

Abstract
Epigenetic agents such as bromodomain and extra terminal region inhibitors (BETi) slow tumor growth via tumor intrinsic alterations; however, their effects on anti-tumor immunity remain unclear. A recent advance is the development of next generation BETi that are potent and display a favorable half-life. Here, we tested the BETi, PLX51107, for immune-based effects on tumor growth in BRAF V600E melanoma syngeneic models. PLX51107 delayed melanoma tumor growth and increased activated, proliferating and functional CD8+ T cells in tumors leading to CD8+ T cell-mediated tumor growth delay. PLX51107 decreased Cox2 expression, increased dendritic cells and lowered PD-L1, FasL, and IDO-1 expression in the tumor microenvironment. Importantly, PLX51107 delayed the growth of tumors that progressed on anti-PD-1 therapy; a response associated with decreased Cox2 levels, decreased PD-L1 expression on non-immune cells and increased intratumoral CD8+ T cells. Thus, next generation BETi represent a potential first-line and secondary treatment strategy for metastatic melanoma by eliciting effects, at least in part, on anti-tumor CD8+ T cells.
Keywords: Melanoma, T cells, BET inhibitor, anti-PD-1 non-responsive, Cox2
Introduction
There is increasing interest in targeting transcriptional programs that drive cancer progression. Bromodomain (BRD) and Extra-Terminal Domain (BET) proteins are evolutionarily conserved epigenetic readers that recruit transcriptional RNA polymerase II to acetylated lysines on histones (Filippakopoulos & Knapp, 2014). BRD/BET proteins widely regulate the expression of genes involved in cell proliferation and survival, as well as eliciting effects on inflammation (Filippakopoulos et al., 2014; Gallagher, Tiffen, & Hersey, 2015). Their action is blocked by BET inhibitors (BETi), which have been investigated in models of leukemia, ovarian cancer and melanoma (Filippakopoulos et al., 2014; Gallagher et al., 2015; Jung, Gelato, Fernandez-Montalvan, Siegel, & Haendler, 2015; Paoluzzi et al., 2016; Segura et al., 2013), as well as immune-mediated diseases (Chen et al., 2016; Klein et al., 2016; Sun et al., 2015). BETi also impact pro- and anti-inflammatory responses by altering cytokine expression, inducing pro-inflammatory macrophages, affecting dendritic cell (DC) activation and regulating T cell activation and differentiation (Adeegbe et al., 2017; Das et al., 2015; Kagoya et al., 2016; Qiao & Ivashkiv, 2015; Toniolo et al., 2015; Xu & Vakoc, 2014). Despite their efficacy, first generation BETi (JQ1, iBET762, and OTX015) have hematologic dose-limiting toxicities at pharmacologically effective doses in mice and humans (Amorim et al., 2016; Berthon et al., 2016; Lee et al., 2016). Thus, new structurally unique BETi have been developed, such as PLX51107, which have improved therapeutic indexes, shorter half-lives, and robust efficacy in preclinical leukemia models (Ozer et al., 2018). PLX51107 is now in phase I dose-escalation clinical trials (NCT02683395).
Melanoma is regarded as an immunogenic tumor and immune therapies, such as anti-CTLA-4 (ipilimumab) and anti-PD-1 (pembrolizumab and nivolumab), have improved overall survival for many melanoma patients (Sharma, Hu-Lieskovan, Wargo, & Ribas, 2017). However, the majority of patients treated with anti-CTLA-4 or anti-PD-1 monotherapy are non-responsive (O’Donnell, Long, Scolyer, Teng, & Smyth, 2017) and combinations of anti-PD-1 with anti-CTLA-4 lead to grade 3–5 adverse events (Boutros et al., 2016). It is crucial to identify new front-line and second-line therapies for metastatic melanoma that target multiple mechanisms of tumor progression, including anti-tumor immunity. BRD/BET proteins are overexpressed in melanoma (Segura et al., 2013) and BETi target tumor growth mechanisms in melanoma (Dar et al., 2015; Gallagher, Mijatov, Gunatilake, Gowrishankar, et al., 2014; Gallagher, Mijatov, Gunatilake, Tiffen, et al., 2014; Gallagher et al., 2015; Paoluzzi et al., 2016). Additionally, first generation BETi alters the tumor immune microenvironment by the decreasing PD-L1 levels and inducing CD8+ T cell-mediated effects in lymphoma and ovarian cancer (Hogg et al., 2017; Zhu et al., 2016). These attributes make BETi a potentially impactful primary and secondary treatment for melanoma; however, their effects on anti-tumor immunity remain unclear.
Our previous work suggests that PLX51107 alters T cells in the tumor immune microenvironment (Nikbakht, Tiago, Erkes, Chernova, & Aplin, 2019), leading to successful combination with PD-L1 blockade. How these changes impact the efficacy of PLX51107 in melanoma remains unclear. Here, we tested PLX51107 in immune-competent BRAF V600E melanoma as a primary and secondary therapy and analyzed effects on the tumor microenvironment and tumor-infiltrating lymphocytes (TIL). We show that PLX51107 slowed the growth of mouse BRAF V600E melanoma tumors. PLX51107 efficacy was CD8+ T cell-mediated and were associated with decreased Cox2 expression, influx of activated DCs, and decreased expression of immune inhibitory molecules in the tumor microenvironment. Additionally, PLX51107 treatment was an effective second-line therapy for tumors that did not respond to anti-PD-1 treatment. Together, this work demonstrates that PLX51107 impacts tumor growth by inducing CD8+ T cell-mediated anti-tumor effects and, thus, represents a potential therapy for metastatic melanoma.
Materials and Methods
Cell lines
D4M3.A cells (donated by Drs. Constance Brinckerhoff and David Mullins, Dartmouth University, 2016) were cultured in DMEM/F12 advanced with 5% FBS and 1% L-Glutamine. YUMM3.3, and YUMM4.1 cells (donated by Dr. Marcus Bosenberg, Yale University, 2017) were cultured in DMEMF-12 50/50 with 10% FBS and 1% non-essential amino acids. Mutational status for mouse cell lines are described in Supplemental Fig. 1A. A375 cells (purchased from ATCC, 2005) were cultured in DMEM with 10% FBS. WM793 cells (donated by Dr. Meenhard Herlyn, The Wistar Institute, 2005) were grown in WM media (MCDB153 with 2% FBS, 10% Leibowtiz’s L-15 medium, 5 µg/ml insulin). M238 cells (donated by Dr. Antoni Ribas, UCLA, 2011) were grown in RPMI with 10% FBS and 1% L-glutamine. PenStrep (1%) was added to all media. CAF cultures isolated from tumors were validated by αSMA and FAP expression. All cell lines were STR analyzed, confirmed for BRAF V600E mutation, and IMPACT III PCR pathogen tested (IDEXX) to authenticate them and determine they were pathogen free.
