

Mycobacteria are common inhabitants of soil, and while most members of this bacterial group are innocuous, some mycobacteria can cause environmentally acquired infections of humans and other animals. Human infections from nontuberculous mycobacteria (NTM) are increasingly prevalent worldwide, and some areas appear to be “hotspots” for NTM disease. While exposure to soil is frequently implicated as an important mode of NTM transmission, the diversity, distributions, and ecological preferences of soil mycobacteria remain poorly understood. We analyzed 143 soils from a range of ecosystems and found that mycobacteria and lineages within the group often exhibited predictable preferences for specific environmental conditions. Soils harbor large amounts of previously undescribed mycobacterial diversity, and lineages that include known pathogens were rarely detected in soil. Together, these findings suggest that soil is an unlikely source of many mycobacterial infections. The biogeographical patterns we documented lend insight into the ecology of this important group of soil-dwelling bacteria.
KEYWORDS: Mycobacterium, mycobacteria, soil microbiology
ABSTRACT
Mycobacteria are a diverse bacterial group ubiquitous in many soil and aquatic environments. Members of this group have been associated with human and other animal diseases, including the nontuberculous mycobacteria (NTM), which are of growing relevance to public health worldwide. Although soils are often considered an important source of environmentally acquired NTM infections, the biodiversity and ecological preferences of soil mycobacteria remain largely unexplored across contrasting climates and ecosystem types. Using a culture-independent approach by combining 16S rRNA marker gene sequencing with mycobacterium-specific hsp65 gene sequencing, we analyzed the diversity, distributions, and environmental preferences of soil-dwelling mycobacteria in 143 soil samples collected from a broad range of ecosystem types. The surveyed soils harbored highly diverse mycobacterial communities that span the full extent of the known mycobacterial phylogeny, with most soil mycobacteria (97% of mycobacterial clades) belonging to previously undescribed lineages. While mycobacteria tended to have higher relative abundances in cool, wet, and acidic soil environments, several individual mycobacterial clades had contrasting environmental preferences. We identified the environmental preferences of many mycobacterial clades, including the clinically relevant Mycobacterium avium complex that was more commonly detected in wet and acidic soils. However, most of the soil mycobacteria detected were not closely related to known pathogens, calling into question previous assumptions about the general importance of soil as a source of NTM infections. Together, this work provides novel insights into the diversity, distributions, and ecological preferences of soil mycobacteria and lays the foundation for future efforts to link mycobacterial phenotypes to their distributions.
IMPORTANCE Mycobacteria are common inhabitants of soil, and while most members of this bacterial group are innocuous, some mycobacteria can cause environmentally acquired infections of humans and other animals. Human infections from nontuberculous mycobacteria (NTM) are increasingly prevalent worldwide, and some areas appear to be “hotspots” for NTM disease. While exposure to soil is frequently implicated as an important mode of NTM transmission, the diversity, distributions, and ecological preferences of soil mycobacteria remain poorly understood. We analyzed 143 soils from a range of ecosystems and found that mycobacteria and lineages within the group often exhibited predictable preferences for specific environmental conditions. Soils harbor large amounts of previously undescribed mycobacterial diversity, and lineages that include known pathogens were rarely detected in soil. Together, these findings suggest that soil is an unlikely source of many mycobacterial infections. The biogeographical patterns we documented lend insight into the ecology of this important group of soil-dwelling bacteria.
INTRODUCTION
Mycobacteria are a diverse and ubiquitous group of actinobacteria that have been well-studied given their potential importance as pathogens and their prevalence in a wide variety of habitats, including many soil and aquatic environments (1). Obligate pathogens Mycobacterium tuberculosis and M. leprae are perhaps the best-studied mycobacteria, but there are almost 200 additionally described mycobacterial species that are collectively referred to as nontuberculous mycobacteria (NTM) (2, 3). The members of this group are phylogenetically diverse, and there is recent genomic evidence to suggest that the genus Mycobacterium should actually be split into 5 different genera (4). Here, we studied the entire group, referring to the group as “NTM” or “mycobacteria” for reasons detailed in Materials and Methods.
Most NTM are innocuous to humans, and some have metabolic capabilities that make them potentially useful for the biodegradation of environmental pollutants (5, 6) or as industrial biocatalysts (7). Likewise, exposures to several NTM strains have been linked to positive human health outcomes via immune system regulation and modulation of serotonin pathways (8, 9). However, a subset of NTM are important pathogens of humans and animals (10, 11). NTM infections are predominantly acquired from the environment and are increasingly recognized as a threat to public health worldwide (12). Whether pathogenic or beneficial mycobacteria, water and soil are widely considered their predominant environmental reservoirs (1, 11, 13). A growing number of studies have focused on NTMs in aquatic systems, including residential plumbing systems (14–18), lakes (19, 20), and other aquatic environments (21–25). However, much less is known about the biodiversity and ecological preferences of NTM in soil and the relevance of soil-derived NTM to human and environmental health.
Mycobacteria are commonly detected across a broad range of soil types using both cultivation-dependent and cultivation-independent methods. We know from cultivation-independent surveys of soil bacterial communities that mycobacteria are nearly ubiquitous in soil and are consistently one of the more abundant genera of soil bacteria, ranging in relative abundance from ∼0.5% to 3.0% of the total community (26–28). Mycobacterial species have been detected in soils from bogs (29), forests (20, 30, 31), croplands (32, 33), and livestock farms (34–36), and even in potting soil (37). However, much less is known about the ecological preferences of soil mycobacteria. Current knowledge derived from a few studies suggests that mycobacteria are particularly abundant in high-latitude boreal forests (20, 31), with some research suggesting higher abundances in more acidic soils (24, 29, 32). However, studies identifying the specific ecological preferences of mycobacteria at both the genus and species or strain level of resolution across a range of soil and ecosystem types are lacking.
There are three main reasons why we lack a comprehensive understanding of mycobacterial diversity and distributions in soils across the globe. First, the majority of studies investigating environmental mycobacteria have relied on cultivation-based approaches to survey abundances and diversity (25, 38–40). While such approaches are clearly useful for addressing specific research questions, typical cultivation-based approaches are likely to underestimate the mycobacterial diversity found in environmental samples due to well-known culturing limitations, including the difficulties associated with growing, isolating, and identifying many mycobacterial colonies in vitro (39, 41, 42). As such, it is likely that many environmental mycobacteria have remained uncharacterized (14, 43). Second, although PCR-based methods can be used to more broadly survey soil mycobacterial diversity (27, 34, 44, 45), most of these cultivation-independent studies have relied on the PCR amplification and sequencing of regions of the 16S rRNA gene, which often do not provide sufficient resolution to differentiate between distinct species or strains of mycobacteria (35, 44, 45). Alternate genetic markers, such as the heat shock protein (hsp65) gene, have proven useful for resolving closely related mycobacterial species, but to date these marker gene sequencing approaches have been used predominantly for clinical isolate identification (46–49). Third, preexisting studies have generally focused on a relatively small number of soil samples with limited geospatial coverage. A more comprehensive understanding of the diversity and distributions of soil mycobacteria will clearly benefit from cultivation-independent analyses that provide greater taxonomic and phylogenetic resolution of those mycobacteria found across a broad range of soil types.
To advance our understanding of the ecology and biogeography of soil-dwelling mycobacteria, we analyzed a broad range of distinct soils collected from a range of ecosystem types across the United States and other sites across the globe (143 locations in total) via cultivation-independent amplicon sequencing of two different marker genes (the 16S rRNA gene for genus-level analyses and the hsp65 gene for more detailed analyses of specific mycobacterial taxa). Using this extensive survey, we addressed the following questions: (i) which soils harbor the highest relative abundances of taxa within the Mycobacterium genus? (ii) how does the diversity of mycobacterial communities vary across contrasting ecosystem types? and (iii) what are the ecological preferences of individual mycobacterial lineages?
