

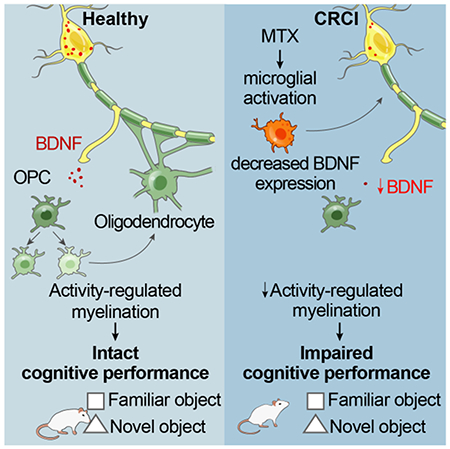
Summary
Activity-dependent myelination is thought to contribute to adaptive neurological function. However, the mechanisms by which activity regulates myelination and the extent to which myelin plasticity contributes to non-motor cognitive functions remain incompletely understood. Using a mouse model of chemotherapy-related cognitive impairment (CRCI), we recently demonstrated that methotrexate chemotherapy induces complex glial dysfunction for which microglial activation is central. Here we demonstrate that remote methotrexate exposure blocks activity-regulated myelination. Methotrexate decreases cortical Bdnf expression, which is restored by microglial depletion. Bdnf-TrkB signaling is a required component of activity-dependent myelination. Oligodendrocyte precursor cell (OPC)-specific TrkB deletion in chemotherapy-naïve mice results in impaired cognitive behavioral performance. A small molecule TrkB agonist rescues both myelination and cognitive impairment after MTX chemotherapy. This rescue after MTX depends on intact OPC-TrkB expression. Taken together, these findings demonstrate a molecular mechanism required for adaptive myelination that is aberrant in CRCI due to microglial inflammation.
Graphical Abstract
eTOC Blurb
Methotrexate chemotherapy results in a microglial-dependent reduction of Bdnf expression and loss of activity-regulated myelination, which requires Bdnf to TrkB signaling. OPC-specific loss of TrkB results in cognitive impairment. Stimulating OPC TrkB signaling restores myelination and rescues cognition after MTX.
Introduction
The role that neuronal activity plays in shaping developing circuitry has long been appreciated (Wiesel and Hubel, 1965). A new dimension along which neuronal activity modulates neural structure, and thus function, is emerging in the myelinated infrastructure of the brain. We previously reported that optogenetic stimulation of frontal cortical projection neuronal activity results in robust oligodendrocyte precursor cell (OPC) proliferation and subsequent oligodendrogenesis in both juvenile and adult mice, with activity-regulated increase in myelination (Gibson et al., 2014). This circuit-specific change in myelin structure results in positive alterations to motor function that depend on the generation of new oligodendrocytes (Gibson et al., 2014). Subsequent work demonstrated that neuronal activity similarly influences myelination in the somatosensory system (Hill et al., 2018; Hughes et al., 2018; Mitew et al., 2018), that oligodendrogenesis is necessary for motor learning (McKenzie et al., 2014; Xiao et al., 2016), and that neuronal activity can influence axon selection for successful developmental myelination (Hines et al., 2015; Mensch et al., 2015). These insights support the emerging concept that plasticity of myelin can contribute to structural changes sculpting adaptive development and ongoing plasticity. However, the extent to which activity-regulated myelination contributes to non-motor cognitive function and the molecular mechanisms mediating myelin plasticity remain to be fully elucidated.
Cancer chemotherapy frequently results in a lasting syndrome of cognitive dysfunction characterized by deficits in attention, memory, speed of information processing, multi-tasking and executive function (Ellenberg et al., 2009; Koppelmans et al., 2012). The commonly used antimetabolite chemotherapeutic agent methotrexate (MTX) is particularly associated with chemotherapy-related cognitive impairment (CRCI), colloquially known as “chemobrain” or “chemofog” (Aukema et al., 2009; Deprez et al., 2011; Kaiser et al., 2014; Pierson et al., 2016). We recently demonstrated that tri-glial dysfunction underlies MTX CRCI, with direct effects of MTX on microglial activation state subsequently inducing neurotoxic astrocyte reactivity and oligodendroglial lineage dysregulation (Gibson et al., 2019). Microglial depletion following MTX exposure decreases astrocyte reactivity, normalizes oligodendroglial lineage dynamics and myelination, and rescues cognitive function in a mouse model of juvenile MTX exposure. Myelination, which continues throughout the lifespan (Hill et al. 2018; Hughes et al., 2018; Tripathi et al., 2017) is regulated both by activity-independent (Bechler et al., 2015; Lee et al., 2013; Mayoral et al., 2018; Rosenberg et al., 2008) and activity-dependent (Gibson et al. 2014; Hughes et al., 2018; Mitew et al., 2018) mechanisms. Whether disruption of activity-dependent, adaptive myelination contributes to cognitive dysfunction following MTX chemotherapy is an important open question that may further elucidate the role of adaptive myelination in non-motor cognition and may implicate loss of activity-regulated myelin plasticity as an etiological factor in a cognitive disease. In the present study, we sought to explore the role of activity-regulated myelination in cognitive function and dysfunction following MTX chemotherapy.
Results
Failure of adaptive myelination in a mouse model of methotrexate chemotherapy-related cognitive impairment
To test the integrity of adaptive myelination after chemotherapy exposure, we utilized our recently developed mouse model of MTX CRCI (Gibson et al., 2019). MTX administered to mice at doses that achieve clinically-relevant serum and brain drug concentrations (Bratlid and Moe, 1978; Janka et al., 1984) results in persistent dysregulation of white matter OPC population dynamics and dysmyelination (Gibson et al., 2019). Mice exposed to MTX at P21, 28, and 35 exhibit lasting deficits in OPC proliferation, oligodendrogenesis and myelin structure following cessation of MTX exposure together with complex alterations of the gliogenic microenvironment and persistent impairment in attention and short-term memory function (Gibson et al., 2019). To ascertain the extent to which oligodendroglial lineage dysregulation after MTX exposure includes disruption of neuronal activity-regulated myelination, we tested the influence of optogenetically stimulated frontal cortical projection neuronal activity in Thy1::ChR2 mice with previous MTX exposure. Thy1::ChR2 mice chiefly express the excitatory opsin channelrhodopsin-2 (ChR2) in cortical layer V projection neurons (Arenkiel et al., 2007). Optical fiber placement in superficial premotor cortex allows for light penetration to mid-cortex, stimulating the apical dendrites of ChR2-expressing layer V projection neurons (Yizhar et al., 2011). The premotor cortex is an association cortical area in the medial prefrontal complex just lateral to the anterior cingulate cortex and is involved in motor planning, learning and goal-directed actions (Barthas and Kwan, 2017). This optogenetic stimulation paradigm results in complex motor behavior that confirms successful stimulation of premotor circuit activity (Gibson et al., 2014; Yizhar et al., 2011). Microglial inflammation occurs chiefly in the superficial cortex around the optical-neural interface, resolves within a week of optical fiber placement and does not involve the corpus callosum at any time point (Gibson et al., 2014). We have previously shown in this model that neuronal activity results in circuit-specific OPC proliferation, oligodendrogenesis and myelin structural changes in the premotor projections in the corpus callosum (Gibson et al., 2014), and we examined the same region of corpus callosum here (Figure 1A).
Fig 1. Failure of adaptive myelination in a mouse model of MTX CRCI.
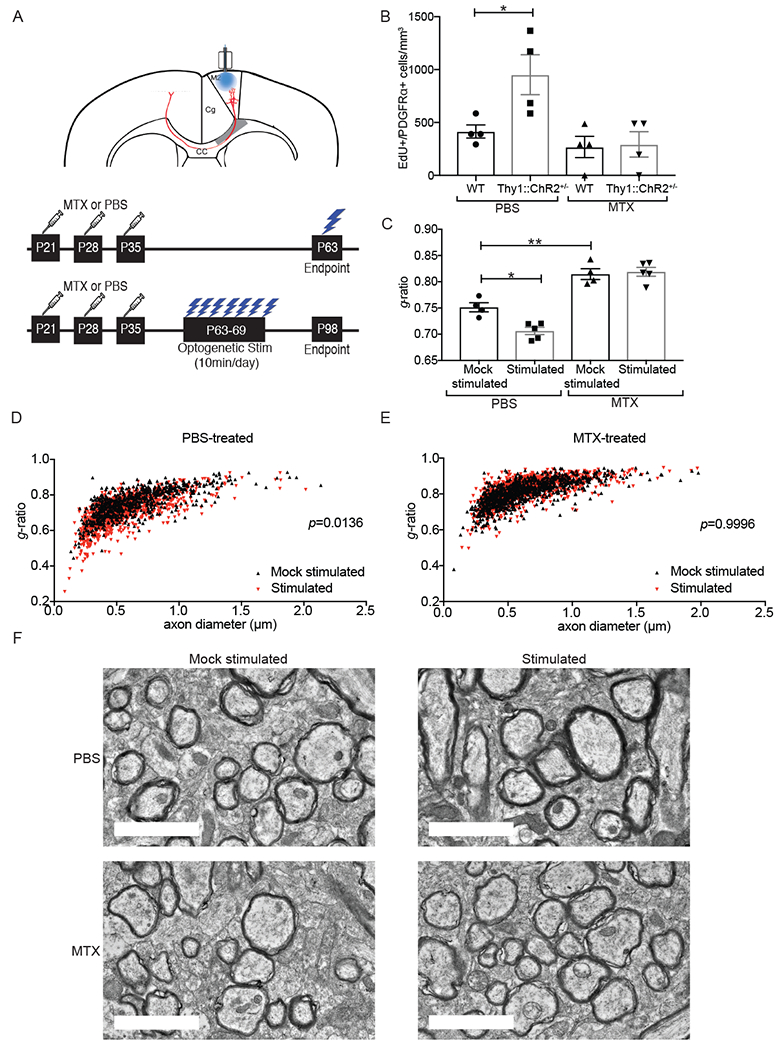
A) (Above) Coronal section of mouse brain, with prefrontal cortex areas labeled (M2 = premotor cortex; Cg = cingulate cortex). Optogenetic stimulation of premotor (M2) projection neurons, with analysis of oligodendroglial lineage cells in the corpus callosum (CC) in the region of premotor projections (shaded grey). (Below) Timelines of methotrexate (MTX) treatment and optogenetic stimulation (single and 7-day stimulation paradigms).
B) MTX abrogates activity-regulated OPC proliferation. Density of EdU-marked OPCs in the corpus callosum of Thy1::ChR2+/− mice and identically manipulated WT (no opsin) mice that were previously exposed to MTX or PBS vehicle control at 3-hours following a single optogenetic stimulation session. n=4 mice per group.
C) MTX abrogates activity-regulated myelination. TEM was performed one month following the end of the 7-day optogenetic stimulation paradigm in Thy1::ChR2+/− mice that were either stimulated or identically manipulated (mock-stimulated controls) that were previously exposed to MTX of PBS vehicle control. Myelin sheath thickness (g-ratio) analyzed at the level of the cingulum of the corpus callosum. n = 4-5 mice/group.
D-E) g-ratio shown as a function of axon caliber in scatterplot of all axons measured in (D) PBS vehicle control-treated, mock-stimulated mice (n = 4; black triangles) compared to PBS vehicle control-treated, optogenetically stimulated mice (n = 5; red triangles) and in (E) MTX-treated, unstimulated mice (n = 4; black triangles) compared to MTX-treated, optogenetically stimulated mice (n = 5; red triangles). A single point indicates the g-ratio for a single axon; ~100 axons quantified. P-values (indicated on plots) determined by comparing the mean g-ratio per mouse between groups.
F) Representative TEM images of premotor projections. Scale bars=2μm.
Data shown as mean ± SEM (B, C). Each point = one mouse (B, C). ns = p > 0.05, *p < 0.05, **p < 0.01, two-way ANOVA with Tukey post-hoc analysis for multiple comparisons. See also Figure S1.
Thy1::ChR2+/− and WT (no opsin) mice were exposed to MTX (100 mg/kg i.p.) or PBS vehicle on P21, 28 and 35 (Figure 1A; Gibson et al., 2019). The optical-neural interface was placed in premotor superficial cortex at P56. Mice underwent a single optogenetic stimulation session at P63 together with administration the thymidine analogue EdU and were sacrificed 3 hours later (Figure 1A). Similar to our previous findings (Gibson et al., 2014), optogenetically stimulated PBS vehicle control-treated mice exhibited the expected increase in EdU-marked, proliferating OPCs compared to identically manipulated WT (no opsin) controls (Figure 1B). In contrast, Thy1::ChR2+/− mice exposed to remote MTX did not exhibit activity-regulated OPC proliferation (Figure 1B), demonstrating a failure of activity-dependent OPC proliferation following remote MTX exposure.
To determine if activity-regulated myelin changes accompany the failure of activity-regulated OPC proliferation following MTX, we exposed Thy1::ChR2+/− mice to MTX or PBS vehicle control and optogenetically stimulated or identically manipulated mice from P63-69 (Figure 1A). Four weeks following the end of the stimulation paradigm (P98; Figure 1A), myelin sheath thickness was assessed using transmission electron microscopy (TEM) at the level of the cingulum of the corpus callosum. As expected (Gibson et al., 2014), optogenetically stimulated Thy1::ChR2+/− mice exposed to PBS vehicle exhibited an increase in myelin sheath thickness (as indicated by a decreased g-ratio) compared to unstimulated PBS-exposed mice (Figures 1C–D,F, S1). This neuronal activity-dependent increase in myelin sheath thickness was completely abrogated in mice exposed to previous MTX (Figures 1C,E,F, S1). Taken together, these data demonstrate a failure of adaptive myelination in this model of MTX CRCI.
Methotrexate chemotherapy reduces Bdnf levels in a microglia-dependent manner
A role for Bdnf in activity-regulated myelination has been postulated (Lundgaard et al., 2013) but not yet directly tested. Prompted by the intriguing observations that BDNF polymorphisms predict cognitive impairment severity in women receiving chemotherapy for breast cancer (Ng et al., 2016) and that other chemotherapy agents can decrease Bdnf expression in rodent models (Mustafa et al., 2008; Park et al., 2018), we examined Bdnf levels following MTX exposure to determine if decreased Bdnf levels accompany the observed failure of adaptive myelination in this mouse model of CRCI.
Bdnf is expressed by layer V projection neurons in the healthy brain (Zeisel et al., 2015; Tasic et al., 2016). We examined Bdnf mRNA expression in the deep layers of frontal cortex using RNAscope in mice exposed to MTX or PBS vehicle at 4 weeks after the final dose. Evaluation of the neuronal marker Rbfox3 (NeuN) and astrocyte marker Slc1a3 (Glast) revealed that Bdnf mRNA puncta are chiefly found in neurons in this region rather than cortical astrocytes (Figure 2A). The fraction of cells expressing Bdnf was markedly decreased in mice previously exposed to MTX compared to PBS vehicle control mice (Figure 2B). Microglial depletion using the CSF1R inhibitor PLX5622 during the 4-week period following the final MTX dose, which rescues myelination and cognitive behavioral performance in this model (Gibson et al., 2019), restored frontal cortex Bdnf mRNA expression (Figure 2B). Concordant with the observed changes in Bdnf mRNA expression, mice with previous MTX exposure exhibited a substantial decrease in Bdnf protein levels compared to PBS vehicle controls in microdissected frontal deep cortex and subjacent corpus callosum tissue (Figure 2C), and microglial depletion restored Bdnf protein levels after MTX exposure (Figure 2D). Bdnf signals through the TrkB receptor (also known as Ntrk2), and MTX exposure decreases TrkB phosphorylation (Figure 2E) as well as signaling events downstream of TrkB such as ERK phosphorylation (Figure 2F). Similar to the effect on Bdnf mRNA and protein, microglial depletion restores both phospho-TrKB and phospho-ERK levels after MTX exposure (Figure 2E–F). Of note, PLX5622 independently diminishes Bdnf protein and pERK levels to a moderate extent (Figure 2 D and F). Taken together, these data indicate that MTX exposure results in decreased neuronal Bdnf expression evident at the mRNA, protein and signaling levels, and that Bdnf expression is largely restored by microglial depletion after MTX.
Figure 2. MTX treatment depletes Bdnf mRNA and protein expression and disrupts TrkB signaling.
