

Abstract
Neuronal circuits regulating hunger and satiety synthesize information encoding the energy state of the animal and translate those signals into motivated behaviors to meet homeostatic needs. Proopiomelanocortin (POMC) neurons in the arcuate nucleus of the hypothalamus are activated by energy surfeits and inhibited by energy deficits. When activated, these cells inhibit food intake and facilitate weight loss. Conversely, decreased activity in POMC cells is associated with increased food intake and obesity. Circulating nutrients and hormones modulate the activity of POMC neurons over protracted periods of time. However, recent work indicates that calcium activity in POMC cells changes in response to food cues on times scales consistent with the rapid actions of amino acid transmitters. Indeed, the frequency of spontaneous IPSCs (sIPSCs) onto POMC neurons increases during caloric deficits. However, the afferent brain regions responsible for this inhibitory modulation are currently unknown. Here, through the use of brain region-specific deletion of GABA release in both male and female mice we show that neurons in the dorsomedial hypothalamus (DMH) are responsible for the majority of sIPSCs in POMC neurons as well as the fasting-induced increase in sIPSC frequency. Further, the readily releasable pool of GABA vesicles and the release probability of GABA is increased at DMH-to-POMC synapses following an overnight fast. Collectively these data provide evidence that DMH-to-POMC GABA circuitry conveys inhibitory neuromodulation onto POMC cells that is sensitive to the animal's energy state.
SIGNIFICANCE STATEMENT Activation of proopiomelanocortin (POMC) cells signals satiety, whereas GABAergic cells in the dorsomedial hypothalamus (DMH) can increase food consumption. However, communication between these cells, particularly in response to changes in metabolic state, is unknown. Here, through targeted inhibition of DMH GABA release, we show that DMH neurons contribute a significant portion of spontaneously released GABA onto POMC cells and are responsible for increased GABAergic inhibition of POMC cells during fasting, likely mediated through increased release probability of GABA at DMH terminals. These data provide important information about inhibitory modulation of metabolic circuitry and provide a mechanism through which POMC neurons could be inhibited, or disinhibited, rapidly in response to food availability.
Keywords: arcuate nucleus, calcium imaging, electrophysiology, optogenetic, proopiomelanocortin, synaptic
Introduction
Proopiomelanocortin (POMC) neurons in the arcuate nucleus (ARC) of the hypothalamus target neurons throughout the brain to modulate satiety, glucose metabolism, and body weight through the release of peptide and amino acid transmitters (Yaswen et al., 1999; Cone, 2005; Xu et al., 2005; Mercer et al., 2013; Dennison et al., 2016). Optogenetic stimulation of POMC cells reduces body weight (Aponte et al., 2011), whereas chemogenetic inhibition of these cells increases feeding (Atasoy et al., 2012). The activity of POMC neurons is regulated in part by the integration of peripheral signals such as leptin, insulin, and glucose (Pinto et al., 2004; Parton et al., 2007; Belgardt et al., 2009; Dodd et al., 2018). These findings have led to the hypothesis that peripheral signals reflect the energy state of that animal and that POMC neurons detect the concentration of these circulating hormones, gradually adjusting their activity to reflect nutritional state (Belgardt et al., 2009; Williams and Elmquist, 2012). However, recent data indicate that amino acid transmitters, particularly GABA, rapidly modulate metabolic brain circuits in response to changes in energy state (Suyama and Yada, 2018).
Anorexigenic POMC neurons receive substantial inhibitory synaptic inputs suggesting that the activity of these cells can be modulated through changes in afferent GABA release. Although the origin of these inputs in not fully understood, changes in synaptic activity onto POMC cells regulates their neuronal excitability and mouse body weight (Vong et al., 2011; Zeltser et al., 2012; Branco et al., 2016; Andermann and Lowell, 2017). Recent studies have also demonstrated that calcium activity in POMC cells is altered immediately upon sensory detection of food (Chen et al., 2015; Mandelblat-Cerf et al., 2015), further suggesting that synaptic transmission onto POMC cells acts to regulate the activity of these cells well before peripheral signals are able to reach the brain. POMC cells receive GABAergic inputs from orexigenic hypothalamic agouti-related protein (AgRP) neurons (Atasoy et al., 2012), as well as from numerous other brain regions (Wang et al., 2015). However, our recent report (Rau and Hentges, 2017) and the work of others (Tong et al., 2008; Vong et al., 2011) indicate that AgRP neurons are not a primary source of GABA onto POMC neurons, nor the source of fasting-induced increases in GABA release onto POMC cells (Rau and Hentges, 2017), indicating that a yet to be identified brain region plays a critical role in synaptic inhibition of POMC cells during caloric deficits.
The dorsomedial hypothalamus (DMH) is a potential source of the fasting-regulated GABA inputs to POMC neurons based on the fact that the DMH contains primarily GABAergic neurons (McIntire et al., 1997; Vong et al., 2011) that project to the ARC (Wang et al., 2015). Lesions to the DMH result in decreased food consumption (Bernardis, 1970; Bellinger and Bernardis, 2002), whereas optogenetic stimulation of DMH neurons promotes food intake (Otgon-Uul et al., 2016), indicating a hypophagic role for the DMH and suggesting that GABAergic cells in the DMH may promote food intake through direct inhibition of anorexigenic POMC cells. However, the extent to which energy-state affects this circuit is unknown.
In this study we assess how energy state in male and female mice affects DMH-to-POMC cell GABA release. Using brain region-specific deletion of the vesicular GABA transporter (VGAT), or expression of channelrhodopsin (ChR2), followed by ex vivo electrophysiology, the present results indicate that neurons in the DMH provide a significant portion of spontaneous GABAergic inhibition onto POMC cells and provide evidence that fasting-induced increases in GABAergic inhibition onto POMC cells is driven by increased GABA release from DMH cells. Additionally, calcium imaging in brain slices revealed that loss of GABA release from the DMH results in increased calcium activity in POMC cells. These studies provide a better understanding of the interaction between two hypothalamic brain regions during changes in energy state and support the hypothesis that synaptic amino acid transmission plays a critical role in modulating satiety circuits.
Materials and Methods
Animals.
All experiments adhered to the Guide for the Care and Use of Laboratory Animals set forth by the National Institutes of Health and were approved by the Colorado State University Animal Care and Use Committee. The following mice were procured from Jackson Laboratories: POMC enhanced green fluorescent protein (eGFP), stock #009593; Cre-dependent ChR2, stock #024109; Slc32a1flox/flox, stock #012987; PomcCre/+, stock #005965; ROSA-tom, stock #007914. Neurons expressing the red fluorescent protein DsRed driven by the Pomc gene promoter (Hentges et al., 2009) were originally a gift from Dr. Malcolm Low (University of Michigan). Transgenes and floxed alleles were detected with standard PCR techniques.
Male and female mice were used for all experiments. Animals were housed on a 12 h light/dark schedule and had ad libitum access to tap water. In some experiments, individual mice were assigned to one of three experimental conditions to determine whether energy state affects DMH-to-POMC GABAergic circuitry: (1) ad libitum, mice had continuous and uninterrupted access to standard rodent chow until kill; 2) overnight fast, 18–20 h before kill mice were transferred to a fresh cage containing the same bedding and enrichment as ad libitum mice, but no chow was present; 3) re-fed, mice were fasted overnight for 18–20 h. At ∼08:00 on the day of kill, these mice were given access to ∼10 g of fresh rodent chow for 2 h before kill. Mice consumed 0.99 ± 0.1 g of chow during this period (data not shown).
Stereotaxic surgery for in vivo gene delivery.
Stereotaxic surgery was performed on 6- to 8-week-old mice. Mice were induced into a deep anesthetic plane with isoflurane and were placed into a stereotaxic apparatus (David Kopf Instruments) fitted with a nose cone for delivery of isoflurane for the entirety of the operation. To express the genetically encoded fluorescent calcium indicator GCaMP6f in POMC neurons, 200 nl (1.52E13 GC/ml) of an adeno-associated virus (AAV) encoding a double-floxed inverse orientation of GCaMP6f (AAV9.CAG.Flex.GCaMP6f.WRPE.SV40; Penn Vector Core, University of Pennsylvania School of Medicine, Philadelphia, PA) was delivered bilaterally into the arcuate nucleus (from bregma, A/P: −1.63, M/L: ±0.32, D/V: −6.00) of PomcCre/+ mice. To drive the expression of Cre-recombinase in specific brain regions of either Slc32a1flox/flox mice or Cre-dependent ChR2 mice, 100 nl (4.37E13 GC/ml) of an AAV encoding Cre-recombinase (AAV1.hSYN.Cre.WPRE.hGH; Penn Vector Core) was injected bilaterally into either the dorsomedial hypothalamus (from bregma, A/P: −1.80, M/L: ±0.35, D/V: −5.42), the bed nucleus of the stria terminalis (from bregma, A/P: +0.30, M/L: ±1.10, D/V: −5.00), or the lateral hypothalamus (from bregma, A/P: −1.50, M/L: ±1.00, D/V: −5.42). For all injections, virus was infused over the course of 1 min and the needle was left in place for 150 s following the end of solution delivery. For dual injection surgeries, 200 nl of the AAV encoding GCaMP6f was first injected bilaterally into the arcuate nucleus followed by a 15 min waiting period, 100 nl of the AAV encoding Cre recombinase was then injected into the DMH. Sham surgeries were conducted in an identical manner with AAV1.EF1a.DIO.eYFP (Addgene) replacing AAV-Cre. Electrophysiology and calcium imaging experiments were conducted 2–6 weeks following surgery.