RESULTS
Global variation in relative abundance of genus Mycobacterium.
We first used universal 16S rRNA gene sequencing to assess the overall bacterial community composition and genus-level relative abundances of Mycobacterium spp. in each of 143 soil samples (described in reference 28). For the following analysis, we elected to maintain the historic definition of the Mycobacterium genus to include all mycobacterial taxa together in our genus-level comparisons. The soil samples were collected from a broad range of ecosystem types, including tropical rainforests, arctic tundra, temperate grasslands, and hot deserts (see Fig. S1 in the supplemental material; see Table S1 in the supplemental material). We found mycobacteria to be widespread and reasonably abundant across the broad range of soil types examined here. Mycobacterial 16S rRNA gene sequences (see Materials and Methods) could be detected in all 143 samples. Out of 398 identifiable named genera, Mycobacterium was the 12th most abundant genus across all soils by rank (top 3% of named genera), ranging from 2nd to 67th across individual soils (see Fig. S2 in the supplemental material). The relative abundance of the genus Mycobacterium was variable across the soils studied, ranging from 0.03% to 2.9% of all bacterial and archaeal 16S rRNA reads, with a median abundance of 0.52% (Fig. 1A).
FIG 1.
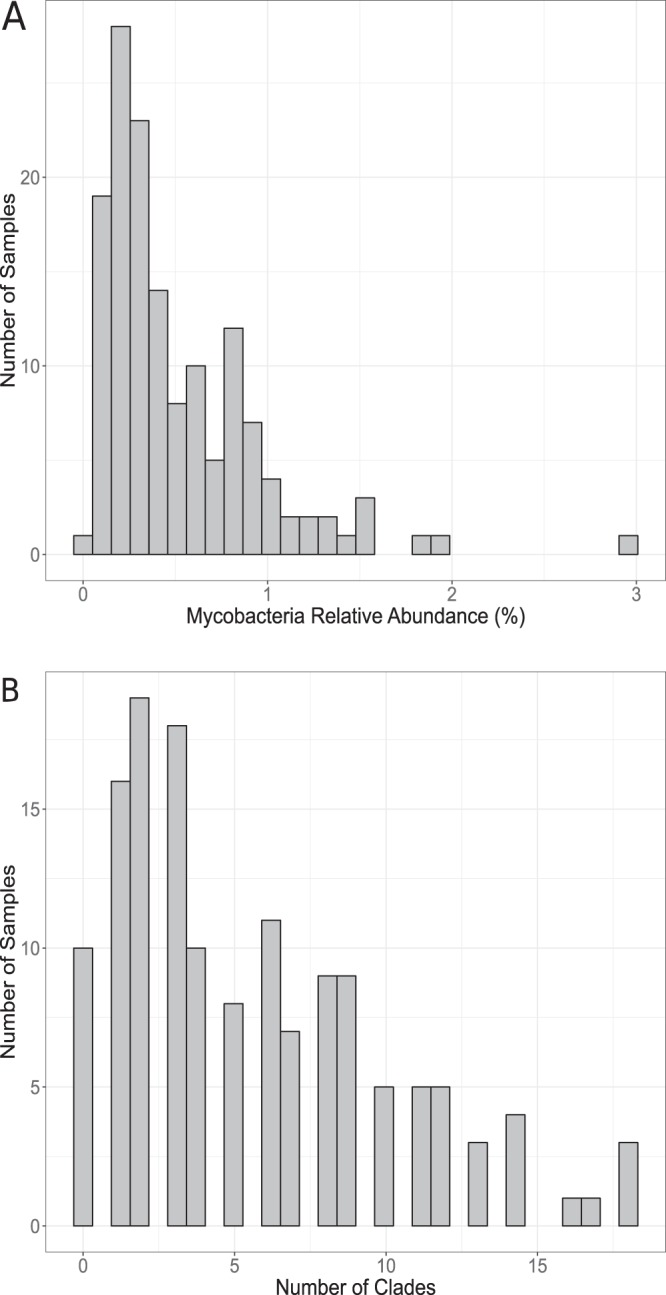
Distribution of mycobacterial relative abundances and clade richness across samples. Bars indicate either the number of samples containing the specified relative abundance of mycobacteria (% of all bacterial or archaeal 16S rRNA reads) (A) or the number of samples containing the specified number of mycobacterial clades (via hsp65 reads), which varies from 0 to 18 clades per sample (B).
The variation in the relative abundances of the genus Mycobacterium was, to some extent, predictable from the measured soil and site variables. We used random forest modeling (50) to identify the most important environmental predictors of the relative abundance of Mycobacterium spp. across our samples. We found that ∼25% of the variation in the relative abundance of Mycobacterium spp. could be explained by the environmental variables measured. Soil pH, aridity, and temperature were the most important predictors of mycobacterial relative abundances (see Table S3 in the supplemental material). Mycobacteria were typically more abundant in soils collected from cold and temperate forests (Fig. 2D). These patterns are consistent with the observation that mycobacteria generally had higher relative abundances in cooler, wetter, and more acidic soils (Fig. 2A to C). Across the 143 samples analyzed, mycobacterial relative abundances were, on average, higher in lower pH soils and in cooler climates (Fig. 2B and C). Mycobacterial relative abundances were lower in drier (i.e., more arid) environments (51, 52) (Fig. 2A).
FIG 2.

Relative abundances of mycobacteria across the 143 soils included in this study. Random forest analyses of 8 soil and site characteristics that described 25% of the variation in mycobacterial 16S rRNA gene relative abundances identified the most important environmental predictors as aridity index (A), soil pH (B), and minimum annual temperature (C). Boxplots show quartile ranges for each environmental category, with black dots indicating exact sample values. Significant differences between groups (determined using pairwise Wilcoxon signed-rank tests) are denoted by the asterisks with corresponding P values. Spearman correlations suggest that mycobacteria are more abundant in wetter, more acidic, and cooler soils. Aridity categories were based on site aridity index values: arid, 0 to 0.2; semiarid, 0.201 to 0.4; semihumid, 0.401 to 0.7; and Humid, 0.701 to 2.5. Categories for minimum annual temperature determined as cool, −35.0 to −9.0°C; temperate, −9.01 to 1.0°C; and warm, 1.0 to 22.0°C. (D) Differences in mycobacterial abundances across general ecosystem categories.
Global diversity of soil mycobacteria.
The aforementioned 16S rRNA analyses do not allow us to comprehensively discriminate between different species or strains of mycobacteria. This is an important limitation given that mycobacteria as a group contain a broad diversity of lineages with distinct phenotypic characteristics (4, 53, 54). Thus, we complemented the 16S rRNA gene analyses with amplicon sequencing of the hsp65 gene, which is widely used for phylogenetic analyses of the genus (55). The targeted region of the hsp65 gene is more variable across mycobacteria than the 16S rRNA gene region, and the hsp65 primers we used were designed to target mycobacteria (although amplification of the hsp65 gene region from other closely related actinobacteria commonly occurs). These complementary amplicon sequencing analyses allowed us to identify specific lineages of mycobacteria and their distributions across the 143 soils studied.
To streamline further analyses and investigate the ecological attributes of mycobacterial lineages, we used a phylogenetic approach to group closely related mycobacterial sequences (exact sequence variants [ESVs]) into phylogenetic clades. Across all soils, the 472 mycobacterial ESVs that met our criteria for inclusion in downstream analyses (see Materials and Methods) fell into 159 distinct clades, as identified by phylogenetic patristic distance. We clustered ESVs into these phylogenetic clades because many sequences uncovered here were divergent from those in existing reference databases, creating challenges in confidently assigning taxonomic classifications.
