

INTRODUCTION TO HEALTH-RELATED OUTCOME MEASURES
There are several aspects of health that can be measured, and these represent different aspects of the disease, from the pathophysiology (eg, antibody titers), to the symptoms and signs, to the effect on the individual and their relation to society.1 Because different outcome measures are aimed at different aspects of the disease process, it is fundamental to understand what a given tool measures, as well as for which purpose it was developed and in which population it was tested. One way to understand these different aspects of the disease is through the International Classification of Functioning, Disability and Health (ICF),2 published by the World Health Organization. The ICF identifies 3 major ways in which a disease or injury affects in individual: impairments of body function or structures, which are basically the signs and symptoms; activity limitations, which are the effects of the disease and its symptoms on activities of daily life; and participation limitations, which are the effects on a patient’s social interactions, such as looking for work or caring for their family. Additionally, these aspects of the disease are also affected by personal and environmental factors (eg, social support, cultural factors, and accessibility). Disability is—according to the ICF—the interaction between symptoms, activities, and participation restrictions and personal and environmental factors.2 Health-related quality of life (HRQoL) goes beyond the concept of disability and it is, by definition, a subjective and multidimensional concept, including physical functioning, mental or psychological well-being, occupational status, and social interactions.3 The impairments of body functions/structures are thought to be less affected by social and environmental factors and, therefore, are typically considered to reflect more directly disease severity. This factor is why most outcome measures aimed at quantifying disease severity capture the signs and symptoms, whereas measures focused on the impact of the symptoms on the individual as a whole are usually disability or HRQoL measures. Putting these concepts into the perspective of a clinical trial, the primary outcome should match the study intervention. For example, a phase II study for a new medication will likely be focused on the effect of signs and symptoms, whereas a psychosocial intervention will probably have more effect on HRQoL or disability than on the symptoms in isolation. Fig. 1 depicts the ICF model in relation to some of the outcome measures specific to myasthenia gravis (MG) that are available.
Fig. 1.
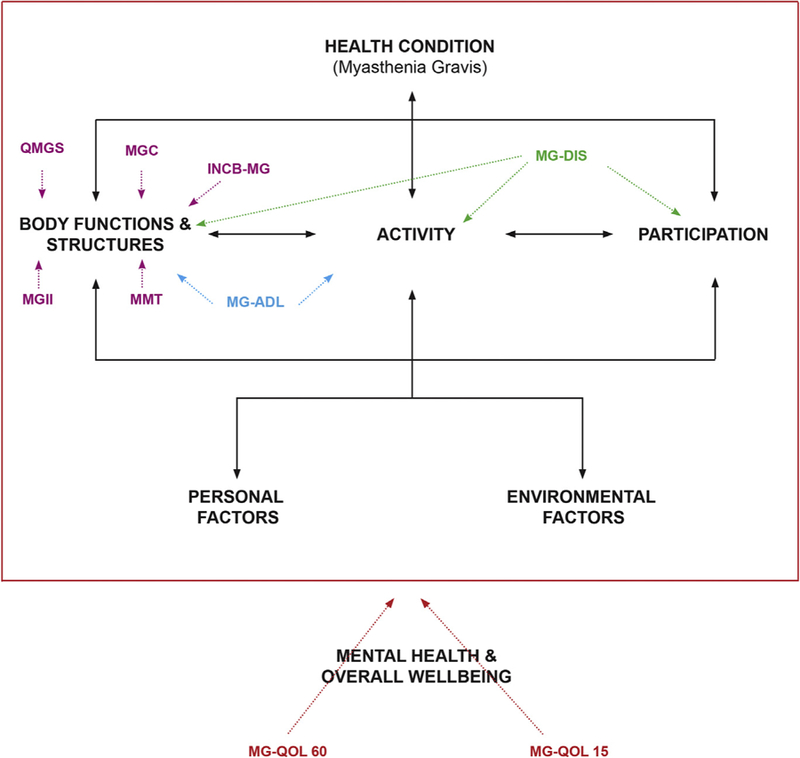
Commonly Used myasthenia gravis (MG) outcomes in relation to the International Classification of Functioning, Disability and Health (ICF). The ICF framework (within the red box) and which aspects of the disease are measured by commonly used MG measures. Measures of health-related quality of life (HRQoL) also incorporate mental health and overall wellbeing. INCB-MG, Besta Neurologic Institute rating scale for MG; MG-ADL, Myasthenia Gravis Activities of Daily Living; MGC, Myasthenia Gravis Composite; MG-DIS, MG Disability Scale; MGII, Myasthenia Gravis Impairment Index; MMT, Manual Muscle Test; QMGS, Quantitative Myasthenia Gravis Score. (Modified from World Health Organization. International classification of functioning, disability and health (ICF). 1st edition. Geneva: World Health Organization; 2002.)
Additionally, when choosing an outcome measure, it is fundamental to recognize that, beyond what they measure, they might have been developed with different purposes, typically discriminative, predictive, and evaluative.4 Discriminative means that a measure can distinguish between individuals that have different degrees of the underlying construct (eg, more or less severe disease). Predictive measures are aimed at classifying individuals such as in a diagnostic test or predicting an outcome. Finally, evaluative measures are aimed at detecting change, which is fundamental to determining treatment response. Additionally, there are several methodologic requirements that need to be met to ensure that the measure is adequate for the intended purpose. All measures have to be valid (ie, measure what they are supposed to measure) and reliable (ie, reproducible). In addition, evaluative measures have to demonstrate responsiveness, or the ability to detect change. To interpret change scores, it is important to know the minimal important difference (MID), which is the smallest change in a measure that is meaningful for patients.5 Additionally, there has been a shift in recent years toward more patient-reported outcomes, considering that patients are the best judges of their disease status and that many symptoms or signs might not be evident in a clinical encounter or—when present—do not affect patient function. The specific standards for the development of outcome measures are beyond the scope of this article, but for those interested, the US Food and Drug Administration6 and the COnsensus-based Standards for the selection of health Measurement INstruments (COSMIN)7 guidelines are excellent resources. Finally, it is important to keep in mind that validity, reliability, and responsiveness are not universal characteristics of a measure, and depend on the populations and interventions tested.8
OUTCOME MEASUREMENT IN MYASTHENIA GRAVIS
MG presents specific challenges for outcome measurement. The classical manifestation of MG—fatigable weakness—results in signs and symptoms that fluctuate. These fluctuations can occur within the same day, because typically patients are more symptomatic as the day goes by, but also from day to day, and even within longer time-frames. Therefore, a clinical assessment that is anchored in a single time-point might be insufficient to cover the breadth of the manifestations of MG. In a qualitative study of patients’ experiences with MG, patients consistently reported fatigability as a major manifestation of the disease.9 Regarding the assessments, one patient said: “It’s [the assessment] just such a quick snapshot of how I’m doing, really, at that very moment. And it seems so variable throughout the day. I could have a good hour where people wouldn’t even know that I have MG at all. I look like I have lots of energy and whatnot. But then, at a moment’s notice, it could completely change.”9 This is why many MG measures include the patients’ report of their symptoms, or other measures of fatigability.10–12
There are several measures that have been used in MG studies, including generic measures, and reviewing all would not be feasible. Therefore, we only cover those measures that have been specifically developed for MG. Additionally, we only include measures that are available in English and where there are enough data on the development and validation process. To further organize the many MG-specific outcomes available, we have divided them by what they measure: signs and symptoms, and disability/HRQoL. Finally, we discuss other outcomes that are relevant for MG that do not directly fit into the disability framework, such as biomarkers and steroid-sparing effects.
