

Abstract
Even though the TET enzymes catalyze the generation of 5-hydroxymethyl-cytosines required for lineage commitment and subsequent differentiation of stem cells into erythroid cells, the mechanisms that link extra-cellular signals to TET activation and DNA hydroxymethylation are unknown. We demonstrate that hematopoietic cytokines phosphorylate TET2 leading to its activation in erythroid progenitors. Specifically, cytokine receptor associated, JAK2, phosphorylates TET2 at tyrosines 1939 and 1964. Phosphorylated TET2 interacts with erythroid transcription factor, KLF1, and this interaction with TET2 is increased upon exposure to erythropoietin. The activating JAK2V617F mutation seen in myeloproliferative disease patient samples and in mouse models is associated with increased TET activity and cytosine hydroxymethylation as well as genome wide loss of cytosine methylation. These epigenetic and functional changes are also associated with increased expression of several oncogenic transcripts. Thus, we demonstrate that JAK2 mediated TET2 phosphorylation provides a mechanistic link between extracellular signals and epigenetic changes during hematopoiesis.
Introduction
Epigenetic changes including DNA cytosine methylation and hydroxymethylation play important roles in regulating differentiation of hematopoietic cells into committed progeny (1–3). The 5- hydroxyl-methyl cytosine residue (5-hmC), is generated from 5-mC through dioxidation by the Ten-eleven translocation (TET) family of proteins (2, 4, 5). TET enzymes, especially TET2 are critical for myeloid and erythroid differentiation and inactivating mutations in this enzyme are associated with development of myeloid malignancies and altered hematopoietic differentiation (2, 4). TET enzymes require cofactors/substrates such as iron, oxygen and alpha-ketoglutarate for their catalytic activity, and also ascorbate, which indirectly increase TET activity by acting on iron oxidation state leading to modulation of hydroxymethylcytosine levels in various models (1, 6, 7). Even though limited data exist on post-translation modification in TET enzymes such as glycosylation and phosphorylation (8), these changes have not yet been linked to its activity or have been shown to be as a result of extracellular stimuli. More importantly, even though TET enzyme catalyzed DNA hydroxymethylation has been a critically important pathway for sustaining normal hematopoiesis, no mechanistic link between hematopoietic cytokine stimuli, TET activity, and DNA hydroxymethylation has been established.
Janus activated kinases (JAKs) are receptor associated tyrosine kinases that are activated by cytokine receptors. Erythropoietin, Stem cell factor and FLT3 ligand are important regulators of hematopoietic stem and progenitors whose cognate receptors utilize JAK2 to transduce signals intracellularly. Thus JAK2 activation is critical for cytokine induced signaling and the activating V617F mutation in the JAK2 pseudo kinase domain can lead to over-activation of signaling leading to the development of myeloproliferative neoplasms (9–11). Accumulated data have established that the vast majority of activation signals downstream of JAK2 are due to STAT activation by JAK2. There have not been any reports linking JAK2 activation directly to modulators of epigenetic regulation.
In this report, we demonstrate that JAK2 is a kinase responsible for phosphorylating TET2 and that the sites of phosphorylation are at tyrosines 1939 and 1964 located within the catalytic core of TET2. We also show that TET2 phosphorylation is associated with increased TET activity resulting in increased DNA hydroxymethylation. The two identified sites of tyrosine phosphorylation within TET2 are important in promoting erythroid differentiation and are absent in TET1 and TET3 family members, suggesting non-overlapping functions for TET2. We also demonstrate that the Jak2V617F activating mutation is associated with increased hydroxymethylation and decreased genome cytosine methylation in primary myeloproliferative neoplasm samples and in transgenic mouse models with the Jak2V617F mutation. These data provide a novel mechanistic link between cytokine induced signaling and epigenetic regulation of hematopoiesis. These data also show the importance of novel post translation modifications of TET2 in hematopoiesis.
Results
TET enzymes are activated in response to extracellular cytokines
Cytokines are important regulators of hematopoiesis and act via their cognate receptors to elicit downstream changes. Since TET enzymes and DNA hydroxymethylation have been shown to be important regulators of hematopoiesis, we wanted to determine whether TET enzymes are capable of responding to extracellular cytokine stimuli. We exposed human primary hematopoietic stem/progenitor cells and erythroid progenitors to the pleotropic cytokines-, FLT3 ligand (FLT3L) and, Stem Cell Factor (SCF), and to the lineage specific cytokine Erythropoietin (EPO) in a time dependent fashion and examined TET enzyme activity and 5-hmC levels. We observed that TET2 activity gradually increased over a 60 minute time period when exposed to FLT3L, whereas TET2 activities peaked at 30 minutes and then declined by 60 minutes when erythroid progenitors were exposed to SCF or EPO (Fig. 1A, B, C). In order to ascertain whether cytokine mediated increased TET activity also result in increased 5-hmC content, we exposed both early human stem/progenitors and erythroid progenitors to each of the three cytokines for 30 minutes prior to DNA isolation and determining the levels of 5-hmC levels by ELISA. We observed that 5-hmC levels were consistently increased following cytokine exposure in three independent biological replicates (Fig. 1D, E, F). These studies were extended by investigating whether thrombopoietin (TPO) and granulocyte colony stimulation factor (G-CSF), are also capable of activating TET2 leading to increased 5-hmC levels, which showed increased TET2 activity and 5hm-C levels upon cytokine stimulation (Fig. S1A-D).
Fig.1. Activation of TET enzymes in response to extracellular cytokines leads to gain of 5-hmC in the genome.

(A) Lineage uncommitted CD34+ hematopoietic stem/early progenitors were exposed to pleotropic cytokine FLT3L for various times and nuclear lysates were used for determining TET enzyme activity. Data are represented as mean +SEM from three biological replicates. (B, C) Primary erythroid progenitors (basophilic stage) derived after differentiation of CD34+ stem/early progenitors were exposed to stem cell factor (SCF) or erythropoietin (EPO) for various times and nuclear lysates were used for determining TET enzyme activity. Data are represented as mean +SEM from three biological replicates. (D, E, F) Lineage uncommitted CD34+ hematopoietic stem/early progenitors or primary erythroid progenitors were exposed to FLT3L, SCF or EPO as indicated for 30 minutes prior to isolating genomic DNA and determining the 5-hmC content. Data are represented as mean +SEM from three biological replicates.
TET2 is Tyrosine phosphorylated in response to cytokine exposure and its activation is mediated by JAK2
In order to determine the mechanism of TET activation and increased 5-hmC levels in response to hematopoietic cytokines, we first interrogated the TET2 protein sequence for potential sites of post-translational modifications. Using multiple algorithms (12) we performed in-silico analysis to and identified multiple phosphorylation consensus sequence motifs for several well-characterized tyrosine and serine/threonine kinases. Many of these potential phosphorylation sites were clustered within the catalytic core of the TET2 protein, including many of the sites predicted to be phosphorylated by JAK2 (Fig. 2A). Therefore we focused on examining tyrosine phosphorylation at these sites, especially since JAK2 is a critical mediator of cytokine signaling cascades in hematopoietic stem and progenitors. First, we exposed human CD34+ early stem/progenitor cells to FLT3L and erythroid progenitors to either EPO or SCF followed by immunoprecipitation of total protein with an anti-TET2 antibody followed by immunoblot analysis using an anti-phospho tyrosine antibody (Fig. 2B). We observed that exposure to each of the cytokines resulted in tyrosine phosphorylation of TET2 (Fig. 2B). To further interrogate this finding we focused on erythroid progenitors and exposed primary human erythroid progenitors to EPO in a time dependent fashion. This experiment showed that TET2 is phosphorylated by EPO and maintains such phosphorylation up to 60 minutes (Fig. 2C). A similar pattern was also observed in UT-7 erythroid cell line further confirming this finding (Fig. S2A). In order to establish whether blocking the actions of FLT3L and EPO lead to abolishing TET2 activity, we exposed primary stem/progenitor cells or erythroid progenitors to FLT3L or EPO in the presence or absence of specific inhibitors of FLT3 and JAK2 (activator of EPOR), which showed that inhibiting these two kinases resulted in abrogation of TET activity to its basal levels suggesting that TET activation is dependent on these two pathways (Fig. 2D, E).
Fig. 2. Phosphorylation of TET2 by JAK2 in response to hematopoietic cytokines.

