

Abstract
Increased synaptic N-methyl-D-aspartate receptor (NMDAR) activity in the hypothalamic paraventricular nucleus (PVN) plays a major role in elevated sympathetic output in hypertension. Although exogenous angiotensin II (AngII) can increase NMDAR activity in the PVN, whether endogenous AT1 receptor–protein kinase C (PKC) activity mediates the augmented NMDAR activity of PVN presympathetic neurons in hypertension is unclear. Here we show that blocking AT1 receptors with losartan or inhibiting PKC with chelerythrine significantly decreased the frequency of NMDAR-mediated miniature excitatory postsynaptic currents (mEPSCs) and the amplitude of puff NMDA currents of retrogradely labeled spinally projecting PVN neurons in spontaneously hypertensive rats (SHRs). Also, treatment with chelerythrine abrogated the potentiating effect of AngII on mEPSCs and puff NMDA currents of labeled PVN neurons in SHRs. In contrast, neither losartan nor chelerythrine had any effect on mEPSCs or puff NMDA currents in labeled PVN neurons in Wistar-Kyoto (WKY) rats. Furthermore, levels of AT1 receptor mRNA and PKC-mediated NMDAR phosphorylation in the PVN were significantly higher in SHRs than in WKY rats. In addition, microinjection of losartan or chelerythrine into the PVN substantially reduced blood pressure and renal sympathetic nerve discharges in SHRs but not in WKY rats. Chelerythrine blocked sympathoexcitatory responses to AngII microinjected into the PVN. Our findings suggest that endogenous AT1 receptor–PKC activity is essential for presynaptic and postsynaptic NMDAR hyperactivity of PVN presympathetic neurons and for the augmented sympathetic outflow in hypertension. This information advances our mechanistic understanding of the interplay between angiotensinergic and glutamatergic excitatory inputs in hypertension.
Keywords: autonomic nervous system, cardiovascular regulation, homeostasis, renin-angiotensin system, synaptic plasticity, sympathetic nervous system
Introduction
Hypertension is a worldwide epidemic and a major risk factor for stroke, myocardial infarction, congestive heart failure, and end-stage kidney disease. However, in the vast majority (>90%) of affected individuals, the etiology of hypertension is unknown. Primary hypertension is a complex, multifactorial disease arising from the combined action of genetic, environmental, and behavioral factors. Although spontaneously hypertensive rats (SHRs) are a commonly used genetic model of hypertension, how the genetic abnormality leads to the development of hypertension remains poorly understood. Enhanced sympathetic outflow from the central nervous system has been recognized as a major contributor to the pathogenesis of hypertension (DiBona, 2013; Dampney et al., 2018). The hypothalamus is intricately involved in the development of hypertension in SHRs (Eilam et al., 1991; Takeda et al., 1991). The paraventricular nucleus (PVN) in the hypothalamus, as the interface between the nervous and endocrine systems, plays a crucial role in coordinating the sympathetic output through its projection to the rostral ventrolateral medulla (RVLM) and intermediolateral cell column in the spinal cord (Pyner & Coote, 1999). The PVN is a major source of excitatory drive to augment sympathetic outflow in hypertension (Allen, 2002; Li & Pan, 2007; Dampney et al., 2018).
The renin-angiotensin system (RAS) plays a predominant role in the pathogenesis of hypertension, and systemically blocking angiotensin AT1 receptors or inhibiting the angiotensin-converting enzyme largely prevents hypertension development in SHRs (Harrap et al., 1990; Oddie et al., 1992). In addition to the peripheral RAS, a local RAS may exist in the brain (Ganten et al., 1983; Lee-Kirsch et al., 1999; Nakagawa & Sigmund, 2017), and angiotensin-like immunoreactivity is densely present in the hypothalamus of SHRs (Weyhenmeyer & Phillips, 1982). Although the presence of the brain RAS remains controversial (van Thiel et al., 2017), circulating angiotensin II (AngII) can access the PVN via circumventricular organs, which do not have a well-formed blood-brain barrier (Ferguson, 2009). Furthermore, circulating AngII may enter the PVN directly via the compromised blood-brain barrier in SHRs (Zhang et al., 2010; Biancardi et al., 2014). The AT1 receptor has two subtypes: AT1A (encoded by Agtr1a) and AT1B (encoded by Agtr1b). The Agtr1a mRNA level is high in the circumventricular organs and PVN, whereas Agtr1b mRNA is undetectable in these brain areas (Lenkei et al., 1997, 1998). Receptor autoradiography also shows that AT1 receptors are distributed in the circumventricular organs and PVN (Vieira et al., 2015). AngII administered systemically or locally into the PVN can augment sympathetic output (Li et al., 2003; Gabor & Leenen, 2013; Glass et al., 2015; Ma et al., 2018a). However, it is unclear whether AT1 receptors in the PVN are endogenously activated to sustain the elevated sympathetic outflow in SHRs.
The glutamate N-methyl-D-aspartate receptor (NMDAR) activity of PVN presympathetic neurons is increased, and this activity sustains elevated sympathetic vasomotor activity in SHRs (Li & Pan, 2007; Li et al., 2008; Qiao et al., 2017). But the upstream signaling mechanism leading to NMDAR hyperactivity in the PVN in SHRs is uncertain. Genetically ablating NMDARs in the PVN reduces AngII-induced hypertension (Glass et al., 2015), and inhibiting NMDAR activity in the PVN blocks the sympathoexcitatory responses to AngII (Ma et al., 2018a). Nevertheless, our understanding of the intrinsic relationship between the RAS and NMDARs in the PVN in hypertension remains fragmented. In the present study, we determined whether AT1 receptors in the PVN are endogenously activated and play a role in augmented presynaptic and postsynaptic NMDAR activity in SHRs. We demonstrated for the first time that the increased activity of endogenous AT1 receptors in coordination with protein kinase C (PKC) is essential for presynaptic and postsynaptic NMDAR hyperactivity of PVN presympathetic neurons and for the elevated sympathetic outflow in SHRs. Our study provides new insight into the molecular mechanisms underlying the interaction between the RAS and sympathetic nervous system in hypertension.
Methods
Ethical Approval
The experimental procedures and protocols were approved by the Institutional Animal Care and Use Committee of The University of Texas MD Anderson Cancer Center (approval #919-RN02) and conformed to the National Institutes of Health guidelines on the ethical use of animals. We used four-week-old and thirteen-week-old male spontaneously hypertensive rats (SHRs; Harlan, Indianapolis, IN) and age-matched normotensive Wistar-Kyoto (WKY) rats in this study. Animals were deeply anesthetized with 2–3% isoflurane via a nose cone before any surgery was performed, as described in detail below. At the end of the experiments, animals were anesthetized with 3% isoflurane inhalation and then rapidly decapitated.
Retrograde Labeling of Spinally Projecting PVN neurons
Rats were anesthetized with 2–3% isoflurane, and their spinal cords at the T1–T4 level were exposed through laminectomy. A fluorescent microsphere suspension (FluoSpheres, 0.04 µm; Molecular Probes, Invitrogen, Eugene, OR) was pressure ejected bilaterally into the intermediolateral nucleus of the spinal cord in 4–5 separate 50-nl injections using a microinjector and a micropipette, as described previously (Li et al., 2002, 2003; Chen & Pan, 2007). The wound was closed with sutures after injection. Animals were returned to their cages for 4–7 days to allow the tracer to be transported to the hypothalamus.
Brain Slice Preparation and Electrophysiological Recordings
After the rats were deeply anesthetized with isoflurane, their brain was quickly removed and sliced in an ice-cold artificial cerebrospinal fluid (aCSF) solution containing (in mmol/L) 126 NaCl, 3 KCl, 1.5 MgCl2, 2.4 CaCl2, 1.2 NaH2PO4, 10 glucose, and 26 NaHCO3 saturated with 95% O2 and 5% CO2. Coronal hypothalamic slices (300 μm thick) containing the PVN were obtained from FluoSphere-injected rats using a vibrating microtome. The slices were incubated in the aCSF at 34°C before being transferred to a recording chamber. The hypothalamic slices were continuously perfused with aCSF (saturated by 95% O2 and 5% CO2; speed, 3.0 mL/min) at 34°C.
The labeled neurons in the PVN were identified under an upright microscope with epifluorescent and infrared differential interference contrast optics. Miniature excitatory postsynaptic currents (mEPSCs) were recorded in the presence of 1 μmol/L tetrodotoxin and 20 μmol/L bicuculline at a holding potential of –60 mV (Li et al., 2003; Li et al., 2008; Ye et al., 2011). The recording glass pipette (4–8 MΩ) was filled with internal solution containing (in mmol/L) 135.0 potassium gluconate, 5.0 tetraethylammonium, 2.0 MgCl2, 0.5 CaCl2, 5.0 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 10.0 lidocaine N-ethyl bromide, 5.0 ethylene glycol tetraacetic acid (EGTA), 5.0 Mg-ATP, and 0.5 Na-GTP (adjusted to pH 7.2–7.4 with 1 mol/L KOH; 290–300 mOsmol/L). To record postsynaptic NMDAR currents, we puffed NMDA (100 μmol/L) directly onto the recorded neuron at a holding potential of –60 mV, as described previously (Li et al., 2008; Ye et al., 2011). The puff pipette (10-μm tip diameter) was placed 150 μm away from the recorded neuron. Puff NMDA-induced currents were recorded in Mg2+-free aCSF in the presence of 10 μmol/L glycine and 1 μmol/L tetrodotoxin.
