

Abstract
T cells play important roles in autoimmune diseases and cancers. Following the cloning of the T cell receptor (TCR), the race was on to map signaling proteins that contributed to T cell activation, downstream of the TCR as well as co-stimulatory molecules such as CD28. We term this “canonical TCR signaling” here.
More recent, it has been appreciated that T cells need to accommodate increased metabolic needs that stem from T cell activation in order to function properly. A central role herein has emerged for mammalian target of rapamycin, mTOR.
In this review we briefly cover canonical TCR signaling to set the stage for discussion on mTOR signaling in T cells, mRNA translation, and metabolic adaptation. We also discuss the role of mTOR for in follicular helper T cells, regulatory T cells, and other T cell subsets. Our lab recently uncovered that “tonic signals” known to pass through proximal TCR signaling components are robustly and selectively transduced to mTOR to promote baseline translation of various mRNA targets. We discuss insights on (tonic) mTOR signaling in the context of T cell function in autoimmune diseases such as Lupus as well as in cancer immunotherapy through CAR-T cell or checkpoint blockade approaches.
Keywords: T cell, signaling, mTOR, tonic, autoimmune, cancer
Introduction
T cells are adaptive immune cells that can become rapidly and robustly activated when their T cell receptor (TCR) recognizes cognate foreign antigen. Upon this interaction, a signalling cascade from the TCR is triggered: the signalling events can be roughly divided into proximal kinases and more distal effector kinase pathways (Figure 1). These components are coupled with the aid of several adapter molecules and second messengers. The process is further tuned by costimulatory molecules like CD28 and built-in brakes like CTLA-4 and PD-1. Ultimately, these signal transduction programs lead to full activation of the T cell and, depending on the cytokine milieu, differentiation of T cells into more specialized effector subsets such as Th1 and Th2.
FIGURE 1. Proximal TCR Signaling, Adapters, and Secondary Messengers.
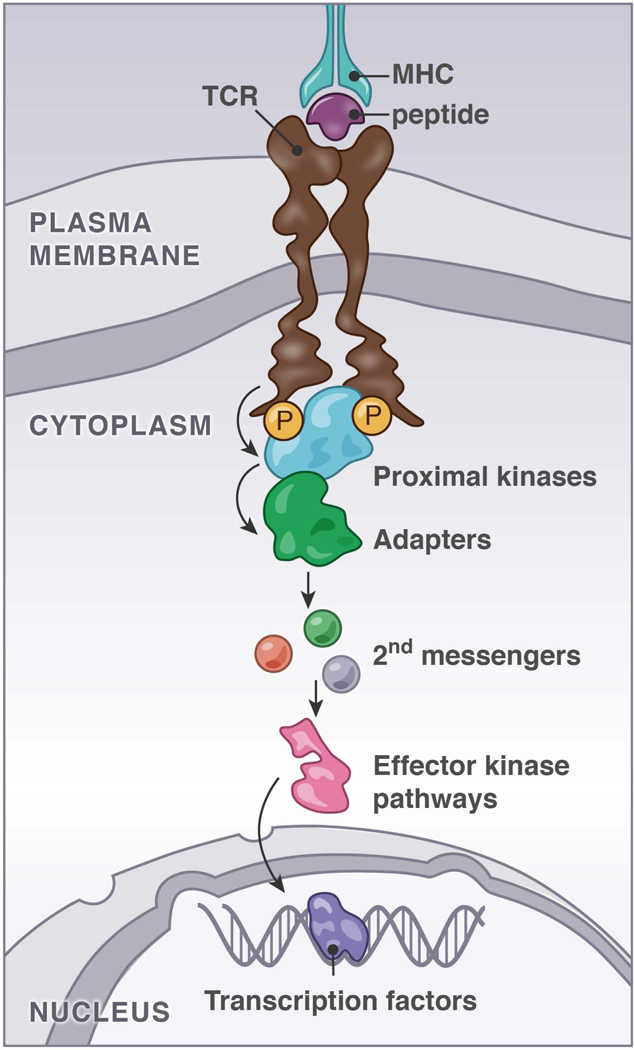
Simplified scheme of the canonical pMHC/TCR signaling pathway viewed as a linear relay system from TCR to transcription factors through proximal kinases, adapters, second messengers, and effector kinase pathways.
Concomitant with T cell activation are changes in gene expression and the synthesis of new proteins, one of which is the cytokine IL-2, a critical cytokine that supports T cell survival. The coordinated activity of three transcription factors, NFAT, NFkB, and AP-1, is required for the production of IL-2 (Figure 2). Additionally, activated, IL-2 secreting T cells are remarkably proliferative, and as such they have a high metabolic demand. Over the past decade, it has become increasingly clear that metabolic reprogramming of the T cell needs to occur to enable the transition from the naïve to the activated state, and these metabolic changes are necessary to support T cell proliferation and effector functions. Furthermore, the metabolic demands of different helper T cell subsets are not identical. The kinase mechanistic/mammalian target of rapamycin (mTOR) plays an important role in the aforementioned processes of helper T cell activation, differentiation, and metabolic reprogramming. At the level of gene expression, the induction and activity of the transcription factor c-Myc appears to be particularly important for T cell metabolic reprogramming.
FIGURE 2. Requirements for induction of T cell activation markers.
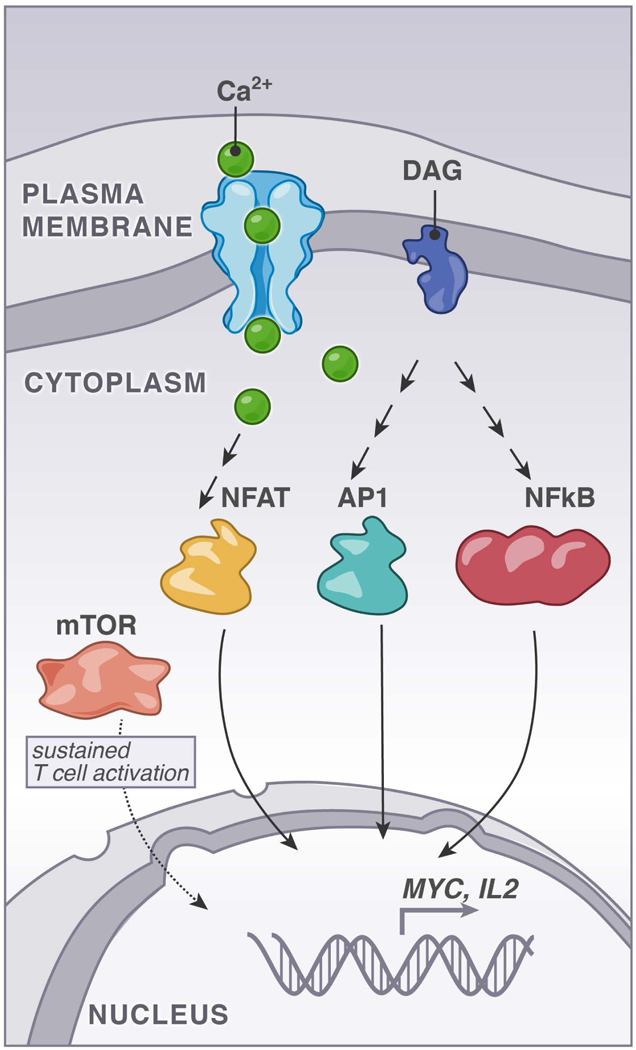
The second messenger molecules calcium and diacylglycerol (DAG) trigger NFAT-, AP1-, and NFκB- transcription factor pathways. At the same time, an activated T cell must induce metabolic changes to enable full and sustained activation and mTOR plays an important role herein.
The culmination of these signalling and transcriptional events is the extensive transformation from a naïve T cell to a proliferative, active cell that drives the immune response. Exactly how T cells respond to triggers from pathogen-infected cells or transformed cancer cells in an exquisitely sensitive manner, yet stay quiescent when encountering normal cells in the body (termed “self”) is still poorly understood. It is clear that to enact the above-mentioned changes, T cells must exist in a “primed” state.
In this review, we aim to review the role of mTOR in transducing and controlling the signals involved in T cell activation, as well as cover the downstream functional consequences of mTOR activity. In the first section of this review we will briefly discuss the well described, canonical TCR signaling cascade, including how the TCR engages proximal kinases, adapter molecules and second messengers to activate the key transcription factors required IL-2 induction. We also refer the reader to additional literature in this first section for a more comprehensive overview of the field. From here we will we discuss the mTOR pathway in more detail and cover how mTOR signals and metabolic changes are essential to accompany the more traditionally studied pathways for T cell activation. We will describe what is known about how proximal TCR signaling events couple to mTOR and will discuss how mTOR integrates signals from the TCR as well as other upstream signal inputs such as nutrient availability to reshape T cell effector functions. We will describe work from our lab in which we show naïve T cells signal to mTOR in the basal state. Rasgrp1, canonically an activator of the Ras pathway, mediates these tonic signals to mTOR. Tonic mTOR signaling impacts helper T cell differentiation and the translation of mRNAs into proteins. Our work points to a role for tonic mTOR signals in priming the translational landscape in naïve T cells for the metabolic changes required upon activation. All this together suggests that mTOR serves to integrate multiple signals in T cells, both in the basal state and upon TCR engagement by foreign cognate antigen. In the basal state, tonic mTOR signals “prime” T cells, and upon TCR stimulation these primed T cells engage mTOR to fully enable their activation, differentiation, and metabolic reprogramming. Lastly, we discuss how perturbations in tonic and inducible mTOR signaling in T cells can lead to disease, focusing on autoimmune disorders and cancer.
Proximal TCR Signaling, Adapters, and Secondary Messengers.
In secondary lymphoid organs, T cells sample their surroundings via their T cell receptor (TCR), making transient interactions with peptide-major histocompatibility complexes (pMHC) presented on the surface of antigen presenting cells (APCs). When pMHC-TCR interactions are of sufficient affinity a TCR signaling pathway is triggered through activation of proximal tyrosine kinases and the phosphorylation and assembly of adaptor complexes, ultimately leading to changes in transcription and translation within the cell. The canonical “TCR signal” that we will briefly describe in the next 4 sections finds its way to the transcription factors nuclear factor of activated T cells (NFAT), nuclear factor kappa B (NFκB), and activator protein 1 (AP-1), which together are required to bind to the promoter of the key T cell survival cytokine IL-2 and induce its transcription (1, 2) (Figure 2). We will briefly describe this canonical TCR signaling pathway to provide the reader with context for our subsequent discussion of the mTOR pathway, tonic signaling, and the cellular functions they control.
Upon TCR ligation, a T cell undergoes vast changes mediated by a diverse array of distinct molecular events. Initiation of TCR signaling involves phosphorylation of the immunoreceptor tyrosine-based activation motifs (ITAMs) of the TCR by the Src kinase Lck. Recruitment of Zeta-associated protein of 70 kDa (Zap70) to the phosphorylated ITAMs sets up the top of the signaling cascade: Activated Zap70 is then able to phosphorylate a number of its effector substrates. For further reading on proximal kinase signaling in T cells we refer you to other literature (3–6) as well as to a review that describes how these proximal TCR signals may relate to kinetic proofreading (7).
One critical substrate of Zap70 is the transmembrane adapter Linker for Activation of T cells (LAT). LAT has nine conserved tyrosines that, when phosphorylated, provide docking sites for other proteins, leading to the assembly of a T cell signaling complex often called the “LAT signalosome.” The LAT signalosome is composed of LAT, the cytosolic adapter SH2-domain containing leukocyte phosphoprotein of 76kDa (SLP-76), growth factor receptor-bound protein 2 (Grb2), and other molecules. The exact identity of the LAT signalosome is unknown and it is not clear if there are different types of LAT signalosomes that are formed under different conditions that the T cell may encounter. Lck binds to LAT and essentially forms a bridge between Zap70 and its substrate, LAT (8). Excellent review articles have discussed the role of LAT and other adapter molecules in T cells (9–11).
One important downstream consequence of forming the LAT signalosome is recruitment and activation of the enzyme PLCγ1 (phospholipase C gamma 1) at tyrosine 136 (murine LAT; Y132 for human LAT). PLCγ1 converts the phospholipid PIP2 (phosphatidylinositol 4,5-bisphosphate) into two key second messenger molecules inositol trisphosphate (IP3) and diacylglycerol (DAG) (12). T cell specific deletion of PLCγ1 leads to autoimmune and inflammatory diseases in the mice. Looking specifically at the T cells, it was demonstrated that thymocyte development was impaired and T cells were unable to perform key functions such as proliferation and IL-2 production (13).
The two second messengers generated by PLCγ1, IP3 and DAG, are important for coupling the above-described proximal TCR signaling molecules to the downstream transcription factors NFAT, NFkB, and AP-1 to trigger T cell activation. Specifically, IP3 triggers calcium (Ca2+) release, which eventually leads to the activation of the transcription factor NFAT. DAG recruits and activates several signaling proteins that play a role in T cell biology: these include the protein kinase C family (PKCs) and the Ras guanine nucleotide releasing protein (RasGRP) family members. We will summarize IP3 and DAG signaling in T cells in the next sections.
Calcium Signals and NFAT Activation
When T cells are activated by pMHC/TCR signals, calcium concentrations rapidly increase 10-fold from 100nM to 1uM, a phenomenon termed a “calcium flux.” Calcium flux can lead to activation of NFAT as well as other molecules (14–16). One manner by which calcium can activate effector proteins is by binding to a type of protein domain called an EF hand. EF hand domains are characterized by a helix-loop-helix structure and often come in pairs in signaling molecules. Binding of a calcium ion to the EF domain results in a repositioning of the directional vectors of the two helices and often to a conformational changed of the entire signaling molecule with the EF hands (17). For more reading on how calcium connects to NFAT via EF hand-containing proteins, please see (14, 15, 18).
