

Abstract
Gain-of-function mutations in TRPC6 cause familial focal segmental glomerulosclerosis, and TRPC6 is upregulated in glomerular diseases including diabetic kidney disease. We studied the effect of systemic TRPC6 knockout in the Akita model of type 1 diabetes. Knockout of TRPC6 inhibited albuminuria in Akita mice at 12 and 16 weeks of age, but this difference disappeared by 20 weeks. Knockout of TRPC6 also reduced tubular injury in Akita mice; however, mesangial expansion was significantly increased. Hyperglycemia and blood pressure were similar between TRPC6 knockout and wild-type Akita mice, but knockout mice were more insulin resistant. In cultured podocytes, knockout of TRPC6 inhibited expression of the calcium/calcineurin responsive gene insulin receptor substrate 2 and decreased insulin responsiveness. Insulin resistance is reported to promote diabetic kidney disease independent of blood glucose levels. While the mechanisms are not fully understood, insulin activates both Akt2 and ERK, which inhibits apoptosis signal regulated kinase 1 (ASK1)-p38-induced apoptosis. In cultured podocytes, hyperglycemia stimulated p38 signaling and induced apoptosis, which was reduced by insulin and ASK1 inhibition and enhanced by Akt or ERK inhibition. Glomerular p38 signaling was increased in TRPC6 knockout Akita mice and was associated with enhanced expression of the p38 gene target cyclooxygenase 2. These data suggest that knockout of TRPC6 in Akita mice promotes insulin resistance and exacerbates glomerular disease independent of hyperglycemia.
Keywords: diabetic nephropathy, insulin resistance, insulin signaling, podocyte
Diabetic nephropathy (DN) is a serious complication of both type 1 and type 2 diabetes mellitus.1 The economic consequences of this disorder are significant because DN is the most common cause of end-stage kidney disease in developed countries.1 Moreover, both the incidence and prevalence of diabetes are increasing worldwide.1,2 Current treatment is focused on optimizing blood pressure and glucose control as well as on reducing proteinuria by using angiotensin-converting enzyme inhibitors and angiotensin II receptor blockers.3,4 Although these strategies slow disease progression,4,5 ~20% of patients with overt nephropathy ultimately develop end-stage kidney disease requiring renal replacement therapy.1,2 As a result, much effort has been devoted to under-standing the mechanisms that promote glomerular damage in diabetic kidney disease (DKD) with the goal of identifying new therapeutic targets and treatment strategies.
Accumulating evidence suggests that glomerular podocytes play a key role in DN.6 These highly differentiated cells are important for maintaining the integrity of the glomerular filtration barrier.7,8 Podocyte injury is a common feature of DKD in both animal models and diabetic humans.6,7 In the later stages of the disease, a reduction in the number of glomerular podocytes is characteristic of the disease process.9–11 Because podocytes are terminally differentiated cells with a limited capacity for replication,8,12 a sufficient loss of podocytes leads to instability of the glomerular tuft and in turn promotes glomerulosclerosis.8
Familial forms of glomerular disease have provided important insights into the pathogenesis of glomerular disease processes.13 Gain-of-function mutations in transient receptor potential cation channel C6 (TRPC6) cause familial forms of focal segmental glomerulosclerosis.14,15 Moreover, TRPC6 is upregulated in both primary and secondary glomerular disease processes including DN.16–18 These observations may be relevant to podocyte injury in diabetes, because angiotensin II receptor blockers potently activates TRPC614,16,19–21 and in turn causes podocyte apoptosis.22–24 In addition, abundant data indicate that reactive oxygen species generation is increased in the diabetic milieu25 and reactive oxygen species directly stimulate TRPC6 activity.21,26,27 Taken together, these data suggest that the upregulation and activation of TRPC6 may play an important role in promoting podocyte damage in DKD.
Accordingly, we used a genetic approach to investigate the role of TRPC6 in DKD using the Akita model of type 1 diabetes.28,29 For experiments, mice with the Akita mutation28 were bred with TRPC6 knockout (KO) mice30 to create Akita mice lacking TRPC6 (KO Akita mice). Using this genetic model, we then investigated the effect of TRPC6 on the functional and histopathological features of DKD.
RESULTS
Effect of TRPC6 KO on blood glucose levels, hemoglobin A1c levels, systemic blood pressure, and kidney hypertrophy
As shown in Figure 1a, fasting blood glucose levels were similarly elevated in both groups of Akita mice compared with nondiabetic mice at 8, 12, 16, and 20 weeks of age. Consistent with blood glucose levels, hemoglobin A1c levels were similarly increased in both diabetic groups at the 20-week time point (Figure 1b). We next evaluated systemic blood pressure. As shown in Figure 1c, there were no significant differences in systolic blood pressure between the diabetic groups at 12- and 20-week time points. Figure 1d shows the effect of genotype and age on body weight. Body weights were reduced in both diabetic groups compared with nondiabetic wild-type (WT) animals (WT controls). There was also a significant reduction in body weight in nondiabetic KO mice (KO controls) compared with WT controls. As shown in Figure 1e and f, diabetes increased kidney weights in both diabetic groups compared with nondiabetic animals when expressed as either total kidney weight (Figure 1e) or kidney weight corrected for body weight (Figure 1f).
Figure 1 |. Effect of transient receptor potential cation channel C6 knockout (KO) on systemic blood pressure (BP), hyperglycemia, body weight, and kidney size.
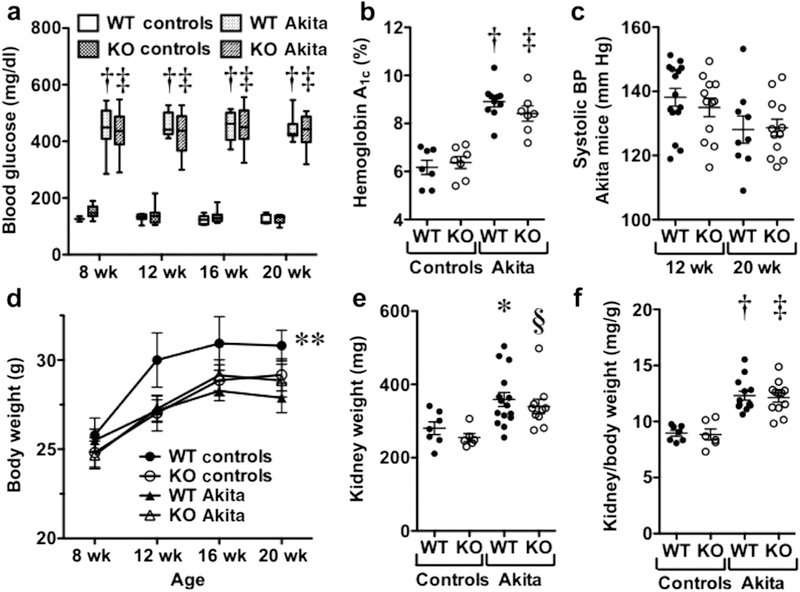
(a) Blood glucose levels were similarly elevated in both groups of Akita mice. (b) Hemoglobin A1c levels were similar in both groups of nondiabetic mice and were similarly increased in wild-type (WT) and KO Akita mice. (c) Systolic BP was similar in WT and KO Akita mice at 12- and 20-week time points. (d) Nondiabetic WT mice (WT controls) had a significantly higher body weight than did nondiabetic KO mice (KO controls). Body weight in WT controls was also increased compared with both groups of Akita mice, but these differences were not statistically significant. (e,f) Kidney weights were increased in both groups of diabetic mice compared with the nondiabetic groups (panel e), and this difference was proportional to body weight (panel f). *P < 0.05 versus nondiabetic WT mice, §P < 0.01 versus nondiabetic KO mice, †P < 0.001 versus WT controls, ‡P < 0.001 versus KO controls, **P < 0.05 versus KO controls.
