

Abstract
Epigenomic mapping of tissue samples generates critical insights into genome-wide regulations of gene activities and expressions during normal development and disease processes. Epigenomic profiling using a low number of cells produced by patient and mouse samples presents new challenges to biotechnologists. In this review, we first discuss the rationale and premise behind profiling epigenomes for precision medicine. We then examine the existing literature on applying microfluidics to facilitate low-input and high-throughput epigenomic profiling, with emphasis on technologies enabling interfacing with next-generation sequencing. We detail assays on studies of histone modifications, DNA methylation, 3D chromatin structures and non-coding RNAs. Finally, we discuss what the future may hold in terms of method development and translational potential.
Graphical Abstract
A review of microfluidic technologies for epigenetic and epigenomic analyses.
1. Background
1.1. Epigenomes and their dynamics
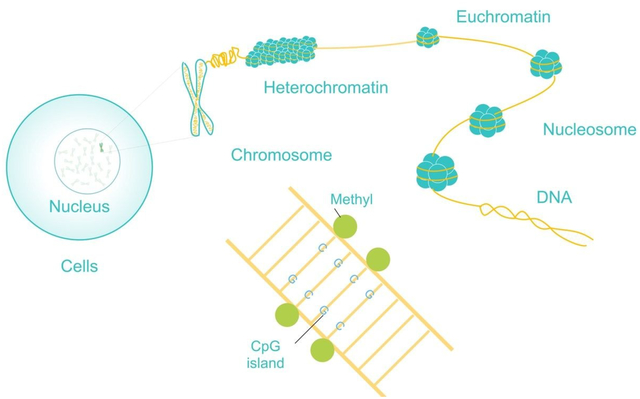
Epigenetics is the study of heritable changes in phenotypes that do not involve alterations to the DNA sequence1. The epigenome is the body of genome-wide chemical structures, nucleic acids, and proteins that manipulates the DNA1. DNA is wrapped around histone proteins, forming nucleosomes, which is then further coiled up to form chromatin2. In contrast to the relatively static genome, the epigenome is highly dynamic and can be altered by environmental influences, lifestyle, and aging. For example, monozygotic twins have indistinguishable epigenetic signatures at birth, yet they have increasingly significant epigenetic differences over their lifespan3. This can cause one twin to become more susceptible to a disease than the other. For instance, schizophrenia only occurs in both twins about 50% of the time4, 5.
There are four main epigenomic mechanisms of altering the genome that are currently being studied: DNA methylation, histone modification, 3D chromatin structures, and non-coding RNAs (Fig. 1)1. DNA methylation occurs when methyl (CH3) groups are added to cytosines in the DNA and, when occurring in the promoter region of a gene, it primarily acts to silence gene transcription6, 7. Similarly, histone modifications occur when acetyl groups (CH2CO), mono-, di-, or trimethyl groups, or one of many other functional groups are added to the amino acid tails present on the histone proteins8, 9. For simplicity, the tri-methylation of the fourth lysine on the tail of histone H3 is denoted as H3K4me3. The position and type of histone modification can both change whether it activates or represses gene expression2.
Figure 1 |. Summary of epigenetic mechanisms and various genome-wide mapping methods.

a. Overview of various epigenetic modifications and profiling methods. b. The timeline on development of NGS-based epigenomic mapping toolkits over the years. “sc” refers to “single cell”. References: ChIP-seq8, 9, 48; RRBS65; MeDIP-seq38, 39; DNase-seq52; Mnase-seq53; WGBS(BS-seq/MethylC-seq)36, 37; Hi-C50; CHIA-PET102; MBD-seq40, 41; MRE-seq42; FAIRE-seq103; Nanopore104–106; ChIP-exo107; 3D-DSL108; CHIRP-seq56, 57; PBAT64; oxBS-seq44; TAB-seq45; SMRT109; sc RRBS71, 72, 81; fCAB-seq46; 5caC-seq47; sc Hi-C77, 78; ATAC-seq51; T-WGBS67; sc PBAT75; sc ChIP-seq (Drop-ChIP)110; scDamID111; sc Dnase-seq112; sc ATAC-seq79, 80; sc WGBS73; MOWChIP-seq113; ChIPmentation114; sc Aba-seq115; sc MT-seq82; sc M&T-seq83; sc Trio-seq84; CLEVER-seq76; sc MAB-seq116; sci-MET117; CUT&RUN61, 62; sc BS-seq74; MID-RRBS118; SurfaceChIP-seq118; ChiL-seq63; sc ChIP-seq119.
The three-dimensional chromatin structure can also affect DNA expression10. Chromatin structures can limit accessibility to the DNA within, such as in packed regions of the chromatin, or promote interactions between two distant locations that are adjacent due to folding. Finally, non-coding RNA (ncRNA) is a subset of RNA that is not translated into protein11, 12. ncRNA is involved in the regulation of epigenetic state and post-transcriptional gene silencing. Long non-coding RNA (lncRNA) regulates chromatin states by interacting with specific chromatin modification complexes. For example, HOTAIR works as a scaffold for PRC2 that modifies H3K27me3. Altogether, epigenetic modifications can activate or repress gene expression, and are essential for many cellular processes, such as genomic imprinting and X-chromosome inactivation2. Sensibly, disruptions to the epigenome can have significant consequences and lead to diseases.
1.2. Epigenetics in cancer
For many years, we have believed that the initiation and progression of cancer was only due to genetic mutations, yet in more recent years, a prevalence of aberrant epigenetic modifications have also been found13–17. Now, the epigenome has a more widespread role and many researchers argue that epigenetic disruption, often connected with genetic mutations, is the central driving force of cancer18. In fact, many of the genes mutated in cancers are genes that directly modify the epigenome, such as DNA methyltransferase genes that control DNA methylation19.
Almost every cancer has abnormal specific or widespread DNA methylation levels20. For instance, CpG (cytosine and guanine separated by one phosphate group) island (CGI) hypermethylation and genome-wide hypomethylation frequently occur in cancer cells. CGIs are thousand base-pair long regions of the genome that are rich in CpGs, have little to no methylation, and often are associated with gene promoters21. Hypermethylation of a CGI in the promoter of a tumor suppressor gene often silences the function of that gene. For example, the BRCA1 gene is inactivated in breast and ovarian carcinomas by CGI hypermethylation22. BRCA1 is part of the protein complex that repairs DNA when it is damaged. Its dysfunction leads to damage building up which can cause cancerous mutations. Meanwhile, global hypomethylation, which mainly affects repetitive regions of the genome and gene promoters that have few CpG dinucleotides, is common in all types of cancer and results in genomic instability20. Furthermore, hypomethylation of gene promoters in cancer frequently activates oncogenes, metastasis related genes, and drug-resistance genes.
Similarly, histone modification aberrations in cancer are also genome-wide and can be tumor-type specific23. For example, global loss of the histone modifications H4K16ac and H4K20me3 is a common hallmark of human cancers24. A recent study shows that mutations in the H3F3A gene, which encodes the histone variant H3.3, are associated with pediatric brain cancers. H3.3 mutation disrupts post-translational modification leading to a loss of the histone mark at lysine 2725. Lastly, non-coding RNAs that regulate the epigenome also express aberrantly in cancer. It’s observed in different cancers that a long noncoding RNA, HOTAIR, is overexpressed and can be used as a prognostic biomarker. HOTAIR interacts with both PRC2 and LSD1 complexes leading to silencing of tumor suppressor genes26.
Interestingly, these epigenomic aberrations are also hallmarks of aging, though it’s unclear whether modifications caused by aging contribute to cancer27. Overall, understanding the epigenome has prognostic and diagnostic significance for both cancer and aging.
1.3. Epigenomic therapy
The reversible nature of the epigenome has opened new avenues for precision medicine, which leverages the genetic and epigenetic information of patients to determine a personalized course of treatment28–30. For example, the use of RG108, a DNA methyltransferase inhibitor, showed demethylation and re-expression of the tumor suppressor gene, p16, in a human cancer cell line31. As such, epigenetic patterns are useful in assessing various chemotherapeutic targets and responses30.
In fact, several epigenome-modifying drugs have been FDA approved as treatments for cancers32–35. One of these drugs is used in the treatment of peripheral T-cell lymphomas which are an aggressive subgroup of non-Hodgkin lymphomas with a high rate of relapse, in part due to few treatments specifically targeted at them35. As hypoacetylation frequently occurs in cancers, Romidepsin, a type of histone deacetylase (HDAC) inhibitor that prevents acetyl groups on histones from being removed, was clinically tested and found to be suitable for treatment in relapsed patients.
1.4. Genome-wide mapping of epigenetic events
In order to gain an understanding of how the epigenome affects the genome-wide molecular biology, many methods have been developed to map the epigenome in recent years with next-generation sequencing.
The principal method of determining DNA methylation is by conducting bisulfite conversion followed by sequencing, known as Whole-Genome Bisulfite Sequencing (WGBS) and sometimes referred to as BS-seq or methylC-seq36, 37. Single-stranded DNA fragments are incubated with bisulfite agents which sulphonates (adds HSO3) and deaminates (removes NH2) the unmethylated cytosine (C) nucleotides to form an intermediate product. Due to differences in the rates of reaction, the methylated cytosines are left unchanged during this process. Next, the fragments are desalted and incubated with a strong base to desulphonate the intermediary to form uracil (U), an RNA nucleotide, which is subsequently converted to its corresponding base pair thymine (T) through polymerase chain reaction (PCR) amplification. The bisulfite-converted DNA is sequenced and compared to the reference genome to determine the unmethylated C’s that are converted to T’s. In addition to WGBS, other methods of analyzing methylated DNA includes using antibodies or proteins that target methylated cytosines to capture and sequence methylated DNA (MeDIP-seq38, 39, MBD-seq40, 41) or utilizing restriction enzymes sensitive and insensitive to methylated cytosine before sequencing and comparison to identify methylated regions (MRE-seq)42.
In the process of DNA demethylation, the methylated cytosine (5mC) can be oxidized to form intermediates such as 5-hydroxylmethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxycytosine(5caC) which could act as epigenetic marks43. In traditional bisulfite sequencing, 5hmC is indistinguishable from 5mC while 5fC and 5caC are similar to cytosine and thus convert to thymine. However, oxidative bisulfite sequencing (oxBS-seq44) oxidizes 5hmC to form 5fC before undergoing bisulfite conversion and sequencing. 5mC can then be differentiated from 5hmC through comparison of oxBS-seq and traditional BS-seq results. Tet-assisted bisulfite sequencing (TAB-seq45) instead oxidizes 5mC to form 5caC while protecting 5hmC through glucosylation. After bisulfite sequencing, only 5hmC is read as a C. To map 5fC at the single-nucleotide level, traditional bisulfite conversion (5fC > T) is compared to bisulfite sequencing where 5fC is chemically labelled to prevent conversion to thymine (5fC > C) in fC assisted bisulfite sequencing (fCAB-seq)46. Lastly, antibodies specific to 5hmC, 5fC, and 5caC have been used to isolate and sequence the nucleotide variants47.
Chromatin immunoprecipitation followed by sequencing (ChIP-seq) is the method of choice for examining histone modifications8, 9, 48. Superparamagnetic beads are bound to antibodies against the histone mark of interest (e.g. anti-H3K4me3) before being exposed to cross-linked chromatin fragments generated by shearing or digesting chromosomes. These beads are washed to remove non-specific binding and the bound chromatin is then eluted from the bead surface. Finally, the ChIP DNA is separated from the histones and sequenced. Thus, the isolated genomic sequences map all locations in the genome marked by the modified histone. This method can also be used to determine transcription factor binding sites.
In order to look at the interactions that occur between various regions of the chromatin, chromosome conformation capture (3C) techniques are used49. The most recent iteration is Hi-C, which allows researchers to look at how all the regions interact50. First, cells are cross-linked to secure the chromatin-chromatin interactions, lysed, and washed before the chromatin is digested. The fragments are labeled with a biotinylated nucleotide at each end, which are then ligated together and the proteins in the chimeric DNA are digested. Finally, the chromatin is sheared and purified to leave only the DNA fragments that have internal biotin which are isolated with anti-biotin beads. Therefore, the remaining DNA fragments consist of the two interacting segments combined end-to-end, which are sequenced by paired-end sequencing.
As for chromatin accessibility and nucleosome positioning, ATAC-seq51, DNase-seq52, and MNase-seq53 have all been utilized. ATAC-seq, or Assay for Transposase Accessible Chromatin followed by high-throughput sequencing, is the most straight-forward and popular method for examining chromatin accessibility51. Cells are lysed and the genomic DNA is isolated and incubated with transposase. The transposase simultaneously fragments and tags the regions of accessible chromatin. The resulting fragments are purified, amplified, and sequenced, with the data yielding a profile of open regions in the genome. DNase-seq focuses on regions that are hypersensitive to DNaseI (primarily accessible regions)52. Chromatin is incubated with DNaseI, purified, and size selected to exclude segments that would be part of a nucleosome. These small segments are then sequenced to identify regions of accessible chromatin. Conversely, chromatin that is digested with micrococcal nuclease (MNase) in MNase-seq provides data that is essentially the inverse of that produced by DNase-seq53. MNase first cleaves the DNA into single-strands, then eats away any exposed DNA. DNA that is wrapped around nucleosomes, or sometimes attached to another DNA-interacting protein, will not be digested. The DNA is purified and sequenced, showing locations of nucleosomes and other regions where the DNA have bound regulatory proteins.