Electrophysiology.
Following an induction of a deep anesthetic plane with isoflurane, mice were decapitated and their brains were removed and placed into ice-cold artificial CSF (aCSF) consisting of the following (in mm): 126 NaCl, 2.5 KCl, 1.2 MgCl2∧6H2O, 2.4 CaCl2∧2H2O, 1.2 NaH2PO4, 11.1 glucose, and 21.4 NaHCO3, and bubbled with 95% O2 and 5% CO2. Sagittal slices containing the ARC were cut at a thickness of 240 μm using a Leica VT1200S vibratome. Brain slices were allowed to recover for at least 1 h at 37°C in aCSF containing the NMDA receptor blocker MK-801 (15 μm). Slices were then transferred to a recording chamber and perfused with oxygenated, 37°C aCSF at a flow rate of ∼2 ml/min. Recording electrodes were pulled with a Narishige PC-10 vertical pipette puller (Narishige International) and had a resistance of 1.6–2.2 MΩ when filled with an internal recording solution containing the following (in mm): 57.5 KCL, 57.5 K-methyl sulfate, 20 NaCl, 1.5 MgCl2, 5 HEPES, 0.1 EGTA, 2 ATP, 0.5 GTP, and 10 phosphocreatine. The pH was adjusted to 7.3. POMC cells were visually identified for recording based on the transgenic expression of eGFP or DsRed. Whole-cell, voltage-clamp recordings were made at a holding potential of −60 mV using an AxoPatch 200B amplifier (Molecular Devices). Electrophysiological data were collected at 10 kHz and filtered at 5 kHz using AxoGraph X software running on a Mac OS X operating system. Recordings were excluded from analysis if the series resistance exceeded 18 MΩ or changed significantly over the course of the experiment.
Spontaneous IPSPs (sIPSCs) were continuously collected in 60 s epochs for 12–15 min and 3–5 epochs were combined for frequency and amplitude analysis. sIPSC events were detected by sliding an event template over the raw data trace and data were then visually inspected to exclude spurious events. Previous work from our laboratory and others has demonstrated that the majority of spontaneous inputs to POMC neurons are insensitive to TTX (Cowley et al., 2001; Pinto et al., 2004; Vong et al., 2011; Pennock and Hentges, 2016), indicating that the sIPCSs recorded herein represent action-potential independent events. Paired-pulse ratio (PPR) experiments were performed by light activating ChR2-expressing cells with a 2 ms 470 nm light pulse with an interstimulus interval of 100 ms through the use of a 470 nm LED (Thorlabs) driven by an LEDD1B driver (Thorlabs) triggered through the TTL output on a ITC-18 computer interface board (HEKA Instruments). PPR was assessed every 20 s for 10 min and 10–20 consecutive sweeps were averaged for data analysis. The stimulus intensity was reduced at the beginning of the experiment so that the first IPSC was ∼50% of the maximal amplitude. Established protocols were used to estimate the readily releasable pool (RRP) (Schneggenburger et al., 2002; Gioia et al., 2016). Trains of 40 2 ms light pulses were applied to ChR2-expressing DMH terminals at 10 Hz every 40 s for 10–15 min. Traces were averaged and a cumulative amplitude plot of the averaged IPSCs was compiled. A linear slope was fit to the steady-state level achieved during the final 14 points on the plot. The y-intercept of the linear slope was used as the RRP. The recycling pool was estimated by subtracting the cumulative amplitude of all 40 IPSCs from the RRP.
Evoked and spontaneous IPSCs were pharmacologically isolated with the addition of 10 μm 6,7-dinitroquinoxaline (DNQX) into the recording solution. IPSCs were confirmed to be GABAA mediated by bath perfusion of (-)-bicuculline methiodide (BIC; 10 μm).
GCaMP6f imaging.
Brain slices were prepared and treated in an identical manner as with electrophysiology experiments. Once in the recording chamber, GCaMP6f was excited with 470 nm light generated by an LED (Thorlabs) through a 40× water-immersion objective (LUMPlanFL, Olympus). Images were captured using CellSens Dimension software (Olympus) and were acquired at ∼10 Hz with a 50 ms exposure time using an electron-multiplying charge-coupled device (Evolve 512 Delta, Photometrics). A value of 1 was assigned to the median fluorescence intensity over a 5 min baseline period that immediately preceded picrotoxin application (F0) to normalize the raw fluorescence intensity of individual POMC neurons. Normalized data were then imported to AxoGraph X software where the average fluorescent intensity over the final 5 min of a 10 min drug application was used to calculate the average change in fluorescence, relative to the normalized baseline fluorescence (ΔF/F0). Calcium events were counted over a 5 min baseline period immediately preceding picrotoxin application. Events were detected by sliding an event template over the raw data trace in AxoGraph. Traces were then visually inspected. For all calcium imaging experiments, DNQX (10 μm) was included in the recording solution for the entirety of the data collection period to exclude potential effects arising from disinhibition of glutamatergic afferents.
Confocal imaging.
After electrophysiological or GCaMP6f recordings, slices were transferred to a multi-well plate and stored overnight in potassium PBS (KPBS) containing 4% paraformaldehyde. The next day, slices were washed in KPBS 3 times for 20 min each and mounted on slides for subsequent imaging and confirmation of injection location and viral spread. Images were acquired on either a Zeiss 800 or 880 confocal microscope.
Drugs.
BIC was purchased from Tocris Bioscience, MK-801, picrotoxin and DNQX were purchased from Sigma-Aldrich. Drugs were prepared in distilled water (BIC) or DMSO (MK-801, picrotoxin, and DNQX) as 1000 or 10,000× concentrates and diluted in aCSF to achieve desired concentration immediately before use.
Experimental design and statistical analysis.
Data were compared using one-way and two-way ANOVAs or paired and unpaired Student's t tests as indicated. Tukey's multiple-comparison tests were used to make comparisons between individual groups following one-way ANOVAs. Sidak's multiple-comparison tests were used following two-way ANOVAs. Normalized GCaMP6f fluorescent intensity analyzed by using a one-way repeated-measures ANOVA following a Geisser–Greenhouse correction. Individual points on each figure represent a recording from a single cell. For electrophysiological experiments, no more than two recordings were made from a single slice, and no more than five recordings were made from a single animal. Both male and female mice were used for all experiments and approximately equal numbers of mice from both sexes were used in each experiment. Data were initially analyzed for sex differences. As no differences were detected, data from both sexes were pooled for final analysis and presentation. All data were analyzed using GraphPad Prism v6. Data are presented as mean ± SEM, and differences were considered significant where p < 0.05.
Results
The activity of POMC cells is regulated by spontaneous GABAergic inhibition
POMC cells in the ARC receive a high-frequency of spontaneous, action potential independent, GABAA receptor-mediated events (Pennock and Hentges, 2011; Vong et al., 2011; Zeltser et al., 2012). Despite extensive evidence indicating that GABAergic activity in hypothalamic circuits affects metabolic activities (Wu et al., 2008; Jennings et al., 2013; Krashes et al., 2013; Garfield et al., 2016; Suyama and Yada, 2018), the extent to which this inhibition regulates POMC neuron activity is unknown. Thus, we designed an initial experiment to determine whether pharmacological blockade of GABAA receptors alters calcium activity in POMC neurons. Calcium imaging in brain slices was chosen as a measure of POMC neuron activity as this technique allows for estimation of cellular activity without disruption of the intracellular milieu inherent with traditional whole-cell patch-clamp techniques. Transgenic mice expressing Cre-recombinase under control of the Pomc gene promoter (PomcCre/+) received bilateral injections into ARC of an AAV encoding Cre-dependent sequence of the genetically encoded calcium indicator GCaMP6f resulting in robust expression of GCaMP6f in POMC cells (Fig. 1A). Bath application of the GABAA receptor antagonist picrotoxin (100 μm) significantly increased GCaMP6f fluorescence in POMC cells (n = 22 cells; F(1.85, 38.33) = 30.0, p < 0.0001; one-way repeated-measures ANOVA; Fig. 1B,C) indicating that spontaneous release of GABA onto POMC neurons inhibits the activity of these cells. Experiments were conducted in the presence of the AMPA receptor antagonist, DNQX (10 μm). Thus, increased activity of POMC cells following GABA receptor antagonism was not because of disinhibition of glutamatergic afferents.