Effect of TRPC6 KO on albuminuria, renal histopathology, and glomerular ultrastructure
Figure 2a shows the effect of TRPC6 KO on albuminuria in diabetic mice. There was a significant decrease in albuminuria in KO Akita mice at 12 and 16 weeks of age, but this difference disappeared by 20 weeks of age. Albuminuria was modest in nondiabetic mice and was not affected by KO of TRPC6 (Table 1). We next evaluated the effect of TRPC6 KO on renal histopathology. Figure 2b shows periodic acid-Schiff-stained sections from WT and KO Akita mice, and Figure 2c shows the effect of TRPC6 KO on mesangial expansion by using a semiquantitative scoring system. Mesangial expansion was significantly increased in KO Akita mice compared with WT Akita mice (P < 0.001). In contrast, KO of TRPC6 significantly decreased tubular injury (P < 0.05) (Figure 2d) and tended to decrease tubulointer- stitial inflammation (P = 0.0604) (Figure 2e). No significant tubulointerstitial fibrosis was detected in diabetic mice by light microscopy. Total collagen content in kidney sections was quantitated using Sirius Red/Fast Green collagen staining (Table 2). There were no significant differences in kidney collagen content between the groups.
Figure 2 |. Effect of transient receptor potential cation channel C6 (TRPC6) knockout (KO) on albuminuria and kidney histopathology.

(a) KO of TRPC6 reduced albuminuria at 12 and 16 weeks of age, but this difference disappeared by 20 weeks of age. (b,c) Mesangial expansion was significantly increased in KO Akita mice compared with wild-type (WT) Akita mice. (d) Tubular injury was significantly decreased in KO Akita mice compared with WT Akita mice. (e) Tubulointerstitial (TI) inflammation also tended to be decreased in KO Akita mice compared with WT Akita mice, but this difference did not reach statistical significance (P = 0.0604). (f–h) Focal areas of foot process effacement were seen in both groups of Akita mice compared with nondiabetic WT and KO mice (nondiabetic controls). (i) Glomerular basement membrane (GBM) width was similar in all groups. For ultrastructural studies, 3–4 mice were studied per group. *P < 0.05 or †P < 0.001 versus WT Akita mice. RBC, red blood cell. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
Table 1 |.
Albuminuria (mg/mg creatinine) in nondiabetic mice
| Experimental group | 12 wk | 16 wk | 20 wk |
|---|---|---|---|
| WT mice (n =12) | 69 ± 15a | 84 ± 27b | 125 ± 24b |
| KO mice (n =11) | 64 ± 9.2 | 54 ± 16b | 134 ± 14b |
KO, knockout; n, number of mice in each group, WT, wild type.
P < 0.05 versus age-matched WT or KO Akita mice.
P < 0.001 versus age-matched WT or KO Akita mice.
Table 2 |.
Effect of TRPC6 knockout on kidney collagen content (μg collagen/mg protein)
| Experimental group | Nondiabetic mice | Diabetic Akita mice |
|---|---|---|
| Wild-type mice | 27.6 ± 0.66 (n = 8) | 27.1 ± 0.38 (n = 6) |
| TRPC6 knockout mice | 28.1 ± 0.69 (n = 10) | 27.3 ± 0.76 (n = 12) |
n, number of mice in each group.
Figure 2f–h shows the effect of diabetes on glomerular ultrastructure. Focal areas of foot process effacement were seen in both groups of Akita mice compared with nondiabetic WT and KO mice (nondiabetic controls), which were qualitatively similar in both groups of diabetic animals. There was no significant difference in glomerular basement membrane width between the groups (Figure 2i).
Lastly, Table 3 shows the effects of TRPC6 KO on glomerular morphology and podocyte number. Similar to body and kidney weights (Figure 1c–e), glomerular volume tended to be lower in nondiabetic TRPC6 KO mice than in nondiabetic WT mice. Diabetes increased the glomerular volume in both groups, which was similarly increased in both groups of diabetic mice when expressed as a percentage of the value in the nondiabetic groups. Podocyte number tended to decrease in the diabetic groups, but these changes were not statistically significant. Podocyte density was increased in diabetic and nondiabetic TRPC6 KO mice, likely because of the lower glomerular volume.
Table 3 |.
Effect of TRPC6 KO on glomerular volume and podocyte number in Akita mice
| Experimental group | Glomerular volume (×105 μm3) |
No. of podocytes per glomerular profile |
No. of podocytes per glomerulus | NV (P/Glom) (×10−5/μm3) |
|---|---|---|---|---|
| Nondiabetic WT mice (n = 7) | 2.54 ± 0.12 | 8.57 ± 0.27 | 51.8 ± 1.7 | 22.3 ± 0.7 |
| Nondiabetic KO mice (n = 6) | 1.92 ± 0.07 | 8.02 ± 0.29 | 52.3 ± 1.9 | 29.5 ± 1.1a |
| WT Akita mice (n = 8) | 3.92 ± 0.31b (153%)c | 6.88 ± 0.16a (80%)c | 50.7± 0.9 (97%)c | 14.2 ± 0.3b (63%)c |
| KO Akita mice (n = 12) | 2.92 ± 0.18d,e (153%)f | 7.55 ± 0.18 (94%)f | 48.6 ± 1.2 (93%)f | 17.9 ± 0.4f,g (60%)f |
KO, knockout; n, number of mice in each group; NV (P/Glom), numerical density of podocytes in glomeruli; WT, wild type.
P < 0.05 versus nondiabetic WT mice.
P < 0.001 versus nondiabetic WT mice.
Percentage of nondiabetic WT mice.
P < 0.05 versus nondiabetic KO mice.
P < 0.01 versus WT Akita mice.
Percentage of nondiabetic KO mice.
P < 0.001 versus nondiabetic KO mice.
TRPC6 KO mice are insulin resistant
Although hyperglycemia is an important determinant of the severity of kidney disease in diabetes mellitus,1 studies in animal models of type 1 diabetes have shown that insulin sensitizers ameliorate DKD independent of blood glucose levels.31–33 This dichotomy between blood glucose levels and severity of DN is particularly striking in mice with a podocyte-specific deletion of the insulin receptor.34 These KO mice develop albuminuria and glomerular features of DKD in the normoglycemic environment.34 We therefore examined TRPC6 KO mice for abnormalities in glucose regulation by performing the glucose tolerance test (GTT) and insulin tolerance test. As shown in Figure 3 a and b, KO Akita mice were insulin resistant compared with WT Akita mice as evidenced by the significant decrease in the area under the curve (AUC) in KO Akita mice (Figure 3b). A similar trend was seen for AUCs in nondiabetic mice, but this difference did not reach statistical significance (Figure 3c and d). Similar to previous reports,35 diabetic mice were also insulin resistant compared with nondiabetic animals (252 ± 30 AUC [nondiabetic WT mice] vs. 132 ± 10 AUC [WT Akita mice]; P < 0.01 and 235 ± 27 AUC [nondiabetic KO mice] vs. 89 ± 11 AUC [KO Akita mice]; P < 0.001). The results of GTTs are shown in Figure 3e and f. There were no statistically significant differences in the GTT curve between WT and KO Akita mice or between nondiabetic WT and KO controls. Glucose levels were similar in nondiabetic WT and KO mice as well as in WT and KO Akita mice at time 0 in the insulin tolerance test and GTT (Table 4).
Figure 3 |. Transient receptor potential cation channel C6 knockout (KO) causes insulin resistance.