Diacylglycerol Connects to NFκB and AP-1 pathways
NFAT alone is insufficient to fully activate T cells. To fully induce T cell activation and production of IL-2, NFAT must synergize with NFκB and AP-1 (Figure 2). DAG is a hydrophobic component of cellular membranes (19, 20) and both a potent signaling molecule as well as a metabolic source of fatty acids, as it is composed of fatty acids and glycerol molecules (19). Illustrative for the importance of DAG as second messenger, there are different manners to metabolize DAG. For example, diacylglycerol kinases (DGKs) can phosphorylate DAG thereby converting it to phosphatidic acid. When two of these enzymes, DGKα and DGKζ, are simultaneously deleted, normal T cell development is perturbed as a result of elevated DAG-driven signaling (21). For more reading on DAG measurements and localization we suggest these studies (12, 22, 23). DAG can induce membrane recruitment of proteins through a conserved 50 amino acid sequence termed “C1 domain”; a hydrophobic cup-like structure that binds DAG (24). Various signaling proteins contain C1 domains, PKCs (25), RasGRPs (26), protein kinase D (PKD) (27), chimaerin family proteins, and munc13 proteins (20). We will focus on the PKCs, which couple to NFkB, and Rasgrp1, which couples to AP-1 via Ras, in this review.
DAG-induced NFκB Activation in T cells
The PKC family of serine/threonine kinases couple DAG production to the activation of NFκB. In T cells, PKCθ is the PKC family member that appears to be dominant in T cells, and it is recruited to the plasma membrane upon TCR ligation and DAG generation (28–31). Binding of PKCθ to DAG at the membrane relieves autoinhibition of the molecule, putting it into an “open/active” conformation that is able to bind and phosphorylate its substrates (32). Experiments utilizing small molecule inhibitors of PKCθ or antisense oligos to revealed its critical role in the activation of the transcription factors NFκB and AP-1 (33, 34). Two independent groups generated PKCθ knockout mice and further corroborated PKCθ’s connection to AP-1 and NFκB pathways in T cells (29, 35). PKCθ was also shown to phosphorylate RasGRP1; this provided a mechanistic link for how PKCθ connects to AP-1, as AP-1 is directly downstream of the RasGRP1-Ras-MAPK pathway (36).
The Ras exchange factor RasGRP1 Couples TCR Stimulus to AP-1 Activation
RasGRP1 is a guanine nucleotide exchange factor (GEF) for the small GTPase Ras. Ras cycles between a GTP-bound “on” state where it signals to activate the mitogen-activated protein kinase (MAPK) pathway, and a GDP-bound “off state” where it is not signaling. Ras activity is a strong driver of cell proliferation and activating mutations are amongst the most frequent mutations in metastatic cancers (37). Non-cancerous cells also GTP load Ras, and interestingly, T cells were the first non-transformed cell type in which this was demonstrated (38). The canonical Ras-ERK pathway consists of Ras-GTP binding the kinase RAF, which phosphorylates of MEK (mitogen-activated protein kinase kinase), culminating in phosphorylation of ERK1/2 (extracellular signal regulated kinase 1/2) (39). Phosphorylation of ERK1/2 activates ERK to act on a number of cytosolic proteins and transcription factors via its kinase activity. ERK signaling leads to the induction of Jun and Fos expression and a heterodimer of the Jun/Fos transcription factors composes AP-1 (2). The importance of ERK is further exemplified by the impaired positive selection of thymocytes when Erk1 and Erk2 are deleted (40). RasGTP has also been shown to recruit the lipid kinase phosphoinositide 3-kinase (41–43), resulting in activation of PI3K, production of PIP3 and signal transduction to Akt, which in turn has many targets including mTOR (43).
Exchange factors for Ras displace GDP from Ras-GDP and release empty (non-nucleotide bound) Ras that subsequently binds either GDP or GTP. Given that intracellular GTP concentrations are higher than GDP, this leads to accumulation of RasGTP in a stochastic manner. T cells utilize two families of RasGEFs: son of sevenless (SOS) and RasGRP1. For a comprehensive review on SOS in T cells, see Jun et al. (44). For further reading on the RasGRP family of GEFs, which includes RasGRP1, see Ksionda et al. (26).
In Rasgrp1, the catalytic module of the REM (Ras exchange motif) and a CDC25 is followed by a pair of EF-hands, a C1 domain, and a coiled-coil domain. Our lab and the Kuriyan lab solved the initial crystal structure of RasGRP1. This structure revealed that Rasgrp1 exists as an autoinhibited dimer in the basal state and provided key insights into how Rasgrp1 activity is regulated. We found that the C1 and EF hands negatively regulate the intrinsic activity of the catalytic domain of RasGRP1 (45). The crystal structure demonstrated that a dimer interface between these domains prevents DAG binding by capping off the C1 domain. Specifically, the C1 domain of one RasGRP1 monomer contacts the EF2 and CDC25 domains of the other, which likely prevents Rasgrp1 from binding to DAG. Additionally, a linker domain between the CDC25 and EF hands runs through the cleft of the catalytic pocket in a configuration that is incompatible with Ras binding to the catalytic pocket (45). Co-crystals of RasGRP2 with Rap1b and RasGRP4 with H-Ras provided insights as to how calcium and DAG result in activation of RasGRP1, and a pH-sensor Histidine residue conserved in all RasGRPs plays a prominent role in the rearrangement from the inactive to active conformation of a RasGRP molecule (46). Lastly, the RasGRP1 protein contains a C-terminal coiled-coil which allows for RasGRP1 homodimerization (45) (and unpublished data from the Roose lab). A mouse model where endogenous Rasgrp1 was genetically modified with a version that lacked the tail domain revealed that the C-terminal tail has an important functional role in vivo. These tail-deficient mice exhibit autoimmune features, and have impaired thymocyte development and T cell proliferation (45).
Fully Rasgrp1-deficient mice revealed an important role for this Ras GEF in T cell development. Thymocytes in Rasgrp1 knockout mice are blocked at the double-positive (DP) stage, and as such very few mature T cells populate the peripheral lymphoid organs (47). Rasgrp1-deficient thymocytes exhibit impaired TCR-induced Erk phosphorylation in vitro, which likely explains the in vivo block at the DP stage, as DP thymocytes require a productive Ras-Erk signal for positive selection (40).
RasGRP1 and disease
Data from human patients as well as genetic mouse and cell line models have demonstrated that Rasgrp1 activity must be properly controlled to prevent disease. For example, Rasgrp1’s normal resting auto-inhibitory state can be overwhelmed by overexpression such that T cell leukemia develops (48–50). We found that overexpression of Rasgrp1 drives T cell acute lymphoblastic leukemia (T-ALL), and that this mechanism of oncogenesis is distinct from mutant Ras, as the two genetic aberrations are mutually exclusive (48, 51).
Rasgrp1 has also been implicated in immunodeficiencies and autoimmune diseases. Loss of Rasgrp1 in patients leads to immunodeficiency, impaired cytotoxic T cell and NK T cell functions, and susceptibility to bacterial and viral infections (52–55). Mice with point mutations in RasGRP1 have also been created in addition to genetic deletions or forced overexpression. One such variant is the Rasgrp1Anaef mouse. These mice harbor a missense point mutation (R519G) in the EF2 domain such that basal autoinhibition is weakened (56). As we will discuss extensively throughout this review, these mice exhibit increased basal mTOR signaling that leads to a lupus-like phenotype (56). Interestingly, splice variants in RasGRP1 have been identified in human systemic lupus erythematosus (SLE) patients. Additionally, variants in RasGRP1 have been associated with type 1 diabetes and Graves’ disease and loss of precise regulation of RasGRP1 expression levels appear to underlie the autoimmune disease in some of these patients (57–60).
T cells harboring the Rasgrp1Anaef variant exhibit increased baseline mTOR signaling, yet the expression of Ras target genes such as the activation marker CD69 remains unchanged (56). This suggests that RasGRP1 may serve dual roles upon TCR stimulation to first activate the classical Ras-Erk pathway, but secondarily engage mTOR signaling.
mTOR Signaling
Now that we set the stage by describing canonical TCR signaling with several pathways converging on induction of IL-2, and discussed RasGRP1-Ras-ERK signaling, we will next focus on the mTOR pathway. mTOR is classically known to be a critical regulator of cell growth, proliferation, and mRNA translation. The biochemical details of this pathway have been largely characterized in cell lines, and the physiological role of mTOR signaling has been widely studied in a number of model organisms and disease states such as cancer. In T cells, it is known that mTOR becomes activated upon TCR ligation, however the molecular details that connect the TCR to mTOR are still being elucidated. Work from our lab and others has implicated a role for Rasgrp1 in mTOR activation, as Rasgrp1-deficient mouse thymocytes exhibit impaired TCR-induced S6 phosphorylation (61). In the next sections, we will provide an overview of the mTOR signaling pathway and the downstream cellular functions it controls, with a particular focus on T cell biology. Additionally, we have found that there is robust basal (tonic) activity through the mTORC1 pathway in naïve T cells, and we identified a role for Rasgrp1 in this tonic mTORC1 signaling (56, 62). We will discuss this work on tonic mTORC1 in T cells and discuss how tonic signals may be altered in disease contexts such as autoimmunity and cancer.
mTOR Signals Through Two Distinct Complexes: mTORC1 and mTORC2
mTOR is a serine/threonine kinase that associates with several cofactors to form two distinct complexes, mTORC1 and mTORC2. The accessory protein regulatory-associated protein of mTOR (RAPTOR) is a critical component of complex 1, whereas rapamycin-insensitive companion of mTOR (RICTOR) has an analogous role in mTORC2. RAPTOR and RICTOR function as scaffolds to bring the complexes together and for binding substrates (63) (Figure 3). mTORC1 can be activated by diverse signal inputs such as nutrients and growth factors from the microenvironment (reviewed in (64–66)), and these receptor inputs have also been shown to couple to the lipid kinase PI3K. PI3K produces phosphatidylinositol (3,4,5)-triphosphate (PIP3), which activates phosphoinositide-dependent protein kinase-1 (PDK1). PDK1 subsequently activates Akt at the plasma membrane. Akt phosphorylates and inhibits the Ras homolog enriched in brain (Rheb) GTPase activating protein (GAP) complex tuberous sclerosis complex 1/2 (TSC1/2). The net result is activation of the small GTPase Rheb (GTP-bound Rheb), which in turn activates mTORC1. In T cells, exactly how TCR signals couple to mTOR activation is not fully understood.
FIGURE 3. mTOR Signaling; mTORC1 and mTORC2.
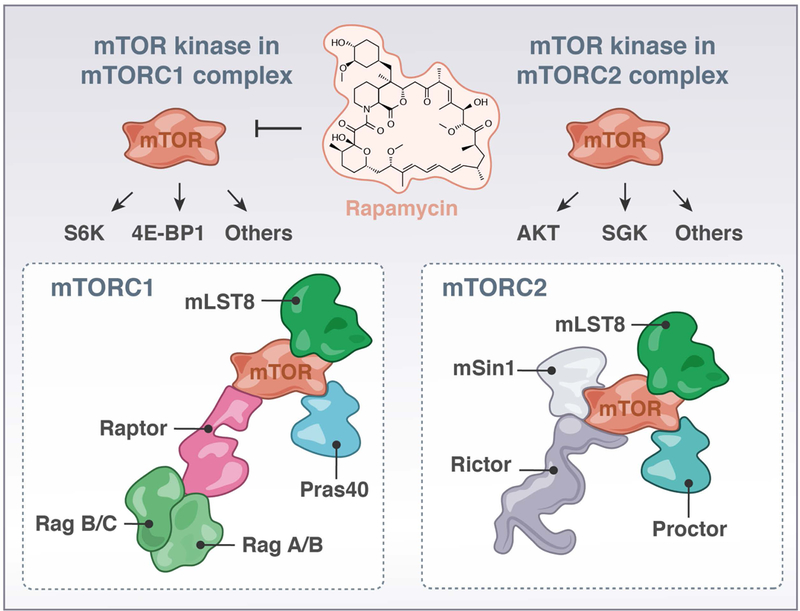
The kinase mTOR can partner with different signaling molecules to form either a mTORC1- or mTORC2- complex that have distinct substrates. In the mTORC1 complex, Raptor can bring mTOR to RagA/B and RagB/C, localizing mTORC1 to the lysosome.
Following activation, mTORC1 directly phosphorylates its substrates: the S6 kinases (S6K1 and S6K2) (which phosphorylates the ribosomal S6 protein, referred to as S6 throughout this review) and the eukaryotic translation initiation factor 4E (eIF4E) binding proteins (4E-BP1, 2, and 3) (Figure 4). It is still relatively unclear what upstream cues activate mTORC2, but it is firmly established that mTORC2 activity results in the phosphorylation and activation of AGC-family kinases such as Akt and SGK1 (64, 65, 67, 68). Interestingly, it seems that T cells may have a distinct pathway for turning on mTORC1, as mTORC1 activation in CD8+ T cells occurs independently of PI3K or Akt. Treatment with the Akt inhibitor Akti-1/2 or with the p110δ inhibitor IC87114 does not impact phosphorylation of downstream mTORC1 effectors S6K, S6, and 4E-BP1 (69).
FIGURE 4. mTORC1 regulates mRNA translation on ribosomes & mTOR-responsive mRNA elements.
