

SUMMARY
Skeletal muscle cell (myofiber) atrophy is a detrimental component of aging and cancer that primarily results from muscle protein degradation via the proteasome and ubiquitin ligases. Transcriptional upregulation of some ubiquitin ligases contributes to myofiber atrophy, but little is known about the role that most other ubiquitin ligases play in this process. To address this question, we have used RNAi screening in Drosophila to identify the function of > 320 evolutionarily conserved ubiquitin ligases in myofiber size regulation in vivo. We find that whereas RNAi for some ubiquitin ligases induces myofiber atrophy, loss of others (including the N-end rule ubiquitin ligase UBR4) promotes hypertrophy. In Drosophila and mouse myofibers, loss of UBR4 induces hypertrophy via decreased ubiquitination and degradation of a core set of target proteins, including the HAT1/RBBP4/RBBP7 histone-binding complex. Together, this study defines the repertoire of ubiquitin ligases that regulate myofiber size and the role of UBR4 in myofiber hypertrophy.
Graphical Abstract
In Brief
Hunt et al. use the fruit fly Drosophila to identify ubiquitin-related enzymes that regulate skeletal muscle cell (myofiber) size, including the ubiquitin ligase UBR4. Loss of UBR4 promotes myofiber hypertrophy in Drosophila and mice via decreased ubiquitination and degradation of a core set of target proteins.
INTRODUCTION
Skeletal muscle wasting is a debilitating condition associated with aging and with many human diseases. Several studies have demonstrated that muscle wasting worsens disease outcome and decreases patient survival whereas preserving skeletal muscle mass and function is protective (Johnston et al., 2015; Tisdale, 2010; Zhou et al., 2010b). For example, preventing muscle wasting prolongs the survival of tumor-bearing mice even without impacting tumor burden (Johnston et al., 2015; Tisdale, 2010). However, despite its importance, the mechanisms that control skeletal muscle wasting are incompletely understood and there are no therapies available.
Muscle mass is postnatally determined primarily by changes in myofiber size due to the balance between protein synthesis and degradation (Bonaldo and Sandri, 2013; Piccirillo et al., 2014). Myofiber atrophy results from protein degradation via the autophagy-lysosome and ubiquitin-proteasome (UPS) systems (Bonaldo and Sandri, 2013; Reid et al., 2014). The UPS relies on a series of enzymes for tagging target proteins for degradation. These are a single E1 ubiquitin-activating enzyme, ~40 E2 ubiquitin-conjugating enzymes, and ~620 E3 ubiquitin ligases. During atrophy, there is transcriptional upregulation of several E3 ubiquitin ligases, including atrogin-1/MAFbx (Fbxo32) and MuRF1 (Trim63), which contribute to poly-ubiquitin tagging and proteasomal degradation of target proteins (Bonaldo and Sandri, 2013; Piccirillo et al., 2014). However, there are > 620 ubiquitin ligases in mice that are each believed to have distinct substrate specificities, but their role in muscle wasting is largely unknown.
Almost two decades ago, it was found that pharmacologic inhibitors of ubiquitin ligases of the N-end rule pathway (UBRs) reduce protein ubiquitination in muscle extracts from cachectic and diabetic mice. This suggests that UBRs have a major role in protein degradation during muscle wasting (Lecker et al., 1999a, 1999b; Solomon et al., 1998). Consistently, subsequent studies demonstrated transcriptional induction of some UBRs during atrophy (Hockerman et al., 2014; Judge et al., 2014; Kwak et al., 2004; Kwon et al., 2001, 2003; Zhang et al., 2013). However, since then, studies on UBRs have been neglected because of the discovery of other ubiquitin ligases such as atrogin-1 and MuRF1. Interestingly, apart from UBR1, UBR2, and UBR3, which are highly related, most other UBRs have different domain compositions and, presumably, distinct substrate specificities (Tasaki et al., 2005). For example, UBR4 carries a unique UBR4 domain (not present in any other protein) that may confer unique substrate specificity (Rinschen et al., 2016). In yeast, UBRs recognize and ubiquitinate proteins with destabilizing N-terminal amino acids that are generated by N-terminal modifications, such as arginylation, and N-terminal cleavage by intracellular proteases, such as caspases (Sriram et al., 2011). In mammals, UBR1–7 are proposed to function similarly (Sriram et al., 2011) but their ubiquitination substrates are largely unknown because no proteome-wide analysis has been done. Therefore, which UBRs regulate myofiber size, the associated E2 conjugating enzyme, and what are their protein targets are unknown.
Here, we have used developmental muscle growth in the fruit fly Drosophila melanogaster (Demontis and Perrimon, 2009; Piccirillo et al., 2014) as an innovative system to assess the function of evolutionarily conserved ubiquitin ligases (323 genes) in myofiber size regulation in vivo. Among the regulators uncovered by this screen, we have found that RNAi for the UBR, UBR4, increases myofiber size in Drosophila. Similarly, UBR4 siRNA and muscle-specific UBR4 knockout induce myofiber hypertrophy and increase muscle mass in mice. By using proteomics and epistatic analyses, we have defined key UBR4-target proteins that are required for myofiber hypertrophy. Moreover, we have identified UBE2B as a key E2 ubiquitin-conjugating enzyme that works together with UBR4 in regulating myofiber size. Together, our study demonstrates an in vivo role of UBR4 in myofiber size determination in Drosophila and mice.
RESULTS
In Vivo High-Throughput Screening in Drosophila Identifies Evolutionarily Conserved Ubiquitin Ligases that Regulate Myofiber Size
In the Drosophila larva, skeletal muscles each comprise a single myofibers and constitute the body wall musculature located beneath the epidermis. During larval development, Drosophila body wall muscles grow ~40-fold in size via myofiber hypertrophy, thus providing a sensitive setup for identifying genetic interventions that regulate myofiber size (Demontis and Perrimon, 2009). Interestingly, skeletal muscle-specific interventions that regulate the size of body wall muscles also correspondingly change the size of the larva (Demontis and Perrimon, 2009).
Here, we have used this visual larval phenotype as a convenient, easily scorable readout to screen for ubiquitin ligases that regulate muscle growth (Figure 1). To this purpose, we used the upstream activating sequence (UAS)/Gal4 system (Brand and Perrimon, 1993), the skeletal muscle-specific Mef2-Gal4 driver (Ranganayakulu et al., 1995), and 647 UAS-RNAi transgenic fly stocks to knock down all evolutionarily conserved ubiquitin ligases and ubiquitin-related genes (323 genes) and assess their role in myofiber growth (Figure 1A). We examined 3rd instar Drosophila larvae expressing RNAi for a given ubiquitin ligase specifically in skeletal muscles and uncovered several ubiquitin ligases and related genes that regulate myofiber size (Figure 1; Table S1). Importantly, as previously demonstrated, transgene expression with Mef2-Gal4 does not affect myoblast fusion and the resulting number of syncytial nuclei (Demontis and Perrimon, 2009), indicating that the changes in myofiber size here observed are not due to changes in myoblast fusion.
Figure 1. In Vivo High-Throughput Screening in Drosophila Identifies Evolutionarily Conserved Ubiquitin Ligases that Regulate Myofiber Size.
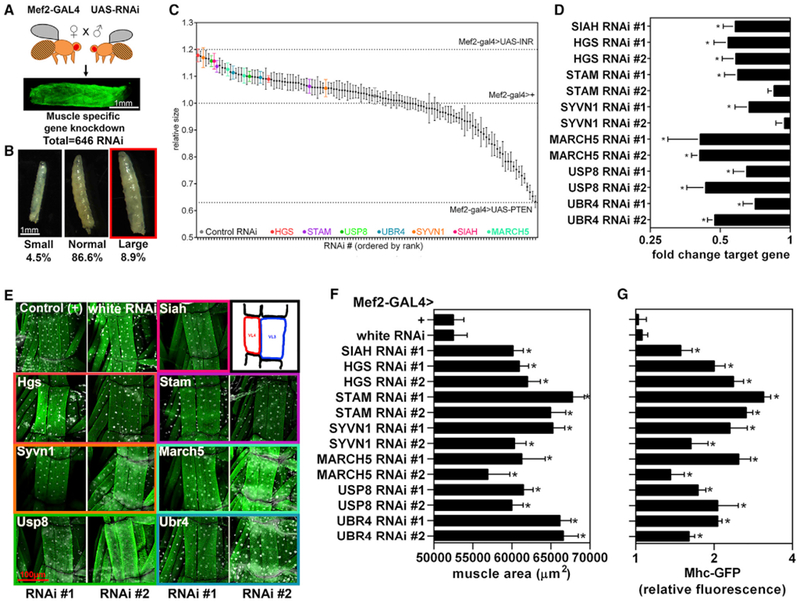
(A) Scheme of the experimental approach to screen for ubiquitin-related genes that regulate myofiber size utilizing Mef2-Gal4 to drive UAS-RNAi expression during larval muscle development. Skeletal muscle-specific interventions that regulate the size of body wall muscles also correspondingly change the size of the larva (Demontis and Perrimon, 2009). On this basis, we have used larval size to identify regulators of muscle growth.
(B) Examples of large and small larvae assessed qualitatively to identify screen hits that may regulate muscle growth.
(C) Quantitative assessment of larval size for screen hits that scored qualitatively. Each data point shows the mean of the pooled larval size at 25°C and 29°C with SEM and n (larvae for each temperature) = 10. Highlighted in color are high-confidence screen hits that consistently increased larval size with multiple UAS-RNAi lines whereas the values for a control RNAi are shown in gray. In addition, dashed lines indicate the values of negative (no overexpression) and positive controls, e.g., insulin receptor (INR) and PTEN overexpression.
(D) qPCR verification of RNAi-mediated knockdown for high-confidence RNAi screen hits that induce myofiber hypertrophy (n = 3 biological replicates per genotype containing 5 larvae).
(E) Representative images of VL3 and VL4 skeletal muscles, each composed by single myofibers, from 3rd instar larvae, which express GFP-tagged myosin heavy chain (Mhc) (green) and with nuclei stained by 4′, 6-diamidino-2-phenylindole (DAPI; white). RNAi for Drosophila homologs of Siah, Hgs, Stam, Syvn, March5, Usp8, and Ubr4 increase myofiber size compared with controls (white RNAi and wild-type).
(F) Quantitation of cumulative VL3+VL4 myofiber areas demonstrate significant increases in myofiber sizes in response to the RNAi interventions shown in (E); n = 20 VL3+VL4 muscles from ≥ 5 larvae/genotype.
(G) Quantitation of Mhc-GFP fluorescence from larval homogenates shows a corresponding increase in sarcomeric protein content; n = 4 biological replicates per genotype containing 5 larvae. In (C–G), SEM is shown with *p < 0.05 compared to a control white RNAi.
Small larval phenotypes were observed for 4.5% of RNAi interventions (Figure 1B) and included UBA1 (the sole E1 ubiquitin-activating enzyme), several E2 ubiquitin-conjugating enzymes (including UBE2D/E/L family members), and E3 ubiquitin ligases (CUL1, CUL2, SKP1, and UBR5), which therefore appear to be essential for skeletal muscle growth.
Large larval phenotypes were found for 8.9% of RNAi lines (Figure 1B), suggesting that these genes are negative regulators of muscle growth. In particular, quantitative assessments pin-pointed seven ubiquitin ligases that increase larval size when targeted in muscle by two distinct RNAi (Figure 1C), and that conversely reduce larval size when overexpressed in muscle (Figures S1A-S1C). These high-confidence negative regulators of muscle growth are sina, Hrs, Stam, sip3, CG17991/CG9855, Usp8, and poe, respectively homologous to human SIAH1/2, HGS, STAM/STAM2, SYVN1, MARCH5, USP8, and UBR4 (full results of the qualitative primary screen and quantitative secondary screen are available in Table S1).
To further test whether these growth-regulating RNAi interventions work by reducing the levels of target ubiquitin ligases, the mRNA levels were assessed by qPCR and found to be lowered in response to RNAi (Figure 1D). To examine whether these ubiquitin ligases indeed regulate myofiber size, body wall muscles of 3rd instar larvae were dissected and immunostained. Measurement of the area of ventral longitudinal VL3 and VL4 muscles (Figure 1E) revealed that, as expected, RNAi for the Drosophila homologs of SIAH, HGS, STAM, SYVN1, MARCH5, USP8, and UBR4 induce myofiber hypertrophy (Figure 1F). As a secondary measure, fluorescence from GFP-tagged myosin heavy chain (Mhc-GFP), a skeletal muscle-specific contractile protein, was assessed in larval homogenates and showed an increase similar to that of the muscle area (Figure 1G), indicating that higher myofibrillar protein content accompanies RNAi-induced myofiber hypertrophy.
