figure 1], under development by Cara Therapeutics (Shelton, CT), exhibits a peripheral-selectivity of 548-fold and has demonstrated efficacy in the treatment of visceral and neuropathic pain without the negative side-effects of centrally acting KOAs. However, CR665 does not have significant oral bioavailability, which limits its potential use.
Figure 1. Structure of CR665 and side-chain structures of proprietary CR665 derivatives.

The position 4 Arg residue of CR665 was converted to derivatives containing the modified D-Lys residues shown above.
We have applied a peptide modification technology developed in our laboratory to CR665, and have generated derivatives demonstrating significantly improved efficacy and PK/PD properties. In this and in previous studies (Hughes et al., 2013), a total of over 30 D-Arg(4) non-natural derivatives of CR665 were synthesized and evaluated for their analgesic potential and peripheral versus central activities. Our lead compound, JT09, binds to the kappa-opioid receptor over the mu- and delta-opioid receptors at a selectivity of >33,400-fold, and binds potently to the kappa-opioid receptor with an EC50 of 29.9 nM. Additionally, JT09 demonstrated a peripheral-selectivity of >938-fold and an oral EC50 at a druggable 4.7 mg/kg. CR665 is not considered a candidate for an oral drug; although, Cara Therapeutics reports a CR665 derivative, CR845 [Figure 2], demonstrating 15% oral bioavailability in a Phase 1 clinical trial when administered in enteric capsules (American Society of Addiction Medicine, 2018). CR845 is currently under phase III clinical trials for the intravenous (i.v.) treatment of post-operative pain and uremic pruritus. We anticipate that JT09 will be sufficiently bioavailable without the need for formulation for oral delivery as, in contrast to the Cara compounds, JT09 exhibits oral activity in rats. In this study, we further evaluated JT09 for central versus peripheral pain mediation and for signs of behavioral toxicity.
Figure 2.
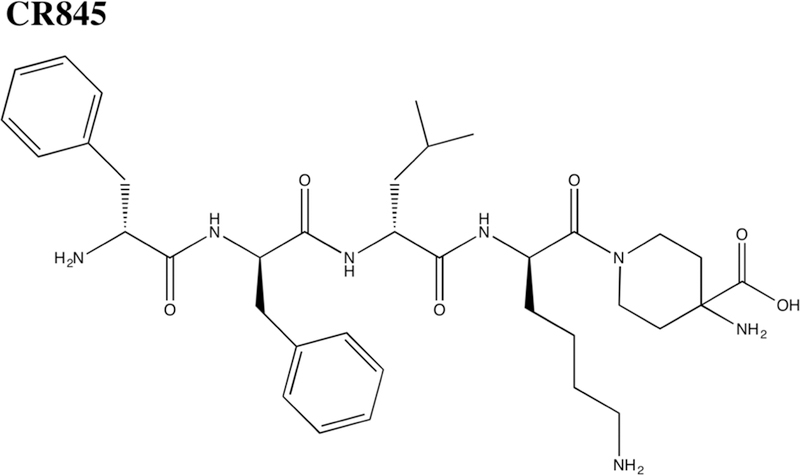
Chemical structure of CR845.
2. Materials and Methods
2.1. Synthesis of Analogs
The Position 4 D-Arg residue of CR665 was converted to derivatives containing modified D-Arg or D-Lys residues (Hughes et al., 2013). In doing so, we hypothesized that slight modifications of CR665 would impart enhanced oral activity and peripheral selectivity. Our laboratory has successfully applied this strategy in the past to other peptides, resulting in identification of leads for psychosis, stroke, and neuropathic pain that feature drastically enhanced PK/PD properties versus the native peptide (DeHaven-Hudkins and Dolle, 2004; Lundquist, 1998; Lundquist, 1999). These compounds are in advanced preclinical trials. Figure 1 illustrates the structure of CR665 and two of the eight second-generation lead peptide derivatives (out of approximately 30 unique compounds) that were synthesized a evaluated. All tested compounds featured modifications to the Position 4 D-Arg residue, and were chosen to provide a range of structures to initially probe potential structure-activity relationships (SARs) at this position. Lead compounds JT07 and JT09 contain modified D-Lys residues. For the D-Lys compounds, we have found in earlier studies that a Lys derivative often improves PK/PD when substituted for an Arg residue in the parent compound. All compounds were assembled using standard Merrifield chemistry to build the peptides while incorporating the non-natural D-Arg and D-Lys residues, synthesized as described previously (Kennedy et al., 2000; Kokko, 2004; Kokko et al., 2001; Kokko et al., 2003; Lundquist et al., 2002; Orwig, 2005). All compounds were HPLC-purified and characterized by MALDI-mass spectrometry, giving measured [M+1]+ molecular weights within 0.1% of theoretical values [Table 1]. This is the standard method of analysis for peptides synthesized through standard Merrifield chemistry, in which the identity of each monomer is pre-defined.
Table 1.