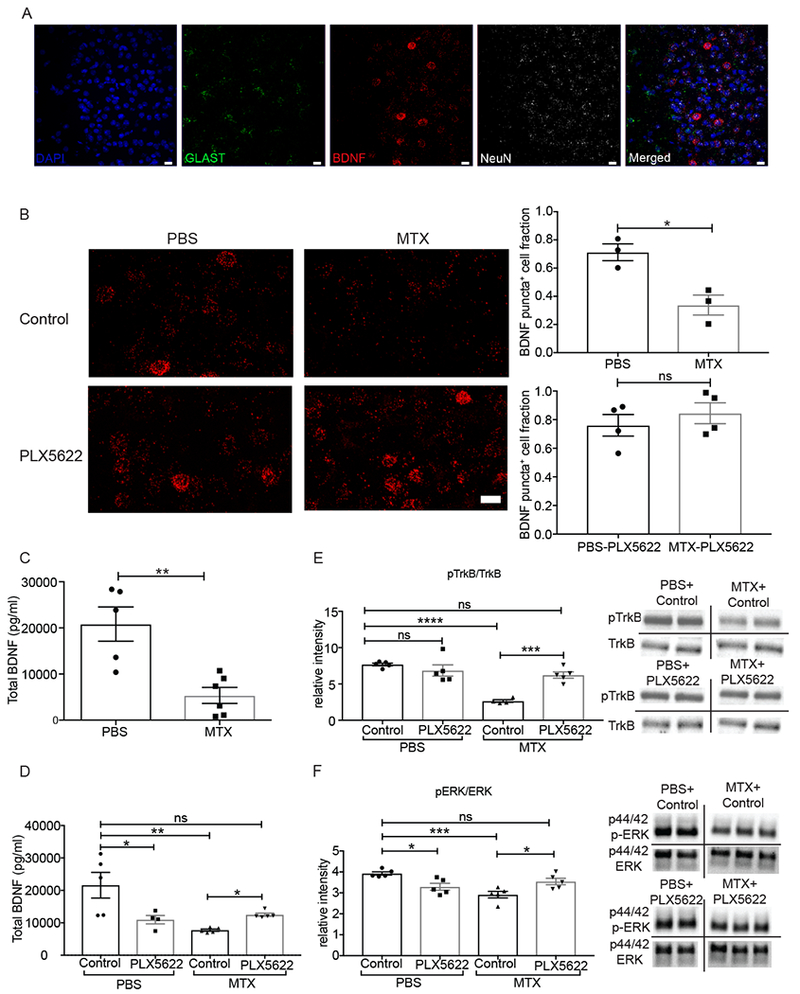
A) Representative image demonstrating RNAscope visualization of frontal cortex deep layer neurons (NeuN, white), astrocytes (Glast, green), and Bdnf mRNA (red). DAPI, blue. Scale bar = 20 μm
B) MTX treatment decreases Bdnf mRNA expression and microglial depletion with PLX5622 rescues Bdnf levels. (Left) Representative images of Bdnf puncta (red) in mice treated with PBS+control chow (n=3 mice) and PBS+PLX5622 chow (n=4 mice), MTX+control chow (n=3 mice) and MTX+PLX5622 chow (n=4 mice). (right) Quantification of DAPI+ cells expressing above threshold Bdnf mRNA puncta in each group. Scale bar = 20 μm
C) Decreased Bdnf protein levels following MTX exposure. Total Bdnf protein levels from frontal deep cortex and corpus callosum tissue microdissected at P63 and measured by ELISA in mice treated with MTX (n = 6 mice) or PBS vehicle control (n = 5 mice).
D) Microglial depletion with PLX5622 restores Bdnf levels following MTX exposure. Total Bdnf protein levels from frontal deep cortex and corpus callosum tissue microdissected at P63 and measured by ELISA in mice treated with PBS+control chow (n = 5 mice), PBS+ PLX5622 chow (n = 4 mice), and MTX+control chow (n = 5 mice), MTX+PLX5622 chow (n = 5 mice).
E) Microglial depletion with PLX5622 restores TrkB signaling following MTX exposure. Ratio of phospho-TrkB/TrkB western blot band intensity from microdissected frontal deep cortex and corpus callosum tissue of mice treated with PBS+control chow (n = 5 mice), PBS+ PLX5622 chow (n = 5 mice), MTX+control chow (n = 4 mice), and MTX+PLX5622 chow (n = 5 mice). Representative images of westerns to the right of the graph.
F) Microglial depletion with PLX5622 restores downstream TrkB signaling following MTX exposure. Ratio of phospho-ERK/ERK western blot band intensity from microdissected frontal deep cortex and corpus callosum tissue of mice treated with PBS+control chow (n = 5 mice), PBS+ PLX5622 chow (n = 5 mice), MTX+control chow (n = 5 mice), and MTX+PLX5622 chow (n = 5 mice). Representative images of westerns to the right of the graph.
Data shown as mean ± SEM. Each point = one mouse. ns = p > 0.05, * p < 0.05, ** p < 0.01, *** p<0.001, **** p<0.0001. Students t-tests (B, C); Two-way ANOVA with Tukey post-hoc analysis for multiple comparisons (D,E,F).
Bdnf signaling to OPC TrkB is necessary for activity-regulated myelination
Given that activity-regulated myelination is dysfunctional in the context of chemotherapy-induced reduction in Bdnf expression, we next set out to test the role of Bdnf to TrkB signaling in neuronal activity-regulated myelination. We optogenetically stimulated neuronal activity as above in genetically engineered mouse models that either lack activity-regulated Bdnf expression, or lack OPC-specific TrkB expression. In the first model, we used a mouse deficient in activity-induced expression of Bdnf due to knockin mutations in the Bdnf promoter IV (BdnfTMKI, TMKI= triple-site mutant (Hong et al., 2008); Figure 3A). These engineered mutations prevent the binding of calcium-regulated factors critical for neuronal activity-induced Bdnf transcription, particularly CREB, and specifically block activity-regulated Bdnf transcription while allowing activity-independent transcription to proceed (Hong et al., 2008). Bdnf secretion is also activity-dependent (Balkowiec and Katz, 2000; Dieni et al., 2012; Hartmann et al., 2001; Kokaia et al., 1998; Park et al., 2014; Wong et al., 2015), and the constitutive loss of activity-regulated Bdnf transcription results in reduced protein levels available for activity-regulated release in this mouse model. In frontal cortex and subjacent corpus callosum tissue microdissected from unmanipulated mice in standard housing conditions, we find a decrease in total Bdnf protein levels by ~50% in TMKI mice compared to WT controls (Figure 3B). In the second model, Pdgfra-CreERr2 mice (Kang et al., 2010) were bred to TrkBfl/fl mice (Grishanin et al., 2008) to conditionally knockout the TrkB receptor specifically from OPCs (Figure 3C). Using this conditional, inducible Pdgfra-CreERT2 driver mouse we find recombination is achieved in ~80% of OPCs following tamoxifen administration (Venkatesh et al., 2017). Accordingly, induction of recombination with tamoxifen (P24-P28) resulted in a robust decrease in OPC expression of TrkB (Figure 3D, S2A).
Fig. 3. BDNF to TrkB signaling is necessary for activity-regulated oligodendrocyte precursor cell proliferation.
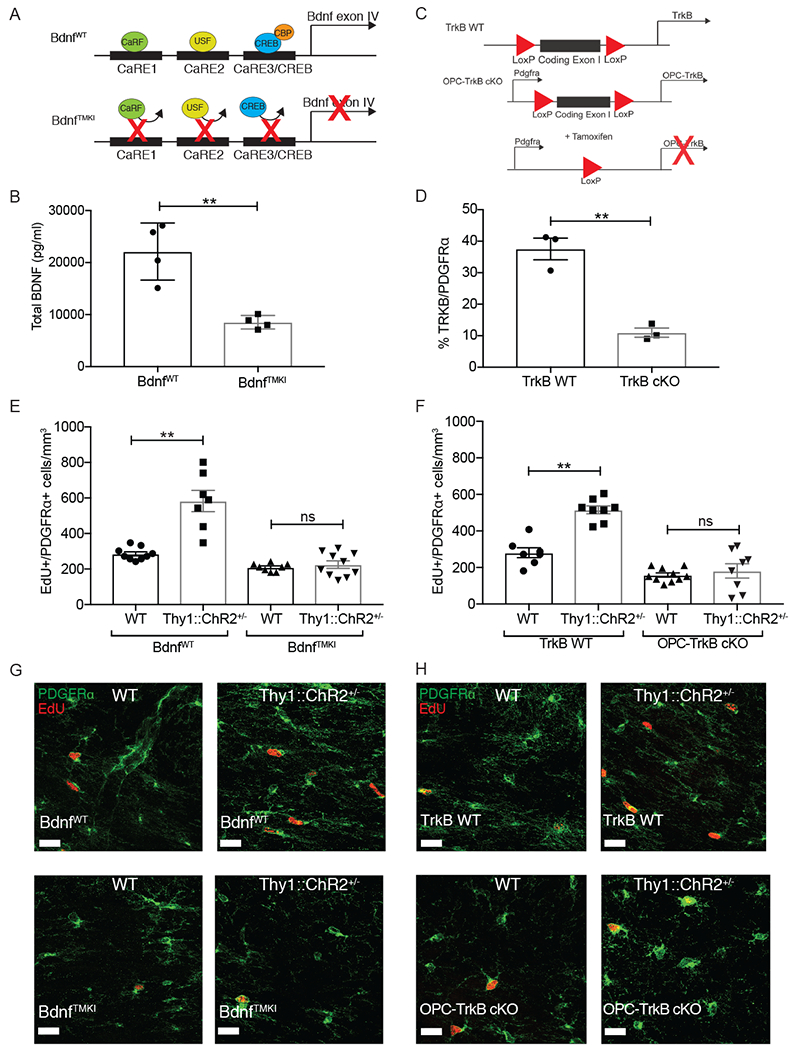
A) Schematic of BdnfTMKI mouse, which lacks activity-regulated Bdnf expression.
B) Bdnf protein is decreased in TMKI animals. Total Bdnf protein levels in frontal deep cortex and corpus callosum tissue are decreased in BdnfTMKI mice compared to BdnfWT control mice (n = 4 mice/group).
C) Schematic of OPC-TrkB cKO genetic mouse model in which exon1 of the TrkB gene is deleted, resulting in loss of both full-length and truncated TrkB receptor in PDGFRα+ cells (OPCs) following tamoxifen administration.
D) Residual OPC TrkB expression following recombination induced by tamoxifen (P24-28), expressed as percentage of PDGFRα+ OPCS co-expressing TrkB (n = 3 mice/group).
E) Loss of activity-dependent Bdnf blocks neuronal activity-regulated OPC proliferation. Density of proliferating OPCs (EdU+/PDGFRα+) in the corpus callosum of BdnfWT;WT (no opsin; n = 9), BdnfWT;Thy1::ChR2+/− (n = 7), BdnfTMKI;WT (no opsin; n = 8), and BdnfTMKI; Thy1::ChR2+/− (n = 10) mice at 3-hr following a single optogenetic stimulation session.
F) Inducible loss of OPC-specific TrkB blocks neuronal activity-regulated OPC proliferation. Density of proliferating OPCs (EdU+/PDGFRα+ cells) in the corpus callosum of TrkB WT;WT (no opsin; n=7), TrkB WT;Thy1::ChR2+/− (n=8), OPC-TrkB cKO;WT (no opsin; n=10), OPC-TrkB cKO;Thy1::ChR2+/− (n=8) mice at 3-hr following a single optogenetic stimulation session.
G) Representative confocal images of PDGFRα+ OPCs (green) co-localized with EdU+ nuclei (red) in the corpus callosum. Scale bars = 20μm.
Data shown as mean ± SEM. Each point = one mouse. ns = p > 0.05, ** p < 0.01. Student’s t-test (B and D); Two-way ANOVA with Tukey post-hoc analysis for multiple comparisons (E and F). See also Figure S2 and S3
To test the role of Bdnf to TrkB signaling in activity-dependent myelination, we performed optogenetic stimulation as above. The BdnfTMKI mouse and the Pdgfra-CreERT2;TrkBfl/fl (henceforth referred to as OPC-TrkB cKO) mouse were each bred to the Thy1::ChR2+/− mouse (Arenkiel et al., 2007). To conditionally delete TrkB specifically in OPCs in the OPC-TrkB cKO model, tamoxifen was administered (P24-28) to all mice in the four experimental groups, including OPC-TrkB cKO mice with and without ChR2 expression and TrkB WT (no Cre driver) littermates with and without ChR2 expression (OPC-TrkB cKO;Thy1::ChR2+/−, OPC-TrkB cKO;WT, TrkB WT; Thy1::ChR2+/− TrkB WT;WT). We have previously demonstrated that optogenetic stimulation of cortical projection neuronal activity in the Thy1::ChR2+/− mouse results in Bdnf secretion (Venkatesh et al., 2015).
Using these two genetic mouse models (BdnfTMKI;Thy1::ChR2+/− and OPC-TrkB cKO;Thy1::ChR2+/−), we sought to determine if loss of activity-dependent Bdnf to TrkB signaling influences OPC proliferation, subsequent oligodendrogenesis and myelin changes in response to optogenetic stimulation of neuronal activity. The optical-neural interface was placed in the superficial premotor cortex at P28 and mice underwent a single optogenetic stimulation at P35. Non-opsin expressing (WT) littermate control mice were identically manipulated to control for any effects of surgery, ferrule placement, or blue light exposure. As above, EdU was administered at the time of stimulation to mark cells proliferating during this time, and mice were sacrificed three hours after the end of a single stimulation session to assess OPC proliferation, or one month following 7 daily stimulation sessions to assess oligodendrogenesis and myelin structure.
Optogenetic stimulation of the premotor cortex in BdnfWT;Thy1::ChR2+/− mice or TrkB WT; Thy1::ChR2+/− mice resulted in the expected increase in proliferating OPCs within the corpus callosum, measured as the density of PDGFRα+ OPCs co-expressing EdU, compared to identically manipulated, non-opsin expressing (WT) littermate controls (Figure 3E–F, S2B). In contrast, optogenetic stimulation of premotor circuit activity in mice that lack activity-regulated Bdnf expression (BdnfTMKI mice) or that lack OPC-specific expression of TrkB (OPC-TrkB cKO;Thy1::ChR2+/− mice) exhibited no increase in OPC proliferation in response to neuronal activity (Figure 3E–F, representative images Figure 3G–H). OPC proliferation was not different between identically manipulated, non-opsin containing groups in either experiment (Figure 3E–H). Thus, loss of activity-regulated Bdnf expression or loss of OPC-specific TrkB expression blocks activity-regulated OPC proliferation in the premotor circuit.
We next examined the role of activity-dependent Bdnf to TrkB signaling in activity-regulated oligodendrogenesis. Oligodendrogenesis can occur through OPC proliferation with subsequent differentiation of the daughter cell, or by direct differentiation of an OPC without antecedent proliferation (Hughes et al., 2013). In both cases, generation of new mature oligodendrocytes depends upon successful differentiation and survival of the cell. Using both the BdnfTMKI and OPC-TrkB cKO mouse models, we quantified EdU-marked and total oligodendrocytes in the same corpus callosum location at one month following the end of the one-week paradigm of daily premotor optogenetic stimulation. Consistent with the loss of OPC proliferation described above, we found no increase in EdU-marked, newly generated oligodendrocytes nor increase in total oligodendrocyte numbers in either BdnfTMKI or OPC-TrkB cKO mouse models following optogenetic stimulation of neuronal activity (Figure S3), in contrast to the increase in oligodendrogenesis observed in Bdnf WT or OPC-TrkB WT animals following optogenetic stimulation (Figure S3).
We sought to determine the influence of Bdnf to TrkB signaling in activity-regulated myelin changes in these mouse models. As above, the week-long optogenetic stimulation paradigm was administered and myelin examined 4 weeks later by TEM, measuring myelin sheath thickness relative to axon caliber in the premotor projection fibers at the level of the cingulum. Optogenetically stimulated BdnfWT;Thy1::ChR2+/− mice exhibited thicker myelin sheathes, consistent with our previous observations (Gibson et al., 2014). In contrast, optogenetically stimulated BdnfTMKI;Thy1::ChR2+/− mice exhibited no change in myelin thickness as compared to either identically manipulated BdnfTMKI;WT (no opsin) and BdnfWT;WT (no opsin) mice (Figures 4A–C and S4A–C). When we repeat this experiment in the OPC-TrkB cKO model, we find that while TrkB WT;Thy1::ChR2+/− mice exhibit the expected increase in myelin sheath thickness following optogenetic stimulation of premotor neuronal activity, this myelin change was blocked in mice lacking OPC-TrkB expression (Figures 4D–F and S4D–F). Bdnf to TrkB signaling is thus required for activity-regulated myelination of cortical projection neurons.
Fig 4. BDNF to TrkB signaling is necessary for activity-regulated myelination.
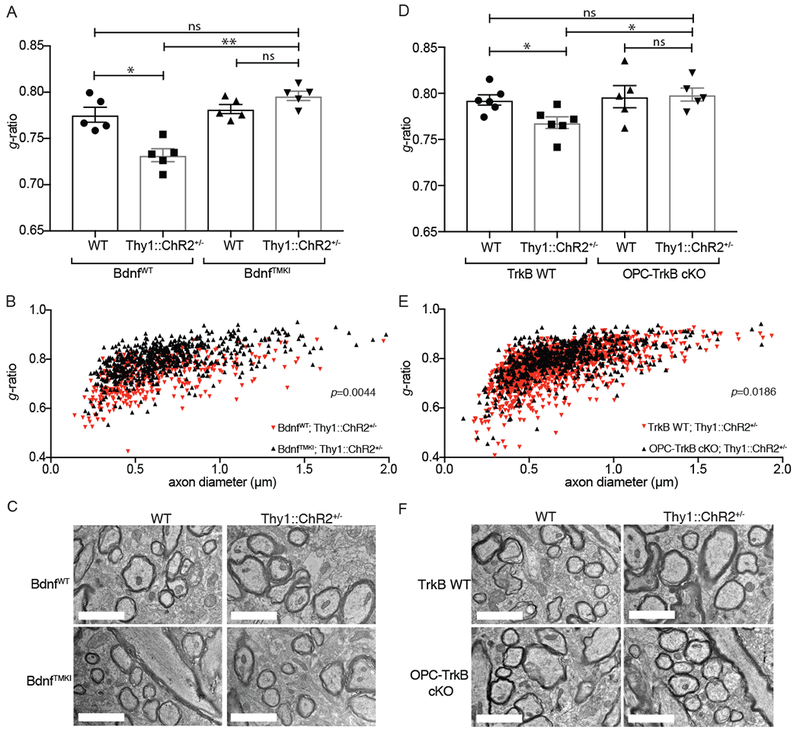
A) Loss of activity dependent Bdnf blocks neuronal activity-regulated myelination. TEM performed 4 weeks following the cessation of the 7-day optogenetic stimulation paradigm. Bar graphs representing the g-ratio data shown in B, expressed as mean g-ratio ± SEM for each group of mice (BdnfWT, WT (no opsin), BdnfWTThy1::ChR2+/−, BdnfTMKI;WT (no opsin), and BdnjTMKI; Thy1::ChR2+/−; n=5 mice/group).