Figure 1.
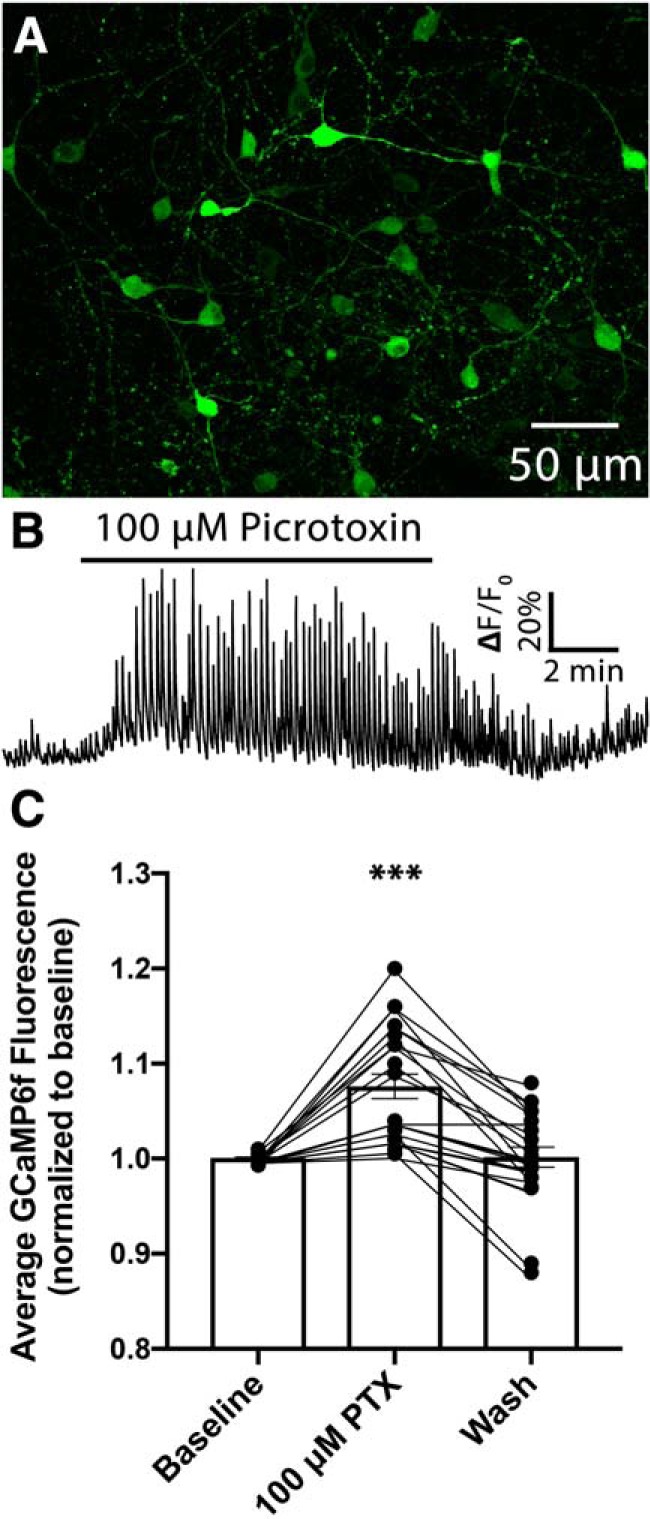
Antagonism of GABAA receptors on POMC neurons increases GCaMP6f fluorescence in these cells. A, Confocal micrograph demonstrating the expression of Cre-recombinase-dependent GCaMP6f in the arcuate nucleus of a PomcCre/+ mouse. B, Representative GCaMP6f fluorescence from a POMC cell in the mouse arcuate nucleus demonstrating the change in calcium activity when GABAA receptors are blocked by the addition of the GABAA receptor antagonist picrotoxin (100 μm) to the recording chamber. For each trial, fluorescence was normalized by assigning a value of 1.0 to the median value of data points within a 5 min window immediately preceding the drug application. C, Quantification of change in normalized fluorescence that occurs following bath application of picrotoxin (***p < 0.0001, one-way repeated-measures ANOVA). Data are presented as mean ± SEM.
GABAA receptors localize to distinct compartments on target neurons, mediating different forms of inhibition based on postsynaptic location (Farrant and Nusser, 2005). Presynaptic release of GABA results in mm concentrations of GABA in the synaptic cleft (Mody et al., 1994), which binds to GABAA receptors located in the adjacent postsynaptic target. This point-to-point communication allows for rapid modulation of the target neuron and is critical for information transfer in the brain. Although GABA is rapidly cleared from the synapse, ambient concentrations of GABA can activate extrasynaptic GABAA receptors (Belelli et al., 2009), resulting in tonic chloride conductance through these channels (Kaneda et al., 1995), and sustained hyperpolarization of the target cell. To distinguish which type of inhibition is present on POMC neurons we made voltage-clamp recordings from POMC cells at a holding potential of −60 mV. Bath application of the GABAA receptor antagonist picrotoxin (100 μm) did not significantly affect the amount of current needed to maintain the −60 mV holding potential (n = 10 cells; baseline: 5.61 ± 6.0 pA; picrotoxin: 7.26 ± 5.6 pA; t(9) = 0.63, p = 0.54; paired t test; Fig. 2A,B) indicating that the increased excitability observed in POMC neurons when GABAA receptors are antagonized (Fig. 1B,C) is because of blockade of postsynaptically located GABAA receptors.
Figure 2.

Increased calcium activity in POMC cells following GABAA receptor antagonism is not because of changes in membrane potential. A, Representative voltage-clamp recording from a POMC neuron demonstrating the effect of bath application of 100 μm picrotoxin. B, Summary data showing that bath application of 100 μm picrotoxin does not change the amount of current needed to maintain a holding potential of −60 mV. (p = 0.544; paired t test). Data are presented as mean ± SEM.
Dorsomedial hypothalamic neurons contribute a significant portion of spontaneous GABAergic inhibition onto POMC cells
Considering the importance of spontaneous GABAergic inhibition onto POMC neurons in regulating the activity of these cells, we went on to determine whether a specific brain region contributes the majority of these GABA inputs. Although previous work has demonstrated that AgRP neurons expressing ChR2 can release GABA onto POMC neurons when light-stimulated (Atasoy et al., 2012), our recent study indicated that AgRP cells are not a significant source of sIPSCs onto POMC cells (Rau and Hentges, 2017). Based on the predominance of GABAergic cells (McIntire et al., 1997; Vong et al., 2011), connection to POMC neurons (Wang et al., 2015; Garfield et al., 2016), and an established role in modulating food intake (Gooley et al., 2006; Yang et al., 2009; Otgon-Uul et al., 2016), we hypothesized that the DMH represents a viable candidate for GABAergic inhibition of POMC cells. To test this hypothesis, we used a Cre/lox strategy where mice homozygous for loxP sites flanking exon 2 of the Slc32a1 gene that encodes the vesicular GABA transporter (VGAT; Slc32a1flox/flox) were injected with an AAV encoding Cre-recombinase (AAV-Cre). These mice also expressed a floxed STOP sequence upstream of a sequence encoding the red fluorescent protein Td-tomato (referred to here as ROSA-tom). Therefore, brain regions injected with AAV-Cre no longer transcribe functional Slc32a1, thus no longer produce functional VGAT (VGAT KO). However, the injected brain regions now express Td-tomato, allowing for confirmation of injection site. Initial experiments targeted AAV-Cre or a sham virus to distinct brain regions (Fig. 3A). In voltage-clamp recordings from POMC neurons from these mice, the frequency of sIPSCs was significantly reduced only when VGAT was deleted from the DMH (F(3,61) = 4.89, p = 0.004; one-way ANOVA; Fig. 3B,C). Post hoc multiple comparisons revealed that the frequency of sIPSCs onto POMC cells was reduced following deletion of VGAT in the DMH relative to sham injections into the DMH (Sham: n = 17 cells; 8.56 ± 1.2 Hz; DMH: n = 19 cells; 4.06 ± 0.7 Hz; p = 0.025), or AAV-Cre injections into the BNST (n = 13 cells; 9.51 ± 1.5 Hz; p = 0.009), or LH (n = 16 cells; 8.60 ± 1.3; p = 0.027; Tukey's multiple comparisons). Together, it appears that the DMH provides a significant source of spontaneous GABAergic input to POMC neurons. The amplitudes of sIPSCs were unaffected by deletion of GABA release from the brain regions tested (F(3,61) = 2.75, p = 0.051; one-way ANOVA; Fig. 3B,D).