(A) In-Silico analysis using KinasePhos 2.0 and PPSP computational program for prediction of sites for tyrosine phosphorylation within the catalytic core of TET2 protein. The cysteine-rich (C-rich) and DSBH domains are depicted as well as the C-terminal region. (B) Total cell lysates were collected from CD34+ stem/early progenitor cells or from erythroid progenitors after stimulation with FLT3L, SCF or EPO as indicated and subjected to immunoprecipitation using an anti-TET2 antibody followed by immunoblot analysis using an anti-phospho-tyrosine antibody. The same blot was then used for immunoblotting against an anti-TET2 antibody as a protein loading control. (C) A time course determination of TET2 phosphorylation in response to EPO (10U/ml) in primary erythroid progenitors. The same blot was then used for immunoblot analysis using tubulin and GAPDH as protein loading controls. (D) Inhibition of TET2 activity by FLT3 inhibitors, Sorafenib (10 nM), and AC2220 (10 nM), in CD34+ stem/early progenitors. Cells were exposed to the inhibitors for 1 hr prior to stimulation with FLT3L for 30 minutes followed by determination of TET activity. Cytokine unstimulated and FLT3L stimulated samples without the presence of inhibitors were used as controls. Data are represented as mean SEM ± from three biological replicates. (E) Inhibition of TET2 activity by JAK2 Inhibitor Ruxolitinib (100 nM) in primary erythroid progenitors. Cells were exposed to the inhibitor for 1 hour prior to stimulation with EPO for 30 minutes followed by determination of TET activity. Unstimulated and EPO stimulated samples without the inhibitor was used as controls. Data are represented as mean SEM ± from three biological replicates. (F) An in vitro kinase assay was developed using recombinant GFP/Flag-tagged TET2 catalytic domain and active JAK2 kinase under cell-free conditions for ascertaining whether TET2 catalytic domain is a direct target for phosphorylation by JAK2. In vitro kinase assays were performed using the GFP/Flag-tagged catalytic domain (1129–2002) of TET2 together with recombinant active JAK2, SYK and p38 MAP kinases as indicated. In vitro kinase reaction products were resolved on a SDS-PAGE gel and immunoblot analysis performed using an anti-phospho-tyrosine antibody. As controls the immune blots were also exposed to anti-FLAG and anti-phospho-serine antibodies to confirm immunoprecipitation reactions in all experimental samples were intact and phosphorylation was tyrosine specific respectively. (G) The in vitro kinase reaction products were analyzed for TET activity in the presence or absence of the JAK2 inhibitor by ELISA. Data are represented as mean SEM ± from three biological replicates. (H) Phosphorylation of TET2 by JAK2 is dose dependent. In vitro kinase assays were performed using various concentrations of recombinant JAK2 followed by analysis of the reaction products by immunoblot analysis using an anti-phospho-tyrosine antibody followed by immunoblot analysis using an anti-FLAG antibody as a protein loading control. (I) TET2 specifically interacts with JAK2 in UT-7 erythroid cells. Total cell lysates from UT-7 erythroid cells transfected with GFP/Flag-tagged TET2 catalytic domain were used to immunoprecipitate JAK2 using and anti-JAK2 antibody and the immunoprecipitates were subjected to immunoblot analysis using an anti-FLAG antibody. As a control the same lysates were subjected to immunoblot analysis using an anti-JAK2 antibody. (J) A reciprocal immunoprecipitation coupled immunoblot analysis was performed using anti-FLAG antibody for immunoprecipitation followed by immunoblot analysis using an anti-JAK2 antibody. As a control the same lysates were subjected to immunoblot analysis using an anti-FLAG antibody.
TET2 is a substrate for JAK2 kinase
We then focused on ascertaining whether TET2 is a direct substrate for JAK2 as suggested by the presence of multiple JAK2 consensus motifs and abolishment of TET2 activity in the presence of the JAK2 inhibitor. In order to determine this we devised a cell-free in vitro kinase assay where reconstituted recombinant GFP/Flag-tagged TET2 protein (catalytic core) was used as a substrate for recombinant active form of JAK2 as described in our methods. In parallel sets of in vitro assays, we also used recombinant SYK and p38 MAP kinases as controls for establishing specificity. The reaction products of these in vitro kinase assays were analyzed by immunoprecipitation with an anti-FLAG antibody (GFP/Flag-tagged TET2 as the target) followed by immunoblot analysis using an anti-phospho-tyrosine antibody. Our data showed that only JAK2 kinase was able to phosphorylate the TET catalytic core and not SYK and p38 MAP kinases (Fig. 2F). Furthermore, the presence of the JAK2 inhibitor in the kinase assay abrogated TET2 phosphorylation re-enforcing the specificity of the in vitro kinase assays. Since p38 MAP kinase (used as a control) is a serine/threonine kinase, we also determined whether the catalytic domain of TET2 is a target for p38 MAP kinase by performing immunoblot analysis with an anti-phospho-serine specific antibody, which showed no such phosphorylation (Fig. 2F). We then determined whether phosphorylation of the TET2 catalytic domain leads to activation of TET2 in a cell-free system. These experiments revealed that the phosphorylated TET2 catalytic domain is active and its activity can be abolished by the presence of the JAK2 inhibitor during the in vitro kinase assay reaction (Fig. 2G). To further interrogate these findings we performed a series of in vitro kinase assays, where we used increasing concentrations of JAK2 to demonstrate that phosphorylation of TET2 by JAK2 is dose-dependent re-enforcing that TET2 is a direct substrate for JAK2 (Fig. 2H). Finally we investigated whether JAK2 physically interacts with TET2 catalytic core by carrying out reciprocal immunoprecipitation coupled immunoblot analysis. These experiments show that JAK2 physically interacts with TET2 in multiple cell lines with erythroid characteristics (Figs. 2I, 2J, S2B, S2C). We then investigated localization and interaction between these two proteins within the cytoplasmic and nuclear compartments using primary erythroid progenitors. These experiments revealed that TET2 is primarily localized within the nucleus, whereas JAK2 was localized both in the cytoplasm and in the nucleus, although the amount of JAK2 protein in the nucleus was less than in the cytoplasm (Fig. S2D). Examination of the nuclear fraction showed that interaction between TET2 and JAK2 occurs within the nucleus and this interaction is reduced in the presence of the JAK2 inhibitor (Fig. S2E).
JAK2 phosphorylates specific tyrosine residues within the catalytic core of TET2
Given that TET2 is phosphorylated by JAK2, we sought to identify specific tyrosine residues within TET2 catalytic domain that are phosphorylated by JAK2. Using the in vitro kinase platform, we used the recombinant TET2 catalytic domain under our assay conditions in the presence or absence of enzymatically active JAK2 prior to subjecting the TET2 catalytic core to mass spectrometry analysis. The results of repeated experiments revealed tyrosine-1939 and tyrosine-1964 are phosphorylated as seen by the 80 Da shift corresponding to gain of a phosphate group in each of the peptides representing this region of the TET2 protein, reflected by the peaks within the chromatograms (Fig. 3A-D). The control samples that were not exposed to JAK2 kinase showed no such shift (Fig. 3A-D). Interestingly, in silico analysis of amino acid sequences of TET1, TET2 and TET3 showed that the two identified tyrosine residues were only present in TET2, but not in TET1 or TET3 (Fig. S3A). Furthermore, sequence alignment of the TET2 catalytic cores of multiple other species showed that Tyr-1964 site was conserved among all the species examined, whereas Tyr-1939 was slightly less conserved (Fig. S3B).
Fig. 3. Identification of phosphorylated tyrosines 1939 and 1964 onTET2 catalytic domain.

TET2 catalytic domain was exposed to active JAK2 in an in vitro Kinase assay and the reaction products were resolved by SDS-PAGE and further processed and used for liquid chromatography coupled mass spectrometry (MS) determination. (A) MS spectra of peptide fragments generated from proteolytic digestion of TET2 following in vitro kinase reactions in the presence of JAK2. The phosphorylated peptide, YGPDpYVPQK is depicted corresponding to tyrosine-1939 along with the series of y- and b-ions, including the phosphorylated residue. (B) MS spectra of TET2 peptide fragments generated from proteolytic digestion in vitro kinase reaction in the absence of JAK2. The peptide fragment surrounding Y1939 residue, EKEEECEKYGPDYVPQK is depicted without phosphorylation of tyrosine, as reflected by no gain in molecular weight of 80 Da, corresponding to the weight of PO4. (C) MS spectra of peptide fragments generated from proteolytic digestion of TET2 following in vitro kinase reactions in the presence of JAK2. The phosphorylated peptide, EPAEPHETSEPTpYLR is depicted corresponding to tyrosine-1964 along with the series of y- and b-ions, including the phosphorylated residue. (D) MS spectra of TET2 peptide fragments generated from proteolytic digestion in vitro kinase reaction in the absence of JAK2. The peptide fragment surrounding Y1964 residue, EPAEPHETSEPTYLRis is depicted without phosphorylation of tyrosine, as reflected by no gain in molecular weight of 80 Da, corresponding to the weight of PO4. (E) Y1939A, Y1964A, and Y1939A + Y1964A mutations were introduced into the GFP/Flag-tagged TET2 cDNA and proteins carrying these mutations were immunoprecipitated directly from HEK 293 cells using an anti-FLAG antibody. These immunoprecipitates were then used in the in vitro kinase assays in the presence or absence of active JAK2 followed by re-Immunoprecipitation with anti-FLAG antibody, resolved by SDS-PAGE and used for immunoblot analysis using an anti-tyrosine antibody. Parallel samples were used for immunoblot analysis using either an anti-TET2 antibody or an anti-FLAG antibody as controls. (F) Samples carrying single or double mutations of GFP/Flag- tagged TET2 or the wild-type catalytic domain of GFP/Flag-tagged TET2 were immunoprecipitated using an anti-FLAG antibody followed by immunoblot analysis against an anti-JAK2 antibody to determine whether TET2 catalytic domain interacts with JAK2. In order to confirm immunoprecipitated GFP/Flag-tagged TET2 samples were also immunoblotted against an anti-TET2 antibody or an anti-FLAG antibody. (G, H, I, J) Binding affinity between TET2 catalytic domain and JAK2 is dependent on phosphorylation status of Y1939 and Y1964. Biotinylated peptides carrying various combinations of Y1939 and Y1964 phosphorylation was used together with recombinant JAK2 as described in methods to determine the binding affinity (KD) between TET2 catalytic domain and JAK2. Graphs corresponding to TET2 and JAK2 interaction (red), between TET2 and p38 MAP kinase (black) and interaction in the presence of the JAK2 inhibitor (green) are depicted in each plot. The data are representative of duplicate determination.