In vivo tumor growth studies
Animal experiments were approved by the IACUC and performed at TJU in a facility accredited by the AAALAC. Male C57BL/6 mice (Jackson, 6–10 weeks) were used unless denoted. Tumors were implanted intradermally in 100 µL HBSS. Tumor volume was tracked with a digital caliper: volume = (length x width2)*0.52. When tumor volume reached ~50mm3, animals were fed either control or PLX51107 (90 ppm PLX51107, AIN-76A) laced chow. For all experiments, mice were supplemented with intraperitoneal PBS injections or ClearH2O DietGel® Recovery to combat weight loss (Supplemental Fig. 1A) and dehydration. For tumor growth curves, X’s indicate when animals had to be euthanized early. For survival curves, circles indicate animals excluded due to early sacrifice or death. PLX51107 was provided by Plexxikon and chow was purchased from Research Diets. The structure for PLX51107 has previously been published (Ozer et al., 2018).
Western blot analysis
Protein lysates were prepared in Laemmli sample buffer, separated by SDS-PAGE, and proteins were transferred to PVDF membranes. Immunoreactivity was detected using HRP-conjugated secondary antibodies (CalBioTech) and chemiluminescence substrate (Thermo Scientific) on a Versadoc Imaging System (Bio-Rad). Primary antibodies were: anti-PD-L1 (mouse, Santa Cruz and human, Cell Signaling Tech), anti-Cox2 (Cell Signaling and AbCam), anti-HMGB-1 (Cell Signaling Tech), Bim/Bod (Enzo Life Sciences), anti-actin and anti-HSP90 (both Cell Signaling Tech).
Flow cytometry of tumor and spleen samples
Samples were analyzed on the BD Fortessa and data quantified with FlowJo software. For all techniques, detailed protocols and antibodies are available in the Supplemental Material. Tumors were removed and dissociated into single cell suspensions. Spleens were processed mechanically using a 70 µm nylon filter and a syringe plunger. For immunogenicity studies, cells were stained with a fixable live/dead stain (Zombie UV, BioLegend) per company instructions, fixed and permeabilized using BD Cytofix/Cytoperm kit (BD Biosciences), followed by antibody staining. For TIL studies, cells were first stained with a fixable live/dead stain followed by surface antibody staining (listed in Supplemental Materials). For Treg staining and Ki67 staining cells were fixed and nuclear permeabilized using the eBioscience™ Foxp3/Transcription factor buffer staining set and an antibody specific for FoxP3 (clone FJK-16s) or Ki67 (clone 16A8) following company instructions. For T cell re-stimulation assays, cytokine production by T cells was assessed after ex vivo stimulation with the eBioscience™ cell stimulation cocktail. ~0.5–4E6 cells were incubated in TCM for 5 hours at 37°C in 5% CO2, with the stimulation cocktail and 1 µg/ml brefeldin A (GolgiPlug; BD Biosciences). Cells were washed with ice-cold FACS, stained with Zombie UV and antibodies specific for surface proteins, and then analyzed for intracellular cytokines.
Reverse-phase protein array (RPPA) analysis
D4M3.A and YUMM3.3 (1E5) were plated in 6 well plates and treated, as indicated. Cells were lysed and proteins analyzed, as described previously (Tibes et al., 2006).
CRISPR
To generate Cox2 CRISPR clones, D4Ms were transfected with an Edit-R Cas9 expression plasmid with a puromycin resistance marker, crRNAs for Cox2 (Dharmacon), and tracrRNA using DharmaFECT Duo Transfection Reagent (GE Life Sciences) following company recommended protocols.
Fluorescence microscopy
Tumors isolated at sacrifice were frozen in Tissue-Plus OCT (Fisher Scientific) and cut into 6–8 µm sections. Samples were stained and images generated. Entire tumor sections were imaged using tiling, followed by quantification of PD-L1+, CD45.2 negative, or CD8+ cells using Fiji imaging software. Full details are described in the Supplemental Material.
In vivo CD8 depletion
To deplete CD8+ T cells, mice were treated with 300 µg of anti-CD8α (clone 53–6.72) or the irrelevant IgG2a antibody control every 3 days for the duration of the experiment, starting 2 days before tumor implantation.
Statistical analysis of tumor growth
The log-transformed tumor volumes were modeled as a low order polynomial function using a linear mixed effects (LME) model. Details on the fitted LME model are included in the Supplemental Material. The fitted model was used to estimate the doubling time of tumor volume.
Results
PLX51107 treatment inhibits BRAF V600E melanoma tumor growth.
The effect of PLX51107 on melanoma tumor growth and TIL in the tumor microenvironment was tested in immune-competent, BRAF V600E mouse melanoma models: D4M3.A and YUMM3.3 (Jenkins et al., 2014; Meeth, Wang, Micevic, Damsky, & Bosenberg, 2016). PLX51107 and the first generation BETi, JQ1, slowed growth of D4M3.A and YUMM3.3 cells in vitro (Fig. 1A and Supplemental Fig. 1B). Similar effects were observed in human A375 and WM793 BRAF V600E melanoma cell lines. Importantly, PLX51107 significantly inhibited D4M3.A and YUMM3.3 tumor growth in vivo (Fig. 1B) and prolonged animal survival in mice bearing tumors (Fig. 1C). PLX51107 treatment caused 10–15% weight loss in tumor-bearing mice (Supplemental Fig. 1C); however, weight was maintained with liquid supplementation (detailed in Materials and Methods). These data suggest that BETi delays the growth of cutaneous melanoma in the presence of an intact immune system.
Figure 1. BETi treatment delays the growth of BRAF V600E melanoma and improves the survival of tumor-bearing mice.
