

Abstract
Human cytomegalovirus (HCMV) establishes latency within incompletely differentiated cells of the myeloid lineage. The viral protein UL138 participates in establishing and maintaining this latent state. UL138 has multiple functions during latency that include silencing productive phase viral gene transcription and modulating intracellular protein trafficking. Trafficking and subsequent downregulation of the multidrug resistance-associated protein 1 (MRP1) by UL138 is mediated by one of four Golgi sorting motifs within UL138. Here we investigate whether any of the Golgi sorting motifs of UL138 are required for the establishment and/or maintenance of HCMV latency in model cell systems in vitro. We determined that a mutant UL138 protein lacking an acidic cluster dileucine sorting motif unable to downregulate MRP1, as well as another mutant lacking all four Golgi sorting motifs still silenced viral immediate early (IE) gene expression and prevented progeny virion formation during latency. We conclude that the Golgi sorting motifs are not required for latency establishment or maintenance in model cell systems in vitro.
1. Introduction
Human cytomegalovirus (HCMV) is a major human pathogen that causes birth defects, the loss of organ and tissue transplants, and is being investigated as a cofactor for glioblastoma multiforme brain tumors. Like all herpesviruses, HCMV replicates productively but also establishes non-productive, latent infections that allow life-long colonization of the infected host (Collins-McMillen et al., 2018; Mocarski ES, Shenk T, RF Pass, 2007). The long-term latent reservoir for HCMV is thought to be CD34+ hematopoietic progenitor cells (Goodrum et al., 2004, 2002; Maciejewski et al., 1992; Mendelson et al., 1996; Sindre et al., 1996; Söderberg-Nauclér et al., 1997), with monocytes likely serving as a short-term reservoir (Söderberg-Nauclér et al., 2001; Taylor-Wiedeman et al., 1994, 1991). Reactivation in response to inflammatory and differentiation signals allows for viral spread (Reeves et al., 2005b; Reeves and Sinclair, 2013; Sinclair, 2008; Sinclair and Reeves, 2014; Taylor-Wiedeman et al., 1994). The currently available anti-HCMV drugs target productive (lytic) infection (Britt and Prichard, 2018). As more about the molecular events of latency is revealed, potential drug targets for latent reservoirs are emerging (B. A. Krishna et al., 2017; Spiess et al., 2015; Weekes et al., 2013).
The UL138 gene, within the UL133-UL138 latency locus of the ULb’ region of the HCMV genome, is one such target. Viruses lacking UL138 generate more infectious progeny virus during in vitro infections of myeloid cells than do wild type viruses (Lee et al., 2015; Petrucelli et al., 2009; Umashankar et al., 2014), indicating that UL138 helps establish and maintain latent infections. The mechanism through which UL138 supports latency is not defined. The UL138 protein is found in the Golgi apparatus but represses transcription from the major immediate early promoter (MIEP) in the cell nucleus (Lee et al., 2016, 2015; Petrucelli et al., 2009). The MIEP launches expression of the immediate early 1 (IE1) protein, which is critical for the initiation and completion of the productive replication cycle (Ahn and Hayward, 1997; Mocarski et al., 1996; Paulus and Nevels, 2009). UL138 achieves this transcriptional silencing by preventing lysine demethylases from removing repressive epigenetic marks from histones associated with the MIEP on latent genomes (Lee et al., 2015). Motifs within UL138 responsible for MIEP silencing have not been identified, and therefore the role that repression of the MIEP plays in latency has not yet been examined.
UL138 also downregulates multidrug resistance-associated protein 1 (MRP1) cell surface expression and steady state levels (Weekes et al., 2013). MRP1 is an ATP-binding cassette transporter and functions as an organic ion transporter across cellular membranes (Cole, 2014). Recently, an acidic cluster dileucine motif within UL138 was identified as required for MRP1 down-regulation (Gelbmann and Kalejta, 2019), but the role of this motif during latency has not been examined. Acidic cluster dileucine motifs direct proteins and their associated cargoes into transport vesicles that shuttle between the Golgi apparatus, the plasma membrane, the lysosome, and other intracellular membranous compartments (Bonifacino and Traub, 2003).
In addition to the acidic cluster dileucine motif, the UL138 protein also has three tyrosine sorting motifs that perform similar functions as dileucine motifs. Furthermore, UL138 has been implicated in the trafficking of additional cellular proteins including the epidermal growth factor receptor (EGFR) and the tumor necrosis factor alpha receptor (TNFR1) (Buehler et al., 2016; Le et al., 2011; Montag et al., 2011). Whether the Golgi sorting motifs participate in trafficking EGFR or TNFR1 and the role UL138-mediated protein trafficking plays during latency are not known.
Here we show that neither the acidic cluster dileucine motif, nor any of the tyrosine sorting motifs are required for repressing MIEP activity and suppressing IE1 transcript and protein accumulation in the THP-1 monocyte model for experimental latency in vitro. Furthermore, we show that none of these Golgi sorting motifs is required for HCMV to efficiently maintain experimental latency in Embryonic Stem Cells (ESCs) in vitro. We conclude that the Golgi sorting motifs of UL138 are not required for MIEP repression or latency maintenance in vitro in the model systems utilized here.
