

SUMMARY
In the current model of endothelial barrier regulation, the tyrosine kinase SRC is purported to induce disassembly of endothelial adherens junctions (AJs) via phosphorylation of VE cadherin, and thereby increase junctional permeability. Here, using a chemical biology approach to temporally control SRC activation, we show that SRC exerts distinct time variant effects on the endothelial barrier. We discovered that the immediate effect of SRC activation is to transiently enhance endothelial barrier function as the result of accumulation of VE cadherin at AJs and formation of morphologically distinct reticular AJs. Endothelial barrier enhancement via SRC required phosphorylation of VE cadherin at Y731. In contrast, prolonged SRC activation induced VE cadherin phosphorylation at Y685 resulting in increased endothelial permeability. Thus, time variant SRC activation differentially phosphorylates VE cadherin and shapes AJs to fine-tune endothelial barrier function. Our work demonstrates important advantages of synthetic biology tools in dissecting complex signaling systems.
eTORC
An eTOC blurb should also be included that is no longer than 50 words describing the context and significance of the findings for the broader journal readership. When writing this paragraph, please target it to non-specialists by highlighting the major conceptual point of the paper in plain language, without extensive experimental detail. The blurb must be written in the third person and refer to “First Author et al.”
Klomp et al. used a chemically inducible kinase system and physiological stimuli to demonstrate SRC kinase regulates the endothelial barrier in a temporally dependent manner. SRC activity initially enhances barrier function via the adherens junction protein VE cadherin while prolonged activity leads to endothelial barrier disruption.
Graphical Abstract
INTRODUCTION
SRC activation plays a key role in regulating endothelial permeability, with overexpression of constitutively active SRC leading to the disruption of cell-cell junctions (Adam et al., 2010). Mediators such as VEGF also increase endothelial permeability through activation of SRC family kinases (SFKs) (Kim and Wong, 1995; Orsenigo et al., 2012; Piedra et al., 2003; Vouret-Craviari et al., 2002; Xu et al., 2004). However, the view that SRC activation solely increases endothelial permeability is contradicted by studies demonstrating that SFKs are activated during barrier enhancement and recovery following increased endothelial permeability (Birukova et al., 2013; Han et al., 2013; Knezevic et al., 2009). For example, reduction in endothelial permeability induced by sphingosine-1-phosphate (S1P), is accompanied by SFK activation (Vouret-Craviari et al., 2002) and inhibition of SFKs prevents endothelial barrier recovery following thrombin-induced disruption (Birukova et al., 2013; Han et al., 2013). However, the specific and temporal effects that SRC activation has on the endothelial barrier remains to be established.
Endothelial barrier is controlled by adherens junctions (AJs), multiprotein complexes formed at cell-cell contacts. VE cadherin, a component of AJs plays an essential role in the regulation of endothelial permeability (Dejana and Orsenigo, 2013). The localization and permeability of AJs is believed to be regulated by SRC-mediated phosphorylation of VE cadherin at Y658, Y685, and Y731 in response to a variety of inflammatory mediators (Dejana and Orsenigo, 2013; Dejana et al., 2008; Komarova and Malik, 2010; Komarova et al., 2007; Lambeng et al., 2005; Lilien and Balsamo, 2005; Lum and Malik, 1994; Orsenigo et al., 2012) leading to endocytosis of VE cadherin and disassembly of AJs (Gavard and Gutkind, 2006; Gavard et al., 2008). However, several studies suggest that the role of VE cadherin phosphorylation in the regulation of AJs is more complex. SRC-mediated phosphorylation of VE cadherin at Y658 and Y685 in the absence of inflammatory mediators failed to induce VE cadherin internalization (Adam et al., 2010; Orsenigo et al., 2012). VE cadherin mutant Y731F failed to prevent disassembly of AJs in endothelial cells, suggesting a different role for this phosphorylation site in barrier regulation (Wessel et al., 2014). In summary, SRC-mediated phosphorylation of VE cadherin may not always lead to disruption of endothelial barrier.