Evolutionarily Conserved Roles of Ubiquitin Ligases in C2C12 Myogenesis and Myofiber Growth Regulation in Mice
To test whether candidate ubiquitin ligases have an evolutionarily conserved role in myofiber growth in higher organisms, we next assessed their function in mouse C2C12 myoblasts.
First, we assessed the involvement of candidate ubiquitin ligases in myoblast fusion, which is an important determinant of C2C12 myotube size (Hunt et al., 2015; Kim et al., 2015). For these experiments, C2C12 cells were transfected with small interfering RNAs (siRNAs), which typically yielded > 70% reduction in target mRNAs (Figures 2A and S2A). Assessment of the amount of fusion that occurred by 4 days of differentiation indicated that knockdown of HGS and MARCH5 inhibits whereas knockdown of USP8 and UBR4 significantly increases myoblast fusion (Figures 2B, 2C, and S2B).
Figure 2. Screen Hits Identified in Drosophila Regulate C2C12 Myogenesis and Myofiber Size in Mice.
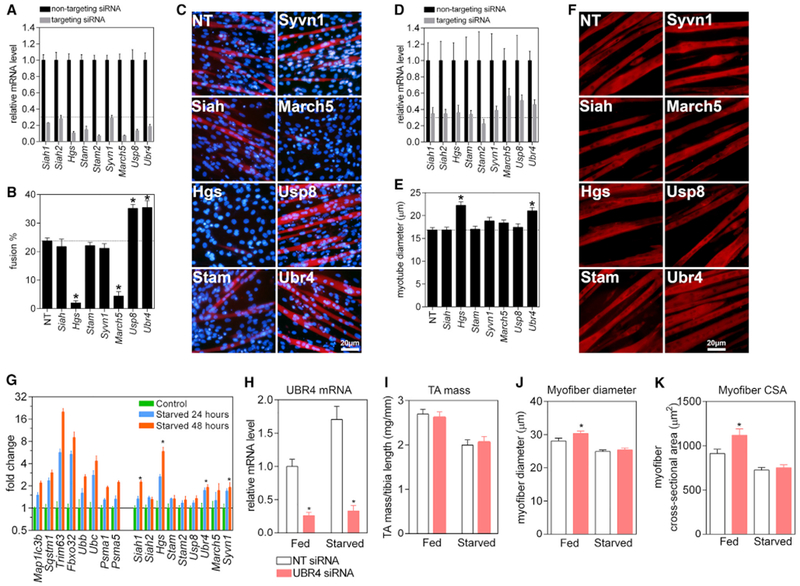
(A) qPCR demonstrating siRNA-mediated knockdown in C2C12 myoblasts for all mouse homologs of Drosophila high-confidence screen hits. RNAi reduced mRNA levels to < 30% of controls (n = 3 biological replicates obtained from separate experiments, which had three technical replicates of parallel cultures each; all p < 0.05 compared to NT siRNA).
(B) Myoblasts with gene knockdown were scored for their capacity to form syncytial myofibers by scoring the percentage of nuclei (blue) within differentiated myotubes (identified by Mhc immunofluorescence, red) after 4 days of differentiation (n = 3 with biological replicates obtained from separate experiments with ± 500 nuclei/replicate from 3 technically replicated cultures). There is significantly decreased myoblast fusion with siRNAs for HGS and MARCH5 where as siRNAs for UBR4 and USP8 siRNAs increase fusion.
(C) Representative images of myoblast fusion assays show results consistent with (B).
(D) qPCR demonstrating siRNA-mediated knockdown in C2C12 myotube-enriched cultures. RNAi reduced mRNA levels to 30%–50% of control values (n = 3 biological replicates obtained from separate experiments which had 3 technical replicates of parallel cultures each; all p < 0.05 compared to NT siRNA).
(E) Measurement of myotube width following siRNA treatment of C2C12 myotube-enriched cultures indicates that HGS and UBR4 knockdown induce significant myofiber hypertrophy (n = 3 biological replicates obtained from separate experiments with ≥ 100 myotubes/replicate).
(F) Representative images of myotubes stained for myosin heavy chain (red) show results consistent with (E).
(G) qPCR from the TA muscles of mice that were starved for 24 and 48 h. Upregulation of known atrogenes (left side) is observed as well as significant upregulation of our screen hits SIAH1, HGS, UBR4, and SYVN1 at 48 h (n = 3 TA muscles from 3 mice).
(H) qPCR for siRNA-mediated knockdown of UBR4 in electroporated TA muscles of 3-month-old C57BL6/J mice that were fed ad libitum or starved for 24 h.
(I) Muscle mass (normalized to tibia bone length) was decreased by starvation but unaffected by UBR4 knockdown.
(J and K) Type 2A and 2X myofiber diameter (J) and cross-sectional area (K) were significantly increased by UBR4 knockdown but myofiber atrophy caused by starvation was not prevented (n = 7 mice for H–K with the mean of myofibers, typically ~2000, represented per animal). The size of type 2B myofibers was not affected by UBR4 siRNAs (not shown).
In (A)–(J), SEM is shown with *p < 0.05 compared to control NT siRNAs.
In order to test the role of candidate ubiquitin ligases in myofiber atrophy and hypertrophy independently from effects on myoblast fusion, we next used myoblast-depleted cultures of myotubes (Rommel et al., 2001). Coincident with > 50% reduction in mRNA levels (Figures 2D and S2C), HGS RNAi and UBR4 RNAi significantly increased the diameter of myotubes (Figures 2E and 2F) and inhibited starvation-induced atrophy (Figure S2D). Therefore, UBR4 inhibition promotes both myogenesis and hypertrophic growth (Figures 2A-2F). Myoblasts with constitutive overexpression of UBR4C (the C-terminal part of UBR4) showed delayed myogenesis (Figure S2F). Moreover, UBR4C rescued the myotube hypertrophy induced by UBR4 siRNAs, indicating that this phenotype is specifically due to UBR4 loss (Figures S2G-S2I).
We next asked whether any of the candidate ubiquitin ligases are transcriptionally regulated by starvation, which is a prominent inducer of atrophy in mice. HGS gene expression was dramatically increased following nutrient deprivation (Figure 2G), similar to established atrogenes (Milan et al., 2015), and to a lesser extent also SIAH1, SYVN1, and UBR4 mRNAs were induced (Figure 2G), suggesting that they might contribute to muscle wasting.
Loss of UBR4 in Mouse Skeletal Muscle Induces Myofiber Hypertrophy
Given the consistent phenotypes obtained with loss of UBR4 across all experimental models (Figures 1 and 2), we next tested its function in mouse muscles in vivo. In mice, muscles are a heterogeneous mix of muscle cells (myofibers) of different types corresponding to differences in metabolism, contraction rate, and fatigability. These myofibers are classified as type I, 2A, 2X, and 2B going from the slowest to fastest rate of contraction. These different myofiber types have distinct susceptibility to atrophic and hypertrophic stimuli (Ciciliot et al., 2013) and so were analyzed separately when assessing the role of UBR4 in myofiber size regulation.
For these experiments, the tibialis anterior (TA) muscle of adult male mice was targeted with siRNA by injection and electroporation, which effectively incorporated siRNA into myofibers (Figure S2E). After 7 days, in parallel with a reduction in UBR4 mRNA levels (Figure 2H), there was a significant increase in the mean size of type 2A and 2X myofibers (Figures 2J and 2K) whereas type 2B myofibers and muscle mass were not affected (data not shown; Figure 2I). Thus, siRNA-mediated partial loss of UBR4 in mouse TA muscles induces hypertrophy of some myofiber types.
To further test these findings and overcome limitations of siRNA-mediated UBR4 removal, homozygous UBR4-floxed mice, with or without ACTA1-CREER (McCarthy et al., 2012), were generated to achieve tamoxifen-inducible, skeletal muscle-specific UBR4 knockout and control treatments.
Tamoxifen-induced recombination in adult 3-month-old male mice and chased for 3 months led to loss of UBR4 at the mRNA (Figure 3A) and protein level (Figure 3B) in muscles (UBR4 muscle-specific knockout [mKO] mice). A significant increase in body mass was observed (Figure 3C) and was not attributable to a generalized increase in growth, as estimated based on the length of a long bone, the tibia (Figure 3D). Muscle growth was induced in both the fast-twitch TA (Figure 3E) and slow-twitch soleus muscles (Figure 3F). Therefore, increased body mass is likely caused by a whole-body increase in muscle mass. There was no significant change in the number of myofibers in the TA (Figure 3I), suggesting that increased mass is caused by myofiber hypertrophy. Coincident with increased mass of the TA muscle, there was significantly higher mean size of type 2A and 2X but not 2B myofibers (Figures 3G-3K and S3A), as observed with siRNA-mediated electroporation (Figures 2J and 2K). There was also a visible increase in the total cross-sectional area of muscles (Figure 3H) but no significant changes in the proportion of myofiber types (Figure 3L). The increase in muscle mass is therefore best explained by induction of myofiber hypertrophy. Both male and female UBR4 mKO mice show muscle hypertrophy (Figure S3C), indicating no sexual dimorphism in UBR4 mKO-induced myofiber hypertrophy. Moreover, analysis of the soleus muscle, which predominantly consists of type I and 2A myofibers, indicated that there was hypertrophy of type 2A but not of type I myofibers upon UBR4 KO (Figure S3B).
Figure 3. Adult Myofiber-Specific Knockout of UBR4 Induces Hypertrophy and Protects from Cancer Cachexia-Induced Atrophy in a Myofiber Type-Dependent Manner in Mice.
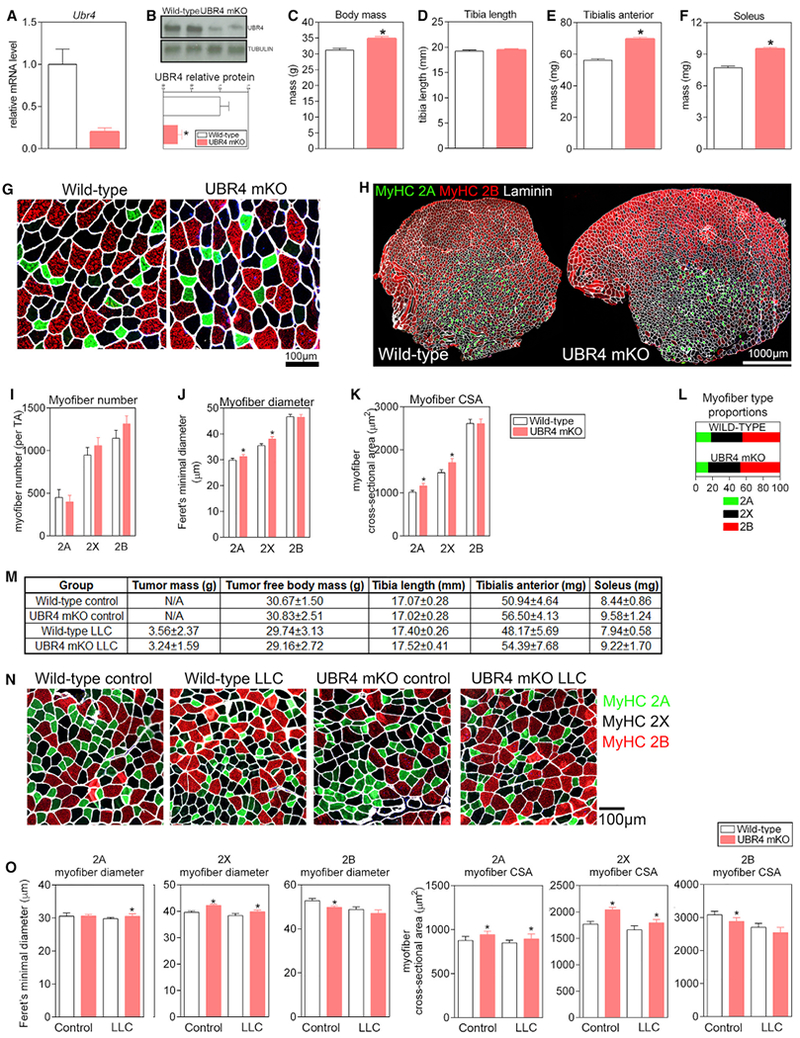
(A and B) Reduced UBR4 mRNA (A) and protein (B) levels in the TA muscles of UBR4 mKO mice (UBR4fl/fl ACTA1-CREER) compared with wild-type controls (UBR4fl/fl). Both UBR4 mKO and control mice were injected with tamoxifen to induce recombination at 3 months of age and were assessed at 6 months of age.
(C–F) UBR4 mKO mice showed a significant increase in body mass (C) with no change in tibia length (D) but significantly increased mass of TA (E) and soleus (F) muscles.