B) Scatterplot of g-ratios as a function of axon caliber for BdnfWT;Thy1::ChR2+/− mice (n=5; red triangles) compared to BdnfTMKI;Thy1::ChR2+/− mice (n=5; black triangles). A single point indicates the g-ratio for a single axon. ~100 axons quantified for each mouse. P-values (indicated on plots) determined by comparing the mean g-ratio per mouse between groups.
C) BdnfTMKI model: Representative TEM images of premotor projections in cross section. Scale bars = 2μm.
D) Inducible loss of OPC-specific TrkB blocks neuronal activity-induced myelination. TEM analyses of g-ratio as described in A, in the OPC-TrkB cKO model. Bar graphs representing the g-ratio data shown in C, expressed as mean g-ratio ± SEM for each group of mice (TrkB WT;WT (no opsin), TrkB WT;Thy1::ChR2+/− OPC-TrkB cKO;WT (no opsin), OPC-TrkB cKO;Thy1::ChR2+−; n=6 TrkB WT mice/group and n = 5 OPC-TrkB cKO mice/group).
E) Scatterplot of g-ratios as a function of axon caliber in TrkB WT;Thy1::ChR2+/− mice− (n=6; red triangles) compared to OPC-TrkB cKO;Thy1::ChR2+/− mice (n=5; black triangles). A single point indicates the g-ratio for a single axon; ~100 axons were quantified for each mouse. P-values (indicated on plots) were determined by comparing the mean g-ratio per mouse between groups.
F) OPC-TrkB cKO model: Representative TEM images of premotor projections in cross section. Scale bars = 2μm.
Data shown as mean ± SEM;Each point = one mouse (A, D). ns = p > 0.05, *p < 0.05. Two-way ANOVA with Tukey post-hoc analysis for multiple comparisons (A, B, D and E). See also Figure S4
Impaired cognitive behavioral function in mice lacking OPC-specific expression of TrkB
As impairments in attention and memory are prominent features of CRCI (Pierson et al., 2016) that are recapitulated in our MTX mouse model (Gibson et al., 2019), we next asked how loss of OPC-TrkB influences these cognitive parameters. Attention and short-term memory function were tested in chemotherapy-naïve, group-housed OPC-TrkB cKO mice one month following tamoxifen induction (compared to tamoxifen-exposed TrkB WT mice as above) using a modified novel object recognition test (NORT) in which the time interval between training and testing phases is shortened to weight the test towards attentional function (Leger et al., 2013; Messier, 1997). After introduction to two identical objects, mice are then exposed to one familiar object and a novel object after a time delay of 5 minutes (Figure 5). Healthy mice spend more time exploring the novel object, while mice experiencing deficits in attention and/or short-term memory do not recognize either object as new and therefore spend equal time with each object (Leger et al., 2013). Discrimination of novel objects over familiar objects is a test that is sensitive to previous environmental enrichment (Loss et al., 2015; Mesa-Gresa et al., 2013), indicating that performance is influenced by mechanisms of plasticity. Distinct from previous work demonstrating a role for active oligodendrogenesis in motor learning (McKenzie et al., 2014; Xiao et al., 2016), here we are instead investigating the cumulative effects of OPC-TrkB loss for one month on cognitive behavioral function. TrkB deletion from OPCs was induced at P35 to mirror the juvenile timepoint of MTX administration in the CRCI model. Because developmental myelination is not complete at this juvenile time point (P35), we also tested a second cohort of mice in which TrkB deletion was induced at P60 to evaluate the effects conferred by loss of TrkB in OPCs after the developmental period for myelination has passed. Mice were assessed one month after induction of OPC-specific TrkB loss in both groups. Loss of OPC TrkB expression does not influence vision (Figure 5A), overall locomotion (Figure 5B) nor rearing behavior (Figure 5C), confirming that this mouse model has the capacity to see the objects and to move around to explore them. As expected, in TrkB WT mice we observed increased exploration of the novel object over the familiar one (Figure 5D–F). In contrast, OPC-TrkB cKO mice performed poorly in the novel object recognition test, failing to discriminate between familiar and novel objects in both age groups (Figure 5E–F). One interpretation of this finding is that ongoing adaptive myelination contributes to these aspects of cognitive function both during the juvenile period and in adulthood. It is important to note that there may be additional mechanisms by which OPC TrkB expression contributes to cognitive function apart from a role in activity-regulated myelination.
Figure 5. OPC-specific TrkB receptor loss results in deficits in cognitive behavioral performance.
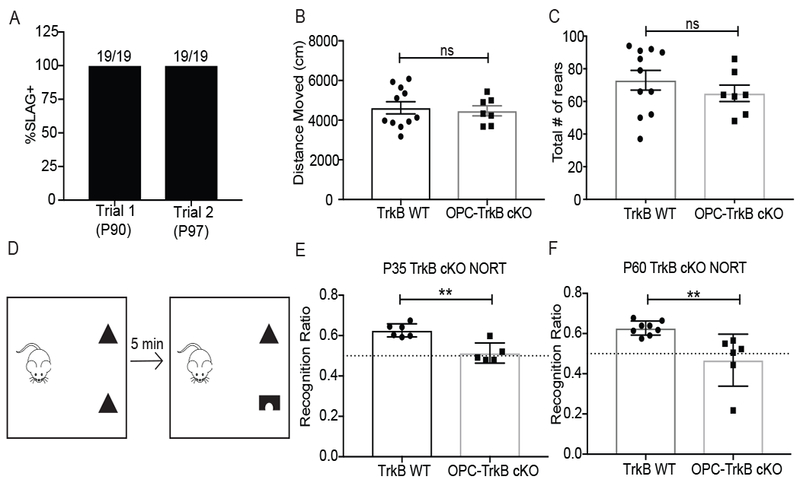
A) Vision is unaffected in OPC-TrkB cKO mice. OPC-TrkB cKO mice underwent slow angled-descent forepaw grasping (SLAG) testing at P90 and again one week later. All mice (n=19) exhibited SLAG+ behavior consistent with intact visual perception of objects.
B) Locomotion is similar between OPC-TrkB cKO mice and WT controls. Total distance moved in Intellicage activity chamber over 10-min testing period in OPC-TrkB cKO mice (n=7) and TrkB WT mice (n=11).
C) Rearing behavior is similar between OPC-TrkB cKO mice and WT controls. Total number of rears counted in the Intellicage activity chamber over ten minutes in OPC-TrkB cKO mice (n=7) and TrkB WT mice (n=11).
D) Schematic illustrating the Novel Object Recognition Test (NORT) of attention and shortterm memory function. Preference for novel object expressed as percent time spent with novel object over the total time spent interacting with either object (recognition ratio).
E) OPC-TrkB cKO mice (juvenile TrkB loss with tamoxifen given to all mice at P35) exhibit a deficit in novel object recognition. NORT performed at P63 in OPC-TrkB ckO mice (n=5 mice) and no Cre, TrkBfl/fl (TrkB WT; n=6) controls.
F) OPC-TrkB cKO mice (adult TrkB loss with tamoxifen given to all mice at P60) exhibit a deficit in novel object recognition. NORT performed at P89 in OPC-TrkB ckO mice (n=6 mice) and no Cre, TrkBfl/fl (TrkB WT; n=8) control mice.
Data shown as mean ± SEM. Each point = one mouse. ns = p > 0.05, ** p < 0.01, Student’s t-test (B,C,E,F)
TrkB partial agonist LM22A-4 normalizes myelination and rescues cognitive function after chemotherapy exposure
As OPC TrkB expression contributes to cognitive behavioral function and Bdnf to TrkB signaling is both required for activity-regulated myelination and dysregulated after MTX, we hypothesized that therapeutic TrkB agonism might improve cognitive behavioral function in CRCI. We thus tested a small molecule TrkB partial agonist (LM22A-4), a compound with a preclinical toxicity and physical chemistry profile suitable for potential therapeutic development. This compound specifically activates the TrkB receptor (Massa et al., 2010) and penetrates the blood-brain-barrier following systemic administration (Simmons et al., 2013). To validate that LM22A-4 functions similarly to Bdnf in the oligodendroglial lineage, we tested it side-by-side with Bdnf in in vitro protocols of OPC proliferation and oligodendrogenesis and also tested its capacity to promote repair after toxic demyelinating injury (Figure S5). In all assays, we found that LM22A-4 functions similarly to Bdnf to promote OPC proliferation, oligodendrogenesis and myelin repair (Figure S5).
Having validated LM22A-4, we next tested the hypothesis that stimulating TrkB signaling could promote myelination and improve cognitive dysfunction after MTX exposure. Mice were subjected to the above MTX protocol, then treated with the TrkB partial agonist LM22A-4 or vehicle control (Figure 6A). Myelin structure and behavioral performance in the novel object recognition test were then assessed. As expected (Gibson et al., 2019), mice that received MTX exhibited decreased myelin sheath thickness (Figure 6B). Chemotherapy-naïve mice treated with LM22A-4 exhibited an increase in myelin sheath thickness as compared with those that received vehicle control (Figures 6B–C and S6A–B). LM22A-4 restored myelin sheath thickness to control levels following MTX exposure (Figures 6B) in small, medium and large caliber axons (Figure S6C).
Fig 6. TrkB partial agonist LM22A-4 normalizes myelination and rescues cognitive behavioral function after methotrexate chemotherapy exposure.
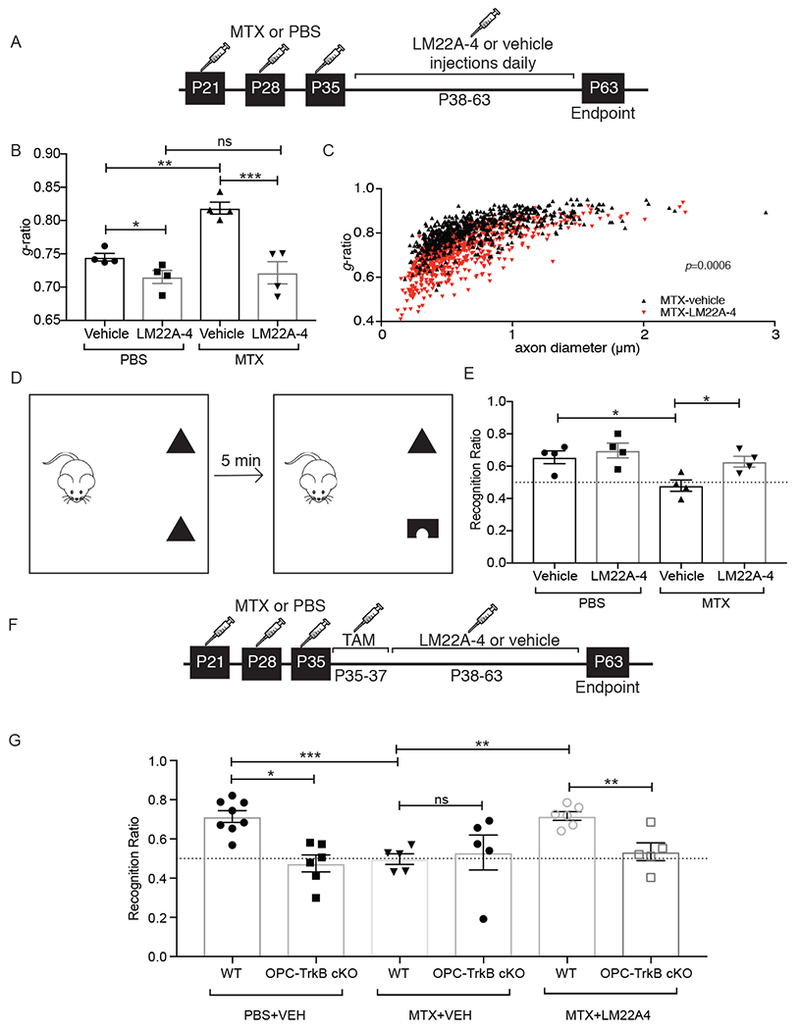
A) Schematic of LM22A-4 treatment in MTX CRCI model, with MTX or PBS vehicle control at P21, 28 and 35 followed by daily LM22A-4 or vehicle control administration from P38-P63.
B) Treatment with LM22A-4 rescues myelin deficits after MTX. TEM performed at P63 and g-ratios measured at the level of the cingulum of the corpus callosum. Bar graphs representing the g-ratio data shown in C and Fig. S6. n=4 mice/group.
C) Scatterplot of g-ratios as a function of axon caliber in MTX+LM22A-4-treated mice (n=4; red triangles) compared to MTX+vehicle control-treated mice (n=4; black triangles). A single point indicates the g-ratio for a single axon; ~100 axons quantified for each mouse. P-values (indicated on plots) determined by comparing the mean g-ratio per mouse between groups.
D) Schematic of Novel Object Recognition Test (NORT). Preference for novel object measured as the percent time spent with novel object over the total time spent with either object (recognition ratio).
E) LM22A-4 rescues the cognitive behavioral impairment observed after MTX. NORT performance in mice previously exposed to MTX or PBS vehicle control and subsequently treated with LM22A-4 or vehicle control. (n=4 mice in each group).
F) Schematic of LM22A-4 treatment in MTX CRCI model, with tamoxifen (TAM) induction of OPC TrkB cKO prior to LM22A-4 exposure.
G) TrkB expression in OPCs is required for LM22A-4 rescue of cognitive behavioral performance after MTX. NORT performance in OPC-TrkB cKO and TrkB WT mice previously exposed to either MTX or PBS vehicle control followed by LM22A-4 or vehicle control, as outlined in F.
Data shown as mean ± SEM; each point = one mouse (B, E, G). ns = p > 0.05, * p < 0.05, ** p < 0.01 *** p<0.001 as determined by two-way ANOVA (B, C, E) or three-way ANOVA (G) with Tukey post-hoc analysis for multiple comparisons. See also Figure S5 and S6
To test the influence of LM22A-4 on attention and short-term memory function, NORT (Figure 6D) was used as above. As expected (Gibson et al., 2019), control mice interact with the novel object significantly more than chance (Figure 6E) while MTX-treated mice demonstrate no preference for the novel object (Figure 6E), similar to performance of OPC-TrkB cKO mice described above. Treatment with LM22A-4 rescues NORT performance in MTX-treated mice (Figure 6E).
Bdnf influences numerous mechanisms of neuroplasticity and the cognitive behavioral improvement observed could thus reflect more than just effects on myelin. To test the relative importance of oligodendroglial effects to this rescue by LM22A-4, we repeated this experiment of LM22A-4 administration after MTX exposure in OPC-TrkB cKO mice (Figure 6F). Strikingly, the rescue of cognitive behavioral performance with LM22A-4 administration in this model of CRCI depends upon OPC expression of TrkB (Figure 6G). Taken together, these data demonstrate that LM22A-4 normalizes myelin sheath thickness and rescues cognitive behavioral deficits in attention and short-term memory function following MTX exposure in a manner dependent on oligodendroglial effects.
Discussion
The present study demonstrates that activity-regulated myelination fails in a model of MTX chemotherapy-related cognitive impairment, a relatively common and clinically consequential syndrome characterized by impaired attention and memory function for which microglia are central to the complex pathophysiology (Gibson et al., 2019). Associated with this failure of adaptive myelination is microglial-dependent reduction in cortical Bdnf expression. Bdnf-TrkB signaling in OPCs is a necessary mechanistic component of cortical projection neuronal activity-regulated myelination. Loss of TrkB signaling in OPCs impairs cognitive behavioral performance in a test of attention and short-term memory function in chemotherapy-naïve mice. Harnessing this mechanistic insight, we find that TrkB agonism with a small molecule therapeutic restores myelin following MTX exposure and rescues the behavioral deficits observed in a CRCI mouse model in a manner dependent on oligodendroglial TrkB signaling. Taken together, these data demonstrate that dysfunction of adaptive myelination is one important component of MTX CRCI and that stimulating OPC TrkB signaling represents a promising strategy to improve cognition following MTX chemotherapy exposure.
The neural substrate of attentional and short-term (<5 min) memory function is diffusely distributed (Cohen and Maunsell, 2011; Zhou and Desimone 2011; Zhang and Desimone, 2011; Luo and Maunsell, 2015; Veldsman et al., 2017). While both attentional and short-term memory function involve the prefrontal cortex, including premotor cortex (Rizzolatti et al., 1987; Bichot and Desimone, 2015; Chen and Svoboda, 2017; Barthas and Kwan 2017; Moore 2006), there is no clearly defined fiber bundle to specifically examine the axons relevant to these cognitive behavioral functions. An important caveat is that the region of frontal cortical projection neuron myelination analyzed for the optogenetic activity-regulated myelination assay contains dense projections of axons involved in motor function (Gibson et al., 2014).
Intrinsic and adaptive myelination
The idea that neuronal activity may influence myelin-forming cell behavior was introduced by Ben Barres and Martin Raff (Barres and Raff, 1993), and the well-described but still somewhat functionally enigmatic discovery that OPCs receive synaptic inputs from neurons (Bergles et al., 2000; Karadottir et al., 2005) stoked interest in the role neuronal activity may play in myelin development and plasticity. Several elegant in vitro studies (Ishibashi et al., 2006; Stevens et al., 1998) and observations linking experience to changes in myelin ultrastructure (Liu et al., 2012; Makinodan et al., 2012; Scholz et al., 2009) supported the concept of adaptive myelination. Controversy surrounded this idea, however, as activity-independent modes of myelination also exist (Bechler et al., 2015; Lee et al., 2013; Mayoral et al., 2018; Rosenberg et al., 2008) and contribute to myelin development and homeostasis.