Western Immunoblotting
Rats were anesthetized with 2–3% isoflurane and were then decapitated. The brain was quickly removed and sectioned 1.08–2.12 mm caudal to the bregma. Hypothalamic tissues containing the PVN were micro-punched bilaterally with a slice puncher (0.5 mm diameter) according to the following stereotactic coordinates: 0.1–0.6 mm lateral to the midline and 7.0–9.0 mm ventral to the surface of the cortex (Ye et al., 2012; Qiao et al., 2017; Ma et al., 2018b). The RVLM tissues were bilaterally punched out in accordance with the stereotactic coordinates: 12.5–13.0 mm caudal to the bregma, 1.9–2.0 mm lateral to the midline, and 7.5–8.0 mm ventral to the dura. Frontal cortex tissues were also obtained at the same time. The tissues were frozen in liquid nitrogen and then transferred to a –80 °C freezer.
Total proteins were extracted with a RIPA lysis/extraction buffer (Thermo Fisher Scientific, Waltham, MA) in the presence of a mixture containing protease and phosphatase inhibitor cocktail (#PPC1010, Sigma-Aldrich, St. Louis, MO). The brain tissues were homogenized and then subjected to centrifugation at 16,000 × g for 15 min to obtain the supernatant. The samples were subjected to 4–15% Tris-HCl sodium dodecyl sulfate–polyacrylamide gel electrophoresis (#456–1086, Bio-Rad; Hercules, CA) and then transferred to a polyvinylidene difluoride membrane (EMD Millipore, Burlington, MA). The membranes were incubated with a rabbit polyclonal anti-phospho-GluN1 Ser890 antibody (1:1000, #3381, Cell signaling; Beverly, MA) (Gomez de Salazar et al., 2018), a rabbit anti-phospho-GluN1 Ser896 antibody (1:500, #ABN88, EMD Millipore; Billerica, MA) (Xie et al., 2016), a rabbit anti-GluN1 antibody (1:2000, #G8913, Sigma) (Ma et al., 2018a), and a rabbit anti-GAPDH antibody (1:1000, #ab37168, Abcam; Cambridge, MA) overnight at 4°C. An ECL kit was used to detect the protein band, which was visualized and quantified with the Odyssey Fc Imager (LI-COR Biosciences, Lincoln, NE) and normalized to the GAPDH band on the same blot.
Quantitative PCR
Total RNAs were extracted from the PVN, frontal cortex, and RVLM tissues using TRIsure (#BIO-38032, Bioline; Taunton, MA). After treatment with RNase-free DNase (#79254, Qiagen; Hilden, Germany), 1 μg RNA was used for reverse-transcription with a RevertAid RT Reverse Transcription Kit (#K1619, Thermo Fisher Scientific, Waltham, MA). Two μL of 5-times diluted cDNA was added to a 20-μLreaction volume with SYBR Green Real-Time PCR Mix (#A25780, Thermo Fisher Scientific). Real-time PCR was performed using a QuantStudio 7 Flex Real-Time PCR System (Applied Biosystems, Waltham, MA). The thermal cycling conditions were as follows: 1 cycle at 95°C for 5 min, 40 cycles at 95 °C for 15 s, and 60°C for 45 s. The following primers were used: rat Agtr1a forward, 5’-CAC AAC CCT CCC AGA AAG TGA TC-3’; rat Agtr1a reverse, GAT GAT GCT GTA GAG GGT AGG GAT C; rat Agtr1b forward, GGG CGT CAT CCA TGA CTG TGA AAT TG; rat Agtr1b reverse, GGG GGA ATA TAT TTC AGA AGC TGG AGG A; rat Gapdh forward, CAT CCC AGA GCT GAA CGG GAA G; rat Gapdh reverse, GTC CTC AGT GTA GCC CAG GATGC.
PVN Microinjection and Recording of Renal Sympathetic Nerve Activity
Rats were anesthetized via intraperitoneal administration of α-chloralose (60 mg/kg) and urethane (800 mg/kg) and mechanically ventilated. The arterial blood pressure (ABP) was continuously monitored via a catheter placed in the left femoral artery, and heart rate (HR) was calculated from the pulsatile pressure wave. A branch of the left renal postganglionic sympathetic nerve was isolated under a microscope, and the renal sympathetic nerve was cut distally. The ABP, HR, and renal sympathetic nerve activity (RSNA) were recorded using a 1401-PLUS analog-to-digital converter and Spike2 system (Cambridge Electronic Design, Cambridge, UK). When the afferent nerve from one kidney was cut (for recording renal efferent nerve activity), the hemodynamic and renal efferent nerve responses to manipulation of neurotransmission in the PVN are still intact (Silva et al., 2005; Zahner & Pan, 2005; Huang et al., 2014; Ma et al., 2018b). The vasomotor nature of the renal nerve activity has been shown previously. For example, the renal nerve activity response to intravenously injected phenylephrine and sodium nitroprusside has been shown (Sun et al., 2009; Hamza & Hall, 2012). Also, the ganglionic blocker hexamethonium inhibits the renal nerve activity (Morrison & Whitehorn, 1984; Hamza & Hall, 2012).
After the brain was exposed, a microinjection pipette was advanced into the PVN according to the following stereotactic coordinates: 1.4–2.1 mm caudal to the bregma, 0.1–0.5 mm lateral to the midline, and 7.0–7.5 mm ventral to the dura. The microinjection was done using Nanoject II (Drummond Scientific) (Li & Pan, 2006, 2007; Qiao et al., 2017). Losartan (100 μmol/L, 50 nl), chelerythrine (100 μmol/L, 50 nl), or AngII (200 μmol/L, 50 nl) was microinjected into the PVN using similar doses as described previously (Khanmoradi & Nasimi, 2017; Ma et al., 2018a). The electrical background noise was measured and subtracted from the nerve recording signal after the proximal end of renal nerve was crushed at the end of each experiment. We included 5% rhodamine-labeled fluorescent microspheres in the microinjection solution to determine the locations of the injection sites at the end of each experiment (Li & Pan, 2006, 2007; Qiao et al., 2017). Two of 21 rats were excluded from the data analysis because the microinjections were misplaced outside the PVN. All drugs were freshly prepared in artificial cerebrospinal fluid before the experiments. AngII (#4474-91-3), losartan (#124750-99-8), and chelerythrine (#3895-92-9) were obtained from Sigma-Aldrich.
Study Design and Statistical Analysis
Data are presented as means ± standard errors of the mean (SEM). For synaptic recording protocols, only one neuron in each brain slice was recorded, and at least four animals were used for each group. The amplitude and frequency of mEPSCs were analyzed with MiniAnalysis program (Synaptosoft Inc., Decatur, GA). Clampfit 10.2 (Molecular Devices, Sunnyvale, CA) was used to measure the peak amplitude of puff NMDA currents. The mean ABP, RSNA, and HR were analyzed using Spike2 software. RSNA was rectified and integrated offline after subtracting the background noise. The integrated RSNA value was calculated and derived from the raw RSNA with an integrating time of 1.0 using Spike2 software (Li & Pan, 2007; Qiao et al., 2017). A two-tailed Student t-test was used to compare two groups, and one-way analysis of variance followed by the Dunnett’s post hoc test was used to compare more than two groups. Statistical analyses were performed using Prism 8 software (GraphPad Software Inc., La Jolla, CA). P < 0.05 was considered statistically significant.
Results
AT1 Receptors and PKC Are Endogenously Activated to Augment Presynaptic NMDAR Activity of PVN Presympathetic Neurons in SHRs
Presynaptic NMDARs in the PVN are quiescent under normotensive conditions, but in hypertension, these NMDARs become tonically activated to augment synaptic glutamate release to PVN presympathetic neurons (Ye et al., 2011; Li et al., 2017; Qiao et al., 2017; Ma et al., 2018a; Ma et al., 2018b). Exogenous AngII application stimulates presynaptic NMDARs via AT1 receptors in the PVN (Ma et al., 2018a). To determine whether the increased presynaptic NMDAR activity in hypertension is associated with endogenous activation of AT1 receptors, we recorded mEPSCs, which reflect the quantal release of glutamate from presynaptic terminals (Sulzer & Pothos, 2000), in SHRs. The baseline frequency of mEPSCs of fluorescence-labeled spinally projecting PVN neurons was significantly higher in SHRs than in WKY rats (WKY, n = 10 neurons; SHR, n = 10 neurons; P < 0.0001, F(11,111) = 9.313; Fig. 1A,D & Fig. 2A,D), whereas the amplitude of those mEPSCs did not differ significantly between the two groups. Bath application of a specific NMDAR antagonist, AP5 (50 μmol/L), for 6 min quickly reduced the frequency of mEPSCs of labeled PVN neurons in SHRs to the level seen in WKY rats (n = 10 neurons per group; Fig. 1A,D). Pre-incubation with an AT1 receptor antagonist, losartan (2 μmol/L, 30 min) (Li et al., 2003; Ma et al., 2018a), normalized the baseline frequency of mEPSCs of labeled PVN neurons in SHRs (n = 12 neurons; Fig. 1B,D,E). Subsequent bath application of AP5 had no effect on mEPSCs in the neurons pretreated with losartan.
Figure 1. AT1 receptors and PKC activity mediate the increased presynaptic NMDAR activity in PVN presympathetic neurons in SHRs.