(G and H) Representative images of myosin heavy chain isotype staining at high magnification (G) and of the whole muscle cross section (H).
(I) Increased mass of the TA muscle did not coincide with a change in myofiber number per muscle.
(J and K) Increased mass of the TA muscle coincided with a significantly increased diameter (J) and cross-sectional area (K) of type 2A and 2X myofibers.; n ≥ 11 mice for all experiments in (A)–(L).
(L) Myofiber type proportions were not changed in UBR4 mKO mice.
(M) Body parameters measured for male wild-type and UBR4 mKO mice 3 weeks after inoculation with LLC cancer cells to induce cachexia. No change in tumor growth or tumor-free body mass is detected but there is a trend toward increased muscle mass in UBR4 mKO regardless of tumor burden.
(N) Representative cross sections of TA muscles from wild-type and UBR4 mKO mice, control treated or injected with LCC cancer cells.
(O) The average size of type 2A and 2X myofibers is significantly increased in UBR4 mKO LLC mice compared to wild-type LLC mice, while type 2B myofibers show a significant decrease (n ≥ 8 mice per group).
In (A)–(N), SEM is shown with *p < 0.05 compared to control.
Loss of UBR4 in Mouse Skeletal Muscle Protects from Cancer Cachexia in a Myofiber Type-Specific Manner
Cachexia associated with cancer is a common cause of muscle wasting that contributes to disease prognosis in humans (Johnston et al., 2015; Tisdale, 2010). We therefore tested whether UBR4 mKO mice are protected from muscle wasting induced by injection of Lewis lung carcinoma (LLC) cells, a common model of cancer cachexia (Puppa et al., 2014). As expected, inoculation of wild-type mice with LLC cells produced tumors that reduced total body and muscle mass (Figure 3M). Although UBR4 mKO mice had larger muscles than wild-type mice with and without tumors, the reduction in muscle mass caused by cachexia was proportionally similar to that of wild-type mice, suggesting that muscle wasting occurs overall at a similar rate. However, as observed before (Figures 3H-3K), there were differential responses of different myofiber types (Figures 3N and 3O). Specifically, in UBR4 mKO mice, type 2A myofibers appeared protected from tumor-induced atrophy whereas type 2X myofibers (although hypertrophic) showed proportionally similar atrophy as observed in wild-type controls. Conversely, type 2B myofibers had significantly reduced size in this cohort of UBR4 mKO mice, and this was further decreased by cachexia. Taken together, loss of UBR4 conveys myofiber type-specific responsiveness to cancer cachexia.
Loss of UBR4 Increases Muscle Mass but Does Not Proportionally Increase Muscle Strength
Although muscle mass is typically proportional to strength, in some instances hypertrophic muscle growth coincides with reduced muscle quality and strength (Mendias et al., 2011). To determine whether loss of UBR4 increases muscle strength proportionally to the increase in muscle mass, we examined the in situ force production of TA muscles from muscle-specific UBR4 knockout mice (UBR4 mKO), compared to wild-type controls, after 1 month of recombination. In male UBR4 mKO mice, the mass of the TA muscle was significantly increased (Figure 4A). However, there was no significant increase in twitch or tetanic force production, which indicates that normalized force production per unit of muscle mass is reduced. Similar results were obtained for females (Figures 4A and S3C). Together, this suggests that the increase in muscle mass does not produce a proportional increase in strength, presumably because of a reduction in the quality of the muscle per unit mass.
Figure 4. Loss of UBR4 Does Not Increase Strength Proportionally to Muscle Mass.
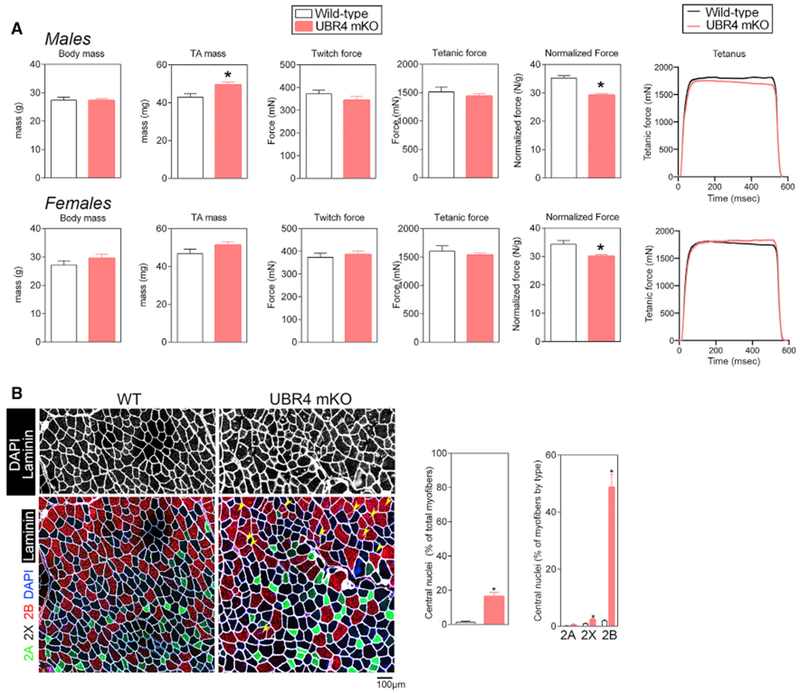
(A) Body mass, muscle weight, and force production measured in situ for the TA muscles of male (n ≥ 8 mice per group) and female (n ≥ 7 mice per group) UBR4 mKO and control wild-type mice, after 1 month of tamoxifen-induced recombination.
(B) Representative images of TA sections from UBR4 mKO mice compared to wild-type controls. Yellow arrowheads indicate centrally located nuclei. n ≥ 11 mice for each genotype.
In (A) and (B), SEM is shown with *p < 0.05 compared to control.
A possible explanation for reduced muscle quality is the increase in the number of myofibers with central nuclei in UBR4 mKO mice (Figure 4B). Central nucleation is indicative of myofiber degeneration followed by myoblast fusion and regeneration, with the newly incorporated nuclei being located centrally. Overall, there was a significant increase in the percentage of all myofibers that were centrally nucleated; however, the majority were type 2B myofibers (Figure 4B). Given that hypertrophy occurred predominantly in type 2A/X myofibers and that central nucleation occurred predominantly in type 2B myofibers, hypertrophy and central nucleation appear to be separate events. Together, this demonstrates differential responses of different myofiber types to UBR4-mediated hypertrophic stimuli.
Identification of UBR4 Protein Targets and Interactome
UBR4 is classified as an E3 ubiquitin ligase of the N-recognin family due to its conserved UBR box domain (Tasaki et al., 2005). However, the E3 ligase activity of UBR4 and its targets are not well characterized. To identify the mechanisms involved in UBR4 mKO-induced myofiber hypertrophy, we searched for UBR4 targets by utilizing multiple methods. First, we determined the transcriptome and proteome of TA muscles with siRNA-induced UBR4 knockdown and controls. For these experiments, two distinct UBR4 siRNAs were utilized and compared to a non-targeting (NT) control by RNA sequencing and tandem mass tag (TMT) mass spectrometry.
At the mRNA and protein levels, both siRNAs decreased UBR4 levels and induced similar changes (Figure 5A; Table S2). Specifically, UBR4 knockdown changed the levels of ~50 proteins (> 50% change with p < 0.05 for both siRNAs; Figure 5A, highlighted in red), which were mostly upregulated. Consistent with a role for UBR4 in protein degradation, UBR4 loss resulted in increased levels of target proteins, whereas the corresponding mRNAs were not significantly regulated. UBR4 knockdown therefore affects these proteins post-transcriptionally.
Figure 5. UBR4 Regulates a Set of Muscle Proteins that Physically Interact with and Are Ubiquitinated by UBR4.
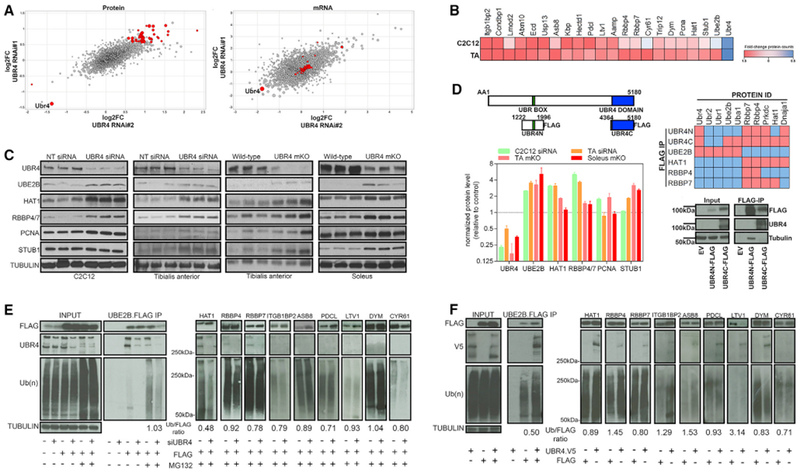
(A) TMT proteomics and RNA-sequencing from TA muscles electroporated with two different UBR4 siRNAs and examined 7 days after electroporation, compared to contralateral muscles electroporated with a non-targeting (NT) control siRNA. log2 fold changes (averaged from three biological replicates) are shown. Increasing circle size represents higher statistical significance. Red highlights putative UBR4 targets, i.e., proteins significantly regulated (> 50% change with p < 0.05) by both siRNAs in proteomics experiments. Note that only few of these are also transcriptionally regulated (n = 3 separate muscle biological replicates).
(B) Putative UBR4 target proteins found to be significantly regulated in TA muscles are overall consistently regulated in C2C12 cells transfected with UBR4 siRNAs (n = 3 separate biological replicates).
(C) Western blots and protein quantification confirm upregulation of the putative UBR4 target proteins UBE2B, HAT1, RBBP4/7, PCNA, and STUB1 in different model systems with loss of UBR4, i.e., C2C12 treated with UBR4 siRNAs, TA muscles electroporated with UBR4 siRNAs, and TA and soleus muscles from UBR4 mKO mice and controls.
(D) Diagram of FLAG-tagged UBR4 N- and C-terminal domains used for co-immunoprecipitation experiments following transfection into HEK293T cells. FLAG affinity purification of UBR4N and UBR4C retrieved associated proteins, which were identified by mass spectrometry, and are indicated in the heatmap as significantly enriched (red; p < 0.05) or not (blue). See Tables S4 for the corresponding primary data.
(E) Co-immunoprecipitation of FLAG-tagged UBR4 target proteins expressed in HEK293T cells treated with either NT control or UBR4 siRNAs and 50-μM MG132 for 4 h. Full western blots with UBE2B-FLAG are shown as an example. Both input and immunoprecipitation (IP) samples are probed with anti-UBR4 antibodies to detect pull-down of endogenous UBR4, and with anti-ubiquitin antibodies to determine whether putative UBR4 target proteins are differentially ubiquitinated in response to NT and UBR4 siRNAs. Similar to UBE2B, IP of the putative UBR4 targets HAT1, RBBP4, and RBBP7 retrieves endogenous UBR4, and UBR4 knockdown reduces the ubiquitination of these proteins, compared to control samples. Ubiquitin (Ub)/FLAG ratios indicate changes in ubiquitination relative to total FLAG protein levels with UBR4 knockdown.
(F) Immunoprecipitation of FLAG-tagged UBR4 target proteins in HEK293T cells that express either full-length human V5-tagged UBR4 or carry a control empty vector. UBE2B and HAT1 co-immunoprecipitate with UBR4-V5. Ub/FLAG ratios indicate changes in ubiquitination relative to total FLAG protein levels with UBR4-V5 expression.
Gene ontology classification indicated that proteins upregulated by UBR4 loss are primarily ubiquitin ligases and related proteins (UBE2B, STUB1, ASB8, KLHL30, TRIP12, and others), suggesting that UBR4 regulates the levels and activity of several UPS components. Nuclear components (including PCNA, PRKDC, HAT1, RBBP4, and RBBP7) were also significantly enriched among UBR4-regulated proteins, suggesting a role for UBR4 in modulating nuclear functions.
To further test the regulation of protein targets by UBR4, the proteome of C2C12 myoblasts treated with UBR4 siRNAs was examined (Table S3). This analysis revealed overlapping regulation of many of the same proteins found to be modulated in TA muscle experiments (Figure 5B). To confirm the accuracy of proteomics, we tested by western blotting the regulation of UBR4-target proteins in C2C12 myoblasts, siRNA-electroporated muscles, and the TA and soleus muscles from UBR4 mKO mice. As expected based on the proteomic analyses (Figures 5A and 5B), UBE2B, HAT1, RBBP4 and 7, PCNA, and STUB1 were upregulated also in these experimental systems coincident with loss of UBR4 (Figure 5C). However, there were also some differences in regulated protein targets when comparing siRNA-treated C2C12 with UBR4 mKO mice, possibly owing to divergence in the differentiation state of muscle cells and differences in the length of UBR4 loss depending on the model.