Finally, sequencing of non-coding RNA requires the isolation of RNA from the cells or tissue. RNA is then converted into complementary DNA (cDNA) and sequenced. The resulting sequences can be aligned to the genome to study previously documented ncRNAs. However, bioinformatic prediction followed by verification is often necessary for identifying novel ncRNAs54, 55. In order to map the locations of lncRNA binding sites, ChIRP-seq (Chromatin Isolation by RNA Purification) was developed56, 57. Briefly, cross-linked chromatin is extracted and fragmented by sonication. Biotinylated oligonucleotide probes are then hybridized to target lncRNAs. These RNA-chromatin-oligo probe complexes are isolated by magnetic streptavidin beads followed by DNA purification and sequencing.
All of these assays provide information critical for understanding their own piece of the epigenome. Unfortunately, most current methods have long reaction times, are generally low-throughput, and require large numbers of cells as input.
1.5. Low-input epigenomic mapping
In the majority of epigenetic assays, more than 10 million cells are required as input due to DNA loss and poor reaction efficiency8, 36, 58, 59. This severely limits the application of these assays, particularly with primary cells taken directly from patients and rare cell samples such as stem cells. In many cases, these cells cannot be effectively cultured, or to do so would alter their epigenome60. In this section, we will give some examples on low-input assays based on strategies that do not use microfluidic devices.
Cleavage Under Targets and Release Using Nuclease (CUT&RUN) maps in situ protein-DNA interaction with low background noise61, 62. After immobilizing unfixed nuclei to magnetic beads, the nuclei are incubated with antibody, followed by Protein A-MNase. The Protein A-MNase binds to the antibody and cleaves the DNA on either side of the target protein. Then, the target protein-DNA complex diffuses out of the nuclei, leaving the rest of the genome behind. CUT&RUN only requires 100 cells for histone modification mapping and 1,000 cells for transcription factor mapping62. Similarly, chromatin integration labelling (ChIL) is another in situ method that obtains the genomic sequence around the histone modification or transcription factor of interest from 100 cells63. Cells are grown, fixed, permeabilized, and stained with a primary antibody within a 96 well plate. Then, the cells are incubated with a secondary antibody probe that is conjugated with a primer for sequencing and a T7 promotor. Next, Tn5 attaches to the probe, cuts and then tags the genomic region bound to the target protein with the conjugated primer and T7 promotor. Linear amplification is then performed to amplify sufficient target fragments for sequencing.
Adjustments to DNA methylation assays aim to either reduce reaction-based loss or to reduce required input. For example, post-bisulfite adapter tagging (PBAT) reduces loss that occurs during bisulfite conversion64. In traditional bisulfite conversion, the DNA is tagged with the library sequences before it undergoes conversion, but the fragments are susceptible to being cut during the conversion. This prevents them from being sequenced, causing substantial loss of data. PBAT shifts the adapter tagging to post-conversion, ensuring a significantly higher percentage of fragments can be sequenced. Meanwhile, reduced representation bisulfite sequencing (RRBS) uses restriction enzymes to digest the DNA before size selection65. These two steps together isolate regions rich in CpG islands and promoter regions, which contain a large portion of the regions of interest. The rest of the protocol is then similar to that of WGBS. Since RRBS does not require the sequencing depth that is traditionally required across an entire genome, RRBS is effective with 100 times less DNA65 – 10ng compared to 5μg for WGBS36, 37.
In addition, the transposase-based ‘tagmentation’ method performed in ATAC-seq has also been adopted in low-input analysis for histone modification (ChIPmentation66) and DNA methylation (T-WGBS)67. ChIPmentation, a combination of traditional ChIP with tagmentation, uses the transposase to add adapters to the chromatin66. The chromatin was immunoprecipitated with beads and, once immobilized, incubated with the tagged transposases. This combined library preparation – adding adapters to samples in order to facilitate sequencing – with the immunoprecipitation, ultimately reducing loss from additional purification steps. Similarly, tagmentation-based whole-genome bisulfite sequencing (T-WGBS) also uses transposases to attach adapters for library preparation but performs WGBS otherwise normally67. In both cases, adding tagmentation reduced the amount of DNA input required; ChIPmentation used 50 ng of DNA66 (compared to 50μg in ChIP8) while T-WBGS used as little as 20 ng of DNA67 (compared to 5μg in WGBS36, 37).
1.6. Single-cell mapping
Single-cell mapping technologies have exhibited unprecedented potential to revolutionize biological investigations by revealing cellular heterogeneity obscured in bulk experiments68, 69. As a result, single-cell methylation and single-cell chromatin accessibility and interactions assays have gained popularity rapidly in recent years.
Multiple groups have published work on single-cell methylation. A locus-specific bisulfite sequencing (SLBS) method converted single-cells and the converted DNA underwent whole genome amplification before specific converted regions were amplified and sequenced70. Since only a few regions were being analyzed, single-cell analysis was possible. Single-cell RRBS (sc RRBS71, 72) sorted a single cell into each tube. Then, almost all steps of the RRBS – lysis, restriction enzyme digestion, end repair, adapter ligation, and bisulfite conversion – were performed in the same tube, sequentially. In between steps, enzymes were heat-inactivated, and DNA was only purified and amplified after bisulfite conversion. The single-tube reaction with reduced purification decreased loss to make single-cell conversion possible. Similar to sc RRBS, sc WGBS used a single-tube method of bisulfite conversion and library prep to reduce loss73. Conversely, sc BS-seq used random priming to pre-amplify the DNA before PCR amplification74, while sc PBAT utilized PBAT to reduce reaction-based loss75. Single-cell methods have also been developed for the analysis of other cytosine modifications. Chemical-labeling-enabled C-to-T conversion sequencing (CLEVER-seq76) uses a single-tube approach where it labels 5fC locations in a single cell with a chemical before PCR amplification. During amplification, the labeled 5fC is read as a T, allowing the 5fC to be identified.
While single-cell chromatin accessibility and single-cell chromatin interaction assays probe different aspects of the epigenome, similar techniques have been used to reduce the cell input requirements. In both single-cell Hi-C77, 78 and single-cell ATAC-seq79, 80, a successive series of barcoding steps was included to add unique barcodes to each cell. The pooled cells were then processed, and the sequenced data was demultiplexed to obtain cell-specific information.
Single-cell multi-omics methods have also been developed that simultaneously profile epigenomics, transcriptomics, and genomics from the same single cell and investigate the association among different omics81–84. For example, single-cell methylome and transcriptome sequencing (scMT-seq82 and scM&T-seq83) analyzed the DNA-methylation and the RNA transcriptome in parallel from the same cell. In both methods, RNA was separated from the DNA before running downstream analysis. In scMT-seq, a single-cell was lysed while leaving the nucleus intact. The nucleus was manually separated and subjected to sc RRBS while the cytosolic fraction was used for single-cell RNA-seq. Similarly, scM&T-seq chose to use beads to isolate the RNA from a lysed cell while the DNA was used for scWGBS. Single-cell triple-omics sequencing (scTrio-seq84) was performed in much the same way as scMT-seq, while including a method of analyzing the copy-number variations (CNVs) by comparing the single-cell data to bulk cell data.
In general, single-cell epigenomic data offer significantly less genome coverage (i.e. genome-wide information) than their competing low-input alternatives, even after pooling the single-cell data sets together. This is primarily due to the barcoding step required by single-cell profiling which tends to be incomplete and loses DNA.
2. Microfluidic techniques for profiling epigenomes
Microfluidics provides flexible platforms for manipulating liquids at pico/nanoliter scale in miniaturized devices. Furthermore, it has shown advantages in performing chemical and biological analysis. Compared to conventional tube-based and manual assays, microfluidic assays are often rapid, automated, and scalable for high-throughput operations. In addition, the small dimensions of microfluidics pre-disposes it to be better equipped for low-input assays and for integrating distinct steps to reduce the sample loss. Altogether, this has earned microfluidics the nickname “lab-on-a-chip” as it is conceptually similar to the integrated circuit.
Over the past decades, the development and commercialization of microfluidic technologies have led to new revolutions in biology research related to single-cell analysis and high-sensitivity molecular detections85. Microfluidics has found widespread applications to cell biology86–92, drug research93–96, microbiology97, 98, immunology99, 100, and physiology101. In the below text, we will focus on how microfluidics facilitates the studies of epigenomes with emphases on histone modifications, DNA methylation, 3D chromatin structures, and non-coding RNAs. Fig. 1 shows an overview of various epigenetic changes and associated technologies developed over the years.
2.1. Microfluidics for profiling histone modifications and transcription factor bindings
2.1.1. Chromatin immunoprecipitation (ChIP)
ChIP is the gold standard technique for mapping histone modifications8, 9, 48. However, conventional ChIP assays have major drawbacks including large sample size (>107 cells) and time-consuming procedures (2–4 days)8. Over the past years, microfluidic technology has been utilized to improve the performance of the ChIP assay by reducing the input amount and the assay times, while maintaining high quality.
Several devices were developed for ChIP followed by quantitative polymerase chain reaction (qPCR) to detect histone modifications at specific loci. AutoChIP decreased antibody-chromatin incubation time from overnight to 2 h and required only 2,000 cells per assay120, 121. The device contained reagent flowing channels and valve-actuating control channels for precise manipulation of liquids. To enhance ChIP efficiency, chromatin solution and bead suspension were mixed in ring channels using valve-actuated rotary mixing. An updated device, HTChIP, included 16 parallel units (Fig. 2a)122. DEM (DNA-Enrichment Microfluidic) utilized a bead reservoir and a dispersion channel to trap antibody-coated agarose beads while allowing chromatin solution flow through123. When compared to a commercial ChIP assay kit, DEM reduced assay time by 25% and reagent use by 50%. We developed a device that utilized partially closed valves to prevent beads from passing while permitting liquid to flow through and obtained ChIP-qPCR results from 50 cells124. The device consisted of sequential chambers separated by valves for cell lysis, immunoprecipitation, and washing. Moreover, we integrated sonication into a microfluidic device by placing an ultrasonic transducer near a microfluidic chamber125. Microscale sonication also facilitated the mixing and washing of magnetic beads in a subsequent ChIP process. Recently, a droplet microfluidic platform was reported that transferred antibody-coated magnetic beads between droplets using magnetic tweezers to accomplish multiple steps involved in a ChIP assay126. The advantages of this droplet platform include nanoliter scale volume, minimized cross contamination, and enhanced mixing.
Figure 2 |. Microfluidic technologies for profiling histone modifications and transcription factor bindings.

a. HTChIP with reagent-containing flow channels (blue) and valve-actuating control channels (red). Reprinted with permission122. Copyright 2012 The Royal Society of Chemistry. b. MOWChIP-seq protocol, its performance on H3K4me3 and H3K27ac, and comparison with competing technologies. Reprinted with permission113. Copyright 2015 Macmillan Publishers Ltd. c. SurfaceChIP-seq (left panel) generated highly correlated data for H3K4me3 from 30–5000 GM12878 cells and for H3K27me3 from 100–10,000 cells (right panel). Reprinted with permission129. Copyright 2018 American Association for the Advancement of Science. d. Drop-ChIP procedure and multidimensional scaling plot showing three distinct ES cell subpopulations. Scale bars are 100 μm. Reprinted with permission110. Copyright 2015 Macmillan Publishers Ltd.
Microfluidic devices that perform ChIP coupled with next-generation sequencing were developed only in recent years. Similar to AutoChIP/HTChIP devices120–122, Shen et al. described dead-end ring-chambers integrated with 3-valve peristaltic pumps to assist with mixing and immunoprecipitation which obtained genome-wide H3K4me3 profile from 1,000 cells127. By taking advantage of gas permeable polydimethylsiloxane (PDMS), dead-end channels can be filled with liquid. In the vast majority of ChIP methods, ChIP DNA was collected at less than 1/100 of its theoretical amount. In order to dramatically increase collection efficiency of ChIP DNA, we developed MOWChIP (Microfluidic Oscillatory Washing based ChIP), a simple single-chambered device, to conduct ChIP-seq using as few as 100 cells (Fig. 2b)113, 128. In MOWChIP, sheared chromatin was flowed through a tightly packed bed of antibody-coated beads to facilitate a fast and complete immunoprecipitation. After ChIP, oscillatory washing was used to remove non-specific binding. Even with the low cell input, MOWChIP was capable of collecting ChIP-DNA within the theoretical limit. We also developed another ultralow-input technology, SurfaceChIP-seq, for profiling genome-wide histone modification from as low as 30 cells129. It was also the only device that did not use antibody-coated beads. Instead, the device had 8 parallel microfluidic channels, where the bottom surface of each channel was coated with antibodies for immunoprecipitation (Fig. 2c). Chromatin was flowed through and non-specific binding was washed away, before the bound DNA was eluted and processed for sequencing. We also developed LIFE-ChIP (Low-Input Fluidized-bed Enabled ChIP), an automated microfluidic platform for high-throughput operations (4 assays in parallel with as few as 50 cells per assay)130. The device consisted of 4 chambers connected to channels for loading reagents. The operation was controlled by syringe pumps and solenoid valves programmed using the software LabVIEW.
Besides bulk experiments mentioned above, ChIP assays have been performed at the single-cell and single-molecule resolution in micro- and nanofluidic platforms. Single-cell ChIP-seq (Drop-ChIP) encapsulated single-cells into droplets, where they were lysed and their chromatin was digested110. The chromatin, still contained within the droplet, was mixed with barcodes that had a unique DNA sequence for each cell. This allowed the identity of each cell to be determined after being pooled for downstream ChIP and sequencing. Drop-ChIP was able to successfully delineate three subpopulations within mouse embryonic stem cells based on various chromatin signatures (Fig. 2d). Similarly, another droplet microfluidic platform was recently reported that performed single-cell ChIP-seq and obtained around 1600 unique reads per cell on average119. The microfluidic system was utilized to label each single cell with a unique barcode in a high-throughput manner. Droplets containing digested chromatin from individual cells were infused with droplets carrying barcode hydrogel bead. Barcoded chromatin was then pooled for immunoprecipitation. The authors profiled single cell H3K27me3 landscape from breast tumors and identified a subpopulation of untreated drug-sensitive tumor cells that shared chromatin signatures with drug-resistant cells.