New techniques such as optogenetics and chemogenetics enabling precise, cell type-specific modulation of neuronal activity facilitated direct demonstration that neuronal activity can promote OPC proliferation, oligodendrogenesis and myelin structural changes in the juvenile and adult forebrain (Gibson et al., 2014; Mitew et al., 2018). Learning a new complex motor task and stimulation of whisker sensation similarly induces OPC proliferation and oligodendrogenesis in the premotor and somatosensory circuits, respectively (Hughes et al., 2018; McKenzie et al., 2014). During development, neuronal activity can influence axon selection (Hines et al., 2015; Mensch et al., 2015), and interneurons can also govern developmental myelination through secreted factors that may be activity-independent (Voronova et al., 2017). Coalescing the available evidence into a cohesive model, ffrench-Constant and colleagues have proposed that an intrinsic program of myelination proceeds independent of activity during development and that myelination can be further sculpted and refined by experience-dependent, adaptive changes (Bechler et al., 2015; Bechler et al., 2018).
Adaptive myelination: unresolved mechanistic and conceptual questions
The neuronal activity threshold level or pattern required to elicit adaptive changes in oligodendroglial lineage cells remains to be fully defined. Not all neuronal activity recruits an oligodendroglial response, as the level of oligodendrogenesis and myelination would be beyond what is observed in the healthy brain. Accordingly, a measurable change was not observed in premotor projection myelination in the non-opsin expressing TrkB WT or OPC TrkB-cKO mice during the month following TrkB loss. These mice remained in the same standard group housing cage environment in which their premotor circuit myelination was established and were not exposed to a motor planning-dependent challenge following TrkB recombination. However, repetitive stimulation of neuronal activity in these premotor projection neurons uncovered a stark difference in myelination between TrkB WT and OPC TrkB cKO mice. Better understanding what defines the threshold for activity to recruit changes in myelin-forming cells represents a critical question for future work.
How new oligodendrocytes incorporate into the myelinated infrastructure is not yet entirely clear, but recent insights have begun to shed light on this question. Myelination of axons in somatosensory cortex is discontinuous (Tomassy et al., 2014), revealing axonal territory available for activity-regulated myelination. Concordantly, cortical myelin accumulates over the lifespan, with new internodes forming on previously unmyelinated axonal regions (Hill et al., 2018; Hughes et al., 2018). Like the neocortex, myelination is also variable and incomplete in subcortical projections, with as many as 30% of corpus callosum fibers remaining unmyelinated (Olivares et al., 2001), creating opportunity for new oligodendrocytes to incorporate. In most regions of the central nervous system (CNS), oligodendrocytes exhibit marked stability until old age in the healthy brain and in the absence of sensory deprivation (Hill et al., 2018; Hughes et al., 2018; Tripathi et al., 2017), arguing against turnover and replacement of older oligodendrocytes. Taken together, the evidence suggests activity-regulated myelination of previously unmyelinated axonal territory, although future work will be needed to understand activity-regulated myelin incorporation in various regions of the CNS. Whether the oligodendrocytes generated as a result of neuronal activity in the corpus callosum myelinate previously unmyelinated axonal territory or remodel existing myelin internodes cannot be conclusively determined in the experimental paradigm used here as previously discussed (Gibson et al., 2014), and this remains an open question for future studies.
While the manner in which newly generated oligodendrocytes incorporate into myelin and how the resultant myelin changes influence neural circuit dynamics remains to be fully determined, it is clear that activity-regulated oligodendrogenesis can influence motor function (Gibson et al., 2014) and motor learning (McKenzie et al., 2014; Xiao et al., 2016). Changes in myelin and the subsequent influence on spike time arrival and neural circuit dynamics could play diverse roles in additional aspects of neural function and cognition. The demonstration here that conditional OPC TrkB deletion results in impaired cognitive behavioral performance, together with failure of activity-regulated myelination in CRCI further suggests a role for myelin plasticity in normal cognitive function and links a neurocognitive disease to dysregulated myelin plasticity. It is important to note that the dysmyelination observed in MTX CRCI is multifactorial (Gibson et al., 2019) and is not solely accounted for by loss of activity-regulated myelination. Additional domains of cognitive behavioral function that may be influenced by adaptive myelination remain to be studied.
Neurotransmitters, neurotrophins and neuron to OPC synapses
Axon-glial synapses provide a direct pathway for communication between neurons and OPCs and have long been suspected to play a role in adaptive myelination, although the functional role of these synaptic connections remains to be fully elucidated. Neurotransmitters influence OPC proliferation and differentiation (Gallo et al., 1996; Wake et al., 2011; Zonouzi et al., 2015), and glutamatergic signaling at the axon-glial synapse promotes oligodendroglial cell survival (Kougioumtzidou et al., 2017). While NMDA receptor activation stimulates in vitro myelination (Lundgaard et al., 2013; Wake et al., 2011), OPC-specific knock out of the requisite NMDA receptor NR1 subunit was not found to influence myelination in vivo (De Biase et al., 2011). However, explicit testing of the activity-regulated response was not probed in that mouse model nor have roles for other forms of glutamatergic neurotransmission been ruled out.
Bdnf influences NMDA receptor expression in OPCs (Lundgaard et al., 2013), suggesting a possible link between neurotrophins and glutamatergic signaling. While the subcellular localization of TrkB in OPCs is not yet known, it is intriguing to note that TrkB can localize to the postsynaptic membrane in neurons (Garber et al., 2018; Lin et al., 1998) and Bdnf can be released in synaptic vesicles (Fawcett et al., 1997; von Bartheld et al., 1996). Vesicular release is responsible for the influence of neuronal activity on axonal selection during development in the zebrafish, although it is not yet clear if the relevant vesicular mechanism involves vesicular glutamate, Bdnf or both (Hines et al., 2015; Mensch et al., 2015). Vesicular release occurs throughout the corpus callosum at axon-glial synapses (Kukley et al., 2007), and this may represent the relevant source of Bdnf. Alternative sources of Bdnf include astrocytes (Fulmer et al., 2014; Zhang et al., 2014) and microglia (Parkhurst et al., 2013). Synaptic localization of TrkB in OPCs would imply that structural synapses between neurons and OPCs may play a crucial role in facilitating activity-regulated myelination. The current working model includes Bdnf to TrkB signaling as one required component of the likely multifaceted mechanism mediating adaptive myelination.
Bdnf in developmental and regenerative myelination
Brain-derived neurotrophic factor (Bdnf), a secreted growth factor robustly regulated by neuronal activity, mediates numerous processes of neurodevelopment and plasticity (Korte et al., 1995; Li et al., 2008; Lohof et al., 1993; Park and Poo, 2013; Patterson et al., 1996). Bdnf plays a role in both developmental myelination (Cellerino et al., 1997; VonDran et al., 2010; Xiao et al., 2010; Wong et al., 2013; Goebbels et al., 2017;) and in remyelination after toxin-induced demyelinating injury (Fulmer et al., 2014; Tsiperson et al., 2015; VonDran et al., 2011). Of note, the oligodendroglial lineage response to Bdnf may be heterogeneous between brain regions (Goebbels et al., 2017). Consistent with the demonstrated role of Bdnf in remyelination following injury (Fulmer et al., 2014; Tsiperson et al., 2015; VonDran et al., 2011), we find that LM22A-4 similarly promotes remyelination after focal demyelinating injury.
LM22A-4 is a small molecule partial agonist developed from in silico screening of small molecule libraries modeled on the loop II domain of Bdnf to identify TrkB agonists (Massa et al., 2010). Currently serving as a lead compound for clinical development in treatment of neurodegenerative disorders, LM22A-4 achieves Bdnf-type-trophic effects including promotion of neuronal survival and neurogenesis, and modulation of neuronal activity (Massa et al., 2010; Han et al., 2012; Li et al., 2017). Preclinical studies utilizing LM22A-4 have thus far focused on its neuronal effects. We now report similarly trophic effects on myelin-forming cells. While TrkB agonism may influence numerous processes of neuroplasticity and regeneration (Korte et al., 1995; Li et al., 2008; Lohof et al., 1993; Park et al., 2014; Patterson et al., 1996), LM22A-4 acts through OPC TrkB to rescue cognitive behavioral function after MTX chemotherapy.
Bdnf levels in frontal cortex and corpus callosum projections are restored by microglial depletion after MTX chemotherapy exposure. How MTX-activated microglia influence Bdnf expression remains to be elucidated, but normalization of Bdnf expression levels following microglial depletion joins a growing list of aberrations rescued by microglial depletion following MTX exposure (Gibson et al., 2019). The cellular consequences of MTX chemotherapy are complex and involve dysregulation of multiple neural cell types in which microglial activation is central (Gibson et al., 2019). TrkB agonism circumvents this complex pathophysiology to restore myelin and rescue cognitive behavioral function. Thus, modulation of microglial activation (Gibson et al., 2019), and TrkB signaling each emerge as potential therapeutic strategies for those suffering long-term cognitive dysfunction following cancer chemotherapy.
In conclusion, Bdnf to TrkB signaling is a necessary component of adaptive myelination and both are dysregulated in MTX chemotherapy-related cognitive impairment. Blocking activity-regulated myelination through OPC-specific loss of TrkB phenocopies key aspects of the cognitive behavioral impairment observed after MTX chemotherapy (Gibson et al., 2019). As future work further elucidates the roles that adaptive myelination may play in healthy cognitive function, implications for dysfunction or dysregulation in a range of neurological diseases may similarly come to light.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michelle Monje (mmonje@stanford.edu).
Experimental Model and Subject Details:
Animal procedures and mouse models
Mouse models
All mouse experiments were conducted in accordance with protocols approved by the Stanford University Institutional Animal Care and Use Committee (IACUC). Mice were housed in group cages (up to 5 mice/cage) according to standard guidelines with ad libitum access to food and water in a 12 h light/dark cycle. Both sexes were used equally in all experiments, with the exception of Bdnf protein, pTrkB and pERK analyses in which male mice were used to avoid effects of the estrous cycle on Bdnf levels (Jezierski and Sohrabji, 2000). No animals were manipulated other than as reported for that experimental group, i.e. there was no history of drug exposures, surgeries or behavioral testing for the animals used other than that reported for the given experimental group. Mice were healthy and tolerated all experimental manipulations well, with the exception of decreased appetite during the period of MTX exposure in the MTX CRCI model.
BdnfTMKI; Thy1::ChR2+/− model
BdnfTMKI mice (C57BL/6J background) with knock-in mutations in three calcium regulatory element binding sites in the Bdnf promoter IV: CaRE1, CaRE2 and CaRE3/CREB (M. Greenberg; Hong et al., 2008) were bred to Thy1::ChR2+/− mice (line 18, The Jackson Laboratory, C57BL/6J background) to produce the BdnfTMKI; Thy1::ChR2+/− genotype. All experiments were performed with both male and female mice either homozygous for the BdnfTMKI mutant or BdnfWT control mice and heterozygous for Thy1::ChR2+/− or WT (no opsin) littermates.
OPC TrkB cKO and OPC TrkB cKO; Thy1::ChR2+/− model
For the TrkB conditional knockout studies, TrkB-flox mice (MMRRC) were bred to either Pdgfra-CreERT2 mice (The Jackson Laboratory) to generate Pdgfra-CreERT2; TrkBfl/fl (OPC-TrkB cKO) mice (C57BL/6J background) or to Thy1::ChR2+/− mice to generate TrkBfl/fl ;Thy1::ChR2+/− mice. These offspring were then bred to each other to create a final genotype of Pdgfra-CreERT2; TrkBfl/fl ;Thy1::ChR2+/−. All experiments were performed with both male and female mice that were heterozygous for both Pdgfra-CreERT2 and Thy1::ChR2 and littermate WTs. Cre+ and Cre− mice were treated with 100 mg/kg tamoxifen intraperitoneally as indicated in specific studies. (As described below, for optogenetic experiments, tamoxifen was given from postnatal day 24 (P24) to P28, for novel object recognition test (NORT) studies, animals were given tamoxifen from P31-35, and then tested on P63 for the P35 group, or given tamoxifen from P56-60 and tested on P89 for the P60 group. For MTX and LM22A4 studies, tamoxifen was given from P35-37 to avoid overlap of administered drugs.) For verification of knockdown (Fig 3D), tamoxifen was administered on days P24-28, and percentage of TrkB-expressing PDGFRα+ was measured by immunohistochemical analysis in deep cortical tissue at P35. Verification of OPC TrkB knockdown was replicated with the 3-day tamoxifen paradigm used in the MTX + LM22A4 experiment and found to be equivalent to the 5-day tamoxifen paradigm.
Lysolecithin demyelination model
Lysolecithin (L-α-Lysophosphatidylcholine from egg yolk, Sigma L4129) was diluted to 1% in PBS, and aliquots were frozen at −20°C for single use. Mice at approximately P28 were anesthetized with 1–4% isoflurane and placed in a stereotactic apparatus. The cranium was exposed via midline incision under aseptic conditions, and a hole was drilled in the skull for injection. A 26s-gauge Hamilton syringe was used to inject 1μl lysolecithin into the cingulum using a digital pump at infusion rate of 0.2 μl/min. Stereotactic coordinates used for cingulum were 1.0 mm lateral to midline, 1.0 mm anterior to bregma, −1.5 mm deep to cranial surface. At the completion of infusion, syringe needle remained in place for a minimum of 5 min to minimize backflow of the injection.
C57BL/6J male and female mice (The Jackson Laboratories) were used in the lysolecithin demyelination/remyelination model. Mice were stereotactically injected with 1% lysolecithin at P28 and were injected intraperitoneally with either 50mg/kg small molecule partial agonist LM22A-4 (F. Longo) or vehicle beginning at P35. Mice did not exhibit overt neurological deficits nor sickness behavior following lysolecithin injection.
Methotrexate chemotherapy-related cognitive impairment model
Mice were treated with 100 mg/kg methotrexate (MTX) or PBS vehicle control i.p. on P21, P28, and P35 (Gibson et al., 2019). For the MTX and LM22A-4 experiments, mice were injected intraperitoneally with either 50mg/kg LM22A-4 or vehicle beginning at P38, administered daily until P63. For the OPC-TrkBcKO MTX experiments, mice were injected intraperitoneally with 100 mg/kg tamoxifen from P35-37. C57BL/6J male mice were bred to CD-1 female mice (Charles River Laboratories) to generate BL6/CD1 mice which were used in all MTX CRCI experiments except where the indicated genetic mouse models (i.e. Thy1::ChR2+/− or OPC TrkB cKO models) were used. Mice exhibited mild sickness behavior (decreased appetite) during the period of MTX exposure, with recovery and normal appetite after MTX cessation.
For microglial depletion experiments, PLX5622, an inhibitor of colony-stimulating factor 1 receptor, was administered to BL6/CD1 mice ad libitum for a month beginning 3 days after the last MTX (or vehicle) injection (P38-63). PLX5622 was provided by Plexxikon Inc. (Berkeley, CA) and formulated in AIN-76A standard chow with 1,200 ppm of PLX5622 by Research Diets Inc. Animals then underwent behavioral testing using the Novel Object Recognition Test (NORT) or perfusion for immunohistochemical analysis, RNAscope or biochemical analyses.
Optogenetic stimulation in BDNFTMKI and OPC-TrkB cKO models
BdnfTMKI; Thy1::ChR2+/− or Pdgfra-CreERT2; TrkB;Thy1::ChR2+/− mouse models (described above) were used. Fiber optic placement was performed at P28 as previously described (Gibson et al., 2014), 7 days prior to optogenetic stimulation. In short, mice were anesthetized with 1–4% isoflurane and placed in a stereotactic apparatus. The cranium was exposed using a midline incision under aseptic conditions. A fiber optic ferrule (Doric Lenses, Quebec, Canada) was placed at the premotor cortex (M2) of the right hemisphere using the following coordinates: 0.5 mm lateral to midline, 1.0 mm anterior to bregma, 0.65-0.7 mm deep to cranial surface in the right hemisphere. For optogenetic stimulation, mice were connected to the laser system with a mono fiber patch cord, which freely permits wakeful behavior of the animal. Pulses of 473 nm wavelengths of light (15-25 mW/mm2 measured at the tip of the patch cord, 1.5–2.5 mW/mm2 in the tissue where stimulation of the layer V projection neuron apical dendrites occurs (Yizhar et al., 2011)), were administered at a frequency of 20 Hz for periods of 30 sec, followed by 120 sec recovery periods, for a total session duration of 30 min for the single stimulation paradigm (on P35) or 10 min per day for 7 consecutive days (P35 – P41) for the repetitive stimulation paradigm. During periods of light administration, all Thy1::ChR2+/− mice responded with unidirectional ambulation to the left for the duration of light exposure, confirming proper ferrule placement at the superficial right premotor cortex. All WT mice demonstrated no change in behavioral output in response to light stimulation. For the single session stimulation, two intraperitoneal injection of 5-ethynyl-20-deoxyuridine (EdU; 40 mg/kg; Invitrogen A10044) were administered to the animal, one five minutes prior to the start of stimulation, and one immediately following stimulation. For the repetitive stimulation paradigm, one 40mg/kg intraperitoneal injection of EdU was administered five minutes prior to the start of stimulation daily. Mice were sacrificed 3h after administration of the second EdU injection in the single stimulation paradigm experiment. For the repetitive stimulation experiment, mice were sacrificed 4 weeks following the final stimulation session.
Optogenetic stimulation in the methotrexate chemotherapy-related cognitive impairment model
Thy1::ChR2+/− male mice (C57BL/6J background) were bred to CD-1 female mice (Charles River Laboratories) to generate CD-1; Thy1::ChR2++/− mice and CD-1; Thy1::ChR2−/− WT mice (single session optogenetic study) or Thy1::ChR2+/+ male mice were bred to CD-1 female mice to generate CD-1; Thy1::ChR2+/− mice (for repetitive stimulation optogenetic study).
Single session optogenetic study:
WT non-opsin containing mice and Thy1::ChR2+/− mice were injected intraperitoneally with 100 mg/kg methotrexate (MTX) or PBS at P21, 28, and 35. At P56, all animals underwent surgery for fiber optic placement (see above) and allowed to recover for one week. At P63, all animals underwent a single, acute 30-min optogenetic stimulation paradigm at 20 Hz and sacrificed 3 h after stimulation (see above). Animals were perfused and brains were cryoprotected for immunohistochemical analysis as described below.