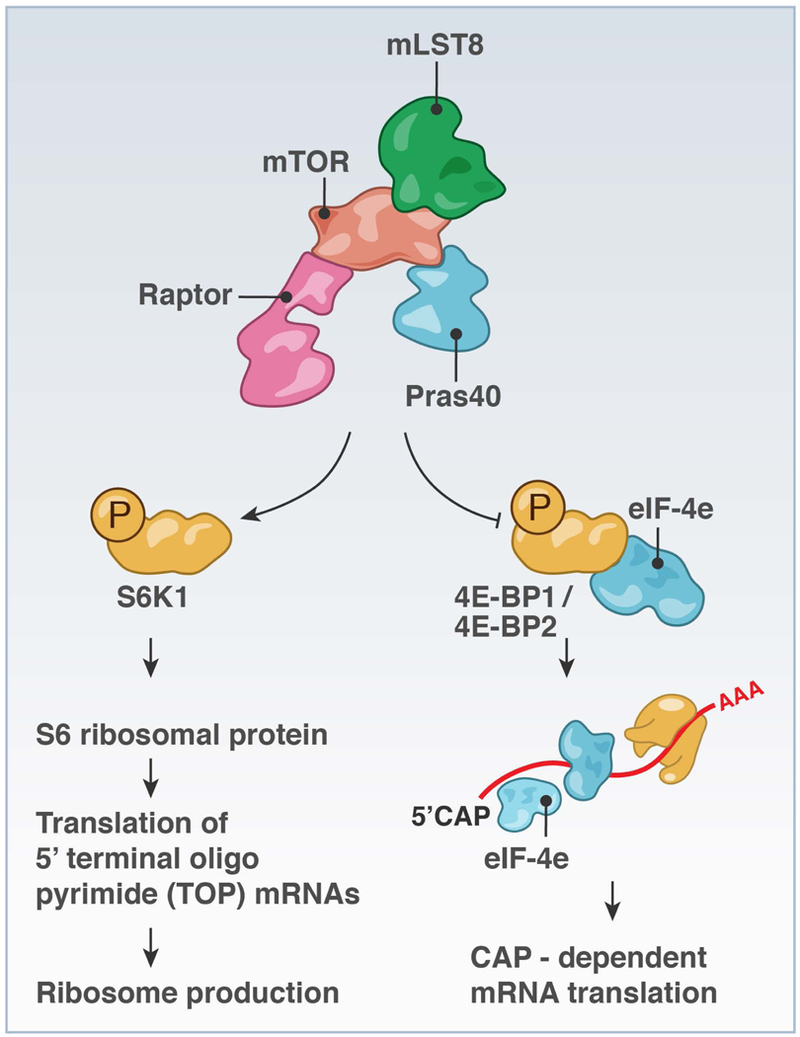
mTORC1 signaling to S6K and to 4E-BP1 or 4E-BP2 to liberate eIF4E can result in increased biogenesis of ribosomes and translation of target mRNAs.
Metabolites that enable mTOR signaling
mTOR activation is contingent upon its localization to the lysosome, a process which is regulated by amino acids and the Rag GTPases. In response to amino acid import, the Rag GTPases exist as heterodimers between either RagA or RagB and either RagC or RagD. In response to amino acid stimulation, RagA/B becomes GTP loaded and RagC/D becomes GDP-loaded, which are the active conformations. Rag GTPases then induce the localization of mTOR to the lysosome, via binding to RAPTOR (Figure 3). mTOR is also able to localize with the GTPase Rheb at the lysosome surface, facilitating its activation independent of the Rag complexes (63, 70, 71). These foundational studies were performed in HEK-293T cells; more recently, it was shown that Rag GTPases also facilitate localization of mTORC1 to the lysosome in T cells (65, 72).
Upstream activation of mTOR signaling is balanced by the AMP-activated protein kinase (AMPK) pathway. AMPK senses when ATP levels in cells drop (and the AMP/ATP ratio rises) and AMPK phosphorylates TSC2. Phospho-TSC2 has increased GAP activity towards Rheb.GTP, effectively shutting off mTOR signaling (73). Overall, AMPK signals to promote catabolic processes instead of the anabolic ones utilized in highly metabolic, glycolytic cells (74). A striking cell biological effect of active mTOR signaling is the alteration in cell metabolism, including the increase in cell growth, cap-dependent translation and elongation, ribosome biogenesis, and switches in metabolic programs (64). Thus, receptor signals, metabolites, and energy state regulate mTOR signaling, and mTOR in turn enables metabolic changes in the cell.
mTOR regulates mRNA translation
When cells are in nutrient-rich environments that support the growth and proliferation described above, cells need to increase their cell size. To meet the biosynthetic demands of this growth, cells need to be able to produce new proteins through a process known as translation (Figure 4). Translation is energetically costly to the cell (each amino acid costs the cell 5 ATP on average (75)), and as such it is very tightly regulated and only occurs under favorable conditions. mTOR and AMPK, as described previously, are key sensors of these environmental cues, and provide the cell a mechanism with which to couple nutrient availability to energetically demanding cell processes such as translation.
Translation is typically divided into four phases: initiation (the rate-limiting step wherein an elongation-competent 80S ribosome is assembled and pairs with an mRNA), elongation (where new amino acids are joined to the nascent polypeptide), termination, and recycling (76, 77). Mechanistically, mTOR regulates translation initiation through its action on the 4E-BPs and the S6 kinases. Active S6K1 stimulates the production of ribosomes by phosphorylating and interacting with several effector proteins that promote translation of mRNAs with 5’ terminal oligopyrimide (5’ TOP) features, including elongation factors and cap-binding proteins (63). Ribosomes do not directly bind to mRNAs, but are recruited to mRNAs by eukaryotic translation initiating factors, or eIFs. One of these eIFs, eIF4E, binds to the 5’ cap present on all transcribed mRNAs. However, in non-activated cells, eIF4E is not bound to the 5’ cap but complexes with hypo-phosphorylated 4E-BPs. When cellular conditions are favorable and mTORC1 signaling is activated, mTORC1 phosphorylates 4E-BP, leading to dissociation of 4E-BP from eIF4E (Figure 4). eIF4E is subsequently free to bind to the 5’ cap of mRNAs. At this point, it also associates with two other proteins: eIF4G (a scaffold) and eIF4A (an RNA helicase that unwinds complex 5’UTR structures in the mRNAs). eIF4G also interacts with eIF3, an important ribosome-initiation factor, and the “ternary complex” (eIF2, Met-tRNA, and GTP) are also recruited to the mRNA (73, 78). Once these effectors are in place, the ribosome can then scan the mRNA in a 5’ to 3’ direction, generating a new polypeptide.
mTOR-responsive mRNA elements
mTORC1-S6K signaling has also been shown to play a role in both translation initiation as well as ribosome biogenesis, which is essential for efficient translation. mTORC1 phosphorylates S6K1 at threonine 389, which results in the formation of a docking site for the kinase PDK1, which phosphorylates S6K1 at T229, fully activating S6K. S6K can phosphorylate another initiation factor, eIF4B, and in turn eIF4B enhances the helicase activity of eIF4A (73). This process is particularly important for the translation of mRNAs that have long and highly structured 5’ untranslated regions (UTRs). A notorious example is c-Myc, which will be discussed a later point in this review. The particular nature of the 5’ UTR structures of potential mTOR targets is an area of active investigation. Some groups previously documented that mTOR targets have long 5’UTRs, whereas a study from the Ruggero lab revealed mTOR-dependent mRNAs have short 5’UTRs (79). Interestingly, in prostate cancer cell lines, 68% of mTOR-responsive mRNAs have these above-mentioned 5’ terminal oligopyrimidine (5’ TOP) features, 63% possess a newly-identified pyrimidine-rich translational element (PRTE), and 89% encompass either a 5’TOP or a PRTE (79) (Figure 4). Whether these elements are indicative of mTOR-responsiveness in cell types other than pancreatic cell lines, such as T lymphocytes, is still unknown.
Target mRNAs of mTOR in T cells
It appears that T cells with different activation states, that is naïve vs. ex vivo TCR-stimulated, and with different effector functions, such as conventional vs. regulatory CD4+ T cells, have different translational programs. Bjur and colleagues compared the translational landscapes of aforementioned populations by isolating polysome-associated mRNAs from ribosomes (80) and documented that the translational programs in these cell populations were distinct from each other and the characterized transcriptional profiles. Thus, mRNA translation is distinct in different T cell populations.
The transcription factor c-Myc has been shown to be regulated at the level of mRNA translation. Biochemically, c-Myc is induced downstream of PI3K, mTOR, and ERK signaling, as its induction can be blocked with inhibitors of these kinases (81). Raptor-deficient CD4+ T cells also do not upregulate c-Myc protein (82). Myc is induced early in T cell activation, prior to upregulation of metabolic genes, suggesting that Myc itself could play a role in inducing metabolic reprogramming (81). Interestingly, c-Myc expression is regulated in a sensitive manner: first, it appears that Myc mRNA and protein are induced in a digital fashion, meaning that Myc is either expressed or not expressed at all in the cell, downstream of TCR signaling, yet c-Myc protein levels are regulated at the post-transcriptional level in a more graded, analog fashion downstream of IL-2R signaling. This was revealed by stimulating OT-1 T cells with agonist peptides of varying affinities and by culturing cells in different amounts of IL-2. These data suggest that TCR-activating signals generate some c-Myc expression in cells, but its exact levels are fine-tuned based on environmental cues such as the presence of cytokines.
Several genes important for Th2 and Tfh biology have been shown to be regulated at the translational level, including Gata3, IL-4, and ICOS (76). Gata3 is the master transcription factor for Th2 cells, which produce IL-4. Naïve T cells differentiate into Th2 cells in the presence of TCR stimulation and IL-4 cytokine input. Gata3 gets induced and then binds to the IL-4, −5, and −13 locus, inducing the transcription of these cytokines (83, 84). Cook and Miller showed that signaling through the IL-4 Receptor in T cells can increase Gata3 mRNA levels somewhat, but not to a degree that is sufficient for Th2 differentiation. Only with TCR stimulation, activating the mTOR pathway, do Gata3 protein levels rise sufficiently. Furthermore, TCR stimulation together with IL-4 increases Gata3 translation in T cells, as Gata3 protein levels increase in a radio-labeled methionine assay, while mRNA levels stay constant (85). IL-4 is under translational control in Th2 cells that are re-activated after initial priming, leading to increased polysomes and increased IL-4 protein production (86). Lastly, the ICOS costimulatory molecule is under translational control. ICOS costimulation augments IL-4 mRNA translation, as IL-4 mRNA was enriched in polysome fractions following TCR + ICOS stimulation compared to stimulation with TCR alone (87). This was dependent on ICOS-PI3K signaling, as no increase in polysome association was observed in mice with a ICOSY181F mutation that abrogates the ability of ICOS to activate PI3K. The ICOS-PI3K loop was suggested to be important for targeted delivery of IL-4 cytokine to B cells by Tfh cells during the germinal center response. In sum, the role of mRNA translation regulating T cell function requires further studies, but results thus far point to an interesting link with Th2 differentiation.
mTOR-dependent growth and proliferation in animal models
mTOR has been well-established as a regulator of cell mass and proliferation. Drosophila with crippling mutations in PI3K, Akt, and mTOR have reduced cell, organ, and organism size, and, conversely, mutations in PTEN, an important negative regulator of PI3K signaling, lead to increased cell growth (summarized in (88)). mTOR is important in embryonic development, as mTOR-deficient Drosophila die in the pupal stage (88), and a whole-body knockout of mTOR in mice is embryonic lethal (89). Mice with reduced mTOR levels, caused by a neo cassette insertion that partially disrupts mTOR transcription, have reduced overall size and weight. We found that mTORchino mice with I205S substitution in the raptor-binding domain of mTOR resulting in a hypomorphic mTOR allele show reduced body mass as well (56). Global deletion of S6K1, a downstream effector of mTOR, also effects growth and proliferation, as S6K1-deficient mice have reduced overall mass and a developmental delay (90).
mTOR-related growth defects in lymphocytes
With respect to the immune system, the mice with a neo insertion in mTOR have reduced spleen size and reduced numbers of T and B cells. T cells exhibited impaired proliferation in response to stimulation with α-CD3 and CD28 antibodies, which activate the T cell receptor and provide costimulation, respectively (91). Another group generated a T-cell specific mTOR knockout mouse by crossing mTORfl/fl animals to CD4-Cre, and T cells from these animals showed impaired TCR-induced proliferation as well (92). Impairing mTOR kinase activity in mouse B or T cells with a kinase-dead knock-in allele crossed to CD19-Cre or CD4-Cre profoundly reduced cell size and impaired proliferation, as did T cell specific knockout of Raptor, one of the critical components of mTORC1 (93). Interestingly, T and B cells from S6K1/2 double-deficient mice grew and proliferated normally, indicating that the S6Ks are not essential for these processes. Instead, this study proposed that the 4E-BPs predominantly regulate growth and proliferation in lymphocytes. This study also revealed that 4E-BP2 is the dominant isoform expressed in lymphocytes, as opposed to 4E-BP1 which is more abundant in other cell types such as fibroblasts (93).
Helper T cell differentiation is controlled by mTORC1 and mTORC2
In T cells, there has been a great deal of recent interest in mTOR signaling and its role in peripheral T cell biology, including helper T cell differentiation (92, 94–96) (Figure 5) and lymphocyte metabolism (74, 97, 98), which we will discuss in the subsequent sections. Although we will not cover it in this review, important work has also revealed a role for mTOR in B:T cell interactions in the germinal center and the expression of adhesion molecules that mediate lymphocyte trafficking. For a discussion of this topic we point the reader to this excellent review (99).
FIGURE 5. mTORC1 and mTORC2 signals and helper T cell differentiation.
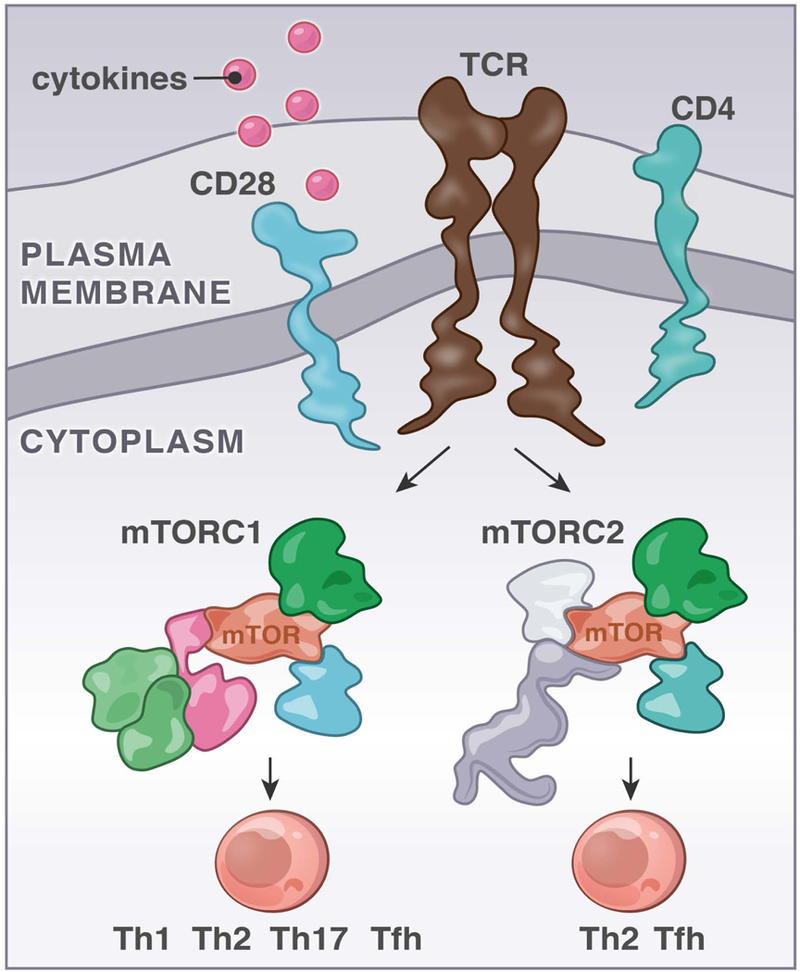
Signals through the mTORC1 or mTORC2 kinase complexes impact the efficiency of CD4 T cells to differentiate into distinct effector T cell populations. Studies with mouse models and deletion of Raptor or Rictor were instrumental to propose the concept depicted in figure 5.
