

SUMMARY
Adult hippocampal neurogenesis has been reported to be decreased, increased, or not changed in Alzheimer’s disease (AD) patients and related transgenic mouse models. These disparate findings may relate to differences in disease stage, or the presence of seizures, which are associated with AD and can stimulate neurogenesis. In this study, we investigate a transgenic mouse model of AD that exhibits seizures similarly to AD patients and find that neurogenesis is increased in early stages of disease, as spontaneous seizures became evident, but is decreased below control levels as seizures recur. Treatment with the antiseizure drug levetiracetam restores neurogenesis and improves performance in a neurogenesis-associated spatial discrimination task. Our results suggest that seizures stimulate, and later accelerate the depletion of, the hippocampal neural stem cell pool. These results have implications for AD as well as any disorder accompanied by recurrent seizures, such as epilepsy.
Graphical Abstract
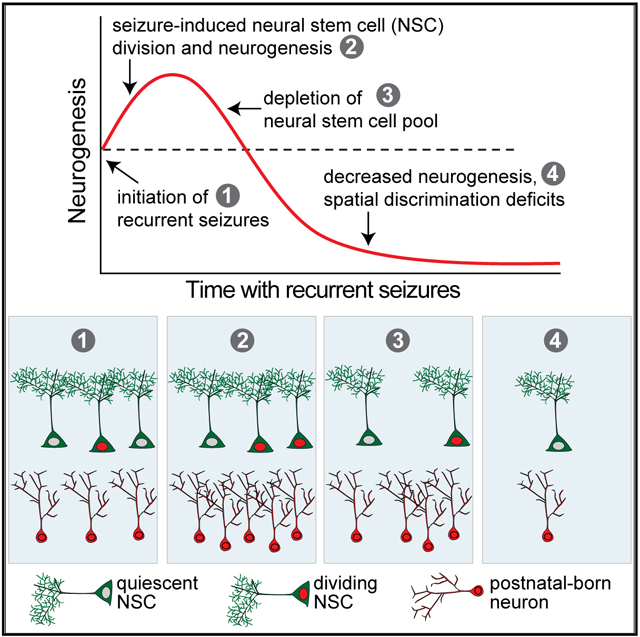
In Brief
The mechanisms that alter hippocampal neurogenesis in AD patients and mice are unclear. Fu et al. show that in a transgenic mouse model, spontaneous seizures stimulate neural stem cell (NSC) proliferation and accelerate depletion of a finite NSC pool. This process leads to neurogenesis that is increased early but decreased later in disease, coinciding with spatial discrimination deficits.
INTRODUCTION
Alzheimer’s disease (AD) is characterized by prominent impairments in memory (Holtzman et al., 2011; Weintraub et al., 2012). Many studies have therefore focused on the hippocampus, which is critical for memory formation and is exquisitely vulnerable to dysfunction (Fjell et al., 2014; Leal and Yassa, 2013; Morrison and Hof, 2002). The hippocampus is also important for mood regulation, which is also affected in AD (Kheirbek et al., 2013; Sala et al., 2004). The amyloid precursor protein (APP) and the amyloid beta (Aβ) peptides cleaved from it play central roles in AD (Bertram et al., 2010; Mucke and Selkoe, 2012); however, the precise mechanisms by which Aβ impairs neuronal function are unclear. One mechanism may be through disruption of adult hippocampal neurogenesis. Aβ affects neural stem cell (NSC) dynamics in vitro (Haughey et al., 2002; Sotthibundhu et al., 2009), and transgenic mice that produce high levels of Aβ exhibit alterations in adult hippocampal neurogenesis (Mu and Gage, 2011; Rodríguez and Verkhratsky, 2011). Importantly, adult neurogenesis is involved in both memory and mood, and it is altered in AD (Aimone et al., 2014; Anacker and Hen, 2017; Christian et al., 2014; Jin et al., 2004b; Miller and Hen, 2015; Moreno-Jiménez et al., 2019).
Neurogenesis in the hippocampal dentate gyrus (DG) continues beyond development, although to diminished levels relative to the developing brain (Bergmann et al., 2015; Knoth et al., 2010). A number of studies have demonstrated evidence of post-natal neurogenesis in adult humans (Boldrini et al., 2018; Eriksson et al., 1998; Ernst et al., 2014; Kempermann et al., 2018; Moreno-Jiménez et al., 2019; Spalding et al., 2013; but see Sorrells et al., 2018). Although it is difficult to examine its function in humans, postnatal neurogenesis has been well studied in rodents, which has advanced our knowledge of the cognitive and psychiatric domains modulated by adult-born neurons. Adult-born granule cells in the DG are important for mood regulation as well as spatial discrimination, the ability to distinguish between similar but different contexts (Aimone et al., 2011; Danielson et al., 2016; Nakashiba et al., 2012). Spatial discrimination is impaired in AD patients and related mouse models (Ally et al., 2013; Richetin et al., 2015; Salmon, 2012; Wesnes et al., 2014), but the underlying mechanisms remain unclear, perhaps due in part to conflicting reports about how adult neurogenesis is altered in AD. Studies have found increased, decreased, or no changes in hippocampal neurogenesis in AD patients (Boekhoorn et al., 2006; Briley et al., 2016; Jin et al., 2004b; Mu and Gage, 2011; Rodríguez and Verkhratsky, 2011). A recent study demonstrated not only the robust presence of adult neurogenesis in older humans, but also consistent decreases in neurogenesis in AD patients (Moreno-Jiménez et al., 2019). Varied alterations in neurogenesis have also been described in transgenic mice that express high levels of Aβ (Chevallier et al., 2005; Donovan et al., 2006; Hamilton et al., 2010; Jin et al., 2004a; Krezymon et al., 2013; López-Toledano and Shelanski, 2007; Rodríguez et al., 2008; Taniuchi et al., 2007; Unger et al., 2016; Verret et al., 2007; Zhang et al., 2007). These disparate results may reflect different stages of disease progression, or the degree to which patients exhibit other symptoms such as seizures, which can directly alter neurogenesis (Mu and Gage, 2011).
Indeed, in experimental models of seizures, hippocampal NSC proliferation increases rapidly after seizure onset, consistent with evidence that increases in neuronal activity increase proliferation (Gray and Sundstrom, 1998; Nakagawa et al., 2000; Parent et al., 2006; Scharfman and Gray, 2007). After severe seizures such as status epilepticus (SE), there is also a rapid increase in proliferation of both NSCs and glia within the first hours to days, and then an increase in newborn neurons starting about 4 days after SE. However, in conditions in which seizures are recurrent, the numbers of newborn neurons are typically reduced (Hattiangady et al., 2004; Ledergerber et al., 2006).
AD patients also exhibit epileptiform activity and spontaneous seizures that begin early in disease progression, often in the fifth or sixth decade of life. Some of these individuals carry familial mutations linked to AD (Hauser et al., 1986; Lozsadi and Larner, 2006; Scarmeas et al., 2009), but subclinical epileptiform activity has also been documented in patients with no known familial mutations (Lam et al., 2017; Vossel et al., 2013, 2016). Transgenic mice that express mutant human APP also have recurrent seizures (Chin and Scharfman, 2013; Kam et al., 2016; Minkeviciene et al., 2009; Palop et al., 2007). Therefore, the recurrent seizures in AD and epilepsy may initiate an acute increase, but subsequent reduction, in neurogenesis in the long term. One mechanism that could explain the early increase and later reduction in neurogenesis is that some populations of NSCs have a finite capability to produce neuronal cells, suggesting that excessive stimulation of NSC division may exhaust the NSC pool (Encinas et al., 2011b; Pilz et al., 2018). Recurrent seizures can lead to an accelerated depletion of the NSC pool (Sierra et al., 2015). However, how the NSC pool is regulated in AD models throughout disease progression, and whether seizure activity plays any role, is not clear. In this study, we tested the hypothesis that the hippocampal NSC pool is prematurely exhausted in a transgenic APP mouse model of AD that exhibits spontaneous recurrent seizures (line J20, Mucke et al., 2000).
RESULTS
The Rate of Adult Hippocampal Neurogenesis in APP Mice Increases after Seizure Activity Starts and then Decreases with Age
Pharmacologically induced seizures acutely increase neurogenesis in the DG (Gray and Sundstrom, 1998; Nakagawa et al., 2000; Parent et al., 2006). However, in chronic stages of epilepsy, neurogenesis is reduced below control levels (Hattiangady et al., 2004), which has been hypothesized to result from the seizure-induced depletion of the NSC pool (Sierra et al., 2015; see also Figure 1A). Because APP mice exhibit spontaneous seizures, we assessed whether neurogenesis follows a similarly biphasic dynamic during disease progression. In this line of APP mice, epileptic spikes are evident by 1 month of age, and seizures are robust by 2 months of age (Figures 1B–1D). To test if neurogenesis is altered as seizures develop in APP mice, we performed immunophenotyping, which identifies cell types based on cell morphology and expression of cell-specific markers (as in Encinas and Enikolopov, 2008). Using doublecortin (DCX) to assess immature neurons (Figure 1E), we found that relative to nontransgenic (NTG) controls, APP mice exhibited similar numbers of immature neurons at 1 month of age, but increased numbers by 2 months of age (Figures 1F and 1G). The number of immature neurons was reduced below NTG levels at 3, 7, and 14 months of age (Figure 1G), as was the number of neuroblasts (Figure S1A). DCX-expressing immature neurons also expressed PSA-NCAM and b3-tubulin, confirming their identity (Figure S2). We also tested whether other APP transgenic lines that exhibit seizures exhibit similar alterations in neurogenesis. We found that the number of immature neurons in transgenic PS1-APP mice was increased at younger ages and decreased at older ages, relative to NTG littermates (Figures S3A and S3B). In addition, aged Tg2576 mice also had fewer immature neurons relative to NTG littermates (Figure S3C). These results are similar to our findings in J20 APP mice and support the hypothesis that seizures alter neurogenesis in APP mice.