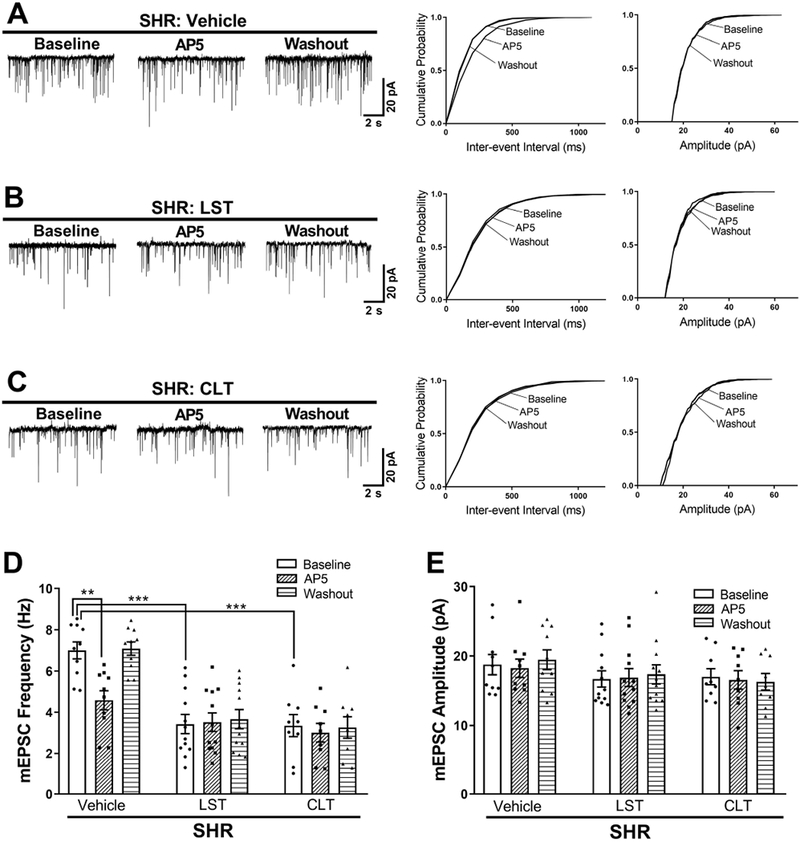
A–C, Original traces and cumulative probability plots show the effect of bath application of 50 μmol/L AP5 on the frequency and amplitude of mEPSCs of labeled PVN neurons pretreated with vehicle, 2 μmol/L losartan, or 1 μmol/L chelerythrine in brain slices from SHRs. D and E, Summary data show the effects of losartan or chelerythrine on the frequency (D) and amplitude (E) of mEPSCs before and after AP5 application in labeled PVN neurons from SHRs (vehicle, n = 10 neurons from 4 rats; losartan, n = 12 neurons from 5 rats; chelerythrine, n = 9 neurons from 4 rats). LST, losartan; CLT, chelerythrine. **P < 0.01, ***P < 0.001 compared with baseline in the vehicle group.
Figure 2. AT1 receptors and PKC activity are not actively involved in regulating presynaptic NMDAR activity in PVN presympathetic neurons in WKY rats.
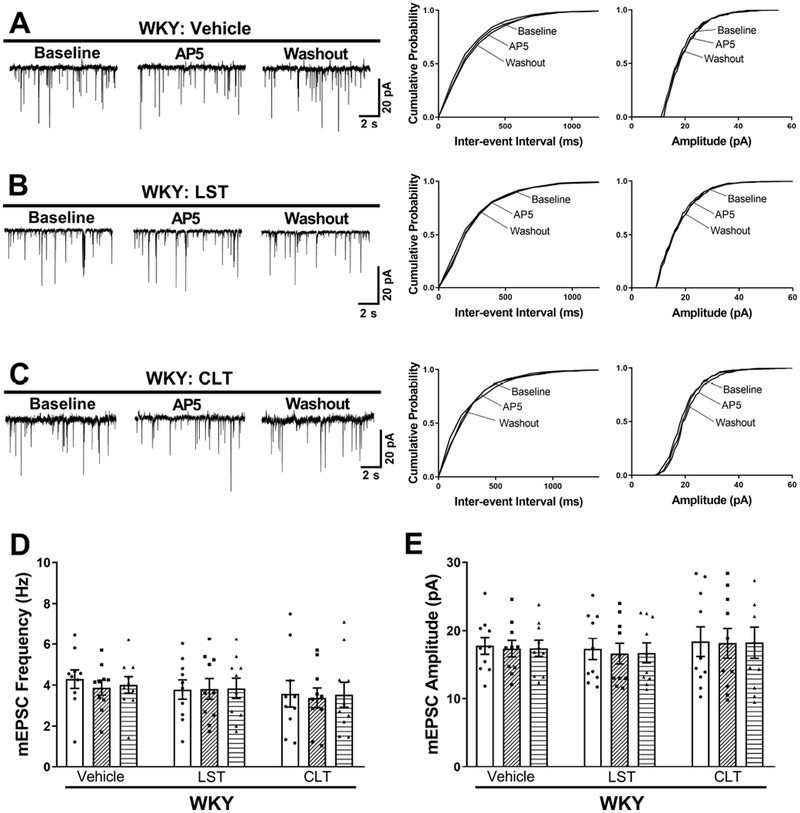
A–C, Original traces and cumulative probability plots show the effect of bath application of 50 μmol/L AP5 on the frequency and amplitude of mEPSCs of labeled PVN neurons pretreated with vehicle, 2 μmol/L losartan, or 1 μmol/L chelerythrine in brain slices from WKY rats. D and E, Summary data show the effects of losartan or chelerythrine on the frequency (D) and amplitude (E) of mEPSCs before and after AP5 bath application in labeled PVN neurons from SHRs (n = 10 neurons from 4 rats per group). LST, losartan; CLT, chelerythrine.
AT1 receptor signaling is mediated largely through its coupling to Gq and PKC (Domazet et al., 2015). To determine whether PKC plays a role in the potentiated presynaptic NMDAR activity of spinally projecting PVN neurons in hypertension, we incubated the brain slices of SHRs with chelerythrine, a specific PKC inhibitor. Pretreatment with chelerythrine (1 µmol/L, 30 min) (Li et al., 2014), also normalized the baseline frequency of mEPSCs of labeled PVN neurons in SHRs to the level observed in WKY rats (n = 9 neurons; P < 0.0001, F(11,111) = 9.313). In SHR brain slices pretreated with chelerythrine, subsequent bath application of AP5 no longer had any effect on the frequency of mEPSCs of labeled PVN neurons (Fig. 1C–E).
We next determined whether endogenous AT1 receptors and PKC play a role in regulating synaptic glutamate release to spinally projecting PVN neurons in WKY rats. Pretreatment with losartan (2 μmol/L, 30 min) had no effect on the frequency or amplitude of mEPSCs of labeled PVN neurons (n = 10 neurons; Fig. 2A,B,D,E). Also, bath application of AP5 had no effect on the frequency of mEPSCs of labeled PVN neurons in vehicle- or losartan-treated slices from WKY rats (n = 10 neurons per group; Fig. 2A,B,D,E). Furthermore, bath incubation with chelerythrine did not significantly affect the frequency or amplitude of mEPSCs of labeled PVN neurons in WKY rats (n = 10 neurons; Fig. 2C–E). Collectively, these findings suggest that endogenous activity of both AT1 receptors and PKC is essential for presynaptic NMDAR activation of PVN presympathetic neurons in SHRs but not in WKY rats.
Endogenous AT1 Receptor–PKC Activity Is Responsible for Increased Postsynaptic NMDAR Activity of PVN Presympathetic Neurons in SHRs
Like the presynaptic NMDAR activity, the postsynaptic NMDAR activity of PVN presympathetic neurons is augmented in SHRs (Li et al., 2008; Qiao et al., 2017; Ma et al., 2018b). We thus investigated whether the endogenous AT1 receptor and PKC activity play a role in the increased postsynaptic NMDAR activity of spinally projecting PVN neurons in SHRs. We examined the effect of losartan or chelerythrine on postsynaptic NMDAR currents elicited by puff application of NMDA (100 μmol/L) directly to labeled PVN neurons in brain slices. In vehicle-treated brain slices from SHRs, puff NMDA induced a large inward current of labeled neurons (Fig. 3). Treatment with losartan (2 μmol/L, 30 min) significantly reduced the amplitude of puff NMDA currents (P < 0.0001, F(5,53) = 7.921; n = 10 neurons; Fig. 3). In a separate group of labeled PVN neurons from SHRs, pretreatment with chelerythrine (1 µmol/L, 30 min) similarly decreased the amplitude of puff NMDA currents to the level seen in the losartan-treated group (Fig. 3).
Figure 3. AT1 receptors and PKC activity are essential for postsynaptic NMDAR hyperactivity of PVN presympathetic neurons in SHRs but not in WKY rats.