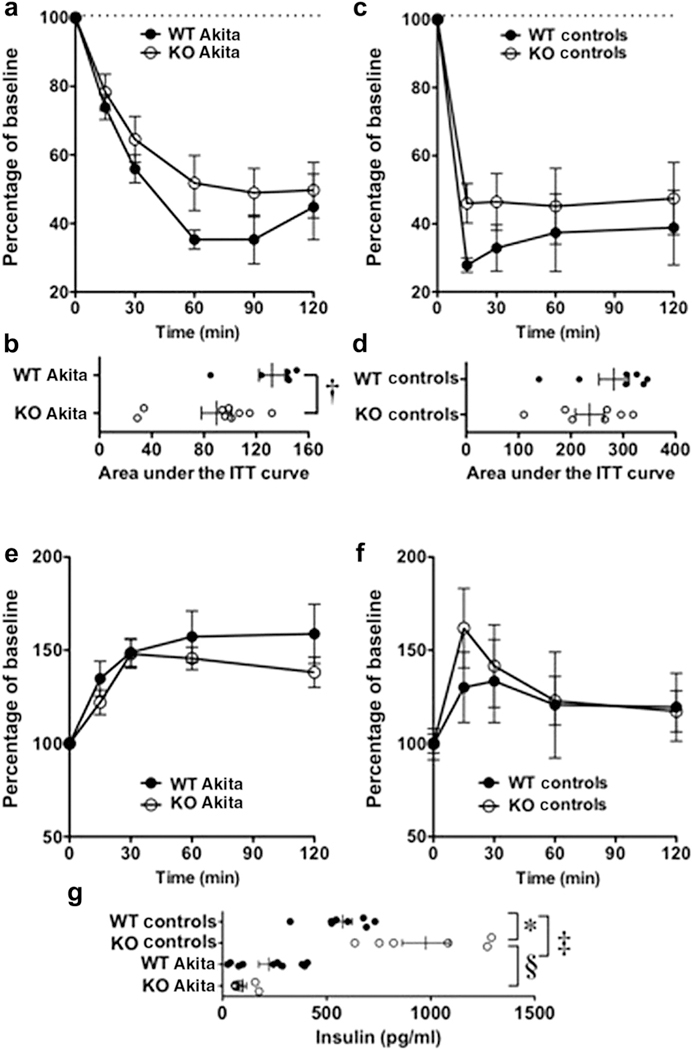
(a,b) The area under the insulin tolerance test (ITT) curves (AUCs) was significantly greater in wild-type (WT) Akita mice than in KO Akita mice. (c,d) The AUCs tended to be greater in nondiabetic WT mice (WT controls) than in nondiabetic KO mice (KO controls), but this difference was not statistically different. (e,f) The glucose tolerance test (GTT) curves were similar in both the Akita groups and the nondiabetic groups. (g) Insulin levels at the end of GTT were significantly elevated in nondiabetic KO mice compared with nondiabetic WT mice. Insulin levels tended to be lower in KO Akita mice than in WT Akita mice, but this difference was not statistically significant. *P < 0.05 or ‡P < 0.01 versus WT controls, †P < 0.025 versus WT Akita mice, §P < 0.001 versus KO controls.
Table 4 |.
Glucose levels (mg/dl) at time 0 in ITT and GTT
| Experimental group | Nondiabetic mice | Diabetic Akita mice |
|---|---|---|
| ITT TRPC6+/+ mice | 129 ± 5 (n = 6) | 476 ± 10a (n = 6) |
| ITT TRPC6−/− mice | 136 ± 9 (n = 6) | 485 ± 18.6b (n = 9) |
| GTT TRPC6+/+ mice | 129 ± 10 (n = 6) | 407 ± 15a (n = 6) |
| GTT TRPC6−/− mice | 136 ± 7 (n = 7) | 406 ± 20b (n = 7) |
GTT, glucose tolerance test; ITT, insulin tolerance test; n, number of mice in each group; WT, wild type.
P < 0.01 versus TRPC6+/+ WT mice.
P < 0.01 versus TRPC6−/− WT mice.
After the completion of GTT, blood was drawn to measure serum insulin levels. As shown in Figure 3g, insulin levels were significantly increased in nondiabetic KO mice compared with nondiabetic WT mice, suggesting insulin resistance. Insulin levels were decreased in both groups of Akita mice compared with nondiabetic animals (Figure 3g), but tended to be lower in KO Akita mice than in WT Akita mice (P = 0.054). Taken together with Figure 1a and b, these data suggest that TRPC6 KO promotes insulin resistance without affecting overall glycemic control.
TRPC6 KO inhibits insulin signaling in cultured podocytes
To investigate the effect of insulin signaling in cultured podocytes, we knocked out TRPC6 in cultured podocytes using CRISPR (clustered regularly interspaced short palindromic repeats)/CAS9 (CRISPR-associated protein 9) technology.36 As shown in Figure 4a, TRPC6 was not detected in TRPC6 KO podocytes (TRPC6−/−). Insulin activates both Akt2 and extra-cellular signal-regulated kinase (ERK) signaling.37,38 As shown in Figure 4b–e, ERK activation was potently inhibited by TRPC6 KO (decreased phospho-ERK or P-ERK) at 1 and 10 nM insulin concentrations. TRPC6 KO also inhibited Akt signaling at the 10 nM concentration as indicated by the decreased phosphorylation of Akt on threonine 308, the downstream target of phosphoinositide 3-kinase (PI3K). Akt is also phosphorylated on serine 473 by mammalian target of rapamycin, and serine 473 phosphorylation was not significantly affected by TRPC6 KO.
Figure 4 |. Insulin signaling in transient receptor potential cation channel C6 (TRPC6) knockout (KO) podocytes.
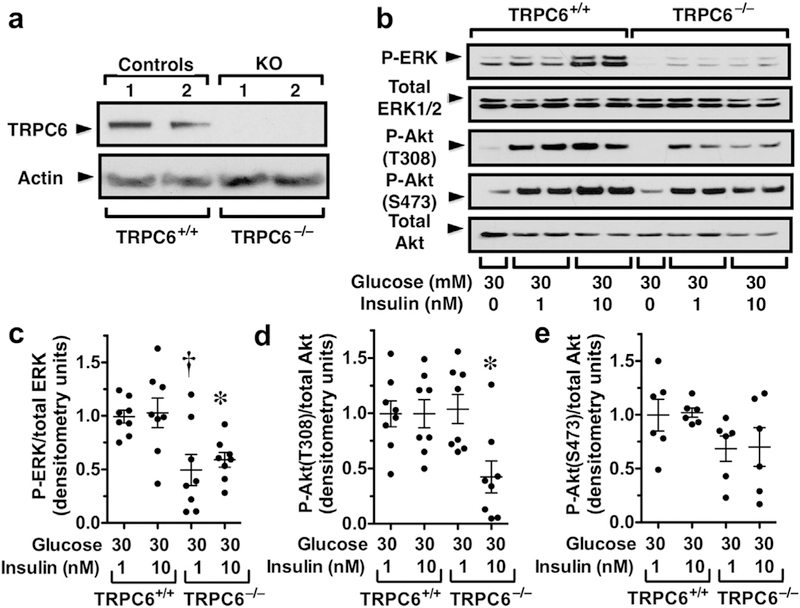
(a) TRPC6 protein was not detected in KO podocytes. (b,c) Extracellular signal-regulated kinase (ERK) activation was inhibited by TRPC6 KO (decreased phospho-ERK [P-ERK]) at 1 and 10 nM insulin concentrations. (b,d) Phosphorylation of Akt (P-Akt) on threonine 308 (T308) was inhibited at the 10 nM insulin concentration. (b,e) Phosphorylation of Akt on serine 473 (S473) was not significantly affected by TRPC6 KO. *P < 0.05 versus 10 nM insulin in TRPC6+/+ podocytes, †P < 0.01 versus 1 nM insulin in TRPC6+/+ podocytes. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
TRPC6 KO inhibits the expression of insulin receptor substrate 2
After activation of the insulin receptor, insulin receptor substrate 1 (IRS1) and IRS2 proteins play a key role in transmitting the signal to the downstream signaling molecules Akt2 and ERK.37,39 In this regard, KO of IRS2 potently inhibits ERK signaling with lesser effects on Akt,37,38 which is similar to the pattern in TRPC6 KO podocytes. Moreover, IRS2 is a calcineurin (CN)-responsive gene40,41 and TRPC6 activation increases intracellular calcium levels and stimulates the calcium-sensitive phosphatase CN.16,42 In support of a role for TRPC6 in promoting IRS2 expression, hyperglycemia induces IRS2 expression by promoting calcium entry into the cell from extracellular sources, which activates CN.40 As shown in Figure 5a–c, we found that hyperglycemia enhanced IRS2 expression, without altering expression or IRS1. Hyperglycemia-induced IRS2 expression is CN dependent because expression of IRS2 protein and mRNAwas attenuated by the CN inhibitor FK506 (Figure 5d–f). In support of a role for TRPC6 in modulating CN activity in vivo, the CN-responsive gene RCAN1 (regulator of CN 1)16,42 was potently induced in Akita mice and this increase in gene expression was blocked by KO of TRPC6 (Figure 5g). TRPC6 is also a CN-responsive gene16,42 and was induced in WT Akita mice (Figure 5h). TRPC6 mRNA and TRPC6 protein were not detected in TRPC KO mice (Figure 5h and Supplementary Figure S1, respectively). We also investigated the expression of other TRPC family members that are thought to contribute to calcium entry in podocytes.43 We therefore focused on TRPC3 and TRPC5 for the study. As shown in Supplementary Figure S2, there were no significant differences in the expression of TRPC3 or TRPC5 in either nondiabetic KO mice or KO Akita mice.