MEASURES OF SIGNS AND SYMPTOMS OF MYASTHENIA GRAVIS
These measures constitute the majority of outcomes specifically developed for MG. For each measure, we provide a brief description of its development process, items, how it is scored, and its basic psychometric properties. We have organized the measures based on the first description of the measure or its direct predecessor. Table 1 depicts the primary characteristics of these measures.
Table 1.
Measures of signs and symptoms severity in myasthenia gravis
| Total Items | Patient Reported Items | Total Score | Interpretation | MID | Instrumentation | |
|---|---|---|---|---|---|---|
| QMGS | 13 | 0 | 0–39 | Higher score, more severe disease | 2 or 3 points | Spirometer, dynamometer |
| MMS | 9 | 0 | 0–100 | Lower score, more severe disease | NA | No |
| INCB-MG | 11 | 2 | 0–427,153 | Higher score, more severe disease | NA | No |
| MG-ADL | 8 | 8 | 0–24 | Higher score, more severe disease | 2 points | No |
| MMT | 30 | 0 | 0–120 | Higher score, more severe disease | NA | No |
| MGC | 10 | 4 | 0–50 | Higher score, more severe disease | 3 points | No |
| OBFR | 5 | 0 | 0–21 | Higher score, more severe disease | NA | Spirometer |
| MGII | 28 | 22 | 0–84 | Higher score, more severe disease | 8.1 groups 5.5 individual | No |
Abbreviations: INCB-MG, Besta Neurologic Institute rating scale for Myasthenia Gravis29; MG-ADL, Myasthenia Gravis Activities of Daily Living19; MGC, Myasthenia Gravis Composite21; MGII, Myasthenia Gravis Impairment Index22; MID, minimal important difference (these are for improvement); MMS, Myasthenia Muscle Score57; MMT, Manual Muscle Test16; NA, not applicable; OBFR, Oculobulbar Facial Respiratory Score41; QMGS, Quantitative Myasthenia Gravis Score.10
The Myasthenia Gravis Foundation of America Classification
This measure is derived from the Osserman score, developed in the 1950s,13 which was one of the first classifications systems in MG. The Myasthenia Gravis Foundation of America (MGFA) classification is aimed at separating patients in groups based on disease severity and the localization of the symptoms, and does not have an evaluative purpose. The MGFA classes are pure ocular (class I), mild generalized (class II), moderate generalized (class III), severe generalized (class IV), and intubation/myasthenic crisis (class V). Within the generalized categories II, III, and IV, patients are subclassified as class A if their symptoms are predominantly generalized or class B if their symptoms are predominantly bulbar.14 The MGFA also has a system to classify patients based on postintervention outcomes and includes remission, defined as 1 year or longer without signs or symptoms and without any symptomatic (pyridostigmine) treatment, and which can be divided in complete (no pharmacologic treatment at all) or pharmacologic remission. Minimal manifestation status is defined as minimal signs or symptoms (no specific time-frame was defined) and pyridostigmine use may be accepted. Additionally, patients can be improved, unchanged, worse, experiencing an MG exacerbation, or have died of MG.14 Because the original MGFA severity classification does not take into account those patients who are asymptomatic, many MG studies use a hybrid, whereby symptomatic patients are classified based on the I to V class system, and asymptomatic or oligosymptomatic patients are classified as remission or minimal manifestation status.15
The Quantitative Myasthenia Gravis Score
The Quantitative Myasthenia Gravis Score (QMGS) was developed in the context of a clinical trial in MG and originally had 8 items16; it was later modified for a trial of cyclosporine,17 increasing the number of items to 13. This measure was modified again by Barohn and colleagues,18 making the 13 items all based on the examination, and this is the version currently in use. The QMGS has several items that measure endurance or fatigability, taking into account the fluctuating nature of the disease. The items are as follows: ptosis, diplopia, orbicularis oculi weakness, swallowing a cup of water, speech, percent predicted forced vital capacity, grip strength (2 items), arm endurance (2 items), leg endurance (2 items), and neck flexion endurance. All items are scored on a scale of 0 to 3, and total scores range from 0 to 39; higher scores indicate greater disease severity. There are 2 studies on interobserver reliability18,19 and one on test–retest reliability20 showing adequate reliability coefficients. Construct validity has been studied by demonstrating correlations between the QMGS scores to other outcome measures like the Manual Muscle Test (MMT)21 and Myasthenia Muscle Score,19 as well as comparing QMGS scores across different MGFA classes, and with electrodiagnostic markers.22 There is study on longitudinal validity where 53 patients were seen on a second visit between 2 and 10 months after initial examination. Using the physician’s impression of change as marker or improvement, the difference in the QMGS score was significantly higher in those improved compared with those who were stable.23
The QMGS also has shown to be responsive to change in several clinical trials. In a randomized study of intravenous immunoglobulin (IVIG)24 and in a cyclosporine trial,17 treated patients had a statistically significant improvement in the QMGS compared with a placebo group. Based on the mean change in the cyclosporine trial and the reliability a studies, a change of greater than 3.5 points has been typically considered as significant.17,18 Using the data from the IVIG versus placebo study, the MID with mild to moderate MG (QMGS ≤16) was calculated to be 2 points,25 although patients with higher baseline values (QMGS >16) had a higher MID of 3 points, which is a well-described phenomenon. Additionally, the performance of the individual items of the QMGS was studied,26 with evidence of floor effect—a high proportion of patients scoring 0 (best score) at baseline—on grip strength, dysarthria, swallowing, and percent forced vital capacity. The items that changed the most after treatment were ptosis, and arm, leg, and neck endurance. The main drawbacks of the QMGS are in terms of feasibility and ease of use, because it requires a dynamometer and spirometer and can take up to 25 minutes to complete. Therefore, it is mostly used in research rather than routine clinical assessments.