To address the basis for the apparent distinct functions of SRC, we employed a protein engineering approach, the RapR-kinase system, which enabled direct activation of SRC in living cells (Karginov et al., 2010, 2014; Klomp et al., 2016). Control of SRC activity was achieved through a small allosteric switch, the iFKBP domain, inserted at a specific site within the SRC catalytic domain. Insertion of the iFKBP domain made the kinase sensitive to rapamycin. RapR-SRC is inactive in the absence of rapamycin, whereas its addition induces interaction of RapR-SRC with the co-expressed FKBP12-rapamycin-binding (FRB) domain leading to its activation. We showed that insertion of iFKBP only regulated catalytic activity without altering protein-protein interactions, intramolecular interactions, and localization of RapR-kinases (Karginov et al., 2010, 2014). Characterization of RapR-SRC showed that it functions similarly to endogenous SRC and shows identical localization (Karginov et al., 2010, 2014). Introduction of Y527F mutation (position corresponds to avian SRC) in RapR-SRC made it insensitive to negative regulation by the endogenous machinery but ensured activation only when rapamycin was applied (Karginov et al., 2010, 2014). Using RapR-SRC, we have previously uncovered transient morphodynamic changes induced by activation of SRC (Karginov et al., 2010, 2014; Klomp et al., 2016) demonstrating the utility of this tool.
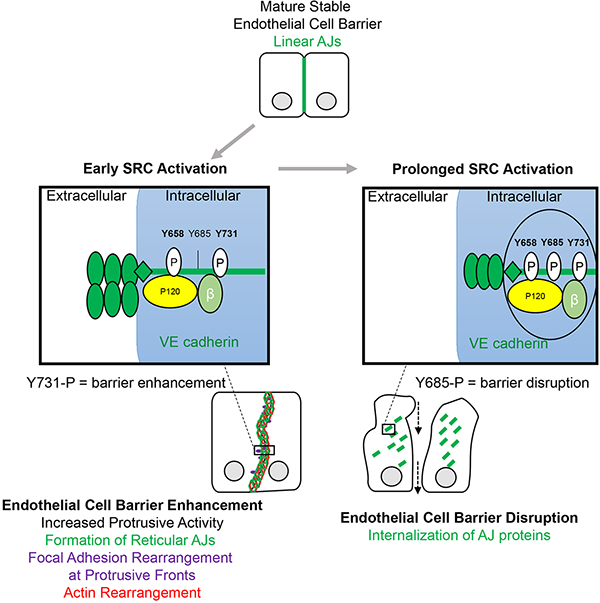
RapR-SRC allowed us to address the complex behavior of SRC signaling in regulating endothelial barrier function. We uncovered a time variant role for SRC activity in regulating the endothelial permeability. Transient activation of SRC reduced endothelial permeability due to organization of broad reticular AJs and VE cadherin phosphorylation on Y731. However, prolonged SRC activation disassembled AJs leading to an increase in endothelial permeability, mimicking inflammation.