Interdependency of Tyr-1964 and Tyr-1939 phosphorylation by JAK2
To further interrogate whether phosphorylation of Tyr-1939 and Tyr-1964 by JAK2 are interdependent, we performed in vitro mutagenesis, where the two sites were mutated individually or together by substituting tyrosine resides with alanine within the TET2 catalytic core. Using these constructs, we produced recombinant TET2 proteins and used them as substrates in the presence or absence of active JAK2 in our in vitro kinase assay. These experiments revealed that even when Tyr-1939 was substituted with alanine, JAK2 was able to phosphorylate the Tyr-1964 site. However, when Tyr-1964 site was replaced with alanine, it abrogated phosphorylation of the Tyr-1939 site (Fig. 3E). As expected in the wild-type TET2 catalytic core, where both these tyrosine residues were intact, we observed a high level of tyrosine phosphorylation in the presence of JAK2 compared to the sample where we omitted the addition of active recombinant JAK2 (Fig. 3E). These results suggest that the presence of Tyr-1964 is important for robust phosphorylation of Tyr-1939 of TET2 by the JAK2 enzyme.
Phosphorylation of Tyr-1939 and Tyr-1964 allows interaction between TET2 and JAK2.
We then determined whether phosphorylation of Tyr-1939 and Tyr-1964 is important for the association of TET2 with JAK2. We transfected each of the two single mutants, double mutant or wild-type TET2 constructs into UT-7 erythroid cell line and performed immunoprecipitation coupled immunoblot analysis. Each of the UT-7 cell lines carrying GFP/Flag-tagged TET2 constructs were immunoprecipitated with an anti-FLAG antibody followed by immunoblot analysis with an anti-JAK2 antibody to detect JAK2. These experiments showed a marked disruption of interaction between JAK2 and TET2 when Tyr-1939 or Tyr-1964 was mutated, whereas the wild-type TET2 sustained JAK2-TET2 interaction suggesting that both Tyr-1939 and Tyr-1964 are critical for maintaining the interaction between JAK2 and TET2 (Fig. 3F).
To further interrogate the nature of the interaction between TET2 and JAK2, we performed surface plasmon resonance (SPR) binding analysis using the Biacore™ instrument. This instrument allows determination of the binding affinity of a given protein to specific peptide motif under dynamic flow conditions. In order to determine the affinity of TET2 phosphorylated motifs to JAK2, we synthesized four different biotinylated peptides surrounding the two tyrosine residues (Y1939 and Y1964) with four different permutations as described in our methods. The first peptide was synthesized without the phosphate moiety on either one of the two tyrosine residues. The second peptide contained the phosphate moiety on both tyrosines, the third peptide contained the phosphate moiety only on Tyr-1939 and the fourth peptide contained the phosphate moiety only on Tyr-1964. These peptides were immobilized on sensor chips via streptavidin as described in our methods in each of the channels available on the chip. Recombinant JAK2 (experimental) and p38 MAP kinase (control) were used as analytes to determine the binding affinity of each of these proteins to the four peptides on the chip. Various concentrations (0–500 nM) of each analyte was applied to the channels in the chip at the flow rate and temperature as specified in our methods to generate sensorgrams, which were then used to plot the response to various concentrations of JAK2 for each of the four peptides being tested (Fig. 3G-J). We also included a series of samples that contained the JAK2 inhibitor (Fig. 3G-J). These plots were then used to determine the binding constants for JAK2 interaction with each of the four peptides. The results of these experiments showed that JAK2 binding to TET2 was most effective (highest binding affinity), when both Tyr-1939 and Tyr-1964 were phosphorylated (KD =127) (Fig. 3H). All the other permutations of phosphorylation or non-phosphorylation exhibited very weak binding (KD= 352, 265, 345) compared to the dual phosphorylated peptide (Fig. 3H). The presence of JAK2 inhibitor abolished all binding (Fig. 3G-J), similar to recombinant p38 MAP kinase, which was used as a negative control (Fig. 3G-J). These data showed that phosphorylation of TET2 by JAK2 within the catalytic domain of TET2 provide docking sites for sustained JAK2 interaction with TET2.
Transcription factor KLF1 binds phosphorylated TET2
Our previous work (5) had demonstrated that several transcription factor binding sites within the genome acquire 5-hmC during erythroid differentiation, implying TET2 is recruited to these sites by transcription factors that normally bind these sites. Our recent published work had demonstrated that the binding site motif for KLF1 was among the top transcription factor binding sites that gained 5-hmC (4, 5). Therefore, we reasoned KLF1 is a likely candidate for interactions with TET2 for potential recruitment to the specific cis regions within the genome. We used the erythroid cell line UT-7, which is known abundantly express KLF1, to ascertain whether TET2 binds KLF1. First, we transfected GFP/Flag-tagged TET2 catalytic domain to UT-7 cells and performed reciprocal immunoprecipitation coupled immunoblot analysis using cells cultured under steady state conditions (Fig. 4A, B). These experiments showed that indeed KLF1 interacts with TET2. In order to determine whether this interaction between TET2 and KLF1 is responsive to EPO, we deprived the UT-7 cells of EPO or exposed the cells to EPO after a period of EPO derivation and performed immunoprecipitation coupled immunoblot analysis, which showed increased association between TET2 and KLF1 upon exposure to EPO (Fig. 4C). We then determined whether TET2 phosphorylation at the JAK2 sites were a prerequisite for KLF1 binding to TET2 by using the Biacore instrument and the methodology we had established as described previously. These experiments showed that double phosphorylated peptides bind with the highest affinity (KD = 48.2) and non-phosphorylated peptides bind with less affinity (KD = 74.7) (Fig. 4D, E). Phosphorylation at either Tyr-1939 or Tyr-1964 site alone showed very low binding affinity (Fig. 4F, G). Once again using p38 MAP kinase as a control showed no binding (Fig. 4D-G).
Fig. 4. TET2 interacts with KLF1 transcription factor.
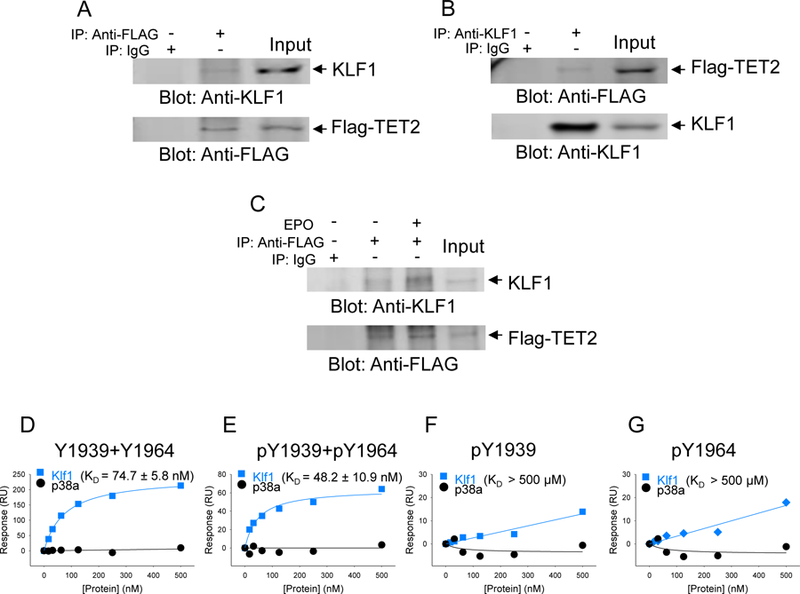
The catalytic domain of TET2 was transfected into UT-7 erythroid cell line and used to determine interaction between TET2 and erythroid specific transcription factor KLF1. (A, B) Reciprocal immunoprecipitations were performed using total cell lysates and performing either immunoprecipitation with an anti-KLF1 antibody followed by immunoblot analysis using an anti-FLAG antibody or immunoprecipitation using an anti-FLAG antibody followed by an anti-KLF1 antibody. Anti-KLF1 or anti-FLAG antibodies were used as controls. (C) KLF1 interaction with TET2 catalytic domain is enhanced in response to stimulation with erythropoietin. Anti-FLAG antibody was used as a control to confirm the presence of equivalent amounts of GFP/Flag-tagged TET2. (D, E ,F, G) Binding affinities between TET2 catalytic protein and KLF1 were determined for each for peptide carrying no phosphorylation, Y1939 +Y1964 double phosphorylation or single residue phosphorylation. p38 MAP kinase was used as a negative control to demonstrate the specificity of the interaction. The data are representative of duplicate determination.
TET2 Tyr-1939 and Tyr-1964 residues are critical in maintaining hematopoietic colony formation and terminal erythroid differentiation
In order to ascertain the functional role played by the two tyrosine residues, we transfected GFP containing plasmids carrying either wild type TET2 catalytic core or, a TET2 construct where both tyrosine residues were replaced by alanine, into an established Tet2 knockout murine cell line (C57Bl6 Tet2 −/−). The Tet2 knockout and Tet2 wild type cell lines had been created from hematopoietic cells derived from Tet2 knockout and control mice after stable transfection with an estrogen-regulated -Hoxb8 transgene, as previously described (13). We demonstrated that the Tet2 ko cells did not express TET2 and that both the KO and control cell lines expressed similar levels of CD34 and CD71 (Fig. 5A, S4A, S4B). Additionally, cells were at early stages in the hematopoietic differentiation program and did not display expression of myeloid markers (Figs S4B). We then transfected the Tet2 KO line either with wild type Tet2 catalytic core or the plasmid carrying the Tet2 catalytic core with replacement of both tyrosines with alanine. Subsequently cells were flow cytometry sorted based on GFP expression and immediately used in colony assays (Fig. S4C). The morphologies of these colonies showed both erythroid and non-erythroid colonies (Fig. 5B). The cell line with Tet2 ko did not lead to any colony formation although there were individual viable single cells. Transfection of unmutated Tet2 catalytic core in the Tet2 ko cells lead to robust colony formation (Fig 5C). The transfection of Tet2 with double tyrosine to alanine mutations led to significantly reduced numbers of total colonies, suggesting a role of these tyrosines in hematopoietic colony formation. We then demonstrated that the two tyrosines are also important in promoting terminal differentiation of erythroid cells by harvesting the cells from the methyl cellulose media and observing a reduction in Ter-119 expression in the mutant containing cells compared to cells expressing the wild type Tet2. (Fig. 5D). These data show that the Tet2 tyrosines 1939 and 1964 are essential for adequate hematopoietic colony formation as well as erythroid differentiation.