Repetitive optogenetic stimulation study:
Thy1::ChR2+/− mice were injected with MTX or PBS at P21, 28, and 35 followed by stereotactic surgery to place the optic fiber in superficial M2 at P56 (see above). Half of the MTX and PBS animals were optogenetically stimulated daily for 7 days starting on P63 as described above. The other half of the MTX and PBS mice were sham stimulated so as to control for surgery and fiber optic placement but were not exposed to 473 nm blue light (mock-stimulated). Four weeks following the last stimulation session, mice were perfused for transmission electron microscopy processing and myelin sheath thickness was assessed (see below).
Generation of mouse OPC cultures
To generate cultures of mouse oligodendrocyte precursor cells for in vitro BDNF and LM22A-4 experiments, BL6/CD1 P6-8 mouse pups were anesthetized and rapidly decapitated, and the full cortex region was dissected in Hibernate-A medium (Thermo Fisher Scientific, A12475-01) for processing. Both male and female pups were included from each litter. Tissue was enzymatic dissociated in buffer containing HEPES-HBSS with DNAse (Worthington Biochemical LS002007) and Liberase (Roche Applied Sciences 05401054001) at 37°C. Cells were then passed through a 100 μm cell strainer and processed through a sucrose gradient to remove myelin. OPCs were isolated using the CD140 (PDGFRα) Microbead Kit (MACS, Miltenyi Biotec 130-101-502) according to the manufacturer’s protocol. Cells were plated at 10,000 cells per well in an 8 well chamber slide (Thermo Fisher Scientific 154534PK) and kept in proliferative media DMEM (Thermo Fisher Scientific 11320082) containing: Glutamax (Invitrogen 35050-061), Sodium pyruvate (Invitrogen 11360070), MEM Non-Essential Amino Acids (Thermo Fisher Scientific, 11140076), antibiotic-antimytotic (Gibco), N21-MAX (R&D systems AR012), Trace Elements B (Corning 25-022-CI), 5μg/ml Insulin (Sigma-Aldrich I9278), 5mg/ml N-acetyl cysteine (Sigma-Aldrich A9165), 10ng/ml PDGFAA (Shenandoah Biosciences 200-54), 10ng/ml CNTF (PeproTech 450-13), and 1ng/ml NT-3 (PeproTech 450-03). For the proliferation studies, after 3 days in proliferative media, cells were treated with various concentrations of either recombinant BDNF (PeproTech 450-02), LM22A-4 (F. Longo), or appropriate vehicles, in the presence of 10 μM EdU for 24 h. This proliferation assay was adapted from (Van’t Veer et al., 2009). Cells were then fixed with 4% PFA for 30 min, incubated with DAPI for 5 min (1:1,000; Thermo Fisher Scientific) and mounted with ProLong Gold mounting medium (Life Technologies).
For the oligodendrogenesis studies, after 3 days in proliferative media, cells were changed to ‘differentiation media’, containing all the same factors as above but from which PDGFAA, CNTF and NT-3 were removed and T3 was added. Cells were then treated with either 50nM BDNF, 100nM LM22A-4 or vehicle for 48 h. This “differentiation” assay was adapted from (Du et al., 2006). Following the final treatment, cells were fixed for 30 min with 4% PFA, incubated with DAPI for 5 min (1:1,000; Thermo Fisher Scientific) and mounted with ProLong Gold mounting medium (Life Technologies). All in vitro experiments were performed in duplicate wells (technical replicates) and then independently replicated (biological replicates).
Generation of hiPSC-derived oligodendrocyte lineage cells in human oligodendrocyte-lineage spheroids
Human oligodendrocyte-lineage spheroids (hOLS) were generated from human induced pluripotent stem cell (hiPS) cells using a variation of the protocol described in (Marton et al., 2019). Cells were plated on vitronectin-coated plates (5 μg/mL, Thermo Fisher Scientific, A14700) in Essential™ 8 media (Thermo Fisher Scientific, A1517001). Cells were passaged every 4 days with 0.5 mM EDTA (Life Technologies, pH 8.0).
For the generation of 3D spheroids, hiPS cells were incubated with Accutase (Innovate Cell Technologies, AT104) at 37°C for 7 min and dissociated into single cells. To obtain uniformly-sized spheroids, AggreWell-800™ (Stemcell Technologies, 34815) containing 300 microwells were used. Approximately 3 × 106 single cells were added per AggreWell-800™ well in Essential™ 8 medium supplemented with the ROCK inhibitor Y-27632 (10 μM, EMD Chemicals, S1049), centrifuged to capture the cells in the microwells and incubated at 37°C with 5% CO2. After 18-24 h, spheroids from each microwell were harvested by pipetting medium in the well up and down and transferred into ultra-low attachment plastic dishes (Thermo Fisher Scientific, 3262) in Essential™ 6 medium (Thermo Fisher Scientific, A1516401) supplemented with two SMAD pathway inhibitors– dorsomorphin (2.5 μM, Sigma-Aldrich, P5499-CONF) and SB-431542 (10 μM, R&D systems, 1614). For the first five days, Essential™ 6 medium was changed every day and supplemented with dorsomorphin and SB-431542.
To generate hOLS, on the sixth day in suspension the spheroids were transferred to glial medium containing DMEM/F12 (Thermo Fisher Scientific, 11330-057), B-27 Supplement without vitamin A (Thermo Fisher Scientific, 12587010), N-2 Supplement (Thermo Fisher Scientific, 17502048), MEM Non-Essential Amino Acids (1:100, Thermo Fisher Scientific, 11140076), GlutaMax (1:100, Thermo Fisher Scientific, 35050079), human insulin solution (25 μg/mL, Sigma-Aldrich, I9278-5ML), and Penicillin-streptomycin (1:100, Thermo Fisher Scientific, 15140163). The glial medium was supplemented with 20 ng/ml EGF (R&D Systems, 236-EG-01M) and 20 ng/ml bFGF (Peprotech, 100-26) for 19 days (until day 24) with daily medium change in the first 10 days, and every other day for the subsequent 9 days. Retinoic acid (RA, 100 nM, Sigma Aldrich, R2625) was added from day 3 until day 24, and the SHH pathway agonist SAG (smoothened agonist, 1 μM, Millipore Sigma, 566660) was added from day 12 to day 24. From day 25 to day 36, hOLS were cultured in glial medium supplemented with T3 (60 ng/mL, Sigma Aldrich, T2877), Biotin (100 ng/mL, Sigma Aldrich, B4639), NT3 (20 ng/mL, Peprotech, 450-03), BDNF (20 ng/mL, Peprotech, 450-02), cAMP (1 μM, Sigma Aldrich, D0627), HGF (5 ng/mL, Peprotech, 315-23), IGF-1 (10 ng/mL, VWR, 100-11), and PDGFaa (10 ng/mL, R&D Systems, 221-AA). Beginning on day 37, hOLs were cultured in complete glial media (glial media supplemented with T3, Biotin, cAMP, and ascorbic acid (AA, 20 μg/mL, Wako Pure Chemical, 323-44822) for the duration of culture. From day 17 to day 43 media changes took place every other day. From day 44 onwards media changes took place every 4 days. All hiPSC experiments were performed in multiple wells (2 technical replicates) and then independently replicated (2 biological replicates). Two hiPSC lines (S. Paca) were used for the two biological replicates of these experiments; one hiPSC line was male (line 8858-3) and one hiPSC line was female (line 0410-1). To authenticate the hiPSC lines used, hiPS cell lines were assessed for pluripotency and genomic integrity (by Cyto-SNP arrays).
Method Details:
Perfusion and immunohistochemistry
Mice were anesthetized with intraperitoneal injections of 2.5% Avertin (tribromoethanol; Sigma T48402) and transcardially perfused with 20ml 0.1M PBS. Brains were fixed in 4% PFA overnight at 4°C, before being transferred to 30% sucrose for cryoprotection. Brains were embedded in Tissue-Tek (Sakura 25608-930) and sectioned coronally at 40mm using a sliding microtome (Microm HM450; Thermo Scientific). For immunohistochemistry, a 1 in 6 series of 40-μm coronal sections was first stained using the Click-iT EdU cell proliferation kit and protocol (Life Technologies C10339), then incubated in 3% normal donkey serum in 0.3% Triton X-100 in TBS blocking solution at room temperature for one hour. Goat anti-PDGFRα (1:500; R&D Systems AF1062), rabbit anti-TrkB (1:100; Santa Cruz Biotechnology sc-8316) mouse anti-CC1 (1:100; EMD Millipore OP80), mouse anti-O4 (1:500; Millipore MAB345), rabbit anti-PDGFRα (1:200, Cell Signaling Technology 5241; for human iPSC analysis) and rat anti-MBP (1:200; Abcam ab7349) were diluted in 1% normal donkey serum in 0.3% Triton X-100 in TBS and incubated overnight at 4°C (CC1 was incubated for 7 days at 4°C as previously described (Gibson et al., 2014; Gibson et al., 2019)). Sections were then rinsed three times in 1X TBS and incubated in secondary antibody solution (Alexa 647 donkey anti-mouse IgG, 1:500 (Jackson Immunoresearch 715-605-150); Alexa 488 donkey anti-rabbit IgG, 1:500 (Jackson Immunoresearch 711-545-152); Alexa 647 donkey anti-goat IgG, 1:500 (Thermo Fisher Scientific A-21447)) in 1% blocking solution at 4°C overnight. The next day, sections were rinsed 3 times in TBS, incubated with DAPI for 5 min (1:1000; Thermo Fisher Scientific 62247) and mounted with ProLong Gold mounting medium (Life Technologies P36930).
Confocal imaging and quantification
All cell counting was performed by experimenters blinded to experimental conditions and genotype on a Zeiss LSM700 scanning confocal microscope (Zeiss). Images were taken at 400X magnification and analyzed using Zen 2011 software. Z-stacks were acquired and a maximum intensity image was generated for each frame. Cells were considered co-labeled when two immunofluorescent markers co-localized in the same plane. For each mouse, 400-600 cells were counted for each immunohistochemical marker analysis. In all optogenetic stimulation studies, three consecutive sections ranging approximately from bregma +1.1 to +0.8 mm surrounding the ferrule placement were selected for imaging. For each section, the cingulum and genu of the corpus callosum were identified, and four standardized 159.9μm × 159.9μm fields were selected in those two areas for quantification (Figure S3B). These standardized regions contain the projections of the layer V premotor projection neurons stimulated optogenetically. All EdU+/PDGFRα+ cells and CC1+ cells with and without EdU were counted in those regions. For lysolecithin studies, three consecutive slices ranging from approximately +1.1 to +0.8 mm anterior to bregma were selected, and two 159.9μm × 159.9μm frames were selected within the lesioned area of the cingulum for quantification. All EdU+/ PDGFRα+ cells and CC1+ cells with and without EdU were counted within those regions. The density of cells was determined by dividing the total number of cells quantified for each lineage by the total volume of the imaged frames (mm3).
Electron Microscopy
Mice were sacrificed by transcardial perfusion with Karnovsky’s fixative: 2% glutaraldehyde (EMS 16000) and 4% paraformaldehyde (EMS 15700) in 0.1M sodium cacodylate (EMS 12300), pH 7.4. Region containing premotor cortex and projections to the corpus callosum was resected from the brain and post-fixed in Karnovsky’s fixative for at least 2 weeks. Transmission electron microscopy was performed in the region of the premotor projections entering the corpus callosum at the level of the cingulum. The samples were post-fixed in 1% osmium tetroxide (EMS 19100) for 1 hr at room temperature, washed 3 times with ultrafiltered water, then en bloc stained for 2 hrs at room temperature. Samples were then dehydrated in ethanol (50%, 75%, and 95%) for 15 min each at 4°C. The samples were then allowed to equilibrate to room temperature and were rinsed in 100% ethanol 2 times, followed by acetonitrile for 15 min. Samples were infiltrated with EMbed-812 resin (EMS 14120) mixed 1:1 with acetonitrile for 2 hrs followed by 2:1 EMbed-812:acetonitrile for 2 hrs. The samples were then placed into EMbed-812 for 2 hrs, then placed into TAAB capsules filled with fresh resin, which were then placed into a 65°C oven overnight. Sections were cut at 75 to 90 nm thickness on a Leica Ultracut S (Leica, Wetzlar, Germany) and mounted on Formvar/carbon coated slot grids (EMS FCF2010-Cu) or 100 mesh Cu grids (EMS FCF100-Cu). Grids were contrast stained for 30 sec in 3.5% uranyl acetate in 50% acetone followed by staining in 0.2% lead citrate for 30 sec. Samples were imaged using a JEOL JEM-1400 TEM at 120kV and images were collected using a Gatan Orius digital camera. With experimenters blinded to sample identity and condition, g-ratios were measured by dividing the axonal diameter by the diameter of the entire fiber (diameter of axon/diameter of axon + myelin sheath) at 4000X using ImageJ software. For each animal, approximately 100 axons were scored. Statistical analyses were calculated using the mean g-ratio per mouse, with each mouse representing one data point.
Enzyme-linked immunosorbent assay (ELISA)
For Bdnf protein analysis, animals were sacrificed at P21 for the TMKI experiment, and at P63 for the MTX chemotherapy experiments. As Bdnf levels vary over the estrus cycle in mice (Jezierski and Sohrabji, 2000), only male mice were used for Bdnf analysis. Animals were rapidly decapitated and the deep frontal cortex and corpus callosum were microdissected in Hibernate-A medium (Thermo Fisher Scientific A12475-01). Tissue was lysed using RIPA buffer and protease inhibitors. Lysates were incubated on ice for 30 min, then centrifuged at 14,000g for 15 min at 4°C. Quantification of Bdnf protein levels were determined by using a Total BDNF Quantikine ELISA kit (R&D Systems DBNT00), performed according to the manufacturer’s instructions. One sample in each experiment was not included in the dataset because it fell below the level of detection of the kit.
Western Blots
Western blot analyses were used to probe for protein levels present in the same microdissected brain homogenate used above in ELISA analysis. All samples were normalized to protein concentration, mixed with NuPAGE Sample Reducing Agent (10X; Invitrogen NP0009) and NuPAGE LDS sample buffer (4X; Invitrogen NP0007), incubated at 98°F for 10 min, and loaded onto 4-15% BioRad Mini-Protean TGX precast gels (Biorad 4561086). Protein was transferred to PVDF membranes (iBlot™ 2 Transfer Stacks, PVDF; Thermo Fisher Scientific IB24001) and blocked with 5% bovine serum albumin (BSA; Sigma A3294) in TBST for 1 h. Rabbit anti phospho-TrkB tyr705 (1:1,100; Signalway Antibody 11328), rabbit anti-TrkB (1:2,000; EMD Millipore 07-225), rabbit anti-p44/42 ERK1/2 (1:1,000; Cell Signaling 9102S) and rabbit anti-phospho-p44/42 ERK1/2 (1:1,000; Cell Signaling 4370S) diluted in 1% BSA/TBST and incubated with the membrane overnight. Secondary anti-rabbit conjugated to horseradish peroxidase (HRP) (BioRad 7074S) was then added for 1 h (1:1,000). Proteins were visualized using Clarity ECL Western Substrate (BioRad 1705061) and quantified and analyzed using Image Studio Lite software (Li-Cor).
RNAscope
Animals were sacrificed at P62-64 and perfused with HBSS. Brains were removed and immediately placed in OCT and frozen in liquid nitrogen. Tissue was stored at −80°C until sectioned on a cryostat (Leica CM3050S) at a thickness of 16 μm. Before performing RNAscope, slides were transferred directly from −80°C to 4% PFA/HBSS on ice for 15 min. Sections were then successively dehydrated in 50% ethanol, 70% ethanol, and twice in 100% ethanol for 5 min each and allowed to dry before treatment for 5 min with Protease IV from the RNAscope Fluorescent Multiplex Reagent Kit (Advanced Cell Diagnostics, 320851). Slides were washed twice with HBSS before proceeding to probe hybridization. RNAscope was performed according to Fluorescent Multiplex Reagent Kit protocol using RNAscope probes against BDNF (Advanced Cell Diagnostics, 424821), RbFox3 (NeuN; Advanced Cell Diagnostics 313311-C2) and Slc1a3 (Glast; Advanced Cell Diagnostics, 320851-C3). Slides were mounted in Prolong Gold and imaged on a Zeiss LSM700 at 400X magnification within 72 h of hybridization. Z-stacks were acquired for four counting frames in the deep cortical levels of the frontal cortex and a maximum intensity image was generated for each image. The number of BDNF puncta per DAPI+ cell was quantified by a blind rater for each image. Bdnf puncta+ cell fraction was determined using CellProfiler software. A DAPI+ cell was considered to have high Bdnf expression if the intensity of Bdnf puncta expression was greater than the median Bdnf intensity of all DAPI+ cells quantified within the imaging frame. All DAPI+ cells that exhibited BDNF puncta intensity lower than the median were considered low Bdnf expressing cells.