Mass spectrometry analysis of JT07 and JT09.
| JT07 |
| Chemical Formula: C43H61N7O4 |
| Exact Mass: 739.48 |
| Molecular Weight: 740.01 |
| m/z: 739.48 (100.0%), 740.48 (46.5%), 741.49 (10.6%), 740.48 (2.6%), 741.48 (1.2%) |
| Elemental Analysis: C, 69.79; H, 8.31; N, 13.25; O, 8.65 |
| JT09 |
| Chemical Formula: C38H53N7O4 |
| Exact Mass: 671.42 |
| Molecular Weight: 671.89 |
| m/z: 671.42 (100.0%), 672.42 (41.1%), 673.42 (8.2%), 672.41 (2.2%) |
| Elemental Analysis: C, 67.93; H, 7.95; N, 14.59; O, 9.52 |
2.2. Animals
All animal work was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at the Medical University of South Carolina. All experimental protocols were performed in accordance with the guidelines set forth in the NIH Guide for the Care and Use of Laboratory Animals, published by the Public Health Service. All protocols were performed on male Sprague-Dawley rats (Harlan, Prattville, AL, USA, 240–280g), which were housed in an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)-approved colony room maintained at a constant temperature and humidity. A total of 110 animals (two per cage; approximately 175–225 g each) were kept on a 12-h light:dark cycle with ad libitum access to food and water. Since many of the protocols are susceptible to learning phenomena, which results in progressive decrease of the physiological response or behavior to be monitored, rats were used no more than three times per analgesic model.
2.3. Acetic Acid-Induced Writhing Model
The acetic acid-induced writhing model is a well-established measure of peripherally-mediated pain (Kennedy et al., 2002; Kokko et al., 2005; Vanderah et al., 2004). Experimental rats were orally gavaged with each test compound (ED90, 20 mg/kg) and rested for 20 min before receiving an intraperitoneal (i.p) injection with 2 ml/kg of 3% acetic acid (Hughes et al., 2013). Control rats were either administered morphine (10 mg/kg, i.p.) or orally gavaged with saline (0.2 ml) and rested for 20 min before receiving an i.p. injection with 2 ml/kg of 3% acetic acid. After receiving acetic acid injections, animals were rested for an additional 10 min before being placed in a 10 × 10 inch Plexiglas chamber. A video of each rat was then recorded for next 20 min. The video was subsequently scored blindly by an investigator who examined each rat every 20 seconds (s) for the entire 20 min (60 individual observations). At each observation, rats were scored on whether they are writhing or not. Writhing is defined as a constriction of the abdominal area, often with extension of the hind legs. At the end of the experiment, the percent of time the animal was writhing was calculated. Data were analyzed and presented using GraphPad Prism (GraphPad Software, Inc., La Jolla, CA, USA).
2.4. Hot Plate Model
The hotplate model evaluates central pain attenuation in a rodent after applying an acute thermal stimuli (Barber and Gottschlich, 1992; Hadden et al., 2005; Hughes et al., 2013). Experimental rats were orally gavaged with lead compounds JT07 or JT09 (20 mg/kg) and rested for 20 min prior to being placed on the hotplate. Control rats either received an i.p. injection of morphine (10 mg/kg) or were orally gavaged with saline (0.2 ml and rested for 20 min prior to be being placed on the hotplate. Rats were then assessed over time on a hotplate analgesia meter (Columbus Instruments, Columbus, OH, USA) at 53.0+/−0.2°C. The time until the rat lifted, nibbled, or shook one of its hind paws was recorded, which is known as the response latency. Animals remained on the hotplate no longer that 30 s to avoid tissue damage. Experiments were scored as the percent of maximal possible effect (%MPE) calculated using the following equation: %MPE = [(post-drug latency – pre-drug latency)/(cut-off pre-drug latency)] × 100%. Data was analyzed and presented using GraphPad Prism.
2.5. Self-Administration Model
Addiction/dependence is a critical side effect of opioids and must be evaluated with any new member of this family. The most reliable test of abuse liability with translational relevance is a contingent drug self-administration model, in which rats are trained to press a lever for intravenous drug delivery (van Ree et al., 1978). Sixteen standard self-administration chambers (30×20×20 cm, Med Associates) were housed inside sound-attenuating cubicles fitted with a fan for airflow and to mask noise. Each chamber contained two retractable levers, two stimulus lights, a speaker for tone delivery, and a house light to provide general illumination. In addition, each chamber was equipped with a balanced metal arm and spring leash attached to a swivel (Instech). Tygon® tubing extended through the leash and was connected to a 10 ml syringe mounted on an infusion pump located outside the sound-attenuating cubicle. Rats were anesthetized with i.p. injections of ketamine (66 mg/kg; VedcoInc, St Joseph, MO, USA), xylazine (1.3 mg/kg; Lloyd Laboratories, Shenandoah, IA, USA), and equithesin (0.5 ml/kg); sodium pentobarbital (4 mg/kg), chloral hydrate (17 mg/kg), and 21.3 mg/kg magnesium sulfate heptahydrate dissolved in 44% propylene glycol, 10 % ethanol solution). Ketorolac (2.0 mg/kg, i.p. Sigma, St. Louis, MO, USA) was given just prior to surgery as an analgesic. One end of a silastic catheter was inserted 33 mm into the external right jugular and secured with 4.0 silk sutures. The other end ran subcutaneously and exited from a small incision just below the scapula. This end is attached to an infusion harness (Instech Solomon, Plymouth Meeting, PA, USA) that provides access to an external port for i.v. drug delivery. Following this surgical procedure, rats were given a subcutaneous injection of antibiotic solution of Cefazolin (10 mg/0.1 ml; Schein Pharmaceuticals, Florham Park, NJ, USA) and allowed to recover for 5 days.