Figure 5 |. Effect of transient receptor potential cation channel C6 (TRPC6) knockout (KO) on the expression of insulin receptor substrate 2 (IRS2).

(a–c) Hyperglycemia-enhanced IRS2 expression, without altering expression or IRS1. (d–f) Hyperglycemia-induced expression of IRS2 protein and mRNA, and the increase in IRS2 protein and mRNA levels was inhibited by the calcineurin (CN) inhibitor FK506. (g) Expression of the CN-responsive gene RCAN1 was enhanced in wild-type (WT) Akita mice, and this increase in gene expression was inhibited in KO Akita mice. (h) Expression of the CN-responsive gene TRPC6 was also increased in WT Akita mice. (i,j) IRS2 protein levels were significantly reduced in cortices from TRPC6 KO mice incubated ex vivo in 30 mM glucose. (k) IRS2 mRNA levels were significantly decreased in KO Akita mice. *P < 0.05 or f P < 0.001 versus nondiabetic WT mice (WT controls), **P < 0.01 versus WT Akita mice, †P < 0.01 versus 5 mM glucose in TRPC6+/+ podocytes, ‡P < 0.05 or §P < 0.01 versus 30 mM glucose in TRPC6+/+ podocytes, ***P < 0.05 versus WT cortices in 5 or 30 mM glucose, ‡‡P < 0.05 versus 5 mM glucose treated with dimethyl sulfoxide (DMSO), ††P < 0.05 versus 30 mM glucose treated with DMSO. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
We next investigated the effect of diabetes on IRS2 expression in vivo, but we were not able to detect IRS2 proteins in either glomerular or cortical lysates from diabetic or nondiabetic animals. Regulation of IRS protein expression is, however, complex and involves both transcriptional and posttranscriptional mechanisms including proteasomal degradation.40,41,44 We therefore incubated kidney cortices from WT and KO mice in low- or high-glucose medium in the presence of the proteasome inhibitor MG132.45 As shown in Figure 5i and j, IRS2 protein levels were significantly reduced in cortices from TRPC6 KO mice. In this ex vivo preparation, IRS2 protein levels tended to increase with hyperglycemia, but this difference was not statistically significant. In vivo, KO of TRPC6 significantly reduced IRS2 mRNA levels in glomerular preparations from KO Akita mice (Figure 5k). We did not detect an increase in IRS2 mRNA levels in WT Akita mice, perhaps because of the multiple pathways that contribute IRS2 transcriptional regulation.46
Hyperglycemia activates apoptosis signal regulated kinase 1-p38 signaling and promotes podocyte apoptosis
As shown in Figure 6a, apoptosis signal regulated kinase 1 (ASK1) is activated by hyperglycemia-induced reactive oxygen species, which stimulates ASK1-p38 signaling and promotes cellular apoptosis in diabetes.47 Insulin activates ERK, which inhibits p38 signaling by stimulating mitogen-activated protein kinase phosphatase 1 and in turn dephosphorylation and inhibition of p38 signaling.50 Insulin also stimulates Akt2,39 which phosphorylates and inhibits ASK1.48,49 We therefore investigated the effect of hyperglycemia on p38 signaling and podocyte apoptosis in cultured mouse podocytes. As shown in Figure 6b, hyperglycemia induced podocyte apoptosis and this apoptotic effect was attenuated by insulin and the ERK inhibitor PD98059 blocked the antiapoptotic effect of insulin. The osmotic control (mannitol) had no significant effect on podocyte apoptosis. Figure 6c–e shows that hyperglycemia activated p38 signaling (phospho-p38 or P-p38), which was associated with an increase in cleaved caspase 3, consistent with stimulation of apoptosis. This apoptotic effect was attenuated by insulin, and the antiapoptotic effect of insulin was inhibited by PD98059. Similarly, inhibition of podocyte apoptosis by insulin was attenuated by the PI3K-Akt inhibitor LY294002 (Supplementary Figure S3). Lastly, we investigated the effect of the ASK1 inhibitor NQD1 on hyperglycemia-induced podocyte apoptosis. As shown in Figure 6f–h, NQD1 (50 μM) inhibited apoptosis induced by 30 mM glucose, similar to the antiapoptotic effect of insulin.
Figure 6 |. Hyperglycemia activates apoptosis signal regulated kinase 1 (ASK1)–p38 signaling in podocytes and promotes apoptosis.
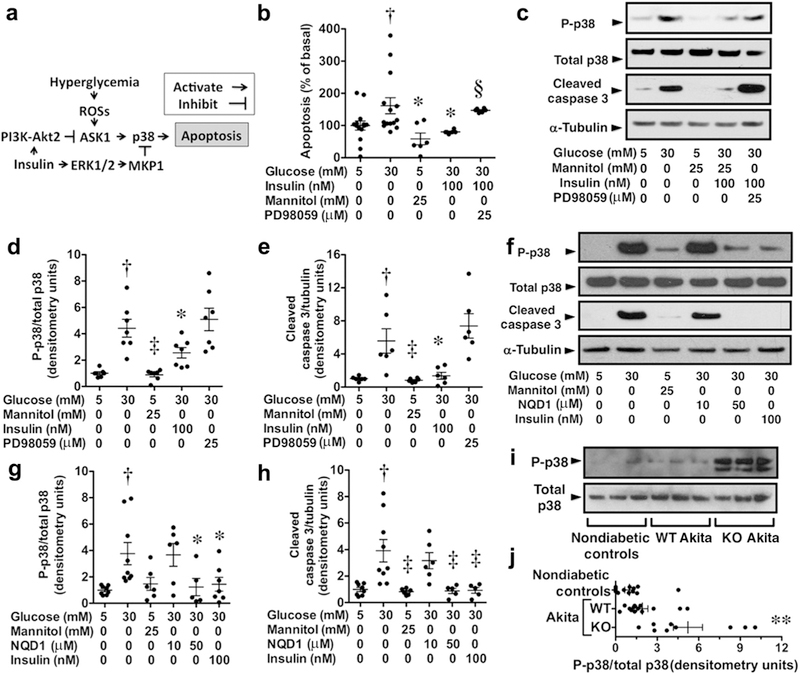
(a) ASK1 is activated by hyperglycemia-induced reactive oxygen species (ROS) generation, which stimulates ASK1-p38 signaling and promotes cellular apoptosis.47 Insulin stimulates both phosphoinositide 3-kinase (PI3K)–Akt2 and extracellular signal-regulated kinase (ERK) signaling, which promotes ERK-dependent inhibition of p38 signaling and PI3K-Akt2–dependent inhibition of ASK1 signaling.39,47,48,49 (b) Hyperglycemia (30 mM glucose) induced podocyte apoptosis, and this apoptotic effect was inhibited by insulin. The ERK inhibitor PD98059 blocked the antiapoptotic effect of insulin. The osmotic control (mannitol) had no significant effect on podocyte apoptosis. (c–e) Hyperglycemia (30 mM glucose) stimulated ASK1-p38 signaling (phospho-p38 or P-p38) in cultured podocytes and enhanced expression of the apoptosis marker cleaved caspase 3. Both p38 signaling and podocyte apoptosis were inhibited by insulin, and the antiapoptotic effect of insulin was inhibited by ERK inhibition with PD98059. (f–h) Hyperglycemia stimulated ASK1-p38 signaling in cultured podocytes and enhanced expression of the apoptosis marker cleaved caspase 3. Both p38 signaling and podocyte apoptosis were inhibited by insulin and the ASK1 inhibitor NQD1 (50 μM). (i,j) ASK1-p38 signaling was enhanced in knockout (KO) Akita mice compared with either nondiabetic controls or wild-type (WT) Akita mice. MKP1, mitogen-activated protein kinase phosphatase 1. †P < 0.05 versus 5 mM glucose, *P < 0.05 or ‡P < 0.01 versus 30 mM glucose, §P < 0.05 versus 30 mM glucose and insulin, **P < 0.05 versus either nondiabetic controls or WT Akita mice. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
To determine whether p38 signaling was enhanced in vivo, we next examined p38 activity in glomerular lysates from diabetic and nondiabetic groups. Because p38 signaling was similar in both WT and KO controls, these data were combined for the analyses (nondiabetic controls). As shown in Figure 6i and j, P-p38 levels tended to increase in WT Akita mice compared with nondiabetic controls (nondiabetic WT and KO mice). In contrast, P-p38 levels were prominently increased in KO Akita mice compared with either nondiabetic controls or WT Akita mice, consistent with enhanced p38 signaling in glomeruli of diabetic mice.