Novel object recognition Test
Cognition was analyzed using a modified version of the novel object recognition task (NORT) that focused on the attentional component of the task. The test was modified so that the duration between the training and testing phase was shortened, to ensure that the test placed a greater cognitive load on short-term (< 5 min) memory, attention and frontal lobe function rather than longer-term memory and hippocampal function. In the OPC-TrkB cKO model, animals were exposed to tamoxifen from P31-35, and then tested on P63 for the P35 group, or given tamoxifen from P56-60 and tested on P89 for the P60 group. In the LM22A-4 studies, all mice were exposed to the same MTX/PBS paradigm at P21-35 as described above, tamoxifen was administered from P35-37 and then mice received either LM22A-4 or vehicle control daily from P38-P63 (100 mg/kg, i.p.). Animals were handled daily for the week leading up to the test for 2 min each day. On the day before testing, the mice were placed in the experimental chamber to acclimatize for 20 min. The set up consisted of an opaque Plexiglas experimental chamber (61cm × 61cm × 61cm), and a camera mounted 115 cm above the chamber. The test was conducted during the animal’s rest phase, in a dark room illuminated with red light. On the day of testing, the mice were handled for 2 min and then placed in the chamber to acclimate for 20 min before being returned to the home cage for another 5 min. Mice were then placed in the experimental chamber with two identical Lego objects (each approximately 5 cm in size). Each time the mouse was placed into the chamber it was facing the opaque wall, with the animal’s tail in the direction of the objects. For the training phase, the mouse was allowed to explore the two identical objects for 5 min and then the mouse was returned to the home cage for 5 min while the experimental chamber and objects were cleaned with 70% ethanol. During this time, one cleaned object from the sample phase was placed back into the experimental chamber along with a new Lego object (of similar size) for the novel object phase. For the novel object testing phase, the mouse was returned to the experimental chamber and allowed to explore for 10 min. The objects used as novel and familiar were counterbalanced, as was the position of the novel object from trial to trial, animal to animal. All of the Lego objects used in the behavioral paradigm were piloted to ensure that there was no bias, or object preference for the animals. The camera footage was then analyzed (using CowLog analysis software), and any exploratory head gesturing within 2 cmof the Lego object, including sniffing and biting, was considered object investigation, but sitting on the object, or casual touching of the object in passing was not considered object investigation (Leger et al., 2013). Only animals that explored the objects for a minimum of 20 s were included in the analysis. After the testing was completed the mice were weighed, as a measure of health.
Slow angled descent forepaw grasping (SLAG)
To measure vision in OPC-TrkB cKO mice (adult recombination paradigm), the SLAG task was utilized. At P90, both TrkB WT and OPC-TrkB cKO mice were held roughly 20 cm above a wire-bar cage-lid by the tail. The mouse is first oriented facing the lid and slowly lowered over and past the lid. Once the mouse begins to descend past the cage lid, a mouse with intact vision will stretch its forepaws towards the lid (SLAG+), while a mouse with impaired or no vision will not (SLAG-). Mice are placed briefly on the lid after the test. The test is immediately repeated, however this time the mouse is facing away from the cage lid. The mouse loses sight of the lid as it descends, and a SLAG+ mouse should twist around towards the lid and reach out their forepaws, while SLAG- mice do not. Determination of SLAG+ or SLAG- was made at the time of the task by an experimenter blinded to the genetic background of the mice. All animals exhibited intact vision.
Intellicage Activity Chamber
To measure distance traveled and number of rears, mice were introduced to the Intellicage activity chamber. The Intellicage is a fully automated testing chamber that uses RFID readers to track tagged mice over a period of time in the chamber, capturing both overall locomotion and rearing. This allows multiple mice to be tested simultaneously, reducing effects of anxiety on behaviors. Mice were handled daily for 2 min for one week, and then were introduced to the chamber and allowed to explore freely for 10 min. All data collection is fully automated and free of experimenter input. OPC-TrkB cKO mice tested in the Intellicage were given tamoxifen at P56-60 and tested on P89.
Quantification and Statistical Analyses:
For all quantifications, experimenters were blinded to sample identity and condition. For EM quantification of g-ratio, g-ratios were measured by dividing the axonal diameter by the diameter of the entire fiber (diameter of axon/diameter of axon + myelin sheath) at 4000X using ImageJ software; approximately 100 axons were scored for each mouse. For fluorescent immunohistochemistry and confocal microscopy, images were taken at 400X and cells considered co-labeled when markers co-localized within the same plane. Three sections per mouse, four frames in standardized locations per section (12 sections per mouse) were counted. For each mouse, 400-600 cells were counted for each immunohistochemical marker analysis. The density of cells was determined by dividing the total number of cells quantified for each lineage by the total volume of the imaged frames (mm3). In all experiments, “n” refers to the number of mice with the exception of in vitro experiments for which “n” refers to number of wells. In every experiment, “n” equals at least 3 mice per group or wells per condition; the exact “n” for each experiment is defined in the figure legends.
All statistics were performed using Prism Software (Graphpad). For all analyses involving comparisons of one, two or three variables, 1-way, 2-way or 3-way ANOVAs, respectively, were used with Tukey’s multiple comparisons post-hoc corrections used to assess main group differences. For analyses involving only two groups, unpaired two-tailed Student’s t-tests were used. Shapiro-Wilk test was used to determine normality for all data sets; all data sets were parametric. A level ofp < 0.05 was used to designate significant differences.
Supplementary Material
Supplemental Fig 1. Failure of adaptive myelination in a mouse model of chemotherapy-induced neurological dysfunction, related to Figure 1
These data represent an independent replicate of the data presented in Figure 1. In this experiment, the tissue processed for transmission electron microscopy (TEM) exhibited more split artifact than typically observed. Given the technical issue, we replicated the experiment entirely (presented in Figure 1). The data presented here in Fig. S1 demonstrated the same failure of activity-regulated myelination following methotrexate as observed in the more technically sound analyses in Fig. 1.
A-B) TEM was performed one month following the cessation of the 7-day optogenetic stimulation paradigm in Thy1::ChR2+/− mice that were either stimulated or identically manipulated with the exception of blue light exposure (mock-stimulated controls). These Thy1::ChR2+/− mice were previously exposed to MTX or PBS vehicle control, resulting in four experimental groups. The g-ratio data are shown as a function of axon caliber as a scatterplot of all axons measured in (A) PBS vehicle control-treated, mock-stimulated mice (n= 6 mice; black triangles) compared to PBS vehicle control-treated, optogenetically stimulated mice (n= 5 mice; red triangles) and in (B) MTX-treated, unstimulated mice (n= 4 mice; black triangles) compared to MTX-treated, optogenetically stimulated mice (n= 3 mice; red triangles). A single point indicates the g-ratio for a single axon. Approximately 100 axons were quantified and the mean g-ratio determined for each mouse. P-values (indicated on plots) were determined by a two-way ANOVA with Bonferoni correction for multiple comparisons, comparing the mean g-ratio per mouse between groups.
C) Representative TEM images of premotor projections entering the subjacent corpus callosum at the level of the cingulum. Scale bars=2μm.
Supplemental Fig 2. Verification of OPC conditional knock out of TrkB and standardized region of cell density quantification, related to Figure 3, S3
A) Representative confocal images of TrkB (red) expression in PDGFRa+ OPCs (green), assessed by immunohistochemistry. TrkB is expressed in TrkB WT OPCs (left), and following tamoxifen induction TrkB expression is lost from OPCs in TrkB cKO mice (right). Scale bar = 10μm
B) Standardized fields (blue squares) sampled in the corpus callosum for all cell density measurements.
Supplemental Fig 3. BDNF to TrkB signaling is necessary for activity-regulated oligodendrogenesis, related to Figures 3, 4 and S2
B) Density of EdU-marked, oligodendrocytes (EdU+/CC1+ cells) in corpus callosum of BdnfTMKI;Thy1::ChR2+/− mice (n=6), BdnfTMKI; WT mice (n=5), BdnfWT;Thy1::ChR2+/− mice (n = 5) and BdnfWT;WT mice (n=6) at 4 weeks after completion of the one-week optogenetic stimulation paradigm.
C) Density of total CC1+ oligodendrocytes in the corpus callosum of BdnfTMKI;Thy1::ChR2+/− mice (n=6), Bdnj™TMKI ;WT mice (n=5), BdnfWT;Thy1::ChR2+/− mice (n = 5) and BdnfWT;WT mice (n=6) at 4 weeks after completion of the optogenetic stimulation paradigm.
D) Density of EdU-marked oligodendrocytes (EdU+/CC1+ cells) in the corpus callosum of OPC TrkB cKO;Thy1::ChR2+/− mice (n=6), OPC TrkB cKO;WT (no opsin) mice (n=6), TrkB WT;Thy1::ChR2+/− mice (n=6) and TrkB WT;WT (no opsin) mice (n=6) at 4 weeks after completion of the optogenetic stimulation paradigm.
E) Density of total CC1+ oligodendrocytes in the corpus callosum of OPC TrkB cKO;Thy1::ChR2+/− mice (n=6), OPC TrkB cKO;WT (no opsin) mice (n=6), TrkB WT;Thy1::ChR2+/− mice (n=6) and TrkB WT;WT (no opsin) mice (n=6) at 4 weeks after completion of the 7-day optogenetic stimulation paradigm.
F) Representative confocal images of oligodendrocytes (CC1+, green) co-localized with EdU (red) in the corpus callosum for each group described above Scale bar = 20μm.
Data shown as mean ± SEM. Each point = one mouse. ns = p > 0.05, *p<0.05, **p < 0.01 ****p<0.0001. Two-way ANOVA with Tukey post-hoc analysis for multiple comparisons.
Supplemental Fig 4. BDNF to TrkB signaling is necessary for activity-regulated myelination, related to Figure 4
Transmission electron microscopy (TEM) was performed 4-weeks following the cessation of the one-week optogenetic stimulation paradigm. Myelin sheath thickness was analyzed at the level of the cingulum of the corpus callosum as the g-ratio (diameter of axon divided by the diameter of axon plus myelin sheath; a smaller g-ratio indicates a thicker myelin sheath). Scatterplots of g-ratio as a function of axon caliber are shown comparing optogenetically stimulated mice vs. identically manipulated WT controls for each genotype studied
A) A scatterplot of all axons measured shows an increase in myelin sheath thickness (reduced g-ratio) in BdnfWT;Thyl::ChR2+/− (n=5 mice; red triangles) mice compared to BdnfWT;WT (n=6 mice; black triangles).
B) A scatterplot of all axons measured show no difference in g-ratio between BdnfTMKI;WT (n=6 mice; black triangles) vs. BdnfTMKI;Thy1::ChR2+/− (n=5 mice; red triangles).
C) G-ratio data of BdnfTMKI model are shown by axon size bins of small (<0.5μm), medium (0.5-1.0 μm) and large (>1.0μm) axons.
D) A scatterplot of all axons measured shows an increase in myelin sheath thickness (reduced g-ratio) in TrkB WT;WT (n=7 mice; black triangles) compared to TrkB WT;Thy1::ChR2+/− (n=6 mice; red triangles).
E) A scatterplot of all axons measured show no difference in g-ratio between OPC-TrkB cKO;WT (n=7 mice; black triangles) compared to OPC-TrkB cKO;Thy1::ChR2+/− mice (n=5 mice; red triangles).
F) G-ratio data of OPC-TrkB cKO model are shown by axon size bins of small (<0.5μm), medium (0.5-1.0 μm) and large (>1.0μm) axons.
A single point indicates the g-ratio for a single axon. Approximately 100 axons were quantified and the mean g-ratio determined for each mouse. P-values (indicated on plots) were determined by two-way ANOVA with Tukey post-hoc analysis for multiple comparisons, comparing the mean g-ratio per mouse between groups.
Supplemental Fig 5. A small molecule TrkB partial agonist recapitulates the effects of BDNF on OPC proliferation and oligodendrogenesis, related to Figure 6
A) Six-point dose response curve demonstrating OPC proliferation index as measured by the fraction of PDGFRα+cells that co-label with EdU in P6-8 mouse OPCs at 24 hours after exposure to recombinant BDNF at a 0 to 1000 nM concentration range (n=6 wells per condition).
B) Four-point dose response curve demonstrating OPC proliferation index as measured by the fraction of PDGFRα+ cells that co-label with EdU in P6-8 mouse OPCs at 24 hours after exposure to LM22A-4 at a 0 to 1000 nM concentration range (n=6 wells per condition).
A-B) Representative confocal images (below) of PDGFRα+ (green), EdU+ (red) and DAPI+ (blue) vehicle control, BDNF- and LM22A-4 -treated cells. Scale bar=50μm.
C) Percent of total DAPI+ cells that express O4+ and do not express PDGFRα+ increases at 48-hours after treatment with BDNF (50nM) or LM22A-4 (100nM) of P6-8 mouse OPCs (n=4 wells per condition). Representative confocal images (below) of PDGFRα+ (red), O4+ (red) and DAPI+ (blue) of vehicle control (left) and LM22A-4-treated (right) cells. Scale bar =20μm
D) Human iPS cells were treated with one of three treatment paradigms from day 25-35 in the OPC induction protocol: no BDNF, 20ng/ml BDNF as per standard protocol, or BDNF was replaced with 1000nM LM22A-4. Cells were fixed at day 100 following induction and the percent of DAPI+ cells expressing the OPC marker PDGFRα was quantified.
E) Representative image of a human iPSC-derived PDGFRα+ OPC. Green = PDGFRα, white = DAPI. Scale bar =10μm.
F) Human iPS cells were treated with one of three treatment paradigms from day 25-35 in the OPC induction protocol: no BDNF, 20ng/ml BDNF as per standard protocol, or BDNF was replaced with 1000nM LM22A-4. Cells were fixed at day 100 following induction and the percent of DAPI+ cells expressing the mature oligodendrocyte marker MBP was quantified.
G) Representative image of a human iPSC-derived MBP+ oligodendrocyte. Red = MBP, white = DAPI. Scale bar = 10μm.
H) Representative image of lysolecithin-induced demyelination in the corpus callosum 10 days after injection. Green = fluoromyelin stain to demonstrate myelinated architecture. White-dashed circle = lesioned area in the corpus callosum. Scale bar = 100μm.
I) Density of EdU+/PDGFRα+ proliferating OPCs in the corpus callosum of mice treated with 50mg/kg LM22A-4 or vehicle control for 7 consecutive days beginning 7 days after lysolecithin injection (n=5 mice in each group). Representative confocal images (at right) of PDGFRα+ (green) and EdU+ (red) in lesioned area of vehicle control (left) and LM22A-4 - treated (right) mice. Scale bar = 20μm.
J) Density of CC1+ oligodendrocyte cells in the corpus callosum of mice treated with 50mg/kg LM22A-4 or vehicle control for 14 consecutive days beginning 7 days after lysolecithin injection (n=5 mice in each group). Representative confocal images (at right) of CC1+ (green) in lesioned area of vehicle control (left) and LM22A-4 -treated (right) mice. Scale bar = 20μm.
Data shown as mean ± SEM. Each point = one mouse (I-J). ns = p > 0.05, * p < 0.05, ** p < 0.01, *** p<0.001, **** p<0.0001 as determined by one-way ANOVA (A-F) or Student’s t-test (I-J).
Supplemental Fig 6. TrkB partial agonist LM22A-4 normalizes myelination after methotrexate chemotherapy exposure, related to Figure 6
A) MTX-treated mice exhibit thinner myelin compared to PBS vehicle-treated controls. g-ratio values as a function of axon caliber are shown as a scatterplot of all axons measured in mice treated with PBS vehicle control+vehicle control (n=4 mice; red triangles) vs. MTX+vehicle control (n=4 mice; black triangle).
B) LM22A-4 increases myelin sheath thickness in control mice. g-ratio values as a function of axon caliber are shown as a scatterplot of all axons measured in mice treated with PBS vehicle control+vehicle control (n=4 mice; black triangle) vs PBS vehicle control +LM22A-4 (n=4 mice; red triangle).
C) G-ratio data shown in Figure 6 are shown by axon size bins of small (<0.5μm), medium (0.5-1.0 μm) and large (>1.0μm) axons. LM22A-4 rescues myelin deficits in mice exposed to juvenile MTX in small caliber axons (< 0.5μm; n=4 mice, p = 0.005), medium caliber axons (0.5μm - 1.0 μm; n= 4, p = 0.006) and large caliber axons (>1.0 μm; n=4 mice; p = 0.033) compared to mice exposed to MTX but treated with vehicle control. Data shown as mean ± SEM, * p < 0.05, ** p < 0.01, *** p<0.001 ns = p > 0.05. Two-way ANOVA with Tukey post-hoc multiple comparisons testing.
A single point indicates the g-ratio for a single axon. Approximately 100 axons were quantified per mouse and the mean g-ratio determined for each mouse. P-values (indicated on plots) were determined two-way ANOVA with Tukey post-hoc multiple comparisons testing comparing the mean g-ratio per mouse between groups.
Highlights.
Methotrexate (MTX) causes a microglia-dependent reduction in Bdnf expression
Activity-regulated myelination requires Bdnf-TrkB signaling and fails after MTX
Conditional, inducible TrkB loss in OPCs impairs cognitive behavioral performance
TrkB agonism rescues cognitive performance after MTX only if OPCs express TrkB
Acknowledgments:
Special thanks to Sigrid Knemeyer for illustrations. We thank Tracy Yuen for her help with the lysolecithin model. Funding: We acknowledge support from the California Institute for Regenerative Medicine (CIRM RB4-06093 and RN3-06510), Anne T. and Robert M. Bass Endowed Faculty Scholarship in Pediatric Cancer and Blood Diseases, National Institute of Neurological Disorders and Stroke (R01NS092597), Unravel Pediatric Cancer, McKenna Claire Foundation, Liwei Wang Research Fund, Virginia and D.K. Ludwig Fund for Cancer Research, the Maternal and Child Health Research Institute at Stanford, Stanford BioX/ Genetech Foundation Postdoctoral Fellowship, McCormick Fellowship, Stanford BioX undergraduate research fellowships.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests: Dr. Longo is listed as an inventor on patents relating to LM22A-4, which are assigned to UNC-Chapel Hill and UCSF. Dr. Longo is eligible for royalties distributed by the assigned universities. Dr. Longo also has financial interest in PharmatrophiX, a company focused on the development of small molecule ligands for neurotrophin receptors, which has licensed several of these patents. Drs. Longo and Monje are inventors on a patent application, coordinated through Stanford University, regarding use of LM22A-4 to promote myelination in disease.