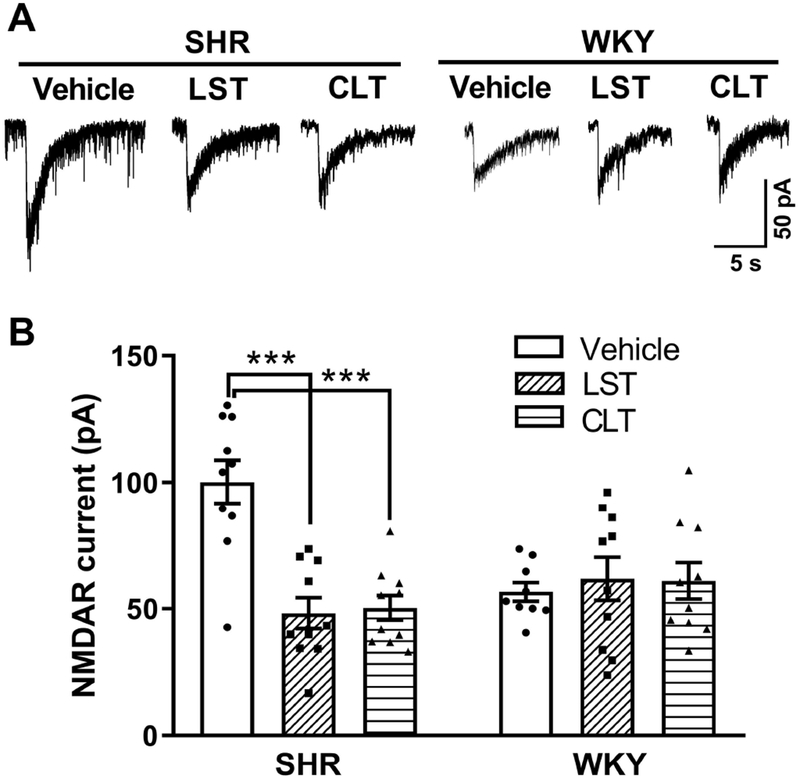
A-B, Original current traces (A) and mean data (B) show the effect of vehicle, 2 μmol/L losartan, or 1 μmol/L chelerythrine on puff NMDA (100 μmol/L)–elicited currents of labeled PVN neurons from SHRs (n = 10 neurons from 4 rats in each group) or WKY rats (vehicle, n = 9 neurons from 4 rats; losartan, n = 10 neurons from 4 rats; chelerythrine, n = 10 neurons from 4 rats). LST, losartan; CLT, chelerythrine. ***P < 0.001 compared with the SHR vehicle group.
To determine whether endogenous AT1 receptors and PKC activity are involved in regulating postsynaptic NMDARs in the PVN in WKY rats, we again examined the effect of losartan or chelerythrine on NMDAR currents elicited by puff application of NMDA (100 μmol/L). Pretreatment with either losartan (2 μmol/L, 30 min) or chelerythrine (1 µmol/L, 30 min) had no effect on the amplitude of puff NMDAR currents of labeled PVN neurons in WKY rats (n = 10 neurons; Fig. 3). These results suggest that both AT1 receptors and PKC are endogenously activated and play a critical role in the augmented postsynaptic NMDAR activity of PVN presympathetic neurons in SHRs but not in WKY rats.
AngII Increases Presynaptic and Postsynaptic NMDAR Activity in Spinally Projecting PVN Neurons via PKC
We have shown that blocking AT1 receptor with losartan abolishes the effect of AngII on presynaptic and postsynaptic NMDARs of PVN presympathetic neurons (Ma et al., 2018a). However, whether PKC is required for AngII to affect the presynaptic and postsynaptic NMDAR activity of spinally projecting PVN neurons remains to be determined. First, we showed that treatment with AngII (2 μmol/L, 30 min) in brain slices from WKY rats significantly increased the baseline frequency, but not the amplitude, of mEPSCs in labeled PVN neurons (P = 0.0013, F(5,57) = 8.856, n = 11 neurons; Fig. 4A,C,D). This increase in mEPSC frequency was rapidly reversed by bath application of the NMDAR antagonist AP5 (50 μmol/L) (P = 0.0098, F(5,54) = 9.801; Fig. 4A,C). Furthermore, the effect of AngII on mEPSCs was completely blocked by pretreatment with 1 μmol/L chelerythrine (n = 9 neurons; Fig. 4B–D).
Figure 4. AngII increases presynaptic and postsynaptic NMDAR activity via PKC in PVN presympathetic neurons in WKY rats.
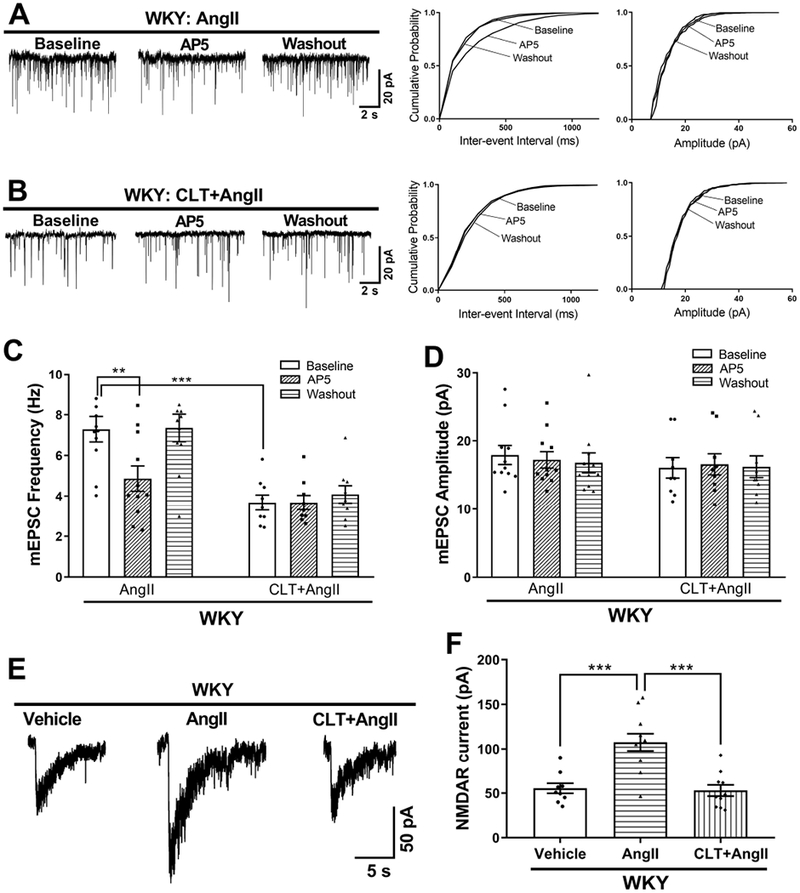
A and B, Original traces and cumulative probability plots show the effect of bath application of 50 μmol/L AP5 on the frequency and amplitude of mEPSCs of labeled PVN neurons pretreated with 2 μmol/L AngII or AngII plus 1 μmol/L chelerythrine (CLT) in brain slices from WKY rats. C and D, Summary data show the effects of AP5 on the frequency (C) and amplitude (D) of mEPSCs in labeled PVN neurons pretreated with AngII or CLT plus AngII in WKY rats (AngII, n = 11 neurons from 5 rats; CLT+AngII, n = 9 neurons from 4 rats). **P < 0.01, ***P < 0.001compared with the baseline in the AngII group. E and F, Original current traces (E) and mean data (F) show the effect of AngII or CLT plus AngII on puff NMDA (100 μmol/L)–elicited currents of labeled PVN neurons recorded from WKY rats (vehicle, n = 9 neurons from 4 rats; AngII, n = 11 neurons from 5 rats; CLT+AngII, n = 10 neurons from 4 rats). ***P < 0.001 compared with the AngII group.
In a separate group of spinally projecting PVN neurons from WKY rats, treatment with AngII (2 μmol/L, 30 min) markedly increased the amplitude of puff NMDA currents (P < 0.0001, F(2,27) = 16.47, n = 11 neurons; Fig. 4E,F). This increased amplitude was abolished by pre-incubation with chelerythrine (1 μmol/L, 30 min; n = 10 neurons; Fig. 4E,F). Collectively, these findings indicate that AT1 receptor–mediated PKC activation is essential for AngII-induced presynaptic and postsynaptic NMDAR hyperactivity of PVN presympathetic neurons.
PKC–Mediated NMDAR Phosphorylation and AT1 Receptor mRNA Are Increased in the PVN in SHRs.
The above electrophysiological data suggest that both AT1 receptors and PKC in the PVN are endogenously activated to potentiate glutamatergic input in SHRs. Because the serine residues 890 and 896 of the NMDAR GluN1 subunit are selectively phosphorylated by PKC (Tingley et al., 1997; Xie et al., 2016), we measured the phosphorylation levels of GluN1 Ser890 and GluN1 Ser896 as an indicator of endogenous PKC activity in the PVN in WKY rats and SHRs. Immunoblotting using both phospho-GluN1 antibodies showed that phosphorylated GluN1 Ser890 (P = 0.0125, t(14) = 2.864) and GluN1 Ser896 (P < 0.0001, t(14) = 5.571) protein levels in the PVN were significantly higher in SHRs than in WKY rats (n = 8 rats per group; Fig. 5A). In contrast, GluN1 Ser890 and GluN1 Ser896 protein levels in the frontal cortex and RVLM did not differ significantly between WKY rats and SHRs (Fig. 5B,C).
Figure 5. PKC-mediated NMDAR phosphorylation and AT1 receptor mRNA levels in the PVN, frontal cortex, and RVLM in SHRs and WKY rats.