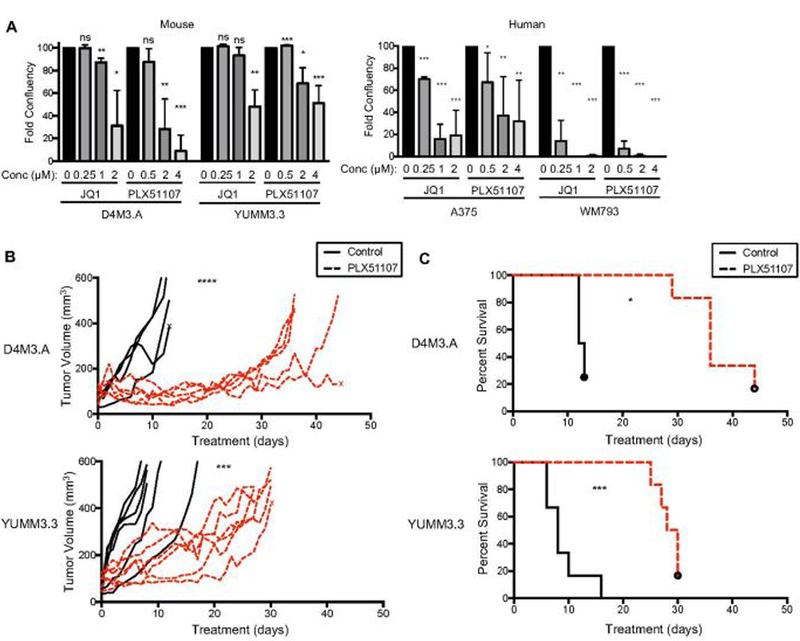
A) Growth of mouse (D4M3.A and YUMM3.3; n=3), and human (A375; n=5 and WM793; n=3) BRAF V600E melanoma cell lines exposed to different concentrations of BETi, JQ1 or PLX51107, for 6 days. Growth was assessed by crystal violet/colony formation assays. Data are displayed as the average percent of control. Significance was determined by unpaired t-test, *p<0.05, **p<0.01, ***p<0.001. B-C) Male C57BL/6 mice were intradermally implanted with mouse BRAF V600E melanoma cell lines, D4M3.A (3E5) or YUMM3.3 (2.5E5). Tumors were grown to ~ 50 mm3 after which animals were given either control (AIN-76A) or PLX51107 (90 ppm PLX51107, AIN-76A) laced chow. Data are combined from two experiments. B) Tumor growth is represented as change of volume (mm3) over time from the start of control or PLX51107 laced chow treatments. Significance ***p<0.001, ****p>0.0001. C) Tumor survival curves of mice ending when tumors were > 450 mm3. Significance was assessed by a logrank test, *p<0.05, ***p<0.001.
BETi treatment efficacy is CD8+ T cell-mediated.
To determine intratumoral T cell alterations induced by PLX51107, tumors were taken after four days of treatment. At this time point, tumor sizes between groups were equivalent. During PLX51107 treatment, CD8+ T cell numbers were increased in D4M3.A tumors (Fig. 2A and Supplemental Fig. 2). Additionally, PLX51107 treatment increased pro-inflammatory cytokine-producing, CD44+ (activated), and Ki67+ (proliferating) CD8+ T cells, and the CD8:Treg ratio in tumors (Fig. 2B-D and Supplemental Fig. 2). These immune cell alterations were mostly localized to TIL and not observed in peripheral immune cells isolated from spleens (Supplemental Fig. 3), suggesting tumor cell intrinsic effects of PLX51107.
Figure 2. BETi treatment elicits CD8+ T cell-mediated effects.

A-D) Data were obtained from D4M3.A tumors from mice after four days from the treatment start with control or PLX51107 chow. Tumors were similar sizes. Data are displayed as the mean (showing individual mice) percent of total cells +/− SD. A) CD8+ T cells cells in tumors. B) Cytokine production (IFNγ, IL-2 and TNFα) from CD8+ T cells in tumors. C) CD44 and Ki67 positivity of CD8 T cells from non-treated and PLX51107-treated tumors. D) The CD8:Treg ratio in tumors. Significance was assessed by unpaired t-test, *p<0.05. E-G) Mice were depleted of CD8a+ cells, as described in the Materials and Methods. Mice were given either control or PLX51107 laced chow when tumors reached ~50 mm3. E) Representative FACS plots showing the depletion of CD8+ cells in the blood on day 19 after beginning the CD8 depletion. F) Tumor growth in control or CD8 depleted mice, represented as change of volume (mm3) over time, shown from the start of control (anti-CD8a alone, n=3, isotype IgG2a alone, n=3) or PLX51107 diet (PLX51107 + anti-CD8a, n=8; PLX51107 + isotype IgG2a, n=8). Significance was assessed as described in Materials and Methods, ****p>0.0001. G) Tumor survival curves of isotype IgG2a or CD8-depleted D4M3.A tumor-bearing mice treated with control or PLX51107 laced chow, ending when tumors were >450 mm3. Dots indicate when animals were censored due to euthanizing when tumors were <450 mm3. Significance was assessed by a logrank test, ****p<0.0001.
To determine the importance of CD8+ T cell infiltration into tumors, we antibody-depleted CD8+ T cells from D4M3.A tumor-bearing mice and analyzed the response to PLX51107 treatment (Fig. 2E). CD8 depletion significantly abrogated PLX51107-induced tumor growth delay and reduced mouse survival (Fig. 2F and G). These data suggest that the influx of functional and activated CD8+ T cells into BRAF V600E melanoma tumors is required for robust responsiveness to PLX51107.
PLX51107 decreases Cox2 expression and induces DC influx.