There have also been several single-molecule devices reported that elongated single chromatin molecules for mapping histone modifications. Generally, a chromatin fiber was confined to a nanochannel and stretched to its full length through rapid squeezing of an elastomeric nanochannel131, pressure driven flow132, electrokinetic flow133, 134, or capillary force135. The chromatin fiber was then incubated with fluorescently-tagged antibodies and imaged to determine the location of histone modifications. Mapping of histone modifications in this manner allows researchers to determine the colocalization of multiple histone modifications. Traditional ChIP methods cannot distinguish whether multiple modifications are colocalizing on the same chromatin strand or if each modification is present on separate strands, which are likely from different cells.
2.1.2. In vitro transcription factor binding analysis
In addition to histone modifications, ChIP assays can also be used to map transcription factor (TF) bindings8. TFs are proteins that bind directly to DNA to either promote or suppress transcription of specific genes136. However, TF binding analysis using conventional ChIP suffers from long assay time and lack of high quality antibody8. As a result, there have been several microfluidic platforms developed with high sensitivity and throughput to study TF binding.
First, the Mechanically Induced Trapping Of Molecular Interactions (MITOMI137) device consisted of an array of 2400 unit cells testing a library of possible binding DNA sequences in vitro. Each unit cell contained one fluorescently labeled DNA sequence spotted onto the glass slide. Next, the device was loaded with His-tagged TFs that would bind to anti-His antibodies on the chamber surface. Then, the spotted DNA was released and a “button” membrane would close on the chamber, locking in the sequences bound to surface-adherent TFs, while the unbound sequences are washed away. Finally, the fluorescent signal in each chamber was measured. This was improved upon in MITOMI 2.0 to increase the capacity to 4,160 wells and expand the testing library to all possible 8bp sequences, as well as include fluorescently labelling on the His-tagged TFs138. The fluorescent signal from the DNA and the TFs can then be recorded separately to build a comprehensive map of TF binding affinity. MITOMI 2.0 has been used to determine conserved and novel target sequences of the TF FOXP2, important in speech, between humans and chimps139 as well as characterize novel transcription factors140.
Two other similar devices include the Quantitative Protein Interaction with DNA (QPID141) device and the MITOMI-based analysis of regulatory elements (MARE142, 143) method. QPID was an array-based device containing 64 by 64 chambers, which allowed up to four different transcription factors to be analyzed at the same time141. Meanwhile, MARE broke down regulatory sequences into fragments that can then be used in a MITOMI device to determine how TFs interact with regulatory elements142, 143.
SELEX Affinity Landscape MAPping on a microfluidic platform (SELMAP)144 also utilized the “button” technology to perform a SELEX assay. In SELEX (systematic evolution of ligands by exponential enrichment), TFs were exposed to a library of DNA sequences. After exposure, unbound sequences were washed away, and bound sequences were eluted and re-exposed to TFs under more and more stringent conditions in order to determine what sequences were most tightly bound by the TF. SELMAP achieved this by immobilizing 16 TFs in parallel on the surface of a microfluidic chip. Once the library has flowed through the device, the button chambers closed off the TFs and unbound sequences are washed away. The chambers were released so that the bound DNA can be eluted, amplified, and reflowed. After two enrichment cycles, the bound DNA was sequenced to determine the most common binding motifs.
2.2. Microfluidics for DNA methylation profiling
2.2.1. Bisulfite conversion
The gold-standard assay for DNA methylation analysis is bisulfite-based detection, which generates methylomic profiles at single-nucleotide resolution36, 37. Microfluidic technology has been used to perform low-input, rapid, and automated bisulfite treatment. Site-specific analysis, such as methylation-specific PCR (MS-PCR)145, has been used as a fast diagnostic tool for detecting DNA hypermethylation in the promoter regions of tumor suppressor genes, which is a biomarker associated with cancer146. LoMA-B integrated an on-chip bisulfite conversion module with a detection module using isothermal DNA amplification for rapid MS-PCR analysis, which can be completed within 80 min, and was capable of detecting down to 1% methylated DNA147. There have also been devices created for multiplexed MS-PCR assays, such as a droplet-in-oil microfluidic device with simple sample handling148. Each functional unit of the device consisted of a central sample input port and multiple peripheral open-surface reaction chambers, with each chamber pre-deposited with primers. After the sample was loaded into the center, mineral oil was added to push the sample into the reaction chambers, where it solubilized and mixed with the primers, and allowed the oil to encapsulate the sample into droplets before PCR was performed. This device can perform 108 assays simultaneously in an array-based manner. In addition to academic devices, one group has reported successfully performing microfluidic PCR after bisulfite conversion with the Access Array system from Fluidigm149–151.
There have also been other methods established to specifically test the methylation of one or more regions by utilizing melting temperatures. The first device utilized a capillary electrophoresis (CE) device152, which contained a cross-shaped channel where an electrical charge was applied to solution in order to separate the molecules by electrophoresis; these were then identified through laser-induced fluorescence (LIF). The CE design was then combined with a slanted radiative heating unit to create a microfluidic temperature gradient capillary electrophoresis (μTGCE) device to differentiate DNA with point mutations from normal DNA153. First, generated DNA homoduplexes, where both strands were normal or mutated, and heteroduplexes, one strand normal and one mutated, were pulled along the thermal gradient. However, the heteroduplexed DNA was less stable and would denature (or ‘melt’) before the normal DNA. The motion of the denatured DNA would then be retarded through the device, allowing for separation as well as detection through LIF. This was adapted for work with bisulfite converted DNA using the same methods154. The bisulfite-μTGCE device was capable of detecting as little as 0.1% methylated DNA from unmethylated.
A more recent droplet microfluidic device also used the lower melting temperature of mismatched sequences for rapid quantification of methylation levels at a target locus155. Methylated and unmethylated DNA of a target region was combined with a probe complementary to the methylated sequence and an intercalating dye. This was flowed in droplets through a temperature gradient and the fluorescence was measured. Since the probe was mismatched to the unmethylated DNA, the unmethylated DNA degraded and lost its fluorescence at a lower temperature than the methylated DNA. A standard curve can be generated to determine the fluorescence of droplets with mixed methylation levels. The device was able to distinguish the distinct melting temperatures of mixed methylation droplets, not possible on a PCR machine, thus enabling a low-cost method of determining the methylation level of a gene without sequencing.
Other devices have used the melting temperature more directly with High Resolution Melting (HRM) based devices. HRM takes advantage of the fact that the temperature needed to break the double stranded DNA depends on the sequence of the strands, including the amount of guanine and cytosine (GC). A microfluidic device with 16 reaction wells atop a thermal plate was developed where each well contained sample DNA, PCR amplified for a locus of interest, and intercalating dye156. The fluorescence of each well was measured before the temperature of the plate beneath was increased and the process was repeated. The device was able to not only distinguish unmethylated from methylated DNA, but could also determine methylation levels in between.
A high-throughput device that utilizes melting temperature, HYPER-Melt, was developed for the detection of methylation with single copy sensitivity using methylation-sensitive high resolution melt (MS-HRM)157. HYPER-Melt is the conversion of the benchtop method DREAMing (Discrimination of Rare EpiAlleles by Melt158) to a microfluidic platform. The DREAMing technique consisted of diluting bisulfite-converted DNA such that, when loaded on a 96 well plate, there were no more than one methylated or heterogeneously methylated epiallele per well among 5–500 unmethylated epialleles. The samples were then PCR amplified, where methylated samples were preferentially amplified in order to increase signal, and subjected to a melt curve. The melt curve was capable of distinguishing methylated and heterogeneously methylated DNA from unmethylated. HYPER-Melt expanded this assay to 4,096 nanowells157. The preferentially amplified DNA was loaded into a straight channel that connected the nanowells in each array before oil was added to isolate the nanowells. The device was then heated incrementally while the fluorescence was measured to determine the melting temperature. HYPER-Melt was able to distinguish 1 methylated DNA molecule from 2 million unmethylated variants at CDKN2A loci, a tumor suppressor gene. It was also capable of detecting methylation in liquid biopsies, notorious for their low DNA amounts, from two cancerous patients that was not found by the current gold standard method of MS-PCR.
A multiplexed microfluidic device for simple and semi-automated bisulfite conversion was reported159. The device consisted of linear arrays of open-surface chambers, each containing different bisulfite reagents (Fig. 3a). DNA was bound to magnetic beads and moved through the chambers by magnetic actuation to undergo bisulfite conversion while small channels between the chambers prevented the reagents from being pulled along with the beads. The on-chip conversion was found to be comparable to tube-based conversion by a qPCR that amplified only converted DNA.
Figure 3 |. Microfluidic technologies for profiling DNA methylation.

a. Steps required for on-chip bisulfite conversion. Droplet merging, separation and mixing are performed through magnetic actuation of magnetic beads. Reprinted with permission159. Copyright 2016 Springer Science+Business Media New York. b. MID-RRBS microfluidic device with a fluidic layer (blue) and a control layer (orange) and a cross-sectional view of the reaction and loading chambers. Pie chart shows MID-RRBS and Zymo commercial kit coverage of gene promoters, CpG islands, CpG island shores, enhancers and 5kb tiles. Reprinted with permission118. Copyright 2018 Macmillan Publishers Ltd. c. SCAN workflow and simultaneous detection of DNA methylation and histone modifications. Reprinted with permission134. Copyright 2013 National Academy of Sciences. d. Immunoreaction between antibody and target cytosine causing SPR angle change. Reprinted with permission161. Copyright 2015 American Chemical Society. e. The microfluidic device for using digital PCR to detect PCDHGB6 for examining methylated DNA with different concentrations. Reprinted with permission162. Copyright 2017 Elsevier.
We recently developed microfluidic diffusion-based RRBS (MID-RRBS) for low-input RRBS118. The microfluidic device consisted of a reaction chamber and two adjoining, symmetric loading chambers (Fig. 3b). For each step of the bisulfite conversion, reagents were flowed into the loading chambers. By leveraging diffusivity differences in DNA and small molecule reagents, the DNA remained within the reaction chamber while the reagents were exchanged with those in the loading chambers. This protocol profiled 2 million CpGs with ≥ 1x coverage (74.5% of maximum) and 1.38 million CpGs with ≥ 10x coverage (49.7% of maximum) from 0.3 ng starting DNA, equivalent to about 60 cells. MID-RRBS outperformed bisulfite conversion performed with a commercial kit using 1 ng of starting DNA (~200 cells), which only profiled 1.85 million CpGs with ≥ 1x coverage and 0.85 million CpGs with ≥ 10x coverage. In addition, MID-RRBS was used to epigenetically profile the effect of the antipsychotic clozapine on mice frontal cortices. Multiple genes associated with mental disorders, such as schizophrenia, were found to be differentially methylated between treated and control mice.
In a departure from previous methods, a microfluidic electrochemical sensor was reported that detected the presence of bisulfite-converted methylated DNA sequences within 20 minutes160. Within the device, hairpin ssDNA probes containing porphyrin modifiers were immobilized on gold microelectrodes. Once the target methylated DNA bound to the DNA probe, changes in the distance of porphyrin to the electrode surface led to surface plasmon resonance (SPR) signal changes. These hairpin probes were not reactive to sequences that they are not specific to, which allowed them to be multiplexed within the chip. In addition, the device was capable of detecting as little as 250 fM target DNA, well below the concentration of biomarkers present in the urine of individuals with bladder cancer. Thus, the device provided a low-cost method for enabling early detection of disease, crucial for effective treatment.
2.2.2. Affinity enrichment assays
Although bisulfite conversion is widely used to study DNA methylation, there are significant drawbacks associated with the approach including long treatment time and severe DNA degradation163. Bisulfite-free methods, such as affinity enrichment assays and restriction enzyme digestions, are used as alternatives. While bisulfite conversion relies on chemically changing the sequence of non-methylated DNA to distinguish it from methylated DNA, affinity enrichment assays rely on antibodies and other proteins that preferentially bind to methylated DNA.
SCAN (Single Chromatin molecule Analysis in Nanochannels) is a single molecule device for detecting a combination of epigenomic modifications simultaneously133. In the first description of the device, either chromatin from HeLa cells with GFP-labelled histone H2B or methylated DNA labelled with fluorescent MBD1 protein (MBD represents methyl CpG binding domain) was incubated with TOTO-3 to stain the DNA red. The DNA was then driven by voltage through the device and fully elongated in a nanochannel (250nm wide by 500 nm deep) before fluorescent emission from the GFP/MBD1 and TOTO-3 was recorded and analyzed (Fig. 3c). SCAN was then used to test the co-occurrence of DNA methylation (mC) with H3K27me3 and H3K9me3 in mouse primary cells134. While the device remained unchanged, they used fluorescent antibody to mark the histone modifications. It was found that H3K9me3 co-occurs with mC, while mC suppresses H3K27me3. Interestingly, it was also observed that in immortalized and transformed cells, which are often derived from cancerous cells, H3K27me3 placement was dependent on mC. This aberrant co-localization between mC and H3K27me3 may reveal a potential role of epigenetics in tumor progression.
A single-molecule optical mapping method was reported that elongated DNA molecules by capillary force on an array-based PDMS stamp before transfer-printing the stretched DNA onto SiO2 surface for optical analysis164, 165. The DNA was incubated with fluorescently-tagged MBD before stamping which allowed the visualization of the methylation state after printing. Another similar device was reported that linearized single chromatin strands in nanochannels and visualized the DNA methylation status through fluorescently labeled MBD166.