RESULTS
SRC activation initially reduces endothelial permeability
The current model implies activation of SRC kinase induces endothelial barrier disruption. However, this model is based on studies which predominantly evaluated either the long-term effects of SRC activity, SRC inhibition, or genetic deletion (Adam et al., 2010; Eliceiri et al., 1999, 2002; Mehta and Malik, 2006; van Nieuw Amerongen and van Hinsbergh, 2002; Okutani et al., 2006; Yuan, 2002). Additionally, the aforementioned studies did not allow for detection of potential transient effects following SRC activation. Thus, we determined the temporal effects of direct SRC activation on endothelial barrier function using the RapR-kinase system (Chu et al., 2014; Karginov et al., 2014; Klomp et al., 2016) in primary human pulmonary arterial endothelial (HPAE) cells. RapR-Src construct contained Y527F mutation (position corresponds to avian SRC) making it insensitive to negative regulation by endogenous signaling. We found that activation of SRC induced a transient increase in transendothelial electrical resistance (Figures 1A and S1A), indicating reduction in endothelial permeability. Transendothelial resistance decreased at later time points demonstrating that prolonged SRC activity increased permeability (Figures 1A and S1A), an observation consistent with previous studies (Adam et al., 2010). Interestingly, activation of LYN kinase, a close SRC homolog, did not induce transient barrier enhancement and only increased endothelial permeability (Figures 1A and S1A), indicating that SFK members induce distinct responses. RapR-SRC and RapR-LYN were expressed at equivalent levels (Figures 1B and S1B–C). Both RapR-SRC and RapR-LYN activation led to phosphorylation of known SFK targets (Figure 1B). However, SRC activation lead to faster phosphorylation of all the evaluated endogenous substrates (Figures 1B and S1C). To confirm that SRC activation can strengthen endothelial barrier, we used the FITC-dextran Transwell endothelial permeability assay. We detected a pronounced enhancement of endothelial barrier consistent with the electrical resistance data (Figure 1C). Rapamycin treatment of cells expressing FRB alone had no effect on paxillin phosphorylation as well as endothelial permeability (Figures 1A, C, and S1A, D), showing that the observed effect was due to activation of engineered SRC. To confirm that SRC induces similar changes in other endothelial cell types, we also tested the response in Human Umbilical Vein Endothelial (HUVE) cells. We observed the same effect as in HPAE cells (Figure S1E). The mechanism of SRC-mediated disruption of endothelial barrier has been studied extensively (Kim and Wong, 1995; Orsenigo et al., 2012; Piedra et al., 2003; Vouret-Craviari et al., 2002; Xu et al., 2004). However, the role of SRC in the strengthening of endothelial barrier is poorly understood. Thus, we focused our further studies on dissecting the mechanism underlying this function of SRC in endothelial cells.
Figure 1: Effect of SRC activation on permeability of endothelial monolayer.

(A) TER analysis of HPAE cell monolayer. HPAE cells co-expressing cerulean-tagged RapR-SRC (SRC) and mCherry-FRB, cerulean-tagged RapR-LYN (LYN) and mCherry-FRB, or mCherry-FRB alone (FRB) were treated with rapamycin (500 nM) at time point 0. The graphs show the average resistance of 3 independent experiments and 90% confidence intervals. (B) Activation of RapR-SRC and RapR-LYN in HPAE cells. Cell lysates were collected at the designated time points and immunoblotted for the indicated proteins. (C) Fluorescein-labeled dextran flux across HPAE cell monolayer. HPAE cells co-expressing cerulean-tagged RapR-SRC and mCherry-FRB or mCherry-FRB only were treated with rapamycin (500 nM) to activate RapR-SRC. FITC-Dextran flux was measured at indicated time points before and after rapamycin treatment. The graph shows the averages and standard deviations from 4 independent experiments. Significance was evaluated using a two-way ANOVA with repeated measures and a post-hoc test with a Bonferroni’s multiple comparisons correction ***p < 0.001, **p < 0.01. All exogenous proteins were expressed using adenoviral transduction. See also figures S1A–E.
SRC activation induces accumulation of VE cadherin at adherens junctions
The endothelial barrier is tightly regulated by VE cadherin and the current model suggests that activation of SRC via numerous external stimuli results in internalization of VE cadherin and disruption of AJs (Dejana, 2004; Dejana and Orsenigo, 2013; Gavard and Gutkind, 2006; Gavard et al., 2008). To assess the initial effect of direct SRC activation on localization of VE cadherin, we employed live cell imaging of GFP-tagged VE cadherin. We observed no disruption of AJs following initial SRC activation (Movie 1, Figure 2A). Staining for endogenous VE cadherin and p120-catenin (another component of AJs) also demonstrated that the junctions remained intact at early time point (30 min following activation) and only disassembled after prolonged SRC activation (Figure S2A). Furthermore, image analysis revealed significant accumulation of endogenous VE cadherin at AJs during barrier enhancement (Figures 2B, C, and S2A). Rapamycin treatment of cells expressing FRB alone showed no accumulation of VE cadherin at 30 min, demonstrating that the effect was due to SRC activation (Figure 2C). Thus, localization of VE cadherin mirrored the permeability results with VE cadherin accumulating in AJs during early activation of SRC, and only being internalized following prolonged SRC activation.