It remained unclear how PLX51107 induced CD8+ T cell-mediated effects. PLX51107 minimally impacted the peripheral immune system (Supplemental Fig 3); thus, we hypothesized that the CD8+ T cell-mediated effects of PLX51107 were due to tumor cell intrinsic changes. We performed reverse phase protein array (RPPA), which showed that BETi decreased Cox2 expression and increased expression of the pro-death protein, Bim-EL in D4M3.A and YUMM3.3 cell lines (Fig. 3A and Supplemental Fig 4A). These findings were validated by western blot analysis of BETi-treated mouse (D4M3.A and YUMM3.3) and human (A375 and M238) BRAF V600E melanoma cell lines in vitro (Fig. 3B-C and Supplemental Fig. 4B). Cox2 expression has been linked to CD8+ T cell function and infiltration (Kalinski, 2011), as well as the influx of activated, immune-stimulatory DC populations responsible for T cell dependent tumor growth delay in melanoma (Zelenay et al., 2015). In vivo, PLX51107-treated D4M3.A tumors displayed decreased Cox2 expression and increased influx of CD103+, CD11b- DCs (Fig. 3D-E). PLX51107 treatment induced release of the toll like receptor (TLR) 4 ligand, HMGB-1 (Kang, 2013) (Supplemental Fig. 4C), which was associated with higher expression of the activation marker MHC-I on tumor infiltrating DCs (Fig. 3F). Additionally, we observed a reduction in the expression of IL-6 and CD62E in D4M3.A cells treated in vitro (Supplemental Fig. 5A), transcripts known to decrease in Cox2-deficient melanomas (Zelenay et al., 2015). Interestingly, transcript levels of Cox2 (ptgs2) were unaltered during BETi suggesting post-translational degradation of Cox2 (Alexanian & Sorokin, 2017) (Supplemental Fig. 5A). Together, these data suggest that PLX51107 decreases Cox2 expression and causes influx of activated DCs in the tumor microenvironment.
Figure 3. PLX51107 decreases Cox2 expression and induces DC influx.
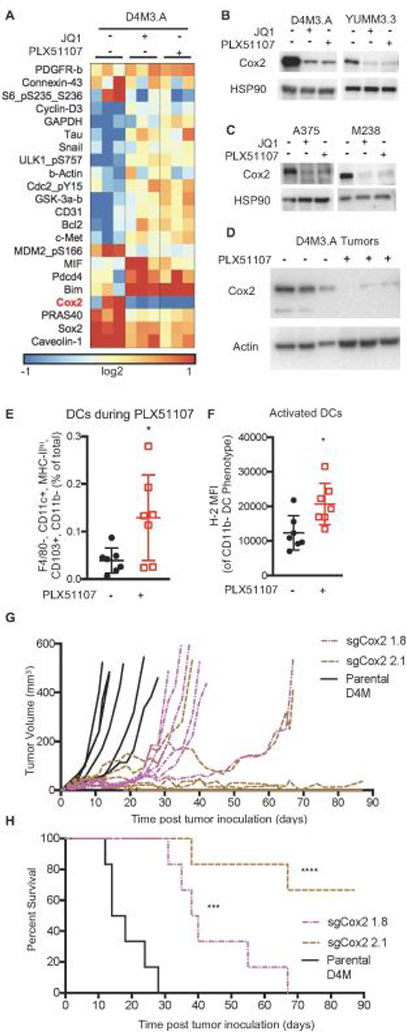
A) RPPA Heat map of median-centered log2-transformed values for each sample for antibodies determined to be significant. RPPA samples were prepared (n=3) after 48 hours of 1 µM JQ1 or 2 µM PLX51107 treatment. B) Confirmation Western blots of Cox2 from mouse D4M.3A and Yumm3.3 BRAF V600E melanoma cell lines after 48 hours of JQ1 or PLX51107 treatment (n=2–3 per line). HSP90 serves as a loading control. C) As for B but in human A375 and M238 cell lines. D) Cox2 levels in D4M3.A tumors at sacrifice after control or PLX51107 treatment (actin as loading control). E) CD103+, CD11b- DCs (F4/80-, CD11c+, MHC-II+) in tumors after 4 days of PLX51107 treatment (analyzed as in Figure 2). F) H-2 levels in intratumoral DCs. Significance for E and F was determined by unpaired t-test, *p<0.01. G) In vivo tumor growth of tumor bearing animals of sgCox2 D4M3.A cells compared to parental. H) In vivo survival of tumor bearing animals of sgCox2 D4M3.A cells compared to parental. Significance was assessed by a logrank test, ***p<0.001, ****p<0.0001.
To analyze the effects of decreased Cox2 expression on melanoma tumor growth, we knocked-out Cox2 from D4M3.A cell lines using CRISPR-Cas9 (sgCox2, Supplemental Fig. 5B). sgCox2 cell lines produced less prostaglandin and grew faster than parental D4M3.A cells in vitro (Supplemental Fig. 4C-D). By contrast in vivo, sgCox2 D4M tumors grew slower than parental D4M tumors and mice bearing Cox2-knock out tumor exhibited improved survival (Fig. 3G-H). Thus, Cox2 expression is reduced by BETi and is required for tumor growth in vivo.
PLX51107 alters the inhibitory microenvironment of BRAF V600E melanoma tumors.
JQ1 has been shown to lower PD-L1 expression in the tumor microenvironment, subsequently improving T cell activity (Hogg et al., 2017; Zhu et al., 2016). Thus, we tested the extent to which PLX51107 lowered expression of markers that inhibit CD8+ T cell responses: PD-L1 (induces T cell dysfunction (Sakuishi et al., 2010)), FasL (kills T cells (Motz et al., 2014)), and IDO-1 (depletes tryptophan to induce T cell anergy (Munn & Mellor, 2007)). PLX51107 significantly decreased the percentage of PD-L1+, FasL+, and IDO-1+ CD45.2 negative, non-immune cells (Fig. 4A and B). Similar to in vivo findings, PD-L1 upregulation by IFNγ was blocked by BETi in mouse (D4M3.A and YUMM3.3) and human (A375 and WM793) BRAF V600E melanoma cell lines in vitro (Fig. 4C and D). In summary, PLX51107 lowers PD-L1 and other immune inhibitory markers in melanoma tumors, suggesting it may offer a therapeutic alternative to checkpoint inhibition.
Figure 4. BETi alters the immunogenicity of BRAF V600E melanoma tumors.