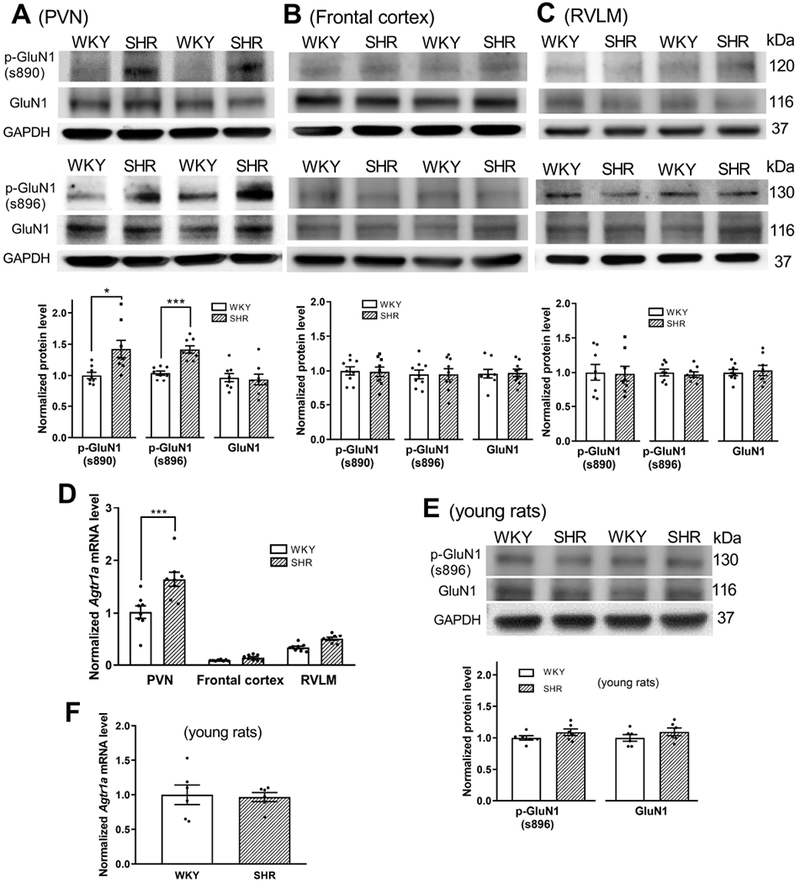
A-C, Representative blotting images and mean changes show the phosphorylation level of GluN1 Ser890 and GluN1 Ser896 in the PVN (A), frontal cortex (B), and RVLM (C) in adult WKY rats and SHRs (PVN, n = 8 rats per group; frontal cortex, n = 9 rats per group; RVLM, n = 8 rats per group; normalized to GAPDH on the same gel). D, Real-time PCR data show the mRNA level of Agtr1a in the PVN, frontal cortex, and RVLM in adult WKY rats and SHRs. Gapdh mRNA level was used as an endogenous control, and the mean value in the PVN of WKY rats was set to 1 (n = 8 rats per group). E, Representative blotting images and mean changes show the phospho-GluN1 Ser896 protein level in the PVN in 4-week-old SHRs and WKY rats (n = 6 rats per group). F, Real-time PCR data show the mRNA level of Agtr1a in the PVN in 4-week-old SHRs and WKY rats (n = 6 rats per group). *P < 0.05, ***P < 0.001 compared with WKY rats.
We then determined the AT1 receptor expression level in the PVN in WKY rats and SHRs. Because commercially available AT1 receptor antibodies are not specific (Benicky et al., 2012; Bouressam et al., 2018), we used real-time PCR to quantify the AT1 receptor mRNA level in the PVN. Quantitative PCR analysis showed that the Agtr1a mRNA level was much higher in the PVN than in the frontal cortex and RVLM (Fig. 5D). The Agtr1b mRNA in these tissues was undetectable under the same PCR assay conditions. Furthermore, the mRNA level of Agtr1a in the PVN was significantly greater in SHRs than in WKY rats (n = 8 rats per group; P < 0.0001, F(5,42) = 64.07; Fig. 5D). However, the Agtr1a mRNA levels in the frontal cortex and RVLM did not differ significantly between WKY rats and SHRs (Fig. 5D). These data suggest that AT1 receptor expression level and PKC activity in the PVN are increased in SHRs.
To determine whether the AT1 receptor mRNA level and PKC activity in the PVN are increased in pre-hypertensive SHRs, we determined levels of Agtr1a mRNA and GluN1 phosphorylation in the PVN in 4-week-old SHRs and age-matched WKY rats. The GluN1 Ser890 protein in the PVN was below detectable levels in the SHRs and WKY rats. Unexpectedly, the Agtr1a mRNA and the GluN1 Ser896 levels in the PVN did not differ significantly between the SHRs and WKY rats (n = 6 rats per group; 5E,F).
Endogenous AT1 Receptor–PKC Activity in the PVN Maintains Elevated Sympathetic Vasomotor Tone in SHRs
We next determined whether endogenous AT1 receptor activity in the PVN plays a role in heightened sympathetic vasomotor tone in SHRs. Microinjection of losartan (100 μmol/L, 50 nL) bilaterally into the PVN significantly decreased the ABP (P < 0.0001, t(9) = 9.626) and RSNA (P = 0.0007, F(9) = 5.067) in SHRs (n = 10 rats; Fig. 6A,B). In contrast, bilateral microinjection of losartan into the PVN had no significant effect on ABP or RSNA in WKY rats (n = 10 rats; Fig. 6C,D).
Figure 6. Endogenous AT1 receptor-PKC activity in the PVN maintains heightened sympathetic outflow in SHRs.
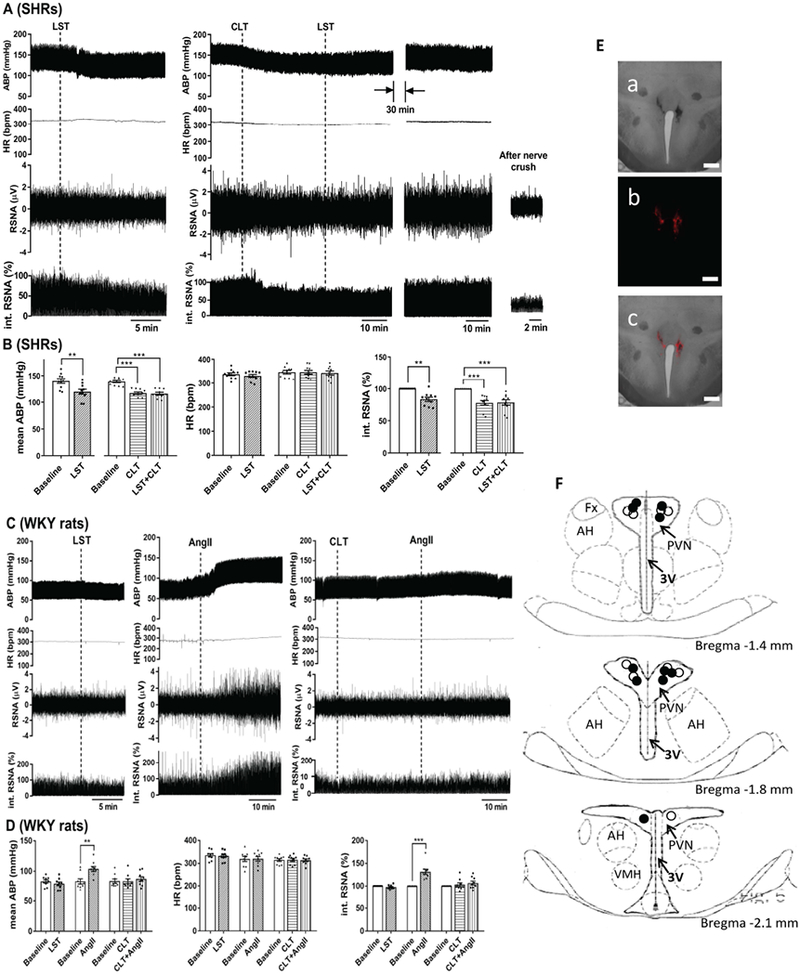
A and B, Original recording traces (A) and summary data (B) show the effects of bilateral microinjection of losartan with or without prior microinjection of chelerythrine into the PVN on arterial blood pressure (ABP), renal sympathetic nerve activity (RSNA), and heart rate (HR) in SHRs (n = 10 rats per group). C and D, Original recording traces (C) and summary data (D) show the effects of bilateral microinjection of losartan alone, and AngII with or without prior microinjection of chelerythrine into the PVN on ABP, RSNA, and HR in WKY rats (n = 9 rats per group). LST, losartan; CLT, chelerythrine. **P < 0.01, ***P < 0.001 compared with the respective baselines. E and F, Representative tissue section images (E) and schematic drawing (F) show the representative microinjection sites in the PVN in SHRs (solid circle) and WKY rats (open circle). a, bright field; b, fluorescence, c, overlay; 3V, third ventricle; AH, anterior hypothalamus; Fx, Fornix; VMH, ventromedial hypothalamus. Scale bar, 500 μm.
We also determined whether endogenous PKC activity in the PVN is involved in the elevated sympathetic outflow in SHRs. The microinjection dose of chelerythrine was originally estimated from a previous study (Schmidt et al., 2015) and confirmed in our preliminary experiments. Microinjection of chelerythrine (100 μmol/L, 50 nl) into the PVN significantly decreased the baseline ABP (P < 0.0001, F(2,27) = 30.39) and RSNA (P < 0.0001, F(2,27) = 15.29) in SHRs (n = 10 rats; Fig. 6A,B) but not in WKY rats (n = 9 rats, Fig. 6C,D).
In addition, we determined a causal relationship between PKC and the action of AngII in the PVN in regulating sympathetic vasomotor activity. In SHRs that received prior chelerythrine microinjection, subsequent microinjection of losartan (100 μmol/L, 50 nL) into the PVN failed to further decrease ABP or RSNA (n = 10 rats, Fig. 6A,B). Microinjection of AngII (200 μmol/L, 50 nl) into the PVN in WKY rats significantly increased the RSNA and ABP. However, when chelerythrine (100 μmol/L, 50 nl) was microinjected into the PVN first, subsequent microinjection of AngII failed to significantly increase ABP or RSNA in WKY rats (n = 9 rats; Fig. 6C,D). These results collectively suggest that endogenous AT1 receptor and PKC activity in the PVN is increased and sustains the elevated sympathetic outflow in SHRs.