Most of these mycobacterial clades were restricted in their distributions, and the number of different clades found in a single soil sample was variable. A median of 4 clades were identified per individual soil sample (range, 0 to 18; mean, 5.5) (Fig. 1B). Mycobacterial hsp65 sequences were absent in 10 out of the 143 soil samples, which could be due to reduced amplification efficiencies or simply because the number of mycobacterial hsp65 reads fell below our threshold for inclusion in downstream analyses (see Materials and Methods). Most soil samples harbored only a few clades (1 to 3 clades), while a few samples harbored a large number of clades (15 to 18 clades) (Fig. 1B). No single clade was identified in all samples, and the most ubiquitous clade was found in 59 of the 143 soil samples (41% of soil samples). Together, these results highlight that a diversity of mycobacterial lineages can be found in individual soil samples and that most soil mycobacteria lineages are restricted in their distributions.
Many well-characterized mycobacterial species, including known pathogens associated with human NTM diseases (including M. abscessus, M. chelonae, M. fortuitum, and M. mucogenicum) (56, 57), were not detected in any of the 143 soil samples investigated here. In fact, the majority (97%) of soil-derived mycobacterial clades were so distantly related to described mycobacteria that they could not be assigned a species or strain identifier. When phylogenetically compared against a comprehensive mycobacterial database (55), only 5 of the 159 soil-derived clades clustered with known strains included in the Dai et al. database (55) (Fig. 3A). Of the five clades that included a described isolate, only one clade included a species frequently implicated in disease (Fig. 3B). This pathogenic clade was comprised of members of the M. avium complex (MAC), which includes M. avium, M. intracellulare, and M. chimaera. Representatives of the MAC clade were found in 15 of the 143 soil samples studied. The most ubiquitous and abundant clade, found in 59 soil samples, comprised ESVs closely related to M. novocastrense and M. rutilum (Fig. 3B).
FIG 3.

Phylogenetic tree representing described and previously undescribed mycobacterial hsp65 sequences. (A) A total of 472 exact sequence variants (ESVs) were identified in soils sampled here, a majority of which represent novel and undescribed taxa. These ESVs span the known phylogenetic diversity of the genus. Colors indicate reference mycobacterial strains (blue) from Dai et al. (55) and sequences recovered from soils in this study (orange). (B) Closely related ESVs were grouped into 159 clades based on patristic distance. The top 20 most ubiquitous clades are highlighted in color, with yellow colors indicating higher ubiquity (clades found in more soil samples). Four of the clades included described members, as indicated by red symbols, namely, clade 10 (M. stomatepiae, M. genavense, M. florentinum, M. lentiflavum, M. montefiorense, and M. triplex), clade 12 (M. novocastrense and M. rutilum), clade 31 (including M. intracellulare, M. avium subsp. paratuberculosis, M. avium subsp. silvaticum, M. colombiense, M. chimaera, and M. avium subsp. avium), and clade 44 (M. doricum and M. monacense). Small black triangles mark sequences from the reference database. Both trees are rooted with the hsp65 sequence from Nocardia farcinica (DSM43665).
We next assessed the geospatial distributions of the dominant mycobacterial clades and investigated the environmental conditions that best predict what clades are found in which soil types.
Biogeography of the dominant mycobacterial lineages.
Although most of the soil mycobacterial lineages identified were not closely related to described mycobacterial taxa, we explored their biogeographical distributions to gain insights into their ecologies. Likewise, by relating the presence of specific mycobacterial clades to soil or site characteristics we identified “hotspots” of specific mycobacterial lineages that include potential pathogens or pollutant degraders. For these biogeographical analyses, we focused on the top 20 most ubiquitous clades (i.e., clades that were found in 10 or more soil samples) and used random forest modeling to identify the most important environmental predictors of presence or absence for each clade. Soil pH, aridity, and temperature were consistently the most important predictors of the distributions of ubiquitous mycobacterial clades (Table S3). Distributions for 13 of the 20 clades could be predicted from the measured variables using random forest modeling, with the majority (12/13) of these predictable clades most likely to be found in soils that are acidic and receive large amounts of precipitation. This finding agrees with the environmental preferences suggested by the genus-level 16S rRNA gene sequence data described above (Fig. 2). However, not all clades shared the same directional relationship with the measured environmental variables, highlighting the diversity and distinct environmental preferences within mycobacteria (Fig. 4; see Table S4 in the supplemental material). For example, the M. rutilum clade (clade 12) was more likely to be present in higher pH soils, while the majority of other tested clades (13/20) were more likely to be present in lower pH soils (Fig. 4). For a comprehensive list of all significant environmental predictors for each clade, see Table S4. The M. avium complex (MAC) clade that includes known human pathogens was more likely to be found in wetter, more acidic soils (Fig. 4). While the number of samples containing members of this clade were too low to allow for robust modeling, Spearman correlations and Wilcoxon rank-sum tests support this apparent pattern (pH, R = −0.41; aridity index, R = 0.39).
FIG 4.

Environmental preferences of selected mycobacterial clades. The environmental conditions associated with the presence of each clade shown here are significantly different from conditions in which the clade was absent, as determined by both Wilcoxon signed-rank tests and Spearman correlations (P < 0.05). Environmental variables that were consistently the most important for predicting clade presence in random forest models were aridity index (A), soil pH (B), and minimum annual temperature (C). Clades with notably strong environmental preferences are visualized in Fig. 1, and a comprehensive list of all significant relationships with supporting statistics is provided in Table S4 and S5. Three of these mycobacterial clades, namely, clade 10, clade 12, and clade 31, included described members (for a detailed list of named species, see Fig. 3 or Table S3).
Geographic location is not independent from the environmental variables tested here, and our sampling scheme was not designed to explicitly test for dispersal limitations. Thus, while we cannot rule out the possibility that the biogeographical patterns are influenced by potential dispersal constraints, we still found multiple associations between environmental factors and clade distributions. To strengthen this message, we conducted further partial correlation analyses to account for any potential influence of geographic location (latitude and longitude) on the reported associations between major environmental predictors (pH, aridity, and temperature) and the distributions of mycobacterial clades. Our results provide further evidence that environmental factors, such as pH, aridity, and temperature, are associated with the distributions of mycobacteria after controlling for geospatial location (see Table S5 in the supplemental material). In fact, our analyses indicate that geospatial location alone did not consistently correlate with clade distributions and that controlling for geospatial location did not typically alter the strength of the relationships between clade distributions and the environmental predictors (Table S5).
DISCUSSION
We found mycobacteria to be ubiquitous and abundant in soil, as determined by our cultivation-independent 16S rRNA gene analyses. When grouped together as one genus, Mycobacterium was typically one of the more abundantly named genera (12th of 398 named genera) of bacteria found in soil, confirming results reported in comparable studies (20, 26, 28). We note that these results based on 16S rRNA gene surveys likely underestimate the relative abundance of mycobacteria, as the 16S rRNA gene copy number for members of this group (∼1) is typically lower than that observed for most other major bacterial groups (58). However, we did not correct for 16S rRNA gene copy number in our analyses, following the recommendations of Louca et al. (59). The relative abundance of mycobacteria was variable across soils ranging from 0.03% to 2.9% of 16S rRNA gene reads, a range similar to that reported previously (26–28). Some of this variation in total mycobacterial abundances could be explained from the measured soil and site characteristics, with mycobacterial relative abundances typically being higher in more acidic, colder, and wetter soils. This observation is consistent with both cultivation-dependent and -independent results published previously which have suggested that mycobacterial abundances tend to be higher in more acidic, wetter environments (23, 24, 29). However, we note that our model only explained 25% of the variance in genus-level mycobacterial abundances across the collected samples. Although such portion of explained variation is considered to be relatively high for comparable global-scale studies (60), unexplained variation could be a product of the fact that we did not measure all possible soil or site characteristics that can influence mycobacterial abundances, including the presence of amoebae (61, 62), specific organic carbon substrates (5), or the presence of livestock (32, 35, 36). Additionally, some of this unexplained variation could be a product of the coarse, genus-level analyses employed, which may not sufficiently capture the phenotypic differences or differences in niche preferences across taxa within the genus Mycobacterium.