Effect of TRPC6 KO on cyclooxygenase 2 expression
In previous studies, we demonstrated that cyclooxygenase 2 (COX2) was a CN-responsive gene in glomerular podocytes.16 Consistent with this response, COX2 mRNA levels tended to increase in WT Akita mice (Figure 7a), but we were surprised to find that COX2 mRNA expression was significantly increased in KO Akita mice. To determine whether COX2 protein expression was also increased in KO Akita mice, we assessed the glomerular expression of COX2 protein by immunoblotting. As shown in Figure 7b and c, there was a prominent increase in COX2 protein in glomerular lysates from KO Akita mice. Immunofluorescence studies found that this increase in glomerular COX2 expression was predominantly localized to glomerular podocytes (Figure 7d). These findings are likely relevant to enhanced glomerular p38 signaling in KO Akita mice because COX2 is an important gene target of p38 signaling.51,52 Moreover, given the key role of COX2 in the pathogenesis of DKD,53–56 these data suggest that COX2 may play a role in exacerbating glomerular disease in KO Akita mice.
Figure 7 |. Effect of diabetes and transient receptor potential cation channel C6 (TRPC6) knockout (KO) on calcineurin target genes.
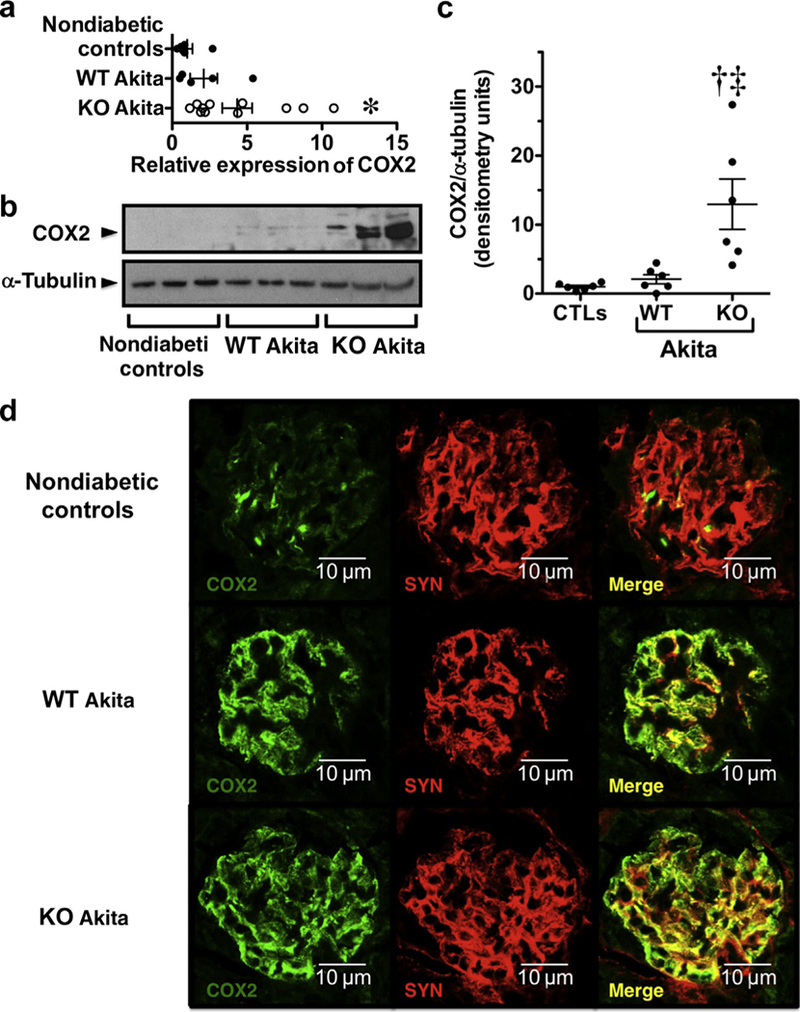
(a) Expression of cyclooxygenase 2 (COX2) mRNA tended to increase in both groups of Akita mice compared with nondiabetic controls, and this difference was statistically significant for the KO Akita group. (b,c) Expression of the COX2 protein was significantly increased in KO Akita mice compared with either nondiabetic controls or wild-type (WT) Akita mice. (d) Tissue sections were stained for COX2 (green) and the podocyte marker synaptopodin (SYN; red) and examined by confocal microscopy. In nondiabetic controls, focal areas COX2 staining were observed, which largely colocalized with the podocyte marker SYN. In both WT and KO Akita mice, COX2 staining also colocalized with the podocyte marker SYN but was more prominent and detected diffusely in the glomerular areas compared with nondiabetic controls. The intensity of staining tended to be more prominent in KO Akita mice, consistent with immunoblotting and quantitative real-time polymerase chain reaction studies. Data for real-time polymerase chain reaction, immunoblotting, and immunofluorescence studies were similar in nondiabetic KO and WT mice (nondiabetic controls), and these data were combined for statistical analyses. For immunofluorescence studies, 4–5 mice were studied per group. *P < 0.05 or †P < 0.025 versus nondiabetic controls, ‡P < 0.05 versus WT Akita mice. CTL, controls. To optimize viewing of this image, please see the online version of this article at www.kidney-international.org.