The Myasthenia Muscle Score
This measure was developed in the context of a clinical trial27 and is extensively used in France. It has 9 items: 3 bulbar (chewing, swallowing, and speech), 1 ocular (combines diplopia and ptosis), 1 on eye closure, 2 axial muscles (neck flexors and sitting up), and 2 on limb endurance (arms/legs). All items are based on clinical examination, with maximal subscore values ranging from 10 to 15, and the total scores ranging from 0 to 100, with lower scores indicating greater disease severity. There is 1 small study on interobserver reliability, with an intraclass correlation coefficient (ICC) of 0.9, which is excellent.19 There are no studies on test–retest reliability. Validity has been studied through correlations with the QMGS (r = −0.87)19 in a small sample. The Myasthenia Muscle Score has been used as primary outcome in a trial of IVIG and plasma exchange,28 and patients who received IVIG or plasma exchange improved by 16 points on average. However, there are no studies on the MID.
The Besta Neurologic Institute Rating Scale for Myasthenia Gravis
This measure was initially developed in 1988 for a clinical trial of azathioprine,29 and has been used extensively in Italy. It was recently modified to improve the reliability of some items, as well as to remove the need for a spirometer or using a water test for swallowing.30 The current version has 11 items: 1 ocular, 5 generalized, and 5 bulbar, all based on the physical examination, with the exception of the items on swallowing and breathing, which are based on the history. The scale is weighted, whereby bulbar items receive more weight than limb and ocular items—427,000 maximum points for bulbar/breathing problems and 153 maximum points for ocular and limb problems—and higher scores indicate more disease severity. The Besta Neurologic Institute rating scale for Myasthenia Gravis also includes 2 endurance tests: one of the arms and one of the legs, which are similar to the QMGS and measured in seconds. These measures are scored independently, rather than being factored into the total score. The total, bulbar, and generalized scores had good internal consistency (Cronbach’s alpha = 0.79, 0.75, and 0.75, respectively), and good interrater reliability (kappa = 0.92).30 Test–retest reliability was not described. In terms of construct validity, it has a high correlation with the MGC (r = 0.83). Responsiveness has not been formally assessed for the sum scores; however, the arm and leg endurance tests are responsive to change, confirming previous studies.26,31
The Myasthenia Gravis Activities of Daily Living
This instrument, like the modified QMGS, was developed for the trial of IVIG by Wolfe and colleagues.32 This is a patient-reported outcome that combines 2 items on daily life activities—ability to brush teeth or comb hair, and limitations in the ability to rise from a chair—with 6 items reflecting other MG symptoms: diplopia, ptosis, chewing, swallowing, voice/speech problems, and respiratory symptoms.10 Each item is scored between 0 and 3 and total scores range from 0 to 24, where higher scores indicate more disease severity. Test–retest reliability was demonstrated in 20 patients, with an ICC of 0.93.33 Construct validity has been studied through correlations with the QMGS,10 the Myasthenia Gravis Composite (MGC),34 and the MG Quality of Life (MG-QOL)15,33 showing moderate correlations with the QMGS (r = 0.58) and high with the MGC and MG-QOL15 (r = 0.85, 0.76). The responsiveness of the Myasthenia Gravis Activities of Daily Living (MG-ADL) was tested in a study of 76 patients who were assessed twice,33 with variable interventions and time intervals between assessments. Patients were considered improved based on an improvement in a quality-of-life measure, the MG-QOL15,35 and in the physician impression of change. The MID to classify an individual patient as a responder was 2 points—using a receiver operator characteristic curve approach—where the MID is the point of greatest sensitivity and specificity. The MG-ADL has been used in several clinical trials, including as a secondary outcome measure in the thymectomy randomized, controlled trial,36 where treated patients and significantly greater improvement than those in the placebo arm (difference between groups, 1.17; P = .008). The main advantages of the MG-ADL are that it is very easy to use, and it is completely patient reported. A drawback is that it does not have a specific recall time frame (eg, 2 or 4 weeks) because it relies on comparing with the last visit, and that it is prone to floor effects.12
The Manual Muscle Test
This was developed in 2003,21 with the aim of having a simple assessment of MG patients, without the need for instrumentation. The MMT evaluates the strength in 12 bilateral muscle groups and 6 ocular or axial (eg, neck flexors) muscles, that are usually affected in MG. Each muscle is scored from 0 (normal strength) to 4 (paralysis), and the total score is the sum of each muscle, where higher scores indicate more strength (less disease severity). Interrater reliability was assessed in 274 patients through Pearson’s correlation, but ICCs were not reported.21 Construct validity was tested through the correlations with the QMGS. The MMT has been used as secondary outcome measure in a clinical trial,37 with low to moderate correlations (r = 0.30–0.59) between the change in MMT scores and change in the QMGS and the MG-ADL.38 The MMT scores changed significantly after treatment (P<.001). Additionally, the MMT change scores were higher in those patients deemed improved (by physician impression of change) than in those who were unchanged. However, there are no studies on the MID. The main advantage of the MMT is the ease of use, because it is based on the routine neurologic examination. Drawbacks are that it measures static strength, so it is likely to be more affected by the natural fluctuations of the disease.