As expected, we detected a diversity of soil mycobacteria by using a higher resolution genetic marker (the hsp65 gene). Across all soils, we identified 472 ESVs that represented 159 distinct phylogenetic lineages, with most of these lineages restricted to a small subset of soils (Fig. 3). Not all mycobacteria were detected in all soils, an observation that could result from either dispersal limitations or, more likely, different mycobacterial strains having distinct environmental preferences. The latter explanation is supported by our results. In particular, we found that soil pH and climatic parameters were often the best predictors of the distributions of individual lineages (Fig. 4; Table S3 and S4), and most (65%) of the lineages preferred low pH soils, as determined by significant Spearman correlations (a result consistent with the genus-level analyses) (Fig. 2). However, we observed three individual lineages that were more commonly detected in high pH soils (Table S4). Likewise, while a majority lineages were mainly found in colder and wetter sites, two clades (clade 12 and clade 44) were more commonly detected in drier ecosystems (Fig. 4; Table S4 and S5). These results highlight that soil mycobacteria can exhibit contrasting, yet often predictable, environmental preferences.
Our findings further indicate that soils harbor a large amount of undescribed mycobacterial diversity. Of the 159 lineages detected in our global survey only 3% of the lineages (5/159) encompassed described isolates. These results contrast to a comparable survey of mycobacteria in household plumbing that used the same hsp65 marker gene sequencing approach, where the majority of the mycobacterial lineages included described taxa (14). The high proportion of undescribed mycobacterial lineages we recovered from soil is slightly higher than that reported previously in soils (27) and lakes (19) via 16S rRNA gene analyses. Of the small subset of soil mycobacterial clades that included described isolates, we found lineages that included M. rutilum and M. avium complex, taxa that have previously been identified from cultivation-dependent analyses of soil mycobacterial communities (5, 33).
Only one of the detected lineages (M. avium complex) included known human pathogens. Members of this group are frequently associated with clinical cases of NTM respiratory disease (24, 63). Interestingly, we found that the lineage including the M. avium complex (clade 31) showed strong preferences for wet and acidic soils, information that may be directly relevant to understanding the epidemiology of mycobacterial disease. However, as mycobacteria related to known pathogens were infrequently detected across the 143 samples studied, we think it is important to reexamine the widespread assumption that exposure to soil is an important mode of transmission of mycobacterial disease in humans. Specifically, outside the M. avium complex, most of the mycobacterial isolates frequently recovered from patients with respiratory NTM disease, such as M. abscessus, M. mucogenicum, M. kansasii, and M. fortuitum (10), were not detected in any of the soils. This differs from a similar investigation showing that mycobacteria frequently implicated in NTM disease are common and relatively abundant in showerhead biofilms (14).
The most ubiquitous clade detected in the soils sampled here included the described members M. rutilum and M. novocastrense. M. rutilum was first identified in soils collected from Hawaii and is known for its ability to degrade polycyclic hydrocarbon pollutants (5). Both species are rapidly growing and are closely related to M. vaccae, which has been well-studied for its potential benefits to human health (64). While M. novocastrense has been found very rarely in infected tissue, it is not generally considered to be pathogenic (65, 66). This result suggests a potentially large reservoir of soil-dwelling mycobacteria that could be beneficial to human or environmental health.
Our study represents a comprehensive investigation of soil mycobacterial diversity and the ecological factors shaping the distributions of individual soil mycobacterial lineages, many of which remain undescribed. We found that soil mycobacterial populations (both the Mycobacterium genus and individual lineages of mycobacteria) are often predictable from information on soil pH and site climatic conditions. Notably, most mycobacteria appear to prefer acidic, colder, and wetter soils; however, contrasting environmental preferences are found for individual clades. More generally, we show that mycobacteria can be abundant members of soil bacterial communities, but much of the soil mycobacterial diversity remains undescribed. Although the cultivation biases inherent in mycobacterial surveys are well-known (14, 43), our results suggest that this cultivation bias is particularly important in soil. We note that those mycobacteria commonly considered to be human pathogens are rarely detected in soils worldwide, challenging widely held expectations that soil is an important source of NTM diseases. Taken together, our study provides novel insights on the distribution and ecological preferences of soil mycobacteria in terrestrial ecosystems across the globe.
MATERIALS AND METHODS
Global soil sample collection.
For detailed soil sample collection methods, see Delgado-Baquerizo et al. (28). Here, we used a subset of 143 locations from Delgado-Baquerizo et al. (28), for which soil was still available. Briefly, soil samples were collected from 143 locations across 6 continents spanning a wide range of ecosystem types (Table S1). Composite samples were collected to roughly 7.5 cm depth and were frozen immediately at −20°C. Global positioning system (GPS) coordinates and ecosystem type were recorded in situ, and climatic variables were determined using the WorldClim database (https://www.worldclim.org/) (51, 67). Soil analyses were performed at the Universidad Rey Juan Carlos (Spain) following standardized protocols (68). Our database included information on key soil properties (organic carbon content, pH, C:N ratio, clay content, and electrical conductivity) and climatic variables (aridity index and annual minimum and maximum temperature) as explained in reference 28. Site aridity index is defined as precipitation/potential evapotranspiration (51, 52). For detailed information on soil and site characteristics, see Table S1.
16S rRNA gene sequencing to characterize the soil bacterial communities.
We characterized the bacterial communities in our soils via 16S rRNA gene sequencing. For detailed sequencing methods, see Delgado-Baquerizo et al. (28). Briefly, DNA was extracted from soils using the DNeasy PowerSoil kit (Qiagen) following the manufacturer’s protocol. The V3-V4 hypervariable region of the bacterial 16S rRNA gene was amplified with 341F/805R primers (341F, 5′-CCTACGGGNGGCWGCAG-3′; 805R, 5′-GACTACHVGGGTATCTAATCC-3′), and sequenced on the Illumina MiSeq platform. Data were processed using the USEARCH10 pipeline (69). Reverse reads were trimmed before 10 bp and after 260 bp, and merged (usearch8 -fastq_mergepairs). Following merging, 17 bp were trimmed off the front of the merged reads to eliminate the possibility of primer barcode contamination. Trimmed reads were quality filtered with a max error rate of 1.0 and a final truncation length at 410 (usearch10 -fastq_filter; 91.4% passed). Unique sequences were identified using usearch10 -fastx_uniques and clustered at 97% identity to operational taxonomic units (OTUs) with usearch10 -cluster_otus uniques.fa. Taxonomy was assigned with the Ribosomal Database Project Classifier (70) against the Greengenes 13_8 database (71), and chloroplast and mitochondrial reads were removed. Read depth at this stage ranged from 3,609 to 103,430 reads per sample, with an average of 59,423 reads per sample (see Table S1 for reads per sample). To correct for differences in sequencing depth, samples were rarified to 10,000 reads per sample in R using the MCtoolsR package (https://github.com/leffj/mctoolsr/), which removed 1 sample with read counts below this threshold. The percentage of mycobacteria in each sample was calculated by summing reads classified to the Mycobacterium genus per sample. Note that throughout the manuscript, we refer to the Mycobacterium genus broadly and have not adopted the revised taxonomic classification scheme proposed by Gupta et al. (4) that splits the genus into multiple genera. We do this to preserve continuity with the preexisting literature and because most of the mycobacteria detected in these soils were not closely related to described strains, making taxonomic reassignment tenuous at this point.
hsp65 gene sequencing to characterize mycobacterial diversity.