Figure 2: Effect of SRC activation on VE cadherin localization.

(A) Localization of overexpressed VE cadherin before and after RapR-SRC activation. HPAE cells expressing, RapR-SRC-cerulean, mCherry-FRB, and VE cadherin-GFP were imaged live. Representative wide-field images show VE cadherin-GFP distribution at the indicated time points before and after rapamycin addition. (B, C) Accumulation of endogenous VE cadherin at cell-cell contacts following SRC activation. HPAE cells expressing RapR-SRC-cerulean and mCherry-FRB or mCherry-FRB alone were treated with rapamycin (500 nM) for the designated amount of time, fixed, and stained for VE cadherin (Alexa488). (B) Representative wide-field images of endogenous VE cadherin before and 30 min after SRC activation. Images were taken using the same settings and adjusted to the same levels of brightness/contrast to show the difference in the amount of VE cadherin at the junctions. (C) Relative average intensity of VE cadherin before and after rapamycin treatment of cells expressing RapR-SRC and control cells (FRB only). (D and E) Broadening of AJs following SRC activation. (D) HPAE cells expressing VE cadherin-GFP, co-expressing RapR-SRC-cerulean and mCherry-FRB were imaged live using 3D-SIM. Images of the same region of AJs where taken at the indicated time points. (E) Change in AJ area following SRC activation. The relative area of AJs was determined for samples in B as described in Materials and Methods. (C and E). Values are normalized to the median value for 0 minutes and analyzed using a blocked ANOVA, ***p<0.001, *p<0.05 to compare across experiments. Box plots represent data quartiles. All exogenous proteins were expressed using adenoviral transduction. See also figure S2A.
Activation of SRC induces formation of reticular adherens junctions exhibiting reduced local permeability
Staining for endogenous VE cadherin as well as live cell imaging of GFP-tagged VE cadherin revealed that barrier enhancement was accompanied by an increase in AJ area and broadening of AJs (Figures 2B, D, E, and S2A). VE cadherin in the broader AJs was rearranged into distinct structures previously described as overlapping or reticular AJs (Cain et al., 2010; Fernández-Martín et al., 2012; Seebach et al., 2015) (Figure 3A). We also observed reticular AJs in live cells (Figure 3B). To determine whether the formation of broader reticular AJs contributed to SRC-mediated enhancement of endothelial barrier, we assessed localized permeability of reticular and linear AJs using the FITC-avidin permeability assay (Dubrovskyi et al., 2013). We observed greater accumulation of FITC-avidin under linear than under reticular junctions, demonstrating that linear junctions were leakier (Figures 3C–D). Therefore, SRC activation induced VE cadherin accumulation and rearrangement into broader reticular AJs that exhibited reduced permeability and contributed to endothelial barrier enhancement.
Figure 3: SRC-mediated formation and structure of reticular AJs.