Fig. 5. Phosphorylation at tyr-1939 and tyr-1964 within the TET2 catalytic domain promote hematopoietic cell colony formation.
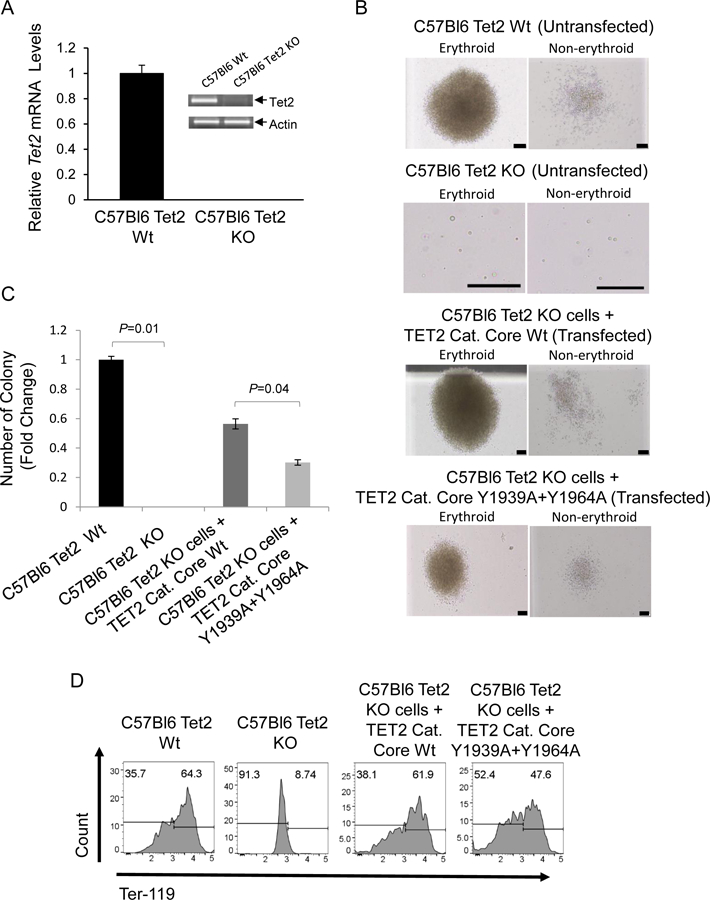
(A) qPCR and RT-PCR were performed in order to confirm absence ofTet2 transcripts in Tet2 Knock out mouse cells. Tet2 wild type cells used as a control (B) Murine C57Bl6 Tet2 KO Cells were transfected with Wild type and Mutant (Y1939A+Y1964A) GFP/Flag-tagged TET2 catalytic core containing constructs. Twenty four hrs post transfection GFP+ cells were sorted by flow cytometry and plated on mouse colony forming cell (CFC) assays. Representative erythroid and non-erythroid colonies are depicted after 19 days in culture (Scale bars, 50 μm) (C) Methyl cellulose colonies were enumerated on day 11 and depicted as fold change in the number of colonies compared to the untransfected Tet2 wild-type cells . Mean values for the number of colonies generated per 5×103 seeded cells per condition (N= 3) (D) Methyl cellulose colony assay samples were harvested on day 19 and analyzed for erythroid differentiation marker Ter-119 by flow cytometry.
JAK2V617F activating mutation leads to increased 5-hmC and genome wide loss of cytosine methylation in primary samples from patients with myeloproliferative neoplasms
Myeloproliferative neoplasms are associated with enhanced erythroid proliferation and contain activating mutations in JAK2. Since patients with the JAK2V617F mutation show the constitutively active form of JAK2, we reasoned that TET2 enzymatic activity as well as levels of 5-hmC might be elevated in these patients based on our results demonstrating TET2 is a substrate for JAK2 kinase. We examined TET activity and 5-hmC levels in erythroid progenitors derived from stem/early progenitors isolated for JAK2V617F patients and observed significantly increased active TET2 as well as increased 5-hmC levels in mutant cells (Fig. 6A, B). Having shown that JAK2V617F mutation leads to TET2 activation and increased levels of 5-hmC, we next wanted to evaluate the functional consequences of this on genome-wide cytosine methylation. Murine hematopoietic FDCP cells were transduced with JAK2V617F mutant or wild type vectors and then used for global cytosine methylation analysis with high density HELP assay. We observed that expression of the JAK2V617F mutation was associated with distinct methylation profile (Fig. 6C) that was characterized by loss of cytosine methylation when compared to cells containing wild type JAK2 (Fig. 6D). Analysis of differentially methylated loci revealed that the loss of methylation was seen to occur throughout the genome and affected all regulatory regions (Fig. S5A). Correlation with parallel transcriptomic analysis revealed a set of genes that were associated with promoter loss of methylation and increased expression in the JAK2V617F containing cells (Table S1) that included important oncogenic transcripts such as Meis1 and Hoxa9 (Fig. 6E, F).
Fig. 6. JAK2V617F leads to increased 5-hmC and genome wide loss of cytosine methylation in primary patient samples.

(A, B) Constitutively active form of JAK2 (JAK2 V617F) in patients with myeloproliferative disorder polycythemia vera exhibit elevated levels of TET activity and 5-hmC content without impacting 5-mC levels. Primary CD34+ cells purified from patient samples or healthy donors were cultured for 6 days and basophilic erythroblasts generated from these patient samples was used to determine TET2 activity and 5-hmC contents. (C) FDCP cells were transfected with JAK2V617F and wildtype JAK2 and genome wide cytosine methylation was determined by the HELP assay. Unsupervised clustering revealed that samples with JAK2V617F mutation were epigenetically distinct and were characterized by more hypomethylated loci. (D) Differentially demethylated loci were significantly more than hypermethylated loci in JAK2V617F FDCP cells. (E, F) Integration with gene expression profiling done in same samples revealed that hypomethylation affecting gene promoters was associated with increased gene expression as shown for Meis1 and Hoxa9 genes. (G) Unsupervised clustering of methylation profiles generated by the HELP assay from 4 myelofibrosis patients with JAK2V617F mutation (without TET2 mutation) and 3 healthy controls shows that the controls are distinct from the JAK2+ cases. (H) Volcano plot shows 911 signficantly hypomethylated loci in JAK2 mutant samples as opposed to 188 hypermethylated loci. (I) Unsupervised clustering of gene expression profiles from 8 myelofibrosis patients with JAK2V617F mutation and 11 healthy controls shows distinct clustering. (J) Volcano plot shows 1986 overexpressed genes in JAK2 mutant samples as opposed to 564 under-expressed genes.
Next, we determined whether JAK2V617F mutation was associated with similar methylation patterns in primary patient samples. Samples from idiopathic myelofibrosis (IMF) patients with JAK2V617F mutation (and wild type TET2) were compared with healthy controls (14) (GEO GSE42721) (Table S2, Fig. 6G). JAK2V617F samples were epigenetically distinct from controls and contained (Fig. 6G) an increased number of demethylated loci (Fig. 6H, Tables S3, S4). The loss of methylation in JAK2V617F samples was abrogated when samples contained an inactivating mutation in TET2 as seen in JAK2V617F/TET2 mutant IMF sample comparison with controls (Fig S5B). Since half of IMF cases do not have the JAK2V617F mutation, we also compared the methylomes of wildtype and mutant samples and again observed greater loss of methylation in the JAK2V617F mutant cases (Fig. S5C).Consistently, a direct comparison of JAK2V617F samples with and without TET2 mutations demonstrated that samples with wild type TET2 contained greater number of hypomethylated loci (Fig S5D). These methylomes demonstrated that JAK2V617F with a wildtype functional TET2 is associated with loss of cytosine methylation in IMF samples. Next, we analyzed the gene expression profiles of IMF cases with JAK2V617F mutations and compared them to control samples, as obtained from an independent Cohort (GEO GSE54646) (15). The JAK2 mutant IMF sample transcriptomes were distinct from controls in unsupervised clustering (Fig. 6I) and revealed a greater number of overexpressed transcripts when compared to controls (Fig. 6J, Tables S5, S6). These data demonstrate increased hydroxymethylation, loss of cytosine methylation and globally increased gene expression in primary MPN samples containing the JAK2V617F mutation.
Jak2V617F leads to 5-hmC gain in vivo that correlates with gene expression
To further determine the effect of Jak2V617F activating mutation on 5hmC in vivo, mice with transgenic expression of constitutively active form of Jak2 (Jak2V617F) (16) and wild-type controls were used to isolate erythroid cells (sorted CD71+ cells) from marrow and spleens and were used for genome wide DNA and RNA analysis. Gene expression profiling by RNA-seq revealed differential expression of transcripts with greater numbers of overexpressed transcripts as well as enhanced expression levels in Jak2V617F samples compared to wild-type samples (Fig 7A,B). We then performed 5-hMe-Seal using genomic DNA from the two cohorts of mice, and observed increased accumulation of 5-hmC peaks in Jak2V617F mice erythroid samples (Fig 7C). In order to evaluate the relationship between 5-hmC and gene expression, a co-localization plot was constructed showing the coverage of the transcripts obtained from RNA-seq samples within the regions defined by the 5-hmc peaks. A positive co-localization of expressed transcripts was seen around 5-hmC peaks and this was more pronounced in Jak2V617F samples (Fig 7D). The transcripts that were overexpressed and associated with gain of 5-hmC in the Jak2V617F mice were found to be associated with important oncogenic functional pathways (Supp Table S7) and the differential 5-hmC sites were significantly enriched in binding sites for regulatory erythroid transcription factors Gata1, Klf1 and others (Fig. S6). Overexpressed transcripts associated with gain of hydroxymethylation in Jak2V617F samples included important erythroid associated proteins Gata1, Cxcr2, Hba1, Klf1 and others (Fig 7E,7F, Fig S7A-C). Finally, we determined whether 5-hmC changes seen in murine Jak2V617F cells were similar to gene expression signatures seen in human MPNs that were obtained from a large transcriptomic database (GEO GSE54646). Differential 5-hmC regions in Jak2V617F samples were correlated with differentially expressed genes in human Jak2V617F samples and a positive correlation was seen between murine and human on GSEA analysis(Fig 7G).