2. Materials and Methods
2.1. Cells, infections and transfections.
Normal human dermal fibroblasts (NHDFs, Clonetics), THP-1 monocytes (TIB-202; ATCC), and human embryonic stem cells (ESC) (WA01; WiCell) were maintained as described previously (Penkert and Kalejta, 2013; Saffert and Kalejta, 2007). An AD169-based virus encoding the Δ4 allele of UL138 (AD138Δ4) was created by replacing the wild type UL138 allele in ADUL138HA (Lee et al., 2015) with the Δ4 allele using a two-step red recombination protocol (Tischer et al., 2010) with the following primers (Forward: 5’-AGTAGCGATGGACGATCTGCCGCTGAAC-3’; Reverse: 5’-TTTCTCATTCAGGCATAGTCAGGCACGTC-3’). Other viruses and viral infection conditions have been previously described (Gelbmann and Kalejta, 2019). UL138 mutant alleles have been previously described and can be found in Table 1 (Gelbmann and Kalejta, 2019). Cells were transfected using TransIT-2020 according to manufacturer’s instructions (Mirus, MIR 5404). THP-1 cells were pretreated with valproic acid (VPA) (Sigma, P4543) at 1 mM for 3 hours and then for the duration of the infection.
Table 1:
UL138 mutant alleles
| Mutant name | Mutation | Specific substitutions | Corresponding mutant virus | Study |
|---|---|---|---|---|
| WT | Wild type; no mutations in UL138 | TB138-HA AD138 |
(Lee et al., 2015) | |
| mY-1 | Mutated first tyrosine motif | Y26LAY to A26AAY | ||
| mY-2 | Mutated second tyrosine motif | Y44RWL to A44AAA | ||
| mY-3 | Mutated third tyrosine motif | Y54GEY to A54GEY | ||
| mLL | Mutated acidic cluster dileucine motif | D142VDLL to A142AAAA | TB138 mLL-HA | |
| Δ4 | All Golgi motifs mutated | The four above mutant alleles combined in a single allele | TB138 Δ4-HA AD138Δ4 |
(Gelbmann and Kalejta, 2019) |
2.2. Antibodies and western blots.
The antibody against IE1 (1B12) has been described previously (Zhu et al., 1995). The following antibodies were obtained from commercial sources GAPDH (6C5, Invitrogen), HA (HA.11, Biolegend), and GM130 (D6B1, Cell Signaling Technology). For western blots equal numbers of cells were lysed in 2% SDS lysis buffer or RIPA buffer and separated by SDS PAGE. Gels were blotted onto nitrocellulose membranes (GE Amersham, Protran). Membranes were blocked in 5% bovine serum albumin (Research Products International Corp.) in TBST (10 mM Tris pH 8, 150 mM NaCl, 0.05% Tween-20), incubated with the indicated primary antibody and washed in TBST, stained with Licor IRDye 680- and 800- secondary antibodies (LI-COR), and imaged on a LI-COR Odyssey FC.
2.3. Luciferase assay.
1×106 THP-1 cells were transfected with 1.5 μg of pSG5-empty vector (EV), pSG5-UL138HA, or pSG5-UL138HA mutant plasmid along with 20 ng pGL-MIEP and 40 ng pRL-TK using Lipofectamine 2000 (ThermoFisher, 11668027). Cells were incubated for 48 hours in antibiotic free media then harvested. Luciferase activity was assayed using a dual luciferase assay reporter system (Promega, E1910) according to manufacturer’s instructions on a Veritas luminometer.
2.4. Latency maintenance assay.
Latency maintenance assays were performed as described previously (Lee et al., 2015). Latency maintenance was quantified by abnormal virion production during latency by plaque assay using permissive fibroblasts co-cultured with latently infected ESCs, fixed and stained with methylene blue.
2.5. Immunofluorescence.
Cells were harvested by centrifugation, allowed to sit on washed coverslips for 1 hour and then fixed with 1% formaldehyde. Cells were then permeabilized with 0.1% Triton X-100 (Sigma) and 0.05% Tween 20 (Sigma) and stained with the indicated antibodies. Secondary antibodies used were Alexa 488 and Alexa 594 (Molecular Probes), and cells were counterstained with Hoechst 33342. Coverslips were mounted on slides using Fluoromount-G (Southern Biotech, 0100–01). Slides were imaged on a Prairie Laser Scanning Confocal Microscope (Prairie Technologies) using a 100X objective. Images were analyzed using FIJI (Schindelin et al., 2012). Co-localization was quantified using the FIJI plugin Colocalization Threshold.
2.6. Quantitative reverse transcriptase and quantitative PCR.
An equal number of cells were harvested by centrifugation. RNA and DNA were extracted using IBI Mini Total RNA Kit (IB47323) and Mini Genomic DNA Kit (IB47202), respectively. 250 ng of total RNA was treated with DNAse and converted to complementary DNA (cDNA) using Maxima™ H minus cDNA Synthesis Master Mix (ThermoFisher, M1682). DNA and cDNA were diluted and amplified using iTaq™ Universal SYBR® Green Supermix (BioRad; 172–5124). Viral transcripts and genomes were compared to cellular glyceraldehyde-3-phosphate dehydrogenase (GAPDH) transcripts and DNA respectively. qPCR primers have been previously described (IE Forward 5’-CGACGTTCCTGCAGACTATG-3’ Reverse 5’- TCCTCGGTCACTTGTTCAAA-3’ and GAPDH Forward 5’-GAGCCAAAAGGGTCATC-3’ Reverse 5’-GTGGTCATGAGTCCTTC-3’) (Hwang et al., 2011; Juckem et al., 2008). Data was compared to untreated AD169 infections using the ΔΔCT method (Livak and Schmittgen, 2001).