DISCUSSION
We investigated the effect of whole body TRPC6 KO on DKD in Akita mice. We found that KO of TRPC6 attenuated tubular injury as well as reduced proteinuria until the later stages of the disease process. In contrast, KO of TRPC6 caused prominent mesangial expansion in the diabetic group. This dissociation between the glomerular and tubular compartments suggests enhanced susceptibility of glomerular cell types to the adverse effects of TRPC6 KO in the setting of hyperglycemia. These observations may be directly relevant to glomerular podocytes because (i) KO of the podocyte insulin receptor promotes albuminuria and histopathological features of diabetic glomerular disease in the normoglycemic environment34 and (ii) KO Akita mice were insulin resistant compared with WT Akita mice. Surprisingly, the adverse effects of TRPC6 KO were observed despite comparable hemoglobin A1c levels and fasting blood glucose levels. These findings are, however, consistent with published studies suggesting that insulin sensitizers ameliorate DKD in type 1 diabetic models with similar glycemic control.31–33 Taken together with published studies,31–34 these data suggest that (i) impaired insulin resistance promotes the development of diabetic glomerular disease and (ii) KO of TRPC6 has adverse effects on kidney disease in a mouse model of type 1 diabetes.
Insulin resistance appears to be mediated, at least in part, by a reduction in the expression of calcium/CN-responsive gene IRS2 in TRPC6 KO mice. These data are supported by the following observations: (i) TRPC6 KO inhibited hyperglycemia-induced IRS expression in vitro and ex vivo, and this increase in IRS2 expression was attenuated by the CN inhibitor FK506 in cultured podocytes (Figure 5). (ii) Similar to IRS2 KO,37 TRPC6 KO reduced ERK signaling to a greater extent than Akt (Figure 4). (iii) Similar to IRS2 KO mice,57 TRPC6 KO mice exhibited mild growth retardation (Figure 1). (iv) IRS2 plays an essential role in β-islet cell proliferation and survival,40,57,58 which is consistent with the trend toward decreased insulin levels in TRPC6 KO Akita mice (Figure 3). Taken together, these suggest an important role for IRS2 in promoting insulin resistance in TRPC6 KO mice. We do, however, acknowledge that systemic KO of TRPC6 might have other important effects that could promote insulin resistance. For example, TRPC6 KO might alter secretion of insulin counterregulatory hormones or affect other downstream insulin-stimulated signaling cascades. These possibilities will require further evaluation in future studies.
The findings of the present study are particularly relevant to patients with type 2 diabetes and to an increasingly common group of patients with type 1 diabetes and insulin resistance.59 In this regard, heterozygous Akita mice are often studied as a model of type 1 diabetes.28,29 These animals have the spontaneous mutation in the insulin 2 gene InsAklta, which causes selective pancreatic β cell failure as a result of proteotoxicity due to misfolding of insulin 2.28 Homozygous Akita mice exhibit failure to thrive and die at ~ 2 months of age.28 In contrast, heterozygous Akita mice retain significant residual insulin secretion and are frequently used to study DKD, but are also insulin resistant.35 Thus, heterozygous Akita mice used in the present study have features of type 2 diabetes, and insulin resistance in these animals is increased by KO of TRPC6.
Insulin has potent prosurvival effects on multiple kidney cell types.34,60,61 For example, insulin inhibits mesangial cell apoptosis after exposure to multiple apoptotic.60 Akt signaling also attenuates renal tubular cell apoptosis by inhibiting the p38-signaling cascade.61 Moreover, insulin-like growth factors have prosurvival effects on podocytes, which are mediated, as least in part, by activating the PI3K-Akt signaling pathway.62 We therefore investigated the effects of insulin on podocyte apoptosis induced by hyperglycemia. We found that hyperglycemia promoted podocyte apoptosis and that this apoptotic effect was inhibited by both insulin and the ASK1 inhibitor NQD1. Moreover, the antiapoptotic effect of insulin was blocked by the ERK inhibitor PD98059 and the PI3K-Akt inhibitor LY294002. Thus, insulin-induced Akt and ERK signaling plays key a role in mediating the prosurvival effects of insulin on glomerular podocytes.
In support of an important role for ASK1-p38 signaling in promoting diabetic glomerular disease, we found that the p38 mitogen-activated protein kinase pathway was potently activated in KO Akita mice. ASK1 inhibitors also ameliorate DKD in animal models,47,63 and ASK1 inhibition is associated with reduced p38 signaling.63 An important gene target of p38 signaling is COX2,51,52 which is induced by both transcriptional and posttranscriptional mechanisms.51,52 In the present study, COX2 expression was upregulated at both the protein and mRNA levels in KO Akita mice. Moreover, COX2 activity plays an important role in the pathogenesis of glomerular injury in DKD.53–55 Indeed, overexpression of COX2 specifically in podocytes exacerbates DKD.53 These observations are directly relevant to the present studies because COX2 expression was potently induced in glomerular preparations from KO Akita mice and localized to glomerular podocytes (Figure 5). Thus, COX2 may be an important mediator of the adverse effects on glomerular injury in KO Akita mice.
Akt is a master regulator of a wide range of physiological functions including cellular metabolism, proliferation, survival, and growth.64 Akt2 is the major isoform activated by insulin,39 and insulin-induced Akt activation is reduced in type 2 diabetic models.65 Moreover, Akt2 signaling plays a key role in maintaining podocyte viability in chronic kidney diseases.66 The multiple cellular functions of Akt have been investigated in isoform-specific Akt KO mice.66,67 In these mice, whole body Akt2 KO causes insulin resistance and mild growth deficiency.67 Consistent with these observations, nondiabetic TRPC6 KO mice were also insulin resistant and have decreased body weight compared with nondiabetic WT mice. Thus, Akt2 may play a role in promoting the pheno-types observed in diabetic and nondiabetic mice lacking TRPC6.
Lastly, several studies have reported beneficial effects of TRPC6 KO in proteinuric kidney diseases.16,68–70 For example, a study from our laboratory16 and a separate study by Dryer and coworkers69 reported that TRPC6 KO ameliorated kidney disease in rodent models of puromycin amino-nucleoside nephrosis,16,69 suggesting favorable effects in nondiabetic kidney disease. In addition, 2 recent studies reported beneficial effects of TRPC6 KO in rodent models of DKD after 11 to 12 weeks of diabetes.68,70 The beneficial effects of TRPC6 KO in these studies were observed at a time point of 12 weeks of diabetes, which was associated with an improvement in albuminuria in the present study (Figure 2a). Thus, a study of longer duration may be necessary to detect the adverse effects of TRPC6 KO on the diabetic phenotype.
In summary, KO of TRPC6 decreased proteinuria and attenuated tubular injury but promoted mesangial expansion in Akita mice. These findings were associated with insulin resistance, impaired insulin signaling, and reduced expression of the CN-responsive gene IRS2, which likely contributes to insulin resistance in KO Akita mice. In cultured podocytes, hyperglycemia activated ASK1-p38 signaling as well as promoted podocyte apoptosis and both p38 signaling and cellular apoptosis were attenuated by insulin-induced activation of either PI3K-Akt or ERK. Lastly, p38 mitogen-activated protein kinase signaling was enhanced in insulin-resistant KO Akita mice and was associated with upregulation of the p38 target gene COX2, which has been implicated in the pathogenesis of DKD.53–55 These data suggest that targeting TRPC6 to treat DN may have harmful effects.
METHODS
Experimental protocol
The experiments used the Akita model of type 1 diabetes.71 Akita mice lacking TRPC6 were created by breeding FVB/NJ Akita mice71 with TRPC6 KO mice30 for >10 generations. All experiments conformed to the Guide for the Care and Use of Laboratory Animals72 and were approved by the Duke University and Durham VA Medical Centers’ Institutional Animal Care and Use Committees. Details of the experimental protocols are given in Supplementary Materials and Methods.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants R01DK087707 (to RFS) from the National Institutes of Health and 1-INO-2015–94-A-B from the Juvenile Diabetes Research Foundation as well as BX002984 (to RFS) from the Veterans Administration Merit Review Award Program. RFS also received salary support through grants RO1 DK094987 and RO1 DK103694 from the National Institutes of Health, National Institute of Diabetes, Digestive and Kidney Diseases as well as P30 DK096493 from the Duke O’Brien Center for Kidney Research. The results presented in this article have not been published previously, in whole or in part, except in abstract format. We thank William Eisner for his technical assistance with the study.