Figure 3.

The frequency of sIPSCs onto POMC cells is significantly reduced when the VGAT is deleted in the DMH. A, Confocal micrographs showing the injection sites of an AAV-Cre. Using the ROSA-tom mouse that expresses the red fluorophore, Td-tomato, in the presence of Cre recombinase, AAV-Cre was targeted to the DMH (left), the bed nucleus of the stria terminalis [BNST; middle, or the lateral hypothalamus (LH), right]. In all images, the far-red channel (gray background) was overexposed to visualize anatomic structures and confirm that injections were localized to the appropriate brain region. To produce mice lacking GABA release from specific brain regions, AAV-Cre was injected into these same brain regions of transgenic mice with both alleles encoding VGAT floxed (Slc32a1flox/flox), followed by voltage-clamp recordings from POMC cells. B, Representative voltage-clamp recordings from POMC neurons following a injection of an AAV encoding enhanced yellow fluorescent protein into the DMH (SHAM; top left), or following injection of AAV-Cre into the DMH (bottom left), BNST (top right), or LH (bottom right) of Slc32a1flox/flox mice. C, Summary data (mean ± SEM) showing the frequency of sIPSCs recorded from POMC cells flowing brain region-specific deletion of GABA release. (**p = 0.004, one-way ANOVA). D, Summary data (mean ± SEM) indicating the effect of brain region-specific VGAT deletion on sIPSC amplitude recorded from POMC cells (p = 0.51, one-way ANOVA).
Deletion of VGAT in the DMH reduces inhibition onto POMC cells, resulting in increased calcium activity in these neurons
To determine whether loss of GABA release from the DMH affects POMC neuronal activity, particularly in response to GABAA receptor antagonism, slice calcium imaging of POMC neurons was used. To accomplish this, mice expressing both PomcCre/+ and Slc32a1flox/flox were used. Importantly, POMC cells do not express Slc32a1 (Jarvie and Hentges, 2012) or VGAT (Vong et al., 2011) suggesting that this transgenic line should not intrinsically affect POMC cells. When these mice were 7–8 weeks of age an AAV expressing GCaMP6f was injected into the ARC, driving GCaMP6f expression in POMC cells. Subsequently, AAV-Cre was injected into the DMH to delete Slc32a1, and thus VGAT expression selectively within this brain region. Following a 2–4 week recovery period, sagittal slices were cut from these mice and calcium imaging in brain slices was performed. In previous experiments where VGAT was intact in the DMH, bath application of picrotoxin increased the activity of POMC cells (Fig. 1B,C). Here, GABAA receptor antagonism again resulted in an increase in POMC calcium activity (n = 17 cells; F(1.989,31.82) = 23.1; p < 0.001; one-way repeated-measures ANOVA; Fig. 4A,B). However, the magnitude to which picrotoxin increased POMC activity was significantly reduced relative to mice where DMH GABA release was intact [DMH VGAT wild-type (WT): n = 22 cells; 7.53 ± 1.3%; DMH VGAT KO: n = 17 cells; 2.28 ± 0.5%; t(37) = 3.46; p = 0.0014; unpaired t test; Fig. 4C]. Next, we measured the number of calcium transients in POMC neurons to determine whether loss of GABA inputs from the DMH affects POMC neuronal activity. Deletion of VGAT in the DMH resulted in a significant increase in POMC calcium transients compared with mice with intact GABA release from DMH terminals (DMH VGAT WT: n = 22 cells; 6.99 ± 0.8; DMH VGAT KO: n = 17 cells; 10.98 ± 0.9; t(37) = 3.33; p = 0.002; unpaired t test; Fig. 4D) indicating that DMH GABA neurons provide an important source of inhibition onto POMC cells.
Figure 4.

Deletion of DMH GABA release increases the excitability of POMC neurons through loss of GABAergic inputs. A, Representative calcium imaging traces showing the effect of 100 μm picrotoxin on GCaMP6f fluorescence. B, Quantification of average change in normalized fluorescence that occurs following bath application of picrotoxin (***p < 0.001, one-way repeated-measures ANOVA). C, Summary data comparing the percentage change in baseline normalized GCaMP6f fluorescence in mice with intact DMH VGAT (DMH VGAT WT; Fig. 1B) to mice with deleted VGAT in the DMH (DMH VGAT KO). Deletion of VGAT in the DMH significantly reduced the increase in normalized fluorescence relative to DMH VGAT WT mice (**p = 0.014; unpaired t test) D, Summary data showing the increase in calcium events per minute recorded from POMC neurons in mice with deleted VGAT in the DMH (**p = 0.002; unpaired t test). Data are presented as mean ± SEM.
Fasting-induced increase in the frequency of sIPSCs onto POMC cells is blocked when VGAT is deleted from the DMH
Consistent with their anorexigenic nature, caloric deficit or fasting is associated with decreased activity of POMC cells (Mizuno et al., 1998; Wu et al., 2014). Although changes in hormone levels contribute to this phenomenon (Belgardt et al., 2009), fasting also increases spontaneous release of GABA onto POMC cells (Vong et al., 2011; Dicken et al., 2015), raising the possibility that fasting-induced increases in GABAergic inhibition onto POMC cells could play an important role in the modulation of POMC activity during caloric deficit. Indeed, sensory detection of food results in immediate enhancements of POMC cellular activity suggesting that synaptic transmission does play a key role in modulating POMC cells (Chen et al., 2015). To test the hypothesis that DMH GABA neurons contribute to fasting-induced increases in spontaneous GABAergic inhibition of POMC cells, DMH GABA release was again deleted by injecting AAV-Cre into the DMH of Slc32a1flox/flox mice, resulting in DMH VGAT KO mice. Following a 3–6 week recovery period, mice were fasted overnight and voltage-clamp recordings were made from POMC cells following kill in the morning. Further validating our findings from Figure 3, there was a significant effect of DMH VGAT deletion on the frequency of sIPSCs onto POMC cells (F(1,99) = 26.71, p < 0.0001, two-way ANOVA; Fig. 5A,C). Consistent with previous results (Vong et al., 2011; Rau and Hentges, 2017), fasting increased sIPSC frequency onto POMC neurons in recordings from WT mice (ad libitum, WT: n = 18 cells, 7.51 ± 0.9 Hz; O/N fast WT: n = 32 cells, 10.81 ± 1.0 Hz; p = 0.034; Tukey's multiple comparisons; Fig. 5A,C). However, this increase was absent in DMH VGAT KO mice (ad libitum, DMH VGAT KO: n = 19 cells, 4.06 ± 0.7 Hz; O/N fast DMH VGAT KO: n = 11 cells, 3.64 ± 0.6 Hz; p = 0.965; Tukey's multiple comparisons; Fig. 5B,C) indicating that DMH GABA neurons play a critical role in synaptic inhibition of POMC cells during fasting. Neither genotype (F(1,99) = 0.079, p = 0.78; two-way ANOVA; Fig. 5D), nor energy state (F(2,99) = 0.312, p = 0.73; two-way ANOVA; Fig. 5D) affected the amplitude of sIPSCs.
Figure 5.

The increase in the frequency of sIPSCs onto POMC cells following an overnight fast is blocked when VGAT is deleted in the DMH. A, Representative voltage-clamp recordings from POMC neurons from WT mice demonstrating the frequency of sIPSCs when mice have been fed ad libitum (top), fasted overnight (middle), or re-fed for 2 h following an overnight fast (bottom). B, Representative voltage-clamp recordings from POMC neurons from mice with deleted GABA release from the DMH demonstrating the frequency of sIPSCs when mice have been fed ad libitum (top), fasted overnight (middle), or re-fed for 2 h following an overnight fast (bottom). C, Summary data (mean ± SEM) showing the frequency of sIPSCs recorded from POMC cells during varying energy states in both WT and DMH VGAT KO mice. Deletion of VGAT in the DMH had a significant effect on sIPSC frequency (F(1,99) = 26.71, p < 0.0001, two-way ANOVA). Fasting resulted in a significant increase in sIPSC frequency in WT mice, relative to ad libitum fed mice (*p = 0.034, Tukey's multiple comparison). This increase was absent following deletion of VGAT in the DMH (p = 0.965, Tukey's multiple comparison). Data reported for ad libitum, DMH VGAT KO condition is replicate data from DMH VGAT KO in Figure 3C. D, Summary data (mean ± SEM) showing that the amplitude of sIPSCs recorded from POMC was not affected by manipulation of energy state (F(2,99) = 0.435, p = 0.648, two-way ANOVA), or deletion of VGAT in the DMH (F(1,99) = 0.154, p = 0.696, two-way ANOVA).