Two similar microfluidic devices were reported for detecting methylated DNA using methylated cytosine recognition molecules167, 168. First, the target fragment was captured by a DNA probe immobilized on a glass surface. Then, biotinylated methylcytosine antibody or methyl binding domain (MBD) protein was bound to methylated cytosine on the target fragment. Finally, fluorescently labeled streptavidin was added and the fluorescence captured to detect the methylated DNA. Both of these devices were capable of detecting ~0.4nM methylated DNA concentration. In addition, one of the devices was capable of performing the assay within 18 minutes, making it more accessible for clinical use167.
Other devices that utilized MBD proteins have also been developed. For instance, a hyper-methylated DNA microfluidic solid phase extraction device (μSPE) was created that used MBD-protein conjugated on micro-pillars as capture agents169. However, the μSPE’s primary function was to enrich methylated DNA for downstream analysis, which it could do in under 5 min with up to 92% efficiency. Similarly, a single-molecule sorting device was developed to sort methylated DNA from unmethylated in real-time170. Methylated and unmethylated DNA were both incubated with red fluorescent stain before the DNA was exposed to MBD1 tagged with a green fluorophore. The DNA was flowed past a fluorescence inspection point, and sequences with two peaks (representing methylated DNA) were sorted to a separate chamber. Like μSPE, the device was capable of detection but its prime use was for enrichment and it was capable of sorting as little as 11 fg of DNA. Both of these devices could be beneficial in enriching rare or dilute clinical samples for downstream disease biomarker testing.
A sequence-specific immuno-assay device was reported for determining methylation status of single cytosine by on-chip surface plasmon resonance (SPR) detection161. A biotinylated DNA probe was used to hybridize with the target sequence, which induced a bulge region and immobilized the DNA on a streptavidin surface (Fig. 3d). The immunoreaction between an anti-methylcytosine antibody and the target cytosine in the bulge region caused a change in the SPR angle, which reported the methylation status. This method allowed rapid assessment within 45 min in a simple microfluidic device with easy operation.
An electrochemical sensor-based chip was developed for accurately measuring the concentration and level of methylated DNA171. The device consisted of a DNA pre-concentration unit and a methylation detection unit. The DNA pre-concentration unit used ion concentration polarization (ICP) to first concentrate the DNA sequences before they were incubated with the detection unit, a gold electrode hybridized with DNA complementary to the target of interest. Next, they were exposed to a silica-nanoparticle (SiNP) conjugated with methyl binding domain 1 (MBD1), which bound only to methylated CpG sites. The MBD1-SiNP blocked the transfer of electrons, such that low signal occurred either due to high methylation level or high concentration of methylated DNA. These two units worked in concert to allow detection of fM-scale methylated DNA concentration and 10% of methylation level.
Recently, we developed a microfluidic device that performed Methylated DNA ImmunoPrecipitation (MeDIP-seq) in a microchamber172. This device utilized a partially closed microvalve to form a packed bed of anti-5mC antibody-coated beads, which facilitated IP efficiency, and oscillatory washing to remove nonspecific binding. This method enabled mapping of the methylome from 0.5 ng of DNA, equivalent to 100 cells.
2.2.3. Restriction enzyme digestion
In addition to the aforementioned affinity enrichment methods, restriction enzyme digestion has also been implemented in microfluidic devices to avoid the use of bisulfite reagents. Restriction enzyme digestion is used to cleave either unmethylated DNA or methylated DNA while the other remains untouched.
In a device using HpaII and MspI digestion, genomic DNA was mixed with beads coated with MBD2a protein (similar to MBD1) and loaded onto the device173. A magnetic array beneath the device was used to immobilize and manipulate the beads. The beads were digested with either MspI or HpaII and the DNA was eluted for amplification and detection on their previously published amplification/detection device174.
Another locus-specific method was capable of performing single-cell restriction analysis of methylation (SCRAM175, 176). Single cells were isolated and lysed before undergoing restriction digestion that specifically cleaved unmethylated target sites. The DNA was amplified with specific primers such that if the DNA was methylated, a large and small product would result. Otherwise, only a small product would be present. SCRAM was then combined with RT-qPCR in a device for single-cell analysis of genotype, expression, and methylation (sc-GEM)177. Utilizing the C1 Auto Prep Platform from Fluidigm, bulk cells were loaded into the integrated fluidic circuit and individually captured in 96 wells. Cell lysis and subsequent enzymatic reactions were then carried out sequentially in micro-chambers automatically to isolate and amplify RNA (as cDNA) and methylated DNA. A Fluidigm qPCR chip was then used to measure gene expression levels and determine the presence of methylation, while genotyping was performed by Sanger sequencing.
A more recent device that contained 10,368 separate chambers utilized only HpaII digestion but was capable of detecting locus-specific methylation level (Fig. 3e)162. Samples were added to the device and oil was used to sequester the samples into individual chambers. DNA was either digested with HpaII or left untreated before being added to the device and amplified. The methylation level was quantitatively determined by detecting the fluorescence intensity in each chamber and comparing the fluorescence of HpaII digested DNA to the fluorescence of untreated DNA. This method enabled the detection of methylation level as low as 0.52%.
2.2.4. Methylation analysis using non-Illumina sequencing technologies
Some non-Illumina sequencing technologies, including Oxford Nanopore104–106 and Pacific Biosciences SMRT109, have the capability to detect base modifications in addition to the DNA sequence. This allows DNA methylation and other modifications to be determined at single base-pair resolution with little sample preparation.
Nanopore used a distinct cylindrical nanopore protein embedded in an electrically charged membrane. An enzyme protein guided DNA through the nanopore and, as the DNA passed through, the nucleotide sequence was determined by measuring the ionic current blockade. Each nucleotide has its own characteristic electronic signature due to their different atomic structure, even methylated cytosine or adenosine, allowing the nucleotide to be sequenced in real-time178–199. Unmethylated CpGs can be quantified using electro-optical nanopore to determine hypomethylation level200. Moreover, DNA bound proteins, such as MBD1201–205, MeCP2, GCD206, methylcytosine antibody207 or TFs208, can also be detected using solid-state nanopores. Similarly, modified bases, such as uracil or 8-oxoguanine, selectively labeled with biotin-dNTP149, 209, streptavidin210, 211 or Fc ⊂ CB212 can be discriminated by nanopores. In fact, microRNA detection was also reported that uses PEG-labeled probe hybridizing with target miRNA to generate ionic current signatures17 and several groups have reported translocating nucleosomal dsDNA to study nucleosome stability213 and histone variants214. Nanopore sequencing has already demonstrated usefulness as a low-cost, same-day method of molecularly classifying tumors by their genetic and epigenetic profiles182.
SMRT sequencing determined DNA sequence by detecting the fluorescence pulse during incorporation of fluorescently labeled nucleotides by DNA polymerase. Additionally, 5mC, 5hmC and other DNA modifications were discriminated by polymerase kinetic signature, including the arrival time and duration of the pulse130, 215–227.
Although still in their infancy, SMRT and Nanopore technologies are powerful tools to analyze the epigenome due to their ability to obtain real-time, ultra-long base calling from single-molecules without tedious sample preparation or PCR amplification.
2.2.5. DNA methyltransferase activity
Aberrant DNA methylation pattern is a hallmark of cancer, where many genes that suppress tumors are silenced by hypermethylation. The inhibition of DNA methyl transferases (DMNTs), which are the enzymes that catalyze the transfer of methyl groups to DNA, has been approved by the FDA as a cancer treatment. Hence, studying DMNT’s catalytic mechanism has great therapeutic significance.
An array-based MITOMI-style microfluidic platform was developed for the assessment of DNMT activity and screening a library of DNMT inhibitors (DNMTi)228. Fluorescently-tagged DNA probes containing one CpG methylation site were immobilized on the surface of each nanowell and closed off by the button chambers. The DNMTi pre-deposited in side chambers was then solubilized and allowed to interact with the DNMT in the nanowell around the button chamber. The button chamber was released, exposing the DNA probes to the DNMT, and then the DNA was subjected to methylation-sensitive HpaII endonuclease digestion. Thus, a successful DNMTi would prevent the DNA methylation so the DNA would be digested and washed off, leading to loss of fluorescent signal. This allowed researchers to perform high-throughput screening of DNMTis while also being able to analyze the level of inhibition.
2.3. Microfluidic for mapping 3D chromatin structures
In eukaryotic cells, the higher-order packing structure of nucleosomes plays crucial roles in gene regulation and cell differentiation. Methods for capturing nucleosome positioning and chromatin accessibility including MNase-seq53, DNase-seq52, ATAC-seq51, chromosome conformation capture (3C) methodology49 and its variants50, 58, 229–231, each of which provides insights into the 3D organization of chromosomes.
Recently, two microfluidic devices have been reported for mapping chromatin structure. A diffusion-based microfluidic platform for 3C assays was developed by us that employed concentration-gradient-driven diffusion for reagents exchanges between steps232. The device contained a main reaction chamber and two loading chambers on each side, similar to the MID-RRBS device118 described previously, and used oscillatory movement to aid in DNA digestion232. The device has been used to investigate chromatin interactions at human β-globin locus from only 104 cells, in contrast to the 107 cells needed by conventional 3C method. A droplet microfluidic device was also reported recently that performed MNase-seq from 2,500 cells for nucleosome positioning analysis233. Cells and reagents were encapsulated into individual droplets for MNase digestion, then EDTA solution was injected into each droplet to stop digestion. This simple device automates conventional on-tube enzymatic digestions and could potentially be used for single cell MNase-seq.
In addition to the aforementioned bulk experiments, several groups have reported devices examining chromatin 3D structure at single-cell and single-molecule resolution. For instance, single-cell ATAC-seq was performed using the C1 Auto Prep System from Fluidigm80. Single cells were first isolated in individual units, and enzymatic reactions were performed sequentially including cell lysis, tagmentation of the chromatin, and PCR amplification before the libraries were removed from the device and sequenced.
Moreover, several microfluidic devices have been reported for non-destructive handling and analysis of native chromatin fibers234–238. An early device for microfluidic manipulation of chromatin strands utilized electroosmotic flow (EOF) to extend the chromatin234. Individual cells were captured in micropockets and lysed before the chromatin was carefully drawn out by an induced EOF. The direction of the EOF was then changed to be perpendicular to the length of the chromatin, causing the chromatin to adsorb to positively charged micropillars on the surface. The device allowed the chromatin to be safely extended and to have that extension maintained after the electric field was turned off. It was also shown that the chromatin could be gently and effectively manipulated or unwound using a set of microhooks and microbobbins manipulated with optical tweezers235. The hooks would grab the DNA without binding it while the bobbins provided a pillar around which DNA could be wrapped.
More recent devices chose instead to utilize antibody-coated microspheres to capture the chromatin236. Optical tweezers were used to guide the microsphere near the chromatin to bind it. For unfolded chromatin, the chromatin was then wrapped around a pillar and the bead was bound to a second point, while the chromatin was pulled away from the pillar through pressure-driven flow. For folded chromatin, the bead was captured between two pillars, and the chromatin was elongated through the small opening in between the pillars by the flow. Similar methods have been used to explore other protein-DNA interactions not covered here239.
In later studies, these structures and methods were used to tether both ends of the chromatin around different micropillars before extension through hydrodynamic flow237, 238. Fluorescently-labeled marker microspheres were added, which randomly bound to the chromatin fibers. The NaCl concentration was then steadily increased, which affected the chromatin structure and could be directly observed by monitoring the distances between the bound fluorescent microspheres. By analyzing chromatin in this way, chromatin from differentiated mouse cells was found to have higher folding stability than undifferentiated mouse cells238.
Using comparable techniques, nucleosome assembly can also be visualized at the single-molecule resolution in a microfluidic flow cell240. One end of a fluorescently-labeled DNA molecule was immobilized on the flow cell surface by biotin-streptavidin interactions, and the other end extended under hydrodynamic flow. Recombinant histones or yeast nucleoplasmic extracts (YNPE) were then loaded for nucleosome assembling and the nucleosome assembly process was recorded in real-time with fluorescence microscopy.
In a device focused more on in vivo chromosomes, an aperture-based array was developed for in situ fluorescence detection of chromosome movement within a single yeast cell241. The device contained a serpentine channel with an array of perpendicular cell trapping sieves to capture yeast cells that carry GFP-labeled Tet-operator (tetO) repeats. The chromatin of the modified yeast cells subsequently expressed TetR-GFP, a fluorophore, at the tetO position on the chromosome. This enabled direct fluorescent imaging of chromosome dynamics during meiosis in a single cell.
2.4. Microfluidics for analyzing non-coding RNAs
Much like DNA methylation and histone modifications, non-coding RNAs are involved in gene regulations and defects in them have also been implicated in cancer11. MicroRNAs (miRNAs) are the most studied subgroup of ncRNAs and are responsible for the regulation of over 60% of protein-coding genes. Multiple miRNAs have been shown to have oncogenic or tumor suppressive effects and are being used as potential therapeutic targets242. Thus, miRNAs also make enticing targets for better understanding disease pathology and have become a recent focus in microfluidics. While ncRNAs can be sequenced in RNA-seq devices, we chose to largely focus on devices that specifically target ncRNAs.
A microfluidic flow cytometry device was developed for miRNA detection using locked nucleic acid flow fluorescent in situ hybridization (LNA-Flow FISH)243. The device, which utilized their previously published chamber designs and microfluidic FISH methods, contains 10 individual holding chambers with a 3-way channel for hydrodynamic focus and laser detection6, 243, 244. The device was coated in surface adhesive before fixed cells were loaded and allowed to bind to the surface. The cells were incubated with fluorescently-tagged CD69 antibody, permeabilized, and incubated with miR155-specific LNA probe. miR155 was chosen for its role in T-cell activation, as the cells used were from a stimulated T-cell model cell line. The miRNA, hybridized to the LNA probe, was then amplified through rolling circle amplification (RCA) along with the fluorophore FITC, resulting in circular amplicons for robust signal detection. Finally, the cells were incubated with Hoechst to stain the nucleus blue and flowed through the fluorescence detection channel.