Discussion
The most significant finding of our study is that increased endogenous AT1 receptor activity is responsible for presynaptic and postsynaptic NMDAR hyperactivity of PVN presympathetic neurons in a genetic model of hypertension. We showed that blocking AT1 receptors with losartan diminished presynaptic NMDAR activity and normalized postsynaptic NMDAR activity of PVN presympathetic neurons in SHRs. Furthermore, blocking AT1 receptors in the PVN profoundly attenuated the elevated ABP and sympathetic nerve discharges activity in SHRs. Strikingly, treatment with losartan produced no effect on NMDAR activity or sympathetic vasomotor activity in WKY rats, suggesting that angiotensinergic input to the PVN is not tonically active under the normotensive condition. Although blocking AT1 receptors in the RVLM also reduces blood pressure in SHRs, the PVN seems to act as a prevailing generator of elevated sympathetic output in hypertension by increasing excitatory input to the RVLM (Pyner & Coote, 1999; Ito et al., 2002). In the PVN, presynaptic NMDAR activation enhances synaptic glutamate release, whereas increased postsynaptic NMDAR activity can directly potentiate neuronal firing activity; both actions are likely involved in the maintenance of elevated sympathetic outflow in SHRs (Li & Pan, 2007; Li et al., 2008; Ye et al., 2011). Our findings suggest that AT1 receptors in the PVN are endogenously activated and serve as an upstream signaling mechanism for the augmented synaptic NMDAR activity and sympathetic vasomotor tone in hypertension. Previous studies have shown that sympathetic outflow generated from the brain increases renin activity and renin release from the kidney (Richardson et al., 1974; Porter, 1988). Conversely, increased circulating AngII could stimulate AT1 receptors in the PVN to potentiate glutamatergic input to augment sympathetic outflow in hypertension. Therefore, our findings support the notion that the NMDAR in the PVN is a key mechanism enabling the dynamic interaction between the RAS and sympathetic nervous systems in hypertension.
Another new finding of our study is that endogenous PKC activity in the PVN plays an essential role in the AT1 receptor–mediated increase in presynaptic and postsynaptic NMDAR activity of PVN presympathetic neurons in SHRs. We showed that treatment with the PKC inhibitor chelerythrine diminished the potentiated presynaptic and postsynaptic NMDAR activities of spinally projecting PVN neurons in SHRs. Also, inhibition of PKC in the PVN abrogated the elevated sympathetic vasomotor activity in SHRs. Importantly, we found that chelerythrine completely blocked the potentiating effect of AngII on presynaptic and postsynaptic NMDAR activities and abolished the sympathoexcitatory response to AngII. Remarkably, PKC inhibition in the PVN had no effect on NMDAR activity and sympathetic vasomotor tone in WKY rats, suggesting that PKC activity in the PVN is preferentially increased in SHRs. Increased PKC activity can promote synaptic trafficking of NMDARs in the PVN (Li et al., 2014) and potentiate NMDAR activity by reducing the Mg2+ block of NMDAR channels (Chen & Huang, 1992). Therefore, endogenous AT1 receptor–mediated PKC activation in the PVN is responsible for the increased synaptic NMDAR activity and sympathetic outflow in hypertension.
An imbalance between phosphorylation and dephosphorylation can dramatically affect NMDAR activity, and many protein kinases, such as Src, CK2, and CaMKII, are involved in synaptic NMDAR hyperactivity of PVN presympathetic neurons in SHRs (Ye et al., 2011; Li et al., 2015; Li et al., 2017; Qiao et al., 2017). How these kinases are linked to PKC and AT1 receptors in the PVN in hypertension remains unclear. PKC mainly regulates the obligatory GluN1 subunit (Lan et al., 2001), whereas CaMKII and CK2 predominantly affect the phosphorylation of GluN2 subunits (Ye et al., 2012; Sanz-Clemente et al., 2013). Furthermore, because PKC, Src, and CaMKII are associated with AT1 receptor–mediated signaling (Zhu et al., 1999; Bali & Jaggi, 2016), the increased activity of these kinases in the PVN may directly or indirectly result from endogenous activation of AT1 receptors in SHRs. It is thus possible that these protein kinases differentially control various phosphorylation sites of NMDARs in the PVN. It has been shown that chelerythrine is a substrate-competitive active-site inhibitor of PKC with an IC50 of 0.66 μM and is selective for PKC over PKA, CaM kinases, and tyrosine kinases (Herbert et al., 1990). Also, the potentiating effect of the Src-activating peptide on NMDAR activity of PVN neurons is not affected by the CK2 inhibitor DRB (Qiao et al., 2017). However, caution should be used for interpreting these pharmacological data because the kinase inhibitors may have off-target effects on other proteins. Additionally, synaptic NMDAR hyperactivity in the PVN is due to the increased α2δ-1–NMDAR coupling in SHRs (Ma et al., 2018a; Ma et al., 2018b), and targeting α2δ-1–bound NMDARs with a specific peptide in the PVN or with systemic gabapentin administration reduces ABP in SHRs (Behuliak et al., 2018; Ma et al., 2018b). Since the protein binding complexes are strengthened by increased protein phosphorylation through changes in their physicochemical properties, stability, and dynamics (Nishi et al., 2013), endogenous AT1 receptor–PKC activity could increase NMDAR activity in the PVN by promoting synaptic trafficking of α2δ-1–bound NMDARs in SHRs.
We showed that the Agtr1a mRNA level was significantly increased in the PVN, but not in the cortex and RVLM, in SHRs. We also found that the substrates of PKC—the phosphorylated GluN1 Ser890 and GluN1 Ser896—were increased primarily in the PVN in SHRs. These biochemical data provide further evidence for the increased AT1 receptor–PKC activity in the PVN in hypertension. Interestingly, the AT1 receptor mRNA level and PKC-mediated NMDAR phosphorylation level in the PVN were similar in pre-hypertensive young SHRs and age-matched WKY rats, suggesting that the increased AT1 receptor expression and PKC activity in the PVN are not inherently associated with genetic abnormalities in SHRs. Nevertheless, blocking AT1 receptors or PKC activity in the PVN substantially reduced sympathetic outflow in adult SHRs, indicating that high levels of AT1 receptor–PKC activity in the PVN likely play a crucial role in the development of neurogenic hypertension. It should be noted that many cellular and molecular mechanisms may interact and account for enhanced sympathetic outflow from the central nervous system in SHRs, and hypertension in SHRs is likely a consequence of changes at several levels of generation and modulation of the sympathetic output beyond PVN presympathetic neurons. It has been shown that denervation of the carotid bodies in SHRs reduces ABP to a similar extent to that in the present study (Abdala et al., 2012), indicating that excitatory input from chemoreceptors plays an important role in sustaining hypertension in SHRs. In addition, blocking orexin receptors in the RVLM attenuates ABP in SHRs (Li et al., 2013), suggesting that orexin receptor activity in the RVLM also contributes to the maintenance of hypertension in SHRs. However, whether heightened inputs from chemoreceptors and orexin receptor hyperactivity in the RVLM converge to PVN presympathetic neurons in SHRs remains to be determined. Similar to our present study, blockade of AT1 receptors in the RVLM of SHRs also profoundly reduces ABP (Ito et al., 2002). Because the PVN neurons can project to the spinal cord directly or indirectly via the RVLM (Pyner & Coote, 1999), it is possible that hypertension in SHRs is dependent on an interaction between inputs to spinal sympathetic neurons arising from both the PVN and RVLM.
Our study also raises issues that should be addressed in future studies. Although AT1 receptors in the PVN are tonically activated in SHRs, the exact sources of AngII that maintain high activity of AT1 receptors are unclear. Also, the AT1 receptor expression level in the PVN is increased in adult SHRs but not in pre-hypertensive young SHRs. It would be important to define the epigenetic mechanism that regulates the AT1 receptor expression in the hypothalamus during the development of hypertension. In addition, further studies are needed to determine which PKC isoforms are altered in the PVN in SHRs and other models of hypertension. Such information will further increase our knowledge about how the glutamatergic and angiotensinergic excitatory inputs are integrated in the hypothalamus to augment sympathetic output in hypertension.
In summary, we present substantial new evidence that the endogenous AT1 receptor–PKC activity is required for the increased presynaptic and postsynaptic NMDAR activity of PVN presympathetic neurons and that this signaling cascade in the PVN is critically involved in elevated sympathetic vasomotor activity in SHRs. Our findings provide new information about the molecular and signaling mechanisms responsible for the interaction between the RAS and sympathetic nervous systems in hypertension. Targeting AT1 receptor–PKC signaling in the hypothalamus may be considered for treating neurogenic hypertension.
Figure 7. Schematic diagram shows the proposed interaction among AT1 receptors, PKC, and synaptic NMDARs of PVN neurons in hypertension.
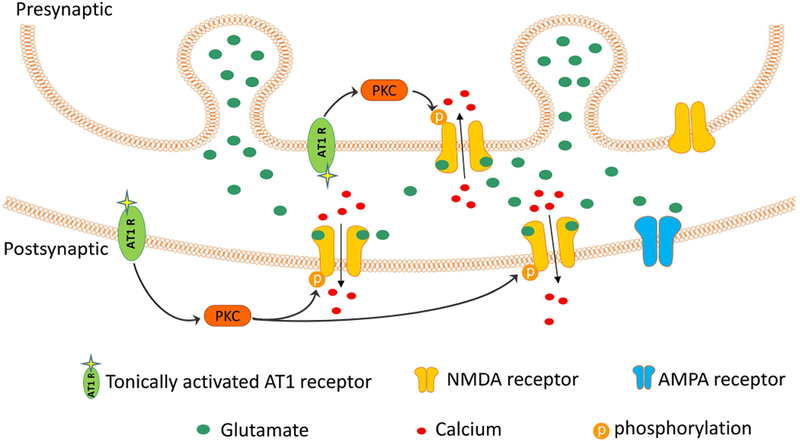
In SHRs, the angiotensin AT1 receptor at the presynaptic terminal and postsynaptic site becomes endogenously activated, which stimulates PKC and increases NMDAR phosphorylation. The resulting NMDAR hyperactivity leads to augmented glutamate release from presynaptic terminals and glutamatergic input to PVN presympathetic neurons.