Figure 1. The Rate of Adult Hippocampal Neurogenesis in APP Mice Increases after Seizure Activity Starts and Then Decreases with Age.

(A) Model illustrating how seizure activity may induce changes in neurogenesis.
(B and C) Representative electroencephalogram (EEG) traces from NTG and APP mice at 1 and 2 months of age, with epileptiform spikes at 1 month of age (B) and a seizure at 2 months of age (C) in APP mice. Electrodes were in left and right frontal cortices (LFC and RFC), hippocampus (HIP), and parietal cortex (PC). Scale bars, 1 mV, 10 s.
(D) The number of epileptic spikes per hour in NTG or APP mice at 1, 2, and 4–6 months of age (n = 3–5 mice per genotype and age).
(E) Immunophenotyping of immature neurons (ImN) and neuroblasts (Nb) by examining morphology of cells that express doublecortin (DCX). Scale bar, 20μm.
(F) DCX staining in NTG and APP mice at 1, 2, and 7 months of age. Scale bar, 100 μm.
(G) DCX expression at 1 month of age (n = 9–12 mice per genotype) and number of DCX+ ImNs at 2 (n = 6 mice per genotype), 3 (n = 8 mice per genotype), 7 (n = 9–10 mice per genotype), and 14 (n = 11–12 mice per genotype) months of age, normalized to NTG at each time point.
*p < 0.05; **p < 0.01; ***p < 0.001; two-tailed unpaired Student’s t test comparing means between NTG and APP mice at each age. Values indicate mean ± SEM. See also Figures S1–S4 and Tables S1 and S2.
Studies of seizure-induced neurogenesis in pharmacological models of epilepsy showed that neurons born postnatally into an epileptic hippocampus can exhibit altered morphology and ectopic migration (Jessberger et al., 2007; Parent, 2007; Scharfman et al., 2000). We found that DCX-expressing immature neurons in a pilocarpine mouse model of epilepsy exhibited altered morphology with disorganized neurites as well as ectopic migration into the hilus (Figure S4A), as previously described (Myers et al., 2013; Scharfman et al., 2000). We did not find obvious morphological differences in DCX-expressing immature neurons in APP mice, although we did find a small increase in the number of ectopic granule neurons in the hilus (Figures S4A–S4C). These results may reflect differences in seizure severity and frequency, which is greater in pilocarpine-induced epilepsy than what occurs spontaneously in APP mice.
APP Mice Exhibit Increased NSC Division Followed by Accelerated NSC Pool Depletion
To assess whether the initial increase and subsequent decrease in neurogenesis in APP mice corresponds to changes in NSC numbers, we quantified NSCs at different ages. In wild-type mice, the number of NSCs declines with aging (Encinas et al., 2011b), but whether this process is the same in APP mice was not clear. We immunophenotyped nestin-positive cells with radial glia-like morphology (Figure 2A; as in Encinas and Enikolopov, 2008) and found that APP and NTG mice had similar numbers of NSCs at 1 month of age, and both genotypes exhibited age-dependent decreases in NSC numbers (Figures 2B and 2C). However, compared to NTG mice, APP mice exhibited an accelerated loss of NSCs, such that by 2 months of age and onward, there were fewer total NSCs in APP mice than in NTG controls (Figures 2B and 2C). NSCs were similarly reduced in PS1-APP and Tg2576 mice (Figures S3D–S3F).
Figure 2. APP Mice Exhibit Increased NSC Division Followed by Accelerated NSC Pool Depletion.

(A) Immunophenotyping of NSCs and neuronal precursors called amplifying neural progenitors (ANP) that express nestin. Scale bar, 20μm.
(B) Nestin immunostaining in NTG and APP mice at 1, 2, and 6 to 7 months of age. Scale bar, 100 μm.
(C) The number of NSCs in NTG and APP mice at 1 (n = 10–11 mice per genotype), 2 (n = 14 mice per genotype), 3 (n = 8 mice per genotype), 6 (n = 9–10 mice per genotype), and 14 (n = 11–12 mice per genotype) months of age. Cell counts were normalized to the average of 1-month-old NTG mice.
(D) Immunophenotyping of dividing NSCs based on nestin expression and presence of BrdU. Scale bar, 20 μm.
(E) Nestin and BrdU staining in NTG and APP mice at 1, 2, and 6–7 months of age. Scale bar, 100 μm.
(F) The number of BrdU+ dividing NSCs in NTG and APP mice at 1 (n = 9–10 mice per genotype), 2 (n = 8 mice per genotype), 3 (n = 8 mice per genotype), 6 (n = 8 mice per genotype), and 14 (n = 11–12 mice per genotype) months of age. Cell counts were normalized to the average of 1-month-old NTG mice. Inset shows the proportion of dividing NSCs to total NSCs at the 2-month time point.
(G) Immunophenotyping of dividing NSCs based on nestin expression and presence of Ki67. Scale bar, 20 μm.
(H) Nestin and Ki67 staining in NTG and APP mice at 2 months of age. Scale bar, 50 μm.
(I) The total number of Ki67+ dividing NSCs in 2-month-old NTG and APP mice. Cell counts were normalized to the average of NTG mice (left; n = 8 mice per genotype). The percentage of dividing NSCs was calculated as the number of Ki67+ Nestin+ dividing NSCs divided by the total number of NSCs in NTG and APP mice (right).
*p < 0.05; **p < 0.01; ***p < 0.001; two-tailed unpaired Student’s t test comparing means between NTG and APP mice at each age. Values indicate mean ± SEM. See also Figures S1, S3, S5, and S6 and Tables S1 and S2.
To test if the accelerated loss of NSCs in APP mice could be due to excessive cell division, leading to a depletion of the NSC pool, we administered the thymidine analog BrdU as a marker of cell division. We assessed the number of BrdU+ NSCs in APP and NTG mice at different ages (Figures 2D–2F). We found an initial increase in the numbers of dividing NSCs in APP mice at 1 month of age (Figures 2E and 2F), no change at 2 months of age, and a reduction by 3 months of age (Figures 2E and 2F; see Figures S1B and S1C for ANPs and dividing ANPs). We noted that at 2 months of age, APP mice had similar levels of dividing NSCs, but fewer total NSCs than NTG mice. Therefore, the rate of division relative to NTG mice was proportionally increased at that time point (Figure 2F inset; see Figure S5 for rates of division at other time points). We obtained similar results using Ki67 as an independent marker of cell division (Figures 2G–2I).
APP Mice Have a Higher Fraction of NSCs Engaged in Consecutive Divisions Early in Life than NTG Mice Do
When NSCs divide, they can divide consecutively 3 or 4 times to produce neuronal precursors before exiting the stem cell pool (Encinas et al., 2011b, 2006). If this process is altered in APP mice such that NSCs do not divide consecutively, the accelerated loss of NSCs in APP mice might be due to fundamental differences in the process of neurogenesis, not due to accelerated usage. To test this possibility, we administered BrdU and EdU 22 h apart and collected brains 2 h after EdU injection. NSCs dividing on day 1 of the injection with BrdU, but not on day 2 with EdU, should only incorporate BrdU labeling. NSCs dividing only on day 2 should only incorporate EdU labeling. NSCs that divided on day 1 and then immediately reentered the cell cycle to divide again on day 2 (“consecutively dividing”) should incorporate both BrdU and EdU (Figure 3A; as in Encinas et al., 2011b). Both APP and NTG mice exhibited age-dependent decreases in numbers of consecutively dividing NSCs (Figures 3B and 3C). However, at 3 weeks of age, APP mice had more consecutively dividing NSCs compared to NTG mice, but fewer than NTG mice by 6 months of age (Figures 3B and 3C). These results were similar to the pattern found with just BrdU labeling (Figure 2F) and indicate that NSCs enter consecutive cycles of cell division in both APP and NTG mice. These data are compatible with the notion that the fundamental process of NSC division is not grossly altered in APP mice, but rather the rate at which they are engaged to enter the cell cycle.
Figure 3. APP Mice Have Higher Fraction of NSCs Engaged in Consecutive Divisions Early in Life than NTG Mice Do.

(A) Administration of BrdU and EdU 22 h apart captures NSCs that were dividing only on day 1 (BrdU+ NSCs), only on day 2 (EdU+ NSCs), or on both days 1 and 2 (BrdU+ EdU+ NSCs; “consecutively dividing NSCs”). Scale bar, 20 μm.
(B and C) Number of consecutively dividing NSCs at 3 weeks (n = 6 mice per genotype) and at 2 (n = 6–8 mice per genotype), 6 (n = 6–8 mice per genotype), and 12 (n = 5–8 mice per genotype) months of age, normalized to the average of the NTG mice at each time point (B) or 3-week-old NTG mice (C).
*p < 0.05; **p < 0.01; two-tailed unpaired Student’s t test comparing means between NTG and APP mice at each age. Values indicate mean ± SEM. See also Figure S6 and Tables S1 and S2.