When a naïve CD4+ T cell receives TCR input in the appropriate cytokine milieu, the cell becomes activated and can begin a program of proliferation and differentiation into effector subsets (100). Th1 cells produce IFNγ, express the transcription factor T-bet, and are important for immunity to intracellular pathogens; Th2 cells (mentioned above for being regulated in part by mRNA translation) express Gata3, produce IL-4 and IL-13, and are important for immunity to helminths (yet are pathogenic in atopic diseases). Th17 cells are pro-inflammatory and produce IL-17. Follicular helper T cells (Tfh) express the transcription factor Bcl6 and important helper cells for B cells in the germinal center response. For example, Tfh-derived IL-4 can promote class switching to IgG1 and IgE in mice (101). Regulatory T cells, which suppress effector T cell responses, can be generated centrally in the thymus (called natural Treg, or nTreg) as well as induced in the periphery (induced Treg, or iTreg) as a means to ensure tolerance.
It has been appreciated that mTOR signaling plays an important role in the above-described helper T cell activation and fate determination. TCR-stimulated mTOR-deficient T cells do not upregulate activation markers or proliferate well. Additionally, mTOR deficient T cells preferentially become regulatory, and not effector T cells (92). Abrogating either mTORC1 signaling (via T cell specific Rheb KO) or mTORC2 signaling (via T cell specific Rictor KO) in CD4+ T cells revealed that mTORC1 is essential for Th1 and Th17 differentiation, whereas Th2 cells require mTORC2 (94). A separate study revealed a role for mTORC1 in Th2 differentiation, using T-cell specific Raptor knockout to eliminate mTORC1 signaling (82). T cell specific deletion of the AGC kinase family member SGK1, which, just like its family member Akt, is downstream of mTORC2, led to a modest reduction in T cell proliferation but a profound defect in the generation of Th2 cells as measured by IL-4, −5, and −13 production (67). Subsequently, two groups independently identified a role for both mTORC1 and mTORC2 signaling in the generation of Tfh cells (PD1hi CXCR5+) and the germinal center reaction, finding that Raptor-, Rictor- and mTOR-deficient T cells could not differentiate to Tfh, had impaired GC B cell formation and reduced production of antibodies by those B cells (95, 96) (Figure 5).
mTOR is also active in regulatory T cells (Tregs) and plays a critical role in their function. Freshly-isolated CD25hi regulatory T cells exhibit higher mTOR activity, read out by P-S6K and P-S6, relative to CD25low effector T cells (83, 102). To determine the functional role of mTORC1 signals in Tregs, mice with a Treg specific deletion of mTORC1 signaling (FoxP3-Cre Raptorfl/fl) were generated (83). Strikingly, these mice exhibited severe inflammation associated with immune cell infiltration in multiple tissue types. Effector T cells in these mice had an effector/memory phenotype (CD44hi CD62Llow) and produced cytokines such as INFγ, which suggests that the Tregs in these mice were unable to effectively suppress conventional T cells. To confirm that Raptor-deficient Tregs have impaired suppressive capacity, the authors utilized the adoptive T cell transfer colitis model. In this model, adoptive transfer of effector T cells into Rag1−/− hosts leads to development of colitis in mice, and this can be prevented by co-transferring WT Tregs. The authors found that co-transfer of Raptor-deficient Tregs did not prevent colitis, consistent with impaired suppressive capacity of Raptor-deficient Treg in vivo (83). In sum, it has become evident that mTOR signaling is closely tied to CD4+ T cell fate decisions, and in the next sections we discuss how this may occur through the effects of mTOR signaling on metabolism.
Metabolic programs in naïve, activated, and regulatory T cell subsets
There has been an outpouring of research into the role of metabolic programs used by CD4+ T cells (and other immune cells, which we will not cover here), and the role of mTOR therein. By now it is well established that T cells in different activation states dynamically adopt different metabolic programs to meet their functional needs. Naïve, quiescent T cells that are not actively dividing or producing cytokines generate most of their energy by converting glucose to pyruvate and oxidizing that pyruvate in the mitochondria (TCA cycle), a process referred to as oxidative phosphorylation or OXPHOS. Once T cells become activated, effector cells, their metabolic demand greatly increases, and, as such, they respond by primarily utilizing a different metabolic pathway: aerobic glycolysis. Glycolysis is not an energy-efficient process as it only yields two molecules of ATP per input molecule of glucose, but it is advantageous to rapidly dividing cells because it generates many biosynthetic precursors for cell growth (103).
Several of the different effector T cell subsets (Th1, Th2, and Th17) have been shown to utilize aerobic glycolysis (104). Additionally, cells can obtain energy through the oxidation of lipids instead of glucose. Tregs primarily gain energy via lipid oxidation and have higher levels of AMPK, though it has been shown that Tregs do utilize glycolysis when proliferating. Interestingly, it seems that there are tradeoffs between metabolism and function, as these proliferating Tregs that use a more glycolytic metabolic program have reduced suppressive capabilities (105). Cholesterol biosynthesis also appears to be regulated by mTORC1 signaling in Tregs, as Raptor-deficient Tregs had reduced expression levels of cholesterol biosynthesis pathway genes, and these cells failed to perform de novo lipid synthesis or maintain levels of intracellular cholesterol upon TCR stimulation (83). At the end of an immune response, memory cells form, and these typically utilize lipid oxidation (97). Inhibiting mTOR with rapamycin, or activating AMPK with metformin, can promote the generation of memory CD8+ T cells, suggesting an additional link between T cell metabolism and function (106, 107).
Costimulatory signals can also control these metabolic changes, in addition to TCR signaling. Cells primed with CD28 in vitro exhibited enhanced glycolysis and also had increased spare respiratory capacity compared to T cells that did not receive CD28 costimulation, indicative of a role for CD28 signaling in mitochondrial metabolism. This appears to be particularly important for the generation of functional cytokine-secreting memory T cells. Mechanistically, CD28 signaling promotes expression of carnitine palmitotransferase 1a (Cpt1a), which facilitates fatty acid oxidation. This CD28-Cpta1 axis was important for memory T cell function in vivo in a tumor model: T cells primed with TCR and CD28 stimulation were able to control the growth of EL4 tumors long-term, whereas T cells primed in the same fashion yet also treated with the Cpt1a inhibitor etomoxir (ETO) could initially control tumors (indicative of an intact effector T cell response), but after several weeks tumors grew out, consistent with an impaired memory T cell response (108).
Reprogramming from OXPHOS to aerobic glycolysis
The reprogramming from OXPHOS to aerobic glycolysis involves several cell-biological changes. First, activated cells upregulate transporters to bring in glucose, amino acids, and other nutrients. These include glucose receptor Glut1 (109), the trophic receptors CD71 (transferrin receptor), CD98 (large neutral amino acid transporter), and Slc7a5, which forms a heterodimer with CD98 (110, 111). Additionally, activated T cells upregulate certain transcription factors, such as ERRα and c-Myc, which in turn enhance expression of metabolic genes (74, 97).
The glucose transporter Glut1 is expressed at low levels in resting T cells, but Glut1 protein expression dramatically increases and is accompanied by increased trafficking to the plasma membrane when T cells are activated in vitro (109). Surface Glut1 is also induced on T cells in vivo in response to immunization (104). Treatment of in vitro activated T cells with PI3K and mTOR inhibitors demonstrated that these kinases are required for Glut1 induction. Interestingly, it seems that Glut1 is important for sustaining mTORC1 signaling once T cells are already activated, as cultured Glut1-deficient T cells exhibit reduced P-S6 levels and increased p-AMPK levels (indicating metabolic stress) compared to WT, suggesting a positive feedback loop (109).
Mouse models of Glut1 overexpression (104) or deficiency (109) revealed an important functional role for Glut1 in T cell proliferation, survival, and cytokine production. Measuring OXPHOS levels via the oxygen consumption rate and aerobic glycolysis via the extracellular acidification rate revealed that Glut1-deficient cells could not undergo metabolic reprogramming from OXPHOS to glycolysis. These studies reveal a critical role for cellular import of glucose in lymphocyte activation, proliferation, and metabolism.
The process of aerobic glycolysis itself is important for T cell effector function, as naïve CD4+ T cells stimulated in media containing galactose (which cannot be metabolized using aerobic glycolysis) were dramatically impaired in their ability to produce the cytokines IFNγ and IL-2. Mechanistically, it was shown that cells utilizing aerobic glycolysis increase the translation of IFNγ mRNA, likely mediated by preventing inhibitory GAPDH binding to IFNγ transcripts. Aerobic glycolysis has also been shown to be important in modulating T cell proliferation. Inhibiting glucose metabolism with 2-deoxyglucose (2-DG) in vivo partially inhibits T cell proliferation in an adoptive transfer experiment (81), and 2-DG treatment was also shown to protect mice from EAE (112). Perhaps unexpectedly, T cells use OXPHOS in addition to aerobic glycolysis to aid their activation and proliferation. Treatment of cells with the ATP synthase inhibitor oligomycin impaired T cell activation and proliferation (113).
In addition to Glut1 and glucose enabling mTOR signals, amino acid signaling also activates mTOR signaling, (63, 70) and several groups uncovered an important role for amino acid uptake in T cell function. T cell-specific deletion of Slc7a5, a protein that hetero-dimerizes with CD98 to form a system L transporter, has profound effects on T cell biology. System-L transporters are critical for the import of leucine and other large neutral amino acids into cells. CD98 is an mTOR target, as Raptor-deficient T cells show impaired TCR-induced CD98 upregulation (82). Slc7a5-deficient T cells cannot proliferate, fail to differentiate into Th1 and Th17 effector subsets, and cannot efficiently undergo metabolic reprogramming to aerobic glycolysis (110). Slc7a5-deficient T cells cannot activate the mTOR pathway or induce c-Myc protein, despite expressing robust c-Myc transcript. Interestingly, mTOR translocates to the lysosome in CD8+ T cells following in vitro TCR stimulation, and mTOR-lysosome colocalization was stronger in cells that expressed higher levels of Slc7a5 (114). Slc7a5 is also a key transporter of the amino acid methionine in T cells, and methionine import was shown to be critical for full T cell activation. This is because T cells use methionine both for the synthesis of new proteins and for generating donor methyl groups for methylation of histones and nucleic acids. Interestingly, naïve T cells express all of the key enzymes required for the metabolism of methionine, suggesting they are primed to carry out these metabolic steps, and the ability to import methionine via Slc7a5 is a rate-limiting step in this process (115). These studies suggest that TCR signals and amino acid uptake are coupled in T cells to promote mTOR signaling, metabolism, and effector function in T cells.
c-Myc as a critical decider
As discussed above, c-Myc is a translational target of mTOR. c-Myc itself also plays a critical role in metabolic reprogramming of T cells. Conditional deletion of Myc using a floxed allele crossed to a tamoxifen-inducible Cre profoundly alters T cell biology (81). Myc-deficient cells showed impaired proliferation in vitro and in vivo and did not increase their cell size upon T cell activation like WT activated lymphocytes do. Myc deficiency has a selective effect on distinct metabolic programs, as glycolysis and glutamine metabolism were impaired in Myc-deficient T cells, but mitochondrial oxidation of glucose (OXPHOS) and fatty acids were not affected. Functionally, Myc expression correlates with and controls upregulation of the transferrin receptor CD71, revealing an intimate link between c-Myc and nutrient import (116).
In two provocative studies, it was shown that the decisive function of c-Myc is inherited in an asymmetric manner in CD8+ T cells. Specifically, in the process of cell division, the daughter cell more proximal to the original parental cell receives more c-Myc protein than the distal daughter cell (114, 117). This finding was extended to mTOR pathway components such as P-S6 and metabolic transporters such as CD98: these were also enriched in the c-Mychigh, proximal daughter cells when compared to the c-Myclow distal daughter cells. Intriguingly, inheritance of c-Myc in the first cell division also appeared to correlate with the differentiation state of the CD8+ T cell. Sorted c-Mychigh cells transferred into recipients that were subsequently immunized with Influenza A dominated the immune response, acting more like effector cells, while Myclow cells dominated the memory response (117). It is still unclear how this effector/memory dichotomy with respect to c-Myc inheritance is explained, as the cells continue to divide during the course of an immune response. Nevertheless, mTOR is likely to play a role in this asymmetric cell division. By transiently inhibiting mTOR via rapamycin treatment, Borsa et al. were able to increase asymmetric divisions and memory cell formation in ex vivo stimulated CD8+ T cells (118).
To this point we have focused on the events that happen after antigen recognition and the role that mTOR signaling has in mediating this response. However, to effectively mediate this response T cells must be poised to do so. This has led to the hypothesis that T cells are primed for activity by tonic signals generated by TCR/pMHC interactions from seeing self-peptides, which lower the threshold for T cell activation (119–122). We will first define tonic or basal signaling and provide a brief overview of the literature exploring tonic signals in lymphocytes. For a more comprehensive overview of tonic signaling in T and B cells, we point the readers to a review from our lab and the Zikherman lab (120). Next, we will detail some of our work that demonstrates that the mTORC1 pathway is particularly active in the basal state in T cells.