The Myasthenia Gravis Composite
The MGC was developed more recently, aiming for a simple yet comprehensive measure of MG severity. It was developed by combining items from other MG measures, based on their performance of 2 clinical trials of mycophenolate in MG.37,39 The measures considered for items were the QMGS, the MG-ADL, and the MMT. Item selection was determined by responsiveness to treatment (defined by a positive physicians’ and patients’ impression of change), as well as with the correlations with the MG-QOL15, a disease-specific quality-of-life measure.35 Final item selection and weights were decided by a large group of MG experts, and bulbar and generalized items have more weight than ocular items. The final measure has 10 items: 2 ocular (diplopia and ptosis) from the QMGS; 4 items (facial, neck, deltoids, and hip flexors strength) from the MMT, and 4 patient-reported items from the MG-ADL (chewing, swallowing, breathing, and speech). Regarding the latter, these are read to the patients and the whole scale is completed by an examiner. Total scores range from 0 to 50 where higher scores indicate greater disease severity. Interrater reliability was high in a study of 38 patients.34 Construct validity was evaluated in 175 patients, and this was based on positive correlations between the MGC and other measures, including the MMT, ADL, and the MG-QOL15.34 Responsiveness was studied in 151 patients who had routine follow-up examinations, with an average time between assessments of 4.7 months, and variable interventions within visits.34 In that study, using a receiver operator characteristic curve and defining improvement based on the physician’s impression of change and the change in the MG-QOL15, the MID for individuals was estimated at 3 points.39 The MGC was recommended as the primary outcome measure of choice in MG trials by the MGFA scientific board,40 and it has been subsequently used as primary or secondary outcome in several trials. The main strength of the MGC is its simplicity and the incorporation of the patient’s history.
The Oculo-Bulbar Facial Score
This measure was developed specifically to quantify the signs and symptoms affecting the extraocular, facial, and respiratory muscles in MG, specifically considering patients with musculoskeletal MG.41 The Oculo-Bulbar Facial Score has 1 item that sums the strength of 5 facial muscles, 1 item on palatal contractility, 1 on tongue appearance, 1 on forced vital capacity, and 1 on swallowing time, based on swallowing 100 mL of water. The total score ranges from 0 to 21, and higher scores indicate worse oculo-bulbar and respiratory function. A modified version removed 2 of the 5 facial muscles tested give low agreement between raters, and has a maximum possible score of 17.41 Overall interrater reliability coefficients were not described. The Oculo-Bulbar Facial Score has moderate correlations with the MGC, MG-ADL, and MG-QoL, more so to bulbar-related items. Its sensitivity to change or its predictive properties have not been studied.
The Myasthenia Gravis Impairment Index
The Myasthenia Gravis Impairment Index (MGII) has been recently developed using a patient-centered approach, whereby patient input was incorporated through the development process.12 This method was grounded in a qualitative study of patients’ experiences with MG,9 where fatigability was a key component of overall MG severity. The scale has 22 patient-reported items (2-week recall time) and 6 examination items that reflect severity and fatigability of ocular, bulbar, and limb/generalized impairments. The total scores range between 0 and 84, but it can also be scored as an ocular (0–23) and a generalized (0–61) score, where higher scores indicate greater disease severity. The MGII was validated in a cohort of 200 MG patients.12 Test–retest reliability for the whole scale and interobserver reliability for the examination component was high (ICCs of 0.93 and 0.90, respectively). Construct validity was tested through predefined hypotheses of the correlations between the MGII and other MG symptoms measures (QMGS, MGC, MG-ADL), an MG-specific HRQoL measure (MG-QoL15), and generic measures (Short Form-36 [SF-36],42 the EQ-5D,43 and the NeuroQoL-Fatigue module44). Additionally, MGII scores were significantly different across MGFA classes and between ocular/generalized patients, indicating gross discriminative validity.12 Responsiveness was studied in 95 patients receiving prednisone or immunomodulation (IVIG or plasma exchange), using stable patients from the reliability study as controls.45 Patients receiving prednisone or immunomodulation changed significantly more than controls. Additionally, patients in the pure ocular group changed significantly in the ocular subscore, without change in generalized items. Overall, patients receiving prednisone changed more in the ocular subscore than those receiving immunomodulation, who changed more in the generalized subscore. Patient-meaningful change was studied using the patient’s impression of change, whereby patients who felt better changed significantly more than those who were unchanged or worse. The MID at a group level—to estimate sample size for a trial—was 8.1 points, and at the individual level—to classify a patient as responder—was 5.5 points. The MGII takes approximately 10 minutes to complete, which is feasible depending on the setting. It has not yet been used in a clinical trial.
Special considerations should be made regarding outcomes in patients in myasthenic crisis. Most of these outcomes have been validated in outpatients, so data on crisis are scarce. Many of the physician-rated scales can be applied to patients in crisis, because they usually include intubation/ventilation within the respiratory items. However, patient-reported outcomes by their nature are not suited for these patients, at least in the acute phase.46 Considering the mortality associated with myasthenic crisis, survival should be a main outcome. Additionally, because MG crisis is defined by the need for intubation, then successful extubation and overall time to extubation have been used to describe the outcomes in this population, because they are associated with the duration of intensive care and hospital stay, medical complications, and overall mortality.47
MEASURES OF DISABILITY AND HEALTH-RELATED QUALITY OF LIFE IN MYASTHENIA GRAVIS
There is one measure specifically aimed at measuring disability in MG using the ICF framework, the MG-DIS. Additionally, there are 2 disease-specific measures of HRQoL: the MG-QOL 60, a 60-item measure, and the MG-QOL 15, which derives from the 60 item questionnaire and that has been recently modified (MG-QOL15r). Table 2 summarizes these measures.
Table 2.
Measures of disability and HRQoL in myasthenia gravis
| Total Items | Score Range | Interpretation | |
|---|---|---|---|
| MG-DIS | 20 | 0–100 | Higher score, more disability |
| MG-QOL 60 | 60 | 0–240 | Higher score, worse HRQoL |
| MG-QOL 15 | 15 | 0–60 | Higher score, worse HRQoL |
| MG- QOL 15 r | 15 | 0–30 | Higher score, worse HRQoL |
Abbreviations: HRQoL, health-related quality of life; MG-DIS, Myasthenia Gravis Disability Scale; MG-QOL, Myasthenia Gravis Quality of Life.