(A and B) Formation of reticular AJs following SRC activation. (A) HPAE cells expressing RapR-SRC-cerulean and mCherry-FRB were treated with rapamycin for 0 or 30 minutes and stained for endogenous VE cadherin (Alexa488). Confocal images demonstrating the difference in the arrangement of VE cadherin at the junctions. Inserts show zoomed in area of the image to demonstrate organization of VE cadherin. (B) HPAE cells expressing RapR-SRC-cerulean-myc, mCherry-FRB, and VE cadherin-GFP were imaged live using a 3D-SIM microscope. VE cadherin-GFP image was taken at 30 minutes, post-rapamycin treatment (500 nM). (C and D) Localized permeability of linear and reticular AJs. HPAE cells were analyzed using a FITC-avidin permeability assay. FITC-avidin was added to cell media for 2 minutes, cells were then washed, fixed, and stained for endogenous VE cadherin (Alexa647). The amount of FITC-avidin detected at the junction reflects its permeability. (C) Representative images of VE cadherin and FITC-avidin. (D) Quantification of local FITC-avidin permeability. The average integrated intensity of FITC-Avidin was measured under 30 reticular (>4.5 μm wide) and 30 linear (<2.5 μm wide) AJs, designated by endogenous VE cadherin staining. Results are collected from 3 independent experiments. A two-sample t-test was used to compare the two types of junctions. ***p < 0.001. Box plots represent data quartiles. (E) Changes in protrusive activity of HPAE cells following activation of SRC. HPAE cells co-expressing RapR-SRC-cerulean, mCherry-FRB, and the membrane marker Stargazin-mVenus were imaged live every 2 minutes. Transient transfection was used to express Stargazin. Rapamycin was added a time point 0 (500 nM, green line). Protrusive activity was calculated and standardized to the average activity for each cell prior to rapamycin treatment (N=30 cells). Error bars represent the 90% confidence interval for each time point. (F-H) Organization of reticular AJs. HPAE cells co-expressing RapR-SRC-cerulean and mCherry-FRB were treated with rapamycin (500 nM) for 30 minutes, fixed, and stained for endogenous VE cadherin (Alexa488) and F-actin (Phalloidin-Alexa647). Immunofluorescence images were collected using a Nikon SIM microscope. (G) Zoomed in area from the indicated white square in B. (H) 3D representation of VE cadherin (green) and actin (purple) organization in the zoomed in area in C. 3D renderings were generated using IMARIS software. All exogenous proteins were expressed using adenoviral transduction unless notated otherwise. See also Figure S2B and Movie 2.
Organization of reticular adherens junctions
Since reticular AJs formed in areas of broad overlap between two neighboring endothelial cells, we hypothesized that the overlap was created by cell protrusions extending on top of or underneath the adjacent cell. In concordance with this hypothesis, we found that transient activation of SRC in confluent endothelial cells induced a sustained increase of protrusive activity with the highest activity coinciding with barrier enhancement (Figure 3E, Movie 2). Since membrane protrusions are driven by actin polymerization and formation of cortical actin meshwork (Bernheim-Groswasser et al., 2002; Mullins et al., 1998), we evaluated organization of actin cytoskeleton in reticular AJs. Structure illumination microscopy (SIM) analysis and confocal microscopy revealed that F-actin formed reticular structures directly underneath VE cadherin structures (Figures 3F–H and S2B). The fact that the actin structures were mostly formed under VE cadherin structures suggests that reticular AJs are formed in the areas where endothelial cell protrudes underneath its neighbor.
Formation of a stable membrane protrusion is accompanied by establishment of focal adhesions that anchor it to extracellular matrix (Deakin and Turner, 2008). SRC activation was shown to induce the formation of focal adhesions (Karginov et al., 2014). Thus, we evaluated organization of focal adhesions in reticular AJs induced by activation of SRC. We stained for endogenous VE cadherin and paxillin (focal adhesion marker) at 30 min following activation of RapR-SRC (during the barrier enhancement phase). We observed formation of focal adhesions at reticular AJs (Figure 4A). To gain insights into the localization of paxillin and VE cadherin in these structures, we employed interferometric photoactivation and localization microscopy (iPALM). Assessment of the vertical distribution of proteins relative to coverslip surface revealed that a significant fraction of VE cadherin overlapped with paxillin within broad AJs (Figures 4B, C, and S2C). Immunoprecipitation of endogenous VE cadherin revealed an increase in the amount of paxillin associated with VE cadherin following SRC activation (Figure 4D). Thus, SRC mediated reticular AJs come in close contact with focal adhesions and their formation is accompanied by increased association of VE cadherin with paxillin.
Figure 4: Organization of focal adhesions in reticular AJs.