A SERS-based microfluidic device was developed using silver coated silicon-PDMS chambers for detection of miR-222, a miRNA involved in multiple cancers245. The device focused on two methods. The one-step method used fluorescently-labeled miRNA that then bound to thiol-capped ssDNA probes immobilized on silver nanoparticles. After hybridization, solid surface enhanced Raman scattering spectra was obtained to quantify the miR-222 level. In contrast, the two-step method only used half of the probe on the nanoparticles. The miRNA bound to the first half of the probe before the second half of the probe, tagged with a Raman label, was then hybridized and quantified. The device could selectively detect miR-222, even in the presence of another miRNA, and had a limit of detection of 0.55nM (one-step) and 1.51nM (two-step). Altogether, this makes it possible to detect miR-222 as a means of early diagnosis for cancer.
In addition to the previously mentioned academic platforms, multiple commercial platforms have been used in miRNA analysis. μParaflo from LC Science was an array-based microfluidic platform for miRNA expression profiling that performed highly parallel RT-PCR to examine a large number of miRNAs246–252. This platform enabled massive custom DNA and RNA probes synthesized on the chamber surface in a photo-programmable manner253. Other microfluidic platforms for miRNA qPCR analysis included the BioMark from Fluidigm254–256, and the TaqMan array microfluidic card257–261. Furthermore, it was also reported that single-nuclei RNA-seq performed in the C1 platform from Fluidigm has been used in identifying differential miRNA expression262.
3. Future directions
Microfluidics has been applied to facilitate high-sensitivity chemical and biological analysis and detection since its inception. However, the majority of previous microfluidic works have been focused on implementing phenotypical examinations (e.g. cell biomechanics) and locus-specific molecular assays (e.g. PCR detection) of cells. Only in recent years, microfluidics started to be increasingly used in conjunction with next-generation sequencing and genome-wide analysis92. A significant fraction of these works included epigenomic assays. Microfluidics provides unique capabilities for processing low-input (including single-cell) samples due to its dramatic sample size reduction and implementing automated and high-throughput operations due to its extraordinary scalability.
Future microfluidic works for epigenomic profiling will largely focus on the below several directions. First, microfluidics will continue to provide solution to low-input epigenomic assays (i.e. analysis based on a low number of cells or single cells). Unique microscale physics and chemistry113, 118, 129 will enable highly efficient sample treatment that is not feasible at large scale. These solutions will be designed to suit the needs of specific epigenomic assays whose working principles can be drastically different. The capability to process low-input samples can be particularly crucial in early screening of cancer for more effective treatment and increased patient survival. Second, automated and high-throughput processing will continue to be a major advantage of microfluidic platform. Decades of work on microfluidics has yielded a significant toolbox for automation and parallel processing. With NGS cost decreasing and potential of precision medicine looming large, the processing of a large number of patient samples is becoming more important than ever and microfluidics has the chance to play a significant role in this process. Furthermore, since microfluidics dramatically reduces costs of reactions through reduction of reagents, these epigenomic tests can be made available to a wide population of patients. Third, the level of integration and complexity on these microfluidic platforms will likely increase and new strategies will be needed. Compared to the vast majority of existing microfluidic assays, most epigenomic profiling requires a much larger number of steps (on both the treatment of genomic DNA and sequencing library preparation). How to seamlessly assemble these steps into an efficient yet manageable microfluidic process will be a new challenge for the community. In the majority of these situations, the complexity may call for use of multiple devices (instead of a highly complex single device with a high probability of failure). New strategies and practices will be needed. Finally, technology development will be closely accompanied by in-depth biomedical studies. Real impact of microfluidic technologies will only be generated by pushing the frontier of biological/clinical knowledge. For example, access to the epigenomic profiles of clinical subjects during treatment and combination of the knowledge with clinical outcomes can provide new breakthroughs in personalized medicine. Given the relatively high implementation barrier of microfluidic devices/assays, microfluidic researchers will need to take it upon themselves to implement the microfluidic tools, often under collaboration with scientists and clinicians, to make major biomedical discoveries.
Acknowledgements
This work was supported by US National Institutes of Health grants CA214176, EB017235, HG009256, HG010195, and seed grants from Center for Engineered Health of Virginia Tech Institute for Critical Technology and Applied Science.
Biography

Chengyu Deng is a Ph.D. candidate in chemical engineering at Virginia Tech working with Professor Chang Lu. She received her B.S. from South China University of Technology in 2015. Her research interests include developing microfluidic-based low-input ChIP-seq and cell-type-specific epigenome profiling.

Lynette Naler is a graduate student in chemical engineering at Virginia Tech. Lynette obtained her B.S. in Chemical and Biomolecular Engineering and B.A. in Computer Science from Johns Hopkins University in 2014. She then spent two years as a research fellow at the National Institute on Aging. Lynette is currently a member of Prof. Chang Lu’s group where she studies epigenetics with the use of microfluidics and bioinformatics.

Chang Lu is the Fred W. Bull Professor at Virginia Tech. He obtained his B.S. in Chemistry from Peking University and PhD in Chemical Engineering from University of Illinois at Urbana-Champaign. He spent two years as a postdoc in Applied Physics at Cornell University. His recent effort has been on profiling cell-type-specific epigenomes using a low number of cells. His lab has published in leading journals such as Nature Methods, Nature Biomedical Engineering, Science Advances, and Nature Protocols on this topic. He received Wallace Coulter Foundation Early Career Award, NSF CAREER Award, and VT Dean’s Award for Research Excellence.
References
- 1.Soshnev AA, Josefowicz SZ and Allis CD, Mol Cell, 2016, 62, 681–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allis CD and Jenuwein T, Nat Rev Genet, 2016, 17, 487–500. [DOI] [PubMed] [Google Scholar]
- 3.Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suner D, Cigudosa JC, Urioste M, Benitez J, Boix-Chornet M, Sanchez-Aguilera A, Ling C, Carlsson E, Poulsen P, Vaag A, Stephan Z, Spector TD, Wu YZ, Plass C and Esteller M, Proc Natl Acad Sci U S A, 2005, 102, 10604–10609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Castillo-Fernandez JE, Spector TD and Bell JT, Genome Med, 2014, 6, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gottesman II and Erlenmeyer-Kimling L, Schizophrenia Research, 2001, 51, 93–102. [DOI] [PubMed] [Google Scholar]
- 6.Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG, Wen B, Wu H, Liu Y, Diep D, Briem E, Zhang K, Irizarry RA and Feinberg AP, Nat Genet, 2011, 43, 768–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Suzuki MM and Bird A, Nature Reviews Genetics, 2008, 9, 465–476. [DOI] [PubMed] [Google Scholar]
- 8.Park PJ, Nature Reviews Genetics, 2009, 10, 669–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collas P, Mol Biotechnol, 2010, 45, 87–100. [DOI] [PubMed] [Google Scholar]
- 10.Pombo A and Dillon N, Nat Rev Mol Cell Biol, 2015, 16, 245–257. [DOI] [PubMed] [Google Scholar]
- 11.Esteller M, Nat Rev Genet, 2011, 12, 861–874. [DOI] [PubMed] [Google Scholar]
- 12.Peschansky VJ and Wahlestedt C, Epigenetics, 2014, 9, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Versteege I, Sevenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, Aurias A and Delattre O, Nature, 1998, 394, 203–206. [DOI] [PubMed] [Google Scholar]
- 14.Pugh TJ, Weeraratne SD, Archer TC, Krummel DAP, Auclair D, Bochicchio J, Carneiro MO, Carter SL, Cibulskis K, Erlich RL, Greulich H, Lawrence MS, Lennon NJ, McKenna A, Meldrim J, Ramos AH, Ross MG, Russ C, Shefler E, Sivachenko A, Sogoloff B, Stojanov P, Tamayo P, Mesirov JP, Amani V, Teider N, Sengupta S, Francois JP, Northcott PA, Taylor MD, Yu FR, Crabtree GR, Kautzman AG, Gabriel SB, Getz G, Jager N, Jones DTW, Lichter P, Pfister SM, Roberts TM, Meyerson M, Pomeroy SL and Cho YJ, Nature, 2012, 488, 106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang JH, Benavente CA, McEvoy J, Flores-Otero J, Ding L, Chen X, Ulyanov A, Wu G, Wilson M, Wang JM, Brennan R, Rusch M, Manning AL, Ma J, Easton J, Shurtleff S, Mullighan C, Pounds S, Mukatira S, Gupta P, Neale G, Zhao D, Lu C, Fulton RS, Fulton LL, Hong X, Dooling DJ, Ochoa K, Naeve C, Dyson NJ, Mardis ER, Bahrami A, Ellison D, Wilson RK, Downing JR and Dyer MA, Nature, 2012, 481, 329–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee RS, Stewart C, Carter SL, Ambrogio L, Cibulskis K, Sougnez C, Lawrence MS, Auclair D, Mora J, Golub TR, Biegel JA, Getz G and Roberts CWM, J Clin Invest, 2012, 122, 2983–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mack SC, Witt H, Piro RM, Gu L, Zuyderduyn S, Stutz AM, Wang X, Gallo M, Garzia L, Zayne K, Zhang X, Ramaswamy V, Jager N, Jones DT, Sill M, Pugh TJ, Ryzhova M, Wani KM, Shih DJ, Head R, Remke M, Bailey SD, Zichner T, Faria CC, Barszczyk M, Stark S, Seker-Cin H, Hutter S, Johann P, Bender S, Hovestadt V, Tzaridis T, Dubuc AM, Northcott PA, Peacock J, Bertrand KC, Agnihotri S, Cavalli FM, Clarke I, Nethery-Brokx K, Creasy CL, Verma SK, Koster J, Wu X, Yao Y, Milde T, Sin-Chan P, Zuccaro J, Lau L, Pereira S, Castelo-Branco P, Hirst M, Marra MA, Roberts SS, Fults D, Massimi L, Cho YJ, Van Meter T, Grajkowska W, Lach B, Kulozik AE, von Deimling A, Witt O, Scherer SW, Fan X, Muraszko KM, Kool M, Pomeroy SL, Gupta N, Phillips J, Huang A, Tabori U, Hawkins C, Malkin D, Kongkham PN, Weiss WA, Jabado N, Rutka JT, Bouffet E, Korbel JO, Lupien M, Aldape KD, Bader GD, Eils R, Lichter P, Dirks PB, Pfister SM, Korshunov A and Taylor MD, Nature, 2014, 506, 445–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feinberg AP, New Engl J Med, 2018, 378, 1323–1334. [DOI] [PubMed] [Google Scholar]
- 19.Feinberg AP, Koldobskiy MA and Gondor A, Nature Reviews Genetics, 2016, 17, 284–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ehrlich M, Epigenomics-Uk, 2009, 1, 239–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deaton AM and Bird A, Gene Dev, 2011, 25, 1010–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E, Bussaglia E, Prat J, Harkes IC, Repasky EA, Gabrielson E, Schutte M, Baylin SB and Herman JG, Jnci-J Natl Cancer I, 2000, 92, 564–569. [DOI] [PubMed] [Google Scholar]
- 23.Chervona Y and Costa M, Am J Cancer Res, 2012, 2, 589–597. [PMC free article] [PubMed] [Google Scholar]
- 24.Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, Iyer NG, Perez-Rosado A, Calvo E, Lopez JA, Cano A, Calasanz MJ, Colomer D, Piris MA, Ahn N, Imhof A, Caldas C, Jenuwein T and Esteller M, Nature Genetics, 2005, 37, 391–400. [DOI] [PubMed] [Google Scholar]