Footnotes
DISCLOSURE
RFS receives funding from Amgen Inc. All the other authors declared no competing interests.
REFERENCES
- 1.Molitch ME, DeFronzo RA, Franz MJ, et al. Nephropathy in diabetes. Diabetes Care. 2004;27:S79–S83. [DOI] [PubMed] [Google Scholar]
- 2.Maahs DM, West NA, Lawrence JM, Mayer-Davis EJ. Epidemiology of type 1 diabetes. Endocrinol Metab Clin North Am. 2010;39:481–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lewis EJ, Hunsicker LG, Bain RP, Rohde RD, The Collaborative Study Group. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. N Engl J Med. 1993;329:1456–1462. [DOI] [PubMed] [Google Scholar]
- 4.Lewis EJ, Hunsicker LG, Clarke WR, et al. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med. 2001;345:851–860. [DOI] [PubMed] [Google Scholar]
- 5.Brenner BM, Cooper ME, de Zeeuw D, et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med. 2001;345:861–869. [DOI] [PubMed] [Google Scholar]
- 6.Wolf G, Chen S, Ziyadeh FN. From the periphery of the glomerular capillary wall toward the center of disease: podocyte injury comes of age in diabetic nephropathy. Diabetes. 2005;54:1626–1634. [DOI] [PubMed] [Google Scholar]
- 7.Jefferson JA, Shankland SJ, Pichler RH. Proteinuria in diabetic kidney disease: a mechanistic viewpoint. Kidney Int. 2008;74:22–36. [DOI] [PubMed] [Google Scholar]
- 8.Wiggins RC. The spectrum of podocytopathies: a unifying view of glomerular diseases. Kidney Int. 2007;71:1205–1214. [DOI] [PubMed] [Google Scholar]
- 9.Dalla Vestra M, Masiero A, Roiter AM, et al. Is podocyte injury relevant in diabetic nephropathy? Studies in patients with type 2 diabetes. Diabetes. 2003;52:1031–1035. [DOI] [PubMed] [Google Scholar]
- 10.Meyer TW, Bennett PH, Nelson RG. Podocyte number predicts long-term urinary albumin excretion in Pima Indians with type II diabetes and microalbuminuria. Diabetologia. 1999;42:1341–1344. [DOI] [PubMed] [Google Scholar]
- 11.White KE, Bilous RW, Marshall SM, et al. Podocyte number in normotensive type 1 diabetic patients with albuminuria. Diabetes. 2002;51:3083–3089. [DOI] [PubMed] [Google Scholar]
- 12.Kriz W, Gretz N, Lemley KV. Progression of glomerular diseases: is the podocyte the culprit? Kidney Int. 1998;54:687–697. [DOI] [PubMed] [Google Scholar]
- 13.Hall G, Gbadegesin RA. Translating genetic findings in hereditary nephrotic syndrome: the missing loops. Am J Physiol Renal Physiol. 2015;309:F24–F28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Winn MP, Conlon PJ, Lynn KL, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308:1801–1804. [DOI] [PubMed] [Google Scholar]
- 15.Reiser J, Polu KR, Moller CC, et al. TRPC6 is a glomerular slit diaphragm- associated channel required for normal renal function. Nat Genet. 2005;37:739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang L, Jirka G, Rosenberg PB, et al. Gq signaling causes glomerular injury by activating TRPC6. J Clin Invest. 2015;125:1913–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moller CC, Wei C, Altintas MM, et al. Induction of TRPC6 channel in acquired forms of proteinuric kidney disease. J Am Soc Nephrol. 2007;18: 29–36. [DOI] [PubMed] [Google Scholar]
- 18.Zhang X, Song Z, Guo Y, Zhou M. The novel role of TRPC6 in vitamin D ameliorating podocyte injury in STZ-induced diabetic rats. Mol Cell Biochem. 2015;399:155–165. [DOI] [PubMed] [Google Scholar]
- 19.Eckel J, Lavin PJ, Finch EA, et al. TRPC6 enhances angiotensin II-induced albuminuria. J Am Soc Nephrol. 2011;22:526–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nijenhuis T, Sloan AJ, Hoenderop JG, et al. Angiotensin II contributes to podocyte injury by increasing TRPC6 expression via an NFAT-mediated positive feedback signaling pathway. Am J Pathol. 2011;179:1719–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Anderson M, Roshanravan H, Khine J, Dryer SE. Angiotensin II activation of TRPC6 channels in rat podocytes requires generation of reactive oxygen species. J Cell Physiol. 2014;229:434–442. [DOI] [PubMed] [Google Scholar]
- 22.Wang L, Chang JH, Paik SY, et al. Calcineurin (CN) activation promotes apoptosis of glomerular podocytes both in vitro and in vivo. Mol Endocrinol. 2011;25:1376–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ding G, Reddy K, Kapasi AA, et al. Angiotensin II induces apoptosis in rat glomerular epithelial cells. Am J Physiol Renal Physiol. 2002;283: F173–F180. [DOI] [PubMed] [Google Scholar]
- 24.Jia J, Ding G, Zhu J, et al. Angiotensin II infusion induces nephrin expression changes and podocyte apoptosis. Am J Nephrol. 2008;28: 500–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stanton RC. Oxidative stress and diabetic kidney disease. Curr Diab Rep. 2011;11:330–336. [DOI] [PubMed] [Google Scholar]
- 26.Roshanravan H, Dryer SE. ATP acting through P2Y receptors causes activation of podocyte TRPC6 channels: role of podocin and reactive oxygen species. Am J Physiol Renal Physiol. 2014;306:F1088–F1097. [DOI] [PubMed] [Google Scholar]
- 27.Kim EY, Anderson M, Dryer SE. Insulin increases surface expression of TRPC6 channels in podocytes: role of NADPH oxidases and reactive oxygen species. Am J Physiol Renal Physiol. 2012;302:F298–F307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Breyer MD, Bottinger E, Brosius FC III, et al. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2005;16:27–45. [DOI] [PubMed] [Google Scholar]
- 29.Brosius FC III, Alpers CE, Bottinger EP, et al. Mouse models of diabetic nephropathy. J Am Soc Nephrol. 2009;20:2503–2512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dietrich A, Mederos YSM, Gollasch M, et al. Increased vascular smooth muscle contractility in TRPC6−/− mice. Mol Cell Biol. 2005;25:6980–6989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Isshiki K, Haneda M, Koya D, et al. Thiazolidinedione compounds ameliorate glomerular dysfunction independent of their insulin-sensitizing action in diabetic rats. Diabetes. 2000;49:1022–1032. [DOI] [PubMed] [Google Scholar]
- 32.Nicholas SB, Kawano Y, Wakino S, et al. Expression and function of peroxisome proliferator-activated receptor-gamma in mesangial cells. Hypertension. 2001;37:722–727. [DOI] [PubMed] [Google Scholar]
- 33.Zhang H, Saha J, Byun J, et al. Rosiglitazone reduces renal and plasma markers of oxidative injury and reverses urinary metabolite abnormalities in the amelioration of diabetic nephropathy. Am J Physiol Renal Physiol. 2008;295:F1071–F1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Welsh GI, Hale LJ, Eremina V, et al. Insulin signaling to the glomerular podocyte is critical for normal kidney function. Cell Metab. 2010;12:329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hong EG, Jung DY, Ko HJ, et al. Nonobese, insulin-deficient Ins2Akita mice develop type 2 diabetes phenotypes including insulin resistance and cardiac remodeling. Am J Physiol Endocrinol Metab. 2007;293: E1687–E1696. [DOI] [PubMed] [Google Scholar]
- 36.Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32:347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thirone AC, Huang C, Klip A. Tissue-specific roles of IRS proteins in insulin signaling and glucose transport. Trends Endocrinol Metab. 2006;17:72–78. [DOI] [PubMed] [Google Scholar]