(A) Presence of focal adhesions in reticular junctions. HPAE cells co-expressing RapR-SRC-cerulean and mCherry-FRB were treated with rapamycin (500 nM) for 0 or 30 minutes, fixed, and stained for VE cadherin (Alexa488) and Paxillin (Alexa647) and imaged via confocal microscopy. Arrowheads indicate focal adhesions in reticular junctions. (B and C) Analysis of VE cadherin and paxillin organization in reticular junctions using iPALM. SRC was activated for 30 minutes in HPAE cells co-expressing RapR-SRC-cerulean, FRB, and td-EOS-paxillin, fixed cells were stained for VE cadherin (Alexa647) and imaged on an iPALM microscope. (B) Representative image showing z-(top image) and x-y-projection (bottom image) of VE cadherin arranged in a reticular structure. (C) Distribution of paxillin (green) and VE cadherin (red) in Z within overlapping reticular junctions (indicated by white rectangle in (B)). The average relative abundance of VE cadherin and paxillin in Z was measured in 3 distinct regions containing reticular junctions, 4–6 sub-regions were analyzed per region. Analyzed regions are shown in Figure SI 2C. (D) Association of paxillin with VE cadherin following SRC activation. SRC was activated for the designated amount of time in HPAE cells co-expressing RapR-SRC-cerulean and mCherry-FRB. VE cadherin was immunoprecipitated from cell lysates and immunoblotted with designated antibodies. (E) Role of SRC SH2 domain in regulation of endothelial barrier. HPAE cells co-expressing RapR-SRC-cerulean (SRC) and mCherry-FRB or RapR-SRC-R175L-cerulean (R175L, SRC SH2 mutant) and mCherry-FRB were analyzed using TER. Rapamycin (500 nM) was added at time point 0. Graphs represent the average resistance and 90% confidence interval from 3 independent experiments. All exogenous proteins were expressed using adenoviral transduction, except for td-EOS-paxillin which was transiently transfected. See also Figure S2D–E.
SRC-induced formation of focal adhesion in membrane protrusions may contribute to SRC-mediated enhancement of endothelial barrier. Our previous studies showed that R175L mutation disrupts the SH2 domain of SRC and abolishes SRC-mediated stimulation of new focal adhesions and membrane protrusions (Karginov et al., 2014). Activation of the SH2 domain mutant R175L-SRC in endothelial cells failed to induce barrier enhancement (Figures 4E and S2D). Interestingly, even though the R175L mutation does not affect catalytic activity of SRC (Karginov et al., 2014), we observed diminished phosphorylation of endogenous paxillin indicating that signaling downstream of this mutant is perturbed (Figure S2E). Thus, interactions of SRC mediated by its SH2 domain are important for regulation of focal adhesion proteins and endothelial barrier enhancement.
Role of SRC-mediated VE cadherin phosphorylation in regulating endothelial barrier
The current model suggests that SRC-mediated phosphorylation of VE cadherin disrupts its interaction with binding partners p120-catenin and β-catenin (Potter et al., 2005) and thereby negatively regulates AJs. Therefore, we investigated the role of SRC-mediated VE cadherin phosphorylation in regulating interactions with p120-and β-catenins and endothelial permeability. Immunoprecipitation of endogenous VE cadherin from endothelial cells revealed a substantial increase in phosphorylation of VE cadherin as early as 15 min following SRC activation (Figure 5A). However, co-immunoprecipitation experiments demonstrated that VE cadherin phosphorylation did not disrupt interaction with p120-catenin up to 4h following SRC activation (Figures S3A–B). We observed a small decrease in interaction with β-catenin but only at later time points (Figures S3A–B). Importantly, there was no change in the expression of VE cadherin, p120-catenin, or β-catenin (Figure S3C). These data are in agreement with previous observations (Adam et al., 2010) showing that phosphorylation of VE cadherin is insufficient to drive the dissociation of p120-and β-catenins, and phosphorylation of VE cadherin alone is also insufficient to induce permeability.