Tonic Signals from seeing self
Tonic signals are characterized by low-level recruitment and activation of the proximal kinases and signaling molecules downstream of the TCR. Early studies indicated that both thymocytes and resting T cells in the lymph node exhibit constitutive phosphorylation of the TCR zeta chain and recruitment of Zap-70 (122–128). Accordingly, tonic signals in T cells are generated through low affinity stimulation of the TCR. In vivo, one source of this low affinity interaction in T cells are self-pMHC, as tonic signaling is dampened by the administration of MHC blocking antibodies (122). Thus, in the blood as T cells circulate they make few cell-cell contacts and receive very little tonic TCR activity. Conversely, upon entering secondary lymphoid organs, which are densely-packed with MHC-expressing cells, T cells make frequent contacts and generate these sub-threshold, tonic signals (62, 122). Intriguingly, transfer of CD4+ T cells into MHCII-deficient recipient mice (119) yields a different result from the experiments utilizing MHCII blocking antibodies. T cells transferred into class II-deficient mice take on an activated phenotype and were reactive against skin grafts, leading the authors to propose that MHCII expression in the periphery is important for dampening T cell activity.
Work from our lab and others suggests that tonic signaling can both prime T cells for activity, consistent with the Stefanova study, while other tonic signaling pathways can promote a quiescent state, consistent with the Bhandoola study. We propose that tonic signals carry out this dual function to maintain T cells in a primed-yet-quiescent state: this enables T cells to be rapidly activated when they do encounter cognate foreign antigen, yet prevents spontaneous activation in response to self, thereby preventing autoimmune damage.
In recent years, two reporters of tonic signaling have been identified: the surface marker CD5, as well as the orphan nuclear hormone receptor Nur77 (read-out by levels of GFP in a reporter mouse). Both are expressed at a level that correlates with the amount of tonic signal a cell receives (62, 129, 130). These reporters have demonstrated that there is a range or continuum of CD5 or GFP expression within the T cell pool, indicating heterogeneity with respect to the level of tonic signal T cells receive. Some cells exhibit high tonic signaling (high CD5 or Nur77-GFP), and others have low tonic signals (low CD5 or Nur77-GFP) (62, 129, 130). Use of these reporters has demonstrated that cells with high tonic signaling out-proliferate their lower tonic signaling counterparts in bacterial and viral infection models, indicating that the level of tonic signal a T cell receives impacts its function.
Despite this recent interest in tonic signaling, we still have a relatively limited understanding of what pathways are active in a tonic fashion in T cells as a result of TCR-self pMHC interactions. This is in contrast to the signaling pathways that are engaged following TCR ligation by cognate antigen, which have been very well described. It will be interesting to define the similarities and differences between antigen-induced signaling pathways and tonic signaling pathways. Recent work from our lab has utilized two mouse models wherein tonic signaling becomes dysregulated to delineate tonic signaling pathways and determine their role in T cell function (62, 121). We will next discuss these two mouse models, one of which points to a mechanism for how tonic signals maintain T cell quiescence, and the other demonstrates that a distinct tonic signaling pathway can prime T cells for activation.
Tonic LAT-HDAC7 signals curb T cell activity by promoting transcription of negative regulators
As discussed, the linker for activation of T cells (LAT) is phosphorylated at conserved tyrosine residues upon TCR ligation. Mutation of one of these residues, tyrosine 136, to phenylalanine (Y136F) leads to an intriguing phenotype: the mice have a partial block in the development of thymocytes into T cells; those T cells that do populate the periphery expand rapidly and spontaneously secrete the Th2 cytokine IL-4 even without ex vivo restimulation. Furthermore, when LAT is inducibly deleted in peripheral T cells or the Y136F point mutation is inducibly knocked-in to the endogenous LAT locus, the mice still exhibit lymphoproliferation and Th2-biased differentiation, indicating that these phenotypes develop independently of thymocyte development (131, 132). Despite the fact that LAT-deficient or –pointmutated T cells aberrantly proliferate and produce cytokines, they are refractory to ex vivo stimulation via the TCR and were unable to flux calcium, produce critical cytokines such as IL-2, and divide (132).
This paradox described above led our group to hypothesize that LAT transmits a tonic signal in T cells, and loss of this tonic signal upon LAT perturbation drives the lymphoproliferation and Th2 differentiation. To uncover the mechanism by which tonic signals through LAT could impact T cell homeostasis, we first turned to LAT-deficient cell lines. We had previously demonstrated that LAT impacts basal transcription, as a Jurkat T cell line deficient for LAT (J.CaM2) exhibited impaired expression of the Rag genes as well as the alpha chain of the T cell receptor (Tcra) (126). This gene expression could be restored by treating the cells with the HDAC inhibitor trichostatin A (TSA), suggesting that tonic LAT-HDAC signals impact gene expression (133). To address this in primary T cells from mice, we performed microarray analysis on WT CD4+ CD44low (naïve) T cells, as well as cells from mice where LAT was inducibly deleted or point-mutated to Y136F (121). We found a cluster of genes was significantly downregulated in LATKO or LATY136F cells, and many of these genes had been previously proposed as targets of the epigenetic regulator histone deacetylase 7 (HDAC7) (134, 135).
We found that tonic signals impact the basal phosphorylation and subcellular localization of HDAC7: freshly isolated CD4 T cells have highly phosphorylated HDAC7, which is localized to the cytoplasm. HDAC7 is dephosphorylated and localized to the nucleus when cells are deprived of tonic signals (via resting in non-stimulatory media at low-density). When LAT is perturbed and the tonic signal through LAT is lost, HDAC7 is dephosphorylated and localized to the nucleus, where it represses its target genes. Functionally, we found that lower expression of one of these targets, Nr4a1, which encodes Nur77, leads to enhanced T cell proliferation. Additionally, mice heterozygous for Irf4 in T cells, which have reduced but not absent Irf4 protein, exhibit a subtle increase in their ability to differentiate to Th2 in vitro. We hypothesize that the Th2-biased lymphoproliferation observed in the LAT-perturbed mouse model is driven in part to reduced expression of these HDAC7 target genes Nr4a1 and Irf4. Thus, in WT cells, a tonic LAT-HDAC7 signal promotes low-level expression of target genes that function to curb T cell proliferation and differentiation.
Selective tonic mTOR signals in T cells
The LAT mouse model demonstrates a context where a loss-of-function tonic signal leads to altered T cell functionality. In contrast, we also recently characterized a mouse model with a gain of function tonic signal, called the Rasgrp1Anaef mouse. Our work points to a novel tonic Rasgrp1-mTORC1-S6 signaling pathway that promotes T cell differentiation and mRNA translation.
The Rasgrp1Anaef mouse was identified in an N-ethyl-N-nitrosourea (ENU) chemical mutagenesis screen for exhibiting activated (CD44high) CD4+ T cells and anti-nuclear antibodies (ANAs) in the serum. The mice developed autoantibodies when T cells harbor the Anaef mutation but B cells are WT, indicating that T cell help to B cells is altered in these animals (56). We also found that the CD4+ T cells in the Rasgrp1Anaef mouse exhibit an autoreactive TCR repertoire, revealed using the Nur77-GFP reporter (62). Given this, we further characterized these mice for features of T cell dysregulation and immunopathology. We found that they had expanded populations of follicular helper-like (Tfh-like, PD1high Helios+) and peripheral helper T cells (Tph, PD-1highICOShighCXCR5lowBcl6low) in the peripheral lymphoid organs, and expansion of conventional Tfh cells (PD1high CXCR5+) in the Peyer’s Patches (PPs) (56, 62). The increase in the activation marker CD44, the expansion of these populations, and the penetrance of ANA production increased with age.
Using phospho flow cytometry and western blotting, we found that Rasgrp1Anaef CD4+ T cells exhibited gain-of-function tonic signaling to mTORC1-S6 relative to WT T cells (Figure 6). As discussed, T cells constantly see self pMHC in the peripheral lymphoid organs, which generates tonic signals, we hypothesized that the accumulation of tonic signals over the lifespan of a T cell led to the observed immunopathology in the Rasgrp1Anaef mice. To test this hypothesis, we treated mice with a low dose of rapamycin for one week to dampen tonic signaling in vivo. We observed a reduction in CD44 expression on Anaef T cells down to WT levels and a contraction of the Tfh population in the PPs (56, 62). We also crossed Rasgrp1Anaef mice to the mTORchino model described previously, which has reduced mTORC1 activity. Mice with this genetic cross had reduced CD44 levels on CD4+ T cells and the ANAs were resolved. These studies indicate that elevated mTORC1 signals drive T cell autoreactivity and immunopathology.
FIGURE 6. Tonic translation in T cells.

Constant recognition of self-peptide/MHC by the TCR leads to a tone of a biochemical signal in T cells that we term “tonic signal”. The Roose lab uncovered that the tonic signal through the TCR is preferentially transduced to the mTORC1-S6 pathway. We also uncovered that resting T cells have a pattern of baseline translation in which there is enrichment for mTOR-target genes. T cells from a mouse model with autoimmune features, Rasgrp1Anaef, have increased basal mTORC1-S6 signaling and display alterations in baseline translation.
As discussed, previous work analyzing tonic signaling in T cells focused on signals very proximal to the T cell receptor, such as phosphorylation of the TCR zeta chains, recruitment of Zap70 (122–127), and the adapter LAT (121, 133). Given that our results from the Rasgrp1Anaef mouse pointed to tonic activity to mTORC1, we explored what other distal, downstream effector kinase pathways might be active in the basal state in T cells. We focused our attention on the mTORC1-S6, mTORC2-Akt, and Ras-Erk pathways and used a barcoding phospho-flow cytometry approach to analyze phosphorylated proteins in different cell subsets immediately upon fixation, where desired. Resting lymph node T cells ex vivo for two hours in non-stimulatory media (RPMI with amino acids and glucose but no serum) at a low cell density dramatically reduced P-S6 levels compared to cells fixed immediately after isolation, with a more modest decrease in P-AktS473, a readout of mTORC2 activity. P-Erk, a readout of Ras pathway activity, did not decrease to a detectable degree in this two hour rest, though P-Erk levels were quite low, nearly comparable with background, in freshly-isolated cells (62). We also analyzed P-S6, P-Akt, and P-Erk levels in freshly-isolated and fixed cells that were subsetted into CD4+ CD5high, which have been shown in the literature to have high levels of proximal tonic TCR signals, and CD4+ CD5low cells, which have low tonic signals. We found that CD5high cells exhibited higher basal P-S6 relative to CD5low cells, indicating the mTORC1 pathway is under tonic control. In contrast, there was no significant difference in P-Akt or P-Erk in CD5high and CD5low T cells (62). Thus, the mTORC1 pathway appears to be selectively active in the basal state in CD4+ T cells.
The Rasgrp1Anaef mice exhibited increased tonic mTORC1 signals in T cells, so we undertook further studies to analyze the role of Rasgrp1 in this process. As discussed in previous sections, Rasgrp1 is a Ras guanine nucleotide exchange factor that acts to facilitate the exchange of GDP for GTP on Ras, thereby activating Ras (26). However, despite its canonical activity on the Ras pathway, we did not observe increases in tonic P-Erk, a downstream readout of Ras activity, in Rasgrp1Anaef T cells (56). Rasgrp1 had been previously demonstrated to be important for TCR-induced mTORC1 activation, as Rasgrp1-deficient thymocytes did not efficiently phosphorylate S6K, S6, or 4EBP1 in response to TCR crosslinking with anti-CD3. Of note, this study also demonstrated that Rasgrp1WT unstimulated (resting) thymocytes exhibited high basal P-S6 compared to Rasgrp1KO cells, indicating a tonic Rasgrp1-mTORC1 signal (61). To further explore the role of Rasgrp1 in tonic signaling, we turned to the chicken B cell line DT40 in which Dr. Kurosaki has made several gene deletions of lymphocyte signaling genes (136). These cells are readily amenable to genetic manipulation and transfection in vitro. We utilized a Rasgrp1/Rasgrp3 double deficient line (referred to as “DKO” cells here). Rasgrp1/3 DKO cells exhibited reduced P-S6 compared to WT cells (62). Transient transfection of a WT Rasgrp1-eGFP construct into DKO cells led to increased basal P-S6 in a Rasgrp1-dose-dependent manner, indicating that Rasgrp1 is important for driving the observed tonic mTORC1 signal. Interestingly, transient transfection of a Rasgrp1R271E-eGFP construct, which encodes a variant of Rasgrp1 that lacks catalytic GEF activity, did not restore basal P-S6. This suggests that the nucleotide exchange activity of Rasgrp1 is required for mTORC1 activity. Consistent with our data from Rasgrp1Anaef T cells, transient transfection of a Rasgrp1R519G-eGFP construct, which encodes the Anaef variant allele, led to a subtle increase in basal P-S6 compared to WT Rasgrp1.
Tonic mTORC1 signals prime T cells for differentiation
As discussed, mTOR signaling is important for a number of cellular functions in T cells, including the differentiation from a naïve CD4+ T cell into a specialized sub-type such as Th1, Th2, and Th17. We found that Anaef CD4+ T cells had an enhanced ability to differentiate to Th2 in vitro and secrete the canonical Th2 cytokine IL-4 (62). This is consistent with published literature showing that Raptor-deficient T cells, which have reduced mTORC1 signaling, are impaired in their ability to differentiate to Th2 (82). We also found that sorted CD5high naïve T cells (CD4+ CD44low CD5high), which have higher tonic mTORC1 signaling, were enhanced in their ability to differentiate to Th2 compared to CD5low cells. Mechanistically, we observed that Rasgrp1Anaef T cells had increased levels of the master transcription factor for Th2 cells, Gata3, early on during in vitro Th2 differentiation. Similarly, CD5high cells had increased Gata3 early in these cultures relative to CD5low cells. We hypothesize that the enhanced ability to differentiate to Th2 by these cells with high tonic mTORC1 signaling may have to do with this early induction and stabilization of Gata3 protein.
Baseline Translation in Resting T Cells
As discussed above, Gata3 has been previously shown to be regulated at the level of mRNA translation in TCR-stimulated CD4+ cells. We confirmed that Gata3 is also regulated at the level of translation in the basal state, as freshly-isolated WT CD5high T cells had reduced Gata3 mRNA yet elevated Gata3 protein relative to CD5low cells. Rasgrp1Anaef CD4+ T cells had similar levels of Gata3 mRNA yet elevated Gata3 protein levels compared to WT T cells (62). Gata3 is likely a translational target of mTOR, as its 5’UTR contains a PRTE, an element shown to correlate with mTOR-dependent translation (79).