3. Results
3.1. The Golgi Sorting Motifs of UL138 are not required for UL138 protein localization to the Golgi apparatus in transfected THP-1 cells.
The simultaneous disruption of all four Golgi sorting motifs within UL138 failed to change its subcellular localization at the Golgi apparatus in fibroblasts where HCMV initiates a productive (lytic) replication program (Gelbmann and Kalejta, 2019). To determine if the Golgi sorting motifs control the localization of UL138 in a cell type that supports latency, we transfected THP-1 cells with expression plasmids for mutant UL138 proteins with each Golgi sorting motif disrupted individually (mY-1, mY-2, mY-3, or mLL), or a mutant UL138 with all four Golgi sorting motifs simultaneously disrupted (Δ4) and visualized their sub-cellular localization by indirect immunofluorescence. Wild type UL138 served as a positive control for Golgi localization as determined by co-localization with the cis-Golgi marker GM130, and the Halo peptide served as a negative control. THP-1 cells were chosen because they have higher transfection efficiency than other in vitro models, and because they faithfully recapitulate all tested parameters of latency as demonstrated in primary monocytes or primary CD34+ hematopoietic progenitor cells, including reactivation (Albright and Kalejta, 2013; Keyes et al., 2012; Benjamin A. Krishna et al., 2017; Lau et al., 2016, p. 20; Saffert et al., 2010; Wagenknecht et al., 2015). Similar to the wild type protein, all the singly substituted mutants, as well as the mutant with the four motifs simultaneously disrupted localized to the Golgi in both qualitative (Fig. 1A) and quantitative (Fig. 1B) assays. Compared to our previous study in fibroblasts we did observe qualitatively more non-specific speckling outside of the Golgi apparatus in THP-1 cells (Gelbmann and Kalejta, 2019). This could be a reflection of the relative paucity of cytoplasm in THP-1 cells compared to fibroblasts. UL138 outside the Golgi apparatus is concentrated into a smaller space in THP-1s giving the appearance of more UL138 outside the Golgi. Quantatively UL138 colocalization with the Golgi apparatus was very similar between THP-1 cells and fibroblasts. We conclude that disruption of all four Golgi sorting motifs is insufficient to displace UL138 from the Golgi. The correct sub-cellular localization of the mutant proteins renders them useful for assays to gauge the role of the Golgi sorting motifs in UL138 function during latency.
Figure 1. The Golgi Sorting Motifs of UL138 are not required for UL138 protein localization to the Golgi apparatus in transfected THP-1 cells.

A. Indirect immunofluorescence of THP-1 cells transfected with plasmids expressing UL138 wild type (WT) or the indicated mutants. Cells were harvested, fixed and stained 48 hours post transfection. GM130 is a Golgi marker. Nuclei were counterstained with Hoechst. HALO serves as a non-Golgi localized control. Representative images of at least three independent biological replicates are shown. B. Images from A were analyzed for co-localization and Pearson’s Correlation Coefficients were determined. Data represent the mean ± standard error of the mean (SEM) from three independent biological replicates, where five cells from each replicate were measured. * p<0.05; ns, not significant (p>0.05) by Student’s t test.
3.2. The Golgi Sorting Motifs of UL138 are not required for UL138-mediated suppression of an MIEP reporter in THP-1 cells.
Transfected UL138 represses an MIEP promoter reporter construct in the THP-1 cells where UL138 expressed from the viral genome attenuates IE1 transcript and protein accumulation during latent infections (Lee et al., 2015). To determine if the Golgi sorting motifs of UL138 control repression of the MIEP in a cell type that supports latency, we transfected THP-1 cells with expression plasmids for mutant UL138 proteins with each Golgi sorting motif disrupted individually (mY-1, mY-2, mY-3, or mLL), or a mutant UL138 with all four Golgi sorting motifs simultaneously disrupted (Δ4) and quantitated the activity of a co-transfected MIEP reporter. Wild type UL138 served as a positive control for MIEP repression and an empty vector was used as a negative control. Similar to the wild type protein, all the singly substituted mutants, as well as the mutant with the four motifs simultaneously disrupted repressed the MIEP reporter (Fig. 2A). Western blots confirmed that the mutants were not overexpressed compared to the wild type protein (Fig. 2B). As before (Gelbmann and Kalejta, 2019; Lee et al., 2016), we observed the expression of both the long and short isoforms of UL138, as well as the anomalous electrophoretic migration of mutants in which the acidic cluster dileucine motif is disrupted. We conclude that disruption of all four Golgi sorting motifs does not affect the ability of UL138 to suppress an MIEP reporter.
Figure 2. The Golgi Sorting Motifs of UL138 are not required for UL138-mediated suppression of an MIEP reporter in THP-1 cells.

A. THP-1 cells were transfected with an MIEP firefly luciferase reporter along with a Renilla TK reporter and the indicated UL138 expression vector for 48 h, then luciferase activity was measured. Firefly (MIEP) luciferase activity normalized to Renilla luciferase activity was compared to empty vector (EV) control. Data represent the mean ± SEM three independent biological replicates. * p<0.05; ns, not significant (p>0.05) by Student’s t test. B. Lysates from the transfected cells were analyzed by Western blot with the indicated antibodies. Representative images of three independent biological replicates are shown.