Although little is known on the function of UBR4, previous studies have suggested that UBR4 may act as a ubiquitin ligase via its C-terminal UBR4 domain (Lin et al., 2013; Rinschen et al., 2016; Yang et al., 2015). On this basis, proteins upregulated by UBR4 loss may therefore be targets of UBR4 ubiquitin ligase activity.
To test whether UBR4 directly interacts with these target proteins, we first examined the interactome of UBR4 by affinity purification coupled to mass spectrometry. Because of the size of UBR4 (600 kDa), affinity purification was performed with FLAG-tagged truncated variants of UBR4, corresponding to the N-terminal proximal UBR box (UBR4N), necessary for substrate recognition (Tasaki et al., 2009), and the C-terminal UBR4 domain of UBR4 (UBR4C), which has been proposed to be sufficient for ubiquitin ligation of substrates (Lin et al., 2013; Rinschen et al., 2016; Yang et al., 2015). Expression in HEK293 cells revealed that both UBR4N and UBR4C co-purify with several UBR4-regulated proteins identified from muscle proteomics studies (Figures 5A-5C; Table S4), including UBE2B, HAT1, RBBP4/7, PRKDC, and DNAJA1 (Figure 5D). UBR4C interacted with UBE2B and conversely FLAG-tagged UBE2B retrieved endogenous UBR4 as well as UBA1 (E1 activating enzyme), UBR1, and UBR2 (Figure 5D). UBR4C also pulled down HAT1 and the associated histone chaperones RBBP4 and 7 (Figure 5D). Although pulldown of FLAG-tagged HAT1, RBBP4, and RBBP7 did not retrieve endogenous UBR4, many of these nuclear proteins cross-interacted, suggesting they form a complex that may be regulated by UBR4 (Figure 5D).
To test whether UBR4 directly ubiquitinates proteins interacting with it or upregulated by its loss (Figures 5A-5D), we pulled down several target proteins and examined their ubiquitination status in the presence and absence of UBR4.
UBE2B is an E2 ubiquitin-conjugating enzyme that interacts with several E3 ubiquitin ligases of the UBR family, including UBR4 (Hong et al., 2015). Consistently, UBE2B immunoprecipitation retrieves UBR4 (Figure 5E). Moreover, endogenous UBR4 associates with HAT1, RBBP4, and RBBP7, and loss of UBR4 reduces their poly-ubiquitination and increases their protein levels.
In addition, loss of UBR4 increased ITGB1BP2, ASB8, PDCL, and LTV1 total protein levels that, accompanied by no changes in total ubiquitination, indicates a proportional decrease in the ubiquitination and degradation of these UBR4-target proteins (Figure 5E).
In addition to direct targets of UBR4, there were some proteins (e.g., DYM and CYR61) that were upregulated by the loss of UBR4 but showed no interaction with UBR4 and no change in ubiquitination. These findings indicate that some of the UBR4-upregulated proteins are indirectly regulated by UBR4 and not via ubiquitin-mediated degradation (see Figure S4 for all UBR4-regulated proteins tested by immunoprecipitation).
We also probed the interaction of UBR4 with its protein targets via converse experiments whereby V5-tagged, full-length UBR4 was overexpressed in conjunction with FLAG-tagged protein targets. Consistent with previous results (Figures 5D and 5E), full-length UBR4 interacted with UBE2B, HAT1, RBBP4, and RBBP7 (Figure 5F). Moreover, UBR4 overexpression increased their ubiquitination, further indicating that these are indeed direct ubiquitination targets of UBR4. Altogether, these studies identify proteins directly regulated by UBR4 via poly-ubiquitination and proteasomal degradation and that may contribute to myofiber hypertrophy induced by UBR4 loss.
UBR4-Target Proteins Regulate C2C12 Myogenesis and Myofiber Hypertrophy
To determine the key UBR4-target proteins responsible for UBR4 function in muscle homeostasis, we first tested their epistatic interactions with UBR4 in myoblast fusion assays. For these experiments, siRNAs for 47 UBR4-regulated proteins and NT controls were transfected in C2C12 myoblasts in conjunction with either control or UBR4 siRNAs. The myoblasts were then allowed to fuse for 3 days, at which point the relative amount of fusion (compared with NT+NT siRNA) was estimated for each combination (Figure 6A; Table S5). As in Figure 2, UBR4 knockdown induced fusion and so did UBE2B knockdown, consistent with UBE2B acting as an E2 ubiquitin-conjugating enzyme that functions with UBR4. Knockdown of several UBR4-regulated proteins also inhibited the increase in myoblast fusion due to UBR4 siRNAs. These proteins included the UBR4 direct targets HAT1, RBBP4, and RBBP7 (Figure 6B).
Figure 6. UBR4 Target Proteins Regulate C2C12 Myogenesis and Myofiber Hypertrophy Downstream of UBR4.
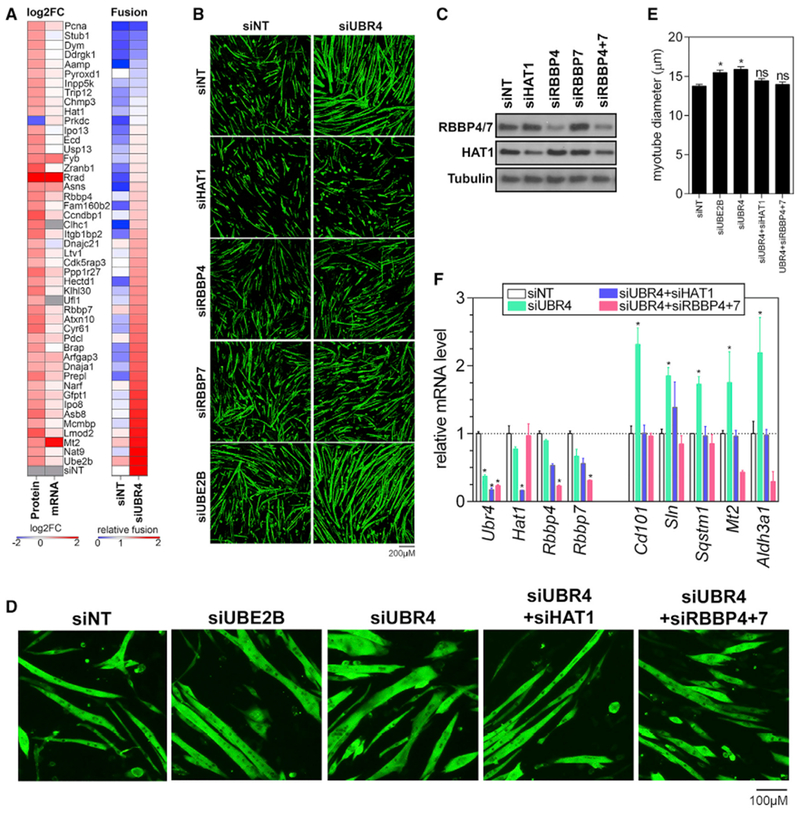
(A) Heatmap showing the protein and mRNA levels (log2 fold change) of 47 UBR4 targets most highly regulated (from Figure 4A). Epistatic interactions between UBR4 and UBR4 target proteins were probed by assessing the relative amount of myoblast fusion that occurs when a UBR4 target gene is silenced by siRNAs in C2C12 myoblasts with concomitant control (siNT) or UBR4 knockdown (siUBR4); n = 3.
(B) Representative images of epistatic interactions during myogenesis between UBR4 and UBR4 target proteins. UBE2B knockdown enhances whereas knockdown of HAT1, RBBP4, and RBBP7 inhibits myoblast fusion induced by UBR4 siRNAs.
(C) Analysis of myotube-enriched cultures to examine the effects of siRNAs on myofiber hypertrophy independently from myoblast fusion. Myotube diameter measurements indicate that UBE2B and UBR4 knockdown increases myotube size. HAT1 and RBBP4+7 knockdown prevents myotube hypertrophy induced by loss of UBR4; n = 3 biological replicates with > 50 myotubes per replicate.
(D) Representative images of myotube-enriched cultures show, consistent with (C), that HAT1 and RBBP4+7 are required for myotube hypertrophy induced by UBR4 siRNAs.
(E) Western blots for HAT1 and RBBP4/7 in C2C12 myotube-enriched cultures confirm that, as expected, siRNA-mediated knockdown reduces the levels of target proteins. Moreover, combined knockdown of RBBP4+7 reduces HAT1 protein levels.
(F) qPCR for transcripts upregulated by UBR4 knockdown (from Figure 4A) demonstrates that HAT1 and RBBP4+7 proteins are required for transcriptional modulation downstream of UBR4; n = 3 biological replicates.
In (A)–(F), SEM is shown with *p < 0.05.
In myoblast-depleted cultures of myotubes, knockdown of HAT1 and RBBP4+7 prevented myotube hypertrophy induced by UBR4 knockdown (Figures 6D and 6E), indicating that HAT1 and RBBP4+7 are key UBR4-target proteins that contribute to both myoblast fusion and to subsequent myofiber hypertrophy.
Because HAT1 and the histone chaperones RBBP4 and RBBP7 are known to form a complex (Verreault et al., 1998), we asked whether their protein levels are cross-regulated. Knockdown of HAT1 did not influence RBBP4 or RBBP7 levels, nor did individual knockdown of RBBP4 or RBBP7 (Figure 6C). However, combined knockdown of RBBP4 and RBBP7 reduced HAT1 proteins levels, indicating that the levels and possibly function of these proteins are interdependent.
Based on their role in chromatin remodeling, we next tested whether the transcriptional changes induced by UBR4 siRNAs depend on HAT1/RBBP4/RBBP7. RNA-sequencing data indicated that UBR4 knockdown increases the expression of many genes, including Mt2, Fyb, Rrad, Plekhh1, Cd101, Sln, Sqstm1, and Aldh3a1 (Figure 5A; Table S2). Interestingly, HAT1 and RBBP4+7 knockdown prevented the transcriptional induction by UBR4 siRNAs of five out of eight genes tested (Figure 6F). Therefore, HAT1 and RBBP4+7 contribute to the transcriptional changes induced by UBR4 knockdown.
To test whether HAT1 regulates muscle growth also in Drosophila, where we first identified UBR4 as a regulator of muscle growth (Figure 1), we examined larvae with muscle-specific HAT1 knockdown. As predicted, HAT1 RNAi reduced larval muscle size (Figure S5B), indicating that HAT1 is a conserved regulator of myofiber size that may account for the hypertrophy induced by UBR4 loss.
We also observed that several other proteins regulated by UBR4 (including PCNA, RRAD, LMOD2, and TRIP12, which were necessary for myoblast fusion) were also required for the growth of muscle and other tissues in Drosophila (Figures S5A and S5B), suggesting that these proteins are important growth regulators that act downstream of UBR4.
DISCUSSION
The UPS and ubiquitin ligases such as atrogin-1 and MuRF1 are important mediators of muscle wasting in response to catabolic stimuli (Bonaldo and Sandri, 2013; Piccirillo et al., 2014) but little is known about the role that the other ubiquitin ligases play in muscle wasting. To assess their function in myofiber size regulation, we have used the fruit fly Drosophila, which is amenable for in vivo transgenic RNAi screening and for identifying genetic pathways responsible for muscle homeostasis (Demontis et al., 2013; Piccirillo et al., 2014). Specifically, we have designed a convenient approach to screen for genes that regulate muscle growth, based on the observation that muscle-specific genetic interventions that modulate the mass of body wall muscles in the Drosophila larva correspondingly affect larval body size (Demontis and Perrimon, 2009), which can be easily scored for multiple animals and genotypes.
By employing muscle-specific RNAi for ubiquitin ligases and related proteins during developmental muscle growth, this screen and follow-up validation have defined the repertoire of ubiquitin ligases that regulate myofiber size (Figure 1). Specifically, this unprecedented in vivo analysis demonstrates that although the UPS typically negatively impacts muscle mass (Bonaldo and Sandri, 2013; Piccirillo et al., 2014), many ubiquitin ligases are necessary for normal muscle growth and maintenance (Figure 1).
Previously, it was known that the loss of a few ubiquitin ligases increases myofiber size (Cohen et al., 2014; Shi et al., 2011; Witt et al., 2008) but that deleting atrogin-1 and individually targeting other ubiquitin ligases that are key for atrophy does not induce hypertrophy (Baehr et al., 2014; Bodine and Baehr, 2014; Milan et al., 2015; Sartori et al., 2013). By identifying several ubiquitin ligases that when lost induce myofiber hypertrophy (Figure 1), our study supports the notion that ubiquitin ligases are copious regulators of myofiber hypertrophy. Altogether, our screen reveals unanticipated diverse roles of ubiquitin ligases in myofiber size regulation.