During self-administration, rats received an i.v. infusion (0.1 ml) of 10 U/ml heparinized saline before each session. After each session, catheters were flushed with cefazolin and 0.1 ml 70 U/ml heparinized saline. Catheter patency was periodically verified with methohexital sodium (10 mg/ml dissolved in 0.9% physiological saline), a short-acting barbiturate that produces a rapid loss of muscle tone when administered i.v.. Daily 2-h sessions occurred in a fixed ratio schedule of reinforcement. The house light signaled the beginning of a session and remained on throughout the session. During the sessions, a response on the active lever resulted in activation of the pump for a 2-s infusion (50 μl bolus infusion) and presentation of a stimulus complex consisting of a 5-s tone (78 dB, 4.5 kHz) and a white stimulus light over the active lever, followed by a 20-s time out. Responses occurring during the time out and on the inactive lever were recorded, but had no scheduled consequences. Rats were trained initially to press a lever for a sucrose pellet (45mg) in a single 6 h session. The following day, a response on the lever no longer resulted in sucrose reward, but instead produced an i.v. infusion of JT09 (20 mg/kg/inf). After five days, JT09 was replaced with cocaine (50ug/50ul bolus infusion) and the lever replaced with a nose poke aperture. We chose cocaine to assess reward processing because it is readily administered to rats and is not considered an opioid compound. This way we can ensure that lever responding for cocaine was not impacted by prior experience with opioids (i.e., JT09).
2.6. Conditioned Place Preference
To test whether JT09 was able to condition appetitive behaviors, we used a conditioned place procedure (Prus A.J., 2009). On day one, rats were habituated to a three-compartment apparatus for 10 min. One compartment was black with a grid floor and the other white with a rod floor. A smaller center compartment was gray with a solid floor. The amount of time spent in each compartment was recorded. The side in which the rats spent the least amount of time on habituation day was paired with JT09, in that rats receive the compound via oral gavage immediately before compartment placement. On alternating days, rats were treated with saline and confined in the opposite compartment. Rats were confined to each compartment for 25 min. Test rats were orally gavaged with saline and given access to the entire apparatus for 10 min. The amount of time spent in each compartment was recorded.
2.7. Forced Swim Assay
The most common reason for discontinuing development of kappa opioid agonists is the induction of depressant-like effects at elevated doses, which is mediated through kappa opioid receptors found in the CNS (Braida et al., 2009; Carlezon et al., 2006). To test for this side effect, we used an established model: the repeated forced swim assay (Paul et al., 1990; Porsolt et al., 1979). Rats were placed in a container with 30°C water without an escape avenue. Time spent immobile during the last 4 min of each trial was recorded. Immobility was defined by a posture in which the forelimbs were motionless in front of the body, hind legs display limited motion, and the tail is directed outward (Pattinson et al., 2009). On day one, rats were placed in the water for 15 min. On the following day, placements consisted of four trials lasting 6 min each, separated by 10 mins. Salvorinorin A (1 mg/kg, i.p.), saline (0.2 ml, p.o.), or JT09 (20 mg/kg, p.o.) were administered immediately before the first placement. Time spent immobile is a diagnostic of depressant activity and typically increases with each trial. Salvinorin A is a centrally-available kappa agonist was used as a positive control due to its known depressant-like effects at high dose levels, such as dysphoria (Carlezon et al., 2006; Curzon et al., 2009).
2.8. Locomotor Open Field Test
Sedation and a loss of muscle coordination are common CNS-mediated side effects of opioids. To assess the sedative effects of JT09, rats were given JT09 (20 mg/kg, p.o.), saline (0.2 ml, p.o.), or morphine (10 mg/kg, i.p.) 20 min before placement in an automated (68 l × 21 w × 21 h cm) activity chambers for a 30 min test. Morphine was used as a comparison group due to its well-known sedative effects.