In addition to Gata3, we found that naïve CD4+ T cells perform low-level translation of many mRNAs, more than half of which have been established as mTOR translational targets in an independent study with mouse embryonic fibroblasts (137). We uncovered this using ribosome profiling, a method where ribosomes are isolated from a cell population of interest and the mRNAs that are actively bound to those ribosomes (ribosome protected fragments or RPFs) are subjected to deep sequencing (138). In parallel, the total RNA in the cell is sequenced, and DESeq2 can be used to identify transcripts that are translational targets, defined as those preferentially expressed in the RPF sequencing data set relative to the total mRNA in the cell. It would be interesting to perform ribosome profiling on WT mice treated with rapamycin to fully determine whether the genes enriched in the RPF data set are bona fide mTORC1 translational targets.
From our dataset, the mRNAs that were most highly translated in naïve T cells were enriched for genes in pathways related to mitochondrion and oxidative phosphorylation, processes linked to mTOR signaling (62). An independent publication utilized an alternate approach to analyze translation, namely transcriptomic and proteomic analysis of T cells, and found that naïve cells accumulate mRNAs for glycolysis and fatty acid synthesis, leading to the hypothesis that these genes contribute to the ability of T cells to rapidly undergo metabolic reprogramming upon TCR stimulation (139). We also analyzed Rasgrp1Anaef T cells from young mice, which were just at the cusp of the immunopathology becoming penetrant, in this ribosome profiling study. There was a high degree of overlap in terms of translationally regulated mRNAs between WT and Anaef T cells (Figure 6). Glutathione metabolism was one functional annotation cluster identified that was unique to Anaef T cells (62). Whether alterations in this metabolic process are due to elevated tonic mTORC1 signaling, or whether they contribute to the immunopathology in Anaef T cells, remains an exciting open research question.
mTOR and autoimmune disease
Autoimmune disease results when immune cells incorrectly recognize self-antigens as foreign. This leads to T cell activation and immune-mediated damage of what are otherwise normal, healthy tissues and many patients with autoimmune diseases are treated with steroids, which broadly dampen the immune system. Data from the LATY136F and Rasgrp1Anaef mouse models points to a role for altered tonic T cell signaling in immunopathology (56, 62, 121, 131, 132). A better understanding of the signaling pathways that are active in the basal state, and whether dysregulation of these pathways contributes to disease, will be an important area of future research.
T cells from Rasgrp1Anaef mice exhibit constitutively higher activation of the mTORC1 signaling pathway in naïve T cells. This elevated tonic mTORC1 signal drives the development of lupus-like immunopathology in these animals, as pharmacological inhibition of the mTORC1 pathway or genetically crossing in a hypomorphic mTOR allele resolves the disease features. As delineated in the previous section, one mechanism by which mTORC1 signaling contributes to immunopathology may be its effects on helper T cell differentiation and translation. Other studies have also demonstrated a role for mTOR activity in mediating autoimmune disease (Figure 7). Oaks et al. demonstrated that rapamycin treatment of lupus-prone mice can block the production of anti-phospholipid antibodies and preserve liver function (140). In addition to mouse models, importantly, the role of mTOR in autoimmune disease has also been established in human lupus patients (141).
FIGURE 7. mTOR and autoimmune disease.
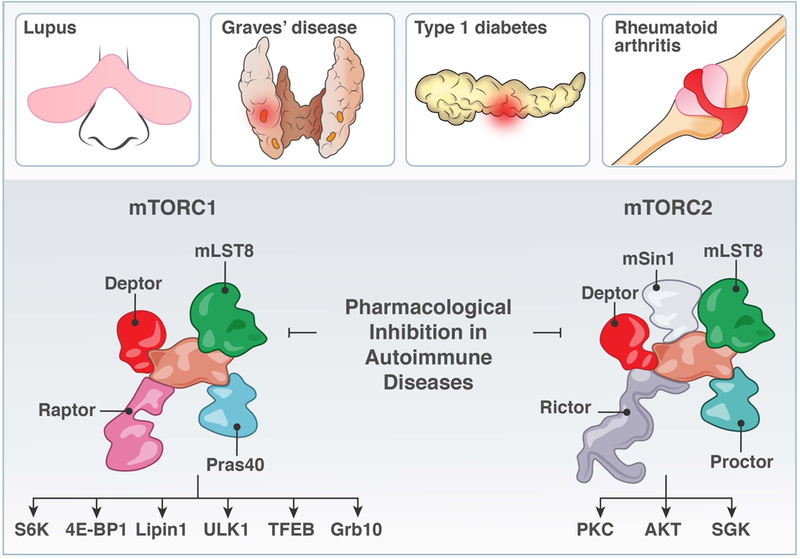
The realization that T cells from autoimmune patients as well as from genetic mouse models with autoimmune features have altered metabolic characteristics has stimulated the field on mTOR signaling and T cell metabolism in the context of autoimmune diseases.
Expanding beyond mTOR, metabolic pathways are also altered in lupus-like disease. In a stimulating study it was revealed that both glycolysis and mitochondrial oxidative metabolism were elevated in T cells from a lupus-prone mouse model. Furthermore, combined inhibition of these metabolic alterations with 2-deoxy-d-glucose (2DG) to dampen glycolysis and metformin to target the mitochondrion reversed lupus biomarkers in the mice (142). 2DG treatment also reduced joint inflammation and other disease features in a moues model of rheumatoid arthritis (143). The connection between mTOR signaling and the presence of autoimmune features such as self-reactive antibodies and increased inflammatory helper T cell subsets such as Tfh portends an exciting direction for the field. Given the central node metabolism plays in T cell activation, it will be important for future studies to understand and connect mTOR signaling and T cell metabolism to autoimmune disease pathology, with the goal of developing novel treatments (Figure 7). We point the readers to this review by Laurence Morel for a broader view of the clinical possibilities of metabolic inhibition (144)
Outlook: Tonic signals and Cancer Immunotherapy?
It is now well appreciated that the immune system is important for the control and elimination of cancer cells. Cancer cells utilize a number of mechanisms to evade the detection and activity of the immune system, including the upregulation of checkpoint molecules that engage immune inhibitory receptors such as PD-1 and CTLA-4, and the downregulation of MHC molecules that present antigen to T cells (145). A number of groundbreaking cancer immunotherapies have been developed that aim to bolster the effector functions of T cells and other immune cells to kill cancer cells, or to dampen the activity of regulatory, suppressive immune cells. We will next discuss the literature supporting a role for tonic signals in T cells in the context of cancer immunotherapy, and how a greater understanding of tonic signaling pathways in T cells could be leveraged to design novel and effective therapies (Figure 8). We will discuss both Chimeric Antigen Receptor (CAR) T cell therapies as well as immune checkpoint blockade (ICB) therapies.
FIGURE 8. Checkpoint blockade and CAR-T cells and Tonic signals?

Checkpoint blockade in the form of anti-CTLA4 or anti-PD1 as well as generation or CAR-T cells to fight tumors may lead to increased tonic mTORC1 signaling in T cells. Questions such as the impact of T cell therapy approaches on mTOR signaling in T cells will be important to address.
An area of intense recent research has been in the development and improvement of CAR T cells. To date, the FDA has approved two CAR-T therapies targeting CD19-expressing B cell malignancies. To generate CAR T cells, T cells are isolated from a patient, an engineered receptor is inserted, and that genetically modified cell is re-infused into the patient. The engineered receptor is composed of several domains: an extracellular portion that recognizes a tumor associated antigen (such as CD19 for the FDA-approved CARs), a hinge region, a transmembrane domain, and intracellular, ITAM-containing signaling domains such as the TCR-CD3 zeta chain and/or the 4–1BB domain, among others (146). The exact consequences on signaling in T cells expressing a CAR is an area of active research with many unknowns particularly in the application for solid tumors (147). As Long et al. showed, some CAR constructs exhibited baseline phosphorylation of the CD3-zeta chain due to CAR mediated, antigen independent tonic signals (148). Further, they show that these tonic signals correlated with over-activation and early exhaustion, thus decreasing the anti-tumor efficacy in vivo. The authors were able to rescue this effect by including an inhibitory 4–1BB domain in the CAR. Intriguingly, when they compared the inhibitory CAR to their previous model they found increased transcription of metabolic genes. A separate study illustrated that CARs containing 4–1BB domains persisted better in vivo (149). In addition, they showed enhanced glycolytic metabolism in the poorly persistent cells, and that 4–1BB inclusion in the CAR drives mitochondrial biogenesis and increased oxidative phosphorylation. These studies primarily assess cellular metabolism at the functional level, but, as we have discussed in this review, mTOR signaling is a crucial regulator of these processes. Therefore, future studies will be important to understand whether and how the CAR may tonically engage mTOR, and how modulating mTOR signaling or metabolic pathways in CAR T cells may enhance their in vivo efficacy.
Beyond engineering cells and receptors, as is the case with CAR-T cells, immune checkpoint blockade has proved to be one of the most important developments in cancer treatments to date. T cells express several checkpoint molecules on their surface, including CTLA-4 and PD-1, which are important for transducing inhibitory signals as a counterbalance to activating signals. Several of these immune checkpoints are upregulated on exhausted or anergic T cells (150, 151). Some work has begun to show that these molecules may also signal in a tonic tonic fashion, which may regulate T cell function. For instance, Previte and colleagues investigated the role of the inhibitory lymphocyte activation gene-3 (LAG-3) in controlling naive T cell metabolism (152). In their work, the authors found that genetic deletion of LAG-3 induced T cells to be more metabolically active and more proliferative under homeostatic conditions in an adoptive transfer model. These tonic signals to LAG-3 were found to inhibit STAT5 and Akt activation, and enhancement of these pathways upon LAG-3 genetic ablation lead to increased aerobic glycolysis, Glut1 expression, and c-myc induction. Thus, this study provides another interesting link between tonic signals, here through an immune checkpoint molecule as opposed to through the TCR, and T cell metabolism.
Similar studies have been conducted with PD-1. Using a lymphopenia-induced proliferation model, Ellestad et al. demonstrated T cells lacking PD-1 repopulated Rag−/− recipient mice better than co-injected WT cells (153). This proliferative break was shown to be dependent upon tonic pMHC interactions as both PD-1 deficient and WT T cells both fail to proliferate in Rag−/− recipients that also lack MHC. The authors also observed increased CD44 expression in the PD-1 deficient T cells, indicating a more activated T cell phenotype. Whether the increased CD44 on PD-1-deficient T cells is driven my increased tonic mTORC1 signals, as in the Rasgrp1Anaef T cells, remains unknown.
Lastly, studies have also implicated the possibility for CTLA-4 negative co-stimulation in the regulation of tonic signals. Knockout of CTLA-4 in mice leads to aberrant T cell differentiation, indicated by the presence of non-canonical CD4+ T helper cell subtypes only in the knockout mice (154). Intriguingly, data from this study illustrate the possibility that CTLA-4 restricts tonic signaling by raising the threshold for differentiation of promiscuous clones. T cells from CTLA-4 deficient mice may be more responsive to self-antigens as well, as CTLA-4 deletion was correlated with decreased N region insertions, which can contribute to self-reactivity (154, 155). How exactly CTLA-4 may receive or transduce tonic signals remains to be defined, but these recent data provide an important starting point. Whether checkpoint inhibitors could be enhanced by co-administration of small molecule inhibitors or agonists that target tonic signaling effector molecules remains an exciting open research question. It is also still unknown whether tonic signals are altered in the highly-suppressive tumor microenvironment and across different tumor histotypes. Gaining a better understanding of tonic signaling pathways in T cells, both downstream of the TCR and from other surface receptors such as immune checkpoints or nutrient transporters, as well as determining how these tonic signaling pathways alter T cell metabolism and function, could enable us to optimize treatments to broaden their efficacy (Figure 8).
Acknowledgments
We thank support through grants from the NSF-GRFP (1650113 to DRM) and the NIH-NIAID (R01-AI104789 and P01-AI091580 to JPR).
Footnotes
Declarations of interests.
Jeroen Roose is a co-founder and scientific advisor of Seal Biosciences, Inc. and on the scientific advisory committee for the Mark Foundation for Cancer Research.