Fig. 7. JAK2V617F leads to 5-hmC gain in vivo that correlates with gene expression.

(A, B) Mice with transgenic expression of constitutively active form of Jak2 (Jak2V617F) and wild-type controls were used to isolate erythroid progenitor cells (sorted CD71+) from marrow and spleen. RNA-seq was performed and bioinformatics analysis revealed a greater numbers of differential expression of transcripts as well as overexpressed transcripts in Jak2V617F samples compared to the wild-type mice. (C) 5-hMe-Seal assay was performed for whole genome 5-hmC analysis, which revealed increased accumulation of 5-hmC peaks in Jak2V617F mice erythroid samples. Differential 5-hmC gain was calculated in 2 Jak2V617F and 2 WT mice. (D) A co-localization plot combining 5-hmc and gene expression was constructed showing the coverage of the RNA-seq samples within the regions defined by the 5-hmc peaks. Positive co-localization of expressed transcripts was seen around 5hmC peaks and this was more pronounced in Jak2V617F samples. (E, F) Plots showing increased expression of Gata1 and Cxcr2 in Jak2V617F samples with increased gain of 5-hmC at their promoters in Jak2V617F samples is seen. (G) Differential 5-hmC regions in Jak2V617F samples were correlated with differentially expressed genes in human Jak2V617F samples. A positive correlation was seen between murine and human on GSEA.
Discussion
In this report, we describe a novel post-translational modification (phosphorylation) of the TET2 enzyme and that such modification results in TET2 activation leading to a global increase in 5-hmC levels in the genome. We also demonstrate that JAK2 is the kinase responsible for that phosphorylation and that association between TET2 and JAK2 is modulated by phosphorylation of the two identified phosphorylated residues. These changes are downstream of various cytokines that regulate hematopoiesis and provide a novel mechanistic link between cytokine mediated signaling and epigenetic changes that have been shown to be important in hematopoiesis.
These findings reveal a novel role of JAK2 kinase as a direct regulator of DNA methylation. JAK over-activation has been shown to globally disrupt heterochromatin to cause blood-tumor formation in drosophila (17). This observation was independent of JAK2-mediated canonical STAT activation and could not be explained by upregulation of STAT targets. Our data demonstrate that JAK2 mediated TET activation potentially regulates gene transcription via direct epigenetic changes. TET2 serine phosphorylation has recently been reported to occur by AMPK and result in stabilization of the protein (18). Furthermore, a report demonstrated that the mutant JAK2V617F can be present in the nucleus of hematopoietic cells and can directly phosphorylate Tyr 41 (Y41) on histone H3 thus inhibiting the binding of Heterochromatin protein 1α (HP1α) on the histones, thereby affecting transcription via regulation of the histone code (19). The JAK2V617F mutant kinase has also been shown to phosphorylate the arginine methyltransferase, PRMT5, and affect oncogenic transcription and cause myeloproliferation (20). Our data now demonstrate another novel kinase activity of JAK2 that influences the DNA hydroxymethylation status of cells. Recent studies have shown a strong association of increased gene expression especially when regulatory elements within the genome are hydroxymethylated (4, 21). Our previous report demonstrated that increased global cytosine hydroxymethylation occurs during blood stem/early progenitor commitment to the erythroid lineage and is associated with upregulation of important erythroid lineage specific genes (4). Our data now demonstrate this increased 5-hmC observed in that study is potentially due to EPO and/or SCF induced JAK2 activation leading to TET enzyme activation.
We also demonstrate that TET2 associates with KLF1 during EPO mediated activation. KLF1 is a master transcription factor that is important for erythroid differentiation program (22, 23). We had previously shown that hydroxymethylation during erythroid differentiation occurred at KLF1 transcription factor binding sites but could not provide a mechanistic link for this observation (4). The association of KLF1 with TET2 raises the possibility that TET2 recruitment to specific cis elements in the DNA may occur via lineage specific transcription factors. Recently published reports have shown TET2 binds transcription factors such as early B-cell factor-1 (EBF1) (24) and WT1 (25). Based on our data it is possible that TET2 is recruited to KLF1 binding sites through their interaction resulting in hydroxymethylation of regulatory elements leading to the activation of specific set of genes critical for the erythroid differentiation program (4). Furthermore this recruitment may be enhanced or dependent on cytokine stimuli such as EPO since we observed enhanced interaction between TET2 and KLF1 upon EPO stimulation. These results provide evidence in support of a mechanism that is CXXC domain-independent in contrast to what has been observed for TET1 and TET3.
We also demonstrate that primary samples of myeloproliferative disease with activating JAK2V617F mutations exhibit increased TET activity and increased hydroxymethylation. Especially in samples from primary myelofibrosis, we observed that in samples with JAK2V617F mutation but without any TET2 mutations, there was significant decrease in cytosine methylation. These epigenetic and transcriptomic changes were lost in samples that contained a TET2 mutation in addition to the JAK2V617F mutation. The decreased cytosine methylation seen in samples with JAK2V617F was associated with upregulation of many oncogenic transcripts such as Meis1 and Hoxa9 that are associated with development of myeloproliferation and myeloid malignancies (26). Furthermore, we saw similar increase in 5hmC in a murine model containing the JAK2V617F mutation. Thus, taken together, the Jak2V617F mediated direct effects on TET activation lead to epigenetic and oncogenic changes that play potential roles in the pathogenesis of myeloproliferation.
Materials and Methods
Human Primary Cell Culture and Cell Lines
Human primary hematopoietic cells were derived from CD34+ stem/early progenitors purified from growth-mobilized peripheral blood (purchased from Fred Hutchinson Cancer Center) from healthy donors. Cells were purified using the CliniMACS® device. In experiments where lineage non-committed early stage cells were used culture conditions consisted of Iscove’s modified Dulbecco’s medium (IMDM), 20% human serum, 1% penicillin/streptomycin, Thrombopoietin (20 ng/mL, TPO), FLT-3 ligand (20 ng/mL, FLT3L), recombinant human interleukin-3 (20 ng/mL, IL-3), recombinant human interleukin-6 (20 ng/mL, IL-6), stem-cell factor (100 ng/mL, SCF). Cells were cultured for 4 days prior to using them in experiments, where TET activity and phosphorylation were determined. Cells were washed with media devoid of cytokines and incubated for 4 hrs in IMDM with 1% BSA prior to stimulation with FLT3L (100 ng/ml). UT-7 cell line was obtained from ATCC and verified by microsatellite analysis.
In experiments where cells were cultured to generate erythroid progenitors CD34+ cells (day 0) were cultured until day 6 according to the methods described previously (27). Erythroid progenitors were then washed with media devoid of cytokines and incubated for 4.5 hrs in IMDM with 1% BSA prior to stimulation with either EPO or SCF. In experiments where inhibitors were used cells were exposed to JAK2-kinase inhibitor Ruxolitinib (INCB018424) at 100 nM or treated with the FLT3 inhibitor, Sorafenib, and AC2220 (10 nM) prior to stimulation with cytokines. UT-7 EPO-dependent subline cells (28) were cultured in 10% fetal bovine serum, IMDM, 1U/ml EPO. In experiments where cells where cells were deprived of EPO prior to stimulation the time period of EPO deprivation was 18 hours followed by EPO stimulation for 30 minutes.
Determination of Total TET Activity
Total TET activity was estimated by fluorescence-based ELISA kit according to the instructions provided by the manufacturer (Epigentek, NY, USA). This assay involves the conversion of 5-mC substrates coated onto the microplate wells by the TET enzymes in the sample and results in the conversion of 5-mC to 5-hmC, which is then detected fluorometrically using a specific antibody against 5-hmC. Nuclear lysates were prepared using a Nuclear Extraction kit (Novagen, CA, USA) and protein concentrations were measured using the Bradford assay. The amount of hydroxymethylated product in the samples was measured using a standard curve (0–2 ng) constructed using the regents provided in the ELISA and TET activity was calculated as ng/min/mg.
Determination of Total 5-hmC content by ELISA
The 5-hmC levels were assessed by antibody-dependent colorimetric assay using the MethylFlash Hydroxymethylated DNA Quantification Kit (Epigentek, NY, USA). DNA was extracted from primary erythroid progenitor cells (day 6 of culture) derived from in vitro culture of CD34+ cells purified from healthy donors used in the assay following the manufacture’s recommendation. A total of 200 ng of DNA was used in each assay. The 5-hmC content was expressed as percentages of total DNA.