Neither the input resistance (F(1,99) = 1.196; p = 0.28, two-way ANOVA) nor the membrane potential (F(1,99) = 0.282; p = 0.59, two-way ANOVA) of POMC cells was affected by knock-out of VGAT in the DMH. Further, manipulation of energy state did not affect input resistance (F(2,99) = 0.369; p = 0.69, two-way ANOVA) or membrane potential (F(2,99) = 0.016; p = 0.98, two-way ANOVA) of POMC cells (Table 1).
Table 1.
POMC neuron recording parameters are not affected by energy state or deletion of VGAT in the DMH
| DMH VGAT WT |
DMH VGAT KO |
|||||
|---|---|---|---|---|---|---|
| Ad libitum | O/N Fast | Re-fed | Ad libitum | O/N Fast | Re-fed | |
| Input resistance, MΩ | 809 ± 52 | 797 ± 48 | 758 ± 33 | 735 ± 46 | 776 ± 59 | 728 ± 48 |
| Membrane potential, mV | −59 ± 2.3 | −62 ± 2.8 | −63 ± 2.5 | −60 ± 3.4 | −59 ± 5.6 | −58 ± 2.9 |
The input resistance measured from POMC cells was not altered following deletion of VGAT in the DMH (F(1,99) = 1.196, p = 0.28; two-way ANOVA) or by challenges to the animal's energy state (F(2,99) = 0.369, p = 0.69; two-way ANOVA). Deletion of VGAT in the DMH (F(1,99) = 0.282, p = 0.59; two-way ANOVA), or energy state challenges (F(2,99) = 0.016, p = 0.98; two-way ANOVA) did not affect the membrane potential of POMC cells. Data are presented as mean ± SEM.
Fasting increases GABA release from DMH neurons onto POMC cells
To determine whether fasting increases the strength of DMH-to-POMC GABA synapses, optogenetic activation of neurons located in the DMH was examined in mice following exposure to various energy states. To drive expression of ChR2 specifically in the DMH, AAV-Cre was stereotaxically injected into the DMH of mice expressing Cre-dependent ChR2 fused to eYFP (Fig. 6A). Light stimulation of DMH terminals reliably produced GABAA receptor-mediated IPSCs in POMC cells (Fig. 7B). The strength of functional coupling between DMH GABA neurons and POMC cells was assessed by a change in the PPR in recordings from POMC cells while light-stimulating DMH terminals during various energy states. Consistent with altered release probability, energy state had a significant effect on PPR (F(2,31) = 3.970, p = 0.029; one-way ANOVA; Fig. 6C,D). Indicating an increase in GABA release probability, an overnight fast significantly decreased the PPR (ad libitum: n = 11 cells, 0.85 ± 0.1 Hz; O/N Fast: n = 13 cells, 0.56 ± 0.06 Hz; p = 0.032; Tukey's multiple comparisons; Fig. 6C,D). In previous experiments conducted on WT mice, 2 h of uninterrupted access to food following an overnight fast returned the frequency of sIPSCs to a value similar to that of mice fed ad libitum (ad libitum: n = 18 cells, 7.51 ± 0.9 Hz; Re-fed: n = 13 cells, 7.99 ± 1.6 Hz; p = 0.95; Tukey's multiple comparison; Fig. 5A,C). To determine whether the functional connectivity of DMH-to-POMC neurons also returns to ad libitum values, the PPR of GABA-mediated currents at DMH-to-POMC synapses was assessed in re-fed mice. After 2 h of refeeding, the release probability of DMH GABA onto POMC cells returned to a value similar to that of mice fed ad libitum (ad libitum: n = 11 cells, 0.85 ± 0.1; Re-fed: n = 10 cells, 0.79 ± 0.05; p = 0.86; Tukey's multiple comparisons; Fig. 6C,D) indicating that this circuit is rapidly modulated by the animal's energy state.
Figure 6.

Fasting increases the release probability of GABA from DMH terminals onto POMC neurons. A, Confocal micrograph of a sagittal hypothalamic slice demonstrating the expression of ChR2eYFP in the dorsomedial hypothalamus and transgenically expressed Td-tomato in POMC neurons in the arcuate nucleus. B, Individual IPSCs (gray) and an average of these events (black) recorded from a POMC cell while light stimulates ChR2-expressing DMH terminals in the presence of DNQX (left). IPSCs were confirmed to be GABAA receptor-mediated by the addition of BIC to the recording solution (right). C, Representative voltage-clamp recordings demonstrating the PPR of light-evoked IPSCs recorded from POMC neurons. ChR2-expressing DMH terminals were light stimulated, as indicated by the blue dash above the trace, at an interstimulus interval of 100 ms. Mice were fed ad libitum (top), fasted overnight (middle), or re-fed for 2 h following an overnight fast (bottom). Representative traces are the average of 10–20 consecutive traces. D, Summary data showing that an overnight fast increases the release probability of GABA from DMH neurons onto POMC cells and that a 2 h re-feed is sufficient to return release probability to that of animals that had ad libitum access to standard rodent chow (F(2,31) = 3.970, p = 0.029, one-way ANOVA; *p = 0.032, Tukey's multiple comparison; NS = not significant, p = 0.865, Tukey's multiple comparison).
Figure 7.

Fasting increases the RRP and recycling pool of GABA vesicles in DMH neuron terminals. A, Average of 10 consecutive traces recorded from POMC neurons from mice fed ad libitum (top) or fasted overnight (bottom). Light stimuli (40 stimulations at 10 Hz; indicated by blue dashes above trace) were applied to DMH terminals expressing ChR2 while recording from POMC cells in voltage-clamp mode. B, Cumulative amplitude plot of a 10 Hz train recorded from DMH-to-POMC synapses from mice fed ad libitum or following an overnight fast. C, Summary data demonstrating that fasting increases the RRP of GABA at DMH-to-POMC synapses (*p = 0.04, unpaired t test). The RRP was estimated by calculating the y-intercept of a linear regression fit to the final 14 points of an individual recording (dashed gray lines). D, Summary data demonstrating that fasting increases the recycling pool of GABA at DMH-to-POMC synapses (**p = 0.008, unpaired t test). The recycling pool was estimated by subtracting the RRP from the cumulative amplitude of all events.
As an additional assessment of DMH-to-POMC synaptic strength, the RRP of GABA was examined in slices from mice fed ad libitum or following an overnight fast. To determine the RRP, 40 light-stimuli were applied at 10 Hz to ChR2-expressing DMH terminals while recording from POMC cells (Fig. 8A). The RRP can be estimated by constructing a cumulative amplitude plot of the IPSC amplitudes resulting from this stimulation protocol and extrapolating the y-intercept from a linear fit to the steady-state level achieved during the final 14 points of stimulation (Schneggenburger et al., 2002). An overnight fast increased the RRP of GABA at DMH terminals relative to mice fed ad libitum (ad libitum: n = 12 cells, 0.71 ± 0.1 nA; O/N Fast: n = 12 cells, 1.25 ± 0.2 nA; t(22) = 2.12, p = 0.045, unpaired t test; Fig. 7B,C). The recycling pool of vesicles can also be estimated from cumulative amplitude plots by subtracting the RRP size from the cumulative amplitude total. Using this strategy, we found that an overnight fast increased the recycling pool of GABA vesicles at DMH-to-POMC synapses (ad libitum: n = 12 cells, 2.97 ± 0.4 nA; O/N Fast: n = 12 cells, 5.23 ± 0.7 nA; t(22) = 2.89, p = 0.008, unpaired t test; Fig. 7D). Together, the results indicate that fasting increases both spontaneous and evoked GABA release from DMH neurons onto POMC neurons and does so, at least in part, by increasing the vesicular pool of GABA in DMH-originating terminals.
Figure 8.

Deletion of VGAT in the DMH transiently reduces body weight. A, Summary plot showing normalized body weight in mice following virally-mediated deletion of VGAT in the DMH or following a sham injection into the DMH (Sham: n = 10; DMH VGAT KO: n = 8; *p = 0.002; Sidak's multiple comparison).