2.9. Maximum Tolerated Dose Determination
In order to determine the maximum dose of JT09 that can be tolerated, its dose level was increased until the maximum tolerated dose (MTD) was identified or, if MTD dose was not reached, the maximum-administered dose (MAD) (Le Tourneau et al., 2010). The MTD is a dose that does not produce mortality, a loss of more than 10% body weight, or overt signs of toxicity. Six rats were included at each dose level (50, 70, and 90 mg/kg, p.o.). The animals were observed twice daily; body weights and detailed clinical observations for behavioral toxicity (porphyrin staining, changes in activity levels, changes in grooming habits, convulsions, catalepsy, muscular rigidity, and excessive vocalization) were made daily for 4 days after dosing at each level. Doses were chosen based on the compound’s ED90 for analgesic potency.
2.10. Multiple Dose Study
Rats were orally treated with JT09 daily at its ED90 (20 mg/kg) for 14 consecutive days. Tolerance to the analgesic effects of the lead compounds was be assessed using the acetic acid-induced writhing assay (described above). Daily clinical observations for behavioral toxicity (described above) were made during the course of the experiment and body weight and food consumption recorded on study days 3, 5, 7, 9, 11 and 13. On day 15, animals were killed in their home cages by carbon dioxide inhalation from a compressed gas tank. A gross necroscopic analysis was immediately performed following euthanasia.
2.11. Statistics
To test for significant differences in the acetic acid-induced rat writhing assays and hot plate assays (Figure 3) a Student’s t-test lead compounds JT09 and JT07 to the control groups using Graphpad Prism software. Results were considered significantly different if P<0.05. Conditioned Place Preference (CPP) was analyzed with a Student’s t-test to compare the amount of time rats spent in each compartment. Self-administration, locomotor, and forced swim data were analyzed by two-way mixed analysis of variance (ANOVA) with group JT09 and control as a between subjects variable and session/time as within subjects variables. Post hoc comparison to control for family wise error were conducted with Dunnet’s or Sidak’s when appropriate.
Figure 3. Acetic Acid-Induced Writhing Assay.

Screen of lead compounds in the acetic acid-induced writhing assay in rats. Results are shown as the mean ± S.E.M. with n = 6–8 for each compound. Asterisk indicates that the value is not significantly different from the morphine control value by Student’s T-Test, *P<0.05.
2.12. Data Availability
Individual animal data is publicly available (Beck 2019).
3. Results
3.1. Screening of Potential Orally-Available KOAs
As a biological screen, the potential for oral availability of the eight new peptides in our library were evaluated in the acetic acid-induced writhing assay for peripheral pain. Rats were administered the various compounds by oral gavage at a screening dose of 20 mg/kg 20 min before i.p. injection with 2 ml/kg of 3% acetic acid. Ten mins later, animals were assessed for writhing, as described in the methods section. As shown in Figure 3, the JT Pharma compounds JT07 and JT09 were able to significantly block outward physical signs of peripheral pain in this model. Thus, several of the Arg modifications can impart CR665 with the ability to cross the gut barrier. In addition, JT07 and JT09 were statistically significantly different from saline when assessing the percentage of time writhing (Student’s t-test, P<0.05), and were indistinguishable in potency when compared to morphine (Student’s t-test, P<0.05). Manipulation of lead compounds JT07 and JT09 did not lead to the development of improved second-generation leads [Figure S1A]. Thus, JT07 and JT09 were selected for further analysis.
3.2. Screening of CNS-Mediated Pain in KOAs
As an initial screen for analgesic activity, JT07 and JT09 were evaluated in rats using the hotplate analgesic model using a standard oral dose of 20 mg/kg. Percent maximum possible effect was analyzed for each compound and compared to morphine (10 mg/kg, i.p.). JT07 and JT09 did not show analgesic activity and were significantly different from morphine (Student’s t-test, P<0.05) [Figure S1B].
3.3. Screening of CNS-Mediated Side Effects
3.3.1. Self-administration
In an operant self-administration procedure in which rats are required to press a lever to receive an intravenous drug infusion, JT09 failed to maintain lever responding in rats over a five-day period [Figure 4]. The number of infusions decreased on all 4 days compared to day one of JT09 administration [F(4,28)=9.04, P<0.0001, and Dunnett post hoc, P<0.05]. Further, to ensure that these rats were not deficient in reward processing, we replaced JT09 with cocaine using a nose-poke operandi and the number of cocaine infusions increased over the 7 days [F(6,42)=4.6, P<0.0012] with significantly more infusion on days 6 and 7 (Dunnett post hoc, P<0.05).
Figure 4. Self-Administration Procedure.

Mean active lever presses (n=8) during two-hour session operant self-administration sessions. JT09 failed to maintain lever responding in rats over a five-day period [Figure 3]. The number of infusions decreased on all 4 days compared to day one of JT09 administration [F(4,28)=9.04, P<0.0001, and Dunnett post hoc, P<0.05]. Further, to ensure that these rats were not deficient in reward processing, we replaced JT09 with cocaine using a nose-poke operandi and the number of cocaine active lever presses increased over the 7 days [F(6,42)=4.6, P<0.0012] with significantly more active lever presses on days 6 and 7 (Dunnett post hoc, P<0.05).