Western Blot and Co-immunoprecipitation
Cell lysis, immunoprecipitation, and western blot analysis were performed according to the protocols from Pierce. Cell lysis and immunoprecipitation were carried out in a buffer containing 25 mM Tris•HCl pH 7.4, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 5% glycerol, and the protease inhibitor cocktail (Thermo Scientific, IL, USA). The following antibodies were used for immunoblotting and immunoprecipitations: mouse monoclonal anti-TET2 (Diagenode, NJ, USA), rabbit polyclonal anti-TET2 (Abiocode, CA, USA), rabbit polyclonal anti-JAK2 (Abcam, MA, USA), mouse monoclonal anti-EKLF 6B3 (N) (23), anti-FLAG® M2 Magnetic Beads (Sigma, MO, USA), mouse monoclonal anti-FLAG (Sigma, MO, USA), mouse monoclonal anti-GAPDH (Santacruz, TX, USA), mouse monoclonal anti-Tubulin (Santa Cruz, TX, USA) and mouse monoclonal anti-Lamin (Santa Cruz, TX, USA). The specificity of mouse monoclonal anti-TET2 antibody (Diagenode) used in imunnoprecipitation studies were confirmed by mass spectrometry by using an aliquot from the immunoprecipitated protein complex.
UT-7 cells were transduced with GFP/Flag-TET2-Catalytic cDNA plasmid and forty eight hours after transduction, cells were harvested and lysed as described earlier, and the protein concentration was determined by Bradford (Bio-Rad, IL, USA). Lysates were immunoprecipitated using the anti-FLAG magnetic beads. Following SDS-PAGE and blotting, the membranes were incubated with either an anti-JAK2 antibody or with an anti-KLF1 antibody. Infrared dye-labeled secondary antibodies and an Odyssey® Infrared Imaging System (LI-COR, USA) used to detect and quantitate the JAK2 and KLF1 bands.
Subcloning of cDNAs and generation of point mutations within the TET2 catalytic core
Recombinant wild-type cDNA construct containing the TET2 catalytic domain (aa 1129–2002) tagged with and without the GFP epitope at the N-terminus or with and without the GFP epitope tagged TET2 catalytic domain carrying tyrosine-1939, tyrosine-1964 or double mutations were generated using Q5 site-directed mutagenesis kit (NEB, MA, USA). These DNA constructs were subcloned into pCMV6-ENTRY plasmid (Origene, MD, USA). These plasmids were then transfected into HEK 293 and recombinant TET2 proteins were produced and used in in vitro kinase assays. The JAK2 and KLF1 (N-terminal 10x His- tag) were subcloned into the pET15b vector (Novagen, Darmstadt, Germany).
In vitro Kinase assay
The in vitro kinase assays contained 1.5 milligrams of protein containing GFP/Flag-tagged TET2 catalytic domain (130 KD) immunoprecipitated with an anti-FLAG M2 magnetic beads, 200 ng of active recombinant JAK2 (Abcam, MA, USA), in vitro kinase buffer (25 mM Hepes pH 7.5, 10 mM MgCl2, 0.5 mM EGTA, 0.01% Triton X-100, 2.5 mM DTT, 0.5 mM sodium orthovanadate, 5 mM glycerophosphate), and 200 μM ATP in a volume of 100 micro liters. The kinase assay was initiated by incubating the kinase assay reactants for 1 h at 30 °C followed by diluting the reaction mix with 500 microliters of phosphate buffered saline prior to collecting the immune precipitate using a magnetic selection method. Then the immunoprecipitates were resolved by SDS-PAGE on a 4–15% SDS-polyacrylamide gel, transferred onto a PVDF membrane, and then probed with an anti-phospho-tyrosine specific mouse monoclonal antibody (Millipore, CA, USA) or a phospho-serine specific mouse monoclonal antibody (Qiagen, Hilden, Germany). A similar approach was taken in in vitro kinase assays that involved TET2 catalytic domain carrying specific tyrosine mutations.
Identification of phosphorylated residues by mass spectrometry
HEK293 cells were transfected with TET2-Catalytic-Flag. Immunoprecipitated TET2 phosphorylated by active recombinant JAK2 for 1 hr. The TET2-Catalytic-Flag protein was captured by anti-FLAG magnetic beads and separated by SDS-PAGE. A band corresponding to the phosphorylated TET2 was excised from the gel and subjected to further separation on a 4–15% SDS-PAGE gel followed by staining with Coomassie blue Protein Stain. The TET2 band was identified by the size, and was excised from the gel and submitted to MS Bioworks, LLC (Ann Arbor, MI) for PTM profiling, which was done by nano-liquid chromatography (LC)-MS/MS with a Waters NanoAcquity high-pressure liquid chromatography (HPLC) system (Thermo Scientific). Peptide identifications were performed using Scaffold™ software together with the MASCOT server for searching against the Swiss-Prot human database. The Mascot was searched with a fragment ion mass tolerance of 0.02 Da and a parent ion tolerance of 10.0 ppm. Carbamidomethyl of cysteine was specified in Mascot as a fixed modification. Deamination of asparagine and glutamine, oxidation of methionine, acetylation of the N-terminus, and phosphorylation of serine, threonine, and tyrosine were specified in Mascot as variable modifications.
Measurement of 5-hydroxymethyl cytosine content in JAK2V617F patient samples by mass spectrometry and analysis
Patient and healthy human samples were collected under an approved IRB protocol and written informed consent at University of Chicago and Albert Einstein College of Medicine. In experiments where 5-hmC levels were quantified in JAK2V617F patient samples or healthy counterparts mass spectrometry approach was used. Briefly, DNA hydrolysis was performed as previously described by Gan et al.(29) with minor modifications. Two micrograms of genomic DNA was first denatured by heating at 100°C for 3 minutes. Five units of Nuclease P1 (Sigma, MO, USA), were added and the mixture incubated at 37°C for 2 h. A 1/10 volume of 2 M ammonium bicarbonate and 0.002 units of venom phosphodiesterase 1 (Sigma, MO, USA) were added to the mixture and the incubation continued for 2 h at 37°C. Next, 0.5 units of calf intestine alkaline phosphatase (Invitrogen, CA, USA) was added, and the mixture was incubated for 1 h at 37°C. Samples were then diluted to 200 microliters with water and filtered through 0.22-micron syringe filters (Millipore, MA, USA). Samples were run on an Agilent 1290 Series liquid chromatography machine in tandem with the Agilent 6460 Triple Quad Mass Spectrometer. LC separation was performed at a flow rate of 300 µL/min through ACQUITY UPLC Oligonucleotide BEH C18 Column (Waters) with a mixture of 100% methanol and 0.1% formic acid in water. The effluent from the column was directed to an electrospray ion source (Agilent Jet Stream) connected to a triple quadrupole mass spectrometer (Agilent 6460 QQQ) operating in the positive ion multiple reaction monitoring mode using previously optimized conditions, and the intensity of specific MH+→fragment ion transitions were recorded (dC (228.1/112.1); 5mC (242.1/126.1); 5hmC (258.1/142.1)). The measured percentage of 5-mC and 5-hmC in each experimental sample were calculated from the MRM peak area divided by the combined peak areas for 5-mC plus 5-hmC plus C (total cytosine pool). To correct for the different detection sensitivity of different nucleosides, observed percentages were then normalized by the standard curves, which were constructed from measuring standard samples with known 5-mC and 5-hmC percentages.
Determination of binding affinity between TET2 and JAK2 and KLF1 by Surface Plasmon Resonance (SPR) binding analysis.
The binding analyses were performed on a BiacoreT200 instrument (GE Healthcare, Uppsala, Sweden) according to the manufacturer’s instructions and previously published methods. Amino-acid sequences of the peptides immobilized on S sensor chips SA (streptavidin) (GE Healthcare, Uppsala, Sweden). Consisted of Y1939Y1964;Biotin-1934KYGPDYVPQKSHGKKVKREPAEPHETSEPTYLRFIK1969; pY1939pY1964;Biotin-1934KYGPD(pY)VPQKSHGKKVKREPAEPHETSEPT(pY)LRFIK1969, pY1939; Biotin-1939KYGPD(pY)VPQK1943, pY1964; Biotin-1958SEPT(pY)LRFIK1969) were loaded on a Series S Sensor Chip SA (streptavidin) (GE Healthcare). The peptides were initially prepared as 1 mM stocks in 100% DMSO and diluted to 25 µg/mL with HBS-EP (10 mM HEPES, pH 7.4, 150 mM NaCl, 3 mM EDTA, and 0.05% surfactant P20) for immobilization. Three analyte proteins (JAK2, KLF1, and p38 α) were first buffer exchanged with SPR binding buffer (HBS-EP + 10% glycerol). A series of increasing concentrations (0–1 μM at 2-fold dilution) of each analyte were applied to all four channels in SPR binding buffer at a 30 μL/min flow rate at 25 °C. Sensorgrams were referenced with zero concentration signals, and analyzed using the Biacore T200 evaluation software v3.0. Response units at each concentration were measured during the equilibration phase for steady-state affinity fittings, and the KD values were determined by fitting the data to the equation is shown below, where y is the response, ymax is the maximum response and x is the analyte protein concentration.
SYBR Green real-time quantitative and RT-PCR PCR assays.
Total RNA was extracted from C57Bl6 Tet2 Wt and C57Bl6 Tet2 KO cells using the RNeasy Mini kit (Qiagen, Hilden, Germany). First-strand cDNA was synthesized by the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, CA, USA). Real-time PCR (ViiA7, Applied Biosystems) was performed using Power SYBR Green PCR Kit (Applied Biosystems, UK). PCR amplifications were performed in triplicate for Tet2 along with parallel measurements of β-actin cDNA (an internal control). To confirm specific amplification of the desired PCR product, melting curves were analyzed and PCR products were separated on a 1.5% agarose gel. Primers used were as follows: Tet2_F:5’-CCAAGACCAAGAAAGCAGCTCG-3’, Tet2_R:5’-CCGAAAGCTGCGGTTGTGC-3’, Actin_F:5’-TGAACCCTAAGGCCAACCGTGAAA-3’, Acrin_R:5’-CAGGATGGCGTGAGGGAGAGCATAG-3’. cDNA was also used for RT-PCR assay.