In support of this notion, when we challenged 8–10-month-old APP and NTG mice with a single injection of kainic acid (15 mg/kg), we found that although there are many fewer remaining NSCs in APP mice relative to NTG mice at this age, the kainic acid induced a proportionally similar increase in NSC division (Figure S6). This result suggests that the capacity of remaining NSCs to divide is unchanged in APP mice despite the accelerated depletion of the NSC pool.
Chronic Levetiracetam Treatment Normalizes Neurogenesis in APP Mice
One possible mechanism for the alterations in neurogenesis in APP mice is that the seizure activity that occurs early in disease progression aberrantly stimulates NSC division and accelerates depletion of the NSC pool (as in Figure 1A). In APP mice at 1 month of age, seizures are not obvious, but bursts of seizure-associated activity called interictal spikes or epileptiform activity do occur (see Figures 1B and 1D). By 2 months of age, APP mice exhibit robust seizures (see Figures 1C and 1D). To test if early seizure activity plays a causal role, we used the antiseizure drug levetiracetam (LEV), which effectively reduces spikes and seizures in APP mice (Corbett et al., 2017; Sanchez et al., 2012).
We previously found that a single injection of LEV (75 mg/kg) reduces epileptiform spikes in APP mice for up to 7 h and that 2 weeks of LEV treatment suppresses seizures (Corbett et al., 2017). We therefore treated 1.5-month-old APP and NTG mice with either LEV or saline (as a control) for 2 weeks. At this age, at which treatment was initiated, epileptiform spikes have begun in APP mice, but the number of NSCs was not yet altered (see Figures 1E–1G and 2A–2C).
Mice were sacrificed at the end of the 2-week treatment, when they were 2 months of age. The LEV efficacy was assessed by verifying that the expression of a seizure-induced transcription factor was reduced (Figure S7A; see Corbett et al., 2017). Similar to the naive APP mice described above, saline-treated APP mice had an increased proportion of dividing NSCs at 2 months of age, compared to saline-treated NTG mice (Figure 4A). However, this increase was prevented by the LEV treatment (Figure 4A). To test if the reduction in NSC division in the LEV-treated APP mice was sufficient to preserve the NSC pool, we assessed the total numbers of NSCs after LEV treatment. Saline-treated APP mice had fewer NSCs than NTG mice, as expected, but there was no difference in total NSCs between the LEV-treated APP and NTG mice (Figure 4B), suggesting a preservation of the NSC pool. These results demonstrate that chronic LEV treatment prevents the accelerated loss of NSCs in APP mice and normalizes the NSC pool. To assess whether LEV treatment also restored neurogenesis, we quantified DCX expression. At 2 months of age, saline-treated APP mice exhibited increased levels of DCX expression, compared to saline-treated NTG mice; this increase was prevented by the LEV treatment (Figure 4C). Taken together, these results suggest that reducing seizure activity in APP mice early in the disease progression can prevent or delay alterations in adult neurogenesis.
Figure 4. Chronic Levetiracetam Treatment Normalizes Neurogenesis in APP Mice.

(A) After 2 weeks of treatment with saline or levetiracetam (LEV; 75 mg/kg), 2-month-old NTG and APP mice (n = 9–11 mice per genotype and treatment) were sacrificed, and the percentage of Ki67+ Nestin+ dividing NSCs was quantified. Two-way ANOVA: genotype (p < 0.01), treatment (p < 0.05). Scale bar, 100 μm.
(B) The total number of Nestin+ NSCs in NTG and APP mice treated with saline or LEV. Two-way ANOVA: genotype (p < 0.05), treatment and genotype interaction (p < 0.05). Scale bar, 100 μm.
(C) DCX expression in NTG and APP mice treated with saline or LEV. Two-way ANOVA: treatment (p < 0.0001), treatment and genotype interaction (p < 0.05). Scale bar, 100 μm.
*p < 0.05; **p < 0.01; ***p < 0.001; Holm Sidak post hoc test. Values indicate mean ± SEM. See also Figures S7 and S8 and Tables S1 and S2.
To test if the alterations in neurogenesis in APP mice might also be influenced by cell-autonomous processes in the NSCs, we prepared neurospheres from hippocampi of NTG and APP mice. NSCs from these neurospheres exhibited comparable rates of division from both NTG or APP mice (Figure S8). These results indicate that the alterations in neurogenesis and NSC dynamics in APP mice are unlikely to be due to cell-autonomous effects and suggest that the increases in NSC division early in the disease in APP mice was more likely due to circuit or network level factors, such as seizure activity.
Chronic LEV Treatment Improves Spatial Discrimination in APP Mice
Adult-born hippocampal neurons are critical for spatial discrimination (Aimone et al., 2011; Sahay et al., 2011). To determine whether spatial discrimination is impaired in APP mice–and, if so, whether restoring neurogenesis with LEV is sufficient to improve behavior–we used a spatial discrimination task modified from the object-location memory test (You et al., 2017). This spatial discrimination task assesses the ability of mice to distinguish incremental distances in object displacement (Figure 5A), which is dependent on the DG and contributes to the ability to discriminate between contexts (Danielson et al., 2016; Gonçalves et al., 2016).
Figure 5. Chronic Levetiracetam Treatment Improves Spatial Discrimination in APP Mice.

(A) Spatial discrimination task. Mice were trained with two identical objects placed on one side of cage, then tested with one object displaced to one of four positions (P1–P4).
(B) Percentage of time spent with the displaced object (DO) in 3–3.5-month-old NTG (top) and APP (bottom) mice at each of the four positions (P1, P2, P3, P4) during training and test trials (n = 6–8 mice per genotype and position, total 57 mice). NTG mice (top), two-way ANOVA: test phase (p < 0.0001), position and test phase interaction (p < 0.001). APP mice (bottom), two-way ANOVA: test position (p < 0.01), phase (p < 0.001), position and test phase interaction (p < 0.001).
(C) Percentage of time spent with the DO at P2 in 3-month-old NTG and APP mice after 4 weeks of treatment with saline or levetiracetam (n = 6–8 per genotype/treatment, total 30 mice). Two-way ANOVA: treatment (p < 0.001), test phase (p < 0.0001), treatment and test phase interaction (p < 0.001).
*p < 0.05; **p < 0.01; ***p < 0.001; Holm-Sidak post hoc test. Values indicate mean ± SEM. See also Figures S7 and S9 and Table S2.
We examined mice at 3–3.5 months of age, when the level of neurogenesis in APP mice first becomes markedly reduced compared to NTG controls (see Figure 1G). NTG mice were unable to discriminate a very short displacement distance (to position 1), as they spent roughly equal time exploring the displaced and nondisplaced objects. (Figure 5B). However, NTG mice spent more time with the displaced object when it was displaced to positions 2, 3, and 4, suggesting that they were able to discriminate those displacement distances. APP mice did not spend more time with the displaced object until it was displaced to position 3 (Figure 5B), indicating that a greater displacement is necessary for APP mice to discriminate changes in distance. Thus, the displacement distance to position 2 highlighted a key difference in discrimination ability between the NTG and APP mice. Indeed, the NTG mice spent more time exploring the displaced object at position 2 than did the APP mice (see Figures S9A and S9B).
To test if the LEV-induced restoration of neurogenesis dynamics in APP mice also improved their spatial discrimination ability, NTG and APP mice were treated with LEV or saline for 4 weeks via Alzet micro-osmotic pumps prior to spatial discrimination testing. This method of LEV administration effectively reduces epileptiform activity in APP mice (Figure S7; see also Sanchez et al., 2012) and reduced the expression of a seizure-induced transcription factor in the current study (Figures S7B–S7D). We implanted the micro-osmotic pumps into mice at just over 2 months of age and tested them at 3 months of age. Both saline- and LEV-treated NTG mice spent more time with the displaced object at position 2, whereas saline-treated APP mice did not, as expected (Figure 5C). Notably, the APP mice treated with LEV spent more time with the displaced object at position 2, indicating an improved spatial discrimination ability (Figure 5C). We noted that the time spent with the displaced object in the testing phase was slightly less in LEV-treated NTG mice than in saline-treated NTG mice (Figure 5C), so we calculated the spatial discrimination index for each group of mice and compared the magnitude of discrimination between groups. The LEV treatment did not affect the magnitude of spatial discrimination in the NTG mice, but it markedly improved the spatial discrimination of the APP mice (Figures S9C and S9D). These results do not prove that the improvement in spatial discrimination was due to the restoration of neurogenesis, but together they demonstrate that treatment of APP mice with an antiseizure drug restores neurogenesis dynamics and improves spatial discrimination.
DISCUSSION
We have shown that the hippocampal NSC pool in APP mice undergoes increased proliferation early in the disease progression, followed by accelerated age-dependent depletion. The early increase is associated with aberrant epileptiform activity, and the depletion is associated with the development of recurrent seizures.
This biphasic pattern of neurogenesis may help explain the divergent reports of the direction of change in neurogenesis in AD. Studies that found increased neurogenesis in AD generally investigated earlier stages of the disease progression, whereas those that found reduced neurogenesis generally examined later stages (Chevallier et al., 2005; Donovan et al., 2006; Hamilton et al., 2010; Jin et al., 2004a; Krezymon et al., 2013; López-Toledano and Shelanski, 2007; Rodríguez et al., 2008; Taniuchi et al., 2007; Unger et al., 2016; Verret et al., 2007; Zhang et al., 2007). A similar biphasic change in neurogenesis was previously found in the line of APP mice used in our studies, although the mechanism underlying such changes was not clear (López-Toledano and Shelanski, 2007). Notably, a recent study demonstrated that adult neurogenesis occurs throughout even the ninth decade of life in humans, and the extent of neurogenesis is diminished in individuals with AD (Moreno-Jiménez et al., 2019). Although the presence of seizures was not assessed in that study, it has been found in AD patients, as well as in numerous mouse models of AD as described previously, suggesting that alterations in neurogenesis described in those patients and models could potentially have been similarly affected by recurrent seizures. Indeed, non-monotonic alterations in neurogenesis with aging have been described in studies of aging mice, which supports the hypothesis that the neurogenic niche responds to changes in the local environment over time (Apostolopoulou et al., 2017).