Figure 5: The role of VE cadherin phosphorylation in SRC-mediated regulation of endothelial cell-cell barrier.

(A-C) Phosphorylation of VE cadherin following activation of SRC. HPAE cells co-expressing RapR-SRC-cerulean and mCherry-FRB were treated with rapamycin (500 nM) for the designated amount of time. Endogenous VE cadherin was immunoprecipiated and analyzed by western blot. (A) Total VE cadherin Tyrosine phosphorylation. (B-C) Phosphorylation of VE cadherin at specific Tyrosine sites: Y658 (n=6), Y685 (n=5), and Y731 (n=6). For each experiment, all time points were normalized to 4h time point. Error bars represent standard deviation. Statistical significance was evaluated using a two-way ANOVA with repeated measures and a post-hoc test with a Bonferroni’s multiple comparisons test. *p < 0.05, ***p < 0.001, ****p < 0.0001. (D) Inactivation of RapR-SRC resulted in a return to basal VE cadherin phosphorylation levels. RapR-SRC-cerulean-as2 and mCherry-FRB cells were activated with rapamycin (500 nM) for 60 min, at which time either DMSO or the allele specific inhibitor 1NA-PP1 (500 nM) was added for an additional 30 min. Lysates were collected at 90 min post rapamycin treatment, VE cadherin was immunoprecipitated and probed for Y658, Y685, and Y731 phosphorylation. See also figure S4A–B. (E) The role of Y658 and Y731 in VE cadherin localization. HPAE cells co-expressing RapR-Src-cerulean, mCherry-FRB, and the indicated VE cadherin-GFP construct (wild-type (WT), Y658F, or Y731F mutant) were imaged live and the amount of cytoplasmic VE cadherin was calculated for each time point. Rapamycin (500 nM) was added at 0min time point. All values were normalized to the average cytoplasmic VE cadherin intensity prior to rapamycin treatment. Error bars show 90% confidence intervals. (F) The role of VE cadherin phosphorylation on Y658 and Y731 in Src-mediated regulation of endothelial barrier. HPAE cells co-expressing RapR-Src-cerulean, mCherry-FRB, and the indicated VE cadherin-GFP construct (wild-type (WT), Y658F, or Y731F mutant) were analyzed by TER and were treated with rapamaycin (500 nM) at time 0. Graph represents the average resistance from 3 independent experiments and error bars show 90% confidence intervals. All exogenous proteins were expressed using adenoviral transduction. See also figures S5A–D.
Phosphorylation of VE cadherin at Y658, Y685, and Y731 is proposed to regulate AJs (Adam et al., 2010; Dejana et al., 2008; Wallez et al., 2006). Immunoprecipitation of endogenous VE cadherin showed that phosphorylation of Y658 and Y731 saturated within 15–30 min following SRC activation, while phosphorylation of Y685 gradually increased over time (Figures 5B–C). Specificity of the phospho-Y685 antibody used has been confirmed previously (Orsenigo et al., 2012). Moreover, specificity of the phospho-Y658 and –Y731 antibodies was verified by probing Y658F and Y731F mutants of VE cadherin following SRC activation (Figure S3D). Inactivation of RapR-SRC by 1NA-PP1 inhibitor that targets only engineered kinase containing T338A gatekeeper residue mutation (Klomp et al., 2016) phosphorylation of VE cadherin, paxillin and p130Cas back to basal levels (Figures 5D and S4A–B). This further demonstrates that phosphorylation of analyzed proteins was mediated by activation of RapR-SRC. Activation of the SH2 mutant R175L-RapR-SRC lead to no significant change in phosphorylation of VE cadherin confirming that SH2 domain is required for proper signaling (Figures S4C).