Flow Cytometry and Colony forming assay
Tet2 knock out cell lines were created using C57BL/6J wild type tet2 mice (stock# 0006641) and the tet2 knockout (stock # 0233591) mice purchased from Jackson Labs using published methodology in Dr. Alan List’s laboratory (13). Briefly, C57B16 tet2−/− bone marrow hematopoietic stem/progenitor cells (HSPC) were stably transfected with an estrogen-regulated -Hoxb8 transgene to immortalize both wt and KO HSPC. Flow cytometry on these established cell lines were performed using fluorochrome-conjugated antibodies against Cd34-PE, Cd71-PE and Ter-119-PE to access for surface protein expression levels. C57Bl6 Tet2 KO cells were transfected with 5 μg of wt or double mutation (Y1939A+Y1964A) GFP/Flag-tagged Tet2 catalytic core plasmid in 5 × 105 cells per electroporation using Neon® Transfection System 10 μL Kit (Invitrogen, Carlsbad, CA). Following transfection cells expressing WT or phospho mutant Tet2 (substituting tyr-1939 and tyr-1964 with alanine) catalytic core were purified by FACS sorting for GFP positive cells. These purified cells were used immediately for performing colony assays. GFP-positive populations cells (GFP/Flag-Tet2-Catalytic) were plated at 5,000 cells/plate in MethoCult SF M3436 (Stem Cell Technologies, Vancouver, Canada) containing methylcellulose in Iscove’s MDM, recombinant human insulin, BSA, 2-mercaptoethanol, human transferrin, murine SCF, beta-estradiol and erythropoietin. The colony assay media was optimum for growing BFU-Es and not myeloid colonies, which were maintained in culture for 19-days, prior to analyses. Cells were harvested for evaluation of morphology and flow cytometry analysis for Ter-119 expression as a marker of terminal erythroid differentiation.
Genomewide methylation analysis on JAK2V617F and Patient samples
Patients samples with MPN were collected at either Johns Hopkins University (Baltimore, MD) or Albert Einstein School of Medicine (Bronx, NY) under IRB approved protocol and written consent. Genomic DNA from neutrophils isolated from peripheral blood via Ficoll gradient density separation and used for methylation analysis by the HELP assays (HD17). HELP microarray data have been submitted to the Gene Expression Omnibus (GEO) database for public access (GSE42721) (14). FDCP cells were stably transfected with JAK2V617F or JAK2 WT retroviral plasmids and used for genome wide methylation analysis with HELP assay (HD18, GEO accession pending).
Gene expression analysis from MPN patient samples
MPN Patient neutrophil gene expression data was obtained from GSE54646 (15). Patients with IMF with JAK2V617F were compared to controls.
5-hMe-Seal assay
Mice with transgenic expression of constitutively active form of Jak2 (Jak2V617F) (16) and wild-type controls were used to isolate erythroid lineage cells (sorted Cd71+ cells) from marrow and spleen. All animal experiments were approved by the IACUC of Northwestern university. One hundred nano grams of genomic DNA per sample was fragmented in 50 μL Tagmentation buffer at 55°C. Fragmented DNA was purified by Zymo DNA clean&concentrator Kit (Zymo Research, CA, USA). Then, the selective 5-hmC chemical labeling was performed in 25 μL glucosylation buffer (50 mM HEPES buffer pH 8.0, 25 mM MgCl2) containing above fragmented DNA, 100 μM N3-UDP-Glc, 1 μM β-GT, and incubated at 37°C 2 hours. After purified in 45 μL nuclease-free water, 1.5 μL DBCO-PEG4-Biotin (Click Chemistry Tools, 4.5 mM stored in DMSO) was added and incubated at 37°C 2 hours. The biotin-labeled DNA was pulled down by 5 µL C1 Streptavidin beads (Life Technologies, CA, USA) for 15 min at room temperature. Next, the C1 beads captured DNA fragments were subjected to 12 cycles of PCR using Nextera DNA sample preparation kit (Illumina, CA, USA). The resulting amplified product was purified by 0.9X AMPure XP beads (Beckman Coulter). Input library was made by direct PCR (4 cycles) from fragmented DNA without chemical labeling and pull-down. All these libraries were sequenced using Illumina NextSeq 500 PE42 platform. Peak calling for each sample was performed using MACS (version 1.4.2) with default parameters as performed previously (21)
RNA-Seq library preparation
Mice with transgenic expression of constitutively active form of Jak2 (Jak2V617F) (16) and wildtype controls were used to isolate erythroid lineage cells (sorted Cd71+ cells) from marrow and spleen. 50ng mRNA per sample was used for RNA-Seq library preparation (KAPA BIOSYSTEMS, KAPA RNA HyperPrep kit, KK8540). Followed the manufacturer’s protocol, RNA in 10 μL nuclease-free water was mixed with 10 μL 2X Fragment, Prime and Elute Buffer. The reaction was heated at 94°C for 8 min and quenched on an ice-water bath. The 1st strand synthesis was performed by adding 10 μL master mix (KAPA Script enzyme and buffer) and incubated at 25°C 10 min, 42°C 15 min, 70°C 15 min, 4°C hold. Then, 30 μL 2nd strand synthesis and A-tailing mixture was added to the reaction and incubated at 16°C 30 min, 62°C 10 min, 4°C hold. The next, Illumina adaptors were added and ligated at 20°C for 15 min. After two rounds of post-ligation clean-up using KAPA Pure Beads, the elution was PCR amplified and purified by 1X KAPA Pure Beads. Finally, the library was subjected to Illumina NextSeq 500 PE42 platform. Sequence reads from RNA-sequencing were aligned to genomic sequences using TopHat. Gene expression analysis was performed using Cufflinks as previously (21).
Supplementary Material
Statement of Significance:
Identification of the TET2 phosphorylation and activation by cytokine stimulated JAK2 links extra-cellular signals to chromatin remodeling during hematopoietic differentiation. This provides potential avenues to regulate TET2 function in the context of myeloproliferative disorders and myelodysplastic syndromes associated with the JAK2V617F activating mutation.
Acknowledgements
We thank Dr. Michael Ford at MS Bioworks for providing excellent technical support and helpful discussions. We also wish to thank Dr. Subhradip Karmakar for technical support with immunoprecipitation studies, Dr. Hyun Lee at the Biophysics Core Facility at University of Illinois and Dr. Michael Thirman for providing patient samples. SS is supported by NCI F99/K00 (CA223044) award. This work was supported in part by MSKCC Support Grant/Core Grant P30 CA008748 (RLL). This work was supported in part by the Evans Foundation, Harith Foundation, Suhith Wickrema Memorial Fund, and the Leinbach Family Fund.
Footnotes
Conflict of Interest Statement
No relevant direct conflicts exist for this study. AV has received research funding from GSK, Incyte, Medpacto and Eli Lilly; is a scientific advisor for Stelexis, Novartis, Acceleron and Celgene and has equity interest in Stelexis. R.L.L. is on the supervisory board of Qiagen and is a scientific advisor to Loxo, Imago, C4 Therapeutics and Isoplexis, which each include an equity interest. He receives research support from and consulted for Celgene and Roche, he has received research support from Prelude Therapeutics, and he has consulted for Incyte, Novartis, Astellas, Morphosys and Janssen. He has received honoraria from Lilly and Amgen for invited lectures and from Gilead for grant reviews.