All these are patient-reported outcomes.
The Myasthenia Gravis Disability Scale
This is a measure specifically developed to quantify disability in patients with MG. The MG-DIS was developed as a patient-reported outcome, using the ICF framework of disability.48 For this, the authors linked items from available measures to different ICF codes, using previous studies where patients had self-reported their MG-related problems using this classification system.49 Additionally, the authors conducted semistructured interviews in a group of MG patients, whereby the different codes previously identified where explored in depth, to determine if those symptoms or limitations in activities or participation were directly related to MG. These steps resulted in retaining 42 ICF categories, with a total of 44 preliminary items covering body functions, activities/participation, and environmental factors.48 The 31 items reflecting impairments and restrictions of activities and participation were further studied for validation.50 Item reduction was based on interitem correlations and factor analysis, in a cohort of 109 patients, and 20 items were retained.
These items reflect generalized impairment problems, bulbar-related problems, mental health and fatigue problems, and vision-related problems. All items have a 30-day recall period, and are graded on a scale of 1 to 5; total scores are transformed to a scale of 0 to 100, where higher scores reflect greater disability. Cronbach’s alpha was 0.92, indicating good internal consistency. The test–retest reliability was tested in 21 patients, with high r coefficients. Construct validity was studied through correlations with the MGC as well as fatigability indices, and discriminative validity was tested by comparing MG-DIS scores between ocular/remission patients and patients with generalized disease.50 Longitudinal data were available for 75 patients, and MG-DIS scores changed according to the patients’ impression of change. However, there are no data on the MID and no other studies on responsiveness in relationship to specific interventions.
The Myasthenia Gravis Quality of Life 60
Before this measure was developed, all studies on HRQoL for MG patients used generic measures, such as the SF-36.42 The aim of the developers was to obtain a measure that would also include the specific effects of MG in HRQoL. Item development was based in a Multiple Sclerosis HRQoL instrument, were experts selected items that were relevant for MG patients.51 Patients’ interviews were conducted and 100 preliminary items were obtained, including physical, social, emotional, and functional problems. The authors obtained feedback from physicians and patients to further reduced the measure to 60 items—each scored in a Likert scale from 0 to 4—resulting in a total score ranging from 0 to 240; higher scores indicate worse HRQoL. The 60-item measure was tested in 80 patients participating in a clinical trial of mycophenolate.39 Internal consistency was excellent (Cronbach’s alpha, 0.94), test–retest reliability was not assessed, given that patients were actively receiving interventions. Construct validity was studied through correlations between the MG-QOL60 and the SF-36 (r = −8.0), the MG-ADL (r = 0.72), the QMGS (r = 0.53), and the MMT (r = 0.43).51 The MG-QOL60 was used as a secondary outcome in a randomized, controlled study comparing IVIG and plasma exchange.52 In this study, the MG-QOL60 scores significantly improved in patients receiving both treatments, changing more in responders—who improved by 22 points—compared with those who were unchanged—who changed only by 7 points.53 However, the MID was not specifically studied.
The Myasthenia Gravis Quality of Life 15 and the Myasthenia Gravis Quality of Life 15 r
The MG-QOL15 was developed with the aim of simplifying the MG-QOL60. First, 20 candidate items were flagged for retention, based on their responsiveness to change in the mycophenolate study.35 Additionally, data on 13 patients followed in routine clinical visits who reported worsening was assessed, and based on this input the items were reduced to 15: mobility (9 items), symptoms (3 items), and contentment and emotional well-being (3 items). Items are scored in a Likert scale from 0 to 4 and the total sum score ranges from 0 to 60, where higher scores indicate worse HRQoL.35 The MG-QOL15 has good internal consistency (Cronbach’s alpha, 0.89). In terms of construct validity, the MG-QOL15 correlated with the physical and mental components of the SF-36, as well as with MG-specific measures (QMGS, MG-ADL, and MMT).35 Longitudinal validity was studied in 138 patients with routine follow-up at different centers. Patients were considered improved if they had a 3 or greater point improvement in the MGC and the MG-QOL changed more in those improved compared with those not improved, and the change in QOL-15 scores correlated with the change in MGC (r = 0.53).54 In the same study, test–retest reliability was excellent (ICC, 0.98). The MG-QOL15 also demonstrated to be responsive in a randomized, controlled study of IVIG versus plasma exchange, where responders to treatment improved in average by 9 points compared with nonresponders, who changed by 2 points, thereby suggesting that a decrease MG QOL-15 of 7 or more points is correlated with improvement in the subgroup with moderate to severe MG (QMGS ≤11).53 The MID has not been fully determined. The MG-QOL15 has been translated and validated in several languages, such as Japanese and French,55,56 and it is widely used around the world. Based on its extensive use, it has been recently modified, to improve the performance of certain items.57 The modified version (MG-QOL15r) retained the 15 items with some wording changes, and these were rescored from a 0 to 4 to a 0 to 2 scale, based on Rasch analysis. The resulting measure scores ranges from 0 to 30, and higher scores indicate worse HRQoL. When compared with the original scale, the modified version had better psychometric properties than the original and it is very easy to use.57 Responsiveness of the MG-QOLr has not yet been studied.
STEROID-SPARING EFFECTS AND OTHER OUTCOMES
Beyond the symptoms and their effect on the patients’ lives, other outcomes have been incorporated in clinical trials of MG, often as biomarkers or surrogate outcomes. Because of the side effects of long-term steroid treatment, it is of interest to measure the overall exposure of prednisone as an outcome, whereby a treatment is superior if it can reduce the use of prednisone over time with good symptomatic control. This steroid-sparing effect has been usually studied using the area under the curve of prednisone over time, for example, in the thymectomy randomized, controlled trial,36 where the prednisone dose was time weighted and the treated group had a lower prednisone exposure over time than the control arm. Recently, the results of the methotrexate for MG study were published. In this 12-month study, the prednisone area under the curve between months 3 and 12 was the primary outcome.58 There was no difference in the prednisone area under the curve between the methotrexate and placebo groups; however, more subjects in the placebo arm withdrew from the study and all patients that withdrew owing to worsening MG had received placebo. Finally, some of the secondary measures showed a trend favoring methotrexate. More studies are needed to better understand what difference in overall prednisone exposure is clinically significant. Recently, a study of rituximab on musculoskeletal MG used a composed outcome mixing the MGFA postintervention status and the dose of immunosuppressants, called the Myasthenia Gravis Status and Treatment Intensity Score.59 Further studies are needed to validate this measure. For more details, the reader is referred to the article by Michael K. Hehir and Nicholas J. Silvestri’s article, “Generalized Myasthenia Gravis: Classification, Clinical Presentation, Natural History, and Epidemiology,” in this issue.