Seizures appear to be intimately linked to alterations in neurogenesis and cognitive function both in AD and epilepsy. AD and epilepsy patients share many cognitive and psychiatric symptoms, pointing to possible common underlying mechanisms (Chin and Scharfman, 2013). Some of these overlapping symptoms, such as impairments in spatial discrimination and mood regulation, have both been associated with aberrant postnatal neurogenesis. Impairments in cognition and mood regulation are also observed in other diseases and disorders in which seizures have been reported, such as Parkinson’s disease (Cooney and Stacy, 2016; Gruntz et al., 2018), Down syndrome (Menéndez, 2005), schizophrenia (Cascella et al., 2009), Rett syndrome (Chahrour and Zoghbi, 2007; Dolce et al., 2013), and others. It is therefore possible that any condition associated with recurrent seizures may similarly be affected by premature depletion of the hippocampal NSC pool, thus giving rise to similar cognitive and psychiatric symptoms.
Strategies that alter NSCs’ fates and prevent them from exiting the pool after division may help preserve the NSC pool and function. To develop such strategies, it is necessary to understand what regulates NSCs as they go through the stages of division as well as entry and exit from the cell cycle. Some clues already exist. Using a combination of Nestin and Gli1 reporter lines, Encinas et al. (2011b) found that a population of NSCs did not self-renew, but instead underwent a few rapid asymmetric divisions to produce ANPs before terminally differentiating into astrocytes. A separate study using in vivo clonal analysis with Nestin-CreERT2 mice with Z-EG reporter to track NSCs did find evidence of self-renewal, however, underscoring the heterogeneity of NSCs in the DG (Bonaguidi et al., 2012, 2016, 2011). These studies focused on wild-type mice; it is not clear if these processes are regulated similarly in the context of disease. A recent study using an intrahippocampal kainic acid rodent model of temporal lobe epilepsy found that NSCs divide symmetrically before both mother and daughter cells convert into astrocytes, suggesting that the process can differ in at least some disease conditions (Sierra et al., 2015). Notably, using a weaker kainic acid stimulus that induces epileptiform spikes but not seizures, the authors did not find an increase in NSC conversion into astrocytes. Thus, heterogeneity exists not only in the ability of NSCs to renew themselves, but also in their fate after activation in different conditions. Alterations in long-range GABA-ergic signaling may also contribute to varying effects on NSCs in different conditions, as impairments lead to increased NSC activation and subsequent depletion (Bao et al., 2017; Song et al., 2012). Additional therapeutic opportunities may lie in inducing remaining NSCs to self-renew and replenish the pool. Targeting the transcription factor REST may be beneficial, as its loss of function in induced pluripotent stem cells (iPSCs) derived from AD patients altered neural differentiation and depletion of NSCs (Meyer et al., 2019).
Our studies indicate that in APP mice, aberrant network activity is a primary driver of alterations in neurogenesis dynamics. Reducing epileptiform activity in APP mice normalized both neurogenesis and an associated behavior, whereas isolation and growth of NSCs from APP mice in vitro did not reveal altered dynamics in intrinsic cell division. These results do not preclude the possibility that APP or its cleavage products might yet have cell-autonomous effects on neurogenic processes, as reported (Lazarov and Demars, 2012). However, the robust effect of seizures on NSC division may overshadow other factors, or these factors may have differential impacts on NSC division, differentiation, or neuronal maturation.
Neurogenesis can also modulate network excitability. Newborn neurons promote inhibition in local hippocampal circuitry and may protect against neuronal injury after severe seizures (Drew et al., 2016; Iyengar et al., 2015; Jain et al., 2019). Thus, reduced neurogenesis in later stages of disease may exacerbate the excitation–inhibition imbalance. Seizures can also induce abnormalities in newborn neurons, such as mossy fiber sprouting, ectopic neuronal migration, and hilar basal dendrites, which may disrupt the circuitry and further promote seizures (Hester and Danzer, 2013; Jessberger et al., 2007). Additional studies are required to assess whether or how aberrant neurogenesis affects epileptogenesis in the APP mice in our study.
Neurogenesis is a multi-stage process that is influenced by many variables. Stimuli that affect neurogenesis may target distinct aspects of this process (Encinas et al., 2011a; Enikolopov et al., 2015; Lugert et al., 2010; Song et al., 2016). Deep brain stimulation in the anterior thalamic nucleus, physical exercise, and fluoxetine (Prozac) increase neurogenesis by stimulating the division of ANPs to increase neurogenic output (Encinas et al., 2011a, 2006), which is speculated to be how they exert their beneficial effects on cognition and mood. However, seizures and traumatic brain injuries also acutely increase neurogenesis but have negative long-term effects on function, which may be related to the fact that excitotoxic stimuli appear to activate the normally quiescent NSCs to divide (Gao et al., 2009; Lugert et al., 2010). Likewise, an ischemic brain injury, which initially induces a surge in NSC division in the subgranular zone (SGZ), ultimately leads to the long-term impairment of proliferation and neurogenesis (Lin et al., 2018). Our data suggest that a detrimental consequence of aberrantly activating quiescent NSCs is the premature exhaustion of a finite NSC pool. Treatment with antiepileptic drugs effectively controls seizures and restores the dynamics of neurogenesis, but is not necessarily optimal for long-term use due to possible adverse effects (Eddy et al., 2011; Schoenberg et al., 2017). Thus, therapeutic strategies to enhance neurogenesis may hold great promise for the treatment of cognitive and/or mood disorders, but care must be taken to stimulate the right cell types to increase neurogenic output but not deplete the NSC pool.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jeannie Chin (Jeannie.Chin@bcm.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Heterozygous transgenic mice (line J20) expressing human amyloid precursor protein (APP) carrying Swedish (K670N, M671L) and Indiana (V717F) familial AD (FAD) mutations (hAPP770 numbering) were used in this study (Mucke et al., 2000). Transgene expression in this line is directed by the platelet-derived growth factor b chain promoter. This line has been crossed for > 10 generations onto a C57BL/6J background using nontransgenic (NTG) C57BL/6J mice from The Jackson Laboratory (Bar Harbor, ME). APP mice are bred, maintained, and analyzed as heterozygous transgenic mice, and age-matched NTG littermates from the same line are used as controls. Male and female mice between the ages of 3 weeks old to 14 months old were used; see Figure Legends for the specific ages used in each figure. Mice are housed in group-housing with ad libitum access to food and water, and maintained on a regular 12/12 light/dark cycle. No specific method of randomization was used, but mice were semirandomly assigned to experimental groups on the basis of birth order after balancing for age, sex, and genotype. No sex differences were observed. Experiments were performed by investigators who were blinded to the genotype and treatment of the mice. For harvesting of brains, mice were deeply anesthetized and flush-perfused transcardially with phosphate-buffered saline. Brains were post-fixed in 4% phosphate-buffered paraformaldehyde. All experiments were approved by the Institutional Animal Care and Use Committee of Thomas Jefferson University and Baylor College of Medicine.
METHOD DETAILS
Immunohistochemistry
Preparation of brains and brain sections from line J20 mice was performed as previously described (Corbett et al., 2017; You et al., 2017). Immunohistochemistry was performed on brain sections from line J20, as well as on brain sections from PSAPP mice and Tg2576 mice as previously described. PSAPP mice (Holcomb et al., 1998) express hAPP with the Swedish mutation and presenilin-1 with FAD mutation M146L, whereas Tg2576 mice (Hsiao et al., 1996) express human APP (hAPP) carrying the Swedish FAD mutation. Fixed brains were cryoprotected in 30% sucrose in phosphate-buffered saline, and coronal sections (30 mm) were cut on a sliding microtome (Microm). Sections were distributed into ten subseries, each containing every tenth section throughout the rostral-caudal extent of the brain. Each immunostain was performed on one full subseries of sections. For avidin-biotin/immunoperoxidase immunohistochemistry, sections were immunostained using mouse-anti-nestin (Millipore) or rabbit-anti-doublecortin (Cell Signaling) primary antibodies followed by biotinylated donkey anti-mouse or goat anti-rabbit secondary antibodies (Vector Laboratories). Diaminobenzidine was used as the chromagen. For immunofluorescence, goat anti-mouse secondary antibody (Jackson) was used. Immunoreactive neurons in the subgranular zone of the hippocampus were counted in every tenth coronal section throughout the rostral-caudal extent of the hippocampus and summed by an experimenter blinded to genotype and treatment. Neural stem cells, amplifying neural progenitors, neuroblasts, and immature neurons were identified by immunophenotyping on the basis of expression of nestin or doublecortin, and on morphology, following criteria previously published (Encinas and Enikolopov, 2008; Encinas et al., 2011b). For only the 1 month time point in Figures 1G, and 4C, DCX expression was assessed by taking the sum of the percent area of the subgranular zone and granule cell layer that was covered by a predesignated optical density threshold of DCX staining (modified from Jain et al., 2019). Data are shown normalized to control groups to illustrate genotype and/or treatment-specific differences; raw values are listed in Table S1. DFosB immunoreactivity quantification was performed by assessing the mean pixel intensity using ImageJ software. DFosB immunoreactivity was calculated as the ratio of the intensity of the granule cell layer and the stratum radiatum.