To resolve mycobacterial taxonomic diversity beyond the genus level, we performed marker gene analysis of a region of the hsp65 gene specifically targeting mycobacteria. For detailed PCR and sequence methods, see Gebert et al. (14). Briefly, a two-step PCR approach was used with mycobacterium-specific primers (72) and with the resulting amplicons (∼400 bp) sequenced on the Illumina MiSeq platform. The sequence reads were processed using the uSEARCH pipeline (69). Raw reads were processed by trimming reverse reads to 250 bp (usearch8 -fastq_filter), merging (usearch8 -fastq_mergepairs), and quality filtering at a max error rate of 0.005 (usearch8 -fastq_filter -fastq_maxee_rate 0.005). Exact sequence variants (ESVs) were identified using uNOISE3 (73) (usearch10 -fastx_uniques and usearch10 -unoise3).
The hsp65 gene-targeting PCR primers do not exclusively amplify this marker gene from mycobacteria alone, as it can also amplify sequences representing other groups of actinobacteria, such as Nocardia (74). To restrict our analyses to mycobacterial taxa alone, we filtered ESVs against a mycobacterial reference database (55), removing any reads that were <94% similar to the mycobacterial sequences in the reference database (usearch10 -usearch_global). Due to the large number of exact sequence variants remaining (2,836 mycobacterial ESVs out of 1,225,518 reads from all 143 samples), we only included those ESVs represented by at least 500 reads across all samples to focus on the more abundant and ubiquitous mycobacterial ESVs. After applying this threshold, 75% of all mycobacterial reads remained (926,258 reads, 472 ESVs). At this stage, the number of reads per sample ranged from 1 to 46,627 and averaged 5,208 reads per sample (see Table S1 for exact read counts per sample).
Phylogenetic tree construction and characterization of mycobacterial diversity.
A majority of the soil-derived mycobacterial hsp65 sequences were highly divergent from reference sequences (see Results). Thus, to characterize the diversity of soil mycobacteria and for conducting downstream ecological analyses, we collapsed the ESVs into distinct phylogenetic clades. Phylogenetic relationships for the 472 ESVs were determined via maximum likelihood with RaxML (75). First, soil-derived ESVs were combined with the reference sequences from Dai et al. (55), and these sequences were aligned using MUSCLE v.3.8.31 (76). Aligned reads were used to construct a tree with RaxML (raxmlHPC -f a -m GTRGAMMA -p 12345 -x 12345 -number 100), including Nocardia farcinica (DSM43665) as an outgroup for tree rooting. Sample and reference sequences were clustered into phylogenetic clades using RAMI (77), with a patristic distance threshold of 0.05 (5%). With the soil-derived ESVs combined with the reference sequences, this produced 242 clades containing 630 ESVs (472 mycobacterial sequences from our soils, 158 reference sequences). Many of the 242 clades included only reference sequences, as only 159 clades included soil-derived ESVs from this study. The tree was visualized and annotated using iTOL (78). Only five clades contained both reference sequences and soil-derived sequences. Sample sequences in these clades were confirmed to be relatives of the named taxa via BLAST (79), with all ESVs within a given clade sharing >99% identity with the corresponding reference strain. We determined the presence/absence of each clade in each sample, with absence defined as a clade represented by less than 50 hsp65 reads in a given sample. Information regarding clade assignments for each ESV can be found in the publicly available hsp65 ESV table (https://figshare.com/projects/Global_survey_of_mycobacteria_in_soil/63812).
Statistical analysis and modeling.
We used random forest analysis via the R package rfPermute (80) to identify the most important ecological predictors of genus-level mycobacterial abundance and mycobacterial clade distributions (functions rfPermute, rp.importance). Previous to these analyses, we preselected environmental variables from reference 28 that were not highly correlated (Pearson's R < 0.7) (see Table S2 in the supplemental material). Based on these correlations and the relevance of remaining variables, the following environmental variables were included in downstream analyses: soil organic carbon content, pH, site aridity index, annual minimum and maximum temperature at the site, soil C:N ratio, soil clay content, and soil electrical conductivity. An independent random forest model was run separately for each clade of interest in addition to that built with the genus-level proportional abundance data set (16S rRNA gene sequence data). Only models that described more than 20% of the variation were evaluated. “Important” environmental predictors in these models were defined as those that increased the mean standard error of the model (MSE) more than 15% when excluded with P values of <0.01.
To further understand the strength and directionality of the relationships between important environmental predictors and mycobacterial relative abundances or mycobacterial linage presence, we used Spearman rank correlations as we did not necessarily expect a linear relationship between the environmental variables and mycobacterial response. We then determined whether these correlations were associated with significant differences in mycobacterial relative abundances or clade presence for the environmental variables using the Wilcoxon signed-rank test. We performed partial correlations using the ppcor package (81) to assess Spearman correlations between environmental factors and mycobacteria distributions, controlling for geospatial location.
The statistical analyses to determine which soil or site characteristics were predictive of mycobacterial abundances (genus level analyses, 16S rRNA gene sequencing) or the presence/absence of individual mycobacterial clades (hsp65 gene sequencing) were performed in the R environment v.3.4.3 (82) with all plots generated using ggplot (83) and sample maps created with the maps package (84).
Data availability.
All data used in this study are publicly available in Figshare (https://figshare.com/projects/Global_survey_of_mycobacteria_in_soil/63812).
Supplementary Material
ACKNOWLEDGMENTS
We thank Hannah Holland-Moritz, Angela Oliverio, and Tess Brewer for their input and assistance in bioinformatics processing and statistical analyses. We also thank Victoria Ochoa and Beatriz Gozalo for their help with the laboratory analyses.
Funding for this work was provided by grants to N.F. from the High Plains Intermountain Center for Agricultural Health & Safety and the Innovative Research Program of the Cooperative Institute for Research in Environmental Sciences and to C.M.W. by the NSF IGERT grant number 1144807 via the BioFrontiers Institute. The work of F.T.M. and the global drylands database were supported by the European Research Council (ERC grant agreements 242658 [BIOCOM] and 647038 [BIODESERT]).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.01180-19.