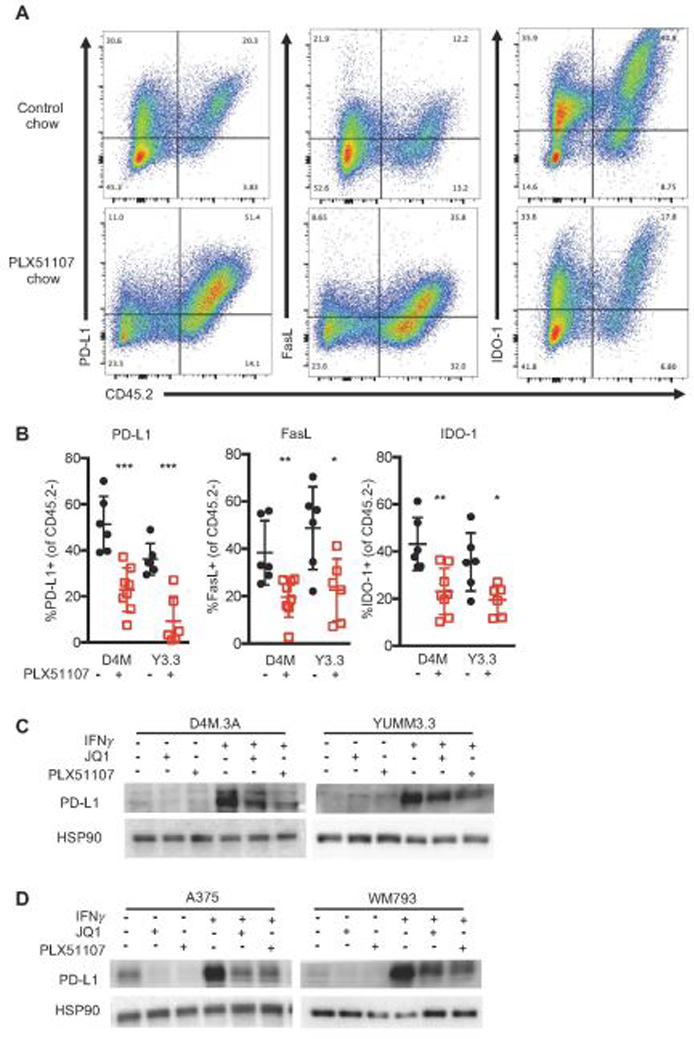
A-B) Data were obtained from D4M3.A (D4M) or YUMM3.3 (Y3.3) tumors at sacrifice from mice in Fig. 1. Tumors were collected, made into single suspensions, and phenotyped using FACS analysis. A) Representative FACS plots of PD-L1, FasL, or IDO-1 expression by CD45.2+. B) Mean expression +/− SD of PD-L1, FasL, and IDO-1 on non-hemaetopoetic (CD45.2 negative) cells in tumors. Significance was assessed by unpaired t-test comparing BETi treatments compared to its relative control, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Mouse (C) and human (D) BRAF V600E melanoma cell lines (n=3 for all cells) were treated with 1 µM JQ1, 2 µM PLX51107, and/or 100 ng IFNγ treatment for 48 hours and analyzed for PD-L1 expression by Western blot. HSP90 serves as loading control.
PLX51107 profoundly impacts melanoma tumors that were non-responsive to anti-PD-1.
A high proportion of melanoma patients do not respond to anti-PD-1 therapy and have limited treatment options; thus, salvage therapies are required (Boutros et al., 2016; Hugo et al., 2016; O’Donnell et al., 2017). PLX51107 may be an effective salvage therapy for anti-PD-1 non-responsive tumors as it maintains inhibition of the PD-1/PD-L1 pathway by lowering PD-L1 while inducing the influx of functional CD8+ T cells. To determine this, we treated D4M3.A tumor-bearing mice with anti-PD-1 until tumors progressed on therapy (> 300mm3, Supplemental Fig. 6A), at which point mice were switched to either control or PLX51107-laced diet. PLX51107 slowed the growth and improved survival of anti-PD-1 non-responsive BRAF V600E D4M3.A tumors (Fig. 5A-B). Similar effects albeit less pronounced were observed in wild-type BRAF/wild-type NRAS, YUMM4.1, tumors (Supplemental Fig. 6B-C). We also extended and observed similar results in a PDX model derived from a BRAF V600E melanoma tumor that did not respond to anti-PD-1 therapy (TJU-MEL 50, Supplemental Fig.6D-E).
Figure 5. PLX51107 delays growth and improves survival in anti-PD-1 non-responsive melanoma.
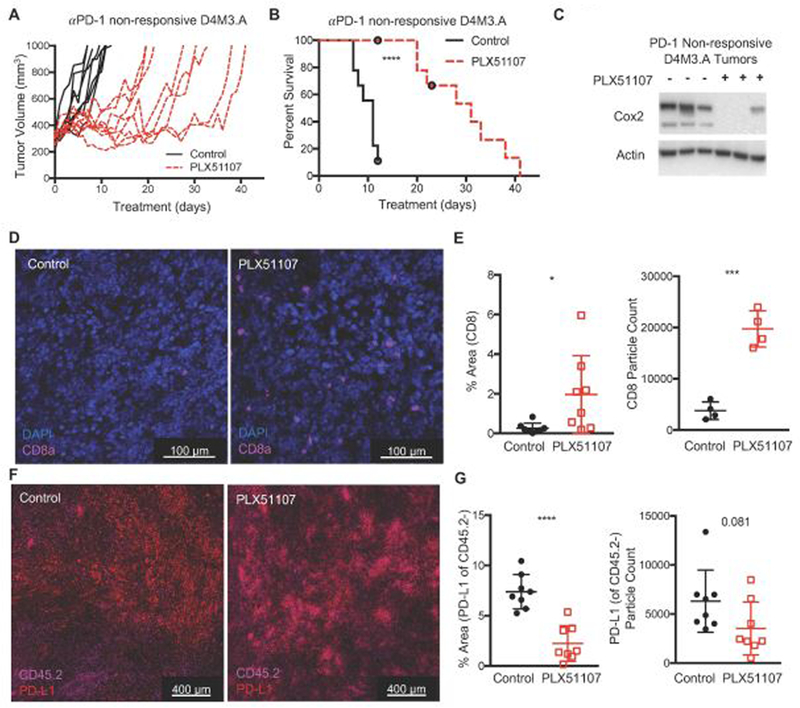
D4M3.A cells were implanted intradermally into C57BL/6 mice. When tumors reached >50 mm3, mice were treated with anti-PD-1 (200 µg of anti-PD-1, RMP1–14, every 3 days) until tumors grew beyond 300 mm3. Then mice were switched to either control (D4M3.A, n=8) or PLX51107 (D4M3.A, n=9) laced chow. Mice were sacrificed when tumors reached 1000 mm3. Tumor growth (A) and percent survival (B) of anti-PD-1 non-responsive, D4M3.A tumor-bearing mice. Significance for survival was assessed with a log-rank test, ****p<0.0001. C) Decreased Cox2 expression in PD-1 non-responsive D4M3.A tumors treated with PLX51107. Actin served as loading control. D) Representative immunofluorescent images of CD8+ cells from D4M3.A tumors at sacrifice (n=4 tumors per group). E) Quantitation of CD8+ cells. The percent area that cells covered [left panel] or the marker particle count [right panel]. Mean +/− SD. F) Representative immunofluorescent images of PD-L1+, CD45.2 negative cells from D4M3.A tumors at sacrifice (n=4 tumors per group). F) Quantitation of PD-L1 staining. Significance was assessed by unpaired t-test, *p<0.05, ***p<0.001, ****P<0.0001.