3.3.2. Conditioned Place Preference
To further establish that JT09 does not have rewarding properties, rats were tested for the amount of time spent in an environment previously associated with JT09 compared to a saline paired compartment. After receiving alternate treatment of JT09 and saline over the course of eight days, rats did not develop a preference for JT09 over saline [Figure 5].
Figure 5. Conditioned Place Preference Procedure.

JT09 (20 mg/kg, p.o.) had no effect on compartment placement in a conditioned place preference procedure. Baseline preferences for each compartment (black bar) were assessed prior to conditioning and did not change following drug treatment (*P < 0.05).
3.3.3. Forced Swim
As an initial screening for depressant-like effects, rats were evaluated in the forced swim model. Saline and JT09 were statistically indistinguishable in all time bins (Student’s t-test, P<0.05). There was a significant interaction between salvinornin A (Sal A), a centrally active kappa KOA that induces depressant-like effects such as dysphoria, and JT09 [Figure 6, F(3,30)=117, P<0.0001], specifically rats treated with JT09 had lower amounts of time spent immobile relative to Sal A during all trials (Sidak’s multiple comparison, P<0.05). Further, the main effect of treatment [F(1,10)=947, P<0.0001] and time [F(3,30)=418, P<0.0001] were also significant.
Figure. 6. Forced Swim Assay.

Immobility time (s) during the last trial of the forced swim test, 30 min after a single dose of JT09 (20 mg/kg, p.o.), salvinorin A (1 mg/kg, i.p.) or saline (2 ml, p.o.). Saline and JT09 were statistically indistinguishable in each trial (Student’s t-test, P<0.05). There was a significant interaction between Sal A and JT09 [Figure 5, F(3,30)=117, P<0.0001], specifically rats treated with JT09 had lower amounts of time spent immobile relative to Sal A during all trials (Sidak’s multiple comparison, P<0.05). Further, the main effect of treatment [F(1,10)=947, P<0.0001] and time [F(3,30)=418, P<0.0001] were also significant. Data are expressed as mean ± S.E.M. with n = 8.
3.3.4. Locomotor Activity
As a measure of sedation, rats were placed in activity chambers and distance traveled was evaluated. Saline and JT09 were statistically indistinguishable in all time bins (Student’s t-test, P<0.05). There was a significant interaction between morphine and JT09 [Figure 7, F(5,70)=7.0, P<0.0001], specifically JT09 had higher locomotor activity relative to morphine during time bins 1, 2, and 5 (Sidak’s multiple comparison, P<0.05). Further, the main effect of treatment [F(1,14)=18.6, P<0.0007] and time [F(5,70)=84, P<0.0001] were also significant.
Figure 7. Locomotor Activity.

Distance Traveled time (in centimeters) during activity tests, 30 min after a single dose of JT09 (20 mg/kg, p.o.), morphine (10 mg/kg, i.p.) or saline (2 ml, p.o.). Saline and JT09 were statistically indistinguishable in all time bins (Student’s t-test, P<0.05). There was a significant interaction between morphine and JT09 [Figure 6, F(5,70)=7.0, P<0.0001], specifically JT09 had higher locomotor activity relative to morphine during time bins 1, 2, and 5 (Sidak’s multiple comparison, P<0.05). Further, the main effect of treatment [F(1,14)=18.6, P<0.0007] and time [F(5,70)=84, P<0.0001] were also significant. Data are expressed as mean ± S.E.M.
3.4. Screening of Toxicity
3.4.1. Maximum Tolerated Dose Determination
The maximum tolerated dose (MTD) is used to determine the highest dose of JT09 that can be administered without promoting unacceptable side-effects. In rats, a maximum administered dose (MAD) of more than four times the EC90 of JT09 (90 mg/kg) did not produce mortality, a loss of more than 10% body weight, or overt signs of toxicity. These experiments are ongoing at higher dose levels.
3.4.2. Multiple Dose Study
Following the 14-day multiple dose study, six rats were submitted for necropsy. Upon necropsy, the lungs displayed a slight grey discoloration. The grey discoloration of the lungs can be due to hemorrhage, which was seen histologically. Hemorrhage is a common change seen due to euthanasia with CO2. The heart, stomach, intestines, liver, spleen, pancreas, kidneys, adrenal glands, skeletal muscle, bone, reproductive organs, and brain were all grossly normal. To further validate these data, we plan to repeat this study using morphine and saline controls for histological comparison.
4. DISCUSSION
Application of our laboratory’s peptide-modification strategy to the intravenously available, peripherally-restricted, kappa-opioid agonist (KOA) CR665 yielded derivatives with enhanced PK/PD properties; stability in biological fluids; and oral bioavailability. In a previous study, one particular lead, JT09, demonstrated remarkable results. JT09 displayed a potent kappa-opioid receptor selectivity of >33,400 fold; a peripheral selectivity of >938 fold; an oral EC50 of 4.7 mg/kg; and an intravenous (i.v.) EC50 of 0.032 mg/kg. While further modification of JT09 could be contemplated, one difficulty in the design of future compounds is that a structure-activity (SAR) relationship is not apparent for the set of compounds evaluated (Hughes et al., 2013). However, the efficacy of orally delivered JT09 for the treatment of peripherally-derived pain (which is of similar magnitude to morphine) supports further evaluation of its general characteristics and potential side effects that make morphine a less than ideal analgesic.