Data and materials availability: All data is available in the main text or the supplementary materials. LM22A-4 is available through a standard material transfer agreement (MTA) with Pharmatrophix.
Data and Software Availability:
Data Resources: Raw data are available through Mendeley Data: http://dx.doi.org/null/rbv4hwkvc6.1
References
- Arenkiel BR, Peca J, Davison IG, Feliciano C, Deisseroth K, Augustine GJ, Ehlers MD, and Feng G (2007). In vivo light-induced activation of neural circuitry in transgenic mice expressing channelrhodopsin-2. Neuron 54, 205–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aukema EJ, Caan MW, Oudhuis N, Majoie CB, Vos FM, Reneman L, Last BF, Grootenhuis MA, and Schouten-van Meeteren AY (2009). White matter fractional anisotropy correlates with speed of processing and motor speed in young childhood cancer survivors. Int J Radiat Oncol Biol Phys 74, 837–843. [DOI] [PubMed] [Google Scholar]
- Balkowiec A, and Katz DM (2000). Activity-dependent release of endogenous brain-derived neurotrophic factor from primary sensory neurons detected by ELISA in situ. J Neurosci 20, 7417–7423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA, and Raff MC (1993). Proliferation of oligodendrocyte precursor cells depends on electrical activity in axons. Nature 361, 258–260. [DOI] [PubMed] [Google Scholar]
- Barthas F, and Kwan AC (2017). Secondary Motor Cortex: Where ‘Sensory’ Meets ‘Motor’ in the Rodent Frontal Cortex. Trends Neurosci 40, 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechler ME, Byrne L, and Ffrench-Constant C (2015). CNS Myelin Sheath Lengths Are an Intrinsic Property of Oligodendrocytes. Current Biology 25, 2411–2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechler ME, Swire M, and Ffrench-Constant C (2018). Intrinsic and adaptive myelination-A sequential mechanism for smart wiring in the brain. Dev Neurobiol 78, 68–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergles DE, Roberts JD, Somogyi P, and Jahr CE (2000). Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature 405, 187–191. [DOI] [PubMed] [Google Scholar]
- Bichot NP, Heard MT, DeGennaro EM, and Desimone R (2015). A Source for Feature-Based Attention in the Prefrontal Cortex. Neuron 88, 832–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratlid D, and Moe PJ (1978). Pharmacokinetics of high-dose methotrexate treatment in children. European journal of clinical pharmacology 14, 143–147. [DOI] [PubMed] [Google Scholar]
- Cellerino A, Carroll P, Thoenen H, and Barde YA (1997). Reduced size of retinal ganglion cell axons and hypomyelination in mice lacking brain-derived neurotrophic factor. Mol Cell Neurosci 9, 397–408. [DOI] [PubMed] [Google Scholar]
- Chen TW, Li N, Daie K, and Svoboda K (2017). A Map of Anticipatory Activity in Mouse Motor Cortex. Neuron 94, 866–879 e864. [DOI] [PubMed] [Google Scholar]
- Cohen MR, and Maunsell JH (2011). Using neuronal populations to study the mechanisms underlying spatial and feature attention. Neuron 70, 1192–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Biase L, Kang S, Baxi E, Fukaya M, Pucak M, Mishina M, Calabresi P, and Bergles D (2011). NMDA receptor signaling in oligodendrocyte progenitors is not required for oligodendrogenesis and myelination. J Neurosci 31, 12650–12662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deprez S, Amant F, Yigit R, Porke K, Verhoeven J, Van den Stock J, Smeets A, Christiaens MR, Leemans A, Van Hecke W, et al. (2011). Chemotherapy-induced structural changes in cerebral white matter and its correlation with impaired cognitive functioning in breast cancer patients. Hum Brain Mapp 32, 480–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieni S, Matsumoto T, Dekkers M, Rauskolb S, Ionescu MS, Deogracias R, Gundelfinger ED, Kojima M, Nestel S, Frotscher M, et al. (2012). BDNF and its pro-peptide are stored in presynaptic dense core vesicles in brain neurons. The Journal of cell biology 196, 775–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Lercher LD, Zhou R, and Dreyfus CF (2006). Mitogen-activated protein kinase pathway mediates effects of brain-derived neurotrophic factor on differentiation of basal forebrain oligodendrocytes. J Neurosci Res 84, 1692–1702 [DOI] [PubMed] [Google Scholar]
- Ellenberg L, Liu Q, Gioia G, Yasui Y, Packer RJ, Mertens A, Donaldson SS, Stovall M, Kadan-Lottick N, Armstrong G, et al. (2009). Neurocognitive status in long-term survivors of childhood CNS malignancies: a report from the Childhood Cancer Survivor Study. Neuropsychology 23, 705–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett JP, Aloyz R, McLean JH, Pareek S, Miller FD, McPherson PS, and Murphy RA (1997). Detection of brain-derived neurotrophic factor in a vesicular fraction of brain synaptosomes. J Biol Chem 272, 8837–8840. [DOI] [PubMed] [Google Scholar]
- Fulmer CG, VonDran MW, Stillman AA, Huang Y, Hempstead BL, and Dreyfus CF (2014). Astrocyte-derived BDNF supports myelin protein synthesis after cuprizone-induced demyelination. J Neurosci 34, 8186–8196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo V, Zhou JM, McBain CJ, Wright P, Knutson PL, and Armstrong RC (1996). Oligodendrocyte progenitor cell proliferation and lineage progression are regulated by glutamate receptor-mediated K+ channel block. J Neurosci 16, 2659–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber C, Vasek MJ, Vollmer LL, Sun T, Jiang X, and Klein RS (2018). Astrocytes decrease adult neurogenesis during virus-induced memory dysfunction via IL-1. Nature immunology 19, 151–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson EM, Nagaraja S, Ocampo A, Tam LT, Wood LS, Pallegar PN, Greene JJ, Geraghty AC, Goldstein AK, Ni L, et al. (2019). Methotrexate Chemotherapy Induces Persistent Tri-glial Dysregulation that Underlies Chemotherapy-Related Cognitive Impairment. Cell 176, 43–55 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson EM, Purger D, Mount CW, Goldstein AK, Lin GL, Wood LS, Inema I, Miller SE, Bieri G, Zuchero JB, et al. (2014). Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science 344, 1252304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebbels S, Wieser GL, Pieper A, Spitzer S, Weege B, Yan K, Edgar JM, Yagensky O, Wichert SP, Agarwal A, et al. (2017). A neuronal PI(3,4,5)P3-dependent program of oligodendrocyte precursor recruitment and myelination. Nat Neurosci 20, 10–15. [DOI] [PubMed] [Google Scholar]
- Grishanin RN, Yang H, Liu X, Donohue-Rolfe K, Nune GC, Zang K, Xu B, Duncan JL, Lavail MM, Copenhagen DR, et al. (2008). Retinal TrkB receptors regulate neural development in the inner, but not outer, retina. Mol Cell Neurosci 38, 431–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Pollak J, Yang T, Siddiqui MR, Doyle KP, Taravosh-Lahn K, Cekanaviciute E, Han A, Goodman JZ, Jones B, et al. (2012). Delayed administration of a small molecule tropomyosin-related kinase B ligand promotes recovery after hypoxic-ischemic stroke. Stroke 43, 1918–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann M, Heumann R, and Lessmann V (2001). Synaptic secretion of BDNF after high-frequency stimulation of glutamatergic synapses. EMBO J 20, 5887–5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill RA, Li AM, and Grutzendler J (2018). Lifelong cortical myelin plasticity and agerelated degeneration in the live mammalian brain. Nat Neurosci 21, 683–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hines JH, Ravanelli AM, Schwindt R, Scott EK, and Appel B (2015). Neuronal activity biases axon selection for myelination in vivo. Nat Neurosci 18, 683–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong EJ, McCord AE, and Greenberg ME (2008). A biological function for the neuronal activity-dependent component of Bdnf transcription in the development of cortical inhibition. Neuron 60, 610–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes EG, Kang SH, Fukaya M, and Bergles DE (2013). Oligodendrocyte progenitors balance growth with self-repulsion to achieve homeostasis in the adult brain. Nat Neurosci 16, 668–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes EG, Orthmann-Murphy JL, Langseth AJ, and Bergles DE (2018). Myelin remodeling through experience-dependent oligodendrogenesis in the adult somatosensory cortex. Nat Neurosci. 21 (5), 696–706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi T, Dakin KA, Stevens B, Lee PR, Kozlov SV, Stewart CL, and Fields RD (2006). Astrocytes promote myelination in response to electrical impulses. Neuron 49, 823–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janka GJ, Mack R, Helmig M, Haas RJ, and Bidlingmaier F (1984). Prolonged methotrexate infusions in children with acute leukemia in relaspe and in remission and with medulloblastoma: pharmacokinetics, toxicity and clinical results. Oncology 41, 225–232 [DOI] [PubMed] [Google Scholar]
- Jezierski MK and Sohrabji F (2000) Region and peptide-specific regulation of the neurotrophins by estrogen. Brain Res Mol Brain Res 85(1-2), 77–84 [DOI] [PubMed] [Google Scholar]
- Kaiser J, Bledowski C, and Dietrich J (2014). Neural correlates of chemotherapy-related cognitive impairment. Cortex 54, 33–50. [DOI] [PubMed] [Google Scholar]
- Kang S, Fukaya M, Yang J, Rothstein J, and Bergles D (2010). NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron 68, 668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karadottir R, Cavelier P, Bergersen LH, and Attwell D (2005). NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature 438, 1162–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokaia Z, Andsberg G, Yan Q, and Lindvall O (1998). Rapid alterations of BDNF protein levels in the rat brain after focal ischemia: evidence for increased synthesis and anterograde axonal transport. Exp Neurol 154, 289–301. [DOI] [PubMed] [Google Scholar]
- Koppelmans V, Breteler MM, Boogerd W, Seynaeve C, Gundy C, and Schagen SB (2012). Neuropsychological performance in survivors of breast cancer more than 20 years after adjuvant chemotherapy. J Clin Oncol 30, 1080–1086. [DOI] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, and Bonhoeffer T (1995). Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci U S A 92, 8856–8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kougioumtzidou E, Shimizu T, Hamilton NB, Tohyama K, Sprengel R, Monyer H, Attwell D, and Richardson WD (2017). Signalling through AMPA receptors on oligodendrocyte precursors promotes myelination by enhancing oligodendrocyte survival. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukley M, Capetillo-Zarate E, and Dietrich D (2007). Vesicular glutamate release from axons in white matter. Nat Neurosci 10, 311–320. [DOI] [PubMed] [Google Scholar]
- Lee S, Chong SYC, Tuck SJ, Corey JM, and Chan JR (2013). A rapid and reproducible assay for modeling myelination by oligodendrocytes using engineered nanofibers. Nature protocols 8, 771–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leger M, Quiedeville A, Bouet V, Haelewyn B, Boulouard M, Schumann-Bard P, and Freret T (2013). Object recognition test in mice. Nature protocols 8, 2531–2537. [DOI] [PubMed] [Google Scholar]
- Li W, Bellot-Saez A, Phillips ML, Yang T, Longo FM, and Pozzo-Miller L (2017). A small-molecule TrkB ligand restores hippocampal synaptic plasticity and object location memory in Rett syndrome mice. Dis Model Mech 10, 837–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Luikart BW, Birnbaum S, Chen J, Kwon CH, Kernie SG, Bassel-Duby R, and Parada LF (2008). TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron 59, 399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SY, Wu K, Levine ES, Mount HT, Suen PC, and Black IB (1998). BDNF acutely increases tyrosine phosphorylation of the NMDA receptor subunit 2B in cortical and hippocampal postsynaptic densities. Brain Res Mol Brain Res 55, 20–27. [DOI] [PubMed] [Google Scholar]
- Liu J, Dietz K, DeLoyht JM, Pedre X, Kelkar D, Kaur J, Vialou V, Lobo MK, Dietz DM, Nestler EJ, et al. (2012). Impaired adult myelination in the prefrontal cortex of socially isolated mice. Nat Neurosci 15, 1621–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohof AM, Ip NY, and Poo MM (1993). Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature 363, 350–353. [DOI] [PubMed] [Google Scholar]
- Loss CM, Binder LB, Muccini E, Martins WC, de Oliveira PA, Vandresen-Filho S, Prediger RD, Tasca CI, Zimmer ER, Costa-Schmidt LE, et al. (2015). Influence of environmental enrichment vs. time-of-day on behavioral repertoire of male albino Swiss mice. Neurobiol Learn Mem 125, 63–72. [DOI] [PubMed] [Google Scholar]
- Lundgaard I, Luzhynskaya A, Stockley JH, Wang Z, Evans KA, Swire M, Volbracht K, Gautier HO, Franklin RJ, Charles F-C, et al. (2013). Neuregulin and BDNF induce a switch to NMDA receptor-dependent myelination by oligodendrocytes. PLoS Biol 11, e1001743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo TZ, and Maunsell JH (2015). Neuronal Modulations in Visual Cortex Are Associated with Only One of Multiple Components of Attention. Neuron 86, 1182–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinodan M, Rosen KM, Ito S, and Corfas G (2012). A critical period for social experience-dependent oligodendrocyte maturation and myelination. Science 337, 1357–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marton RM, Miura Y, Sloan AE, Li Q, Revah O, Levy R, Huguenard JR, Paşca SP (2019). Differentiation and maturation of oligodendrocytes in human three-dimensional neural cultures. Nature Neuroscience (early online publication, January 28, 2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massa SM, Yang T, Xie Y, Shi J, Bilgen M, Joyce JN, Nehama D, Rajadas J, and Longo FM (2010). Small molecule BDNF mimetics activate TrkB signaling and prevent neuronal degeneration in rodents. J Clin Invest 120, 1774–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayoral SR, Etxeberria A, Shen YA, and Chan JR (2018). Initiation of CNS Myelination in the Optic Nerve Is Dependent on Axon Caliber. Cell Rep 25, 544–550 e543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenzie IA, Ohayon D, Li H, de Faria JP, Emery B, Tohyama K, and Richardson WD (2014). Motor skill learning requires active central myelination. Science 346, 318–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mensch S, Baraban M, Almeida R, Czopka T, Ausborn J, El Manira A, and Lyons DA (2015). Synaptic vesicle release regulates myelin sheath number of individual oligodendrocytes in vivo. Nat Neurosci 18, 628–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesa-Gresa P, Perez-Martinez A, and Redolat R (2013). Environmental enrichment improves novel object recognition and enhances agonistic behavior in male mice. Aggress Behav 39, 269–279. [DOI] [PubMed] [Google Scholar]
- Messier C (1997). Object recognition in mice: improvement of memory by glucose. Neurobiol Learn Mem 67, 172–175. [DOI] [PubMed] [Google Scholar]
- Mitew S, Gobius I, Fenlon LR, McDougall SJ, Hawkes D, Xing YL, Bujalka H, Gundlach AL, Richards LJ, Kilpatrick TJ, et al. (2018). Pharmacogenetic stimulation of neuronal activity increases myelination in an axon-specific manner. Nature communications 9, 306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore T (2006). The neurobiology of visual attention: finding sources. Curr Opin Neurobiol 16, 159–165. [DOI] [PubMed] [Google Scholar]
- Mustafa S, Walker A, Bennett G, and Wigmore PM (2008). 5-Fluorouracil chemotherapy affects spatial working memory and newborn neurons in the adult rat hippocampus. Eur J Neurosci 28, 323–330 [DOI] [PubMed] [Google Scholar]
- Ng T, Teo SM, Yeo HL, Shwe M, Gan YX, Cheung YT, Foo KM, Cham MT, Lee JA, Tan YP, et al. (2016). Brain-derived neurotrophic factor genetic polymorphism (rs6265) is protective against chemotherapy-associated cognitive impairment in patients with early-stage breast cancer. Neuro Oncol 18, 244–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivares R, Montiel J, and Aboitiz F (2001). Species differences and similarities in the fine structure of the mammalian corpus callosum. Brain Behav Evol 57, 98–105. [DOI] [PubMed] [Google Scholar]
- Park H, and Poo MM (2013). Neurotrophin regulation of neural circuit development and function. Nature reviews Neuroscience 14, 7–23. [DOI] [PubMed] [Google Scholar]
- Park H, Popescu A, and Poo MM (2014). Essential role of presynaptic NMDA receptors in activity-dependent BDNF secretion and corticostriatal LTP. Neuron 84, 1009–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park HS, Kim CJ, Kwak HB, No MH, Heo JW, and Kim TW (2018). Physical exercise prevents cognitive impairment by enhancing hippocampal neuroplasticity and mitochondrial function in doxorubicin-induced chemobrain. Neuropharmacology 133, 451–461 [DOI] [PubMed] [Google Scholar]
- Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR 3rd, Lafaille JJ, Hempstead BL, Littman DR, and Gan WB (2013). Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, and Kandel ER (1996). Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron 16, 1137–1145. [DOI] [PubMed] [Google Scholar]
- Pierson C, Waite E, and Pyykkonen B (2016). A meta-analysis of the neuropsychological effects of chemotherapy in the treatment of childhood cancer. Pediatr Blood Cancer 63, 1998–2003. [DOI] [PubMed] [Google Scholar]
- Rizzolatti G, Riggio L, Dascola I, and Umilta C (1987). Reorienting attention across the horizontal and vertical meridians: evidence in favor of a premotor theory of attention. Neuropsychologia 25, 31–40. [DOI] [PubMed] [Google Scholar]