Drosophila is also considered an organism particularly amenable for genetic screens because of the low genetic redundancy (i.e., the lack of gene paralogs with highly related gene sequences), which increases the chance of uncovering gene function compared to mice. Consistent with this notion, we found that some ubiquitin ligases with prominent roles in myofiber size regulation in Drosophila do not score in C2C12 mouse myotubes, presumably because of genetic redundancy or insufficient knockdown of the mouse paralogs. For example, this is the case for mouse SIAH1A, SIAH1B, and SIAH2, corresponding to a single Drosophila sina gene. Moreover, the stereotypical developmental growth of Drosophila larval muscles, which entails a ~40-fold increase in myofiber size (Demontis and Perrimon, 2009; Piccirillo et al., 2014), provides a more sensitive setting for uncovering myofiber growth regulators than mouse C2C12 myotubes, which experience limited post-fusion hypertrophy (~3-fold; data not shown).
Despite these technical differences between Drosophila and mouse experimental models, we find that some of the ubiquitin ligases that regulate myofiber size in Drosophila similarly regulate the size of mouse C2C12 myotubes (Figure 2). These include HGS and UBR4, which are single mouse genes with no related paralogs.
By further testing UBR4 in experimental models of muscle wasting, we find that UBR4 loss induces myofiber hypertrophy and, although atrophy still occurs, it maintains greater muscle mass in a tumor-induced atrophy model (Figure 3). This hypertrophy is myofiber-type dependent: type 2A and 2X myofibers readily showed hypertrophy whereas larger, more glycolytic type 2B myofibers (which are rare in humans) did not undergo hypertrophy and more commonly displayed centralized nuclei, indicative of myofiber degeneration and regeneration. These findings highlight myofiber-type specific responses to UBR4 loss. More generally, hypertrophic stimuli in type 2B myofibers may be detrimental due to overt activation of growth signaling pathways in these myofibers (which have the largest size), similar to the degeneration and reduced muscle function that that occurs with AKT (Wu et al., 2017) and mTORC1 (Castets et al., 2013) activation in skeletal muscle. Differential effects of UBR4-target proteins in distinct myofiber types could also play a role in this regulation.
TMT mass spectrometry and follow-up testing have revealed a set of proteins that are upregulated by UBR4 loss and that physically interact with UBR4 and are poly-ubiquitinated in a UBR4-dependent manner (Figures 5 and 6). Interestingly, many of these proteins are components of the UPS, such as the E3 ubiquitin ligases STUB1, TRIP12, HECTD1, BRAP, ASB8, and KLHL30. This suggests that UBR4 may regulate the UPS by governing the levels of several E3 ubiquitin ligases. In line with this hypothesis, UBR4 is one of the few E3 ubiquitin ligases that physically associates with the 26S proteasome complex (Besche et al., 2009,2014). Although its function at the proteasome is unknown, this is suggestive of a prominent role of UBR4 in regulating the UPS.
Affinity purification of UBR4 retrieved the E2 ubiquitin-conjugating enzyme UBE2B, which in turn engages the E1 ubiquitin-activating enzyme UBA1, thus providing evidence for the participation of UBR4 in a ubiquitin chain editing complex. Although several E3 ligases were associated with UBE2B, only one other E3 ligase co-purified with UBR4 (with a significant Significance Analysis of Interactome [SAINT] score under proteasomal inhibition), which was the proteasome-associated HUWE1 (Figure 4D; Tables S4). Interestingly, HUWE1 acts as a cytoplasmic protein quality control ligase, which degrades unassembled proteins destined for the nucleus (Xu et al., 2016). Proteomics from HUWE1-deficient cells also showed regulation of UBE2B, RBBP4, TRIP12, and PRKDC, indicating that UBR4 and HUWE1 may share common substrates and possibly function together in targeting nuclear proteins.
We have found that UBR4 modulates the levels of many protein targets, and it is likely that several of them contribute to the induction of myofiber hypertrophy upon UBR4 loss. However, the nuclear proteins HAT1 and the associated chaperones RBBP4/7 appear key as they are required for the effect of UBR4 loss on myoblast fusion and myofiber hypertrophy.
Consistent with possible epigenetic roles of HAT1 and RBBP4/7 downstream of UBR4, there are several transcriptional changes induced by UBR4 loss that are prevented by RNAi for HAT1 and RBBP4/7 (Figure 6). Some of these gene expression changes likely contribute to myofiber hypertrophy. For example, sarcolipin (SLN) expression increases during and is necessary for overload-induced hypertrophy (Fajardo et al., 2017). Consistent with a possible role in hypertrophy downstream of UBR4/HAT1, SLN expression increases upon UBR4 loss in a HAT1-dependent manner. We conclude that UBR4 regulates muscle growth through multiple protein targets and diverse mechanisms, including the HAT1-dependent expression of genes that sustain hypertrophy (Figure 7).
Figure 7. RNAi Screening for Ub Ligases that Regulate Myofiber Size Identifies a Key Role for UBR4 in Myofiber Hypertrophy in Drosophila and Mice.
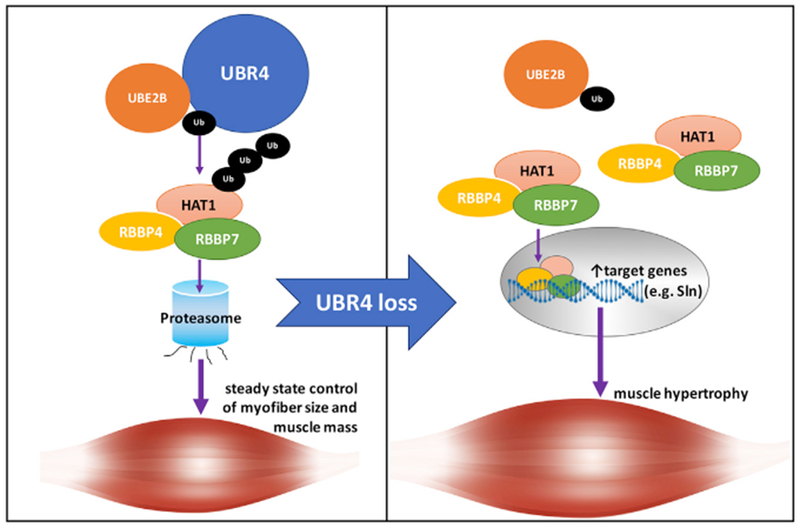
The steady-state control of myofiber size depends on the modulation of the levels of UBR4 protein targets. The E2 Ub conjugating enzyme UBE2B acts in concert with the E3 Ub ligase UBR4 to mediate ubiquitination and proteasomal degradation of UBR4 substrates. UBR4 loss induces myofiber hypertrophy through increased levels of multiple UBR4 protein targets (such as the HAT1/RBBP4/RBBP7 histone-binding complex) and diverse mechanisms, including the HAT1-dependent expression of genes that sustain hypertrophy, such as sarcolipin (Sln).
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Fabio Demontis (Fabio.Demontis@stjude.org). There are no restrictions to the availability of tools generated in this study.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Drosophila
Flies were maintained at 25°C with 60% humidity, unless otherwise specified, in tubes containing cornmeal/soy flour/yeast fly food. Fly stocks were obtained from either the Bloomington Drosophila Stock Center (BDSC) or the Vienna Drosophila Resource Center (VDRC), and are listed in provided tables and supplementary table describing the full results of larval screening for muscle growth regulators.
Cell Culture
C2C12, LLC and HEK293T cells were obtained from the ATCC and screened regularly to ensure the absence of mycoplasma. Cells were maintained at 37°C with 5% CO2 in DMEM containing 10% fetal bovine serum, Glutamax and penicillin/streptomycin. C2C12 cells were maintained as myoblasts with media containing 10% fetal bovine serum and switched to 2% horse serum-containing media to induce differentiation into myotubes when near confluence. C2C12 myoblasts were transfected with 50 μM siRNAs targeting the specified gene or with non-targeting (NT) siRNAs, using a ratio of 2 μL Lipofectamine 2000 to 50 pmol of siRNA in OptiMEM.
Myotube enriched cultures were generated by adding media containing 4 μg/mL Ara-C after 4 days of differentiation for a further 1-2 days at which point the remaining myotubes were transfected as done with myoblasts. Cells were assayed 3 days after transfection. For co-immunoprecipitation experiments, HEK293T cells were transfected with 10 μL Lipofectamine 2000, 100 pmol UBR4 siRNAs or non-targeting siRNA combined with 1 μg of pCDH-EF1-MCS-T2A-GFP plasmids encoding for FLAG-tagged UBR4 target proteins, which were cloned by PCR from mouse cDNA. Cells were assayed 3 days after transfection. When multiple paralogs of the Drosophila gene were present, siRNAs for individual mouse genes were tested alone and in combination with siRNAs.
Mice
All mice were handled following animal ethics guidelines with IACUC approval.
For experiments involving electroporation of UBR4 siRNAs or control siRNAs into tibialis anterior (TA) muscles, adult C57BL6/J male mice were utilized at 6 months of age, when post-natal skeletal muscle growth has halted.
The tibialis anterior (TA) muscles of male C57BL6/J mice were electroporated with either UBR4 or NT siRNA (Dharmacon) by first anaesthetizing mice with isoflurane, removing hair from the hind legs, and then injecting the tibialis anterior with 30 μL of 0.4U/L Hyaluronidase using a 29G1/2 insulin syringe. After recovering for two hours, the mice were anesthetized again and 500 pmol of siRNAs in 50 μL of Dharmacon’s recommended siRNA resuspension buffer (60 mM KCl, 6 mM HEPES-pH 7.5, and 0.2 mM MgCl2) were injected into the tibialis anterior muscle (one leg injected with NT siRNAs and the contralateral leg with UBR4 siRNA) followed by electroporation by using a BTX ECM 830 apparatus with metal electrodes placed parallel to the tibia orientation. Specifically, 4 pulses at 200V/cm with 20 msec length at 1Hz were delivered, followed by another 4 pulses after the orientation of the electrodes was switched to perpendicular to the tibia. After 7 days from electroporation, mice were sacrificed and TA muscles dissected and immediately snap frozen. Subsequently, the transcript levels, protein levels, and histological parameters of the tibialis anterior muscles were examined.
UBR4 floxed mice were bred together with ACTA1-Cre mice to yield homozygous UBR4fl/fl alleles either with (UBR4 mKO) or without ACTA1-Cre (wild-type controls). At 3 months of age, mice received daily intraperitoneal injections of 1 mg/kg of tamoxifen for 5 days to induce Cre-mediated recombination (UBR4 mKO mice). The wild-type siblings were used as matched controls that were injected with tamoxifen but where no recombination occurred due to the lack of the Cre recombinase.
For cancer cachexia experiments, 106 LLC cells were injected into the right flank of wild-type or UBR4 mKO mice at 6 months of age (3 months after inducing Cre-mediated recombination at 3 months of age). Tumors were monitored and allowed to grow for 3 weeks at which point the mice were euthanized and the tumors removed and weighed alongside the analysis of skeletal muscle wasting.
In addition to weighing body mass, which is influenced by muscle mass and by other factors, the length of the tibia was measured as normalization factor. Tibia length is one of the most widely-used measurements to normalize for muscle mass as bone and muscle sizes are usually proportional. Importantly, tibia length does not change in response to atrophic or hypertrophic stimuli in adult mice (Puppa et al., 2014; Shavlakadze et al., 2010; Winbanks et al., 2016), which makes it an ideal normalization parameter for these experiments.
METHOD DETAILS
Drosophila Larval Screening and Body Wall Skeletal Muscle Analysis
UAS-RNAi lines were selected by defining a list of evolutionarily conserved ubiquitin-related genes. All Drosophila genes containing the keyword “ubiquitin” were identified in Flybase and their homology to human genes probed using DIOPT (Hu et al., 2011). Only genes with weighted homology scores of 2 or more were selected. Subsequently, virgin Mef2-GAL4 females (10 flies for each cross) were mated with males (5 flies for each cross) of each RNAi line and transferred to new food tubes every 3 days to avoid overcrowding. Breeders were maintained at both 25°C and 29°C, to screen for the effect of a given RNAi at 25°C and 29°C respectively. The sizes of 3rd instar larvae were assessed qualitatively as either bigger or smaller than control larvae. As secondary validation, the RNAi lines which produced bigger or smaller larvae were selected and > 10 larvae/genotype were imaged under a stereomicroscope to quantify their body size.