Both JT09 and morphine exhibit comparable analgesic activity in the acetic acid-induced writhing model for peripheral pain. However, in the hot plate model of centrally mediated pain, morphine was a potent analgesic, while JT09 exhibited no analgesic effects at the highest concentrations we were able to test. This indicates that administration of JT09 does not significantly attenuate centrally-mediated pain, as it is unable to cross the blood brain barrier. Other potential centrally-mediated side effects of morphine, that combine to make it a less than an ideal drug, were evaluated with JT09. As previously mentioned, addiction is a major issue associated with centrally-acting analgesics, such as morphine. Two different testing methods were utilized to examine the abuse liability of JT09. The most reliable test of abuse liability is a contingent drug self-administration model, in which rats are trained to press a lever for intravenous drug delivery. JT09 failed to maintain lever responding in rats over a five-day period, with the number of infusions decreasing over the last four days in comparison to day one. Positive controls demonstrated that the rats were not deficient in reward processing, as cocaine administration resulted in the expected increase in the number of lever presses over the seven-day period of the procedure. In addition, we employed a conditioned place preference model to ensure that JT09 does not exhibit rewarding properties. Rats did not develop a preference for JT09 over saline, thus further establishing that JT09 lacks rewarding properties.
Evaluation of early KOAs, including non-peptidic compounds U50,488, enadoline, ADL 10–0101, and ADL 10–0116, demonstrated inadequate peripheral versus central distribution that led to centrally-mediated dysphoria and sedation, which forced discontinuation of development. Thus, we employed two experiments to test for the induction of these properties. To test for depressant-like effects such as dysphoria, we examined rats in the forced swim assay, comparing JT09 to salvinorin A (a centrally-active KOA). Depressant effects are measured in the amount of time spent immobile during the last four mins of each test trial. Administration of JT09 did not induce more than a baseline time of immobility, in contrast to rats administered salvinornin A. As a measure of sedation, we used locomotor boxes to determine the activity levels of rats after receiving JT09 and morphine, which is highly sedative as a result of its ability to act centrally. There was a significant difference between morphine- and JT09-treated rats; specifically, JT09 showed no sedation, whereas morphine was highly sedative. Thus, JT09 does not appear to elicit sedation due to its high peripheral selectivity, resulting in a compound that improves in this critical property over early KOAs and morphine. The compounds being developed by our lab and Cara Therapeutics are the only KOAs so far reported that are sufficiently peripherally-restricted, in contrast to other potential KOA drugs, morphine, and their analogs.
In conclusion, this study provides data to demonstrate that JT09 is orally active and peripherally restricted, in contrast to morphine and other KOAs that were previously evaluated. The oral EC90 of JT09 in rats demonstrates a similar efficacy to morphine in alleviating peripheral pain and appears to be of sufficient potency to be druggable when extrapolated to human doses. Our key finding is that JT09 does not promote the negative CNS-mediated effects associated with opioids, including sedation, dysphoria, tolerance, and addiction. Thus, we suggest that JT09 is a candidate for development as a novel analgesic for chronic peripheral pain that would not have the negative side-effects of currently used analgesics.
Supplementary Material
Acknowledgement
Disclosure: This work was supported by NIH grants DA-036398, NS-090629 and MH-65099 to Thomas A. Dix and a grant from SCE&G to Tyler C. Beck. Tyler Beck, Carmela Reichel, Kristi Helke, and Sanat Bhadsavle do not declare a conflict of interest. Thomas A. Dix is the Chief Scientific Officer of JT Pharmaceuticals, Inc. All experiments and data analysis were performed at the Medical University of South Carolina, independent of JT Pharmaceuticals, Inc.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement
Tyler Beck, Carmela Reichel, Kristi Helke, and Sanat Bhadsavle do not declare a conflict of interest. Thomas A. Dix is the Chief Scientific Officer of JT Pharmaceuticals, Inc. All experiments and data analysis were performed at the Medical University of South Carolina, independent of JT Pharmaceuticals, Inc.
References
- American Society of Addiction Medicine, 2019. Opioid addiction 2016: Facts and figures. Opioid Overdose Crisis. National Institute on Drug Abuse. https://www.asam.org/docs/default-source/advocacy/opioid-addiction-disease-facts-figures.pdf. (accessed 21 December 2018).