Electrophysiology testing, such as repetitive nerve stimulation and single fiber electromyography have also been used as surrogate markers of improvement.60 However, although the decrement on repetitive nerve stimulation and jitter can improve with treatment, their correlation with clinical change is only mild to moderate.22 Because these tests can be painful and time consuming, they are not routinely used in the follow-up of patients with MG. The titers of acetylcholine receptor antibodies have also been used as biomarker; however, they do not correlate well with clinical change.61
SUMMARY
There are several outcome measures available for MG and there is no single, perfect measure that works for every scenario. Because standards for outcome measure development have changed in recent years,6,7 newer outcomes tend to incorporate more of the patients’ input and have more studies on their psychometric properties. Things to take into account when choosing a measure are what it measures, in which population it was validated, what purpose(s) does it serve, and what measurement properties it has. Additionally, feasibility has to be considered—for a busy clinical setting shorter measures might be preferable—whereas in a clinical trial more comprehensive measures might be needed to demonstrate treatment benefits. The validation of measures is an ongoing process, so current measures should be studied in different populations and their responsiveness to different interventions tested.
Finally, more information is needed regarding the interpretation of change scores, to understand what magnitude of change is meaningful. The MIDs should be studied in different settings (ie, groups vs individuals), in patient who are worsening—whose MID is often different than from improvement—and based on baseline severity.5 Additionally, research into Patient Acceptable Symptom States is needed, aiming to find thresholds for different outcomes were patients are not only a “little better” but “good enough.”62 These Patient Acceptable Symptom States thresholds could eventually serve as therapeutic targets both for clinical and research settings.
KEY POINTS.
Newer outcome measures incorporate more input from patients and have undergone more rigorous psychometric analysis.
Ideal measures in clinical care are brief to administer, whereas in clinical trials more comprehensive and overlapping measures are needed to demonstrate a positive effect.
Minimal clinically important differences are available in very few of the outcome measures but can help to inform clinical trial design and sample size estimation.
Acknowledgments
Disclosure Statement: C. Barnett participated in the development of the Myasthenia Gravis Impairment Index (MGII) and might receive royalties in the future. She has provided consultancy to UCB regarding outcomes for myasthenia gravis studies. Dr R. Barohn is on the speaker’s bureau for NuFactor, Grifols Therapeutics Inc, and Plan 365 Inc. He is on the advisory board for CSL Behring GmbH, and has received an honorarium from Option Care. He has received research grants from the NIH, FDA/OOPD, NINDS, Novartis, Sanofi/Genzyme, Biomarin, IONIS, Teva, Cytocenetics, Eli Lilly, and PTC. Dr M. Dimachkie is on the speaker’s bureau or is a consultant for Alnylam, Baxalta, Catalyst, CSL-Behring, Mallinckrodt, Novartis, and NuFactor. He has also received grants from Alexion, Biomarin, Catalyst, CSL-Behring, the FDA/OPD, GSK, Grifols, MDA, the NIH, Novartis, Sanofi, and TMA. This work was supported by a CTSA grant from NCATS awarded to the University of Kansas for Frontiers: University of Kansas Clinical and Translational Science Institute (# UL1TR002366) The contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or NCATS.
REFERENCES
- 1.Jette AM. Toward a common language for function, disability, and health. Phys Ther 2006;86(5):726–34. [PubMed] [Google Scholar]
- 2.World Health Organization. International classification of functioning, disability and health (ICF) 1st edition. Geneva: World Health Organization; 2002. Available at: http://www.who.int/classifications/icf/icfbeginnersguide.pdf?ua=1. Accessed June, 2017. [Google Scholar]
- 3.Guyatt GH, Bombardier C, Tugwell PX. Measuring disease-specific quality of life in clinical trials. Can Med Assoc J 1986;134:889. [PMC free article] [PubMed] [Google Scholar]
- 4.Kirshner B, Guyatt G. A methodological framework for assessing health indices. J Chronic Dis 1985;38(1):27–36. [DOI] [PubMed] [Google Scholar]
- 5.King MT. A point of minimal important difference (MID): a critique of terminology and methods. Expert Rev Pharmacoecon Outcomes Res 2011;11(2):171–84. [DOI] [PubMed] [Google Scholar]
- 6.U.S. Department of Health and Human Services FDA Center for Drug Evaluation and Research, U.S. Department of Health and Human Services FDA Center for Biologics Evaluation and Research, U.S. Department of Health and Human Services FDA Center for Devices and Radiological Health. Guidance for industry: patient-reported outcome measures: use in medical product development to support labeling claims: draft guidance. Health Qual Life Outcomes 2006;4:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mokkink LB, Terwee CB, Knol DL, et al. The COSMIN checklist for evaluating the methodological quality of studies on measurement properties: a clarification of its content. BMC Med Res Methodol 2010;10(1):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Vet HCW, Terwee CB, Mokkink LB, et al. Measurement in medicine New York: Cambridge University Press; 2011. [Google Scholar]