Deletion of DMH GABA release results in a transient reduction in body weight
Our data clearly indicate that GABAergic cells in the DMH contribute a significant portion of GABA onto POMC cells, and that this release is regulated by energy state and affects the activity of POMC neurons. To determine whether loss of DMH GABA release affects metabolic endpoints we injected the DMH of Slc32a1flox/flox mice with either AAV-Cre or a sham virus encoding eYFP. Although there was no overall effect of DMH VGAT deletion on mouse body weight over the 6 weeks of testing (Sham: n = 10 mice; DMH VGAT KO: n = 8 mice; F(1,16) = 0.23, p = 0.64; two-way ANOVA; Fig. 8), there was a significant reduction during the first week (p = 0.002; Sidak's multiple-comparisons test) indicating that GABA release from the DMH is orexigenic but also suggesting that a compensatory adaptation occurs to prevent starvation.
Discussion
The DMH has an established role as an orexigenic nucleus (Bernardis, 1970), whereas POMC neurons in the arcuate nucleus drive anorexigenic behaviors and metabolic activity (Millington, 2007). Given the GABAergic nature of cells within the DMH and the high frequency of spontaneous GABA inputs to POMC cells, we hypothesized that the DMH drives orexigenic activity in part through inhibition of POMC cells. To address this hypothesis we used transgenic mice, brain region-specific deletion of GABA release or expression of ChR2 or GCaMP6f, followed by ex vivo patch-clamp electrophysiology or calcium imaging in brain slices to determine the functional connectivity of the DMH and POMC cells.
Calcium activity in POMC neurons is regulated by synaptic GABAA receptors
Initial experiments were designed to assess the role of spontaneous GABA inputs onto POMC neurons in regulating the activity of these cells. Following expression of the genetically encoded calcium indicator GCaMP6f in POMC neurons, we recorded the fluorescent intensity of POMC neurons in the absence and presence of the GABAA receptor antagonist picrotoxin. Bath application of picrotoxin dramatically increased the activity of POMC neurons indicating that the activity of POMC cells it highly regulated by afferent GABA release. Further, our results indicate that presynaptic GABA transmission targets postsynaptic GABAA receptors, and does not spill over to activate extrasynaptic GABA receptors. However, small ionic currents can have large effects on neurons, particularly electronically compact cells such as POMC neurons. Thus, our data cannot exclude the possibility that small tonic conductance through extrasynaptic GABA receptors regulates POMC neuronal activity.
Multiple studies have recently described an important role of GABA in modulating metabolic activity (Wu et al., 2009; Atasoy et al., 2012; Garfield et al., 2016; Suyama and Yada, 2018) collectively indicating that hunger and satiety circuits are regulated not just by hormones and peptide neurotransmitters but also by fasting-acting amino acid transmitters released from afferent neurons (Zeltser et al., 2012). However, the extent to which GABA regulates POMC cells was unknown. Considering the increased calcium activity in POMC neurons when GABAA receptors were blocked (Fig. 1B,C), our data provide compelling evidence that changes in GABA release onto POMC neurons could rapidly and dramatically change the overall excitability of these cells. Indeed, loss of GABA release from the DMH resulted in a significant increase in calcium activity within POMC cells (Fig. 4D) suggesting that fluctuations in the activity of inhibitory afferents can facilitate changes in POMC excitability. These data may help explain how the activity of POMC neurons can change immediately following sensory detection of food (Chen et al., 2015; Mandelblat-Cerf et al., 2015).
DMH neurons modulate the calcium activity of POMC cells though GABAergic innervation
Considering the regulatory actions of GABA on POMC calcium activity, determining the afferent location of the neurons releasing this GABA will provide key insights into the neural circuitry underlying metabolic behaviors. Following region-specific deletion of GABA release in the DMH, sIPSC frequency in POMC neurons was reduced by 53% (Fig. 3B,C) indicating that the DMH is a key regulator of POMC cells. In brain slice calcium imaging experiments, we confirmed that DMH cells play an important role in POMC cell calcium activity by showing that the frequency of calcium transients recorded from POMC neurons was increased following deletion of VGAT in the DMH. Although calcium transients detected using GCaMP6f florescence can resolve single action potentials (Chen et al., 2013), the calcium transients described here likely represent bursts of action potentials (Fox and Hentges, 2017). These findings shed new light on upstream regulators of POMC neurons and provide a novel mechanism through which the DMH may produce hypophagic responses. Indeed, stimulation of ChR2-expressing DMH neurons promotes food intake (Jeong et al., 2017) and our results suggest that it is likely that inhibition of POMC neurons contributes to this observation. Further, the discovery that orexigenic AgRP neurons release GABA led many to hypothesize that these neurons directly inhibit POMC cells through the release of GABA (Cone, 2005; Nuzzaci et al., 2015). Indeed, the reduction in AgRP calcium activity and coincident rise in POMC calcium activity following food presentation has been attributed to reduced GABA release from AgRP cells onto POMC cells (Chen et al., 2015). Although AgRP cells can release GABA onto POMC neurons (Atasoy et al., 2012), they do not contribute a significant portion of spontaneously released GABA onto POMC cells, nor do they contribute in increased spontaneous GABA release onto POMC cells during fasting (Rau and Hentges, 2017). Combined with our present data, it appears as though the DMH plays a large role in modulating the excitability of POMC neurons through spontaneous GABA, particularly when an animal transitions from a fasted to a fed state. Considering the importance of obtaining food during caloric deficits, it seems likely that multiple cell types, including AGRP, may coordinate activity to maximally inhibit POMC cells during times of extreme hunger. Indeed, the release probability of GABA from AgRP neurons onto POMC cells is increased during fasting (Dicken et al., 2015). However, deletion of GABA release from AgRP neurons does not block fasting induced increases in sIPSC frequency onto POMC cells (Rau and Hentges, 2017) indicating that other neurons drive this inhibition.
Dorsomedial hypothalamus neurons drive increased inhibition onto POMC cells during fasting
To investigate whether DMH GABA cells contribute to fasting-induced inhibition of POMC cells, we again used DMH-specific deletion of VGAT as well as DMH-specific expression of ChR2. Our results indicate that DMH neurons increase their functional coupling with POMC cells during fasting and are responsible to fasting-induced increases in sIPSC frequency onto POMC cells. These data collectively indicate that neurons in the DMH respond to energy deficits to increase inhibition onto POMC cells, thereby promoting orexigenic behaviors. Importantly, the DMH is a highly heterogeneous nucleus with multiple subnuclei (Zhang et al., 2011; Soga et al., 2014) and the neuronal subtype(s) within the DMH responsible for inhibiting POMC neurons is presently unknown. However, fasting increases the activity of cholinergic cells in the DMH (Groessl et al., 2013) and optogenetic stimulation of DMH cholinergic neurons promotes food intake (Jeong et al., 2017) suggesting that these cells may contribute to fasting-induced increases in GABA onto POMC cells. Interestingly, subpopulations of leptin receptor- or prodynorphin-expressing neurons in the ventral DMH project to and inhibit AgRP neurons, but not POMC cells (Garfield et al., 2016). These data provide evidence that the diversity of cells in the DMH coordinate disparate metabolic behaviors and suggest that further dissection of DMH-to-ARC circuitry is warranted to more fully understand GABAergic modulation of metabolic behaviors within hypothalamic brain regions.
The DMH plays a critical role in multiple circadian behaviors (Chou et al., 2003; Gooley et al., 2006; Mieda et al., 2006) and is innervated by brains regions that drive circadian activity including the suprachiasmatic nucleus and cells in the subventricular zone (Thompson and Swanson, 1998). Mouse food seeking and consumption follows discrete circadian rhythms, consuming the majority of their food during their nocturnal period (Possidente and Birnbaum, 1979). Thus, our data provide a circuit through which changes in DMH activity in response to time of day can alter the excitability of POMC neurons, coordinating feeding times to coincide with the nocturnal period.
Although our data do not provide evidence that loss of GABA release from the DMH affects body weight over protracted periods of time, we did find evidence that DMH VGAT deletion results in a transient reduction in body weight (Fig. 8). In line with previous work (Bernardis, 1970), the immediate loss in body weight following surgery suggests that DMH GABA release is predominately orexigenic, whereas the normalization of body weight in the subsequent weeks indicates that compensatory mechanism may take over to prevent starvation. Indeed, the physiological changes we observed persist weeks past the loss of body weight indicating that compensation is likely. Considering that we did not observe significant changes in sIPSCs amplitude (Fig. 5) or POMC cellular parameters (Table 1), homeostatic plasticity directly within POMC cells seems unlikely, although excitatory inputs to POMC cells was not assessed. Thus, it is possible that afferent glutamatergic inputs to POMC cells could be downregulated in response to loss of GABA inputs to POMC cells to prevent further reductions in body weight. Additionally, POMC cells release a number of peptides involved in feeding behavior (Millington, 2007), and can also release GABA and glutamate (Hentges et al., 2004, 2009). The downstream targets of POMC cells are poorly understood, and volume transmission of neuropeptides can reach multiple brain regions. Therefore, it is currently challenging to assess how changes in GABA release onto POMC cells affects the release of neurotransmitters and neuropeptides from POMC cells and whether release of these products undergoes compensation to prevent starvation following DMH VGAT KO. However, other studies have also observed transient reductions in mouse body weight following conditional genetic manipulations (Sainsbury et al., 2002; Meng et al., 2016) underscoring the vital role of adaptation to maintain homeostatic processes.