Consistent with data from anti-PD1 naive tumors, PLX51107 treatment of anti-PD1 progressing D4M3.A tumors was accompanied by an intratumoral decrease of Cox2 (Fig. 5C). Additionally, we detected decreased PD-L1+, CD45.2- cells and an increase CD8+ cells in anti-PD-1 non-responsive D4M3.A tumors (Fig. 5D-G). Together, these data suggest that PLX51107 treatment is an effective second-line therapy for melanoma tumors that are non-responsive to anti-PD-1 treatment through both tumor intrinsic as well as tumor immune micro-environmental effects.
Discussion
In this manuscript, we characterize the effects of the next generation BETi, PLX51107, on BRAF V600E melanoma tumors. We demonstrate that PLX51107 slows the growth of melanoma by influencing the tumor immune microenvironment. PLX51107 efficacy was CD8+ T cell-mediated and was linked to PLX51107-induced decrease of tumoral Cox2 and increase of intratumoral activated DCs. Additionally, PLX51107 lowered the expression of immune inhibitory markers in tumors, which allows intratumoral CD8+ T cells to function more productively. Lastly, PLX51107 can serve as a salvage therapy for melanomas that are non-responsive to anti-PD-1 therapy. These data are important as BETi are currently being tested in the clinic as a monotherapy (NCT02683395) and in combination with checkpoint inhibitors (NCT02959437).
Research studies on BETi in melanoma have mainly investigated tumor intrinsic alterations (Dar et al., 2015; Fontanals-Cirera et al., 2017; Gallagher, Mijatov, Gunatilake, Gowrishankar, et al., 2014; Gallagher, Mijatov, Gunatilake, Tiffen, et al., 2014; Gallagher et al., 2015; Paoluzzi et al., 2016). Our study underscores the importance of the immune system and specifically CD8+ T cells in BETi efficacy. Others have shown that the first generation BETi, I-BET (GSK525762A), alters inflammatory gene expression, improving immune responses to bacteria (Nicodeme et al., 2010), and that JQ1 is more efficacious against Eu-Myc lymphoma in immune-competent mice compared to Rag−/− (lacking B and T cells) mice (Hogg et al., 2017). Our previous work suggested that PLX51107 altered intratumoral T cell responses, but how this occurred was unclear (Nikbakht et al., 2019). Here, we have observed that increased CD8+ T cell levels are required for PLX51107 efficacy. Depleting CD8+ T cells in mice abrogated PLX51107 efficacy in melanoma, consistent with previously published data demonstrating the CD8+ T cell-mediated effect of JQ1 in ovarian cancer (Zhu et al., 2016). In line with an important role for the immune system, we observed only modest PLX51107 efficacy in PDX tumors grown in immune-deficient mice, suggesting a functional immune system is crucial for BETi efficacy. Thus, our data reveal a novel role for CD8+ T cells during PLX51107 treatment and underscore the use of immune-competent tumor models to test BETi effects.
The effects of PLX51107 on TIL correlated with efficacy of PLX51107 in mouse BRAF V600E melanomas and are likely multifactorial. First, the tumor microenvironment has decreased expression of immune inhibitory PD-L1, FasL, and IDO-1 during PLX51107, providing CD8+ T cells with a more favorable environment. These data are in line with previous publications establishing that BETi improves CD8+ T cell responses by lowering PD-L1 on tumor cells (Hogg et al., 2017; Zhu et al., 2016). Additionally, we observed a decrease in tumoral Cox2 and an associated increase in activated DCs. Decreased Cox2 leads to increased CD8+ T cell function and infiltration, an influx of activated DCs, and T cell-dependent control of tumor growth (Kalinski, 2011; Zelenay et al., 2015); thus, decreased Cox2, in part, induces the CD8+ T cell mediated efficacy of PLX51107. Treatment with PLX51107 also increased expression of the pro-death marker, Bim-EL, and the release of HMGB1. These data suggest that cells underwent immunogenic cell death during PLX51107 treatment, likely contributing to the DC activation and subsequent CD8+ T cell responses observed (Galluzzi, Buque, Kepp, Zitvogel, & Kroemer, 2017). Together, our data demonstrate that PLX51107 alters multiple aspects of the tumor immune microenvironment, allowing CD8+ T cells to induce anti-tumor effects. We hypothesize that BETi-induced Cox2 reduction mainly contributes to the CD8+ T cell-mediated tumor growth delay observed by inducing the influx of activated DCs, allowing for better antigen presentation and more T cell activation. It remains unclear how PLX51107 modulates Cox2 expression. Cox2 transcript levels remain unchanged during BETi and Cox2 has previously been shown to be proteasomally degraded after ubiquitylation (Alexanian et al., 2017). Determining if BETi induces Cox2 post-translational modifications and subsequent degradation requires further study.
Many melanoma patients treated with checkpoint inhibitors undergo incomplete responses and ultimately have disease progression (Boutros et al., 2016; Hugo et al., 2016; O’Donnell et al., 2017). Developing second-line therapies is key as the treatment options for these patients are ineffective. For example, melanoma patients treated with anti-CTLA-4 as a second-line therapy for anti-PD-1 non-responsive disease have poor response rates (Aya et al., 2016; Bowyer et al., 2016; Jacobsoone-Ulrich et al., 2016; Zimmer et al., 2017). We showed that PLX51107 profoundly reduced the growth of mouse melanomas in vivo that were non-responsive to anti-PD-1 treatment. The effects were associated with decreased Cox2 expression and numbers of PD-L1+ CD45.2- cells, as well as increased CD8+ T cells in tumors. Thus, PLX51107 may represent a promising salvage therapy for anti-PD-1 non-responsive tumors by re-engaging anti-tumor immune responses by increasing CD8+ T cell influx into tumors, and maintaining PD-1/PD-L1 blockade by decreasing PD-L1 expression in the non-immune cells of the microenvironment.