BrdU and EdU labeling
For neurogenesis time course experiments, BrdU (150 mg/kg, Sigma, St. Louis, MO) was dissolved in saline and administered twice intraperitoneally with injections spaced 2 hours apart. Mice were sacrificed 2 hours after the last BrdU injection. For levetiracetam treatment experiments, BrdU (100 mg/kg) was administered intraperitoneally on the final day of levetiracetam treatment. For kainic acid induction of neurogenesis experiments, BrdU (100 mg/kg) was administered intraperitoneally once per day for 7 days following treatment with kainic acid. Mice were sacrificed 24 hours after the last BrdU injection. Brain sections were stained with rat-anti-BrdU antibody (Accurate Chemical) followed by donkey-anti-rat secondary antibody conjugated to AlexaFluor-594 (Invitrogen). Prolong Gold antifade mounting medium with DAPI (Invitrogen) was used to allow visualization of nuclei. Labeling was visualized with epifluorescence microscopy. BrdU/nestin-positive cells in the subgranular zone of the hippocampus were counted in every tenth coronal section throughout the rostral-caudal extent of the hippocampus and summed by an experimenter blinded to genotype.
For double-labeling of BrdU and EdU, BrdU was first injected as described above (100mg/kg), and EdU (82mg/kg, Invitrogen) was dissolved in saline and administered intraperitoneally 22 hours later. Mice were sacrificed 2 hours after the EdU injection. BrdU and EdU were visualized with mouse anti-BrdU (Sigma), followed by goat anti-mouse Rhodamine (Jackson), and Click-iT EdU AlexaFluor 647 Imaging Kit (Invitrogen), respectively.
Neurosphere Experiments
Neurosphere experiments were performed as previously described (Bennett et al., 2009). APP mice were sacrificed at P3 or at P30 by decapitation. Both hippocampi from each mouse were dissected in cold PBS and incubated at 37°C for 10 minutes in 1ml enzyme (1mg/ml Papain, Roche and 1mg/ml DNase I, Roche). The hippocampi were then dissociated in fresh medium centrifuged at 200 g, at room temperature, for 5 minutes. The pellet was re-suspended into 2ml medium, filtered through a 70um cell strainer into 6-well ultra-low attachment plate. Cells were cultured in 3 mL complete medium containing 1xN2 (Life Technologies), 1xB27 (Life Technologies), 0.36U/ml Heparin (Sigma), 20ng/ml bFGF (R&D Systems) and 20ng/ml EGF (R&D Systems) at 37°C. The cell density was 2–6×105 per well. Cells were passaged every 5–6 days a total of four times before BrdU incorporation.
For BrdU incorporation, neurospheres were dissociated into single cells and plated into 24-well plate pre-coated with Geltrex (Life Technology) at 2–5×105 cells per well. The cells were kept in complete medium containing 2 uM BrdU (Sigma) for 24 hours. The cells were then fixed with 4% paraformaldehyde in PBS for 30min at 4°C and then treated with 2N HCl for 10 minutes at room temperature. They were then neutralized with sodium tetraborate and incubated with primary antibodies against BrdU (Accurate Chemical) and nestin (Millipore) overnight at 4°C. The cells were then incubated with Alexa Fluor 594-conjugated anti-rat IgG (Molecular Probe) and FITC-conjugated anti-mouse IgG (Jackson Immune) at room temperature, as well as with Hoechst 33342 to stain nuclei. Pictures were taken from five random fields per well. BrdU-positive cells and total cells were counted by an experimenter blinded to genotype.
EEG Recordings
Mice were stereotaxically implanted with a six-electrode array headcap for EEG monitoring. Teflon-coated silver wire (0.005 in diameter) attached to a 6-pin Delran pedestal (Plastics One) was wrapped around screws implanted bilaterally into the subdural space over frontal and temporal cortices (from Bregma:1.0 mm A-P, 1.5 mm M-L; −2.2 mm A-P, 2 mm M-L) along with a hippocampal depth electrode (−2.2 mm A-P, 2 mm M-L,1.8 mm from brain surface (DV)). All implants in 1 month old mice were depth electrodes. Ground and reference electrodes were implanted directly behind Lambda on either side of the midline. Mice were allowed to recover for at least 4 days before recordings were conducted. EEG recordings were performed in the home cage of the mice on at least two different days for a minimum of 8 hours per trial using a Stellate Harmonie acquisition system (version 7.0a, Natus Medical, Pleasonton, CA) with a sampling rate of 2000 Hz for data acquisition. Native Stellate and Lab Chart Pro (AD Instruments Inc.) software were used for EEG signal processing and spike count analyses.
Pharmacological Treatments
For assessment of the effect of levetiracetam on neurogenesis, levetiracetam (Sequoia Research Products, Pangbourne, United Kingdom) was dissolved in saline and injected intraperitoneally at a dose of 75 mg/kg, 3 times per day for 2 weeks. Control groups were administered with the equivalent volume of saline. Two APP mice that received levetiracetam treatment were observed still having seizures, and were excluded from analysis. For assessment of the effect of levetiracetam on spatial discrimination behavior, levetiracetam was delivered via Alzet micro-osmotic pumps (model 1004) at a dose of 75 mg/kg/day. Micro-osmotic pumps were filled with saline or levetiracetam per manufacturer’s instructions, and primed for 2 days prior to implantation subcutaneously into the intrascapular region. Model 1004 micro-osmotic pumps delivered fluid at a rate of 0.11 μL/hr for 28 days.
For kainic acid seizures, kainic acid (Sigma) was dissolved in saline and injected intraperitoneally at a dose of 15 mg/kg. Control groups were administered with the equivalent volume of saline. Seizures were behaviorally monitored and scored for the first two hours post injection.
Spatial Discrimination Task
The experimental design was based on previously published protocols (Wimmer et al., 2012; You et al., 2017). The experimental apparatus consisted of an empty mouse housing cage placed within a three-sided white enclosure, directly touching one side. To provide visual cues for spatial orientation, the back wall of the enclosure was striped with black tape, the side wall adjacent to the cage had an A4-sized picture taped to it, and a small box was placed to the left of the mouse cage. Two 25 mL Erlenmeyer flasks were placed equidistant to the two corners of the cage facing the striped wall (see Figure 5A). For the training phase, mice were individually placed in the center of the cage and allowed to freely explore for three, 3-minute training sessions separated by 3-minute rest periods in their home cages. For the test trial, which took place 3 minutes after the last training trial, one of the two flasks was displaced to varying distances (one, two, three, or four flask lengths) from its original location before the mice were placed back in the cage for the single 3-minute testing session. During each trial, the amount of time spent exploring each of the two Erlenmeyer flasks was measured by an experimenter blinded to genotype/treatment.
QUANTIFICATION AND STATISTICAL ANALYSIS
Bias Elimination and Randomization
No specific method of randomization of mice was used, but mice were semirandomly assigned to experimental groups on the basis of birth order after balancing for age, sex, and genotype. Experiments were performed and quantified by investigators who were blinded to the genotype and treatment of the mice, and were unblinded once summary data was ready to be prepared.