REFERENCES
- 1.Hruska K, Kaevska M. 2013. Mycobacteria in water, soil, plants and air: a review. Veterinarni Medicina 57:623–679. doi: 10.17221/6558-VETMED. [DOI] [Google Scholar]
- 2.Tortoli E. 2019. The taxonomy of the genus Mycobacterium, p 1–10. In Velayati AA, Farnia P (ed), Nontuberculous mycobacteria (NTM). Academic Press, Cambridge, MA. doi: 10.1016/B978-0-12-814692-7.00001-2. [DOI] [Google Scholar]
- 3.Parte AC. 2018. LPSN—List of Prokaryotic Names with Standing in Nomenclature (bacterio.net), 20 years on. Int J Syst Evol Microbiol 68:1825–1829. doi: 10.1099/ijsem.0.002786. [DOI] [PubMed] [Google Scholar]
- 4.Gupta RS, Lo B, Son J. 2018. Phylogenomics and comparative genomic studies robustly support division of the genus Mycobacterium into an emended genus Mycobacterium and four novel genera. Front Microbiol 9:67. doi: 10.3389/fmicb.2018.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hennessee CT, Seo J-S, Alvarez AM, Li QX. 2009. Polycyclic aromatic hydrocarbon-degrading species isolated from Hawaiian soils: Mycobacterium crocinum sp. nov., Mycobacterium pallens sp. nov., Mycobacterium rutilum sp. nov., Mycobacterium rufum sp. nov. and Mycobacterium aromaticivorans sp. nov. Int J Syst Evol Microbiol 59:378–387. doi: 10.1099/ijs.0.65827-0. [DOI] [PubMed] [Google Scholar]
- 6.Govindaswami M, Feldhake DJ, Kinkle BK, Mindell DP, Loper JC. 1995. Phylogenetic comparison of two polycyclic aromatic hydrocarbon-degrading mycobacteria. Appl Environ Microbiol 61:3221–3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van Beilen JB, Holtackers R, Lüscher D, Bauer U, Witholt B, Duetz WA. 2005. Biocatalytic production of perillyl alcohol from limonene by using a novel Mycobacterium sp. cytochrome P450 alkane hydroxylase expressed in Pseudomonas putida. Appl Environ Microbiol 71:1737–1744. doi: 10.1128/AEM.71.4.1737-1744.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reber SO, Siebler PH, Donner NC, Morton JT, Smith DG, Kopelman JM, Lowe KR, Wheeler KJ, Fox JH, Hassell JE, Greenwood BN, Jansch C, Lechner A, Schmidt D, Uschold-Schmidt N, Füchsl AM, Langgartner D, Walker FR, Hale MW, Lopez Perez G, Van Treuren W, González A, Halweg-Edwards AL, Fleshner M, Raison CL, Rook GA, Peddada SD, Knight R, Lowry CA. 2016. Immunization with a heat-killed preparation of the environmental bacterium Mycobacterium vaccae promotes stress resilience in mice. Proc Natl Acad Sci U S A 113:E3130–E3139. doi: 10.1073/pnas.1600324113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lowry CA, Hollis JH, de Vries A, Pan B, Brunet LR, Hunt JRF, Paton JFR, van Kampen E, Knight DM, Evans AK, Rook GAW, Lightman SL. 2007. Identification of an immune-responsive mesolimbocortical serotonergic system: potential role in regulation of emotional behavior. Neuroscience 146:756–772. doi: 10.1016/j.neuroscience.2007.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Falkinham JO., III. 1996. Epidemiology of infection by nontuberculous mycobacteria. Clin Microbiol Rev 9:177–215. doi: 10.1128/CMR.9.2.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Halstrom S, Price P, Thomson R. 2015. Review: environmental mycobacteria as a cause of human infection. Int J Mycobacteriol 4:81–91. doi: 10.1016/j.ijmyco.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 12.Adjemian J, Olivier KN, Seitz AE, Holland SM, Prevots DR. 2012. Prevalence of nontuberculous mycobacterial lung disease in U.S. Medicare beneficiaries. Am J Respir Crit Care Med 185:881–886. doi: 10.1164/rccm.201111-2016OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Ingen J, Boeree MJ, Dekhuijzen PNR, van Soolingen D. 2009. Environmental sources of rapid growing nontuberculous mycobacteria causing disease in humans. Clin Microbiol Infect 15:888–893. doi: 10.1111/j.1469-0691.2009.03013.x. [DOI] [PubMed] [Google Scholar]
- 14.Gebert MJ, Delgado-Baquerizo M, Oliverio AM, Webster TM, Nichols LM, Honda JR, Chan ED, Adjemian J, Dunn RR, Fierer N. 2018. Ecological analyses of mycobacteria in showerhead biofilms and their relevance to human health. mBio 9:e01614-18. doi: 10.1128/mBio.01614-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Falkinham JO. 2011. Nontuberculous mycobacteria from household plumbing of patients with nontuberculous mycobacteria disease. Emerg Infect Dis 17:419–424. doi: 10.3201/eid1703.101510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu R, Yu Z, Guo H, Liu M, Zhang H, Yang M. 2012. Pyrosequencing analysis of eukaryotic and bacterial communities in faucet biofilms. Sci Total Environ 435–436:124–131. doi: 10.1016/j.scitotenv.2012.07.022. [DOI] [PubMed] [Google Scholar]
- 17.Falkinham JO, Hilborn ED, Arduino MJ, Pruden A, Edwards MA. 2015. Epidemiology and ecology of opportunistic premise plumbing pathogens: Legionella pneumophila, Mycobacterium avium, and Pseudomonas aeruginosa. Environ Health Perspect 123:749. doi: 10.1289/ehp.1408692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feazel LM, Baumgartner LK, Peterson KL, Frank DN, Harris JK, Pace NR. 2009. Opportunistic pathogens enriched in showerhead biofilms. Proc Natl Acad Sci U S A 106:16393–16399. doi: 10.1073/pnas.0908446106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roguet A, Therial C, Saad M, Boudahmane L, Moulin L, Lucas FS. 2016. High mycobacterial diversity in recreational lakes. Antonie Van Leeuwenhoek 109:619–631. doi: 10.1007/s10482-016-0665-x. [DOI] [PubMed] [Google Scholar]
- 20.Niva M, Hernesmaa A, Haahtela K, Salkinoja-Salonen M, Sivonen K, Haukka K, Haahtela A, Sivonen M. 2006. Actinobacterial communities of boreal forest soil and lake water are rich in mycobacteria. Boreal Environ Res 11:45–53. [Google Scholar]
- 21.Jacobs J, Rhodes M, Sturgis B, Wood B. 2009. Influence of environmental gradients on the abundance and distribution of Mycobacterium spp. in a coastal lagoon estuary. Appl Environ Microbiol 75:7378–7384. doi: 10.1128/AEM.01900-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pickup RW, Rhodes G, Bull TJ, Arnott S, Sidi-Boumedine K, Hurley M, Hermon-Taylor J. 2006. Mycobacterium avium subsp. paratuberculosis in lake catchments, in river water abstracted for domestic use, and in effluent from domestic sewage treatment works: diverse opportunities for environmental cycling and human exposure. Appl Environ Microbiol 72:4067–4077. doi: 10.1128/AEM.02490-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iivanainen EK, Martikainen PJ, Väänänen PK, Katila ML. 1993. Environmental factors affecting the occurrence of mycobacteria in brook waters. Appl Environ Microbiol 59:398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kirschner RA, Parker BC, Falkinham JO. 1992. Epidemiology of infection by nontuberculous mycobacteria: Mycobacterium avium, Mycobacterium intracellulare, and Mycobacterium scrofulaceum in acid, brown-water swamps of the southeastern United States and their association with environmental variables. Am Rev Respir Dis 145:271–275. doi: 10.1164/ajrccm/145.2_Pt_1.271. [DOI] [PubMed] [Google Scholar]