- 9.Barnett C, Bril V, Kapral M, et al. A conceptual framework for evaluating impairments in myasthenia gravis. PLoS One 2014;9(5):e98089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolfe GI, Herbelin L, Nations SP, et al. Myasthenia gravis activities of daily living profile. Neurology 1999;52(7):1487–9. [DOI] [PubMed] [Google Scholar]
- 11.Burns TM, Conaway MR, Cutter GR, et al. , Muscle Study Group. Construction of an efficient evaluative instrument for myasthenia gravis: the MG composite. Muscle Nerve 2008;38(6):1553–62. [DOI] [PubMed] [Google Scholar]
- 12.Barnett C, Bril V, Kapral M, et al. Development and validation of the myasthenia gravis impairment index. Neurology 2016;87(9):879–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Osserman KE, Kornfeld P, Cohen E, et al. Studies in myasthenia gravis; review of two hundred eighty-two cases at the Mount Sinai Hospital, New York City. AMA Arch Intern Med 1958;102(1):72–81. [DOI] [PubMed] [Google Scholar]
- 14.Jaretzki A, Barohn RJ, Ernstoff RM, et al. Myasthenia gravis: recommendations for clinical research standards. Task Force of the Medical Scientific Advisory Board of the Myasthenia Gravis Foundation of America. Neurology 2000;55(1): 16–23. [DOI] [PubMed] [Google Scholar]
- 15.Baggi F, Mantegazza R, Antozzi C, et al. Patient registries: useful tools for clinical research in myasthenia gravis. Ann N Y Acad Sci 2012;1274(1):107–13. [DOI] [PubMed] [Google Scholar]
- 16.Besinger UA, Toyka KV, Homberg M, et al. Myasthenia gravis: long-term correlation of binding and bungarotoxin blocking antibodies against acetylcholine receptors with changes in disease severity. Neurology 1983;33(10):1316–21. [DOI] [PubMed] [Google Scholar]
- 17.Tindall RS, Rollins JA, Phillips JT, et al. Preliminary results of a double-blind, randomized, placebo-controlled trial of cyclosporine in myasthenia gravis. N Engl J Med 1987;316(12):719–24. [DOI] [PubMed] [Google Scholar]
- 18.Barohn RJ, McIntire D, Herbelin L, et al. Reliability testing of the quantitative myasthenia gravis score. Ann N Y Acad Sci 1998;841:769–72. [DOI] [PubMed] [Google Scholar]
- 19.Sharshar T, Chevret S, Mazighi M, et al. Validity and reliability of two muscle strength scores commonly used as endpoints in assessing treatment of myasthenia gravis. J Neurol 2000;247(4):286–90. [DOI] [PubMed] [Google Scholar]
- 20.Barnett C, Merkies ISJ, Katzberg H, et al. Psychometric properties of the quantitative myasthenia gravis score and the myasthenia gravis composite scale. J Neuromuscul Dis 2015;2(3):301–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanders DB, Tucker-Lipscomb B, Massey J. A simple manual muscle test for myasthenia gravis: validation and comparison with the QMG score. Ann N Y Acad Sci 2003;998:440–4. [DOI] [PubMed] [Google Scholar]
- 22.Barnett C, Katzberg H, Navabi M, et al. The quantitative myasthenia gravis score: comparison with clinical, electrophysiological, and laboratory markers. J Clin Neuromuscul Dis 2012;13(4):201–5. [DOI] [PubMed] [Google Scholar]
- 23.Bedlack RS, Simel DL, Bosworth H, et al. Quantitative myasthenia gravis score: assessment of responsiveness and longitudinal validity. Neurology 2005;64(11): 1968–70. [DOI] [PubMed] [Google Scholar]
- 24.Zinman L, Ng E, Bril V. IV immunoglobulin in patients with myasthenia gravis: a randomized controlled trial. Neurology 2007;68(11):837–41. [DOI] [PubMed] [Google Scholar]
- 25.Katzberg HD, Barnett C, Merkies ISJ, et al. Minimal clinically important difference in myasthenia gravis: outcomes from a randomized trial. Muscle Nerve 2014; 49(5):661–5. [DOI] [PubMed] [Google Scholar]
- 26.Barnett TC, Bril V, Davis AM. Performance of individual items of the quantitative myasthenia gravis score. Neuromuscul Disord 2013;23(5):413–7. [DOI] [PubMed] [Google Scholar]
- 27.Gajdos P, Simon N, de Rohan-Chabot P, et al. Long-term effects of plasma exchange in myasthenia. Results of a randomized study. Presse Med 1983; 12(15):939–42 [in French]. [PubMed] [Google Scholar]
- 28.Gajdos P, Chevret S, Clair B, et al. Clinical trial of plasma exchange and high-dose intravenous immunoglobulin in myasthenia gravis. Myasthenia Gravis Clinical Study Group. Ann Neurol 1997;41(6):789–96. [DOI] [PubMed] [Google Scholar]
- 29.Mantegazza R, Antozzi C, Peluchetti D. Azathioprine as a single drug or in combination with steroids in the treatment of myasthenia gravis. J Neurol 1988;235: 449–53. [DOI] [PubMed] [Google Scholar]
- 30.Antozzi C, Brenna G, Baggi F, et al. Validation of the Besta Neurological Institute Rating Scale for Myasthenia Gravis. Muscle Nerve 2015;53(1):32–7. [DOI] [PubMed] [Google Scholar]
- 31.Lashley D, Palace J, Jayawant S, et al. Ephedrine treatment in congenital myasthenic syndrome due to mutations in DOK7. Neurology 2010;74(19):1517–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wolfe GI, Barohn RJ, Foster BM, et al. Randomized, controlled trial of intravenous immunoglobulin in myasthenia gravis. Muscle Nerve 2002;26(4):549–52. [DOI] [PubMed] [Google Scholar]
- 33.Muppidi S, Wolfe GI, Conaway M, Burns T and the MG Composite and MG-QOL15 Study Group. MG-ADL: still a relevant outcome measure. Muscle Nerve 2011;44(5):727–31. [DOI] [PubMed] [Google Scholar]
- 34.Burns TM, Conaway M, Sanders DB, MG Composite and MG-QOL15 Study Group. The MG composite: a valid and reliable outcome measure for myasthenia gravis. Neurology 2010;74(18):1434–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burns TB, Conaway MR, Cutter GR, et al. , The Muscle Study Group. Less is more, or almost as much: a 15-item quality-of-life instrument for myasthenia gravis. Muscle Nerve 2008;38(2):957–63. [DOI] [PubMed] [Google Scholar]