In conclusion, GABA neurons in the DMH send dense projections to POMC neurons in the ARC nucleus and are responsible for a significant portion of spontaneous GABAergic inhibition of POMC cells. Additionally, loss of GABA release results in increased calcium activity in POMC cells. The functional coupling of this circuit is enhanced following an overnight fast and is normalized by 2 h of refeeding. These data provide evidence of neuronal circuit from the DMH onto POMC cells that may play a critical role in reducing POMC neuronal activity during caloric deficit. These data also suggest that GABAergic activity onto POMC neurons dramatically regulates calcium activity within these cells, providing a mechanism through which POMC cellular activity can be rapidly modulated in response to food or other homeostatic cues.
Footnotes
This work was supported by National Institute of Health Grants DK078749 and DA032562 (S.T.H.). We thank Connie King for her assistance with animal colony management and genotyping.
The authors declare no competing financial interests.
References
- Andermann ML, Lowell BB (2017) Toward a wiring diagram understanding of appetite control. Neuron 95:757–778. 10.1016/j.neuron.2017.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aponte Y, Atasoy D, Sternson SM (2011) AGRP neurons are sufficient to orchestrate feeding behavior rapidly and without training. Nat Neurosci 14:351–355. 10.1038/nn.2739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atasoy D, Betley JN, Su HH, Sternson SM (2012) Deconstruction of a neural circuit for hunger. Nature 488:172–177. 10.1038/nature11270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belelli D, Harrison NL, Maguire J, Macdonald RL, Walker MC, Cope DW (2009) Extrasynaptic GABAA receptors: form, pharmacology, and function. J Neurosci 29:12757–12763. 10.1523/JNEUROSCI.3340-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgardt BF, Okamura T, Brüning JC (2009) Hormone and glucose signalling in POMC and AgRP neurons. J Physiol 587:5305–5314. 10.1113/jphysiol.2009.179192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellinger LL, Bernardis LL (2002) The dorsomedial hypothalamic nucleus and its role in ingestive behavior and body weight regulation: lessons learned from lesioning studies. Physiol Behav 76:431–442. 10.1016/S0031-9384(02)00756-4 [DOI] [PubMed] [Google Scholar]
- Bernardis LL. (1970) Participation of the dorsomedial hypothalamic nucleus in the “feeding center” and water intake circuitry of the weanling rat. J Neurovisc Relat 31:387–398. 10.1007/BF02312740 [DOI] [PubMed] [Google Scholar]
- Branco T, Tozer A, Magnus CJ, Sugino K, Tanaka S, Lee AK, Wood JN, Sternson SM (2016) Near-perfect synaptic integration by Nav1.7 in hypothalamic neurons regulates body weight. Cell 165:1749–1761. 10.1016/j.cell.2016.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, Looger LL, Svoboda K, Kim DS (2013) Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499:295–300. 10.1038/nature12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Lin YC, Kuo TW, Knight ZA (2015) Sensory detection of food rapidly modulates arcuate feeding circuits. Cell 160:829–841. 10.1016/j.cell.2015.01.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TC, Scammell TE, Gooley JJ, Gaus SE, Saper CB, Lu J (2003) Critical role of dorsomedial hypothalamic nucleus in a wide range of behavioral circadian rhythms. J Neurosci 23:10691–10702. 10.1523/JNEUROSCI.23-33-10691.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cone RD. (2005) Anatomy and regulation of the central melanocortin system. Nat Neurosci 8:571–578. 10.1038/nn1455 [DOI] [PubMed] [Google Scholar]
- Cowley MA, Smart JL, Rubinstein M, Cerdán MG, Diano S, Horvath TL, Cone RD, Low MJ (2001) Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 411:480–484. 10.1038/35078085 [DOI] [PubMed] [Google Scholar]
- Dennison CS, King CM, Dicken MS, Hentges ST (2016) Age-dependent changes in amino acid phenotype and the role of glutamate release from hypothalamic proopiomelanocortin neurons. J Comp Neurol 524:1222–1235. 10.1002/cne.23900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dicken MS, Hughes AR, Hentges ST (2015) Gad1 mRNA as a reliable indicator of altered GABA release from orexigenic neurons in the hypothalamus. Eur J Neurosci 42:2644–2653. 10.1111/ejn.13076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd GT, Michael NJ, Lee-Young RS, Mangiafico SP, Pryor JT, Munder AC, Simonds SE, Brüning JC, Zhang ZY, Cowley MA, Andrikopoulos S, Horvath TL, Spanswick D, Tiganis T (2018) Insulin regulates POMC neuronal plasticity to control glucose metabolism. eLife 7:e38704. 10.7554/eLife.38704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrant M, Nusser Z (2005) Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat Rev Neurosci 6:215–229. 10.1038/nrn1625 [DOI] [PubMed] [Google Scholar]
- Fox PD, Hentges ST (2017) Differential desensitization observed at multiple effectors of somatic mu-opioid receptors underlies sustained agonist-mediated inhibition of proopiomelanocortin neuron activity. J Neurosci 37:8667–8677. 10.1523/JNEUROSCI.1030-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garfield AS, Shah BP, Burgess CR, Li MM, Li C, Steger JS, Madara JC, Campbell JN, Kroeger D, Scammell TE, Tannous BA, Myers MG Jr, Andermann ML, Krashes MJ, Lowell BB (2016) Dynamic GABAergic afferent modulation of AgRP neurons. Nat Neurosci 19:1628–1635. 10.1038/nn.4392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gioia DA, Alexander NJ, McCool BA (2016) Differential expression of Munc13–2 produces unique synaptic phenotypes in the basolateral amygdala of C57BL/6J and DBA/2J mice. J Neurosci 36:10964–10977. 10.1523/JNEUROSCI.1785-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooley JJ, Schomer A, Saper CB (2006) The dorsomedial hypothalamic nucleus is critical for the expression of food-entrainable circadian rhythms. Nat Neurosci 9:398–407. 10.1038/nn1651 [DOI] [PubMed] [Google Scholar]
- Groessl F, Jeong JH, Talmage DA, Role LW, Jo YH (2013) Overnight fasting regulates inhibitory tone to cholinergic neurons of the dorsomedial nucleus of the hypothalamus. PLoS One 8:e60828. 10.1371/journal.pone.0060828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentges ST, Nishiyama M, Overstreet LS, Stenzel-Poore M, Williams JT, Low MJ (2004) GABA release from proopiomelanocortin neurons. J Neurosci 24:1578–1583. 10.1523/JNEUROSCI.3952-03.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentges ST, Otero-Corchon V, Pennock RL, King CM, Low MJ (2009) Proopiomelanocortin expression in both GABA and glutamate neurons. J Neurosci 29:13684–13690. 10.1523/JNEUROSCI.3770-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvie BC, Hentges ST (2012) Expression of GABAergic and glutamatergic phenotypic markers in hypothalamic proopiomelanocortin neurons. J Comp Neurol 520:3863–3876. 10.1002/cne.23127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings JH, Rizzi G, Stamatakis AM, Ung RL, Stuber GD (2013) The inhibitory circuit architecture of the lateral hypothalamus orchestrates feeding. Science 341:1517–1521. 10.1126/science.1241812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong JH, Lee DK, Jo YH (2017) Cholinergic neurons in the dorsomedial hypothalamus regulate food intake. Mol Metab 6:306–312. 10.1016/j.molmet.2017.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda M, Farrant M, Cull-Candy SG (1995) Whole-cell and single-channel currents activated by GABA and glycine in granule cells of the rat cerebellum. J Physiol 485:419–435. 10.1113/jphysiol.1995.sp020739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krashes MJ, Shah BP, Koda S, Lowell BB (2013) Rapid versus delayed stimulation of feeding by the endogenously released AgRP neuron mediators GABA, NPY, and AgRP. Cell Metab 18:588–595. 10.1016/j.cmet.2013.09.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelblat-Cerf Y, Ramesh RN, Burgess CR, Patella P, Yang Z, Lowell BB, Andermann ML (2015) Arcuate hypothalamic AgRP and putative POMC neurons show opposite changes in spiking across multiple timescales. eLife 4:e07122. 10.7554/eLife.07122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntire SL, Reimer RJ, Schuske K, Edwards RH, Jorgensen EM (1997) Identification and characterization of the vesicular GABA transporter. Nature 389:870–876. 10.1038/39908 [DOI] [PubMed] [Google Scholar]