- 25.Yuen BTK and Knoepfler PS, Cancer Cell, 2013, 24, 567–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hajjari M and Salavaty A, Cancer Biol Med, 2015, 12, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zane L, Sharma V and Misteli T, Trends Cell Biol, 2014, 24, 686–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heerboth S, Lapinska K, Snyder N, Leary M, Rollinson S and Sarkar S, Genet Epigenetics, 2014, 6, 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hamm CA and Costa FF, Pharmacol Therapeut, 2015, 151, 72–86. [DOI] [PubMed] [Google Scholar]
- 30.Yan W, Herman JG and Guo M, Epigenomics, 2016, 8, 119–133. [DOI] [PubMed] [Google Scholar]
- 31.Brueckner B, Boy RG, Siedlecki P, Musch T, Kliem HC, Zielenkiewicz P, Suhai S, Wiessler M and Lyko F, Cancer Res, 2005, 65, 6305–6311. [DOI] [PubMed] [Google Scholar]
- 32.Kaminskas E, Farrell AT, Wang YC, Sridhara R and Pazdur R, Oncologist, 2005, 10, 176–182. [DOI] [PubMed] [Google Scholar]
- 33.Mann BS, Johnson JR, He K, Sridhara R, Abraham S, Booth BP, Verbois L, Morse DE, Jee JM, Pope S, Harapanhalli RS, Dagher R, Farrell A, Justice R and Pazdur R, Clin Cancer Res, 2007, 13, 2318–2322. [DOI] [PubMed] [Google Scholar]
- 34.Santos FPS, Kantarjian H, Garcia-Manero G, Issa JP and Ravandi F, Expert Rev Anticanc, 2010, 10, 9–22. [DOI] [PubMed] [Google Scholar]
- 35.Iyer SP and Foss FF, Oncologist, 2015, 20, 1084–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cokus SJ, Feng SH, Zhang XY, Chen ZG, Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M and Jacobsen SE, Nature, 2008, 452, 215–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Laird PW, Nature Reviews Genetics, 2010, 11, 191–203. [DOI] [PubMed] [Google Scholar]
- 38.Jacinto FV, Ballestar E and Esteller M, Biotechniques, 2008, 44, 35–43. [DOI] [PubMed] [Google Scholar]
- 39.Down TA, Rakyan VK, Turner DJ, Flicek P, Li H, Kulesha E, Graf S, Johnson N, Herrero J, Tomazou EM, Thorne NP, Backdahl L, Herberth M, Howe KL, Jackson DK, Miretti MM, Marioni JC, Birney E, Hubbard TJ, Durbin R, Tavare S and Beck S, Nat Biotechnol, 2008, 26, 779–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Serre D, Lee BH and Ting AH, Nucleic Acids Res, 2010, 38, 391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li N, Ye M, Li Y, Yan Z, Butcher LM, Sun J, Han X, Chen Q, Zhang X and Wang J, Methods, 2010, 52, 203–212. [DOI] [PubMed] [Google Scholar]
- 42.Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D’Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C, Zhao Y, Turecki G, Delaney A, Varhol R, Thiessen N, Shchors K, Heine VM, Rowitch DH, Xing X, Fiore C, Schillebeeckx M, Jones SJ, Haussler D, Marra MA, Hirst M, Wang T and Costello JF, Nature, 2010, 466, 253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Song CX, Yi C and He C, Nat Biotechnol, 2012, 30, 1107–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Booth MJ, Branco MR, Ficz G, Oxley D, Krueger F, Reik W and Balasubramanian S, Science, 2012, 336, 934–937. [DOI] [PubMed] [Google Scholar]
- 45.Yu M, Hon GC, Szulwach KE, Song CX, Jin P, Ren B and He C, Nat Protoc, 2012, 7, 2159–2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Song CX, Szulwach KE, Dai Q, Fu Y, Mao SQ, Lin L, Street C, Li Y, Poidevin M, Wu H, Gao J, Liu P, Li L, Xu GL, Jin P and He C, Cell, 2013, 153, 678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shen L, Wu H, Diep D, Yamaguchi S, D’Alessio AC, Fung HL, Zhang K and Zhang Y, Cell, 2013, 153, 692–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ 3rd, Gingeras TR, Schreiber SL and Lander ES, Cell, 2005, 120, 169–181. [DOI] [PubMed] [Google Scholar]
- 49.Dekker J, Rippe K, Dekker M and Kleckner N, Science, 2002, 295, 1306–1311. [DOI] [PubMed] [Google Scholar]
- 50.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES and Dekker J, Science, 2009, 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Buenrostro JD, Giresi PG, Zaba LC, Chang HY and Greenleaf WJ, Nat Methods, 2013, 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Crawford GE, Holt IE, Whittle J, Webb BD, Tai D, Davis S, Margulies EH, Chen Y, Bernat JA and Ginsburg D, Genome research, 2006, 16, 123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schones DE, Cui K, Cuddapah S, Roh TY, Barski A, Wang Z, Wei G and Zhao K, Cell, 2008, 132, 887–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kozomara A and Griffiths-Jones S, Nucleic Acids Res, 2011, 39, D152–D157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Veneziano D, Nigita G and Ferro A, Frontiers in bioengineering and biotechnology, 2015, 3, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chu C, Qu K, Zhong FL, Artandi SE and Chang HY, Molecular cell, 2011, 44, 667–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chu C, Quinn J and Chang HY, J Vis Exp, 2012, e3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Belton JM, McCord RP, Gibcus JH, Naumova N, Zhan Y and Dekker J, Methods, 2012, 58, 268–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Song L and Crawford GE, Cold Spring Harb Protoc, 2010, 2010, pdb prot5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nestor CE, Ottaviano R, Reinhardt D, Cruickshanks HA, Mjoseng HK, McPherson RC, Lentini A, Thomson JP, Dunican DS and Pennings S, Genome biology, 2015, 16, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Skene PJ and Henikoff S, Elife, 2017, 6, e21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Skene PJ, Henikoff JG and Henikoff S, Nature protocols, 2018, 13, 1006. [DOI] [PubMed] [Google Scholar]
- 63.Harada A, Maehara K, Handa T, Arimura Y, Nogami J, Hayashi-Takanaka Y, Shirahige K, Kurumizaka H, Kimura H and Ohkawa Y, Nat Cell Biol, 2019, 21, 287–296. [DOI] [PubMed] [Google Scholar]
- 64.Miura F, Enomoto Y, Dairiki R and Ito T, Nucleic Acids Res, 2012, 40, e136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES and Jaenisch R, Nucleic Acids Research, 2005, 33, 5868–5877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schmidt C, Rendeiro AF, Sheffield NC and Bock C, Nat Methods, 2015, 12, 963–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang Q, Gu L, Adey A, Radlwimmer B, Wang W, Hovestadt V, Bahr M, Wolf S, Shendure J, Eils R, Plass C and Weichenhan D, Nature Protocols, 2013, 8, 2022–2032. [DOI] [PubMed] [Google Scholar]
- 68.Clark SJ, Lee HJ, Smallwood SA, Kelsey G and Reik W, Genome Biol, 2016, 17, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schmid A, Kortmann H, Dittrich PS and Blank LM, Curr Opin Biotech, 2010, 21, 12–20. [DOI] [PubMed] [Google Scholar]
- 70.Gravina S, Ganapathi S and Vijg J, Nucleic Acids Res, 2015, 43, e93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Guo HS, Zhu P, Guo F, Li XL, Wu XL, Fan XY, Wen L and Tang FC, Nature Protocols, 2015, 10, 645–659. [DOI] [PubMed] [Google Scholar]
- 72.Guo HS, Zhu P, Wu XL, Li XL, Wen L and Tang FC, Genome Res, 2013, 23, 2126–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Farlik M, Sheffield NC, Nuzzo A, Datlinger P, Schonegger A, Klughammer J and Bock C, Cell Reports, 2015, 10, 1386–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Clark SJ, Smallwood SA, Lee HJ, Krueger F, Reik W and Kelsey G, Nat. Protoc, 2017, 12, 534–547. [DOI] [PubMed] [Google Scholar]
- 75.Smallwood SA, Lee HJ, Angermueller C, Krueger F, Saadeh H, Peat J, Andrews SR, Stegle O, Reik W and Kelsey G, Nat Methods, 2014, 11, 817–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhu C, Gao Y, Guo H, Xia B, Song J, Wu X, Zeng H, Kee K, Tang F and Yi C, Cell Stem Cell, 2017, 20, 720–731. e725. [DOI] [PubMed] [Google Scholar]
- 77.Ramani V, Deng X, Qiu R, Gunderson KL, Steemers FJ, Disteche CM, Noble WS, Duan Z and Shendure J, Nat Methods, 2017, 14, 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, Laue ED, Tanay A and Fraser P, Nature, 2013, 502, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cusanovich DA, Daza R, Adey A, Pliner HA, Christiansen L, Gunderson KL, Steemers FJ, Trapnell C and Shendure J, Science, 2015, 348, 910–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Buenostro JD, Wu BJ, Litzenburger UM, Ruff D, Gonzales ML, Snyder MP, Chang HY and Greenleaf WJ, Nature, 2015, 523, 486–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Macaulay IC, Ponting CP and Voet T, Trends Genet, 2017, 33, 155–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hu Y, Huang K, An Q, Du G, Hu G, Xue J, Zhu X, Wang CY, Xue Z and Fan G, Genome Biol, 2016, 17, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Angermueller C, Clark SJ, Lee HJ, Macaulay IC, Teng MJ, Hu TX, Krueger F, Smallwood S, Ponting CP, Voet T, Kelsey G, Stegle O and Reik W, Nat Methods, 2016, 13, 229–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hou Y, Guo HH, Cao C, Li XL, Hu BQ, Zhu P, Wu XL, Wen L, Tang FC, Huang YY and Peng JR, Cell Res, 2016, 26, 304–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Volpatti LR and Yetisen AK, Trends Biotechnol, 2014, 32, 347–350. [DOI] [PubMed] [Google Scholar]
- 86.Kim SM, Lee SH and Suh KY, Lab on a Chip, 2008, 8, 1015–1023. [DOI] [PubMed] [Google Scholar]
- 87.El-Ali J, Sorger PK and Jensen KF, Nature, 2006, 442, 403–411. [DOI] [PubMed] [Google Scholar]
- 88.Andersson H and van den Berg A, Sensor Actuat B-Chem, 2003, 92, 315–325. [Google Scholar]
- 89.Salieb-Beugelaar GB, Simone G, Arora A, Philippi A and Manz A, Analytical Chemistry, 2010, 82, 4848–4864. [DOI] [PubMed] [Google Scholar]
- 90.Mazutis L, Gilbert J, Ung WL, Weitz DA, Griffiths AD and Heyman JA, Nature Protocols, 2013, 8, 870–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Murphy TW, Zhang Q, Naler LB, Ma S and Lu C, Analyst, 2018, 143, 60–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ma S, Murphy TW and Lu C, Biomicrofluidics, 2017, 11, 021501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dittrich PS and Manz A, Nature Reviews Drug Discovery, 2006, 5, 210–218. [DOI] [PubMed] [Google Scholar]
- 94.Wu MH, Huang SB and Lee GB, Lab on a Chip, 2010, 10, 939–956. [DOI] [PubMed] [Google Scholar]
- 95.Pihl J, Karlsson M and Chiu DT, Drug Discov Today, 2005, 10, 1377–1383. [DOI] [PubMed] [Google Scholar]
- 96.Kleinstreuer C, Li J and Koo J, Int J Heat Mass Tran, 2008, 51, 5590–5597. [Google Scholar]
- 97.Saleh-Lakha S and Trevors JT, J Microbiol Meth, 2010, 82, 108–111. [DOI] [PubMed] [Google Scholar]
- 98.Weibel DB, DiLuzio WR and Whitesides GM, Nat Rev Microbiol, 2007, 5, 209–218. [DOI] [PubMed] [Google Scholar]
- 99.Ng AHC, Uddayasankar U and Wheeler AR, Anal Bioanal Chem, 2010, 397, 991–1007. [DOI] [PubMed] [Google Scholar]
- 100.Baratchi S, Khoshmanesh K, Sacristan C, Depoil D, Wlodkowic D, McIntyre P and Mitchell A, Biotechnol Adv, 2014, 32, 333–346. [DOI] [PubMed] [Google Scholar]
- 101.Squires TM and Quake SR, Rev Mod Phys, 2005, 77, 977–1026. [Google Scholar]
- 102.Fullwood MJ, Liu MH, Pan YF, Liu J, Xu H, Mohamed YB, Orlov YL, Velkov S, Ho A and Mei PH, Nature, 2009, 462, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gaulton KJ, Nammo T, Pasquali L, Simon JM, Giresi PG, Fogarty MP, Panhuis TM, Mieczkowski P, Secchi A, Bosco D, Berney T, Montanya E, Mohlke KL, Lieb JD and Ferrer J, Nat Genet, 2010, 42, 255–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Derrington IM, Butler TZ, Collins MD, Manrao E, Pavlenok M, Niederweis M and Gundlach JH, Proc Natl Acad Sci U S A, 2010, 107, 16060–16065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, Tyson JR, Beggs AD, Dilthey AT, Fiddes IT, Malla S, Marriott H, Nieto T, O’Grady J, Olsen HE, Pedersen BS, Rhie A, Richardson H, Quinlan AR, Snutch TP, Tee L, Paten B, Phillippy AM, Simpson JT, Loman NJ and Loose M, Nat Biotechnol, 2018, 36, 338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Garalde DR, Snell EA, Jachimowicz D, Sipos B, Lloyd JH, Bruce M, Pantic N, Admassu T, James P, Warland A, Jordan M, Ciccone J, Serra S, Keenan J, Martin S, McNeill L, Wallace EJ, Jayasinghe L, Wright C, Blasco J, Young S, Brocklebank D, Juul S, Clarke J, Heron AJ and Turner DJ, Nat Methods, 2018, 15, 201–206. [DOI] [PubMed] [Google Scholar]