- 25.Bland CS, Ireland JM, Lozano E, Alvarez ME, Primm TP. 2005. Mycobacterial ecology of the Rio Grande. Appl Environ Microbiol 71:5719–5727. doi: 10.1128/AEM.71.10.5719-5727.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karimi B, Terrat S, Dequiedt S, Saby NPA, Horrigue W, Lelièvre M, Nowak V, Jolivet C, Arrouays D, Wincker P, Cruaud C, Bispo A, Maron P-A, Bouré NCP, Ranjard L. 2018. Biogeography of soil bacteria and archaea across France. Sci Adv 4:eaat1808. doi: 10.1126/sciadv.aat1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pontiroli A, Khera TT, Oakley BB, Mason S, Dowd SE, Travis ER, Erenso G, Aseffa A, Courtenay O, Wellington E. 2013. Prospecting environmental mycobacteria: combined molecular approaches reveal unprecedented diversity. PLoS One 8:e68648. doi: 10.1371/journal.pone.0068648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Delgado-Baquerizo M, Oliverio AM, Brewer TE, Benavent-González A, Eldridge DJ, Bardgett RD, Maestre FT, Singh BK, Fierer N. 2018. A global atlas of the dominant bacteria found in soil. Science 359:320–325. doi: 10.1126/science.aap9516. [DOI] [PubMed] [Google Scholar]
- 29.Kopecky J, Kyselkova M, Omelka M, Cermak L, Novotna J, Grundmann G, Moënne-Loccoz Y, Sagova-Mareckova M. 2011. Environmental mycobacteria closely related to the pathogenic species evidenced in an acidic forest wetland. Soil Biol Biochem 43:697–700. doi: 10.1016/j.soilbio.2010.11.033. [DOI] [Google Scholar]
- 30.Eiila K, Martikanien PJ, Räisänem ML, Katila M-L. 1997. Mycobacteria in boreal coniferous forest soils 1997.pdf. FEMS Microbiol Ecol 23:325–332. [Google Scholar]
- 31.Nieminen T, Pakarinen J, Tsitko I, Salkinoja-Salonen M, Breitenstein A, Ali-Vehmas T, Neubauer P. 2006. 16S rRNA targeted sandwich hybridization method for direct quantification of mycobacteria in soils. J Microbiol Methods 67:44–55. doi: 10.1016/j.mimet.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 32.Norby B, Fosgate GT, Manning EJB, Collins MT, Roussel AJ. 2007. Environmental mycobacteria in soil and water on beef ranches: association between presence of cultivable mycobacteria and soil and water physicochemical characteristics. Vet Microbiol 124:153–159. doi: 10.1016/j.vetmic.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 33.Rhodes G, Henrys P, Thomson BC, Pickup RW. 2013. Mycobacterium avium subspecies paratuberculosis is widely distributed in British soils and waters: implications for animal and human health. Environ Microbiol 15:2761–2774. doi: 10.1111/1462-2920.12137. [DOI] [PubMed] [Google Scholar]
- 34.Chilima BZ, Clark IM, Floyd S, Fine PEM, Hirsch PR. 2006. Distribution of environmental mycobacteria in Karonga District, northern Malawi. Appl Environ Microbiol 72:2343–2350. doi: 10.1128/AEM.72.4.2343-2350.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Young JS, Gormley E, Wellington E. 2005. Molecular detection of Mycobacterium bovis and Mycobacterium bovis BCG (Pasteur) in soil. Appl Environ Microbiol 71:1946–1952. doi: 10.1128/AEM.71.4.1946-1952.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eisenberg SWF, Koets AP, Hoeboer J, Bouman M, Heederik D, Nielen M. 2010. Presence of Mycobacterium avium subsp. paratuberculosis in environmental samples collected on commercial Dutch dairy farms. Appl Environ Microbiol 76:6310–6312. doi: 10.1128/AEM.00998-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Groote MA, Pace NR, Fulton K, Falkinham JO. 2006. Relationships between Mycobacterium isolates from patients with pulmonary mycobacterial infection and potting soils. Appl Environ Microbiol 72:7602–7606. doi: 10.1128/AEM.00930-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kamala T, Paramasivan CN, Herbert D, Venkatesan P, Prabhakar R. 1994. Evaluation of procedures for isolation of nontuberculous mycobacteria from soil and water. Appl Environ Microbiol 60:1021–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Iivanainen E. 1995. Isolation of mycobacteria from acidic forest soil samples: comparison of culture methods. J Appl Bacteriol 78:663–668. doi: 10.1111/j.1365-2672.1995.tb03113.x. [DOI] [PubMed] [Google Scholar]
- 40.Thorel M-F, Falkinham JO, Moreau RG. 2004. Environmental mycobacteria from alpine and subalpine habitats. FEMS Microbiol Ecol 49:343–347. doi: 10.1016/j.femsec.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 41.Falkinham JO. 2014. The Mycobacterium avium complex and slowly growing mycobacteria In Molecular medical microbiology, 2nd ed Elsevier Ltd., Amsterdam, Netherlands. [Google Scholar]
- 42.Neumann M, Schulze-Robbecke R, Hagenau C, Behringer K. 1997. Comparison of methods for isolation of mycobacteria from water. Appl Environ Microbiol 63:547–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hussein Z, Landt O, Wirths B, Wellinghausen N. 2009. Detection of non-tuberculous mycobacteria in hospital water by culture and molecular methods. Int J Med Microbiol 299:281–290. doi: 10.1016/j.ijmm.2008.07.004. [DOI] [PubMed] [Google Scholar]
- 44.Mendum TA, Chilima BZ, Hirsch PR. 2000. The PCR amplification of non-tuberculous mycobacterial 16S rRNA sequences from soil. FEMS Microbiol Lett 185:189–192. doi: 10.1111/j.1574-6968.2000.tb09060.x. [DOI] [PubMed] [Google Scholar]
- 45.McVeigh HP, Munro J, Embley TM. 1996. Molecular evidence for the presence of novel actinomycete lineages in a temperate forest soil. J Ind Microbiol Biotechnol 17:197–204. doi: 10.1007/BF01574693. [DOI] [Google Scholar]
- 46.Ringuet H, Akoua-Koffi C, Honore S, Varnerot A, Vincent V, Berche P, Gaillard JL, Pierre-Audigier C. 1999. hsp65 sequencing for identification of rapidly growing mycobacteria. J Clin Microbiol 37:852–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dai J, Chen Y, Dean S, Morris JG, Salfinger M, Johnson JA. 2011. Multiple-genome comparison reveals new loci for Mycobacterium species identification. J Clin Microbiol 49:144–153. doi: 10.1128/JCM.00957-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim H, Kim S-H, Shim T-S, Kim M, Bai G-H, Park Y-G, Lee S-H, Chae G-T, Cha C-Y, Kook Y-H, Kim B-J. 2005. Differentiation of Mycobacterium species by analysis of the heat-shock protein 65 gene (hsp65). Int J Syst Evol Microbiol 55:1649–1656. doi: 10.1099/ijs.0.63553-0. [DOI] [PubMed] [Google Scholar]
- 49.McNabb A, Eisler D, Adie K, Amos M, Rodrigues M, Stephens G, Black WA, Isaac-Renton J. 2004. Assessment of partial sequencing of the 65-kilodalton heat shock protein gene (hsp65) for routine identification of Mycobacterium species isolated from clinical sources. J Clin Microbiol 42:3000–3011. doi: 10.1128/JCM.42.7.3000-3011.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Breiman L. 2001. Random forests. Mach Learn 45:5–32. doi: 10.1023/A:1010933404324. [DOI] [Google Scholar]
- 51.Zomer RJ, Trabucco A, Bossio DA, Verchot LV. 2008. Climate change mitigation: a spatial analysis of global land suitability for clean development mechanism afforestation and reforestation. Agric Ecosyst Environ 126:67–80. doi: 10.1016/j.agee.2008.01.014. [DOI] [Google Scholar]
- 52.Zomer R, Bossio D, Trabucco A, Yuanjie L, Gupta D, Singh V. 2007. Trees and water: smallholder agroforestry on irrigated lands in Northern India. International Water Management Institute, Colombo, Sri Lanka. [Google Scholar]
- 53.Falkinham JO., III 2009. The biology of environmental mycobacteria. Environ Microbiol Rep 1:477–487. doi: 10.1111/j.1758-2229.2009.00054.x. [DOI] [PubMed] [Google Scholar]
