

If you want something done, you’ve got to do it yourself
Sebastian, The Little Mermaid
How do we sense and fight infections? This question has been at the forefront of immunology research since the birth of the field (1). Although scientists from diverse disciplines have attempted to explain our ability to detect microbial encounters, our endeavor here is far from complete. In principle, one may expect that the oldest fields of immunology would be the least interesting to study, because the most fundamental aspects of these biological processes should have already been established. On the basis of this logic, the biology of interleukin-1 (IL-1), the first discovered cytokine (2), should have been uncovered long ago, and the study of this pyrogen should no longer be of interest. In this issue of Science Immunology, LaRock et al. (3) challenge this idea with their discovery of a new mechanism by which IL-1 is activated during a bacterial infection. Their study suggests that IL-1 can serve as both a sensor of infection and an inducer of inflammation, thus revealing the simplest innate immune signaling mechanism known.
IL-1 is an unusual cytokine in that it acts extracellularly but is not released from cells by the secretory pathway (4). IL-1 family members (such as IL-1α, IL-1β, and IL-18) do not contain an N-terminal signal sequence that promotes protein secretion. As such, these proteins are present in the cytosol and are released into the extracellular space either upon cell death or by unclear mechanisms from living cells. In the case of IL-1β, this protein contains an N-terminal prodomain that must be cleaved for it to be rendered capable of activating inflammation by binding the IL-1 receptor (IL-1R) (2).
Because of the importance of IL-1β processing in promoting its inflammatory activities, the study of enzymes that can cleave IL-1β has been an active area of research for decades. Caspase-1, which was originally known as IL-1–converting enzyme (ICE), is the best-studied factor that processes IL-1β. Caspase-1 activity is intimately tied to the functions of inflammasomes, which are supramolecular organizing centers that are assembled in the cytosol to promote caspase-1 activation (5). Inflammasome assembly, and hence caspase- 1 activity, is triggered by a diverse set of proteins that survey the cell for infectious agents or disruptions in homeostasis. Consequently,IL-1processing and release from cells are dependent on the actions of caspase-1 and the structural components of the inflammasome that was assembled.
The regulation of IL-1 processing and release by inflammasomes has been validated by numerous studies using infectious and noninfectious activators of inflammation. However, there were hints in the literature that the process of IL-1 activation may be more complex. For example, several other ICE-like proteins have been shown to cleave and activate IL-1β through inflammasome-independent mechanisms. These include caspase-8, neutrophil elastase, proteinase 3, cathepsin G, chymase, chymotrypsin, and meprin α or β (6). The relative roles of these proteases in various inflammatory conditions have been established, thus raising the question of why IL-1β can be activated by so many enzymes.
LaRock et al. began their study of IL-1 with an interest in understanding the unusual clinical phenomena that patients receiving therapies that neutralize IL-1β were ~330 times more likely to develop group A Streptococcus (GAS) infections. In contrast, immunosuppressive therapies that target other key immune regulators besides IL-1 did not result in such sensitivity to GAS. In mouse models of GAS infection, the authors could reproduce these clinical findings, in that IL-1R–deficient animals were highly sensitive to infection. Wild type mice and those lacking caspase-1 were protected from GAS infection, suggesting an inflammasome-independent mechanism of IL-1β activation and release. To understand how IL-1β was processed during GAS infections, the authors used an in vitro assay of IL-1β activity, where macrophages were infected with GAS, and supernatants from these cultures were used to stimulate the IL-1R pathway in reporter cells. The authors found that GAS elicited the release of bioactive IL-1β from infected caspase-1–deficient macrophages, which was consistent with their in vivo observations. To define this unusual processing of IL-1β, broad-spectrum protease inhibitors were included during the infections. The authors found that these inhibitors blocked IL-1β bioactivity. In contrast, pan-caspase inhibitors could not prevent activation of IL-1β in the context of GAS infection, suggesting that a protease other than caspases was involved. This protease was revealed as SpeB, a protein produced by GAS. Consistent with this conclusion, SpeB-deficient bacteria were unable to elicit IL-1β activity from caspase-1–deficient macrophages, and recombinant SpeB was capable of cleaving IL-1β. Moreover, cotransfection of human 293 cells with plasmids encoding SpeB and IL-1β caused the release of IL-1 activity, but cotransfection with an enzymatically inactive SpeB did not.