3.3. The Golgi Sorting Motifs of UL138 are not required for UL138-mediated suppression of IE1 transcript and protein accumulation in HCMV-infected THP-1 cells.
The AD169 strain of HCMV is missing the UL138 gene (and several others) due to a genomic deletion that occurred during serial passage in fibroblasts (Cha et al., 1996; Wilkinson et al., 2015). Upon infection of cells that support latency (e.g. primary CD34+ cells and THP-1 cells) with AD169, IE1 transcripts and protein fail to accumulate, unless a histone deacetylase inhibitor such as valproic acid (VPA) is added (Albright and Kalejta, 2013; Qin et al., 2014, 2013; Saffert et al., 2010). Re-introduction of UL138 back into AD169 prevents this VPA-induced accumulation of IE1 transcripts and protein in latently infected cells, leading in part to the conclusion that UL138 participates in the silencing of IE1 transcription that is a defining characteristic of latency (Lee et al., 2015). Clinical strains like TB40/E retain unidentified restrictions in addition to UL138 that prevent MIEP activation in the presence of VPA (Lee et al., 2015) and could not be used in these assays. To determine if the Golgi sorting motifs of UL138 control IE1 transcript and protein accumulation in a cell type that supports latency, we created an AD169 virus that encodes a mutant UL138 protein with all four Golgi sorting motifs simultaneously disrupted (Δ4), infected THP-1 cells, and quantitated IE1 transcript and protein accumulation. AD169 encoding wild type UL138 served as a positive control and the parental AD169 virus was used as a negative control. Similar to virus encoding the wild type protein, the virus encoding the UL138 mutant with the four motifs simultaneously disrupted significantly reduced VPA-induced IE transcript accumulation (Fig. 3A) as well as VPA-induced IE1 protein accumulation (Figs. 3C and 3D). The defects in IE1 transcript and protein accumulation in UL138 (wild type or mutant) expressing cells was not due to inefficient infection because they displayed similar genome levels as cells infected with the parental AD169 virus (Fig. 3B). There was also no delay in IE gene expression during lytic infection of fibroblasts in either the wild type or mutant UL138 expressing viruses compared to the parental AD169 virus (Fig. 3E). We conclude that disruption of all four Golgi sorting motifs does not affect the ability of UL138 to suppress IE1 transcript and protein accumulation during latency in THP-1 cells.
Figure 3. The Golgi Sorting Motifs of UL138 are not required for UL138-mediated suppression of IE1 transcript and protein accumulation in HCMV-infected THP-1 cells.

A. IE transcripts from THP-1 cells infected with WT AD169 (AD) or UL138 expressing recombinant AD169 (AD138) or the UL138 quadruple Golgi mutant recombinant AD169 (AD138Δ4) at an MOI of one for 18 h in the presence (+) or absence (−) of VPA were quantitated by qRT-PCR. Data represent the mean ± SEM of three independent biological replicates. * p<0.05; ns, not significant (p>0.05) by Student’s t test. B. Lysates from THP-1 cells infected with the indicated viruses at an MOI of one for 18 h were analyzed for viral genomes by qPCR. Results are plotted relative to WT virus infection. Data represent the mean ± SEM of three independent biological replicates. ns, not significant (p>0.05) by Student’s t test. C. Lysates from THP-1 cells infected with the indicated viruses at an MOI of one for 18 h in the presence (+) or absence (−) of VPA were analyzed by western blot with the indicated antibodies. Representative images of three independent biological replicates are shown. D. Protein from experiments in panel C was quantified by densitometry and compared relative to WT AD169 (AD) infection. Data represent the mean ± SEM of three independent biological replicates. *** p<0.001; * p<0.05 by Student’s t test. E. Lysates from fibroblasts infected with the indicated virus at an MOI of one harvested at the indicated time point post infection were analyzed by western blot with the indicated antibodies. Representative images of three independent biological replicates are shown.
3.4. The Golgi Sorting Motifs of UL138 are not required for the maintenance of latency in ESCs.
The TB40/E strain of HCMV contains the UL138 gene. During in vitro latency assays in CD34+ cells and ESC, TB40/E produces very few infectious virus progeny during the establishment and maintenance phases prior to a reactivation stimulus. Following a reactivation stimulus, infectious progeny virus production is increased (Goodrum et al., 2007; Reeves et al., 2005b, 2005a). During these in vitro latency assays in CD34+ cells and ESCs, viruses lacking UL138 generate more infectious virions prior to a reactivation stimulus than wild type viruses (Lee et al., 2016, 2015; Petrucelli et al., 2009), indicating that UL138 is required for the proper establishment and/or maintenance of latency.