References
- 1.Navarro MN, Cantrell DA. Serine-threonine kinases in TCR signaling. Nat Immunol. 2014;15(9):808–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klammt C, Novotna L, Li DT, Wolf M, Blount A, Zhang K, et al. T cell receptor dwell times control the kinase activity of Zap70. Nat Immunol. 2015;16(9):961–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Katz ZB, Novotna L, Blount A, Lillemeier BF. A cycle of Zap70 kinase activation and release from the TCR amplifies and disperses antigenic stimuli. Nat Immunol. 2017;18(1):86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xie J, Han X, Zhao C, Canonigo-Balancio AJ, Yates JR 3rd, Li Y, et al. Phosphotyrosine-dependent interaction between the kinases PKCtheta and Zap70 promotes proximal TCR signaling. Science signaling. 2019;12(577). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Courtney AH, Lo WL, Weiss A. TCR Signaling: Mechanisms of Initiation and Propagation. Trends Biochem Sci. 2018;43(2):108–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chakraborty AK, Weiss A. Insights into the initiation of TCR signaling. Nat Immunol. 2014;15(9):798–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lo WL, Shah NH, Ahsan N, Horkova V, Stepanek O, Salomon AR, et al. Lck promotes Zap70-dependent LAT phosphorylation by bridging Zap70 to LAT. Nat Immunol. 2018;19(7):733–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balagopalan L, Kortum RL, Coussens NP, Barr VA, Samelson LE. The linker for activation of T cells (LAT) signaling hub: from signaling complexes to microclusters. J Biol Chem. 2015;290(44):26422–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tomlinson MG, Lin J, Weiss A. Lymphocytes with a complex: adapter proteins in antigen receptor signaling. Immunol Today. 2000;21(11):584–91. [DOI] [PubMed] [Google Scholar]
- 11.Lindquist JA, Simeoni L, Schraven B. Transmembrane adapters: attractants for cytoplasmic effectors. Immunol Rev. 2003;191:165–82. [DOI] [PubMed] [Google Scholar]
- 12.Almena M, Merida I. Shaping up the membrane: diacylglycerol coordinates spatial orientation of signaling. Trends Biochem Sci. 2011;36(11):593–603. [DOI] [PubMed] [Google Scholar]
- 13.Fu G, Chen Y, Yu M, Podd A, Schuman J, He Y, et al. Phospholipase C{gamma}1 is essential for T cell development, activation, and tolerance. J Exp Med. 2010;207(2):309–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang YH, Sauer K. Lipid signaling in T-cell development and function. Cold Spring Harbor perspectives in biology. 2010;2(11):a002428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hogan PG, Lewis RS, Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu Rev Immunol. 2010;28:491–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Feske S Calcium signalling in lymphocyte activation and disease. Nat Rev Immunol. 2007;7(9):690–702. [DOI] [PubMed] [Google Scholar]
- 17.Gifford JL, Walsh MP, Vogel HJ. Structures and metal-ion-binding properties of the Ca2+-binding helix-loop-helix EF-hand motifs. Biochem J. 2007;405(2):199–221. [DOI] [PubMed] [Google Scholar]
- 18.Feske S, Wulff H, Skolnik EY. Ion channels in innate and adaptive immunity. Annu Rev Immunol. 2015;33:291–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carrasco S, Merida I. Diacylglycerol, when simplicity becomes complex. Trends Biochem Sci. 2007;32(1):27–36. [DOI] [PubMed] [Google Scholar]
- 20.Krishna S, Zhong X. Role of diacylglycerol kinases in T cell development and function. Crit Rev Immunol. 2013;33(2):97–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guo R, Wan CK, Carpenter JH, Mousallem T, Boustany RM, Kuan CT, et al. Synergistic control of T cell development and tumor suppression by diacylglycerol kinase alpha and zeta. Proc Natl Acad Sci U S A. 2008;105(33):11909–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kunkel MT, Newton AC. Calcium transduces plasma membrane receptor signals to produce diacylglycerol at Golgi membranes. J Biol Chem. 2010;285(30):22748–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sato M, Ueda Y, Umezawa Y. Imaging diacylglycerol dynamics at organelle membranes. Nature methods. 2006;3(10):797–9. [DOI] [PubMed] [Google Scholar]
- 24.Carrasco S, Merida I. Diacylglycerol-dependent binding recruits PKCtheta and RasGRP1 C1 domains to specific subcellular localizations in living T lymphocytes. Mol Biol Cell. 2004;15(6):2932–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spitaler M, Cantrell DA. Protein kinase C and beyond. Nat Immunol. 2004;5(8):785–90. [DOI] [PubMed] [Google Scholar]
- 26.Ksionda O, Limnander A, Roose JP. RasGRP Ras guanine nucleotide exchange factors in cancer. Front Biol (Beijing). 2013;8(5):508–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spitaler M, Emslie E, Wood CD, Cantrell D. Diacylglycerol and protein kinase D localization during T lymphocyte activation. Immunity. 2006;24(5):535–46. [DOI] [PubMed] [Google Scholar]
- 28.Melowic HR, Stahelin RV, Blatner NR, Tian W, Hayashi K, Altman A, et al. Mechanism of diacylglycerol-induced membrane targeting and activation of protein kinase Ctheta. J Biol Chem. 2007;282(29):21467–76. [DOI] [PubMed] [Google Scholar]
- 29.Pfeifhofer C, Kofler K, Gruber T, Tabrizi NG, Lutz C, Maly K, et al. Protein kinase C theta affects Ca2+ mobilization and NFAT cell activation in primary mouse T cells. J Exp Med. 2003;197(11):1525–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quann EJ, Merino E, Furuta T, Huse M. Localized diacylglycerol drives the polarization of the microtubule-organizing center in T cells. Nat Immunol. 2009;10(6):627–35. [DOI] [PubMed] [Google Scholar]
- 31.Monks CR, Kupfer H, Tamir I, Barlow A, Kupfer A. Selective modulation of protein kinase C-theta during T-cell activation. Nature. 1997;385(6611):83–6. [DOI] [PubMed] [Google Scholar]
- 32.Wang X, Chuang HC, Li JP, Tan TH. Regulation of PKC-theta function by phosphorylation in T cell receptor signaling. Frontiers in immunology. 2012;3:197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coudronniere N, Villalba M, Englund N, Altman A. NF-kappa B activation induced by T cell receptor/CD28 costimulation is mediated by protein kinase C-theta. Proc Natl Acad Sci U S A. 2000;97(7):3394–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lin X, O’Mahony A, Mu Y, Geleziunas R, Greene WC. Protein kinase C-theta participates in NF-kappaB activation induced by CD3-CD28 costimulation through selective activation of IkappaB kinase beta [In Process Citation]. Mol Cell Biol. 2000;20(8):2933–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun Z, Arendt CW, Ellmeier W, Schaeffer EM, Sunshine MJ, Gandhi L, et al. PKC-theta is required for TCR-induced NF-kappaB activation in mature but not immature T lymphocytes. Nature. 2000;404(6776):402–7. [DOI] [PubMed] [Google Scholar]
- 36.Roose JP, Mollenauer M, Gupta VA, Stone J, Weiss A. A Diacylglycerol-Protein Kinase C-RasGRP1 Pathway Directs Ras Activation upon Antigen Receptor Stimulation of T Cells. Mol Cell Biol. 2005;25(11):4426–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1989;49(17):4682–9. [PubMed] [Google Scholar]
- 38.Downward J, Graves JD, Warne PH, Rayter S, Cantrell DA. Stimulation of p21ras upon T-cell activation. Nature. 1990;346(6286):719–23. [DOI] [PubMed] [Google Scholar]
- 39.Ahearn IM, Haigis K, Bar-Sagi D, Philips MR. Regulating the regulator: post-translational modification of RAS. Nature reviews. 2011;13(1):39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fischer AM, Katayama CD, Pages G, Pouyssegur J, Hedrick SM. The role of erk1 and erk2 in multiple stages of T cell development. Immunity. 2005;23(4):431–43. [DOI] [PubMed] [Google Scholar]
- 41.Downward J Cancer biology: signatures guide drug choice. Nature. 2006;439(7074):274–5. [DOI] [PubMed] [Google Scholar]
- 42.Schubbert S, Bollag G, Shannon K. Deregulated Ras signaling in developmental disorders: new tricks for an old dog. Curr Opin Genet Dev. 2007;17(1):15–22. [DOI] [PubMed] [Google Scholar]
- 43.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2(7):489–501. [DOI] [PubMed] [Google Scholar]
- 44.Jun JE, Rubio I, Roose JP. Regulation of Ras Exchange Factors and Cellular Localization of Ras Activation by Lipid Messengers in T Cells. Frontiers in immunology. 2013;4:239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Iwig JS, Vercoulen Y, Das R, Barros T, Limnander A, Che Y, et al. Structural analysis of autoinhibition in the Ras-specific exchange factor RasGRP1. eLife. 2013;2:e00813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vercoulen Y, Kondo Y, Iwig JS, Janssen AB, White KA, Amini M, et al. A Histidine pH sensor regulates activation of the Ras-specific guanine nucleotide exchange factor RasGRP1. eLife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dower NA, Stang SL, Bottorff DA, Ebinu JO, Dickie P, Ostergaard HL, et al. RasGRP is essential for mouse thymocyte differentiation and TCR signaling. Nat Immunol. 2000;1(4):317–21. [DOI] [PubMed] [Google Scholar]
- 48.Hartzell C, Ksionda O, Lemmens E, Coakley K, Yang M, Dail M, et al. Dysregulated RasGRP1 Responds to Cytokine Receptor Input in T Cell Leukemogenesis. Science signaling. 2013;6(268):ra21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klinger MB, Guilbault B, Goulding RE, Kay RJ. Deregulated expression of RasGRP1 initiates thymic lymphomagenesis independently of T-cell receptors. Oncogene. 2005;24(16):2695–704. [DOI] [PubMed] [Google Scholar]
- 50.Oki T, Kitaura J, Watanabe-Okochi N, Nishimura K, Maehara A, Uchida T, et al. Aberrant expression of RasGRP1 cooperates with gain-of-function NOTCH1 mutations in T-cell leukemogenesis. Leukemia. 2012;26(5):1038–45. [DOI] [PubMed] [Google Scholar]
- 51.Ksionda O, Melton AA, Bache J, Tenhagen M, Bakker J, Harvey R, et al. RasGRP1 overexpression in T-ALL increases basal nucleotide exchange on Ras rendering the Ras/PI3K/Akt pathway responsive to protumorigenic cytokines. Oncogene. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Platt CD, Fried AJ, Hoyos-Bachiloglu R, Usmani GN, Schmidt B, Whangbo J, et al. Combined immunodeficiency with EBV positive B cell lymphoma and epidermodysplasia verruciformis due to a novel homozygous mutation in RASGRP1. Clinical immunology. 2017;183:142–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roose JP. Lost GRP on cytotoxicity? Nat Immunol. 2016;17(12):1339–40. [DOI] [PubMed] [Google Scholar]
- 54.Salzer E, Cagdas D, Hons M, Mace EM, Garncarz W, Petronczki OY, et al. RASGRP1 deficiency causes immunodeficiency with impaired cytoskeletal dynamics. Nat Immunol. 2016;17(12):1352–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Winter S, Martin E, Boutboul D, Lenoir C, Boudjemaa S, Petit A, et al. Loss of RASGRP1 in humans impairs T-cell expansion leading to Epstein-Barr virus susceptibility. EMBO Mol Med. 2018;10(2):188–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Daley SR, Coakley KM, Hu DY, Randall KL, Jenne CN, Limnander A, et al. Rasgrp1 mutation increases naive T-cell CD44 expression and drives mTOR-dependent accumulation of Helios+ T cells and autoantibodies. eLife. 2013;2:e01020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Plagnol V, Howson JM, Smyth DJ, Walker N, Hafler JP, Wallace C, et al. Genome-wide association analysis of autoantibody positivity in type 1 diabetes cases. PLoS genetics. 2011;7(8):e1002216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Qu HQ, Grant SF, Bradfield JP, Kim C, Frackelton E, Hakonarson H, et al. Association of RASGRP1 with type 1 diabetes is revealed by combined follow-up of two genome-wide studies. Journal of medical genetics. 2009;46(8):553–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yasuda S, Stevens RL, Terada T, Takeda M, Hashimoto T, Fukae J, et al. Defective expression of Ras guanyl nucleotide-releasing protein 1 in a subset of patients with systemic lupus erythematosus. J Immunol. 2007;179(7):4890–900. [DOI] [PubMed] [Google Scholar]
- 60.Molineros JE, Singh B, Terao C, Okada Y, Kaplan J, McDaniel B, et al. Mechanistic Characterization of RASGRP1 Variants Identifies an hnRNP-K-Regulated Transcriptional Enhancer Contributing to SLE Susceptibility. Frontiers in immunology. 2019;10:1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gorentla BK, Wan CK, Zhong XP. Negative regulation of mTOR activation by diacylglycerol kinases. Blood. 2011;117(15):4022–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Myers DR, Norlin E, Vercoulen Y, Roose JP. Active Tonic mTORC1 Signals Shape Baseline Translation in Naive T Cells. Cell reports. 2019;27(6):1858–74 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nature reviews. 2011;12(1):21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Powell JD, Delgoffe GM. The mammalian target of rapamycin: linking T cell differentiation, function, and metabolism. Immunity. 2010;33(3):301–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kim J, Guan KL. mTOR as a central hub of nutrient signalling and cell growth. Nat Cell Biol. 2019;21(1):63–71. [DOI] [PubMed] [Google Scholar]
- 67.Heikamp EB, Patel CH, Collins S, Waickman A, Oh MH, Sun IH, et al. The AGC kinase SGK1 regulates TH1 and TH2 differentiation downstream of the mTORC2 complex. Nat Immunol. 2014;15(5):457–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ebner M, Sinkovics B, Szczygiel M, Ribeiro DW, Yudushkin I. Localization of mTORC2 activity inside cells. J Cell Biol. 2017;216(2):343–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Finlay DK, Rosenzweig E, Sinclair LV, Feijoo-Carnero C, Hukelmann JL, Rolf J, et al. PDK1 regulation of mTOR and hypoxia-inducible factor 1 integrate metabolism and migration of CD8+ T cells. J Exp Med. 2012;209(13):2441–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Perera RM, Zoncu R. The Lysosome as a Regulatory Hub. Annual review of cell and developmental biology. 2016;32:223–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, et al. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science. 2008;320(5882):1496–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pollizzi KN, Powell JD. Regulation of T cells by mTOR: the known knowns and the known unknowns. Trends Immunol. 2015;36(1):13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nature reviews. 2009;10(5):307–18. [DOI] [PubMed] [Google Scholar]
- 74.Maciver NJ, Michalek RD, Rathmell JC. Metabolic Regulation of T Lymphocytes. Annu Rev Immunol. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Piques M, Schulze WX, Hohne M, Usadel B, Gibon Y, Rohwer J, et al. Ribosome and transcript copy numbers, polysome occupancy and enzyme dynamics in Arabidopsis. Molecular systems biology. 2009;5:314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Piccirillo CA, Bjur E, Topisirovic I, Sonenberg N, Larsson O. Translational control of immune responses: from transcripts to translatomes. Nat Immunol. 2014;15(6):503–11. [DOI] [PubMed] [Google Scholar]
- 77.Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nature reviews. 2010;11(2):113–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gingras AC, Raught B, Sonenberg N. Regulation of translation initiation by FRAP/mTOR. Genes Dev. 2001;15(7):807–26. [DOI] [PubMed] [Google Scholar]