References:
- 1.Cimmino L, Dolgalev I, Wang Y, Yoshimi A, Martin GH, Wang J, Ng V, Xia B, Witkowski MT, Mitchell-Flack M, Grillo I, Bakogianni S, Ndiaye-Lobry D, Martin MT, Guillamot M, Banh RS, Xu M, Figueroa ME, Dickins RA, Abdel-Wahab O, Park CY, Tsirigos A, Neel BG, Aifantis I. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 2017;170(6):1079–95 e20. Epub 2017/08/22. doi: 10.1016/j.cell.2017.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ko M, Huang Y, Jankowska AM, Pape UJ, Tahiliani M, Bandukwala HS, An J, Lamperti ED, Koh KP, Ganetzky R, Liu XS, Aravind L, Agarwal S, Maciejewski JP, Rao A. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature 2010;468(7325):839–43. Epub 2010/11/09. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rasmussen KD, Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev 2016;30(7):733–50. Epub 2016/04/03. doi: 10.1101/gad.276568.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Madzo J, Liu H, Rodriguez A, Vasanthakumar A, Sundaravel S, Caces DB, Looney TJ, Zhang L, Lepore JB, Macrae T, Duszynski R, Shih AH, Song CX, Yu M, Yu Y, Grossman R, Raumann B, Verma A, He C, Levine RL, Lavelle D, Lahn BT, Wickrema A, Godley LA. Hydroxymethylation at gene regulatory regions directs stem/early progenitor cell commitment during erythropoiesis. Cell reports 2014;6(1):231–44. Epub 2014/01/01. doi: 10.1016/j.celrep.2013.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mariani CJ, Vasanthakumar A, Madzo J, Yesilkanal A, Bhagat T, Yu Y, Bhattacharyya S, Wenger RH, Cohn SL, Nanduri J, Verma A, Prabhakar NR, Godley LA. TET1-mediated hydroxymethylation facilitates hypoxic gene induction in neuroblastoma. Cell reports 2014;7(5):1343–52. Epub 2014/05/20. doi: 10.1016/j.celrep.2014.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carey BW, Finley LW, Cross JR, Allis CD, Thompson CB. Intracellular alpha-ketoglutarate maintains the pluripotency of embryonic stem cells. Nature 2015;518(7539):413–6. doi: 10.1038/nature13981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Agathocleous M, Meacham CE, Burgess RJ, Piskounova E, Zhao Z, Crane GM, Cowin BL, Bruner E, Murphy MM, Chen W, Spangrude GJ, Hu Z, DeBerardinis RJ, Morrison SJ. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 2017;549(7673):476–81. Epub 2017/08/22. doi: 10.1038/nature23876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bauer C, Gobel K, Nagaraj N, Colantuoni C, Wang M, Muller U, Kremmer E, Rottach A, Leonhardt H. Phosphorylation of TET proteins is regulated via O-GlcNAcylation by the O-linked N-acetylglucosamine transferase (OGT). J Biol Chem 2015;290(8):4801–12. Epub 2015/01/09. doi: 10.1074/jbc.M114.605881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, Boggon TJ, Wlodarska I, Clark JJ, Moore S, Adelsperger J, Koo S, Lee JC, Gabriel S, Mercher T, D’Andrea A, Frohling S, Dohner K, Marynen P, Vandenberghe P, Mesa RA, Tefferi A, Griffin JD, Eck MJ, Sellers WR, Meyerson M, Golub TR, Lee SJ, Gilliland DG. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer cell 2005;7(4):387–97. Epub 2005/04/20. doi: 10.1016/j.ccr.2005.03.023. [DOI] [PubMed] [Google Scholar]
- 10.Scott LM, Tong W, Levine RL, Scott MA, Beer PA, Stratton MR, Futreal PA, Erber WN, McMullin MF, Harrison CN, Warren AJ, Gilliland DG, Lodish HF, Green AR. JAK2 exon 12 mutations in polycythemia vera and idiopathic erythrocytosis. N Engl J Med 2007;356(5):459–68. Epub 2007/02/03. doi: 10.1056/NEJMoa065202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, Garcon L, Raslova H, Berger R, Bennaceur-Griscelli A, Villeval JL, Constantinescu SN, Casadevall N, Vainchenker W. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature 2005;434(7037):1144–8. Epub 2005/03/29. doi: 10.1038/nature03546. [DOI] [PubMed] [Google Scholar]
- 12.Huang HD, Lee TY, Tzeng SW, Horng JT. KinasePhos: a web tool for identifying protein kinase-specific phosphorylation sites. Nucleic Acids Res 2005;33:W226–9. Epub 2005/06/28. doi: 10.1093/nar/gki471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang GG, Calvo KR, Pasillas MP, Sykes DB, Hacker H, Kamps MP. Quantitative production of macrophages or neutrophils ex vivo using conditional Hoxb8. Nat Methods 2006;3(4):287–93. Epub 2006/03/24. doi: 10.1038/nmeth865. [DOI] [PubMed] [Google Scholar]
- 14.Nischal S, Bhattacharyya S, Christopeit M, Yu Y, Zhou L, Bhagat TD, Sohal D, Will B, Mo Y, Suzuki M, Pardanani A, McDevitt M, Maciejewski JP, Melnick AM, Greally JM, Steidl U, Moliterno A, Verma A. Methylome profiling reveals distinct alterations in phenotypic and mutational subgroups of myeloproliferative neoplasms. Cancer Res 2013;73(3):1076–85. doi: 10.1158/0008-5472.CAN-12-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rampal R, Al-Shahrour F, Abdel-Wahab O, Patel JP, Brunel JP, Mermel CH, Bass AJ, Pretz J, Ahn J, Hricik T, Kilpivaara O, Wadleigh M, Busque L, Gilliland DG, Golub TR, Ebert BL, Levine RL. Integrated genomic analysis illustrates the central role of JAK-STAT pathway activation in myeloproliferative neoplasm pathogenesis. Blood 2014;123(22):e123–33. Epub 2014/04/18. doi: 10.1182/blood-2014-02-554634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mullally A, Lane SW, Ball B, Megerdichian C, Okabe R, Al-Shahrour F, Paktinat M, Haydu JE, Housman E, Lord AM, Wernig G, Kharas MG, Mercher T, Kutok JL, Gilliland DG, Ebert BL. Physiological Jak2V617F expression causes a lethal myeloproliferative neoplasm with differential effects on hematopoietic stem and progenitor cells. Cancer cell 2010;17(6):584–96. Epub 2010/06/15. doi: 10.1016/j.ccr.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi S, Calhoun HC, Xia F, Li J, Le L, Li WX. JAK signaling globally counteracts heterochromatic gene silencing. Nat Genet 2006;38(9):1071–6. Epub 2006/08/08. doi: 10.1038/ng1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu D, Hu D, Chen H, Shi G, Fetahu IS, Wu F, Rabidou K, Fang R, Tan L, Xu S, Liu H, Argueta C, Zhang L, Mao F, Yan G, Chen J, Dong Z, Lv R, Xu Y, Wang M, Ye Y, Zhang S, Duquette D, Geng S, Yin C, Lian CG, Murphy GF, Adler GK, Garg R, Lynch L, Yang P, Li Y, Lan F, Fan J, Shi Y, Shi YG. Glucose-regulated phosphorylation of TET2 by AMPK reveals a pathway linking diabetes to cancer. Nature 2018;559(7715):637–41. Epub 2018/07/20. doi: 10.1038/s41586-018-0350-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dawson MA, Bannister AJ, Gottgens B, Foster SD, Bartke T, Green AR, Kouzarides T. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature 2009;461(7265):819–22. Epub 2009/09/29. doi: nature08448 [pii] 10.1038/nature08448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu F, Zhao X, Perna F, Wang L, Koppikar P, Abdel-Wahab O, Harr MW, Levine RL, Xu H, Tefferi A, Deblasio A, Hatlen M, Menendez S, Nimer SD. JAK2V617F-mediated phosphorylation of PRMT5 downregulates its methyltransferase activity and promotes myeloproliferation. Cancer cell 2011;19(2):283–94. Epub 2011/02/15. doi: 10.1016/j.ccr.2010.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bhattacharyya S, Pradhan K, Campbell N, Mazdo J, Vasantkumar A, Maqbool S, Bhagat TD, Gupta S, Suzuki M, Yu Y, Greally JM, Steidl U, Bradner J, Dawlaty M, Godley L, Maitra A, Verma A. Altered hydroxymethylation is seen at regulatory regions in pancreatic cancer and regulates oncogenic pathways. Genome Res 2017;27(11):1830–42. doi: 10.1101/gr.222794.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gregory RC, Taxman DJ, Seshasayee D, Kensinger MH, Bieker JJ, Wojchowski DM. Functional interaction of GATA1 with erythroid Kruppel-like factor and Sp1 at defined erythroid promoters. Blood 1996;87(5):1793–801. [PubMed] [Google Scholar]
- 23.Zhang W, Bieker JJ. Acetylation and modulation of erythroid Kruppel-like factor (EKLF) activity by interaction with histone acetyltransferases. Proc Natl Acad Sci U S A 1998;95(17):9855–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guilhamon P, Eskandarpour M, Halai D, Wilson GA, Feber A, Teschendorff AE, Gomez V, Hergovich A, Tirabosco R, Fernanda Amary M, Baumhoer D, Jundt G, Ross MT, Flanagan AM, Beck S. Meta-analysis of IDH-mutant cancers identifies EBF1 as an interaction partner for TET2. Nat Commun 2013;4:2166 Epub 2013/07/19. doi: 10.1038/ncomms3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rampal R, Alkalin A, Madzo J, Vasanthakumar A, Pronier E, Patel J, Li Y, Ahn J, Abdel-Wahab O, Shih A, Lu C, Ward PS, Tsai JJ, Hricik T, Tosello V, Tallman JE, Zhao X, Daniels D, Dai Q, Ciminio L, Aifantis I, He C, Fuks F, Tallman MS, Ferrando A, Nimer S, Paietta E, Thompson CB, Licht JD, Mason CE, Godley LA, Melnick A, Figueroa ME, Levine RL. DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell reports 2014;9(5):1841–55. Epub 2014/12/09. doi: 10.1016/j.celrep.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mohr S, Doebele C, Comoglio F, Berg T, Beck J, Bohnenberger H, Alexe G, Corso J, Strobel P, Wachter A, Beissbarth T, Schnutgen F, Cremer A, Haetscher N, Gollner S, Rouhi A, Palmqvist L, Rieger MA, Schroeder T, Bonig H, Muller-Tidow C, Kuchenbauer F, Schutz E, Green AR, Urlaub H, Stegmaier K, Humphries RK, Serve H, Oellerich T. Hoxa9 and Meis1 Cooperatively Induce Addiction to Syk Signaling by Suppressing miR-146a in Acute Myeloid Leukemia. Cancer cell 2017;31(4):549–62 e11. Epub 2017/04/12. doi: 10.1016/j.ccell.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sundaravel S, Duggan R, Bhagat T, Ebenezer DL, Liu H, Yu Y, Bartenstein M, Unnikrishnan M, Karmakar S, Liu TC, Torregroza I, Quenon T, Anastasi J, McGraw KL, Pellagatti A, Boultwood J, Yajnik V, Artz A, Le Beau MM, Steidl U, List AF, Evans T, Verma A, Wickrema A. Reduced DOCK4 expression leads to erythroid dysplasia in myelodysplastic syndromes. Proc Natl Acad Sci U S A 2015;112(46):E6359–68. Epub 2015/11/19. doi: 10.1073/pnas.1516394112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Komatsu N, Yamamoto M, Fujita H, Miwa A, Hatake K, Endo T, Okano H, Katsube T, Fukumaki Y, Sassa S, et al. Establishment and characterization of an erythropoietin-dependent subline, UT-7/Epo, derived from human leukemia cell line, UT-7. Blood 1993;82(2):456–64. [PubMed] [Google Scholar]
- 29.Gan H, Wen L, Liao S, Lin X, Ma T, Liu J, Song CX, Wang M, He C, Han C, Tang F. Dynamics of 5-hydroxymethylcytosine during mouse spermatogenesis. Nat Commun 2013;4:1995 Epub 2013/06/14. doi: 10.1038/ncomms2995. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.