Specific features of UL138 required for latency maintenance remain unknown. To determine if the Golgi sorting motifs of UL138 control the establishment or maintenance of latency, we infected ESCs with a TB40/E virus that encodes either a mutant UL138 protein with a disruption in the acidic cluster dileucine motif (mLL) or with all four Golgi sorting motifs simultaneously disrupted (Δ4) for ten days and quantitated infectious virion production during the establishment and maintenance phases of latency in the absence of a reactivation stimulus by plating the infected ESCs on fibroblasts and counting plaques (Lee et al., 2016, 2015). TB40/E (TB138-HA) and TB40/EΔUL138 (TBΔ138-HA) served as controls. We utilized ESCs for this assay because they provide a more robust readout of latency maintenance and broader dynamic range than do primary CD34+ cells while producing identical phenotypes (Lee et al., 2015; Penkert and Kalejta, 2013). For example, UL138-deficient viruses produce, on average, 4.6-fold more infectious virus than wild type viruses during latency maintenance assays in CD34+ cells (Goodrum et al., 2007; Lee et al., 2016; Petrucelli et al., 2009). However, in ESCs, the UL138-null virus produced, on average, 14.3-fold more virus (Lee et al., 2016). This increased dynamic range, combined with the ready supply of cells, allows subtle phenotypes to be quantitated and statistically analyzed with increased rigor and reproducibility than typically achieved using CD34+ cells.
Similar to the parental TB40/E virus, the recombinant TB40/E viruses encoding the mLL or Δ4 allele of UL138 generated significantly fewer infectious progeny virions during the establishment and maintenance phases of latency than did the TB40/E UL138-null virus that produced ~22-fold more virus than the parental TB40/E strain (Fig 4). Interestingly, the acidic cluster dileucine motif mutant (TBmLL) and the Δ4 mutant (TBΔ4) produced ~4- and ~6-fold (respectively) more virus than wild type, but these differences were not statistically significant. In total, we conclude that disruption of the acidic cluster dileucine motif or all four Golgi sorting motifs does not affect the ability of UL138 establish or maintain latency in ESCs in a statistically significant manner.
Figure 4. The Golgi Sorting Motifs of UL138 are not required for the maintenance of latency in ESCs.
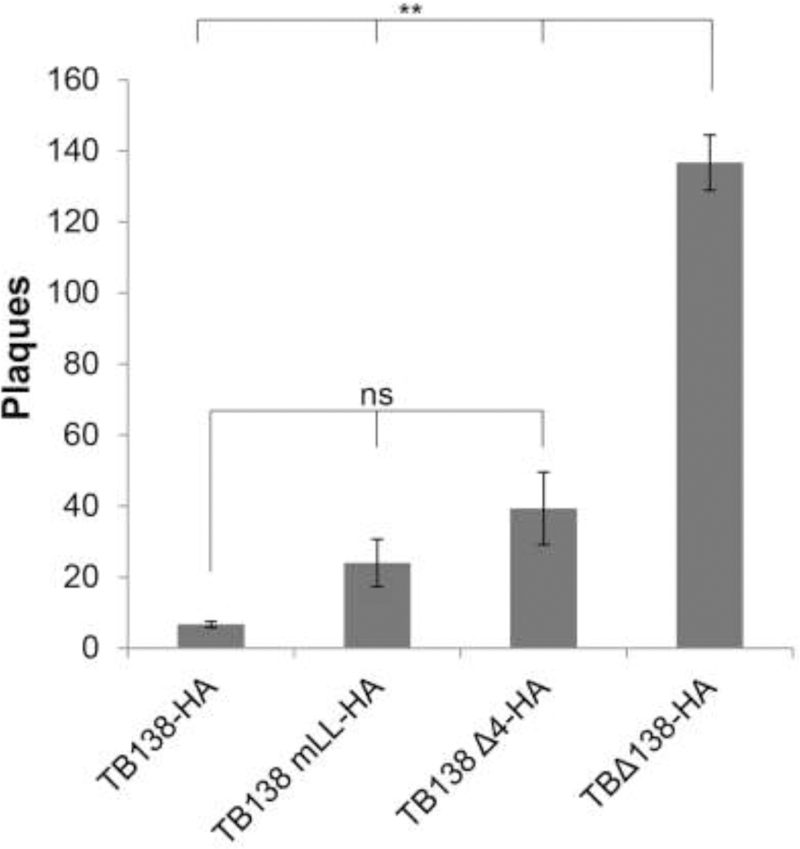
ESCs were infected at an MOI of three for 10 days with the indicated virus then plated on permissive fibroblasts. Plaques were counted 10 days later. Data represent the mean ± SEM of three independent biological replicates, ** p<0.01; ns, not significant (p>0.05) by Student’s t test.
4. Discussion
The Golgi sorting motifs within UL138 are not necessary to suppress the MIEP in THP-1 cells or to maintain long term latency in ESCs, two in vitro models of HCMV latency. The only known role for any of these motifs is the requirement of the acidic cluster dileucine motif for UL138-mediated downregulation of MRP1 (Gelbmann and Kalejta, 2019). Although UL138 has been shown to downregulate MRP1 during latency (Weekes et al., 2013), it would appear from the phenotype of the mLL and Δ4 mutant viruses tested here that UL138-mediated MRP1 downregulation is not required to support in vitro latency. Because viruses with mutations in the acidic cluster dileucine motif grow in fibroblasts without a growth defect (Gelbmann and Kalejta, 2019), this function of UL138 does not appear necessary for productive replication either. Thus, the role of UL138-mediated MRP1 downregulation during HCMV infection remains an enigma.