- 54.Tortoli E, Fedrizzi T, Meehan CJ, Trovato A, Grottola A, Giacobazzi E, Serpini GF, Tagliazucchi S, Fabio A, Bettua C, Bertorelli R, Frascaro F, De Sanctis V, Pecorari M, Jousson O, Segata N, Cirillo DM. 2017. The new phylogeny of the genus Mycobacterium: the old and the news. Infect Genet Evol 56:19–25. doi: 10.1016/j.meegid.2017.10.013. [DOI] [PubMed] [Google Scholar]
- 55.Dai J, Chen Y, Lauzardo M. 2011. Web-accessible database of hsp65 sequences from Mycobacterium reference strains. J Clin Microbiol 49:2296–2303. doi: 10.1128/JCM.02602-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.García-Agudo L, García-Martos P. 2011. Clinical significance and antimicrobial susceptibility of rapidly growing mycobacteria, p 363–377. In Méndez-Vilas A. (ed), Science against microbial pathogens: communicating current research and technological advances. Formatex Research Center, Badajoz, Spain. [Google Scholar]
- 57.Thomson R, Tolson C, Carter R, Coulter C, Huygens F, Hargreaves M. 2013. Isolation of nontuberculous mycobacteria (NTM) from household water and shower aerosols in patients with pulmonary disease caused by NTM. J Clin Microbiol 51:3006–3011. doi: 10.1128/JCM.00899-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Větrovský T, Baldrian P. 2013. The variability of the 16S rRNA gene in bacterial genomes and its consequences for bacterial community analyses. PLoS One 8:e57923. doi: 10.1371/journal.pone.0057923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Louca S, Doebeli M, Parfrey LW. 2018. Correcting for 16S rRNA gene copy numbers in microbiome surveys remains an unsolved problem. Microbiome 6:41. doi: 10.1186/s40168-018-0420-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moles AT, Flores-Moreno H, Bonser SP, Warton DI, Helm A, Warman L, Eldridge DJ, Jurado E, Hemmings FA, Reich PB, Cavender-Bares J, Seabloom EW, Mayfield MM, Sheil D, Djietror JC, Peri PL, Enrico L, Cabido MR, Setterfield SA, Lehmann CER, Thomson FJ. 2012. Invasions: the trail behind, the path ahead, and a test of a disturbing idea. J Ecol 100:116–127. doi: 10.1111/j.1365-2745.2011.01915.x. [DOI] [Google Scholar]
- 61.Delafont V, Mougari F, Cambau E, Joyeux M, Bouchon D, Héchard Y, Moulin L. 2014. First evidence of amoebae–mycobacteria association in drinking water network. Environ Sci Technol 48:11872–11882. doi: 10.1021/es5036255. [DOI] [PubMed] [Google Scholar]
- 62.Drancourt M. 2014. Looking in amoebae as a source of mycobacteria. Microb Pathog 77:119–124. doi: 10.1016/j.micpath.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 63.Nishiuchi Y, Iwamoto T, Maruyama F. 2017. Infection sources of a common non-tuberculous mycobacterial pathogen, Mycobacterium avium complex. Front Med (Lausanne) 4:27. doi: 10.3389/fmed.2017.00027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fox JH, Hassell JE, Siebler PH, Arnold MR, Lamb AK, Smith DG, Day HEW, Smith TM, Simmerman EM, Outzen AA, Holmes KS, Brazell CJ, Lowry CA. 2017. Preimmunization with a heat-killed preparation of Mycobacterium vaccae enhances fear extinction in the fear-potentiated startle paradigm. Brain Behav Immun 66:70–84. doi: 10.1016/j.bbi.2017.08.014. [DOI] [PubMed] [Google Scholar]
- 65.Shojaei H, Goodfellow M, Magee JG, Freeman R, Gould FK, Brignall CG. 1997. Mycobacterium novocastrense sp. nov., a rapidly growing photochromogenic mycobacterium. Int J Syst Bacteriol 47:1205–1207. doi: 10.1099/00207713-47-4-1205. [DOI] [PubMed] [Google Scholar]
- 66.Simner PJ, Khare R, Wengenack NL. 2014. Rapidly growing mycobacteria, p 1679–1690. In Tang Y-W, Sails A (ed), Molecular medical microbiology, 2nd ed Elsevier Ltd., Amsterdam, Netherlands. [Google Scholar]
- 67.Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A. 2005. Very high resolution interpolated climate surfaces for global land areas. Int J Climatol 25:1965–1978. doi: 10.1002/joc.1276. [DOI] [Google Scholar]
- 68.Maestre FT, Quero JL, Gotelli NJ, Escudero A, Ochoa V, Delgado-Baquerizo M, Garcia-Gomez M, Bowker MA, Soliveres S, Escolar C, Garcia-Palacios P, Berdugo M, Valencia E, Gozalo B, Gallardo A, Aguilera L, Arredondo T, Blones J, Boeken B, Bran D, Conceicao AA, Cabrera O, Chaieb M, Derak M, Eldridge DJ, Espinosa CI, Florentino A, Gaitan J, Gatica MG, Ghiloufi W, Gomez-Gonzalez S, Gutierrez JR, Hernandez RM, Huang X, Huber-Sannwald E, Jankju M, Miriti M, Monerris J, Mau RL, Morici E, Naseri K, Ospina A, Polo V, Prina A, Pucheta E, Ramirez-Collantes DA, Romao R, Tighe M, Torres-Diaz C, Val J, Veiga JP, Wang D, Zaady E. 2012. Plant species richness and ecosystem multifunctionality in global drylands. Science 335:214–218. doi: 10.1126/science.1215442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 70.Wang Q, Garrity GM, Tiedje JM, Cole JR. 2007. Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P. 2012. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6:610–618. doi: 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Telenti A, Marchesi F, Balz M, Bally F, Böttger EC, Bodmer T. 1993. Rapid identification of mycobacteria to the species level by polymerase chain reaction and restriction enzyme analysis. J Clin Microbiol 31:175–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Edgar RC. 2016. UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv doi: 10.1101/081257. [DOI]
- 74.Rodríguez-Nava V, Couble A, Devulder G, Flandrois J-P, Boiron P, Laurent F. 2006. Use of PCR-restriction enzyme pattern analysis and sequencing database for hsp65 gene-based identification of Nocardia species. J Clin Microbiol 44:536–546. doi: 10.1128/JCM.44.2.536-546.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stamatakis A, Ludwig T, Meier H. 2005. RAxML-III: a fast program for maximum likelihood-based inference of large phylogenetic trees. Bioinformatics 21:456–463. doi: 10.1093/bioinformatics/bti191. [DOI] [PubMed] [Google Scholar]
- 76.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pommier T, Canbäck B, Lundberg P, Hagström Å, Tunlid A. 2009. RAMI: a tool for identification and characterization of phylogenetic clusters in microbial communities. Bioinformatics 25:736–742. doi: 10.1093/bioinformatics/btp051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Letunic I, Bork P. 2007. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23:127–128. doi: 10.1093/bioinformatics/btl529. [DOI] [PubMed] [Google Scholar]
- 79.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 80.Archer E. 2017. rfPermute: estimate permutation p-values for random forest importance metrics. 2.1.5. GitHub https://github.com/EricArcher/rfPermute.
- 81.Kim S. 2015. ppcor: partial and semi-partial (part) correlation. https://rdrr.io/cran/ppcor/man/spcor.html.
- 82.R Development Core Team. 2014. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 83.Wickham H. 2016. ggplot2: elegant graphics for data analysis. Springer-Verlag New York, New York, NY. [Google Scholar]
- 84.Deckmyn A. 2018. maps: draw geographical maps. R-3.3.0. R Foundation for Statistical Computing, Vienna, Austria: https://cran.r-project.org/package=maps. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data used in this study are publicly available in Figshare (https://figshare.com/projects/Global_survey_of_mycobacteria_in_soil/63812).