Further mechanistic analysis of the activity of SpeB toward IL-1β by Edman degradation revealed that this enzyme cleaves its substrate at a site different from that cleaved by caspase-1. Whereas caspase-1 cleaves IL-1β at the aspartic acid at amino acid position 116, SpeB cleaved this protein at the phenylalanine at position 105. This difference in cleavage site preference is not without precedent, because neutrophil elastase cleaves IL-1β at a site different from that cleaved by caspase-1. This flexibility of sites that release bioactive IL-1β is intriguing and suggests a mechanism by which this cytokine evolved to detect the activity of a spectrum of proteases. What benefit would ensue from IL-1β evolving to become such a promiscuous protease substrate? The authors make the provocative suggestion that this promiscuity of IL-1β sensitivity to cleavage is not a mistake. Rather, IL-1β may have evolved to serve as a sensor of tissue damage and infection. In the case of GAS infections, this idea appears to be correct, in that IL-1β can sense infection by the presence of SpeB activity and serve as its own effector by engagement of the IL-1R.
The past 20 years of immunological research have revealed several dynamic signaling pathways that link pathogen detection to the initiation of inflammation. Some of these pathways contain dozens of signaling intermediates that are subject to multiple layers of regulation. As such, we often think of innate immune signaling pathways as highly complex entities. In this regard, the finding that IL-1β can serve as a sensor and an effector to promote inflammation leads us to conclude that the simplest innate immune signaling pathway may have finally been revealed—consisting of a single protein (Fig. 1).
Fig. 1. GAS SpeB cleaves IL-1β to promote inflammation.
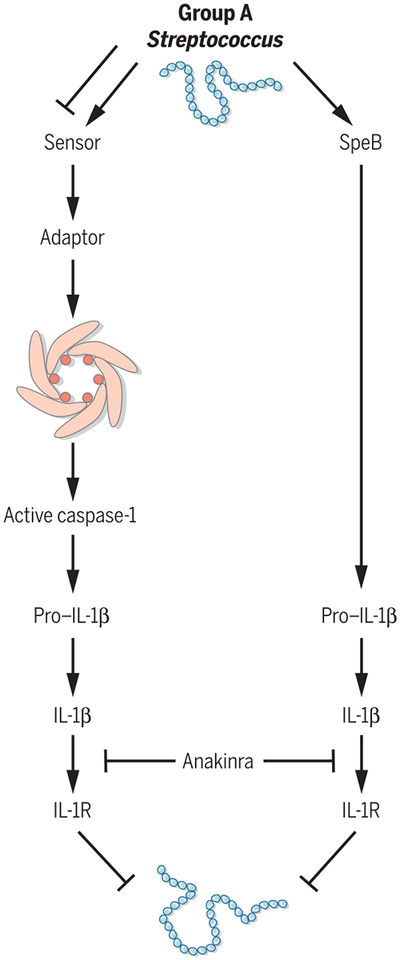
Canonical activation of IL-1β occurs through formation of an inflammasome and subsequent activation of the cellular protease caspase-1. In the absence of inflammasome signaling, GAS secretes SpeB, which cleaves and activates IL-1β. Treatment with anakinra blocks the inflammatory activity of IL-1β and promotes bacterial replication and spread.
Although these ideas are indeed provocative, several questions remain. Are other bacterial infections sensed by a similar mechanism? If IL-1β evolved as a general sensor of tissue damage and infection, one would expect that IL-1β would sense and be cleaved by additional bacterial proteases to promote inflammation. Although LaRock et al. report that only GAS infection is overrepresented in patients treated with anakinra, a neutralizer of IL-1β activity, further studies are warranted to examine the role of IL-1R signaling in controlling other bacterial infections. Given the increased risk of GAS-associated necrotizing fasciitis (NF), but no other GAS-associated diseases in these patients, it is possible that the antibacterial activities of IL-1β are particularly important for controlling infections in the skin and soft tissue. The authors of this study observe that anakinra increased the susceptibility of mice to GAS infection after intradermal inoculation. However, the role of IL-1R signaling in protecting against other GAS-mediated pathologies remains to be fully defined. Additional studies will be necessary to determine whether other bacterial proteases activate IL-1β through similar mechanisms as SpeB and at what sites of infection IL-1β is most important.