Statistical Analysis
GraphPad Prism 6.0 was used for statistical analyses. For comparisons between two experimental groups, unpaired two-tailed Student t tests were used. With respect to neurogenesis or neural stem cell markers, t tests were used to compare NTG and APP mice at each age, as the experiments were designed and powered to assess the difference between genotypes at each individual age. One-tailed unpaired Student t tests were used when there were a priori assumptions made about direction of change (Figure S3). For comparisons between more than two experimental groups, a two-way ANOVA test (when there was normal sample distribution) or a Kruskal–Wallis test (when normality could not be assumed) was used. When two-way ANOVA and Kruskal-Wallis tests were statistically significant, multiple-comparison post hoc analyses were performed to compare the differences between individual groups. A p value of less than 0.05 was considered statistically significant. The statistical tests, n (number of animals), and p values for each dataset are provided in the figure legend that accompanies the data. Detailed results of all statistical analyses are listed in Table S2.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-doublecortin | Cell Signaling Technologies | Cat# 4604; RRID: AB_561007 |
| Mouse anti-nestin | Millipore-Sigma | Cat# MAB353; RRID: AB_94911 |
| Chicken anti-nestin | Novus Biologicals | Cat# NB100–1604; RRID: AB_2282642 |
| Rat anti-BrdU | Accurate Chemical | Cat# OBT0030G; RRID: AB_609567 |
| Mouse anti-BrdU | Sigma-Aldrich | Cat# B8434; RRID: AB_476811 |
| Rabbit anti-Ki67 | ThermoFisher Scientific | Cat#RM-9106-S1; RRID: AB_149792 |
| Mouse anti-PSA-NCAM | Millipore-Sigma | Cat# MAB5324; RRID: AB_95211 |
| Rabbit anti-β3-tubulin | Cell Signaling Technologies | Cat#: 5568; RRID: AB_10694505 |
| Mouse anti-Prox1 | PhosphoSolutions | Cat#: 1685-Prox1; RRID: AB_2492217 |
| Rabbit anti-ΔFosB | Cell Signaling Technologies | Cat#: 14695; RRID: AB_2798577 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 5-bromo-2′-deoxyuridine (BrdU) | Sigma-Aldrich | Cat# B5002 |
| 5-ethynyl-2′-deoxyuridine (EdU) | Invitrogen | Cat# A10044 |
| Kainic acid | Sigma-Aldrich | Cat#K0250–10MG |
| 3,3-diaminobenzidine (DAB) | Sigma-Aldrich | Cat# D5905–50TAB |
| Levetiracetam | Sequoia Research Products | Cat#SRP013811 |
| Papain | Roche | Cat# 10108014001 |
| DNaseI | Roche | Cat# 04716728001 |
| L-cystein | Sigma-Aldrich | Cat# C7352 |
| bFGF | R&D Systems | Cat# P15655 |
| EGF | R&D Systems | Cat# 2028-EG |
| Heparin Sodium | Sigma-Aldrich | Cat# H4784 |
| N2 supplement | Invitrogen | Cat# 17502048 |
| B27 supplement | Invitrogen | Cat# 17504044 |
| Geltrex | Invitrogen | Cat# A1413302 |
| Critical Commercial Assays | ||
| Click-iT EdU Alexa Fluor 647 Imaging Kit | Invitrogen | Cat# C10340 |
| Experimental Models: Organisms/Strains | ||
| B6.Cg-Zbtb20Tg(PDGFB-APPSwInd)20Lms/2Mmjax (J20) | The Jackson Laboratory | MMRRC stock #34836 |
| Software and Algorithms | ||
| Prism 6 | GraphPad | https://www.graphpad.com/scientific-software/prism/; RRID: SCR_002798 |
| MetaMorph software | Molecular Devices | https://www.moleculardevices.com/systems/metamorph-research-imaging/metamorph-microscopy-automationand-image-analysis-software; RRID:SCR_002368 |
| ZEN software | Zeiss Microscope | https://www.zeiss.com/microscopy/int/products/microscope-software/zen.html; RRID: SCR_013672 |
| FIJI (ImageJ) | NIH | RRID: SCR_002285 |
| Adobe Illustrator CC | Adobe | http://www.adobe.com/products/illustrator.html; RRID: SCR_010279 |
| Adobe Photoshop CC | Adobe | https://www.adobe.com/products/photoshop.html; RRID:SCR_014199 |
| Other | ||
| Micro-osmotic pump | Alzet Osmotic Pumps | Model 1004 |
Highlights.
Seizure-induced NSC proliferation in AD mice accelerates the depletion of the finite NSC pool
Hippocampal neurogenesis is increased early, but decreased later, in disease
Reduced neurogenesis coincides with deficits in spatial discrimination in AD mice
Antiseizure drug normalizes neurogenesis and improves spatial discrimination in AD mice
ACKNOWLEDGMENTS
This work was supported by NIH grants NS086965 and NS085171 (J.C.), NS075839 (L.I.), NS086965 (H.E.S.), and AG040209 and NS086965 (G.E.) and by the New York State Office of Mental Health (H.E.S.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.05.101.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Aimone JB, Deng W, and Gage FH (2011). Resolving new memories: a critical look at the dentate gyrus, adult neurogenesis, and pattern separation. Neuron 70, 589–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aimone JB, Li Y, Lee SW, Clemenson GD, Deng W, and Gage FH (2014). Regulation and function of adult neurogenesis: from genes to cognition. Physiol. Rev 94, 991–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ally BA, Hussey EP, Ko PC, and Molitor RJ (2013). Pattern separation and pattern completion in Alzheimer’s disease: evidence of rapid forgetting in amnestic mild cognitive impairment. Hippocampus 23, 1246–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anacker C, and Hen R (2017). Adult hippocampal neurogenesis and cognitive flexibility - linking memory and mood. Nat. Rev. Neurosci 18, 335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolopoulou M, Kiehl TR, Winter M, Cardenas De La Hoz E, Boles NC, Bjornsson CS, Zuloaga KL, Goderie SK, Wang Y, Cohen AR, and Temple S (2017). Non-monotonic Changes in Progenitor Cell Behavior and Gene Expression during Aging of the Adult V-SVZ Neural Stem Cell Niche. Stem Cell Rep. 9, 1931–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao H, Asrican B, Li W, Gu B, Wen Z, Lim SA, Haniff I, Ramakrishnan C, Deisseroth K, Philpot B, et al. (2017). Long-Range GABAergic Inputs Regulate Neural Stem Cell Quiescence and Control Adult Hippocampal Neurogenesis. Cell Stem Cell 21, 604–617.e605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett L, Yang M, Enikolopov G, and Iacovitti L (2009). Circumventricular organs: a novel site of neural stem cells in the adult brain. Mol. Cell. Neurosci 41, 337–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann O, Spalding KL, and Frisén J (2015). Adult Neurogenesis in Humans. Cold Spring Harb. Perspect. Biol 7, a018994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram L, Lill CM, and Tanzi RE (2010). The genetics of Alzheimer disease: back to the future. Neuron 68, 270–281. [DOI] [PubMed] [Google Scholar]
- Boekhoorn K, Joels M, and Lucassen PJ (2006). Increased proliferation reflects glial and vascular-associated changes, but not neurogenesis in the presenile Alzheimer hippocampus. Neurobiol. Dis 24, 1–14. [DOI] [PubMed] [Google Scholar]
- Boldrini M, Fulmore CA, Tartt AN, Simeon LR, Pavlova I, Poposka V, Rosoklija GB, Stankov A, Arango V, Dwork AJ, et al. (2018). Human Hippocampal Neurogenesis Persists throughout Aging. Cell Stem Cell 22, 589–599.e585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaguidi MA, Wheeler MA, Shapiro JS, Stadel RP, Sun GJ, Ming GL, and Song H (2011). In vivo clonal analysis reveals self-renewing and multipotent adult neural stem cell characteristics. Cell 145, 1142–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaguidi MA, Song J, Ming GL, and Song H (2012). A unifying hypothesis on mammalian neural stem cell properties in the adult hippocampus. Curr. Opin. Neurobiol 22, 754–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaguidi MA, Stadel RP, Berg DA, Sun J, Ming GL, and Song H (2016). Diversity of Neural Precursors in the Adult Mammalian Brain. Cold Spring Harb. Perspect. Biol 8, a018838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briley D, Ghirardi V, Woltjer R, Renck A, Zolochevska O, Taglialatela G, and Micci MA (2016). Preserved neurogenesis in non-demented individuals with AD neuropathology. Sci. Rep 6, 27812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascella NG, Schretlen DJ, and Sawa A (2009). Schizophrenia and epilepsy: is there a shared susceptibility? Neurosci. Res 63, 227–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour M, and Zoghbi HY (2007). The story of Rett syndrome: from clinic to neurobiology. Neuron 56, 422–437. [DOI] [PubMed] [Google Scholar]
- Chevallier NL, Soriano S, Kang DE, Masliah E, Hu G, and Koo EH (2005). Perturbed neurogenesis in the adult hippocampus associated with presenilin-1 A246E mutation. Am. J. Pathol 167, 151–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin J, and Scharfman HE (2013). Shared cognitive and behavioral impairments in epilepsy and Alzheimer’s disease and potential underlying mechanisms. Epilepsy Behav. 26, 343–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian KM, Song H, and Ming GL (2014). Functions and dysfunctions of adult hippocampal neurogenesis. Annu. Rev. Neurosci 37, 243–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooney JW, and Stacy M (2016). Neuropsychiatric Issues in Parkinson’s Disease. Curr. Neurol. Neurosci. Rep 16, 49. [DOI] [PubMed] [Google Scholar]
- Corbett BF, You JC, Zhang X, Pyfer MS, Tosi U, Iascone DM, Petrof I, Hazra A, Fu C-H, Stephens GS, et al. (2017). DFosB Regulates Gene Expression and Cognitive Dysfunction in a Mouse Model of Alzheimer’s Disease. Cell Rep. 20, 344–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielson NB, Kaifosh P, Zaremba JD, Lovett-Barron M, Tsai J, Denny CA, Balough EM, Goldberg AR, Drew LJ, Hen R, et al. (2016). Distinct contribution of adult-born hippocampal granule cells to context encoding. Neuron 90, 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolce A, Ben-Zeev B, Naidu S, and Kossoff EH (2013). Rett syndrome and epilepsy: an update for child neurologists. Pediatr. Neurol 48, 337–345. [DOI] [PubMed] [Google Scholar]