A corollary to these findings is that MRP1 downregulation does not appear to be necessary for UL138-mediated silencing of productive phase transcription during latency. Although it is understood that the general means used by UL138 to suppress viral transcription is the preservation of heterochromatic epigenetic signatures on the viral genome (Lee et al., 2015), a detailed molecular mechanism remains elusive. Furthermore, as the Golgi sorting motifs are dispensable for both MIEP repression and latency maintenance, it remains likely, or at the very least possible, that the mechanism through which UL138 helps maintain latency is through the repression of the MIEP and the subsequent restriction of IE1 transcript and protein accumulation.
Interestingly, viruses with disruptions in either the acidic cluster dileucine motif or all four Golgi sorting motifs trended toward higher infectious virion production during latency than wild type virus, although the increases seen did not reach statistical significance (Fig. 4). However, the fold increases (4- and 6-fold respectively) were modest compared to the UL138-null virus (22-fold). It is unclear how these mutants might perform in the more physiologically relevant but less dynamic CD34+ cell assay of latency maintenance. Thus, it remains possible that UL138 impacts on intracellular trafficking may affect latency maintenance. Because the trafficking of at least three cellular proteins is known to be modified (MRP1, EGFR1, TNFR1), it will likely require UL138 mutants defective for trafficking individual proteins, not a mutant generally deficient in trafficking, to reveal the roles of UL138-dependent modulation of these proteins for HCMV latency.
Highlights.
UL138 Golgi motifs are dispensable for latency maintenance
UL138 Golgi motifs are not required for IE gene silencing during latency
Golgi localization of UL138 is independent of Golgi motifs
Acknowledgements
We thank the members of our laboratory for helpful comments and suggestions. This work was supported by grants from the NIH to RFK (AI130089 and AI139180), and to CBG (AI131449). CBG and RFK designed the experiments, analyzed the data, and wrote the paper. CBG performed all the experiments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn JH, Hayward GS, 1997. The major immediate-early proteins IE1 and IE2 of human cytomegalovirus colocalize with and disrupt PML-associated nuclear bodies at very early times in infected permissive cells. J. Virol 71, 4599–4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albright ER, Kalejta RF, 2013. Myeloblastic Cell Lines Mimic Some but Not All Aspects of Human Cytomegalovirus Experimental Latency Defined in Primary CD34+ Cell Populations. J. Virol 87, 9802–9812. 10.1128/JVI.01436-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacino JS, Traub LM, 2003. Signals for Sorting of Transmembrane Proteins to Endosomes and Lysosomes. Annu. Rev. Biochem 72, 395–447. 10.1146/annurev.biochem.72.121801.161800 [DOI] [PubMed] [Google Scholar]
- Britt WJ, Prichard MN, 2018. New therapies for human cytomegalovirus infections. Antiviral Res 159, 153–174. 10.1016/j.antiviral.2018.09.003 [DOI] [PubMed] [Google Scholar]
- Buehler J, Zeltzer S, Reitsma J, Petrucelli A, Umashankar M, Rak M, Zagallo P, Schroeder J, Terhune S, Goodrum F, 2016. Opposing Regulation of the EGF Receptor: A Molecular Switch Controlling Cytomegalovirus Latency and Replication. PLoS Pathog 12, e1005655 10.1371/journal.ppat.1005655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha TA, Tom E, Kemble GW, Duke GM, Mocarski ES, Spaete RR, 1996. Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J. Virol 70, 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole SPC, 2014. Multidrug Resistance Protein 1 (MRP1, ABCC1), a “Multitasking” ATP-binding Cassette (ABC) Transporter. J. Biol. Chem 289, 30880–30888. 10.1074/jbc.R114.609248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins-McMillen D, Buehler J, Peppenelli M, Goodrum F, Collins-McMillen D, Buehler J, Peppenelli M, Goodrum F, 2018. Molecular Determinants and the Regulation of Human Cytomegalovirus Latency and Reactivation. Viruses 10, 444 10.3390/v10080444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelbmann CB, Kalejta RF, 2019. The membrane-spanning peptide and acidic cluster dileucine sorting motif of UL138 are required to downregulate MRP1 drug transporter function in human cytomegalovirus infected cells. J. Virol. JVI 00430–19. 10.1128/JVI.00430-19 [DOI] [PMC free article] [PubMed]
- Goodrum F, Jordan CT, Terhune SS, High K, Shenk T, 2004. Differential outcomes of human cytomegalovirus infection in primitive hematopoietic cell subpopulations. Blood 104, 687–695. 10.1182/blood-2003-12-4344 [DOI] [PubMed] [Google Scholar]