- 79.Hsieh AC, Liu Y, Edlind MP, Ingolia NT, Janes MR, Sher A, et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature. 2012;485(7396):55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bjur E, Larsson O, Yurchenko E, Zheng L, Gandin V, Topisirovic I, et al. Distinct translational control in CD4+ T cell subsets. PLoS genetics. 2013;9(5):e1003494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35(6):871–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yang K, Shrestha S, Zeng H, Karmaus PW, Neale G, Vogel P, et al. T cell exit from quiescence and differentiation into Th2 cells depend on Raptor-mTORC1-mediated metabolic reprogramming. Immunity. 2013;39(6):1043–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zeng H, Yang K, Cloer C, Neale G, Vogel P, Chi H. mTORC1 couples immune signals and metabolic programming to establish T(reg)-cell function. Nature. 2013;499(7459):485–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ho IC, Tai TS, Pai SY. GATA3 and the T-cell lineage: essential functions before and after T-helper-2-cell differentiation. Nat Rev Immunol. 2009;9(2):125–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cook KD, Miller J. TCR-dependent translational control of GATA-3 enhances Th2 differentiation. J Immunol. 2010;185(6):3209–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Scheu S, Stetson DB, Reinhardt RL, Leber JH, Mohrs M, Locksley RM. Activation of the integrated stress response during T helper cell differentiation. Nat Immunol. 2006;7(6):644–51. [DOI] [PubMed] [Google Scholar]
- 87.Gigoux M, Lovato A, Leconte J, Leung J, Sonenberg N, Suh WK. Inducible costimulator facilitates T-dependent B cell activation by augmenting IL-4 translation. Molecular immunology. 2014;59(1):46–54. [DOI] [PubMed] [Google Scholar]
- 88.Gingras AC, Kennedy SG, O’Leary MA, Sonenberg N, Hay N. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev. 1998;12(4):502–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Murakami M, Ichisaka T, Maeda M, Oshiro N, Hara K, Edenhofer F, et al. mTOR is essential for growth and proliferation in early mouse embryos and embryonic stem cells. Mol Cell Biol. 2004;24(15):6710–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shima H, Pende M, Chen Y, Fumagalli S, Thomas G, Kozma SC. Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J. 1998;17(22):6649–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang S, Readinger JA, DuBois W, Janka-Junttila M, Robinson R, Pruitt M, et al. Constitutive reductions in mTOR alter cell size, immune cell development, and antibody production. Blood. 2011;117(4):1228–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B, et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity. 2009;30(6):832–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.So L, Lee J, Palafox M, Mallya S, Woxland CG, Arguello M, et al. The 4E-BP-eIF4E axis promotes rapamycin-sensitive growth and proliferation in lymphocytes. Science signaling. 2016;9(430):ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol. 2011;12(4):295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zeng H, Cohen S, Guy C, Shrestha S, Neale G, Brown SA, et al. mTORC1 and mTORC2 Kinase Signaling and Glucose Metabolism Drive Follicular Helper T Cell Differentiation. Immunity. 2016;45(3):540–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yang J, Lin X, Pan Y, Wang J, Chen P, Huang H, et al. Critical roles of mTOR Complex 1 and 2 for T follicular helper cell differentiation and germinal center responses. eLife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pearce EL, Poffenberger MC, Chang CH, Jones RG. Fueling immunity: insights into metabolism and lymphocyte function. Science. 2013;342(6155):1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Powell JD, Pollizzi KN, Heikamp EB, Horton MR. Regulation of immune responses by mTOR. Annu Rev Immunol. 2012;30:39–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Finlay D, Cantrell DA. Metabolism, migration and memory in cytotoxic T cells. Nat Rev Immunol. 2011;11(2):109–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.O’Shea JJ, Paul WE. Mechanisms underlying lineage commitment and plasticity of helper CD4+ T cells. Science. 2010;327(5969):1098–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Crotty S Follicular helper CD4 T cells (TFH). Annu Rev Immunol. 2011;29:621–63. [DOI] [PubMed] [Google Scholar]
- 102.Procaccini C, De Rosa V, Galgani M, Abanni L, Cali G, Porcellini A, et al. An oscillatory switch in mTOR kinase activity sets regulatory T cell responsiveness. Immunity. 2010;33(6):929–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Finley LWS, Thompson CB The Metabolism of Cell Growth and Proliferation. The Molecular Basis of Cancer 2015;Fourth Edition:Pages 191–208.e2. [Google Scholar]
- 104.Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186(6):3299–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gerriets VA, Kishton RJ, Johnson MO, Cohen S, Siska PJ, Nichols AG, et al. Foxp3 and Toll-like receptor signaling balance Treg cell anabolic metabolism for suppression. Nat Immunol. 2016;17(12):1459–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, et al. mTOR regulates memory CD8 T-cell differentiation. Nature. 2009;460(7251):108–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature. 2009;460(7251):103–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Klein Geltink RI, O’Sullivan D, Corrado M, Bremser A, Buck MD, Buescher JM, et al. Mitochondrial Priming by CD28. Cell. 2017;171(2):385–97 e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, et al. The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 2014;20(1):61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sinclair LV, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol. 2013;14(5):500–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Edinger AL, Thompson CB. Akt maintains cell size and survival by increasing mTOR-dependent nutrient uptake. Mol Biol Cell. 2002;13(7):2276–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med. 2011;208(7):1367–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O’Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153(6):1239–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Pollizzi KN, Sun IH, Patel CH, Lo YC, Oh MH, Waickman AT, et al. Asymmetric inheritance of mTORC1 kinase activity during division dictates CD8(+) T cell differentiation. Nat Immunol. 2016;17(6):704–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sinclair LV, Howden AJ, Brenes A, Spinelli L, Hukelmann JL, Macintyre AN, et al. Antigen receptor control of methionine metabolism in T cells. eLife. 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Preston GC, Sinclair LV, Kaskar A, Hukelmann JL, Navarro MN, Ferrero I, et al. Single cell tuning of Myc expression by antigen receptor signal strength and interleukin-2 in T lymphocytes. EMBO J. 2015;34(15):2008–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Verbist KC, Guy CS, Milasta S, Liedmann S, Kaminski MM, Wang R, et al. Metabolic maintenance of cell asymmetry following division in activated T lymphocytes. Nature. 2016;532(7599):389–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Borsa M, Barnstorf I, Baumann NS, Pallmer K, Yermanos A, Grabnitz F, et al. Modulation of asymmetric cell division as a mechanism to boost CD8(+) T cell memory. Sci Immunol. 2019;4(34). [DOI] [PubMed] [Google Scholar]
- 119.Bhandoola A, Tai X, Eckhaus M, Auchincloss H, Mason K, Rubin SA, et al. Peripheral expression of self-MHC-II influences the reactivity and self-tolerance of mature CD4(+) T cells: evidence from a lymphopenic T cell model. Immunity. 2002;17(4):425–36. [DOI] [PubMed] [Google Scholar]
- 120.Myers DR, Zikherman J, Roose JP. Tonic Signals: Why Do Lymphocytes Bother? Trends Immunol. 2017;38(11):844–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Myers DR, Lau T, Markegard E, Lim HW, Kasler H, Zhu M, et al. Tonic LAT-HDAC7 Signals Sustain Nur77 and Irf4 Expression to Tune Naive CD4 T Cells. Cell reports. 2017;19(8):1558–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Stefanova I, Dorfman JR, Germain RN. Self-recognition promotes the foreign antigen sensitivity of naive T lymphocytes. Nature. 2002;420(6914):429–34. [DOI] [PubMed] [Google Scholar]
- 123.van Oers NS, Killeen N, Weiss A. ZAP-70 is constitutively associated with tyrosine-phosphorylated TCR zeta in murine thymocytes and lymph node T cells. Immunity. 1994;1(8):675–85. [DOI] [PubMed] [Google Scholar]
- 124.van Oers NS, Tao W, Watts JD, Johnson P, Aebersold R, Teh HS. Constitutive tyrosine phosphorylation of the T-cell receptor (TCR) zeta subunit: regulation of TCR-associated protein tyrosine kinase activity by TCR zeta. Mol Cell Biol. 1993;13(9):5771–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hogquist KA, Starr TK, Jameson SC. Receptor sensitivity: when T cells lose their sense of self. Curr Biol. 2003;13(6):R239–41. [DOI] [PubMed] [Google Scholar]
- 126.Roose JP, Diehn M, Tomlinson MG, Lin J, Alizadeh AA, Botstein D, et al. T Cell Receptor-Independent Basal Signaling via Erk and Abl Kinases Suppresses RAG Gene Expression. PLoS Biol. 2003;1(2):E53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zikherman J, Jenne C, Watson S, Doan K, Raschke W, Goodnow CC, et al. CD45-Csk phosphatase-kinase titration uncouples basal and inducible T cell receptor signaling during thymic development. Immunity. 2010;32(3):342–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Nakayama T, Singer A, Hsi ED, Samelson LE. Intrathymic signalling in immature CD4+CD8+ thymocytes results in tyrosine phosphorylation of the T-cell receptor zeta chain. Nature. 1989;341(6243):651–4. [DOI] [PubMed] [Google Scholar]
- 129.Azzam HS, Grinberg A, Lui K, Shen H, Shores EW, Love PE. CD5 expression is developmentally regulated by T cell receptor (TCR) signals and TCR avidity. J Exp Med. 1998;188(12):2301–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zikherman J, Parameswaran R, Weiss A. Endogenous antigen tunes the responsiveness of naive B cells but not T cells. Nature. 2012;489(7414):160–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mingueneau M, Roncagalli R, Gregoire C, Kissenpfennig A, Miazek A, Archambaud C, et al. Loss of the LAT adaptor converts antigen-responsive T cells into pathogenic effectors that function independently of the T cell receptor. Immunity. 2009;31(2):197–208. [DOI] [PubMed] [Google Scholar]
- 132.Shen S, Zhu M, Lau J, Chuck M, Zhang W. The essential role of LAT in thymocyte development during transition from the double-positive to single-positive stage. J Immunol. 2009;182(9):5596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Markegard E, Trager E, Yang CW, Zhang W, Weiss A, Roose JP. Basal LAT-diacylglycerol-RasGRP1 Signals in T Cells Maintain TCRalpha Gene Expression. PloS one. 2011;6(9):e25540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dequiedt F, Kasler H, Fischle W, Kiermer V, Weinstein M, Herndier BG, et al. HDAC7, a thymus-specific class II histone deacetylase, regulates Nur77 transcription and TCR-mediated apoptosis. Immunity. 2003;18(5):687–98. [DOI] [PubMed] [Google Scholar]
- 135.Kasler HG, Verdin E. Histone deacetylase 7 functions as a key regulator of genes involved in both positive and negative selection of thymocytes. Mol Cell Biol. 2007;27(14):5184–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Oh-hora M, Johmura S, Hashimoto A, Hikida M, Kurosaki T. Requirement for Ras guanine nucleotide releasing protein 3 in coupling phospholipase C-gamma2 to Ras in B cell receptor signaling. J Exp Med. 2003;198(12):1841–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature. 2012;485(7396):109–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS. Genome-wide annotation and quantitation of translation by ribosome profiling. Curr Protoc Mol Biol. 2013;Chapter 4:Unit 4 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ricciardi S, Manfrini N, Alfieri R, Calamita P, Crosti MC, Gallo S, et al. The Translational Machinery of Human CD4(+) T Cells Is Poised for Activation and Controls the Switch from Quiescence to Metabolic Remodeling. Cell Metab. 2018;28(6):895–906 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Oaks Z, Winans T, Caza T, Fernandez D, Liu Y, Landas SK, et al. Mitochondrial Dysfunction in the Liver and Antiphospholipid Antibody Production Precede Disease Onset and Respond to Rapamycin in Lupus-Prone Mice. Arthritis Rheumatol. 2016;68(11):2728–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Fernandez D, Perl A. mTOR signaling: a central pathway to pathogenesis in systemic lupus erythematosus? Discovery medicine. 2010;9(46):173–8. [PMC free article] [PubMed] [Google Scholar]
- 142.Yin Y, Choi SC, Xu Z, Perry DJ, Seay H, Croker BP, et al. Normalization of CD4+ T cell metabolism reverses lupus. Science translational medicine. 2015;7(274):274ra18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Abboud G, Choi SC, Kanda N, Zeumer-Spataro L, Roopenian DC, Morel L. Inhibition of Glycolysis Reduces Disease Severity in an Autoimmune Model of Rheumatoid Arthritis. Frontiers in immunology. 2018;9:1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Morel L Immunometabolism in systemic lupus erythematosus. Nature reviews Rheumatology. 2017;13(5):280–90. [DOI] [PubMed] [Google Scholar]
- 145.Rodig SJ, Gusenleitner D, Jackson DG, Gjini E, Giobbie-Hurder A, Jin C, et al. MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Science translational medicine. 2018;10(450). [DOI] [PubMed] [Google Scholar]
- 146.Lim WA, June CH. The Principles of Engineering Immune Cells to Treat Cancer. Cell. 2017;168(4):724–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Watanabe K, Kuramitsu S, Posey AD Jr., June CH Expanding the Therapeutic Window for CAR T Cell Therapy in Solid Tumors: The Knowns and Unknowns of CAR T Cell Biology. Frontiers in immunology. 2018;9:2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4–1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21(6):581–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kawalekar OU RS OC, Fraietta JA, Guo L, McGettigan SE, Posey AD, Jr., et al. Distinct Signaling of Coreceptors Regulates Specific Metabolism Pathways and Impacts Memory Development in CAR T Cells. Immunity. 2016;44(3):712. [DOI] [PubMed] [Google Scholar]
- 150.Farhood B, Najafi M, Mortezaee K. CD8(+) cytotoxic T lymphocytes in cancer immunotherapy: A review. J Cell Physiol. 2019;234(6):8509–21. [DOI] [PubMed] [Google Scholar]
- 151.Dyck L, Mills KHG. Immune checkpoints and their inhibition in cancer and infectious diseases. Eur J Immunol. 2017;47(5):765–79. [DOI] [PubMed] [Google Scholar]
- 152.Previte DM, Martins CP, O’Connor EC, Marre ML, Coudriet GM, Beck NW, et al. Lymphocyte Activation Gene-3 Maintains Mitochondrial and Metabolic Quiescence in Naive CD4(+) T Cells. Cell reports. 2019;27(1):129–41 e4. [DOI] [PubMed] [Google Scholar]
- 153.Ellestad KK, Lin J, Boon L, Anderson CC. PD-1 Controls Tonic Signaling and Lymphopenia-Induced Proliferation of T Lymphocytes. Frontiers in immunology. 2017;8:1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wei SC, Sharma R, Anang NAS, Levine JH, Zhao Y, Mancuso JJ, et al. Negative Co-stimulation Constrains T Cell Differentiation by Imposing Boundaries on Possible Cell States. Immunity. 2019;50(4):1084–98 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gavin MA, Bevan MJ. Increased peptide promiscuity provides a rationale for the lack of N regions in the neonatal T cell repertoire. Immunity. 1995;3(6):793–800. [DOI] [PubMed] [Google Scholar]