Uncovering new front-line and second-line therapies is key to building on recent advances in the treatment of metastatic melanoma. This study characterizes the extensive effects of the next generation BETi, PLX51107, as a primary and secondary therapy for BRAF V600E melanoma tumors in mouse syngeneic models. PLX51107 slowed tumor growth, altered tumor microenvironment immunogenicity, and engaged anti-tumor immune responses. Thus, PLX51107 represents a potential treatment strategy for metastatic melanoma patients.
Supplementary Material
Significance.
Metastatic melanoma is the deadliest skin cancer, with only 2–16% of patients surviving long-term. Disturbingly, the incidence of melanoma has been steadily increasing over the past few decades. In recent years, the onset of new FDA-approved therapeutic strategies targeting melanoma specific mutations and the immune system have offered hope to metastatic melanoma patients. Despite their success, there are large percentages of patients who do not respond or become resistant to these therapies. Thus, drugs like BET inhibitors that can target melanoma directly and the immune system may offer alternatives to or second line treatments for FDA approved therapies.
Acknowledgments and Grant Support
We thank Dr. Gordon Mills and Dr. Michael Davies (MD Anderson) for RPPA, which was performed at the Function Proteomics RPPA core facility and supported by NCI Cancer Center Support Grant CA16672. We acknowledge Plexxikon Inc. (Berkley, CA) for generously providing us with PLX51107. We also thank Dr. Chris M. Snyder (Thomas Jefferson University) and Dr. Erica Stone (The Wistar Institute) for their comments on this manuscript. This work is supported by grants from National Institutes of Health (NIH) R01 CA196278, R01 CA160495), Department of Defense (W81XWH-18-1-0224), and the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation to A.E. Aplin. D. A. Erkes, PhD is supported by an American Cancer Society - CEO’s Against Cancer - PA Chapter Postdoctoral Fellowship (PF-18-096-01-LIB). E. Hartsough was supported by NIH K99 CA207855. The Sidney Kimmel Cancer Center MetaOmics, Flow Cytometry and Translational Pathology core facilities are supported by NIH/NCI (P30 CA056036). We thank Drs. Paolo Fortina and Adam Ertel for their assistance with the RNA-seq analysis.
Footnotes
Conflict of interest: A.E. Aplin reports receiving a commercial research grant from Pfizer Inc. (2013–2017) and has ownership interest in patent number 9880150. No potential conflicts of interest were disclosed by the other authors
References
- Adeegbe DO, Liu Y, Lizotte PH, Kamihara Y, Aref AR, Almonte C, et al. (2017). Synergistic Immunostimulatory Effects and Therapeutic Benefit of Combined Histone Deacetylase and Bromodomain Inhibition in Non-Small Cell Lung Cancer. Cancer Discov, 7(8), 852–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexanian A, & Sorokin A (2017). Cyclooxygenase 2: protein-protein interactions and posttranslational modifications. Physiol Genomics, 49(11), 667–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amorim S, Stathis A, Gleeson M, Iyengar S, Magarotto V, Leleu X, et al. (2016). Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: a dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet Haematol, 3(4), e196–e204. [DOI] [PubMed] [Google Scholar]
- Aya F, Gaba L, CVictoria I, Fernandez-Martinez A, Tosca M, Prat A, et al. (2016). Ipilimumab after progression on anti-PD-1 treatment in advanced melanoma. Fut Med, 12(23), 2683–2688. [DOI] [PubMed] [Google Scholar]
- Berthon C, Raffoux E, Thomas X, Vey N, Gomez-Roca C, Yee K, et al. (2016). Bromodomain inhibitor OTX015 in patients with acute leukaemia: a dose-escalation, phase 1 study. Lancet Haematol, 3(4), e186–e195. [DOI] [PubMed] [Google Scholar]
- Boutros C, Tarhini AA, Routier E, Lambotte O, Ladurie FL, Carbonnel F, et al. (2016). Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat Rev Clin Oncol, 12, 473–486. [DOI] [PubMed] [Google Scholar]
- Bowyer S, Prithviraj P, Lorigan P, Larkin J, McArthur G, Atkinson V, et al. (2016). Efficacy and toxicity of treatment with the anti-CTLA-4 antibody ipilimumab in patients with metastatic melanoma after prior anti-PD-1 therapy. Br J Cancer, 114(10), 1084–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Campfield BT, Wenzel SE, McAleer JP, Kreindler JL, Kurland G, et al. (2016). Antiinflammatory effects of bromodomain and extraterminal domain inhibition in cystic fibrosis lung inflammation. JCI Insight, 1(11), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar AA, Nosrati M, Bezrookove V, de Semir D, Majid S, Thummala S, et al. (2015). The role of BPTF in melanoma progression and in response to BRAF-targeted therapy. J Natl Cancer Inst, 107(5), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Chai JC, Yang CS, Lee YS, Das ND, Jung KH, et al. (2015). Dual transcriptome sequencing reveals resistance of TLR4 ligand-activated bone marrow-derived macrophages to inflammation mediated by the BET inhibitor JQ1. Sci Rep, 5(16932), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, & Knapp S (2014). Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov, 13(5), 337–356. [DOI] [PubMed] [Google Scholar]
- Fontanals-Cirera B, Hasson D, Vardabasso C, Di Micco R, Agrawal P, Chowdhury A, et al. (2017). Harnessing BET Inhibitor Sensitivity Reveals AMIGO2 as a Melanoma Survival Gene. Mol Cell, 68(4), 731–744 e739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher SJ, Mijatov B, Gunatilake D, Gowrishankar K, Tiffen J, James W, et al. (2014). Control of NF-kB activity in human melanoma by bromodomain and extra-terminal protein inhibitor I-BET151. Pigment Cell Melanoma Res, 27(6), 1126–1137. [DOI] [PubMed] [Google Scholar]
- Gallagher SJ, Mijatov B, Gunatilake D, Tiffen JC, Gowrishankar K, Jin L, et al. (2014). The epigenetic regulator I-BET151 induces BIM-dependent apoptosis and cell cycle arrest of human melanoma cells. J Invest Dermatol, 134(11), 2795–2805. [DOI] [PubMed] [Google Scholar]
- Gallagher SJ, Tiffen JC, & Hersey P (2015). Histone Modifications, Modifiers and Readers in Melanoma Resistance to Targeted and Immune Therapy. Cancers, 7(4), 1959–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Buque A, Kepp O, Zitvogel L, & Kroemer G (2017). Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol, 17(2), 97–111. [DOI] [PubMed] [Google Scholar]