- 107.Rhee HS and Pugh BF, Cell, 2011, 147, 1408–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Harismendy O, Notani D, Song X, Rahim NG, Tanasa B, Heintzman N, Ren B, Fu XD, Topol EJ, Rosenfeld MG and Frazer KA, Nature, 2011, 470, 264–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, Bibillo A, Bjornson K, Chaudhuri B, Christians F, Cicero R, Clark S, Dalal R, Dewinter A, Dixon J, Foquet M, Gaertner A, Hardenbol P, Heiner C, Hester K, Holden D, Kearns G, Kong X, Kuse R, Lacroix Y, Lin S, Lundquist P, Ma C, Marks P, Maxham M, Murphy D, Park I, Pham T, Phillips M, Roy J, Sebra R, Shen G, Sorenson J, Tomaney A, Travers K, Trulson M, Vieceli J, Wegener J, Wu D, Yang A, Zaccarin D, Zhao P, Zhong F, Korlach J and Turner S, Science, 2009, 323, 133–138. [DOI] [PubMed] [Google Scholar]
- 110.Rotem A, Ram O, Shoresh N, Sperling RA, Goren A, Weitz DA and Bernstein BE, Nat. Biotechnol, 2015, 33, 1165–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kind J, Pagie L, de Vries SS, Nahidiazar L, Dey SS, Bienko M, Zhan Y, Lajoie B, de Graaf CA and Amendola M, Cell, 2015, 163, 134–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jin W, Tang Q, Wan M, Cui K, Zhang Y, Ren G, Ni B, Sklar J, Przytycka TM, Childs R, Levens D and Zhao K, Nature, 2015, 528, 142–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cao ZN, Chen CY, He B, Tan K and Lu C, Nat Methods, 2015, 12, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schmidl C, Rendeiro AF, Sheffield NC and Bock C, Nat. Methods, 2015, 12, 963–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mooijman D, Dey SS, Boisset JC, Crosetto N and van Oudenaarden A, Nat Biotechnol, 2016, 34, 852–856. [DOI] [PubMed] [Google Scholar]
- 116.Wu X, Inoue A, Suzuki T and Zhang Y, Genes Dev, 2017, 31, 511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Mulqueen RM, Pokholok D, Norberg SJ, Torkenczy KA, Fields AJ, Sun D, Sinnamon JR, Shendure J, Trapnell C, O’Roak BJ, Xia Z, Steemers FJ and Adey AC, Nat Biotechnol, 2018, 36, 428–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ma S, Revenga MD, Sun ZX, Sun C, Murphy TW, Xie HH, Gonzalez-Maeso J and Lu C, Nat Biomed Eng, 2018, 2, 183–194. [PMC free article] [PubMed] [Google Scholar]
- 119.Grosselin K, Durand A, Marsolier J, Poitou A, Marangoni E, Nemati F, Dahmani A, Lameiras S, Reyal F and Frenoy O, Nature genetics, 2019, 51, 1060. [DOI] [PubMed] [Google Scholar]
- 120.Wu AR, Hiatt JB, Lu R, Attema JL, Lobo NA, Weissman IL, Clarke MF and Quake SR, Lab on a Chip, 2009, 9, 1365–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wu AR and Quake SR, Cold Spring Harbor Protocols, 2016, 2016, pdb.prot084996. [DOI] [PubMed] [Google Scholar]
- 122.Wu AR, Kawahara TLA, Rapicavoli NA, van Riggelen J, Shroff EH, Xu LW, Felsher DW, Chang HY and Quake SR, Lab on a Chip, 2012, 12, 2190–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Oh HJ, Park JY, Park SE, Lee BY, Park JS, Kim SK, Yoon TJ and Lee SH, Anal Chem, 2009, 81, 2832–2839. [DOI] [PubMed] [Google Scholar]
- 124.Geng T, Bao N, Litt MD, Glaros TG, Li LW and Lu C, Lab on a Chip, 2011, 11, 2842–2848. [DOI] [PubMed] [Google Scholar]
- 125.Cao ZN and Lu C, Analytical Chemistry, 2016, 88, 1965–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Teste B, Champ J, Londono-Vallejo A, Descroix S, Malaquin L, Viovy JL, Draskovic I and Mottet G, Lab on a Chip, 2017, 17, 530–537. [DOI] [PubMed] [Google Scholar]
- 127.Shen J, Jiang DQ, Fu YS, Wu XL, Guo HS, Feng BX, Pang YH, Streets AM, Tang FC and Huang YY, Cell Res, 2015, 25, 143–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cox MC, Deng C, Naler L, Lu C and Verbridge SS, ACS Biomaterials Science & Engineering, 2019, 5, 1544–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ma S, Hsieh YP, Ma J and Lu C, Sci Adv, 2018, 4, eaar8187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Murphy TW, Hsieh YP, Ma S, Zhu Y and Lu C, Analytical Chemistry, 2018, 90, 7666–7674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Matsuoka T, Kim BC, Huang JX, Douville NJ, Thouless MD and Takayama S, Nano Lett, 2012, 12, 6480–6484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lim SF, Karpusenko A, Blumers AL, Streng DE and Riehn R, Biomicrofluidics, 2013, 7, 64105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cipriany BR, Zhao RQ, Murphy PJ, Levy SL, Tan CP, Craighead HG and Soloway PD, Analytical Chemistry, 2010, 82, 2480–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Murphy PJ, Cipriany BR, Wallin CB, Ju CY, Szeto K, Hagarman JA, Benitez JJ, Craighead HG and Soloway PD, P Natl Acad Sci USA, 2013, 110, 7772–7777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cerf A, Tian HC and Craighead HG, Acs Nano, 2012, 6, 7928–7934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Farnham PJ, Nat Rev Genet, 2009, 10, 605–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Maerkl SJ and Quake SR, Science, 2007, 315, 233–237. [DOI] [PubMed] [Google Scholar]
- 138.Fordyce PM, Gerber D, Tran D, Zheng JS, Li H, DeRisi JL and Quake SR, Nat Biotechnol, 2010, 28, 970–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Nelson CS, Fuller CK, Fordyce PM, Greninger AL, Li H and DeRisi JL, Nucleic Acids Research, 2013, 41, 5991–6004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lohse MB, Hernday AD, Fordyce PM, Noiman L, Sorrells TR, Hanson-Smith V, Nobile CJ, DeRisi JL and Johnson AD, P Natl Acad Sci USA, 2013, 110, 7660–7665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Glick Y, Orenstein Y, Chen D, Avrahami D, Zor T, Shamir R and Gerber D, Nucleic Acids Res, 2016, 44, e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hens K, Feuz JD, Isakova A, Iagovitina A, Massouras A, Bryois J, Callaerts P, Celniker SE and Deplancke B, Nat Methods, 2011, 8, 1065–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Gubelmann C, Waszak SM, Isakova A, Holcombe W, Hens K, Iagovitina A, Feuz JD, Raghav SK, Simicevic J and Deplancke B, Mol Syst Biol, 2013, 9, 682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Chen D, Orenstein Y, Golodnitsky R, Pellach M, Avrahami D, Wachtel C, Ovadia-Shochat A, Shir-Shapira H, Kedmi A, Juven-Gershon T, Shamir R and Gerber D, Sci. Rep, 2016, 6, 33351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Herman JG, Graff JR, Myohanen S, Nelkin BD and Baylin SB, P Natl Acad Sci USA, 1996, 93, 9821–9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Chan MWY, Chan LW, Tang NLS, Tong JHM, Lo KW, Lee TL, Cheung HY, Wong WS, Chan PSF, Lai FMM and To KF, Clin Cancer Res, 2002, 8, 464–470. [PubMed] [Google Scholar]
- 147.Yoon J, Park MK, Lee TY, Yoon YJ and Shin Y, Lab on a Chip, 2015, 15, 35303539. [DOI] [PubMed] [Google Scholar]
- 148.Zhang Y, Bailey V, Puleo CM, Easwaran H, Griffiths E, Herman JG, Baylin SB and Wang TH, Lab on a Chip, 2009, 9, 1059–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Heng JF, Zhang F, Guo XW, Tang LL, Peng LM, Luo XP, Xu XX, Wang SM, Dai LZ and Wang J, Oncotarget, 2017, 8, 25442–25454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Li ZB, Guo XW, Wu YP, Li SY, Yan JH, Peng LM, Xiao Z, Wang SM, Deng ZP, Dai LZ, Yi WJ, Xia K, Tang LL and Wang J, Breast Cancer Res Tr, 2015, 149, 767–779. [DOI] [PubMed] [Google Scholar]
- 151.Li ZB, Heng JF, Yan JH, Guo XW, Tang LL, Chen M, Peng LM, Wu YP, Wang SM, Xiao Z, Deng ZP, Dai LZ and Wang J, Breast Cancer Res Tr, 2016, 160, 371–383. [DOI] [PubMed] [Google Scholar]
- 152.Fang G, Munera D, Friedman DI, Mandlik A, Chao MC, Banerjee O, Feng Z, Losic B, Mahajan MC and Jabado OJ, Nature biotechnology, 2012, 30, 1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Zhang HD, Zhou J, Xu ZR, Song J, Dai J, Fang J and Fang ZL, Lab Chip, 2007, 7, 1162–1170. [DOI] [PubMed] [Google Scholar]
- 154.Zhang HD, Shan LF, Wang XN, Ma Q and Fang J, Biosens Bioelectron, 2013, 42, 503–511. [DOI] [PubMed] [Google Scholar]
- 155.Liu FW, Liao HF, Lin SP and Lu YW, Lab on a Chip, 2018, 18, 514–521. [DOI] [PubMed] [Google Scholar]
- 156.Athamanolap P, Shin DJ and Wang TH, Jala-J Lab Autom, 2014, 19, 304–312. [DOI] [PubMed] [Google Scholar]
- 157.O’Keefe CM, Pisanic TR 2nd, Zec H, Overman MJ, Herman JG and Wang TH, Sci Adv, 2018, 4, eaat6459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Pisanic TR 2nd, Athamanolap P, Poh W, Chen C, Hulbert A, Brock MV, Herman JG and Wang TH, Nucleic Acids Res, 2015, 43, e154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Stark A, Shin DJ, Pisanic T 2nd, Hsieh K and Wang TH, Biomed Microdevices, 2016, 18, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Pursey JP, Chen Y, Stulz E, Park MK and Kongsuphol P, Sens. Actuators B-Chem, 2017, 251, 34–39. [Google Scholar]
- 161.Kurita R, Yanagisawa H, Yoshioka K and Niwa O, Analytical Chemistry, 2015, 87, 11581–11586. [DOI] [PubMed] [Google Scholar]
- 162.Wu ZH, Bai YN, Cheng ZL, Liu FM, Wang P, Yang DW, Li G, Jin QH, Mao HJ and Zhao JL, Biosens Bioelectron, 2017, 96, 339–344. [DOI] [PubMed] [Google Scholar]
- 163.Tanaka K and Okamoto A, Bioorg Med Chem Lett, 2007, 17, 1912–1915. [DOI] [PubMed] [Google Scholar]
- 164.Cerf A, Cipriany BR, Benitez JJ and Craighead HG, Analytical Chemistry, 2011, 83, 8073–8077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Cerf A, Alava T, Barton RA and Craighead HG, Nano Lett, 2011, 11, 42324238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lim SF, Karpusenko A, Sakon JJ, Hook JA, Lamar TA and Riehn R, Biomicrofluidics, 2011, 5, 34106–341068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Hasegawa K, Matsumoto M, Hosokawa K and Maeda M, Anal Sci, 2016, 32, 603606. [DOI] [PubMed] [Google Scholar]
- 168.Heimer BW, Shatova TA, Lee JK, Kaastrup K and Sikes HD, Analyst, 2014, 139, 3695–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.De A, Sparreboom W, van den Berg A and Carlen ET, Biomicrofluidics, 2014, 8, 054119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Cipriany BR, Murphy PJ, Hagarman JA, Cerf A, Latulippe D, Levy SL, Benítez JJ, Tan CP, Topolancik J, Soloway PD and Craighead HG, Proc. Natl. Acad. Sci. U. S. A, 2012, 109, 8477–8482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Hong SA, Kim YJ, Kim SJ and Yang S, Biosens Bioelectron, 2018, 107, 103110. [DOI] [PubMed] [Google Scholar]
- 172.Zhu Y, Cao Z and Lu C, Analyst, 2019, 144, 1904–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Lee TY, Shin Y and Park MK, Lab on a Chip, 2014, 14, 4220–4229. [DOI] [PubMed] [Google Scholar]
- 174.Shin Y, Perera AP, Kim KW and Park MK, Lab Chip, 2013, 13, 2106–2114. [DOI] [PubMed] [Google Scholar]
- 175.Lorthongpanich C, Cheow LF, Balu S, Quake SR, Knowles BB, Burkholder WF, Solter D and Messerschmidt DM, Science, 2013, 341, 1110–1112. [DOI] [PubMed] [Google Scholar]
- 176.Cheow LF, Quake SR, Burkholder WF and Messerschmidt DM, Nature Protocols, 2015, 10, 619–631. [DOI] [PubMed] [Google Scholar]
- 177.Cheow LF, Courtois ET, Tan Y, Viswanathan R, Xing Q, Tan RZ, Tan DS, Robson P, Loh YH, Quake SR and Burkholder WF, Nat Methods, 2016, 13, 833–836. [DOI] [PubMed] [Google Scholar]
- 178.Alvarez JR, Skachkov D, Massey SE, Kalitsov A and Velev JP, Front. Genet, 2015, 6, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Rand AC, Jain M, Eizenga JM, Musselman-Brown A, Olsen HE, Akeson M and Paten B, Nat Methods, 2017, 14, 411–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Wang Y, Zhang Y, Guo Y and Kang XF, Sci Rep, 2017, 7, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Johnson RP, Fleming AM, Perera RT, Burrows CJ and White HS, Journal of the American Chemical Society, 2017, 139, 2750–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Euskirchen P, Bielle F, Labreche K, Kloosterman WP, Rosenberg S, Daniau M, Schmitt C, Masliah-Planchon J, Bourdeaut F, Dehais C, Marie Y, Delattre JY and Idbaih A, Acta Neuropathol, 2017, 134, 691–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Craig JM, Laszlo AH, Derrington IM, Ross BC, Brinkerhoff H, Nova IC, Doering K, Tickman BI, Svet MT and Gundlach JH, PLOS One,2015, 10, e0143253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Carson S, Wilson J, Aksimentiev A, Weigele PR and Wanunu M, Nucleic Acids Research, 2016, 44, 2085–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Zhang X, Price NE, Fang X, Yang Z, Gu L-Q and Gates KS, Acs Nano, 2015, 9, 11812–11819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Ding Y, Fleming AM, White HS and Burrows CJ, Acs Nano, 2015, 9, 11325–11332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Ahmed T, Haraldsen JT, Zhu JX and Balatsky AV, J Phys Chem Lett, 2014, 5, 2601–2607. [DOI] [PubMed] [Google Scholar]