Skeletal muscles from selected candidates were analyzed by dissecting larvae into filets and by exposing the ventral lateral muscles VL3 and VL4 (each consisting of a single myofiber) from abdominal segments 2-4, which were then fixed for 30 minutes with 4% EM-grade PFA in PBS without Ca2+ and Mg2+ (Demontis and Perrimon, 2009). Following washes, larval body wall muscles were stained with DAPI and imaged to detect the endogenous fluorescence of a Mhc-GFP fusion protein. A minimum of 20 VL3/4 muscles were measured by combining the area of VL3/4 muscles from > 10 larvae.
Mhc-GFP levels were measured by homogenizing 10 larvae in 200 μL of 10 mM Tris/EDTA buffer, by pelleting the homogenate, and by reading the GFP fluorescence of the supernatant in 96-well black plates with excitation at 488 nm and emission at 510 nm using a Tecan Infinite 200 Pro reader.
C2C12 Myoblast Fusion and Myotube Size Analysis
To determine the rate of myoblast fusion and the myotube size, cultures of differentiated C2C12 myotubes were fixed by adding an equal volume of 4% PFA to the medium for 10 minutes. Cells were then washed with PBS and blocked for 30 minutes in blocking buffer containing PBS with 0.1% Triton X-100, 5% normal horse serum, and 1% BSA and then incubated with the pan-reactive anti-myosin heavy chain antibody (MF20) overnight at 4°C. The cells were then stained with a fluorescent anti-mouse secondary antibody to detect myosin heavy chain and with DAPI, to visualize the myotube area and the nuclei respectively. The fusion index was determined by counting the percentage of total nuclei present within myosin heavy chain-positive myotubes. The size of myotubes was measured by taking the average width across a myotube at three separate points along it, with ~100 myotubes measured per group.
UBR4 mKO Mouse Genotyping
Genotyping for UBR4flox mice was performed from genomic DNA extracted using sodium hydroxide followed by PCR. Specifically, mouse tissue samples were incubated with 150 μL of 50 Mm NaOH in 0.2 mM EDTAfor lysis at 95°C for 1 hour. Subsequently, the samples were vortexed, neutralized by adding 12.5 μL of 1M Tris-HCl (pH 8), vortexed, and centrifuged. 5 μL of supernatant were used for PCR with the following oligos 5′-TGCTGCGATGGCTACTAATG-3′ and 5′-CAGCCATTCTAGCGCTTACC-3′. The amplicons generated were 233 bp for the wild-type allele, and 450 bp for the floxed allele, and were visualized by agarose gel electrophoresis. PCR was also used to detect the presence or absence of the Cre allele with oligos 5′-GCGGTCTGGCAGTAAAAACTATC-3′ and 5′-GTGAAACAGCATTGCTGTCACTT-3′ for a ~100 bp amplicon.
Analysis of Mouse Myofiber Type, Size, and Number
Tibialis anterior muscles were frozen in liquid nitrogen-cooled isopentane and mounted for cryosectioning at a thickness of 10 μm. Unfixed slides holding the sections were incubated with blocking buffer (PBS with 1% BSA and 2% horse serum) for 1 hour before incubation with primary antibodies against type IIA (SC-71) and IIB myosin heavy chain (BF-F3) and laminin α2 overnight at 4°C. The sections were then washed and incubated with secondary antibodies for type 2A (anti-mouse IgG1 Alexa488), type 2B (anti-mouse IgM Alexa555) and laminin (anti-rat IgG Alexa633), (Bloemberg and Quadrilatero, 2012). The whole tibialis anterior section was imaged on a Nikon C2 confocal microscope with a 10X objective and stitched to compile an overview of the muscle. The fiber types and sizes were analyzed with the Nikon Elements software using the inverse threshold of laminin α2 staining to determine myofiber boundaries. The myosin heavy chain staining was used to classify type IIB fibers (red), type IIA (green) and presumed IIX fibers (black) that were not stained for IIB or IIA. After myofibers were classified and parameters measured, the Feret’s minimal diameter was used as the measurement of myofiber size due to greater accuracy with unevenly shaped or cut objects. For the analysis of soleus muscles, the BA-F8 antibody for type I myosin heavy chain was used with a corresponding secondary antibody for the IgG2b isotype.
For the quantification of the number of myofibers, all fibers in the cross sections of entire tibialis anterior muscles were counted based on the myofiber borders identified by laminin immunostaining.
The size and number of myofibers was measured from the inverse images of laminin immunostaining (for identifying myofiber borders), excluding myofibers with diameters < 5 and >100 μm. To categorize myofiber types, the intersections of the inverse images of laminin and myosin heavy chain-specific staining was used. These analyses were performed using the Nikon Elements software and the “Object count” function.
Real-Time qPCR and RNA-Sequencing
Total RNA was extracted using Trizol and 500 μg of RNA were used for reverse transcription. SYBR Green-based qPCR was performed on the cDNA using the oligonucleotides listed in the Key Resources Table. αTub84B was used for normalization with Drosophila samples. For mouse samples, Ppia was used for normalization with C2C12 samples whereas Hprt was used for normalization with muscle samples.
KEY RESOURCES TABLE
| REAGENT OR RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-Myosin heavy chain (not isoform specific) | DSHB | MF-20 |
| Mouse IgG2b anti-Myosin heavy chain type I | DSHB | BA-F8 |
| Mouse IgG1 anti-Myosin heavy chain type IIA | DSHB | SC-71 |
| Mouse IgM anti-Myosin heavy chain type IIB | DSHB | BF-F3 |
| Rat anti-laminin α-2 (4H8-2) | Santa Cruz | Sc-59854 |
| Mouse anti-ubiquitin (P4D1) | Santa Cruz | Sc-8017 |
| Rabbit anti-V5 tag (D3H8Q) | Cell Signaling | 13202S |
| Rabbit anti-UBR4 | Abcam | Ab86738 |
| Rabbit anti-UBE2B | Proteintech | 10773-1-AP |
| Rabbit anti-HAT1 | Abcam | Ab194296 |
| Rabbit anti-RBBP4/7 (D4F8) | Cell Signaling | 9067S |
| Mouse anti-PCNA (PC10) | Cell Signaling | 2586S |
| Rabbit anti-STUB1 (C3B6) | Cell Signaling | 2080S |
| Rabbit anti-LC3 | Sigma | L7543 |
| Rabbit anti-alpha- tubulin (11H10) | Cell Signaling | 2125S |
| Mouse anti-FLAG (M2) | Sigma | F3165 |
| Rabbit anti-ATG8 | Abcam | Ab109364 |
| Rabbit anti-GFP (D5.1) | Cell Signaling | 2956S |
| Chicken anti-GFP | Aves | GFP-1020 |
| AlexaFluor555 anti-mouse | Life Technologies | A28180 |
| AlexaFluor488 anti-rabbit | Life Technologies | A11034 |
| AlexaFluor488 anti-mouse | Life Technologies | A11001 |
| AlexaFluor555 anti-rabbit | Life Technologies | A21428 |
| AlexaFluor488 anti-mouse IgG1 | Life Technologies | A21121 |
| AlexaFluor555 anti-mouse IgM | Life Technologies | A21426 |
| AlexaFluor647 anti-rat | Life Technologies | A21247 |
| AlexaFluor633 Phalloidin | Life Technologies | A22284 |
| Anti-mouse IgG, HRP-linked Antibody | Cell Signaling | 7076S |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling | 7074S |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DAPI | Roche | 10236276001 |
| Trizol | Ambion | 15596018 |
| IQ Sybr Green supermix | Bio-rad | 170-8885 |
| Black 96-well plates | Greiner | 655209 |
| Lipofectamine 2000 | Invitrogen | 11668027 |
| Opti-MEM | GIBCO | 31985062 |
| High glucose DMEM, with glutamax | GIBCO | 10566016 |
| Fetal bovine serum | GIBCO | 10437-028 |
| Penicillin streptomycin (10,000 U/mL) | GIBCO | 15140122 |
| NP40 cell lysis buffer | Invitrogen | FNN0021 |
| PBS | GIBCO | 10010023 |
| Anti-FLAG M2 affinity gel | Sigma | A2220 |
| FLAG peptide | Sigma | F3290 |
| Blue loading buffer pack | Cell Signaling | 7722 |
| Precision Plus protein standard | Bio-rad | 1610374 |
| 4-20% Mini-PROTEAN TGX Pre-cast gels | Bio-rad | 4561096 |
| Immobilon-P PVDF membrane | Millipore | IPVH00010 |
| Hyaluronidase | Sigma | 4272 |
| Ara-C | Sigma | C1768 |
| Critical Commercial Assays | ||
| iScript reverse transcriptase | Bio-Rad | 1708840 |
| Deposited Data | ||
| RNA-seq data | This paper | GEO: GSE126922 |
| Experimental Models: Cell Lines | ||
| Mouse: C2C12 cells | ATCC | CRL-1772 |
| Mouse: LLC cells | ATCC | CRL-1642 |
| Human: HEK293T cells | ATCC | CRL-3216 |
| Experimental Models: Organisms/Strains | ||
| Drosophila: Mef2-GAL4 | BDSC | 27390 |
| Drosophila: Mhc-GFP | BDSC | 50881 |
| Drosophila: Mhc(F3.580)-GAL4 | BDSC | 38464 |
| Drosophila: GMR-GAL4 | Dr. Sheng Zhang, UTHealth | N/A |
| Drosophila: UAS-UBR4 RNAi | BDSC | 32945 |
| Drosophila: UAS-UBR4 RNAi | VDRC | 108296 |
| Drosophila: UAS-UBE2B RNAi | BDSC | 42631 |
| Drosophila: UAS-UBE2B RNAi | VDRC | 23290 |
| Drosophila: UAS-HAT1 RNAi | BDSC | 34730 |
| Drosophila: UAS-PCNA RNAi | VDRC | 51252 |
| Drosophila: UAS-RRAD RNAi | VDRC | 30101 |
| Drosophila: UAS-LMOD2 RNAi | VDRC | 32602 |
| Drosophila: UAS-TRIP12 RNAi | VDRC | 48022 |
| Drosophila: UAS-TRIP12 RNAi | VDRC | 48117 |
| Drosophila: UAS-Hrs | BDSC | 42692 |
| Drosophila: UAS-sina | BDSC | 30931 |
| Drosophila: UAS-usp8/ubpy | Dr. Satoshi Goto, Rikkyo University (Japan) | N/A |
| Mouse: C57BL6/J | JAX | 000664 |
| Mouse: Ubr4tm1.2Nkt | JAX | 024844 |
| Mouse: Tg ACTA-1 Cre | JAX | 025750 |
| Oligonucleotides | ||
| ON-TARGET Plus Non-targeting siRNA pool | Dharmacon | D-001810-10 |
| ON-TARGET Plus mouse siRNAs, see Table S7 | Dharmacon | N/A |
| qRT-PCR oligonucleotides, see Table S8 | IDT | N/A |
| Recombinant DNA | ||
| pCDNA6.2 pCDNA6.2-UBR4.V5 | Tasaki et al., 2013 | N/A |
| pCDH-EF1-MCS-T2A-GFP | SBI | CD527A-1 |
| Software and Algorithms | ||
| GraphPad Prism | Graphpad | https://www.graphpad.com/ |
| Nikon elements | Nikon | https://www.microscope.healthcare.nikon.com/products/software |
| Photoshop CS6 | Adobe | https://www.adobe.com/products/photoshop.html |
| ImageJ | https://imagej.nih.gov/ij/ |
For RNA sequencing, biological replicates were prepared with the TruSeq stranded mRNA library prep kit (Illumina) and sequenced on the Illumina HiSeq 2000 platform. FASTQ sequences were mapped to the mouse mm9 genome by STRONGARM, developed for the PCGP (Downing et al., 2012). Mapped reads were counted with HTSEQ (Anders et al., 2015) and gene level FPKM values were computed. All sample data was collated into a matrix using R (3.0.1) and log start transformed (log2(FPKM +1)) in STATA/MP 11.2. Genes were statistically tested by class and false discovery rate was determined using the limma package in R.
Western Blotting
For mouse muscles, ~10-20 mg of tissue was homogenized in NP40 buffer (Life Technologies) using zirconium beads in a NextAdvance bullet blender and the protein extracts were quantified. Protein samples were prepared by addition of SDS-Blue loading buffer and DTT (dithiothreitol; to a final concentration of 0.1 M) and heating at 95°C for 5 minutes. Samples were then run on 4%-20% gradient gels with a molecular weight ladder and transferred to PVDF membranes, which were blocked with either 5% milk powder or 5% BSA (according to manufacturer’s instructions) for 1 hour and then incubated with the primary antibodies listed in the Key Resources Table overnight at 4°C. After washing, the appropriate HRP-conjugated secondary antibodies were incubated at 4°C for 2 hours, washed again, and then probed with ECL reagents to detect the protein of interest. Ponceau S staining was also used to confirm even loading of protein samples.