- Hogg SJ, Vervoort SJ, Deswall S, Ott CJ, Cluse LA, Beavis PA, et al. (2017). BET-Bromodomain Inhibitors Engage the Host Immune System and Regulate Expression of the Immune Checkpoint Ligand PD-L1. Cell Rep, 18, 2162–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. (2016). Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell, 165(1), 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsoone-Ulrich A, Jamme P, Alkeraye S, Dzwiniel V, Faure E, Templier C, et al. (2016). Ipilimumab in anti-PD1 refractory metastatic melanoma: a report of eight cases. Melanoma Res, 26(2), 153–156. [DOI] [PubMed] [Google Scholar]
- Jenkins MH, Steinberg SM, Alexander MP, Fisher JL, Ernstoff MS, Turk MJ, et al. (2014). Multiple murine BRAF melanoma cell lines with sensitivity to PLX4032. Pigment Cell Melanoma Res, 27(3), 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung M, Gelato KA, Fernandez-Montalvan A, Siegel S, & Haendler B (2015). Targeting BET bromodomains for cancer treatment. Epigenomics, 7(3), 487–501. [DOI] [PubMed] [Google Scholar]
- Kagoya Y, Nakatsugawa M, Yamashita Y, Ochi T, Guo T, Anczurowski M, et al. (2016). BET bromodomain inhibition enhances T cell persistence and function in adoptive immunotherapy models. J Clin Invest, 126(9), 3479–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinski P (2011). Regulation of Immune Responses by Prostaglandin E2. J. Immunol, 188(1), 21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang R, Zhang Q, Zeh HJ 3rd, Lotze MT, & Tang D (2013). HMGB1 in cancer: good, bad, or both? Clin Cancer Res, 19(15), 4046–4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein K, Kabala PA, Grabiec AM, Gay RE, Kolling C, Lin LL, et al. (2016). The bromodomain protein inhibitor I-BET151 suppresses expression of inflammatory genes and matrix degrading enzymes in rheumatoid arthritis synovial fibroblasts. Ann Rheum Dis, 75(2), 422–429. [DOI] [PubMed] [Google Scholar]
- Lee DU, Katavolos P, Palanisamy G, Katewa A, Sioson C, Corpuz J, et al. (2016). Nonselective inhibition of the epigenetic transcriptional regulator BET induces marked lymphoid and hematopoietic toxicity in mice. Toxicol Appl Pharmacol, 300, 47–54. [DOI] [PubMed] [Google Scholar]
- Meeth K, Wang JX, Micevic G, Damsky W, & Bosenberg MW (2016). The YUMM lines: a series of congenic mouse melanoma cell lines with defined genetic alterations. Pigment Cell Melanoma Res, 29(5), 590–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS, et al. (2014). Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med, 20(6), 607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn DH, & Mellor AL (2007). Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J Clin Invest, 117(5), 1147–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicodeme E, Jeffrey KL, Schaefer U, Beinke S, Dewell S, Chung CW, et al. (2010). Suppression of inflammation by a synthetic histone mimic. Nature, 468(7327), 1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikbakht N, Tiago M, Erkes DA, Chernova I, & Aplin AE (2019). BET inhibition modifies melanoma infiltrating T cells and enhances response to PD-L1 blockade. J. Invest Dermatol [DOI] [PMC free article] [PubMed]
- O’Donnell JS, Long GV, Scolyer RA, Teng MW, & Smyth MJ (2017). Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev, 52(2017), 71–81. [DOI] [PubMed] [Google Scholar]
- Ozer HG, El-Gamal D, Powell B, Hing ZA, Blachly JS, Harrington B, et al. (2018). BRD4 profiling identifies critical Chronic Lymphocytic Leukemia oncogenic circuits and reveals sensitivity to PLX51107, a novel structurally distinct BET inhibitor. Cancer Discov 8(4),458–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoluzzi L, Hanniford D, Sokolova E, Osman I, Darvishian F, Wang J, et al. (2016). BET and BRAF inhibitors act synergistically against BRAF-mutant melanoma. Cancer Med, 5(6), 1183–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Y, & Ivashkiv LB (2015). Effect and mechanism of BET bromodomain inhibition in macrophage transcriptional programming. Inflam Cell Signal, 2(e600), 1–5. [Google Scholar]
- Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, & Anderson AC (2010). Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med, 207(10), 2187–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segura MF, Fontanals-Cirera B, Gaziel-Sovran A, Guijarro MV, Hanniford D, Zhang G, et al. (2013). BRD4 sustains melanoma proliferation and represents a new target for epigenetic therapy. Cancer Res, 73(20), 6264–6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Hu-Lieskovan S, Wargo JA, & Ribas A (2017). Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell, 168(4), 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Wang Y, Toubai T, Oravecz-Wilson K, Liu C, Methweson N, et al. (2015). BET bromodomain inhibition suppresses graft-versus-host disease after allogeneic bone marrow transplantation in mice. Blood, 125(17), 2725–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibes R, Qiu Y, Lu Y, Hennessy B, Andreeff M, Mills GB, et al. (2006). Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol Cancer Ther, 5(10), 2512–2521. [DOI] [PubMed] [Google Scholar]
- Toniolo PA, Liu S, Yeh JE, Moraes-Vieira PM, Walker SR, Vafaizadeh V, et al. (2015). Inhibiting STAT5 by the BET bromodomain inhibitor JQ1 disrupts human dendritic cell maturation. J Immunol, 194(7), 3180–3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, & Vakoc CR (2014). Brd4 is on the move during inflammation. Trends Cell Biol, 24(11), 615–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelenay S, van der Veen AG, Bottcher JP, Snelgrove KJ, Rogers N, Acton SE, et al. (2015). Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell, 162(6), 1257–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Bengsch F, Svoronos N, Rutkowski MR, Bitler BG, Allegrezza MJ, et al. (2016). BET Bromodomain Inhibition Promotes Anti-tumor Immunity by Suppressing PD-L1 Expression. Cell Rep, 16(11), 2829–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer L, Apuri S, Eroglu Z, Kottschade LA, Forschner A, Gutzmer R, et al. (2017). Ipilimumab alone or in combination with nivolumab after progression on anti-PD-1 therapy in advanced melanoma. Eur J Cancer, 75, 47–55. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.