- 188.Kang I, Wang Y, Reagan C, Fu Y, Wang MX and Gu LQ, Sci Rep, 2013, 3, 2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Laszlo AH, Derrington IM, Brinkerhoff H, Langford KW, Nova IC, Samson JM, Bartlett JJ, Pavlenok M and Gundlach JH, Proceedings of the National Academy of Sciences of the United States of America, 2013, 110, 18904–18909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Wanunu M, Cohen-Karni D, Johnson RR, Fields L, Benner J, Peterman N, Zheng Y, Klein ML and Drndic M, J Am Chem Soc, 2011, 133, 486–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Wallace EVB, Stoddart D, Heron AJ, Mikhailova E, Maglia G, Donohoe TJ and Bayley H, Chem Commun, 2010, 46, 8195–8197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Clarke J, Wu HC, Jayasinghe L, Patel A, Reid S and Bayley H, Nat Nanotechnol, 2009, 4, 265–270. [DOI] [PubMed] [Google Scholar]
- 193.Simpson JT, Workman RE, Zuzarte P, David M, Dursi L and Timp W, Nature methods, 2017, 14, 407. [DOI] [PubMed] [Google Scholar]
- 194.Mirsaidov U, Timp W, Zou X, Dimitrov V, Schulten K, Feinberg AP and Timp G, Biophysical Journal, 2009, 96, L32–L34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Tsutsui M, Matsubara K, Ohshiro T, Furuhashi M, Taniguchi M and Kawai T, J Am Chem Soc, 2011, 133, 9124–9128. [DOI] [PubMed] [Google Scholar]
- 196.Lu X and He C, ChemBioChem, 2013, 14, 1289–1290. [DOI] [PubMed] [Google Scholar]
- 197.Yu J, Cao C and Long YT, Analytical Chemistry, 2017, 89, 11685–11689. [DOI] [PubMed] [Google Scholar]
- 198.Rauf S, Zhang L, Ali A, Ahmad J, Liu Y and Li JH, Analytical Chemistry, 2017, 89, 13252–13260. [DOI] [PubMed] [Google Scholar]
- 199.Schreiber J, Wescoe ZL, Abu-Shumays R, Vivian JT, Baatar B, Karplus K and Akeson M, P Natl Acad Sci USA, 2013, 110, 18910–18915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Gilboa T, Torfstein C, Juhasz M, Grunwald A, Ebenstein Y, Weinhold E and Meller A, Acs Nano, 2016, 10, 8861–8870. [DOI] [PubMed] [Google Scholar]
- 201.Sarathy A, Qiu H and Leburton JP, J Phys Chem B, 2017, 121, 3757–3763. [DOI] [PubMed] [Google Scholar]
- 202.Qiu H, Sarathy A and Schulten K and Leburton JP, npj 2D Materials and Applications, 2017, 1, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Shim J, Kim Y, Humphreys GI, Nardulli AM, Kosari F, Vasmatzis G, Taylor WR, Ahlquist DA, Myong S and Bashir R, Acs Nano, 2015, 9, 290–300. [DOI] [PubMed] [Google Scholar]
- 204.Shim J, Humphreys GI, Venkatesan BM, Munz JM, Zou X, Sathe C, Schulten K, Kosari F, Nardulli AM, Vasmatzis G and Bashir R, Sci. Rep, 2013, 3, 1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Shim J, Banerjee S, Qiu H, Smithe KKH, Estrada D, Bello J, Pop E, Schulten K and Bashir R, Nanoscale, 2017, 9, 14836–14845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Sarathy A, Athreya NB, Varshney LR and Leburton JP, Journal of Physical Chemistry Letters, 2018, 9, 5718–5725. [DOI] [PubMed] [Google Scholar]
- 207.Healey MJ, Rowe W, Siati S, Sivakumaran M and Platt M, Acs Sensors, 2018, 3, 655–660. [DOI] [PubMed] [Google Scholar]
- 208.Squires A, Atas E and Meller A, Sci Rep, 2015, 5, 11643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Riedl J, Ding Y, Fleming AM and Burrows CJ, Nature communications, 2015, 6, 8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Carlsen AT, Zahid OK, Ruzicka JA, Taylor EW and Hall AR, Nano Lett., 2014, 14, 5488–5492. [DOI] [PubMed] [Google Scholar]
- 211.Zahid OK, Zhao BS, He C and Hall AR, Scientific reports, 2016, 6, 29565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Zeng T, Liu L, Li T, Li Y, Gao J, Zhao Y and Wu H-C, Chemical science, 2015, 6, 5628–5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Langecker M, Ivankin A, Carson S, Kinney SRM, Simmel FC and Wanunu M, Nano Lett, 2015, 15, 783–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Nechemia-Arbely Y, Fachinetti D, Miga KH, Sekulic N, Soni GV, Kim DH, Wong AK, Lee AY, Nguyen K and Dekker C, J Cell Biol, 2017, 216, 607–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, Korlach J and Turner SW, Nat. Methods, 2010, 7, 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Song CX, Clark TA, Lu XY, Kislyuk A, Dai Q, Turner SW, He C and Korlach J, Nat Methods, 2011, 9, 75–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Lluch-Senar M, Luong K, Lloréns-Rico V, Delgado J, Fang G, Spittle K, Clark TA, Schadt E, Turner SW, Korlach J and Serrano L, PLOS Genet., 2013, 9, e1003191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Clark TA, Lu X, Luong K, Dai Q, Boitano M, Turner SW, He C and Korlach J, BMC Biol, 2013, 11, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Bendall ML, Luong K, Wetmore KM, Blow M, Korlach J, Deutschbauer A and Malmstrom RR, Journal of Bacteriology, 2013, 195, 4966–4974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Chen L, Li HC, Chen T, Yu L, Guo HX, Chen YH, Chen M, Li ZY, Wu ZH, Wang XZ, Zhao J, Yan HM, Wang XC, Zhou L and Zhou J, International Journal of Clinical and Experimental Pathology, 2018, 11, 3036–3045. [PMC free article] [PubMed] [Google Scholar]
- 221.Kang X, Hu L, Shen P, Li R and Liu D, Front. Microbiol, 2017, 8, 1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Wang Y, Chen X, Sheng Y, Liu Y and Gao S, Nucleic acids research, 2017, 45, 11594–11606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Couturier M and Lindas AC, Front Microbiol, 2018, 9, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Beaulaurier J, Zhang XS, Zhu S, Sebra R, Rosenbluh C, Deikus G, Shen N, Munera D, Waldor MK, Chess A, Blaser MJ, Schadt EE and Fang G, Nat Commun, 2015, 6, 7438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Chavez L, Huang Y, Luong K, Agarwal S, Iyer LM, Pastor WA, Hench VK, Frazier-Bowers SA, Korol E, Liu S, Tahiliani M, Wang Y, Clark TA, Korlach J, Pukkila PJ, Aravind L and Rao A, Proc. Natl. Acad. Sci. U. S. A, 2014, 111, E5149–E5158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Kondratyev N, Golov A, Alfimova M, Lezheiko T and Golimbet V, Clin. Epigenet, 2018, 10, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Kozdon JB, Melfi MD, Luong K, Clark TA, Boitano M, Wang S, Zhou B, Gonzalez D, Collier J, Turner SW, Korlach J, Shapiro L and McAdams HH, Proceedings of the National Academy of Sciences of the United States of America, 2013, 110, E4658–E4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Ronen M, Avrahami D and Gerber D, Lab on a Chip, 2014, 14, 2354–2362. [DOI] [PubMed] [Google Scholar]
- 229.Zhao Z, Tavoosidana G, Sjolinder M, Gondor A, Mariano P, Wang S, Kanduri C, Lezcano M, Sandhu KS, Singh U, Pant V, Tiwari V, Kurukuti S and Ohlsson R, Nature Genetics, 2006, 38, 1341–1347. [DOI] [PubMed] [Google Scholar]
- 230.Simonis M, Klous P, Splinter E, Moshkin Y, Willemsen R, De Wit E, Van Steensel B and De Laat W, Nature genetics, 2006, 38, 1348. [DOI] [PubMed] [Google Scholar]
- 231.Dostie J, Richmond TA, Arnaout RA, Selzer RR, Lee WL, Honan TA, Rubio ED, Krumm A, Lamb J, Nusbaum C, Green RD and Dekker J, Genome Research, 2006, 16, 1299–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Sun C and Lu C, Analytical Chemistry, 2018, 90, 3714–3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Xu Y, Lee JH, Li ZY, Wang LG, Ordog T and Bailey RC, Lab on a Chip, 2018, 18, 2583–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Terao K, Kabata H and Washizu M, Journal of Physics: Condensed Matter, 2006, 18, S653–S663. [Google Scholar]
- 235.Terao K, Washizu M and Oana H, Lab on a Chip, 2008, 8, 1280–1284. [DOI] [PubMed] [Google Scholar]
- 236.Oana H, Nishikawa K, Matsuhara H, Yamamoto A, Yamamoto TG, Haraguchi T, Hiraoka Y and Washizu M, Lab on a Chip, 2014, 14, 696–704. [DOI] [PubMed] [Google Scholar]
- 237.Mori H, Okeyo KO, Washizu M and Oana H, Biotechnology journal, 2018, 13, 1700245. [DOI] [PubMed] [Google Scholar]
- 238.Takahashi T, Okeyo KO, Ueda J, Yamagata K, Washizu M and Oana H, Scientific reports, 2018, 8, 13684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Forget AL, Dombrowski CC, Amitani I and Kowalczykowski SC, Nature Protocols, 2013, 8, 525–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Chen XQ, Zhao ES and Fu YV, Sci Bull, 2017, 62, 399–404. [DOI] [PubMed] [Google Scholar]
- 241.Jin SH, Jang SC, Lee B, Jeong HH, Jeong SG, Lee SS, Kim KP and Lee CS, Lab on a Chip, 2016, 16, 1358–1365. [DOI] [PubMed] [Google Scholar]
- 242.Kasinski AL and Slack FJ, Nature Reviews Cancer, 2011, 11, 849–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Wu M, Piccini M, Koh C-Y, Lam KS and Singh AK, PloS one, 2013, 8, e55044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Wu MY, Perroud TD, Srivastava N, Branda CS, Sale KL, Carson BD, Patel KD, Branda SS and Singh AK, Lab on a Chip, 2012, 12, 2823–2831. [DOI] [PubMed] [Google Scholar]
- 245.Novara C, Chiado A, Paccotti N, Catuogno S, Esposito CL, Condorelli G, De Franciscis V, Geobaldo F, Rivolo P and Giorgis F, Faraday Discuss, 2017, 205, 271–289. [DOI] [PubMed] [Google Scholar]
- 246.Zhou X, Zhu Q, Eicken C, Sheng N, Zhang X, Yang L and Gao X, in NextGeneration MicroRNA Expression Profiling Technology, Springer, 2012, vol. 822, pp. 153–182. [DOI] [PubMed] [Google Scholar]
- 247.Chen Y, Gelfond JA, McManus LM and Shireman PK, BMC Genomics, 2009, 10, 407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Pogue AI and Lukiw WJ, Cellular and Molecular Neurobiology, 2018, 38, 1021–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Wei C, Huang HT, Cong W, Li ZG, Zhang XH, Liu H, Wang R and Xiao J, J Hard Tissue Biol, 2018, 27, 243–249. [Google Scholar]
- 250.Zhou W, Du J, Jiang D, Wang X, Chen K, Tang H, Zhang X, Cao H, Zong L and Dong C, Molecular medicine reports, 2018, 18, 1253–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Lazar B, Anand M, Toth R, Varkonyi EP, Liptoi K and Gocza E, Stem Cells Int, 2018, 2018, 1780679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Mar-Aguilar F, Trevino V, Salinas-Hernández JE, Taméz-Guerrero MM, Barrón-González MP, Morales-Rubio E, Treviño-Neávez J, Verduzco-Martínez JA, Morales-Vallarta MR and Reséndez-Pérez D, Plos One, 2013, 8, e68202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Tian JD, Gong H, Sheng NJ, Zhou XC, Gulari E, Gao XL and Church G, Nature, 2004, 432, 1050–1054. [DOI] [PubMed] [Google Scholar]
- 254.Takousis P, Methods Mol Biol, 2018, 1750, 283–292. [DOI] [PubMed] [Google Scholar]
- 255.Jang JS, Simon VA, Feddersen RM, Rakhshan F, Schultz DA, Zschunke MA, Lingle WL, Kolbert CP and Jen J, BMC genomics, 2011, 12, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Kara M, Yumrutas O, Ozcan O, Celik OI, Bozgeyik E, Bozgeyik I and Tasdemir S, Gene, 2015, 567, 81–86. [DOI] [PubMed] [Google Scholar]
- 257.Mensah M, Borzi C, Verri C, Suatoni P, Conte D, Pastorino U, Orazio F, Sozzi G and Boeri M, JoVE (Journal of Visualized Experiments), 2017, e56326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Rizzo S, Cangemi A, Galvano A, Fanale D, Buscemi S, Ciaccio M, Russo A, Castorina S and Bazan V, Oncotarget, 2017, 8, 71924–71932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Kanaan Z, Roberts H, Eichenberger MR, Billeter A, Ocheretner G, Pan JM, Rai SN, Jorden J, Williford A and Galandiuk S, Ann Surg, 2013, 258, 400–408. [DOI] [PubMed] [Google Scholar]
- 260.Joly-Tonetti N, Viñuelas J, Gandrillon O and Lamartine J, BMC genomics, 2013, 14, 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Kanaan Z, Rai SN, Eichenberger MR, Roberts H, Keskey B, Pan J and Galandiuk S, Annals of surgery, 2012, 256, 544–551. [DOI] [PubMed] [Google Scholar]
- 262.Zeng W, Jiang S, Kong X, El-Ali N, Ball AR Jr., Ma CI, Hashimoto N, Yokomori K and Mortazavi A, Nucleic Acids Res, 2016, 44, e158. [DOI] [PMC free article] [PubMed] [Google Scholar]