- Meng F, Han Y, Srisai D, Belakhov V, Farias M, Xu Y, Palmiter RD, Baasov T, Wu Q (2016) New inducible genetic method reveals critical roles of GABA in the control of feeding and metabolism. Proc Natl Acad Sci U S A 113:3645–3650. 10.1073/pnas.1602049113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer AJ, Hentges ST, Meshul CK, Low MJ (2013) Unraveling the central proopiomelanocortin neural circuits. Front Neurosci 7:19. 10.3389/fnins.2013.00019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mieda M, Williams SC, Richardson JA, Tanaka K, Yanagisawa M (2006) The dorsomedial hypothalamic nucleus as a putative food-entrainable circadian pacemaker. Proc Natl Acad Sci U S A 103:12150–12155. 10.1073/pnas.0604189103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millington GW. (2007) The role of proopiomelanocortin (POMC) neurones in feeding behaviour. Nutr Metab (Lond) 4:18. 10.1186/1743-7075-4-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno TM, Kleopoulos SP, Bergen HT, Roberts JL, Priest CA, Mobbs CV (1998) Hypothalamic pro-opiomelanocortin mRNA is reduced by fasting and [corrected] in ob/ob and db/db mice, but is stimulated by leptin. Diabetes 47:294–297. 10.2337/diab.47.2.294 [DOI] [PubMed] [Google Scholar]
- Mody I, De Koninck Y, Otis TS, Soltesz I (1994) Bridging the cleft at GABA synapses in the brain. Trends Neurosci 17:517–525. 10.1016/0166-2236(94)90155-4 [DOI] [PubMed] [Google Scholar]
- Nuzzaci D, Laderrière A, Lemoine A, Nédélec E, Pénicaud L, Rigault C, Benani A (2015) Plasticity of the melanocortin system: determinants and possible consequences on food intake. Front Endocrinol 6:143. 10.3389/fendo.2015.00143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otgon-Uul Z, Suyama S, Onodera H, Yada T (2016) Optogenetic activation of leptin- and glucose-regulated GABAergic neurons in dorsomedial hypothalamus promotes food intake via inhibitory synaptic transmission to paraventricular nucleus of hypothalamus. Mol Metab 5:709–715. 10.1016/j.molmet.2016.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parton LE, Ye CP, Coppari R, Enriori PJ, Choi B, Zhang CY, Xu C, Vianna CR, Balthasar N, Lee CE, Elmquist JK, Cowley MA, Lowell BB (2007) Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature 449:228–232. 10.1038/nature06098 [DOI] [PubMed] [Google Scholar]
- Pennock RL, Hentges ST (2011) Differential expression and sensitivity of presynaptic and postsynaptic opioid receptors regulating hypothalamic proopiomelanocortin neurons. J Neurosci 31:281–288. 10.1523/JNEUROSCI.4654-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennock RL, Hentges ST (2016) Desensitization-resistant and -sensitive GPCR-mediated inhibition of GABA release occurs by Ca2+-dependent and -independent mechanisms at a hypothalamic synapse. J Neurophysiol 115:2376–2388. 10.1152/jn.00535.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto S, Roseberry AG, Liu H, Diano S, Shanabrough M, Cai X, Friedman JM, Horvath TL (2004) Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science 304:110–115. 10.1126/science.1089459 [DOI] [PubMed] [Google Scholar]
- Possidente B, Birnbaum S (1979) Circadian rhythms for food and water consumption in the mouse, Mus musculus. Physiol Behav 22:657–660. 10.1016/0031-9384(79)90226-9 [DOI] [PubMed] [Google Scholar]
- Rau AR, Hentges ST (2017) The relevance of AgRP neuron-derived GABA inputs to POMC neurons differs for spontaneous and evoked release. J Neurosci 37:7362–7372. 10.1523/JNEUROSCI.0647-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sainsbury A, Schwarzer C, Couzens M, Fetissov S, Furtinger S, Jenkins A, Cox HM, Sperk G, Hökfelt T, Herzog H (2002) Important role of hypothalamic Y2 receptors in body weight regulation revealed in conditional knockout mice. Proc Natl Acad Sci U S A 99:8938–8943. 10.1073/pnas.132043299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R, Sakaba T, Neher E (2002) Vesicle pools and short-term synaptic depression: lessons from a large synapse. Trends Neurosci 25:206–212. 10.1016/S0166-2236(02)02139-2 [DOI] [PubMed] [Google Scholar]
- Soga T, Kitahashi T, Clarke IJ, Parhar IS (2014) Gonadotropin-inhibitory hormone promoter-driven enhanced green fluorescent protein expression decreases during aging in female rats. Endocrinology 155:1944–1955. 10.1210/en.2013-1786 [DOI] [PubMed] [Google Scholar]
- Suyama S, Yada T (2018) New insight into GABAergic neurons in the hypothalamic feeding regulation. J Physiol Sci 68:717–722. 10.1007/s12576-018-0622-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RH, Swanson LW (1998) Organization of inputs to the dorsomedial nucleus of the hypothalamus: a reexamination with FluoroGold and PHAL in the rat. Brain Res Brain Res Rev 27:89–118. 10.1016/S0165-0173(98)00010-1 [DOI] [PubMed] [Google Scholar]
- Tong Q, Ye CP, Jones JE, Elmquist JK, Lowell BB (2008) Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat Neurosci 11:998–1000. 10.1038/nn.2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vong L, Ye C, Yang Z, Choi B, Chua S Jr, Lowell BB (2011) Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron 71:142–154. 10.1016/j.neuron.2011.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, He X, Zhao Z, Feng Q, Lin R, Sun Y, Ding T, Xu F, Luo M, Zhan C (2015) Whole-brain mapping of the direct inputs and axonal projections of POMC and AgRP neurons. Front Neuroanat 9:40. 10.3389/fnana.2015.00040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KW, Elmquist JK (2012) From neuroanatomy to behavior: central integration of peripheral signals regulating feeding behavior. Nat Neurosci 15:1350–1355. 10.1038/nn.3217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Howell MP, Cowley MA, Palmiter RD (2008) Starvation after AgRP neuron ablation is independent of melanocortin signaling. Proc Natl Acad Sci U S A 105:2687–2692. 10.1073/pnas.0712062105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Boyle MP, Palmiter RD (2009) Loss of GABAergic signaling by AgRP neurons to the parabrachial nucleus leads to starvation. Cell 137:1225–1234. 10.1016/j.cell.2009.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Lemus MB, Stark R, Bayliss JA, Reichenbach A, Lockie SH, Andrews ZB (2014) The temporal pattern of cfos activation in hypothalamic, cortical, and brainstem nuclei in response to fasting and refeeding in male mice. Endocrinology 155:840–853. 10.1210/en.2013-1831 [DOI] [PubMed] [Google Scholar]
- Xu AW, Kaelin CB, Morton GJ, Ogimoto K, Stanhope K, Graham J, Baskin DG, Havel P, Schwartz MW, Barsh GS (2005) Effects of hypothalamic neurodegeneration on energy balance. PLoS Biol 3:e415. 10.1371/journal.pbio.0030415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Scott KA, Hyun J, Tamashiro KL, Tray N, Moran TH, Bi S (2009) Role of dorsomedial hypothalamic neuropeptide Y in modulating food intake and energy balance. J Neurosci 29:179–190. 10.1523/JNEUROSCI.4379-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaswen L, Diehl N, Brennan MB, Hochgeschwender U (1999) Obesity in the mouse model of pro-opiomelanocortin deficiency responds to peripheral melanocortin. Nat Med 5:1066–1070. 10.1038/12506 [DOI] [PubMed] [Google Scholar]
- Zeltser LM, Seeley RJ, Tschöp MH (2012) Synaptic plasticity in neuronal circuits regulating energy balance. Nat Neurosci 15:1336–1342. 10.1038/nn.3219 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Kerman IA, Laque A, Nguyen P, Faouzi M, Louis GW, Jones JC, Rhodes C, Münzberg H (2011) Leptin-receptor-expressing neurons in the dorsomedial hypothalamus and median preoptic area regulate sympathetic brown adipose tissue circuits. J Neurosci 31:1873–1884. 10.1523/JNEUROSCI.3223-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]