- Goodrum F, Reeves M, Sinclair J, High K, Shenk T, 2007. Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 110, 937–945. 10.1182/blood-2007-01-070078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrum FD, Jordan CT, High K, Shenk T, 2002. Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: a model for latency. Proc. Natl. Acad. Sci. U. S. A 99, 16255–16260. 10.1073/pnas.252630899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang J, Saffert RT, Kalejta RF, 2011. Elongin B-Mediated Epigenetic Alteration of Viral Chromatin Correlates with Efficient Human Cytomegalovirus Gene Expression and Replication. mBio 2, e00023–11. 10.1128/mBio.00023-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juckem LK, Boehme KW, Feire AL, Compton T, 2008. Differential Initiation of Innate Immune Responses Induced by Human Cytomegalovirus Entry into Fibroblast Cells. J. Immunol 180, 4965–4977. 10.4049/jimmunol.180.7.4965 [DOI] [PubMed] [Google Scholar]
- Keyes LR, Hargett D, Soland M, Bego MG, Rossetto CC, Almeida-Porada G, Jeor S St, 2012. HCMV Protein LUNA Is Required for Viral Reactivation from Latently Infected Primary CD14+ Cells. PLoS ONE 7 10.1371/journal.pone.0052827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna Benjamin A., Poole EL, Jackson SE, Smit MJ, Wills MR, Sinclair JH, 2017. Latency-Associated Expression of Human Cytomegalovirus US28 Attenuates Cell Signaling Pathways To Maintain Latent Infection. mBio 8 10.1128/mBio.01754-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna BA, Spiess K, Poole EL, Lau B, Voigt S, Kledal TN, Rosenkilde MM, Sinclair JH, 2017. Targeting the latent cytomegalovirus reservoir with an antiviral fusion toxin protein. Nat. Commun 8, 14321 10.1038/ncomms14321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau B, Poole E, Van Damme E, Bunkens L, Sowash M, King H, Murphy E, Wills M, Van Loock M, Sinclair J, 2016. Human cytomegalovirus miR-UL112–1 promotes the down-regulation of viral immediate early-gene expression during latency to prevent T-cell recognition of latently infected cells. J. Gen. Virol 97, 2387–2398. 10.1099/jgv.0.000546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le VTK, Trilling M, Hengel H, 2011. The Cytomegaloviral Protein pUL138 Acts as Potentiator of Tumor Necrosis Factor (TNF) Receptor 1 Surface Density To Enhance ULb′-Encoded Modulation of TNF-α Signaling. J. Virol 85, 13260–13270. 10.1128/JVI.06005-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Albright ER, Lee J-H, Jacobs D, Kalejta RF, 2015. Cellular defense against latent colonization foiled by human cytomegalovirus UL138 protein. Sci. Adv 1, e1501164 10.1126/sciadv.1501164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Caviness K, Albright ER, Lee J-H, Gelbmann CB, Rak M, Goodrum F, Kalejta RF, 2016. Long and Short Isoforms of the Human Cytomegalovirus UL138 Protein Silence IE Transcription and Promote Latency. J. Virol 90, 9483–9494. 10.1128/JVI.01547-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD, 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods San Diego Calif 25, 402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Maciejewski JP, Bruening EE, Donahue RE, Mocarski ES, Young NS, Jeor SS, 1992. Infection of hematopoietic progenitor cells by human cytomegalovirus. Blood 80, 170–178. [PubMed] [Google Scholar]
- Mendelson M, Monard S, Sissons P, Sinclair J, 1996. Detection of endogenous human cytomegalovirus in CD34+ bone marrow progenitors. J. Gen. Virol 77 ( Pt 12), 3099–3102. 10.1099/0022-1317-77-12-3099 [DOI] [PubMed] [Google Scholar]
- Mocarski ES, Kemble GW, Lyle JM, Greaves RF, 1996. A deletion mutant in the human cytomegalovirus gene encoding IE1(491aa) is replication defective due to a failure in autoregulation. Proc. Natl. Acad. Sci 93, 11321–11326. 10.1073/pnas.93.21.11321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mocarski ES, Shenk T, Pass RF, 2007. Cytomegaloviruses, in: Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (Ed.), Fields Virology Lippincott Williams & Wilkins, Philadelphia, PA, pp. 2701–2772. [Google Scholar]
- Montag C, Wagner JA, Gruska I, Vetter B, Wiebusch L, Hagemeier C, 2011. The latency-associated UL138 gene product of human cytomegalovirus sensitizes cells to tumor necrosis factor alpha (TNF-alpha) signaling by upregulating TNF-alpha receptor 1 cell surface expression. J. Virol 85, 11409–11421. 10.1128/JVI.05028-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulus C, Nevels M, 2009. The Human Cytomegalovirus Major Immediate-Early Proteins as Antagonists of Intrinsic and Innate Antiviral Host Responses. Viruses 1, 760–779. 10.3390/v1030760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penkert RR, Kalejta RF, 2013. Human Embryonic Stem Cell Lines Model Experimental Human Cytomegalovirus Latency. mBio 4, e00298–13. 10.1128/mBio.00298-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrucelli A, Rak M, Grainger L, Goodrum F, 2009. Characterization of a novel Golgi apparatus-localized latency determinant encoded by human cytomegalovirus. J. Virol 83, 5615–5629. 10.1128/JVI.01989-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Q, Lee SH, Liang R, Kalejta RF, 2014. Insertion of myeloid-active elements into the human cytomegalovirus major immediate early promoter is not sufficient to drive its activation upon infection of undifferentiated myeloid cells. Virology 0 10.1016/j.virol.2013.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Q, Penkert RR, Kalejta RF, 2013. Heterologous Viral Promoters Incorporated into the Human Cytomegalovirus Genome Are Silenced during Experimental Latency. J. Virol 87, 9886–9894. 10.1128/JVI.01726-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves MB, Lehner PJ, Sissons JGP, Sinclair JH, 2005a. An in vitro model for the regulation of human cytomegalovirus latency and reactivation in dendritic cells by chromatin remodelling. J. Gen. Virol 86, 2949–2954. 10.1099/vir.0.81161-0 [DOI] [PubMed] [Google Scholar]