- 36.Wolfe GI, Kaminski HJ, Aban IB, et al. Randomized trial of thymectomy in myasthenia gravis. N Engl J Med 2016;375(6):511–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanders DB, Hart IK, Mantegazza R, et al. An international, phase III, randomized trial of mycophenolate mofetil in myasthenia gravis. Neurology 2008;71(6):400–6. [DOI] [PubMed] [Google Scholar]
- 38.Wolfe GI, Barohn RJ, Sanders DB, et al. Comparison of outcome measures from a trial of Mycophenolate mofetil in myasthenia gravis. Muscle Nerve 2008;38(5):1429–33. [DOI] [PubMed] [Google Scholar]
- 39.Muscle Study Group. A trial of mycophenolate mofetil with prednisone as initial immunotherapy in myasthenia gravis. Neurology 2008;71(6):394–9. [DOI] [PubMed] [Google Scholar]
- 40.Benatar M, Sanders DB, Burns TM, et al. Recommendations for myasthenia gravis clinical trials. Muscle Nerve 2012;45:909–17. [DOI] [PubMed] [Google Scholar]
- 41.Farrugia ME, Harle HD, Carmichael C, et al. The oculobulbar facial respiratory score is a tool to assess bulbar function in myasthenia gravis patients. Muscle Nerve 2011;43(3):329–34. [DOI] [PubMed] [Google Scholar]
- 42.Ware JE, Snow KK, Kosinski M, et al. SF-36 health survey Boston: Manual and Interpretation Guide. New England Medical Center Hospital. Health Institute; 1993. [Google Scholar]
- 43.Rabin R, de Charro F. EQ-SD: a measure of health status from the EuroQol Group. Ann Med 2001;33(5):337–43. [DOI] [PubMed] [Google Scholar]
- 44.Cook KF, Victorson DE, Cella D, et al. Creating meaningful cut-scores for NeuroQOL measures of fatigue, physical functioning, and sleep disturbance using standard setting with patients and providers. Qual Life Res 2014;24(3):575–89. [DOI] [PubMed] [Google Scholar]
- 45.Barnett C, Vera B, Kapral M, et al. Myasthenia Gravis Impairment Index: Responsiveness, meaningful change, and relative efficiency. Neurology 2017;89(23):2357–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heyland DK, Guyatt G, Cook DJ, et al. Frequency and methodologic rigor of quality-of-life assessments in the critical care literature. Crit Care Med 1998; 26(3):591–8. [DOI] [PubMed] [Google Scholar]
- 47.Ramos-Fransi A, Rojas-Garćıa R, Segovia S, et al. Myasthenia gravis: descriptive analysis of life-threatening events in a recent nationwide registry. Eur J Neurol 2015;22(7):1056–61. [DOI] [PubMed] [Google Scholar]
- 48.Raggi A, Schiavolin S, Leonardi M, et al. Development of the MG-DIS: an ICF-based disability assessment instrument for myasthenia gravis. Disabil Rehabil 2014;36(7):546–55. [DOI] [PubMed] [Google Scholar]
- 49.Leonardi M, Raggi A, Antozzi C, et al. Identification of international classification of functioning, disability and health relevant categories to describe functioning and disability of patients with myasthenia gravis. Disabil Rehabil 2009;31(24): 2041–6. [DOI] [PubMed] [Google Scholar]
- 50.Raggi A, Leonardi M, Schiavolin S, et al. Validation of the MG-DIS: a disability assessment for myasthenia gravis. J Neurol 2016;263(5):871–82. [DOI] [PubMed] [Google Scholar]
- 51.Mullins LL, Carpentier MY, Paul RH, Sanders DB and the Muscle Study Group. Disease-specific measure of quality of life for myasthenia gravis. Muscle Nerve 2008;38(2):947–56. [DOI] [PubMed] [Google Scholar]
- 52.Barth D, Nabavi Nouri M, Ng E, et al. Comparison of IVIg and PLEX in patients with myasthenia gravis. Neurology 2011;76(23):2017–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barnett C, Wilson G, Barth D, et al. Changes in quality of life scores with intravenous immunoglobulin or plasmapheresis in patients with myasthenia gravis. J Neurol Neurosurg Psychiatry 2013;84(1):94–7. [DOI] [PubMed] [Google Scholar]
- 54.Burns TM, Grouse CK, Wolfe GI, et al. The MG-QOL15 for following the health-related quality of life of patients with myasthenia gravis. Muscle Nerve 2011; 43(1):14–8. [DOI] [PubMed] [Google Scholar]
- 55.Masuda M, Utsugisawa K, Suzuki S, et al. The MG-QOL15 Japanese version: validation and associations with clinical factors. Muscle Nerve 2012;46(2): 166–73. [DOI] [PubMed] [Google Scholar]
- 56.Birnbaum S, Ghout I, Demeret S, et al. Translation, cross-cultural adaptation, and validation of the French version of the 15-item myasthenia gravis quality of life scale. Muscle Nerve 2017;55(5):639–45. [DOI] [PubMed] [Google Scholar]
- 57.Burns TM, Sadjadi R, Utsugisawa K, et al. An international clinimetric evaluation of the MG-QOL15, resulting in slight revision and subsequent validation of the MG-QOL15r. Muscle Nerve 2016;54(6):1015–22. [DOI] [PubMed] [Google Scholar]
- 58.Pasnoor M, He J, Herbelin L, et al. A randomized controlled trial of methotrexate for patients with generalized myasthenia gravis. Neurology 2016;87(1):57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hehir M, Hobson-Webb LD, Benatar M, et al. Rituximab as treatment for anti-MuSK myasthenia gravis: multicenter blinded prospective review. Neurology 2017;89(10):1069–77. [DOI] [PubMed] [Google Scholar]
- 60.Zinman L, Baryshnik D, Bril V. Surrogate therapeutic outcome measures in patients with myasthenia gravis. Muscle Nerve 2008;37(2):172–6. [DOI] [PubMed] [Google Scholar]
- 61.Sanders DB, Burns TM, Cutter GR, et al. Does change in acetylcholine receptor antibody level correlate with clinical change in myasthenia gravis? Muscle Nerve 2014;49(4):483–6. [DOI] [PubMed] [Google Scholar]
- 62.Wijeysundera DN, Johnson SR. How much better is good enough? Patient-reported outcomes, minimal clinically important differences, and patient acceptable symptom states in perioperative research. Anesthesiology 2016;125(1): 7–10. [DOI] [PubMed] [Google Scholar]