- Barber A, Gottschlich R, 1992. Opioid agonists and antagonists: an evaluation of their peripheral actions in inflammation, Med. Res. Rev. 12, 525–62. 10.1002/med.2610120505 [DOI] [PubMed] [Google Scholar]
- [dataset] Beck TC, 2019. “Raw Data and Data Analysis Assessing a Non Addictive Orally Active Kappa Opioid Agonist (JT09) in Pain and Behavioral Models in Rats.” Mendeley Data, v1. [DOI]
- Braida D, Capurro V, Zani A, Rubino T, Vigano D, Parolaro D, Sala M, 2009. Potential anxiolytic- and antidepressant-like effects of salvinorin A, the main active ingredient of Salvia divinorum, in rodents, Br. J. Pharmacol. 157, 844–53. 10.1111/j.1476-5381.2009.00230.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA Jr., Beguin C, DiNieri JA, Baumann MH, Richards MR, Todtenkopf MS, Rothman RB, Ma Z, Lee DY, Cohen BM, 2006. Depressive-like effects of the kappa-opioid receptor agonist salvinorin A on behavior and neurochemistry in rats, J. Pharmacol. Exp. Ther. 316, 440–7. 10.1124/jpet.105.092304 [DOI] [PubMed] [Google Scholar]
- Curzon P, Zhang M, Radek RJ Fox GB, 2009. The Behavioral Assessment of Sensorimotor Processes in the Mouse: Acoustic Startle, Sensory Gating, Locomotor Activity, Rotarod, and Beam Walking in Buccafusco JJ (eds.), Methods of Behavior Analysis in Neuroscience, second ed Boca Raton, Florida. [PubMed] [Google Scholar]
- DeHaven-Hudkins DL, Dolle RE, 2004. Peripherally restricted opioid agonists as novel analgesic agents, Curr. Pharm. Des. 10, 743–57. 10.2174/1381612043453036 [DOI] [PubMed] [Google Scholar]
- Gaskin DJ, Richard P, 2011. The economic costs of pain in the United States. Institute of Medicine (US) Committee on Advancing Pain Research, Care and Education., Appendix C. 10.1016/j.jpain.2012.03.009 [DOI] [Google Scholar]
- Goldberg DS, McGee SJ, 2011. Pain as a global public health priority, BMC Public Health. 11, 770 10.1186/1471-2458-11-770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadden MK, Orwig KS, Kokko KP, Mazella J, Dix TA, 2005. Design, synthesis, and evaluation of the antipsychotic potential of orally bioavailable neurotensin (8–13) analogues containing non-natural arginine and lysine residues, Neuropharmacology. 49, 1149–59. https://doi.org/10.1016Zj.neuropharm.2005.06.010 [DOI] [PubMed] [Google Scholar]
- Hughes FM Jr., Shaner BE, Brower JO, Woods RJ, Dix TA, 2013. Development of a Peptide-derived orally-active kappa-opioid receptor agonist targeting peripheral pain, Open Med. Chem. J. 7, 16–22. 10.2174/1874104501307010016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy KJ, Lundquist JT, Simandan TL, Kokko KP, Beeson CC, Dix TA, 2000. Design rationale, synthesis, and characterization of non-natural analogs of the cationic amino acids arginine and lysine, J. Pept. Res. 55, 348–58. 10.1034/j.1399-3011.2000.00688.x [DOI] [PubMed] [Google Scholar]
- Kennedy KJ, Orwig KS, Dix TA, Christopher J, Jaffa AA, 2002. Synthesis and analysis of potent, more lipophilic derivatives of the bradykinin B2 receptor antagonist peptide Hoe 140, J. Pept. Res. 59, 139–48. 10.1034/j.1399-3011.2002.1o987.x [DOI] [PubMed] [Google Scholar]
- Kennedy KJ, Simandan TL, Dix TA, 1997. A facile route to cyclic and acyclic alkyl-arginines, Syn. Comm. 28, 741 10.1080/00397919808005947 [DOI] [Google Scholar]
- Kennedy KJ, Lundquist JT, Simandan TL, Beeson CC, Dix TA, 1997. Asymmetric synthesis of non-natural homologues of lysine, Bioorg. Med. Chem. 7, 1937–40. 10.1016/S0960-894X(97)00335-1 [DOI] [Google Scholar]
- Kokko KP, Arrigoni CE, Dix TA, 2001. Selectivity enhancement induced by substitution of non-natural analogues of arginine and lysine in arginine-based thrombin inhibitors, Bioorg. Med. Chem. Lett. 11, 1947–50. 10.1016/S0960-894X(01)00328-6 [DOI] [PubMed] [Google Scholar]
- Kokko KP, Hadden MK, Orwig KS, Mazella J, Dix TA, 2003. In vitro analysis of stable, receptor-selective neurotensin[8–13] analogues, J. Med. Chem. 46, 4141–8. 10.1021/jm0300633 [DOI] [PubMed] [Google Scholar]