- 38.Huang C, Thirone AC, Huang X, Klip A. Differential contribution of insulin receptor substrates 1 versus 2 to insulin signaling and glucose uptake in l6 myotubes. J Biol Chem. 2005;280:19426–19435. [DOI] [PubMed] [Google Scholar]
- 39.Coward R, Fornoni A. Insulin signaling: implications for podocyte biology in diabetic kidney disease. Curr Opin Nephrol Hypertens. 2015;24:104–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Demozay D, Tsunekawa S, Briaud I, et al. Specific glucose-induced control of insulin receptor substrate-2 expression is mediated via Ca2+-dependent calcineurin/NFAT signaling in primary pancreatic islet β-cells. Diabetes. 2011;60:2892–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Soleimanpour SA, Crutchlow MF, Ferrari AM, et al. Calcineurin signaling regulates human islet β-cell survival. J Biol Chem. 2010;285:40050–40059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuwahara K, Wang Y, McAnally J, et al. TRPC6 fulfills a calcineurin signaling circuit during pathologic cardiac remodeling. J Clin Invest. 2006;116:3114–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ilatovskaya DV, Staruschenko A. TRPC6 channel as an emerging determinant of the podocyte injury susceptibility in kidney diseases. Am J Physiol Renal Physiol. 2015;309:F393–F397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mima A, Ohshiro Y, Kitada M, et al. Glomerular-specific protein kinase C-β-induced insulin receptor substrate-1 dysfunction and insulin resistance in rat models of diabetes and obesity. Kidney Int. 2011;79:883–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tsubuki S, Saito Y, Tomioka M, et al. Differential inhibition of calpain and proteasome activities by peptidyl aldehydes of di-leucine and tri-leucine. J Biochem. 1996;119:572–576. [DOI] [PubMed] [Google Scholar]
- 46.Jameson JL, De Groot L. Endocrinology: Adult and Pediatric. 7th ed Vols 1 and 2 Philadelphia, PA: Saunders; 2016. xvii, 2687, 2677. [Google Scholar]
- 47.Tesch GH, Ma FY, Nikolic-Paterson DJ. ASK1: a new therapeutic target for kidney disease. Am J Physiol Renal Physiol. 2016;311:F373–F381. [DOI] [PubMed] [Google Scholar]
- 48.Kim AH, Khursigara G, Sun X, et al. Akt phosphorylates and negatively regulates apoptosis signal-regulating kinase 1. Mol Cell Biol. 2001;21:893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yuan ZQ, Feldman RI, Sussman GE, et al. AKT2 inhibition of cisplatin-induced JNK/p38 and Bax activation by phosphorylation of ASK1: implication of AKT2 in chemoresistance. J Biol Chem. 2003;278:23432–23440. [DOI] [PubMed] [Google Scholar]
- 50.Wancket LM, Frazier WJ, Liu Y. Mitogen-activated protein kinase phosphatase (MKP)-1 in immunology, physiology, and disease. Life Sci. 2012;90:237–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dean JL, Brook M, Clark AR, Saklatvala J. p38 mitogen-activated protein kinase regulates cyclooxygenase-2 mRNA stability and transcription in lipopolysaccharide-treated human monocytes. J Biol Chem. 1999;274: 264–269. [DOI] [PubMed] [Google Scholar]
- 52.Singer CA, Baker KJ, McCaffrey A, et al. p38 MAPK and NF-kB mediate COX-2 expression in human airway myocytes. Am J Physiol Lung Cell Mol Physiol. 2003;285:L1087–L1098. [DOI] [PubMed] [Google Scholar]
- 53.Cheng H, Fan X, Moeckel GW, Harris RC. Podocyte COX-2 exacerbates diabetic nephropathy by increasing podocyte (pro)renin receptor expression. J Am Soc Nephrol. 2011;22:1240–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Quilley J, Santos M, Pedraza P. Renal protective effect of chronic inhibition of COX-2 with SC-58236 in streptozotocin-diabetic rats. Am J Physiol Heart Circ Physiol. 2011;300:H2316–H2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cheng HF, Wang CJ, Moeckel GW, et al. Cyclooxygenase-2 inhibitor blocks expression of mediators of renal injury in a model of diabetes and hypertension. Kidney Int. 2002;62:929–939. [DOI] [PubMed] [Google Scholar]
- 56.Vogt L, de Zeeuw D, Woittiez AJ, Navis G. Selective cyclooxygenase-2 (COX-2) inhibition reduces proteinuria in renal patients. Nephrol Dial Transplant. 2009;24:1182–1189. [DOI] [PubMed] [Google Scholar]
- 57.Withers DJ, Gutierrez JS, Towery H, et al. Disruption of IRS-2 causes type 2 diabetes in mice. Nature. 1998;391:900–904. [DOI] [PubMed] [Google Scholar]
- 58.Hayes HL, Moss LG, Schisler JC, et al. Pdx-1 activates islet α- and β-cell proliferation via a mechanism regulated by transient receptor potential cation channels 3 and 6 and extracellular signal-regulated kinases 1 and 2. Mol Cell Biol. 2013;33:4017–4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pozzilli P, Guglielmi C, Caprio S, Buzzetti R. Obesity, autoimmunity, and double diabetes in youth. Diabetes Care. 2011;34:S166–S170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hiromura K, Monkawa T, Petermann AT, et al. Insulin is a potent survival factor in mesangial cells: role of the PI3-kinase/Akt pathway. Kidney Int. 2002;61:1312–1321. [DOI] [PubMed] [Google Scholar]
- 61.Rane MJ, Song Y, Jin S, et al. Interplay between Akt and p38 MAPK pathways in the regulation of renal tubular cell apoptosis associated with diabetic nephropathy. Am J Physiol Renal Physiol. 2010;298:F49–F61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bridgewater DJ, Ho J, Sauro V, Matsell DG. Insulin-like growth factors inhibit podocyte apoptosis through the PI3 kinase pathway. Kidney Int. 2005;67:1308–1314. [DOI] [PubMed] [Google Scholar]
- 63.Tesch GH, Ma FY, Han Y, et al. ASK1 inhibitor halts progression of diabetic nephropathy in Nos3-deficient mice. Diabetes. 2015;64:3903–3913. [DOI] [PubMed] [Google Scholar]
- 64.Toker A, Marmiroli S. Signaling specificity in the Akt pathway in biology and disease. Adv Biol Regul. 2014;55:28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tejada T, Catanuto P, Ijaz A, et al. Failure to phosphorylate AKT in podocytes from mice with early diabetic nephropathy promotes cell death. Kidney Int. 2008;73:1385–1393. [DOI] [PubMed] [Google Scholar]
- 66.Canaud G, Bienaime F, Viau A, et al. AKT2 is essential to maintain podocyte viability and function during chronic kidney disease. Nat Med. 2013;19:1288–1296. [DOI] [PubMed] [Google Scholar]
- 67.Garofalo RS, Orena SJ, Rafidi K, et al. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. J Clin Invest. 2003;112:197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu B, He X, Li S, et al. Deletion of diacylglycerol-responsive TRPC genes attenuates diabetic nephropathy by inhibiting activation of the TGFβ1 signaling pathway. Am J Transl Res. 2017;9:5619–5630. [PMC free article] [PubMed] [Google Scholar]
- 69.Kim EY, Yazdizadeh Shotorbani P, Dryer SE.Trpc6 inactivation confers protection in a model of severe nephrosis in rats. J Mol Med (Berl). 2018;96:631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Spires D, Ilatovskaya DV, Levchenko V, et al. The protective role of Trpc6 knockout in the progression of diabetic kidney disease. Am J Physiol Renal Physiol. 2018;315:F1091–F1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chang JH, Paik SY, Mao L, et al. Diabetic kidney disease in FVB/NJ Akita mice: temporal pattern of kidney injury and urinary nephrin excretion. PLoS One. 2012;7:e33942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals. 8th ed Washington, DC: National Academies Press; 2011;xxv, 220. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.