- Donovan MH, Yazdani U, Norris RD, Games D, German DC, and Eisch AJ (2006). Decreased adult hippocampal neurogenesis in the PDAPP mouse model of Alzheimer’s disease. J. Comp. Neurol 495, 70–83. [DOI] [PubMed] [Google Scholar]
- Drew LJ, Kheirbek MA, Luna VM, Denny CA, Cloidt MA, Wu MV, Jain S, Scharfman HE, and Hen R (2016). Activation of local inhibitory circuits in the dentate gyrus by adult-born neurons. Hippocampus 26, 763–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy CM, Rickards HE, and Cavanna AE (2011). The cognitive impact of antiepileptic drugs. Ther. Adv. Neurol. Disorder 4, 385–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encinas JM, and Enikolopov G (2008). Identifying and quantitating neural stem and progenitor cells in the adult brain. Methods Cell Biol. 85, 243–272. [DOI] [PubMed] [Google Scholar]
- Encinas JM, Vaahtokari A, and Enikolopov G (2006). Fluoxetine targets early progenitor cells in the adult brain. Proc. Natl. Acad. Sci. USA 103, 8233–8238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encinas JM, Hamani C, Lozano AM, and Enikolopov G (2011a). Neurogenic hippocampal targets of deep brain stimulation. J. Comp. Neurol 519, 6–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encinas JM, Michurina TV, Peunova N, Park JH, Tordo J, Peterson DA, Fishell G, Koulakov A, and Enikolopov G (2011b). Division-coupled astrocytic differentiation and age-related depletion of neural stem cells in the adult hippocampus. Cell Stem Cell. 8, 566–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enikolopov G, Overstreet-Wadiche L, and Ge S (2015). Viral and transgenic reporters and genetic analysis of adult neurogenesis. Cold Spring Harb. Perspect. Biol 7, a018804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson PS, Perfilieva E, Björk-Eriksson T, Alborn AM, Nordborg C, Peterson DA, and Gage FH (1998). Neurogenesis in the adult human hippocampus. Nat. Med 4, 1313–1317. [DOI] [PubMed] [Google Scholar]
- Ernst A, Alkass K, Bernard S, Salehpour M, Perl S, Tisdale J, Possnert G, Druid H, and Frisén J (2014). Neurogenesis in the striatum of the adult human brain. Cell 156, 1072–1083. [DOI] [PubMed] [Google Scholar]
- Fjell AM, McEvoy L, Holland D, Dale AM, and Walhovd KB; Alzheimer’s Disease Neuroimaging Initiative (2014). What is normal in normal aging? Effects of aging, amyloid and Alzheimer’s disease on the cerebral cortex and the hippocampus. Prog. Neurobiol 117, 20–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Enikolopov G, and Chen J (2009). Moderate traumatic brain injury promotes proliferation of quiescent neural progenitors in the adult hippocampus. Exp. Neurol 219, 516–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves JT, Schafer ST, and Gage FH (2016). Adult Neurogenesis in the Hippocampus: From Stem Cells to Behavior. Cell 167, 897–914. [DOI] [PubMed] [Google Scholar]
- Gray WP, and Sundstrom LE (1998). Kainic acid increases the proliferation of granule cell progenitors in the dentate gyrus of the adult rat. Brain Res. 790, 52–59. [DOI] [PubMed] [Google Scholar]
- Gruntz K, Bloechliger M, Becker C, Jick SS, Fuhr P, Meier CR, and Rüegg S (2018). Parkinson disease and the risk of epileptic seizures. Ann. Neurol 83, 363–374. [DOI] [PubMed] [Google Scholar]
- Hamilton LK, Aumont A, Julien C, Vadnais A, Calon F, and Fernandes KJ (2010). Widespread deficits in adult neurogenesis precede plaque and tangle formation in the 3xTg mouse model of Alzheimer’s disease. Eur. J. Neurosci 32, 905–920. [DOI] [PubMed] [Google Scholar]
- Hattiangady B, Rao MS, and Shetty AK (2004). Chronic temporal lobe epilepsy is associated with severely declined dentate neurogenesis in the adult hippocampus. Neurobiol. Dis 17, 473–490. [DOI] [PubMed] [Google Scholar]
- Haughey NJ, Nath A, Chan SL, Borchard AC, Rao MS, and Mattson MP (2002). Disruption of neurogenesis by amyloid beta-peptide, and perturbed neural progenitor cell homeostasis, in models of Alzheimer’s disease. J. Neurochem 83, 1509–1524. [DOI] [PubMed] [Google Scholar]
- Hauser WA, Morris ML, Heston LL, and Anderson VE (1986). Seizures and myoclonus in patients with Alzheimer’s disease. Neurology 36, 1226–1230. [DOI] [PubMed] [Google Scholar]
- Hester MS, and Danzer SC (2013). Accumulation of abnormal adult-generated hippocampal granule cells predicts seizure frequency and severity. J. Neurosci 33, 8926–8936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, et al. (1998). Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med 4, 97–100. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Morris JC, and Goate AM (2011). Alzheimer’s disease: the challenge of the second century. Sci. Transl. Med 3, 77sr1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, and Cole G (1996). Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274, 99–102. [DOI] [PubMed] [Google Scholar]
- Iyengar SS, LaFrancois JJ, Friedman D, Drew LJ, Denny CA, Burghardt NS, Wu MV, Hsieh J, Hen R, and Scharfman HE (2015). Suppression of adult neurogenesis increases the acute effects of kainic acid. Exp. Neurol 264, 135–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain S, LaFrancois JJ, Botterill JJ, Alcantara-Gonzalez D, and Scharfman HE (2019). Adult neurogenesis in the mouse dentate gyrus protects the hippocampus from neuronal injury following severe seizures. Hippocampus, Published online January 23, 2019. 10.1002/hipo.23062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessberger S, Zhao C, Toni N, Clemenson GD Jr., Li Y, and Gage FH (2007). Seizure-associated, aberrant neurogenesis in adult rats characterized with retrovirus-mediated cell labeling. J. Neurosci 27, 9400–9407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Galvan V, Xie L, Mao XO, Gorostiza OF, Bredesen DE, and Greenberg DA (2004a). Enhanced neurogenesis in Alzheimer’s disease transgenic (PDGF-APPSw,Ind) mice. Proc. Natl. Acad. Sci. USA 101, 13363–13367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Peel AL, Mao XO, Xie L, Cottrell BA, Henshall DC, and Greenberg DA (2004b). Increased hippocampal neurogenesis in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 101, 343–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kam K, Duffy AM, Moretto J, LaFrancois JJ, and Scharfman HE (2016). Interictal spikes during sleep are an early defect in the Tg2576 mouse model of b-amyloid neuropathology. Sci. Rep 6, 20119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kempermann G, Gage FH, Aigner L, Song H, Curtis MA, Thuret S, Kuhn HG, Jessberger S, Frankland PW, Cameron HA, et al. (2018). Human Adult Neurogenesis: Evidence and Remaining Questions. Cell Stem Cell 23, 25–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheirbek MA, Drew LJ, Burghardt NS, Costantini DO, Tannenholz L, Ahmari SE, Zeng H, Fenton AA, and Hen R (2013). Differential control of learning and anxiety along the dorsoventral axis of the dentate gyrus. Neuron 77, 955–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoth R, Singec I, Ditter M, Pantazis G, Capetian P, Meyer RP, Horvat V, Volk B, and Kempermann G (2010). Murine features of neurogenesis in the human hippocampus across the lifespan from 0 to 100 years. PLoS ONE 5, e8809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krezymon A, Richetin K, Halley H, Roybon L, Lassalle JM, Francès B, Verret L, and Rampon C (2013). Modifications of hippocampal circuits and early disruption of adult neurogenesis in the tg2576 mouse model of Alzheimer’s disease. PLoS ONE 8, e76497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam AD, Deck G, Goldman A, Eskandar EN, Noebels J, and Cole AJ (2017). Silent hippocampal seizures and spikes identified by foramen ovale electrodes in Alzheimer’s disease. Nat. Med 23, 678–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarov O, and Demars MP (2012). All in the Family: How the APPs Regulate Neurogenesis. Front. Neurosci 6, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal SL, and Yassa MA (2013). Perturbations of neural circuitry in aging, mild cognitive impairment, and Alzheimer’s disease. Ageing Res. Rev 12, 823–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledergerber D, Fritschy JM, and Kralic JE (2006). Impairment of dentate gyrus neuronal progenitor cell differentiation in a mouse model of temporal lobe epilepsy. Exp. Neurol 199, 130–142. [DOI] [PubMed] [Google Scholar]
- Lin R, Lang M, Heinsinger N, Stricsek G, Zhang J, Iozzo R, Rosenwasser R, and Iacovitti L (2018). Stepwise impairment of neural stem cell proliferation and neurogenesis concomitant with disruption of blood-brain barrier in recurrent ischemic stroke. Neurobiol. Dis 115, 49–58. [DOI] [PubMed] [Google Scholar]
- López-Toledano MA, and Shelanski ML (2007). Increased neurogenesis in young transgenic mice overexpressing human APP(Sw, Ind). J. Alzheimers Dis 12, 229–240. [DOI] [PubMed] [Google Scholar]
- Lozsadi DA, and Larner AJ (2006). Prevalence and causes of seizures at the time of diagnosis of probable Alzheimer’s disease. Dement. Geriatr. Cogn. Disord 22, 121–124. [DOI] [PubMed] [Google Scholar]
- Lugert S, Basak O, Knuckles P, Haussler U, Fabel K, Götz M, Haas CA, Kempermann G, Taylor V, and Giachino C (2010). Quiescent and active hippocampal neural stem cells with distinct morphologies respond selectively to physiological and pathological stimuli and aging. Cell Stem Cell. 6, 445–456. [DOI] [PubMed] [Google Scholar]
- Menéndez M (2005). Down syndrome, Alzheimer’s disease and seizures. Brain Dev. 27, 246–252. [DOI] [PubMed] [Google Scholar]