Protein Digestion and Peptide Isobaric Labeling by Tandem Mass Tags
The experiment was performed according to a previously published protocol (Pagala et al., 2015; Xu et al., 2009) with slight modifications. Tissue samples were extracted in lysis buffer (50 mM HEPES, pH 8.5, 8 M urea and 0.5% sodium deoxycholate). Protein concentration of the lysates was determined by Coomassie staining of a gel whereby samples were run for a short distance, using bovine serum albumin (BSA) as a standard. 125 mg of protein for each sample was digested with LysC (Wako) at an enzyme-to-substrate ratio of 1:100 (w/w) for 2h in the presence of 1 mM DTT. Following this, the samples were diluted to a final 2 M Urea concentration with 50 mM HEPES (pH 8.5), and further digested with Trypsin (Promega) at an enzyme-to-substrate ratio of 1:50 (w/w) for at least 3h. The peptides were reduced by adding 1 mM DTT for 30 min at room temperature (RT) followed by alkylation with 10 mM iodoacetamide (IAA) for 30 min in the dark at RT. The unreacted IAA was quenched with 30 mM DTT for 30 min. Finally, the digestion was terminated and acidified by adding trifluoroacetic acid (TFA) to 1%, desalted using C18 cartridges (Harvard Apparatus), and dried by speed vac. The purified peptides were resuspended in 50 mM HEPES (pH 8.5) and labeled with 10-plex Tandem Mass Tag (TMT) reagents (Thermo Scientific) following the manufacturer’s recommendations.
Two-Dimensional HPLC and Mass Spectrometry
The TMT-labeled samples were mixed equally, desalted, and fractionated on an offline HPLC (Agilent 1220) using basic pH reverse phase liquid chromatography (pH 8.0, XBridge C18 column, 4.6 mm × 25 cm, 3.5 μm particle size, Waters). The fractions were dried and resuspended in 5% formic acid and analyzed by acidic pH reverse phase LC-MS/MS analysis. The peptide samples were loaded on a nanoscale capillary reverse phase C18 column (New objective, 75 μm IDx ~25 cm, 1.9 μm C18 resin from Dr. Maisch GmbH) by a HPLC system (Thermo Ultimate 3000) and eluted by a 180-min gradient. The eluted peptides were ionized by electrospray ionization, and detected by an inline Orbitrap Fusion mass spectrometer (Thermo Scientific). The mass spectrometer is operated in data-dependent mode with a survey scan in Orbitrap (60,000 resolution, 2 × 105 AGC target and 50 ms maximal ion time) and MS/MS high resolution scans (60,000 resolution, 1 × 105 AGC target, 150 ms maximal ion time, 38 HCD normalized collision energy, 1 m/z isolation window, and 20 s dynamic exclusion).
MS Data Analysis
The MS/MS raw files were processed by a newly developed tag-based hybrid search engine, JUMP, which showed better sensitivity and specificity than commercial packages (e.g., Proteome Discoverer), (Wang et al., 2014). The data was searched against the UniProt mouse database concatenated with a reversed decoy database for evaluating false discovery rate. Searches were performed using a 25-ppm mass tolerance for precursor ions and 25-ppm mass tolerance for fragment ions, fully tryptic restriction with two maximal missed cleavages, three maximal modification sites, and the assignment of a, b, and y ions. TMT tags on Lysine residues and N-termini (+229.162932 Da) were used for static modifications and Met oxidation (+15.99492 Da) was considered as a dynamic modification. MS/MS spectra were filtered by mass accuracy and matching scores to reduce protein false discovery rate to ~1%. Proteins were quantified by summing reporter ion counts across all matched PSMs using the JUMP software suite (Pagala et al., 2015).
Co-immunoprecipitation Experiments
Cells were lysed with NP40 cell lysis buffer with protease inhibitors, sonicated for 5 s with a Branson Digital Sonifier at 30% amplitude, centrifuged, and the protein content of the supernatant measured using the Bio-Rad protein assay. 100 μg of total protein for all samples was added to a final volume of 200 μL of NP40 cell lysis buffer. A small aliquot (20 μL) was removed and prepared for blotting as input. The remaining protein sample was mixed with 50 μL of FLAG affinity gel and incubated in rotating microcentrifuge tubes at 4°C overnight. The mix was centrifuged at 2,000 g for 2 minutes, and the supernatant carefully removed, leaving the affinity resin and co-immunoprecipitated proteins that were then washed 3 times with PBS and once with NP40 cell lysis buffer. The bait and co-immunoprecipitating proteins were then eluted with 20 μL of 5 μg/mL FLAG peptide in NP40 cell lysis buffer while rocking at room temperature. SDS blue loading buffer and DTT were added to the input and eluted protein supernatants and heated at 95°C for 5 min for western blotting.
Spectral Counting for Co-immunoprecipitation Experiments
Following the co-immunoprecipitation procedure above, the samples were run for a short distance on a SDS-polyacrylamide gel. The proteins in the gel bands were reduced with DTT to break disulfide bond and the Cys residues were alkylated by iodoacetamide. The gel bands were then washed, dried down in a speed vacuum, and rehydrated with a buffer containing trypsin for overnight proteolysis. The digested samples were acidified and the peptides were extracted multiple times. The extracts were pooled, dried down, and reconstituted in a small volume. The peptide samples were loaded on a nanoscale capillary reverse phase C18 column by a HPLC system (Thermo EasynLC 1000), and eluted by a gradient (~90 min). The eluted peptides were ionized by electrospray ionization, and detected by an inline mass spectrometer (Thermo Elite). The MS spectra were collected first and the 20 most abundant ions were sequentially isolated for MS/MS analysis. This process was cycled over the entire liquid chromatography gradient.
Database searches were performed using Sequest search engine in our in-house SPIDERS software package. All matched MS/MS spectra were filtered by mass accuracy and matching scores to reduce protein false discovery rate to ~1%. Finally, all proteins identified in one gel lane were combined together. The total number of spectra, namely spectral counts (SC), matching to individual proteins may reflect their relative abundance in one sample after the protein size is normalized. The spectral counts were used for the calculation of p values to identify significantly enriched proteins (Zhou et al., 2010a). Finally, a SAINT score was derived to indicate the probability the proteins detected were more abundant for that FLAG-tagged bait compared to a control in which a lysate from non-FLAG-protein-transfected cells that underwent co-immunoprecipitation (Choi et al., 2011).
Measurements of Muscle Force Production
The in situ contractile function of the tibialis anterior muscle was measured as described previously (Liu et al., 2016). Mice were deeply anesthetized via i.p. injection of ketamine-xylazine (80 and 10 mg/kg) and monitored throughout the experiment. The distal tendon of the tibialis anterior was carefully dissected and individually tied with 4.0 braided surgical silk. The sciatic nerve was exposed and all branches were cut except for the common peroneal nerve. The foot was secured to a platform and the knee immobilized using a stainless-steel pin. The body temperature was monitored and maintained at 37°C. The suture from the tendon was individually attached to the lever arm of a 305B dual-mode servomotor transducer (Aurora Scientific, Ontario, Canada). Muscle contractions were then elicited by stimulating the distal part of the sciatic via bipolar electrodes, using supramaximal square-wave pulses of 0.2 msec (701A stimulator; Aurora Scientific). Data acquisition and control of the servomotor were conducted using a Lab-View-based DMC program (version 5.202; Aurora Scientific). Optimal muscle length (Lo) was determined by incrementally stretching the muscle until the maximum isometric twitch force was achieved. Three maximum isometric tetanic forces (Po) were acquired using a train of 150Hz supramaximal electrical pulses for 500 msec at the optimal length in the muscles and highest Po was recorded. A 2-minute resting period was allowed between each tetanic contraction. Lo was measured using digital calipers. The relative force (N/g) was normalized by dividing Po by the muscle weight.
QUANTIFICATION AND STATISTICAL ANALYSIS
All experiments were performed with biological triplicates unless otherwise indicated. The unpaired two-tailed Student’s t test was used to compare the means of two independent groups to each other. One-way ANOVA with Tukey’s post hoc test was used for multiple comparisons of more than two groups of normally distributed data. The “n” for each experiment can be found in the figure legends and represents independently generated samples for all experiments, including cell populations/wells for in vitro assays, and batches of flies and samples from individual mice for in vivo experiments. Bar graphs present the mean ± SEM. Throughout the figures, asterisks indicate the significance of the p value: *p < 0.05. A significant result was defined as p < 0.05. Statistical analyses were done with Excel and GraphPad Prism.
DATA AND CODE AVAILABILITY
The RNA sequencing data generated in this study are available at the Gene Expression Omnibus with accession number GSE126922. Other datasets generated for this study are provided in Tables S1, S2, S3, S4, and S5. All other primary data are provided in Table S6.
Supplementary Material
Highlights.
RNAi screening identifies ubiquitin-related enzymes that regulate myofiber size
The ubiquitin ligase UBR4 regulates myofiber size in Drosophila and mice
Loss of UBR4 induces hypertrophy via decreased ubiquitination of target proteins
The HAT1/RBBP4/RBBP7 histone-binding complex is a UBR4 target key for hypertrophy
ACKNOWLEDGMENTS
We thank Dr. Satoshi Goto, Dr. Sheng Zhang, and the VDRC and Bloomington stock centers for fly stocks; Drs. Yoon Jee Lee and Takafumi Tasaki for plasmids to express V5-UBR4; and the Light Microscopy facility and the Hartwell Center for Bioinformatics and Biotechnology at St. Jude Children’s Research Hospital, supported by NCI P30 CA021765. We also thank Jason Puglise and Michael Matheny of the University of Florida Wellstone Physiological Assessment Core, supported by NIH U54 AR052646, for muscle force measurements. We are grateful to Dr. Melissa Puppa and other Demontis lab members for insightful discussions and help. This work was supported by research grants to F.D. from the Glenn Foundation for Medical Research, the Ellison Medical Foundation (New Scholar in Aging award), the American Federation for Aging Research, the American Parkinson Disease Association, The Hartwell Foundation (Individual Biomedical Research award), and the National Institute on Aging of the NIH (R01AG055532). L.C.H. was supported by a Glenn/AFAR Postdoctoral Fellowship for Translational Research on Aging. The mass spectrometry analysis was performed in the St. Jude Children’s Research Hospital Proteomics Facility, partially supported by NIH Cancer Center Support grant P30CA021765 and by NIH grant R01AG047928 (J.P.). Research at St. Jude Children’s Research Hospital is supported by the ALSAC.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.06.094.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Anders S, Pyl PT, and Huber W (2015). HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baehr LM, Tunzi M, and Bodine SC (2014). Muscle hypertrophy is associated with increases in proteasome activity that is independent of MuRF1 and MAFbx expression. Front. Physiol 5, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besche HC, Haas W, Gygi SP, and Goldberg AL (2009). Isolation of mammalian 26S proteasomes and p97/VCP complexes using the ubiquitin-like domain from HHR23B reveals novel proteasome-associated proteins. Biochemistry 48, 2538–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besche HC, Sha Z, Kukushkin NV, Peth A, Hock EM, Kim W, Gygi S, Gutierrez JA, Liao H, Dick L, and Goldberg AL (2014). Autoubiquitination of the 26S proteasome on Rpn13 regulates breakdown of ubiquitin conjugates. EMBO J. 33, 1159–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloemberg D, and Quadrilatero J (2012). Rapid determination of myosin heavy chain expression in rat, mouse, and human skeletal muscle using multi-color immunofluorescence analysis. PLoS ONE 7, e35273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodine SC, and Baehr LM (2014). Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol. Endocrinol. Metab 307, E469–E484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaldo P, and Sandri M (2013). Cellular and molecular mechanisms of muscle atrophy. Dis. Model. Mech 6, 25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, and Perrimon N (1993). Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118, 401–415. [DOI] [PubMed] [Google Scholar]