- Reeves MB, MacAry PA, Lehner PJ, Sissons JGP, Sinclair JH, 2005b. Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc. Natl. Acad. Sci. U. S. A 102, 4140–4145. 10.1073/pnas.0408994102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves MB, Sinclair JH, 2013. Circulating Dendritic Cells Isolated from Healthy Seropositive Donors Are Sites of Human Cytomegalovirus Reactivation In Vivo. J. Virol 87, 10660–10667. 10.1128/JVI.01539-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffert RT, Kalejta RF, 2007. Human cytomegalovirus gene expression is silenced by Daxx-mediated intrinsic immune defense in model latent infections established in vitro. J. Virol 81, 9109–9120. 10.1128/JVI.00827-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffert RT, Penkert RR, Kalejta RF, 2010. Cellular and Viral Control over the Initial Events of Human Cytomegalovirus Experimental Latency in CD34+ Cells. J. Virol 84, 5594–5604. 10.1128/JVI.00348-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez J-Y, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A, 2012. Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair J, 2008. Human cytomegalovirus: Latency and reactivation in the myeloid lineage. J. Clin. Virol. Off. Publ. Pan Am. Soc. Clin. Virol 41, 180–185. 10.1016/j.jcv.2007.11.014 [DOI] [PubMed] [Google Scholar]
- Sinclair J, Reeves M, 2014. The intimate relationship between human cytomegalovirus and the dendritic cell lineage. Front. Microbiol 5, 389 10.3389/fmicb.2014.00389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindre H, Tjoonnfjord GE, Rollag H, Ranneberg-Nilsen T, Veiby OP, Beck S, Degre M, Hestdal K, 1996. Human cytomegalovirus suppression of and latency in early hematopoietic progenitor cells. Blood 88, 4526–4533. [PubMed] [Google Scholar]
- Söderberg-Nauclér C, Fish KN, Nelson JA, 1997. Reactivation of Latent Human Cytomegalovirus by Allogeneic Stimulation of Blood Cells from Healthy Donors. Cell 91, 119–126. 10.1016/S0092-8674(01)80014-3 [DOI] [PubMed] [Google Scholar]
- Söderberg-Nauclér C, Streblow DN, Fish KN, Allan-Yorke J, Smith PP, Nelson JA, 2001. Reactivation of Latent Human Cytomegalovirus in CD14+ Monocytes Is Differentiation Dependent. J. Virol 75, 7543–7554. 10.1128/JVI.75.16.7543-7554.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiess K, Jeppesen MG, Malmgaard-Clausen M, Krzywkowski K, Dulal K, Cheng T, Hjortø GM, Larsen O, Burg JS, Jarvis MA, Garcia KC, Zhu H, Kledal TN, Rosenkilde MM, 2015. Rationally designed chemokine-based toxin targeting the viral G protein-coupled receptor US28 potently inhibits cytomegalovirus infection in vivo. Proc. Natl. Acad. Sci 112, 8427–8432. 10.1073/pnas.1509392112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor-Wiedeman J, Sissons JGP, Borysiewicz LK, Sinclair JH, 1991. Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J. Gen. Virol 72, 2059–2064. 10.1099/0022-1317-72-9-2059 [DOI] [PubMed] [Google Scholar]
- Taylor-Wiedeman J, Sissons P, Sinclair J, 1994. Induction of endogenous human cytomegalovirus gene expression after differentiation of monocytes from healthy carriers. J. Virol 68, 1597–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischer BK, Smith GA, Osterrieder N, 2010. En passant mutagenesis: a two step markerless red recombination system. Methods Mol. Biol. Clifton NJ 634, 421–430. 10.1007/978-1-60761-652-8_30 [DOI] [PubMed] [Google Scholar]
- Umashankar M, Rak M, Bughio F, Zagallo P, Caviness K, Goodrum FD, 2014. Antagonistic Determinants Controlling Replicative and Latent States of Human Cytomegalovirus Infection. J. Virol 88, 5987–6002. 10.1128/JVI.03506-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenknecht N, Reuter N, Scherer M, Reichel A, Müller R, Stamminger T, 2015. Contribution of the Major ND10 Proteins PML, hDaxx and Sp100 to the Regulation of Human Cytomegalovirus Latency and Lytic Replication in the Monocytic Cell Line THP-1. Viruses 7, 2884–2907. 10.3390/v7062751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weekes MP, Tan SYL, Poole E, Talbot S, Antrobus R, Smith DL, Montag C, Gygi SP, Sinclair JH, Lehner PJ, 2013. Latency-Associated Degradation of the MRP1 Drug Transporter During Latent Human Cytomegalovirus Infection. Science 340, 199–202. 10.1126/science.1235047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson GWG, Davison AJ, Tomasec P, Fielding CA, Aicheler R, Murrell I, Seirafian S, Wang ECY, Weekes M, Lehner PJ, Wilkie GS, Stanton RJ, 2015. Human cytomegalovirus: taking the strain. Med. Microbiol. Immunol. (Berl.) 1–12. 10.1007/s00430-015-0411-4 [DOI] [PMC free article] [PubMed]
- Zhu H, Shen Y, Shenk T, 1995. Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J. Virol 69, 7960–7970. [DOI] [PMC free article] [PubMed] [Google Scholar]