- Rosenberg SS, Kelland EE, Tokar E, De La Torret AR, and Chan JR (2008). The geometric and spatial constraints of the microenvironment induce oligodendrocyte differentiation. P Natl Acad Sci USA 105, 14662–14667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz J, Klein MC, Behrens TE, and Johansen-Berg H (2009). Training induces changes in white-matter architecture. Nat Neurosci 12, 1370–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons DA, Belichenko NP, Yang T, Condon C, Monbureau M, Shamloo M, Jing D, Massa SM, and Longo FM (2013). A small molecule TrkB ligand reduces motor impairment and neuropathology in R6/2 and BACHD mouse models of Huntington’s disease. J Neurosci 33, 18712–18727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Tanner S, and Fields RD (1998). Control of myelination by specific patterns of neural impulses. J Neurosci 18, 9303–9311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic B, Menon V, Nguyen TN, Kim TK, Jarsky T, Yao Z, Levi B, Gray LT, Sorensen SA, Dolbeare T, et al. (2016). Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat Neurosci 19, 335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomassy GS, Berger DR, Chen HH, Kasthuri N, Hayworth KJ, Vercelli A, Seung HS, Lichtman JW, and Arlotta P (2014). Distinct Profiles of Myelin Distribution Along Single Axons of Pyramidal Neurons in the Neocortex. Science 344, 319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi RB, Jackiewicz M, McKenzie IA, Kougioumtzidou E, Grist M, and Richardson WD (2017). Remarkable Stability of Myelinating Oligodendrocytes in Mice. Cell reports 21, 316–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsiperson V, Huang Y, Bagayogo I, Song Y, VonDran MW, DiCicco-Bloom E, and Dreyfus CF (2015). Brain-derived neurotrophic factor deficiency restricts proliferation of oligodendrocyte progenitors following cuprizone-induced demyelination. ASN Neuro 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van’t Veer A, Du Y, Fischer TZ, Boetig DR, Wood MR, and Dreyfus CF (2009). Brain-derived neurotrophic factor effects on oligodendrocyte progenitors of the basal forebrain are mediated through trkB and the MAP kinase pathway. J Neurosci Res 87, 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh HS, Johung TB, Caretti V, Noll A, Tang Y, Nagaraja S, Gibson EM, Mount CW, Polepalli J, Mitra SS, et al. (2015). Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell 161, 803–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh HS, Tam LT, Woo PJ, Lennon J, Nagaraja S, Gillespie SM, Ni J, Duveau DY, Morris PJ, Zhao JJ, et al. (2017). Targeting neuronal activity-regulated neuroligin-3 dependency in high-grade glioma. Nature 549, 533–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldsman M, Mitchell DJ, and Cusack R (2017). The neural basis of precise visual short-term memory for complex recognisable objects. Neuroimage 159, 131–145. [DOI] [PubMed] [Google Scholar]
- von Bartheld CS, Byers MR, Williams R, and Bothwell M (1996). Anterograde transport of neurotrophins and axodendritic transfer in the developing visual system. Nature 379, 830–833. [DOI] [PubMed] [Google Scholar]
- VonDran MW, Clinton-Luke P, Honeywell JZ, and Dreyfus CF (2010). BDNF+/− mice exhibit deficits in oligodendrocyte lineage cells of the basal forebrain. Glia 58, 848–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VonDran MW, Singh H, Honeywell JZ, and Dreyfus CF (2011). Levels of BDNF impact oligodendrocyte lineage cells following a cuprizone lesion. J Neurosci 31, 14182–14190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voronova A, Yuzwa SA, Wang BS, Zahr S, Syal C, Wang J, Kaplan DR, and Miller FD (2017). Migrating Interneurons Secrete Fractalkine to Promote Oligodendrocyte Formation in the Developing Mammalian Brain. Neuron 94, 500–516 e509. [DOI] [PubMed] [Google Scholar]
- Wake H, Lee P, and Fields R (2011). Control of local protein synthesis and initial events in myelination by action potentials. Science 333, 1647–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesel TN, and Hubel DH (1963a). Effects of Visual Deprivation on Morphology and Physiology of Cells in the Cats Lateral Geniculate Body. J Neurophysiol 26, 978–993. [DOI] [PubMed] [Google Scholar]
- Wiesel TN, and Hubel DH (1963b). Single-Cell Responses in Striate Cortex of Kittens Deprived of Vision in One Eye. J Neurophysiol 26, 1003–1017. [DOI] [PubMed] [Google Scholar]
- Wiesel TN, and Hubel DH (1965). Comparison of the effects of unilateral and bilateral eye closure on cortical unit responses in kittens. J Neurophysiol 28, 1029–1040. [DOI] [PubMed] [Google Scholar]
- Wong AW, Xiao J, Kemper D, Kilpatrick TJ, and Murray SS (2013). Oligodendroglial expression of TrkB independently regulates myelination and progenitor cell proliferation. J Neurosci 33, 4947–4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YH, Lee CM, Xie W, Cui B, and Poo MM (2015). Activity-dependent BDNF release via endocytic pathways is regulated by synaptotagmin-6 and complexin. Proc Natl Acad Sci U S A 112, E4475–4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J, Wong AW, Willingham MM, van den Buuse M, Kilpatrick TJ, and Murray SS (2010). Brain-derived neurotrophic factor promotes central nervous system myelination via a direct effect upon oligodendrocytes. Neurosignals 18, 186–202. [DOI] [PubMed] [Google Scholar]
- Xiao L, Ohayon D, McKenzie I, Sinclair-Wilson A, Wright J, Fudge A, Emery B, Li H, and Richardson W (2016). Rapid production of new oligodendrocytes is required in the earliest stages of motor-skill learning. Nat Neurosci 19, 1210–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhar O, Fenno LE, Davidson TJ, Mogri M, and Deisseroth K (2011). Optogenetics in neural systems. Neuron 71, 9–34. [DOI] [PubMed] [Google Scholar]
- Zeisel A, Munoz-Manchado AB, Codeluppi S, Lonnerberg P, La Manno G, Jureus A, Marques S, Munguba H, He L, Betsholtz C, et al. (2015). Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, et al. (2014). An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34, 11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Meyers EM, Bichot NP, Serre T, Poggio TA, and Desimone R (2011). Object decoding with attention in inferior temporal cortex. Proc Natl Acad Sci U S A 108, 8850–8855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, and Desimone R (2011). Feature-based attention in the frontal eye field and area V4 during visual search. Neuron 70, 1205–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zonouzi M, Scafidi J, Li P, McEllin B, Edwards J, Dupree JL, Harvey L, Sun D, Hubner CA, Cull-Candy SG, et al. (2015). GABAergic regulation of cerebellar NG2 cell development is altered in perinatal white matter injury. Nat Neurosci 18, 674–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig 1. Failure of adaptive myelination in a mouse model of chemotherapy-induced neurological dysfunction, related to Figure 1
These data represent an independent replicate of the data presented in Figure 1. In this experiment, the tissue processed for transmission electron microscopy (TEM) exhibited more split artifact than typically observed. Given the technical issue, we replicated the experiment entirely (presented in Figure 1). The data presented here in Fig. S1 demonstrated the same failure of activity-regulated myelination following methotrexate as observed in the more technically sound analyses in Fig. 1.
A-B) TEM was performed one month following the cessation of the 7-day optogenetic stimulation paradigm in Thy1::ChR2+/− mice that were either stimulated or identically manipulated with the exception of blue light exposure (mock-stimulated controls). These Thy1::ChR2+/− mice were previously exposed to MTX or PBS vehicle control, resulting in four experimental groups. The g-ratio data are shown as a function of axon caliber as a scatterplot of all axons measured in (A) PBS vehicle control-treated, mock-stimulated mice (n= 6 mice; black triangles) compared to PBS vehicle control-treated, optogenetically stimulated mice (n= 5 mice; red triangles) and in (B) MTX-treated, unstimulated mice (n= 4 mice; black triangles) compared to MTX-treated, optogenetically stimulated mice (n= 3 mice; red triangles). A single point indicates the g-ratio for a single axon. Approximately 100 axons were quantified and the mean g-ratio determined for each mouse. P-values (indicated on plots) were determined by a two-way ANOVA with Bonferoni correction for multiple comparisons, comparing the mean g-ratio per mouse between groups.
C) Representative TEM images of premotor projections entering the subjacent corpus callosum at the level of the cingulum. Scale bars=2μm.
Supplemental Fig 2. Verification of OPC conditional knock out of TrkB and standardized region of cell density quantification, related to Figure 3, S3
A) Representative confocal images of TrkB (red) expression in PDGFRa+ OPCs (green), assessed by immunohistochemistry. TrkB is expressed in TrkB WT OPCs (left), and following tamoxifen induction TrkB expression is lost from OPCs in TrkB cKO mice (right). Scale bar = 10μm
B) Standardized fields (blue squares) sampled in the corpus callosum for all cell density measurements.
Supplemental Fig 3. BDNF to TrkB signaling is necessary for activity-regulated oligodendrogenesis, related to Figures 3, 4 and S2
B) Density of EdU-marked, oligodendrocytes (EdU+/CC1+ cells) in corpus callosum of BdnfTMKI;Thy1::ChR2+/− mice (n=6), BdnfTMKI; WT mice (n=5), BdnfWT;Thy1::ChR2+/− mice (n = 5) and BdnfWT;WT mice (n=6) at 4 weeks after completion of the one-week optogenetic stimulation paradigm.
C) Density of total CC1+ oligodendrocytes in the corpus callosum of BdnfTMKI;Thy1::ChR2+/− mice (n=6), Bdnj™TMKI ;WT mice (n=5), BdnfWT;Thy1::ChR2+/− mice (n = 5) and BdnfWT;WT mice (n=6) at 4 weeks after completion of the optogenetic stimulation paradigm.
D) Density of EdU-marked oligodendrocytes (EdU+/CC1+ cells) in the corpus callosum of OPC TrkB cKO;Thy1::ChR2+/− mice (n=6), OPC TrkB cKO;WT (no opsin) mice (n=6), TrkB WT;Thy1::ChR2+/− mice (n=6) and TrkB WT;WT (no opsin) mice (n=6) at 4 weeks after completion of the optogenetic stimulation paradigm.
E) Density of total CC1+ oligodendrocytes in the corpus callosum of OPC TrkB cKO;Thy1::ChR2+/− mice (n=6), OPC TrkB cKO;WT (no opsin) mice (n=6), TrkB WT;Thy1::ChR2+/− mice (n=6) and TrkB WT;WT (no opsin) mice (n=6) at 4 weeks after completion of the 7-day optogenetic stimulation paradigm.
F) Representative confocal images of oligodendrocytes (CC1+, green) co-localized with EdU (red) in the corpus callosum for each group described above Scale bar = 20μm.
Data shown as mean ± SEM. Each point = one mouse. ns = p > 0.05, *p<0.05, **p < 0.01 ****p<0.0001. Two-way ANOVA with Tukey post-hoc analysis for multiple comparisons.
Supplemental Fig 4. BDNF to TrkB signaling is necessary for activity-regulated myelination, related to Figure 4
Transmission electron microscopy (TEM) was performed 4-weeks following the cessation of the one-week optogenetic stimulation paradigm. Myelin sheath thickness was analyzed at the level of the cingulum of the corpus callosum as the g-ratio (diameter of axon divided by the diameter of axon plus myelin sheath; a smaller g-ratio indicates a thicker myelin sheath). Scatterplots of g-ratio as a function of axon caliber are shown comparing optogenetically stimulated mice vs. identically manipulated WT controls for each genotype studied
A) A scatterplot of all axons measured shows an increase in myelin sheath thickness (reduced g-ratio) in BdnfWT;Thyl::ChR2+/− (n=5 mice; red triangles) mice compared to BdnfWT;WT (n=6 mice; black triangles).
B) A scatterplot of all axons measured show no difference in g-ratio between BdnfTMKI;WT (n=6 mice; black triangles) vs. BdnfTMKI;Thy1::ChR2+/− (n=5 mice; red triangles).
C) G-ratio data of BdnfTMKI model are shown by axon size bins of small (<0.5μm), medium (0.5-1.0 μm) and large (>1.0μm) axons.
D) A scatterplot of all axons measured shows an increase in myelin sheath thickness (reduced g-ratio) in TrkB WT;WT (n=7 mice; black triangles) compared to TrkB WT;Thy1::ChR2+/− (n=6 mice; red triangles).
E) A scatterplot of all axons measured show no difference in g-ratio between OPC-TrkB cKO;WT (n=7 mice; black triangles) compared to OPC-TrkB cKO;Thy1::ChR2+/− mice (n=5 mice; red triangles).
F) G-ratio data of OPC-TrkB cKO model are shown by axon size bins of small (<0.5μm), medium (0.5-1.0 μm) and large (>1.0μm) axons.
A single point indicates the g-ratio for a single axon. Approximately 100 axons were quantified and the mean g-ratio determined for each mouse. P-values (indicated on plots) were determined by two-way ANOVA with Tukey post-hoc analysis for multiple comparisons, comparing the mean g-ratio per mouse between groups.
Supplemental Fig 5. A small molecule TrkB partial agonist recapitulates the effects of BDNF on OPC proliferation and oligodendrogenesis, related to Figure 6
A) Six-point dose response curve demonstrating OPC proliferation index as measured by the fraction of PDGFRα+cells that co-label with EdU in P6-8 mouse OPCs at 24 hours after exposure to recombinant BDNF at a 0 to 1000 nM concentration range (n=6 wells per condition).
B) Four-point dose response curve demonstrating OPC proliferation index as measured by the fraction of PDGFRα+ cells that co-label with EdU in P6-8 mouse OPCs at 24 hours after exposure to LM22A-4 at a 0 to 1000 nM concentration range (n=6 wells per condition).
A-B) Representative confocal images (below) of PDGFRα+ (green), EdU+ (red) and DAPI+ (blue) vehicle control, BDNF- and LM22A-4 -treated cells. Scale bar=50μm.
C) Percent of total DAPI+ cells that express O4+ and do not express PDGFRα+ increases at 48-hours after treatment with BDNF (50nM) or LM22A-4 (100nM) of P6-8 mouse OPCs (n=4 wells per condition). Representative confocal images (below) of PDGFRα+ (red), O4+ (red) and DAPI+ (blue) of vehicle control (left) and LM22A-4-treated (right) cells. Scale bar =20μm
D) Human iPS cells were treated with one of three treatment paradigms from day 25-35 in the OPC induction protocol: no BDNF, 20ng/ml BDNF as per standard protocol, or BDNF was replaced with 1000nM LM22A-4. Cells were fixed at day 100 following induction and the percent of DAPI+ cells expressing the OPC marker PDGFRα was quantified.
E) Representative image of a human iPSC-derived PDGFRα+ OPC. Green = PDGFRα, white = DAPI. Scale bar =10μm.
F) Human iPS cells were treated with one of three treatment paradigms from day 25-35 in the OPC induction protocol: no BDNF, 20ng/ml BDNF as per standard protocol, or BDNF was replaced with 1000nM LM22A-4. Cells were fixed at day 100 following induction and the percent of DAPI+ cells expressing the mature oligodendrocyte marker MBP was quantified.
G) Representative image of a human iPSC-derived MBP+ oligodendrocyte. Red = MBP, white = DAPI. Scale bar = 10μm.
H) Representative image of lysolecithin-induced demyelination in the corpus callosum 10 days after injection. Green = fluoromyelin stain to demonstrate myelinated architecture. White-dashed circle = lesioned area in the corpus callosum. Scale bar = 100μm.
I) Density of EdU+/PDGFRα+ proliferating OPCs in the corpus callosum of mice treated with 50mg/kg LM22A-4 or vehicle control for 7 consecutive days beginning 7 days after lysolecithin injection (n=5 mice in each group). Representative confocal images (at right) of PDGFRα+ (green) and EdU+ (red) in lesioned area of vehicle control (left) and LM22A-4 - treated (right) mice. Scale bar = 20μm.
J) Density of CC1+ oligodendrocyte cells in the corpus callosum of mice treated with 50mg/kg LM22A-4 or vehicle control for 14 consecutive days beginning 7 days after lysolecithin injection (n=5 mice in each group). Representative confocal images (at right) of CC1+ (green) in lesioned area of vehicle control (left) and LM22A-4 -treated (right) mice. Scale bar = 20μm.
Data shown as mean ± SEM. Each point = one mouse (I-J). ns = p > 0.05, * p < 0.05, ** p < 0.01, *** p<0.001, **** p<0.0001 as determined by one-way ANOVA (A-F) or Student’s t-test (I-J).
Supplemental Fig 6. TrkB partial agonist LM22A-4 normalizes myelination after methotrexate chemotherapy exposure, related to Figure 6
A) MTX-treated mice exhibit thinner myelin compared to PBS vehicle-treated controls. g-ratio values as a function of axon caliber are shown as a scatterplot of all axons measured in mice treated with PBS vehicle control+vehicle control (n=4 mice; red triangles) vs. MTX+vehicle control (n=4 mice; black triangle).
B) LM22A-4 increases myelin sheath thickness in control mice. g-ratio values as a function of axon caliber are shown as a scatterplot of all axons measured in mice treated with PBS vehicle control+vehicle control (n=4 mice; black triangle) vs PBS vehicle control +LM22A-4 (n=4 mice; red triangle).
C) G-ratio data shown in Figure 6 are shown by axon size bins of small (<0.5μm), medium (0.5-1.0 μm) and large (>1.0μm) axons. LM22A-4 rescues myelin deficits in mice exposed to juvenile MTX in small caliber axons (< 0.5μm; n=4 mice, p = 0.005), medium caliber axons (0.5μm - 1.0 μm; n= 4, p = 0.006) and large caliber axons (>1.0 μm; n=4 mice; p = 0.033) compared to mice exposed to MTX but treated with vehicle control. Data shown as mean ± SEM, * p < 0.05, ** p < 0.01, *** p<0.001 ns = p > 0.05. Two-way ANOVA with Tukey post-hoc multiple comparisons testing.
A single point indicates the g-ratio for a single axon. Approximately 100 axons were quantified per mouse and the mean g-ratio determined for each mouse. P-values (indicated on plots) were determined two-way ANOVA with Tukey post-hoc multiple comparisons testing comparing the mean g-ratio per mouse between groups.