- Castets P, Lin S, Rion N, Di Fulvio S, Romanino K, Guridi M, Frank S, Tintignac LA, Sinnreich M, and Rüegg MA (2013). Sustained activation of mTORC1 in skeletal muscle inhibits constitutive and starvation-induced autophagy and causes a severe, late-onset myopathy. Cell Metab. 17, 731–744. [DOI] [PubMed] [Google Scholar]
- Choi H, Larsen B, Lin ZY, Breitkreutz A, Mellacheruvu D, Fermin D, Qin ZS, Tyers M, Gingras AC, and Nesvizhskii AI (2011). SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat. Methods 8, 70–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciciliot S, Rossi AC, Dyar KA, Blaauw B, and Schiaffino S (2013). Muscle type and fiber type specificity in muscle wasting. Int. J. Biochem. Cell Biol 45, 2191–2199. [DOI] [PubMed] [Google Scholar]
- Cohen S, Lee D, Zhai B, Gygi SP, and Goldberg AL (2014). Trim32 reduces PI3K-Akt-FoxO signaling in muscle atrophy by promoting plakoglobin-PI3K dissociation. J. Cell Biol 204, 747–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demontis F, and Perrimon N (2009). Integration of Insulin receptor/Foxo signaling and dMyc activity during muscle growth regulates body size in Drosophila. Development 136, 983–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demontis F, Piccirillo R, Goldberg AL, and Perrimon N (2013). Mechanisms of skeletal muscle aging: insights from Drosophila and mammalian models. Dis. Model. Mech 6, 1339–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downing JR, Wilson RK, Zhang J, Mardis ER, Pui CH, Ding L, Ley TJ, and Evans WE (2012). The Pediatric Cancer Genome Project. Nat. Genet 44,619–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajardo VA, Rietze BA, Chambers PJ, Bellissimo C, Bombardier E, Quadrilatero J, and Tupling AR (2017). Effects of sarcolipin deletion on skeletal muscle adaptive responses to functional overload and unload. Am. J. Physiol. Cell Physiol 313, C154–C161. [DOI] [PubMed] [Google Scholar]
- Hockerman GH, Dethrow NM, Hameed S, Doran M, Jaeger C, Wang WH, and Pond AL (2014). The Ubr2 Gene is Expressed in Skeletal Muscle Atrophying as a Result of Hind Limb Suspension, but not Merg1a Expression Alone. Eur. J. Transl. Myol 24, 3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong JH, Kaustov L, Coyaud E, Srikumar T, Wan J, Arrowsmith C, and Raught B (2015). KCMF1 (potassium channel modulatory factor 1) Links RAD6 to UBR4 (ubiquitin N-recognin domain-containing E3 ligase 4) and lysosome-mediated degradation. Mol. Cell. Proteomics 14, 674–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Flockhart I, Vinayagam A, Bergwitz C, Berger B, Perrimon N, and Mohr SE (2011). An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinformatics 12, 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt LC, Xu B, Finkelstein D, Fan Y, Carroll PA, Cheng PF, Eisenman RN, and Demontis F (2015). The glucose-sensing transcription factor MLX promotes myogenesis via myokine signaling. Genes Dev. 29, 2475–2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston AJ, Murphy KT, Jenkinson L, Laine D, Emmrich K, Faou P, Weston R, Jayatilleke KM, Schloegel J, Talbo G, et al. (2015). Targeting of Fn14 Prevents Cancer-Induced Cachexia and Prolongs Survival. Cell 162, 1365–1378. [DOI] [PubMed] [Google Scholar]
- Judge SM, Wu CL, Beharry AW, Roberts BM, Ferreira LF, Kandarian SC, and Judge AR (2014). Genome-wide identification of FoxO-dependent gene networks in skeletal muscle during C26 cancer cachexia. BMC Cancer 14, 997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Jin P, Duan R, and Chen EH (2015). Mechanisms of myoblast fusion during muscle development. Curr. Opin. Genet. Dev 32, 162–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak KS, Zhou X, Solomon V, Baracos VE, Davis J, Bannon AW, Boyle WJ, Lacey DL, and Han HQ (2004). Regulation of protein catabolism by muscle-specific and cytokine-inducible ubiquitin ligase E3alpha-II during cancer cachexia. Cancer Res. 64, 8193–8198. [DOI] [PubMed] [Google Scholar]
- Kwon YT, Xia Z, Davydov IV, Lecker SH, and Varshavsky A (2001). Construction and analysis of mouse strains lacking the ubiquitin ligase UBR1 (E3alpha) of the N-end rule pathway. Mol. Cell. Biol 21, 8007–8021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YT, Xia Z, An JY, Tasaki T, Davydov IV, Seo JW, Sheng J, Xie Y, and Varshavsky A (2003). Female lethality and apoptosis of spermatocytes in mice lacking the UBR2 ubiquitin ligase of the N-end rule pathway. Mol. Cell. Biol 23, 8255–8271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecker SH, Solomon V, Mitch WE, and Goldberg AL (1999a). Muscle protein breakdown and the critical role of the ubiquitin-proteasome pathway in normal and disease states. J. Nutr 129 (1S, Suppl), 227S–237S. [DOI] [PubMed] [Google Scholar]
- Lecker SH, Solomon V, Price SR, Kwon YT, Mitch WE, and Goldberg AL (1999b). Ubiquitin conjugation by the N-end rule pathway and mRNAs for its components increase in muscles of diabetic rats. J. Clin. Invest 104, 1411–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Tao R, Gao X, Li T, Zhou X, Guan KL, Xiong Y, and Lei QY (2013). Acetylation stabilizes ATP-citrate lyase to promote lipid biosynthesis and tumor growth. Mol. Cell 51, 506–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Hammers DW, Barton ER, and Sweeney HL (2016). Activin Receptor Type IIB Inhibition Improves Muscle Phenotype and Function in a Mouse Model of Spinal Muscular Atrophy. PLoS ONE 11, e0166803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy JJ, Srikuea R, Kirby TJ, Peterson CA, and Esser KA (2012). Inducible Cre transgenic mouse strain for skeletal muscle-specific gene targeting. Skelet. Muscle 2, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendias CL, Kayupov E, Bradley JR, Brooks SV, and Claflin DR (2011). Decreased specific force and power production of muscle fibers from myostatin-deficient mice are associated with a suppression of protein degradation. J. Appl. Physiol 111, 185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milan G, Romanello V, Pescatore F, Armani A, Paik JH, Frasson L, Seydel A, Zhao J, Abraham R, Goldberg AL, et al. (2015). Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun 6, 6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagala VR, High AA, Wang X, Tan H, Kodali K, Mishra A, Kavdia K, Xu Y, Wu Z, and Peng J (2015). Quantitative protein analysis by mass spectrometry. Methods Mol. Biol 1278, 281–305. [DOI] [PubMed] [Google Scholar]
- Piccirillo R, Demontis F, Perrimon N, and Goldberg AL (2014). Mechanisms of muscle growth and atrophy in mammals and Drosophila. Dev. Dyn 243, 201–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puppa MJ, Gao S, Narsale AA, and Carson JA (2014). Skeletal muscle glycoprotein 130’s role in Lewis lung carcinoma-induced cachexia. FASEB J. 28, 998–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganayakulu G, Zhao B, Dokidis A, Molkentin JD, Olson EN, and Schulz RA (1995). A series of mutations in the D-MEF2 transcription factor reveal multiple functions in larval and adult myogenesis in Drosophila. Dev. Biol 171, 169–181. [DOI] [PubMed] [Google Scholar]
- Reid MB, Judge AR, and Bodine SC (2014). CrossTalk opposing view: The dominant mechanism causing disuse muscle atrophy is proteolysis. J. Physiol 592, 5345–5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinschen MM, Bharill P, Wu X, Kohli P, Reinert MJ, Kretz O, Saez I, Schermer B, Höhne M, Bartram MP, et al. (2016). The ubiquitin ligase Ubr4 controls stability of podocin/MEC-2 supercomplexes. Hum. Mol. Genet 25, 1328–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rommel C, Bodine SC, Clarke BA, Rossman R, Nunez L, Stitt TN, Yancopoulos GD, and Glass DJ (2001). Mediation of IGF-1-induced skeletal myotube hypertrophy by PI(3)K/Akt/mTOR and PI(3)K/Akt/GSK3 pathways. Nat. Cell Biol 3, 1009–1013. [DOI] [PubMed] [Google Scholar]
- Sartori R, Schirwis E, Blaauw B, Bortolanza S, Zhao J, Enzo E, Stantzou A, Mouisel E, Toniolo L, Ferry A, et al. (2013). BMP signaling controls muscle mass. Nat. Genet 45, 1309–1318. [DOI] [PubMed] [Google Scholar]
- Shavlakadze T, Chai J, Maley K, Cozens G, Grounds G, Winn N, Rosenthal N, and Grounds MD (2010). A growth stimulus is needed for IGF-1 to induce skeletal muscle hypertrophy in vivo. J. Cell Sci 123 (Pt 6), 960–971. [DOI] [PubMed] [Google Scholar]
- Shi J, Luo L, Eash J, Ibebunjo C, and Glass DJ (2011). The SCF-Fbxo40 complex induces IRS1 ubiquitination in skeletal muscle, limiting IGF1 signaling. Dev. Cell 21, 835–847. [DOI] [PubMed] [Google Scholar]
- Solomon V, Lecker SH, and Goldberg AL (1998). The N-end rule pathway catalyzes a major fraction of the protein degradation in skeletal muscle. J. Biol. Chem 273, 25216–25222. [DOI] [PubMed] [Google Scholar]
- Sriram SM, Kim BY, and Kwon YT (2011). The N-end rule pathway: emerging functions and molecular principles of substrate recognition. Nat. Rev. Mol. Cell Biol 12, 735–747. [DOI] [PubMed] [Google Scholar]
- Tasaki T, Mulder LC, Iwamatsu A, Lee MJ, Davydov IV, Varshavsky A, Muesing M, and Kwon YT (2005). A family of mammalian E3 ubiquitin ligases that contain the UBR box motif and recognize N-degrons. Mol. Cell. Biol 25,7120–7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasaki T, Zakrzewska A, Dudgeon DD, Jiang Y, Lazo JS, and Kwon YT (2009). The substrate recognition domains of the N-end rule pathway. J. Biol. Chem 284, 1884–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasaki T, Kim ST, Zakrzewska A, Lee BE, Kang MJ, Yoo YD, Cha-Molstad HJ, Hwang J, Soung NK, Sung KS, et al. (2013). UBR box N-recognin-4 (UBR4), an N-recognin of the N-end rule pathway, and its role in yolk sac vascular development and autophagy. Proc. Natl. Acad. Sci. USA 110, 3800–3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tisdale MJ (2010). Reversing cachexia. Cell 142, 511–512. [DOI] [PubMed] [Google Scholar]
- Verreault A, Kaufman PD, Kobayashi R, and Stillman B (1998). Nucleosomal DNA regulates the core-histone-binding subunit of the human Hat1 acetyltransferase. Curr. Biol 8, 96–108. [DOI] [PubMed] [Google Scholar]
- Wang X, Li Y, Wu Z, Wang H, Tan H, and Peng J (2014). JUMP: a tagbased database search tool for peptide identification with high sensitivity and accuracy. Mol. Cell. Proteomics 13, 3663–3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winbanks CE, Murphy KT, Bernardo BC, Qian H, Liu Y, Sepulveda PV, Beyer C, Hagg A, Thomson RE, Chen JL, et al. (2016). Smad7 gene delivery prevents muscle wasting associated with cancer cachexia in mice. Sci. Transl. Med 8, 348ra398. [DOI] [PubMed] [Google Scholar]
- Witt CC, Witt SH, Lerche S, Labeit D, Back W, and Labeit S (2008). Cooperative control of striated muscle mass and metabolism by MuRF1 and MuRF2. EMBO J. 27, 350–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C-L, Satomi Y, and Walsh K (2017). RNA-seq and metabolomic analyses of Akt1-mediated muscle growth reveals regulation of regenerative pathways and changes in the muscle secretome. BMC Genomics 18, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Duong DM, and Peng J (2009). Systematical optimization of reverse-phase chromatography for shotgun proteomics. J. Proteome Res 8, 3944–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Anderson DE, and Ye Y (2016). The HECT domain ubiquitin ligase HUWE1 targets unassembled soluble proteins for degradation. Cell Discov. 2, 16040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HB, Xu YY, Zhao XN, Zou SW, Zhang Y, Zhang M, Li JT, Ren F, Wang LY, and Lei QY (2015). Acetylation of MAT IIα represses tumour cell growth and is decreased in human hepatocellular cancer. Nat. Commun 6, 6973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Lin RK, Kwon YT, and Li YP (2013). Signaling mechanism of tumor cell-induced up-regulation of E3 ubiquitin ligase UBR2. FASEB J. 27, 2893–2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou JY, Afjehi-Sadat L, Asress S, Duong DM, Cudkowicz M, Glass JD, and Peng J (2010a). Galectin-3 is a candidate biomarker for amyotrophic lateral sclerosis: discovery by a proteomics approach. J. Proteome Res 9,5133–5141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, Rosenfeld R, Chen Q, Boone T, Simonet WS, et al. (2010b). Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 142, 531–543. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA sequencing data generated in this study are available at the Gene Expression Omnibus with accession number GSE126922. Other datasets generated for this study are provided in Tables S1, S2, S3, S4, and S5. All other primary data are provided in Table S6.