The study by LaRock et al. also raises mechanistic questions that will influence future studies on the SpeB-dependent IL-1β activation. These include the location where SpeB accesses full-length IL-1β after GAS infection and how IL-1β is released from GAS-infected macrophages. The authors report that SpeB colocalizes with intracellular IL-1β, suggesting that this population is the target of SpeB protease activity. However, secreted SpeB is found throughout GAS-infected tissues from patients presenting with NF (7), suggesting that extracellular full-length IL-1β could also be cleaved by SpeB. In support of extracellular IL-1β being a potential target of SpeB activity, the authors observe high amounts of full-length IL-1β in supernatants isolated from GAS-infected macrophages. Independent of whether intracellular or extra-cellular IL-1β is the target of SpeB, how IL-1β is released from GAS-infected macrophages remains unclear. IL-1β is thought to be released from cells through two distinct cell death mechanisms: pyroptosis and necrosis (8). GAS-infected macrophages release lactate dehydrogenase, indicating that GAS infection increases membrane permeability and cell death. However, caspase-1is not required for GAS-inducible IL-1R signaling in this study, arguing against a pyroptosis-mediated mechanism of IL-1β release. Therefore, there is a clear mandate to investigate the role of necrosis in regulating GAS-mediated macrophage death and IL-1β release.
The observations presented in this study have important implications for human health and raise questions of whether patients undergoing treatment with IL-1 inhibitors would benefit from more specific inhibitors of their inflammatory condition. LaRock et al. suggest that therapeutic targeting of NLRP3 or caspase-1 could limit aberrant activation of IL-1β in some patients but would allow IL-1β to be active in response to GAS infection, thus reducing the risk of GAS-associated NF. Although maintaining host protection against infection is a key factor when designing anti-inflammatory therapies for the treatment of autoimmune diseases, this study underscores the importance of postmarketing drug surveillance in obtaining new biological insights to further refine therapeutic strategies.
Acknowledgments: Funding:
J.C.K. is supported by NIH grants AI093589 and P30 DK34854, and a gift from Mead Johnson & Company, and holds an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund. M.H.O. is supported by NIH T32 AI007512 training grant.
Footnotes
Group A Streptococcus protease SpeB directly binds and activates IL-1β (LaRock et al., this issue).
REFERENCES AND NOTES
- 1.Gordon S, Elie Metchnikoff, the Man and the Myth. J. Innate Immun. 8, 223–227 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Auron PE, Webb AC, Rosenwasser LJ, Mucci SF,Rich A, Wolff SM, Dinarello CA, Nucleotide sequence of human monocyte interleukin 1 precursor cDNA. Proc. Natl. Acad. Sci. U.S.A. 81, 7907–7911 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.LaRock CN, Todd J, LaRock DL, Olson J, O’Donoghue AJ, Robertson AAB, Cooper MA,Hoffman HM, Nizet V, IL-1β is an innate immune sensor of microbial proteolysis. Sci. Immunol 1, eaah3539 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rubartelli A, Cozzolino F, Talio M, Sitia R, A novel secretory pathway for interleukin-1b, a protein lacking a signal sequence. EMBO J. 9, 1503–1510 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kagan JC, Magupalli VG, Wu H, SMOCs: Supramolecular organizing centres that control innate immunity. Nat. Rev. Immunol 14, 821–826 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Netea MG, van de Veerdonk FL, van der Meer JWM, Dinarello CA, Joosten LAB, Inflammasome-independent regulation of IL-1-family cytokines. Annu. Rev. Immunol 33, 49–77 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Thulin P, Johansson L, Low DE, Gan BS, Kotb M, McGeer A, Norrby-Teglund A, Viable group A streptococci in macrophages during acute soft tissue infection. PLOS Med. 3, e53 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wallach D, Kang T-B, Dillon CP, Green DR, Programmed necrosis in inflammation: Toward identification of the effector molecules. Science 352, aaf2154 (2016). [DOI] [PubMed] [Google Scholar]