Translational Perspective:
The renin-angiotensin system (RAS) and sympathetic nervous system are both important for the regulation of blood pressure. However, how these two systems interact in hypertension is unclear. The glutamate N-methyl-D-aspartate receptor (NMDAR) activity in the hypothalamus is increased in hypertension, which plays a major role in the augmented sympathetic output. In this study, we tested the hypothesis that the aberrant RAS activity contributes to synaptic NMDAR hyperactivity in the hypothalamus using a rat genetic model of hypertension. Our study provides new evidence that the angiotensin AT1 receptor is upregulated and constitutively activated to cause hyperactivity of protein kinase C (PKC) in the hypothalamus in hypertension. This signaling pathway is responsible for increased NMDAR phosphorylation and activity, at both presynaptic and postsynaptic sites, and leads to augmented vaso-constrictive sympathetic outflow in hypertension. This new information advances our mechanistic understanding of the relationship and interaction between the RAS and sympathetic nervous system in the brain and suggests that the brain RAS is an important target to treat neurogenic hypertension. Further studies are needed to determine the epigenetic mechanism of AT1 receptor upregulation in the brain associated with hypertension and the involvement of AT1 receptor–PKC–NMDAR signaling in other animal models of hypertension. In addition, because excessive brain AT1 receptor activity is associated with exaggerated sympathetic vasomotor tone, the brain RAS could be targeted to treat stress-induced hypertension and related disorders.
Key Points.
The angiotensin AT1 receptor expression and PKC-mediated NMDA receptor phosphorylation levels in the hypothalamus are increased in a rat genetic model of hypertension.
Blocking AT1 receptors or PKC activity normalizes the increased pre- and postsynaptic NMDA receptor activity of hypothalamic presympathetic neurons in hypertensive animals.
Inhibition of AT1 receptor–PKC activity in the hypothalamus reduces arterial blood pressure and sympathetic nerve discharges in hypertensive animals.
AT1 receptors in the hypothalamus are endogenously activated to sustain NMDA receptor hyperactivity and elevated sympathetic outflow via PKC in hypertension.
Acknowledgments:
Funding: This work was supported by the National Institutes of Health (Grants HL131161 and HL142133) and the N.G. and Helen T. Hawkins Endowment (to H.-L.P.).
Footnotes
Competing Interest/Disclosures: None.
References
- Abdala AP, McBryde FD, Marina N, Hendy EB, Engelman ZJ, Fudim M, Sobotka PA, Gourine AV & Paton JF. (2012). Hypertension is critically dependent on the carotid body input in the spontaneously hypertensive rat. The Journal of physiology 590, 4269–4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen AM. (2002). Inhibition of the hypothalamic paraventricular nucleus in spontaneously hypertensive rats dramatically reduces sympathetic vasomotor tone. Hypertension (Dallas, Tex : 1979) 39, 275–280. [DOI] [PubMed] [Google Scholar]
- Bali A & Jaggi AS. (2016). Angiotensin II-triggered kinase signaling cascade in the central nervous system. Rev Neurosci 27, 301–315. [DOI] [PubMed] [Google Scholar]
- Behuliak M, Bencze M, Polgarova K, Kunes J, Vaneckova I & Zicha J. (2018). Hemodynamic Response to Gabapentin in Conscious Spontaneously Hypertensive Rats. Hypertension (Dallas, Tex : 1979) 72, 676–685. [DOI] [PubMed] [Google Scholar]
- Benicky J, Hafko R, Sanchez-Lemus E, Aguilera G & Saavedra JM. (2012). Six commercially available angiotensin II AT1 receptor antibodies are non-specific. Cell Mol Neurobiol 32, 1353–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biancardi VC, Son SJ, Ahmadi S, Filosa JA & Stern JE. (2014). Circulating angiotensin II gains access to the hypothalamus and brain stem during hypertension via breakdown of the blood-brain barrier. Hypertension (Dallas, Tex : 1979) 63, 572–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouressam ML, Lartaud I, Dupuis F & Lecat S. (2018). No answer to the lack of specificity: mouse monoclonal antibody targeting the angiotensin II type 1 receptor AT1 fails to recognize its target. Naunyn Schmiedebergs Arch Pharmacol 391, 883–889. [DOI] [PubMed] [Google Scholar]
- Chen L & Huang LY. (1992). Protein kinase C reduces Mg2+ block of NMDA-receptor channels as a mechanism of modulation. Nature 356, 521–523. [DOI] [PubMed] [Google Scholar]
- Chen Q & Pan HL. (2007). Signaling mechanisms of angiotensin II-induced attenuation of GABAergic input to hypothalamic presympathetic neurons. Journal of neurophysiology 97, 3279–3287. [DOI] [PubMed] [Google Scholar]
- Dampney RA, Michelini LC, Li DP & Pan HL. (2018). Regulation of sympathetic vasomotor activity by the hypothalamic paraventricular nucleus in normotensive and hypertensive states. Am J Physiol Heart Circ Physiol 315, H1200–H1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiBona GF. (2013). Sympathetic nervous system and hypertension. Hypertension (Dallas, Tex : 1979) 61, 556–560. [DOI] [PubMed] [Google Scholar]
- Domazet I, Holleran BJ, Richard A, Vandenberghe C, Lavigne P, Escher E, Leduc R & Guillemette G. (2015). Characterization of Angiotensin II Molecular Determinants Involved in AT1 Receptor Functional Selectivity. Mol Pharmacol 87, 982–995. [DOI] [PubMed] [Google Scholar]
- Eilam R, Malach R, Bergmann F & Segal M. (1991). Hypertension induced by hypothalamic transplantation from genetically hypertensive to normotensive rats. J Neurosci 11, 401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson AV. (2009). Angiotensinergic regulation of autonomic and neuroendocrine outputs: critical roles for the subfornical organ and paraventricular nucleus. Neuroendocrinology 89, 370–376. [DOI] [PubMed] [Google Scholar]
- Gabor A & Leenen FH. (2013). Central mineralocorticoid receptors and the role of angiotensin II and glutamate in the paraventricular nucleus of rats with angiotensin II-induced hypertension. Hypertension (Dallas, Tex : 1979) 61, 1083–1090. [DOI] [PubMed] [Google Scholar]
- Ganten D, Hermann K, Bayer C, Unger T & Lang RE. (1983). Angiotensin synthesis in the brain and increased turnover in hypertensive rats. Science 221, 869–871. [DOI] [PubMed] [Google Scholar]
- Glass MJ, Wang G, Coleman CG, Chan J, Ogorodnik E, Van Kempen TA, Milner TA, Butler SD, Young CN, Davisson RL, Iadecola C & Pickel VM. (2015). NMDA Receptor Plasticity in the Hypothalamic Paraventricular Nucleus Contributes to the Elevated Blood Pressure Produced by Angiotensin II. J Neurosci 35, 9558–9567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez de Salazar M, Grau C, Ciruela F & Altafaj X. (2018). Phosphoproteomic Alterations of Ionotropic Glutamate Receptors in the Hippocampus of the Ts65Dn Mouse Model of Down Syndrome. Front Mol Neurosci 11, 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamza SM & Hall JE. (2012). Direct recording of renal sympathetic nerve activity in unrestrained, conscious mice. Hypertension (Dallas, Tex : 1979) 60, 856–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrap SB, Van der Merwe WM, Griffin SA, Macpherson F & Lever AF. (1990). Brief angiotensin converting enzyme inhibitor treatment in young spontaneously hypertensive rats reduces blood pressure long-term. Hypertension (Dallas, Tex : 1979) 16, 603–614. [DOI] [PubMed] [Google Scholar]
- Herbert JM, Augereau JM, Gleye J & Maffrand JP. (1990). Chelerythrine is a potent and specific inhibitor of protein kinase C. Biochemical and biophysical research communications 172, 993–999. [DOI] [PubMed] [Google Scholar]
- Huang BS, Chen A, Ahmad M, Wang HW & Leenen FH. (2014). Mineralocorticoid and AT1 receptors in the paraventricular nucleus contribute to sympathetic hyperactivity and cardiac dysfunction in rats post myocardial infarct. The Journal of physiology 592, 3273–3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Komatsu K, Tsukamoto K, Kanmatsuse K & Sved AF. (2002). Ventrolateral medulla AT1 receptors support blood pressure in hypertensive rats. Hypertension (Dallas, Tex : 1979) 40, 552–559. [DOI] [PubMed] [Google Scholar]
- Khanmoradi M & Nasimi A. (2017). Functions of AT1 and AT2 angiotensin receptors in the paraventricular nucleus of the rat, correlating single-unit and cardiovascular responses. Brain Res Bull 132, 170–179. [DOI] [PubMed] [Google Scholar]
- Lan JY, Skeberdis VA, Jover T, Grooms SY, Lin Y, Araneda RC, Zheng X, Bennett MV & Zukin RS. (2001). Protein kinase C modulates NMDA receptor trafficking and gating. Nat Neurosci 4, 382–390. [DOI] [PubMed] [Google Scholar]
- Lee-Kirsch MA, Gaudet F, Cardoso MC & Lindpaintner K. (1999). Distinct renin isoforms generated by tissue-specific transcription initiation and alternative splicing. Circ Res 84, 240–246. [DOI] [PubMed] [Google Scholar]