- Kokko KP, Hadden MK, Price KL, Orwig KS, See RE, Dix TA, 2005. In vivo behavioral effects of stable, receptor-selective neurotensin[8–13] analogues that cross the blood-brain barrier, Neuropharmacology. 48, 417–25. 10.1016/j.neuropharm.2004.10.008 [DOI] [PubMed] [Google Scholar]
- Kokko KP, Hooper BH, Dix TA, 2004. Synthesis of cyclic and acyclic Nα-methyl-Nω-alkyl-L-arginine analogues, Tet. Letts. 45, 2151–53. 10.1016/j.tetlet.2004.01.047 [DOI] [Google Scholar]
- Kumar N 2014. Kappa opioid receptor agonists: New targets in treatment of pain, University of Wellington; Retrieved from https://pdfs.semanticscholar.org/aa4e/24a343bdeac410ea20a53dcc815516ed16e8.pdf. [Google Scholar]
- Le Tourneau C, Stathis A, Vidal L, Moore MJ, Siu LL, 2010. Choice of starting dose for molecularly targeted agents evaluated in first-in-human phase I cancer clinical trials, J. Clin. Oncol. 28, 1401–7. 10.1200/JCO.2009.25.9606. [DOI] [PubMed] [Google Scholar]
- Lundquist JT, Bullesbach EE, Golden PL, Dix TA, 2002. Topography of the neurotensin (NT)(8–9) binding site of human NT receptor-1 probed with NT(8–13) analogs, J. Pept. Res. 59, 55–61. 10.1046/j.1397-002x.2001.10946.x [DOI] [PubMed] [Google Scholar]
- Lundquist JT, Dix TA, 1998. Asymmetric synthesis of [omega]-bromo-2(S)-azido acids as precursors for the synthesis of novel amino acids. Tet. Letts. 39, 775 10.1016/S0040-4039(97)10608-6 [DOI] [Google Scholar]
- Lundquist TJ, Orwig KS, Dix TA, 1999. Synthesis of Ethylene-Bridged (Nδ to Nω)Analogues of Arginine, J. Org. Chem. 64, 9265–67. 10.1021/jo990551b [DOI] [Google Scholar]
- Nitu AN, Wallihan R, Skljarevski V, Ramadan NM, 2003. Emerging trends in the pharmacotherapy of chronic pain, Expert Opin. Investig. Drugs. 12, 545–59. 10.1517/13543784.12A545 [DOI] [PubMed] [Google Scholar]
- Orwig KS, Dix TA, 2005. Synthesis of Ca methylated carboxylic acids: Isosteres of arginine and lysine for use as N-terminal capping residues in polypeptides, Tet. Letts. 46, 7007–09. https://doi.org/10.1016Zj.tetlet.2005.08.056 [Google Scholar]
- Pattinson KT, Governo RJ, Macintosh BJ, Russell EC, Corfield DR, Tracey I, Wise RG, 2009. Opioids depress cortical centers responsible for the volitional control of respiration, J. Neurosci. 29, 8177–86. 10.1523/JNEUR0SCI.1375-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul IA, Duncan GE, Kuhn C, Mueller RA, Hong JS, Breese GR, 1990. Neural adaptation in imipramine-treated rats processed in forced swim test: assessment of time course, handling, rat strain and amine uptake, J. Pharmacol. Exp. Ther. 252, 997–1005. PMID: [PubMed] [Google Scholar]
- Pharmaceutical Product Development for Pruritus & Pain, 2018. Cara Therapeutics.https://www.caratherapeutics.com/ (accessed 21 November 2018).
- Porsolt RD, Bertin A, Blavet N, Deniel M, Jalfre M, 1979. Immobility induced by forced swimming in rats: effects of agents which modify central catecholamine and serotonin activity, Eur. J. Pharmacol. 57, 201–10. 10.1016/0014-2999(79)90366-2 [DOI] [PubMed] [Google Scholar]
- Prus AJ, James JR, Rosecrans JA, 2009. Conditioned Place Preference in Buccafusco JJ (eds.), Methods of Behavior Analysis in Neuroscience, second ed. Boca Raton, Florida. [Google Scholar]
- Riviere PJ-M, 2004. Peripheral kappa-opioid agonists for visceral pain, Br. J. Pharmacol. 8, 1331–34. 10.1038/sj.bjp.0705763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein SC, Clark DJ, Oh U, Vasko MR, Wilcox GL, Overland AC, Vanderah TW, Spencer RH, 2008. Peripheral mechanisms of pain and analgesia, Brain Res. Rev. 60, 90 10.1016/j.brainresrev.2008.12.017. http:// [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ree JM, Slangen JL, de Wied D, 1978. Intravenous self-administration of drugs in rats, J. Pharmacol. Exp. Ther. 204, 547–57. PMID: [PubMed] [Google Scholar]
- Vanderah TW, Schteingart CD, Trojnar J, Junien JL, Lai J, Riviere PJ, 2004FE200041 (D-Phe-D-Phe-D-Nle-D-Arg-NH2): A peripheral efficacious kappa opioid agonist with unprecedented selectivity, J. Pharmacol. Exp. Ther. 310, 326–33. 10.1124/jpet.104.065391 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Individual animal data is publicly available (Beck 2019).