- Meyer K, Feldman HM, Lu T, Drake D, Lim ET, Ling KH, Bishop NA, Pan Y, Seo J, Lin YT, et al. (2019). REST and Neural Gene Network Dysregulation in iPSC Models of Alzheimer’s Disease. Cell Rep. 26, 1112–1127.e1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BR, and Hen R (2015). The current state of the neurogenic theory of depression and anxiety. Curr. Opin. Neurobiol 30, 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minkeviciene R, Rheims S, Dobszay MB, Zilberter M, Hartikainen J, Fülöp L, Penke B, Zilberter Y, Harkany T, Pitkänen A, and Tanila H (2009). Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J. Neurosci 29, 3453–3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Jiménez EP, Flor-García M, Terreros-Roncal J, Rábano A, Cafini F, Pallas-Bazarra N, Ávila J, and Llorens-Martín M (2019). Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med 25, 554–560. [DOI] [PubMed] [Google Scholar]
- Morrison JH, and Hof PR (2002). Selective vulnerability of corticocortical and hippocampal circuits in aging and Alzheimer’s disease. Prog. Brain Res 136, 467–486. [DOI] [PubMed] [Google Scholar]
- Mu Y, and Gage FH (2011). Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol. Neurodegener 6, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, and Selkoe DJ (2012). Neurotoxicity of amyloid b-protein: synaptic and network dysfunction. Cold Spring Harb. Perspect. Med 2, a006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, and McConlogue L (2000). High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J. Neurosci 20, 4050–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers CE, Bermudez-Hernandez K, and Scharfman HE (2013). The influence of ectopic migration of granule cells into the hilus on dentate gyrus-CA3 function. PLoS ONE 8, e68208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa E, Aimi Y, Yasuhara O, Tooyama I, Shimada M, McGeer PL, and Kimura H (2000). Enhancement of progenitor cell division in the dentate gyrus triggered by initial limbic seizures in rat models of epilepsy. Epilepsia 41, 10–18. [DOI] [PubMed] [Google Scholar]
- Nakashiba T, Cushman JD, Pelkey KA, Renaudineau S, Buhl DL, McHugh TJ, Rodriguez Barrera V, Chittajallu R, Iwamoto KS, McBain CJ, et al. (2012). Young dentate granule cells mediate pattern separation, whereas old granule cells facilitate pattern completion. Cell 149, 188–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, Yoo J, Ho KO, Yu GQ, Kreitzer A, et al. (2007). Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 55, 697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent JM (2007). Adult neurogenesis in the intact and epileptic dentate gyrus. Prog. Brain Res 163, 529–540. [DOI] [PubMed] [Google Scholar]
- Parent JM, Elliott RC, Pleasure SJ, Barbaro NM, and Lowenstein DH (2006). Aberrant seizure-induced neurogenesis in experimental temporal lobe epilepsy. Ann. Neurol 59, 81–91. [DOI] [PubMed] [Google Scholar]
- Pilz GA, Bottes S, Betizeau M, Jörg DJ, Carta S, Simons BD, Helmchen F, and Jessberger S (2018). Live imaging of neurogenesis in the adult mouse hippocampus. Science 359, 658–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richetin K, Leclerc C, Toni N, Gallopin T, Pech S, Roybon L, and Rampon C (2015). Genetic manipulation of adult-born hippocampal neurons rescues memory in a mouse model of Alzheimer’s disease. Brain 138, 440–455. [DOI] [PubMed] [Google Scholar]
- Rodríguez JJ, and Verkhratsky A (2011). Neurogenesis in Alzheimer’s disease. J. Anat 219, 78–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez JJ, Jones VC, Tabuchi M, Allan SM, Knight EM, LaFerla FM, Oddo S, and Verkhratsky A (2008). Impaired adult neurogenesis in the dentate gyrus of a triple transgenic mouse model of Alzheimer’s disease. PLoS ONE 3, e2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahay A, Scobie KN, Hill AS, O’Carroll CM, Kheirbek MA, Burghardt NS, Fenton AA, Dranovsky A, and Hen R (2011). Increasing adult hippocampal neurogenesis is sufficient to improve pattern separation. Nature 472, 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala M, Perez J, Soloff P, Ucelli di Nemi S, Caverzasi E, Soares JC, and Brambilla P (2004). Stress and hippocampal abnormalities in psychiatric disorders. Eur. Neuropsychopharmacol 14, 393–405. [DOI] [PubMed] [Google Scholar]
- Salmon DP (2012). Neuropsychological features of mild cognitive impairment and preclinical Alzheimer’s disease In Behavioral Neurobiology of Aging, Pardon M-C and Bondi MW, eds. (Springer Berlin Heidelberg; ), pp. 187–212. [DOI] [PubMed] [Google Scholar]
- Sanchez PE, Zhu L, Verret L, Vossel KA, Orr AG, Cirrito JR, Devidze N, Ho K, Yu GQ, Palop JJ, and Mucke L (2012). Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc. Natl. Acad. Sci. USA 109, E2895–E2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarmeas N, Honig LS, Choi H, Cantero J, Brandt J, Blacker D, Albert M, Amatniek JC, Marder K, Bell K, et al. (2009). Seizures in Alzheimer disease: who, when, and how common? Arch. Neurol 66, 992–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, and Gray WP (2007). Relevance of seizure-induced neurogenesis in animal models of epilepsy to the etiology of temporal lobe epilepsy. Epilepsia 48, 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE, Goodman JH, and Sollas AL (2000). Granule-like neurons at the hilar/CA3 border after status epilepticus and their synchrony with area CA3 pyramidal cells: functional implications of seizure-induced neurogenesis. J. Neurosci 20, 6144–6158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenberg MR, Rum RS, Osborn KE, and Werz MA (2017). A randomized, double-blind, placebo-controlled crossover study of the effects of levetiracetam on cognition, mood, and balance in healthy older adults. Epilepsia 58, 1566–1574. [DOI] [PubMed] [Google Scholar]
- Sierra A, Martín-Suárez S, Valcárcel-Martín R, Pascual-Brazo J, Aelvoet SA, Abiega O, Deudero JJ, Brewster AL, Bernales I, Anderson AE, et al. (2015). Neuronal hyperactivity accelerates depletion of neural stem cells and impairs hippocampal neurogenesis. Cell Stem Cell 16, 488–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Zhong C, Bonaguidi MA, Sun GJ, Hsu D, Gu Y, Meletis K, Huang ZJ, Ge S, Enikolopov G, et al. (2012). Neuronal circuitry mechanism regulating adult quiescent neural stem-cell fate decision. Nature 489, 150–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Olsen RH, Sun J, Ming GL, and Song H (2016). Neuronal Circuitry Mechanisms Regulating Adult Mammalian Neurogenesis. Cold Spring Harb. Perspect. Biol 8, a018937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrells SF, Paredes MF, Cebrian-Silla A, Sandoval K, Qi D, Kelley KW, James D, Mayer S, Chang J, Auguste KI, et al. (2018). Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature 555, 377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotthibundhu A, Li QX, Thangnipon W, and Coulson EJ (2009). Abeta(1–42) stimulates adult SVZ neurogenesis through the p75 neurotrophin receptor. Neurobiol. Aging 30, 1975–1985. [DOI] [PubMed] [Google Scholar]
- Spalding KL, Bergmann O, Alkass K, Bernard S, Salehpour M, Huttner HB, Boström E, Westerlund I, Vial C, Buchholz BA, et al. (2013). Dynamics of hippocampal neurogenesis in adult humans. Cell 153, 1219–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniuchi N, Niidome T, Goto Y, Akaike A, Kihara T, and Sugimoto H (2007). Decreased proliferation of hippocampal progenitor cells in APPswe/PS1dE9 transgenic mice. Neuroreport. 18, 1801–1805. [DOI] [PubMed] [Google Scholar]
- Unger MS, Marschallinger J, Kaindl J, Höfling C, Rossner S, Heneka MT, Van der Linden A, and Aigner L (2016). Early Changes in Hippocampal Neurogenesis in Transgenic Mouse Models for Alzheimer’s Disease. Mol. Neurobiol 53, 5796–5806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verret L, Jankowsky JL, Xu GM, Borchelt DR, and Rampon C (2007). Alzheimer’s-type amyloidosis in transgenic mice impairs survival of newborn neurons derived from adult hippocampal neurogenesis. J. Neurosci 27, 6771–6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Beagle AJ, Rabinovici GD, Shu H, Lee SE, Naasan G, Hegde M, Cornes SB, Henry ML, Nelson AB, et al. (2013). Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. 70, 1158–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Ranasinghe KG, Beagle AJ, Mizuiri D, Honma SM, Dowling AF, Darwish SM, Van Berlo V, Barnes DE, Mantle M, et al. (2016). Incidence and impact of subclinical epileptiform activity in Alzheimer’s disease. Ann. Neurol 80, 858–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weintraub S, Wicklund AH, and Salmon DP (2012). The neuropsychological profile of Alzheimer disease. Cold Spring Harb. Perspect. Med 2, a006171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesnes KA, Annas P, Basun H, Edgar C, and Blennow K (2014). Performance on a pattern separation task by Alzheimer’s patients shows possible links between disrupted dentate gyrus activity and apolipoprotein E ç4 status and cerebrospinal fluid amyloid-b42 levels. Alzheimers Res. Ther 6, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wimmer ME, Hernandez PJ, Blackwell J, and Abel T (2012). Aging impairs hippocampus-dependent long-term memory for object location in mice. Neurobiol. Aging 33, 2220–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You JC, Muralidharan K, Park JW, Petrof I, Pyfer MS, Corbett BF, LaFrancois JJ, Zheng Y, Zhang X, Mohila CA, et al. (2017). Epigenetic suppression of hippocampal calbindin-D28k by DFosB drives seizure-related cognitive deficits. Nat. Med 23, 1377–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, McNeil E, Dressler L, and Siman R (2007). Long-lasting impairment in hippocampal neurogenesis associated with amyloid deposition in a knock-in mouse model of familial Alzheimer’s disease. Exp. Neurol 204, 77–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.