- Lenkei Z, Palkovits M, Corvol P & Llorens-Cortes C. (1997). Expression of angiotensin type-1 (AT1) and type-2 (AT2) receptor mRNAs in the adult rat brain: a functional neuroanatomical review. Front Neuroendocrinol 18, 383–439. [DOI] [PubMed] [Google Scholar]
- Lenkei Z, Palkovits M, Corvol P & Llorens-Cortes C. (1998). Distribution of angiotensin type-1 receptor messenger RNA expression in the adult rat brain. Neuroscience 82, 827–841. [DOI] [PubMed] [Google Scholar]
- Li A, Hindmarch CC, Nattie EE & Paton JF. (2013). Antagonism of orexin receptors significantly lowers blood pressure in spontaneously hypertensive rats. The Journal of physiology 591, 4237–4248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DP, Chen SR & Pan HL. (2002). Nitric oxide inhibits spinally projecting paraventricular neurons through potentiation of presynaptic GABA release. J Neurophysiol 88, 2664–2674. [DOI] [PubMed] [Google Scholar]
- Li DP, Chen SR & Pan HL. (2003). Angiotensin II stimulates spinally projecting paraventricular neurons through presynaptic disinhibition. J Neurosci 23, 5041–5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DP & Pan HL. (2006). Plasticity of GABAergic control of hypothalamic presympathetic neurons in hypertension. American journal of physiology Heart and circulatory physiology 290, H1110–1119. [DOI] [PubMed] [Google Scholar]
- Li DP & Pan HL. (2007). Glutamatergic inputs in the hypothalamic paraventricular nucleus maintain sympathetic vasomotor tone in hypertension. Hypertension (Dallas, Tex : 1979) 49, 916–925. [DOI] [PubMed] [Google Scholar]
- Li DP, Yang Q, Pan HM & Pan HL. (2008). Pre- and postsynaptic plasticity underlying augmented glutamatergic inputs to hypothalamic presympathetic neurons in spontaneously hypertensive rats. The Journal of physiology 586, 1637–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DP, Zhou JJ & Pan HL. (2015). Endogenous casein kinase-1 modulates NMDA receptor activity of hypothalamic presympathetic neurons and sympathetic outflow in hypertension. The Journal of physiology 593, 4439–4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DP, Zhou JJ, Zhang J & Pan HL. (2017). CaMKII Regulates Synaptic NMDA Receptor Activity of Hypothalamic Presympathetic Neurons and Sympathetic Outflow in Hypertension. J Neurosci 37, 10690–10699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DP, Zhu LH, Pachuau J, Lee HA & Pan HL. (2014). mGluR5 Upregulation increases excitability of hypothalamic presympathetic neurons through NMDA receptor trafficking in spontaneously hypertensive rats. J Neurosci 34, 4309–4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Chen SR, Chen H, Li L, Li DP, Zhou JJ & Pan HL. (2018a). α2δ-1 Is Essential for Sympathetic Output and NMDA Receptor Activity Potentiated by Angiotensin II in the Hypothalamus. J Neurosci 38, 6388–6398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Chen SR, Chen H, Zhou JJ, Li DP & Pan HL. (2018b). α2δ-1 couples to NMDA receptors in the hypothalamus to sustain sympathetic vasomotor activity in hypertension. The Journal of physiology 596, 4269–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison SF & Whitehorn D. (1984). Enhanced preganglionic sympathetic nerve responses in spontaneously hypertensive rats. Brain research 296, 152–155. [DOI] [PubMed] [Google Scholar]
- Nakagawa P & Sigmund CD. (2017). How Is the Brain Renin-Angiotensin System Regulated? Hypertension (Dallas, Tex : 1979) 70, 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishi H, Fong JH, Chang C, Teichmann SA & Panchenko AR. (2013). Regulation of protein-protein binding by coupling between phosphorylation and intrinsic disorder: analysis of human protein complexes. Mol Biosyst 9, 1620–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddie CJ, Dilley RJ & Bobik A. (1992). Long-term angiotensin II antagonism in spontaneously hypertensive rats: effects on blood pressure and cardiovascular amplifiers. Clin Exp Pharmacol Physiol 19, 392–395. [DOI] [PubMed] [Google Scholar]
- Porter JP. (1988). Electrical stimulation of paraventricular nucleus increases plasma renin activity. Am J Physiol 254, R325–330. [DOI] [PubMed] [Google Scholar]
- Pyner S & Coote JH. (1999). Identification of an efferent projection from the paraventricular nucleus of the hypothalamus terminating close to spinally projecting rostral ventrolateral medullary neurons. Neuroscience 88, 949–957. [DOI] [PubMed] [Google Scholar]
- Qiao X, Zhou JJ, Li DP & Pan HL. (2017). Src Kinases Regulate Glutamatergic Input to Hypothalamic Presympathetic Neurons and Sympathetic Outflow in Hypertension. Hypertension (Dallas, Tex : 1979) 69, 154–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson D, Stella A, Leonetti G, Bartorelli A & Zanchetti A. (1974). Mechanisms of renal release of renin by electrical stimulation of the brainstem in the cat. Circ Res 34, 425–434. [DOI] [PubMed] [Google Scholar]
- Sanz-Clemente A, Gray JA, Ogilvie KA, Nicoll RA & Roche KW. (2013). Activated CaMKII couples GluN2B and casein kinase 2 to control synaptic NMDA receptors. Cell Rep 3, 607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt HD, Kimmey BA, Arreola AC & Pierce RC. (2015). Group I metabotropic glutamate receptor-mediated activation of PKC gamma in the nucleus accumbens core promotes the reinstatement of cocaine seeking. Addict Biol 20, 285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AQ, Santos RA & Fontes MA. (2005). Blockade of endogenous angiotensin-(1–7) in the hypothalamic paraventricular nucleus reduces renal sympathetic tone. Hypertension (Dallas, Tex : 1979) 46, 341–348. [DOI] [PubMed] [Google Scholar]
- Sulzer D & Pothos EN. (2000). Regulation of quantal size by presynaptic mechanisms. Rev Neurosci 11, 159–212. [DOI] [PubMed] [Google Scholar]
- Sun H, Li DP, Chen SR, Hittelman WN & Pan HL. (2009). Sensing of blood pressure increase by transient receptor potential vanilloid 1 receptors on baroreceptors. The Journal of pharmacology and experimental therapeutics 331, 851–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Nakata T, Takesako T, Itoh H, Hirata M, Kawasaki S, Hayashi J, Oguro M, Sasaki S & Nakagawa M. (1991). Sympathetic inhibition and attenuation of spontaneous hypertension by PVN lesions in rats. Brain research 543, 296–300. [DOI] [PubMed] [Google Scholar]
- Tingley WG, Ehlers MD, Kameyama K, Doherty C, Ptak JB, Riley CT & Huganir RL. (1997). Characterization of protein kinase A and protein kinase C phosphorylation of the N-methyl-D-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J Biol Chem 272, 5157–5166. [DOI] [PubMed] [Google Scholar]
- van Thiel BS, Goes Martini A, Te Riet L, Severs D, Uijl E, Garrelds IM, Leijten FPJ, van der Pluijm I, Essers J, Qadri F, Alenina N, Bader M, Paulis L, Rajkovicova R, Domenig O, Poglitsch M & Danser AHJ. (2017). Brain Renin-Angiotensin System: Does It Exist? Hypertension (Dallas, Tex : 1979) 69, 1136–1144. [DOI] [PubMed] [Google Scholar]
- Vieira SM, de Oliveira VH, Valente Rdo C, Moreira Oda C, Fontes CF & Mignaco JA. (2015). Chelerythrine inhibits the sarco/endoplasmic reticulum Ca(2+)-ATPase and results in cell Ca(2+) imbalance. Arch Biochem Biophys 570, 58–65. [DOI] [PubMed] [Google Scholar]
- Weyhenmeyer JA & Phillips MI. (1982). Angiotensin-like immunoreactivity in the brain of the spontaneously hypertensive rat. Hypertension (Dallas, Tex : 1979) 4, 514–523. [DOI] [PubMed] [Google Scholar]
- Xie JD, Chen SR, Chen H, Zeng WA & Pan HL. (2016). Presynaptic N-Methyl-d-aspartate (NMDA) Receptor Activity Is Increased Through Protein Kinase C in Paclitaxel-induced Neuropathic Pain. J Biol Chem 291, 19364–19373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye ZY, Li DP, Li L & Pan HL. (2011). Protein kinase CK2 increases glutamatergic input in the hypothalamus and sympathetic vasomotor tone in hypertension. J Neurosci 31, 8271–8279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye ZY, Li L, Li DP & Pan HL. (2012). Casein kinase 2-mediated synaptic GluN2A up-regulation increases N-methyl-D-aspartate receptor activity and excitability of hypothalamic neurons in hypertension. J Biol Chem 287, 17438–17446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahner MR & Pan HL. (2005). Role of paraventricular nucleus in the cardiogenic sympathetic reflex in rats. American journal of physiology Regulatory, integrative and comparative physiology 288, R420–426. [DOI] [PubMed] [Google Scholar]
- Zhang M, Mao Y, Ramirez SH, Tuma RF & Chabrashvili T. (2010). Angiotensin II induced cerebral microvascular inflammation and increased blood-brain barrier permeability via oxidative stress. Neuroscience 171, 852–858. [DOI] [PubMed] [Google Scholar]
- Zhu M, Gelband CH, Posner P & Sumners C. (1999). Angiotensin II decreases neuronal delayed rectifier potassium current: role of calcium/calmodulin-dependent protein kinase II. J Neurophysiol 82, 1560–1568. [DOI] [PubMed] [Google Scholar]