The role of Y685 phosphorylation in barrier disruption has been established (Wallez et al., 2006; Wessel et al., 2014). Our data agree with the aforementioned role of Y685 phosphorylation. SRC activation induced a gradual phosphorylation of Y685 which coincided with SRC mediated barrier disruption (Figures 1A, 5B–C). Activation of LYN resulted in a more rapid accumulation of Y685 phosphorylation correlating with the lack of barrier enhancement (Figures 1A, S4D–E). Thus, we focused on dissecting the role of VE cadherin Y658 and Y731 phosphorylation sites in SRC-mediated regulation of VE cadherin localization and endothelial barrier function. We assessed SRC-induced changes in localization using GFP tagged wild-type (WT) VE cadherin and VE cadherin Y658F and Y731F mutants (Gonzalez et al., 2016). We observed that VE cadherin Y731F mutant accumulated in the cytoplasm more rapidly following SRC activation than WT VE cadherin (Figures 5E and S5A–B). However, we found less cytoplasmic accumulation of the Y658F VE cadherin mutant following SRC activation when compared to WT VE cadherin (Figures 5E and S5A–B). To determine the role of these two phosphorylation sites in SRC-mediated regulation of endothelial permeability, we expressed VE cadherin Y658F and Y731F mutants as well as WT in endothelial cells and evaluated changes in transendothelial electrical resistance following SRC activation. Cells expressing WT VE cadherin and Y658F mutant demonstrated SRC mediated endothelial barrier enhancement (Figures 5F and S5C–D). However, cells expressing Y731F mutant did not exhibit the transient enhancement of endothelial barrier (Figures 5F and S5C–D). Thus, our results show that SRC-mediated phosphorylation of VE cadherin is insufficient to disrupt AJs, whereas phosphorylation of Y731 is required for endothelial barrier enhancement.
Role of SRC activity and VE cadherin phosphorylation in physiologically stimulated barrier enhancement
The use of engineered RapR-SRC in endothelial cells identified unique effects mediated by SRC, leading us to investigate the role of SRC downstream of physiological stimuli. First, we evaluated the activation of SFKs downstream of sphingosine-1-phosphate (S1P) and thrombin. S1P and thrombin both activate SFKs but exert different effects on endothelial barrier (Birukova et al., 2013; Huang et al., 2008). Our results confirmed these previous observations. S1P caused a rapid increase in SFK activity, as assessed by phosphorylation of SRC on Tyr416 (Figure 6A). Activation of SFKs was sustained for up to 2h and accompanied S1P-mediated enhancement of endothelial barrier (Figures 6A–B and S6A). Co-treatment with S1P and the SFK inhibitor Saracatinib reduced S1P mediated endothelial cell barrier enhancement (Figures 6B and S6A), showing that SFK activity is required. Interestingly, challenge with thrombin caused activation of SFKs both during endothelial barrier disruption (1 min) and barrier recovery (30min and 2h) (Figure 6C). Inhibition of SFKs at 15min post-thrombin treatment significantly reduced the recovery of endothelial barrier function (Figures 6D and S6B). Therefore, physiological stimuli activate SFKs not only upon barrier disruption, but also during barrier enhancement. Furthermore, SFK activity is required for both efficient barrier enhancement and recovery. This led us to ask whether acute activation of SRC following thrombin disruption could enhance the rate of recovery. Indeed, we observed that activation of RapR-SRC 15min after thrombin treatment facilitated barrier recovery (Figures 6E and S6C). Activation of RapR-LYN however did not have a significant effect on barrier recovery (Figure 6F), again demonstrating the distinct role of SFKs in endothelial barrier regulation. Treatment of cells expressing FRB alone with rapamycin did not affect barrier recovery verifying that the observed changes in endothelial barrier function were mediated by the engineered kinase and not by rapamycin alone (Figure S6D). Overall these data demonstrate that activity of SFKs is required for physiologically stimulated barrier enhancement, and that additional activation of SRC facilitates this process.
Figure 6: The role of SRC activation in regulation of endothelial barrier mediated by physiological stimuli.

(A) Activation of SFKs following S1P stimulation. HPAE cells were serum starved (1 hour) and treated with 1 μM S1P. Cell lysates were collected at the indicated time points and analyzed with the indicated antibody. (A and C) Average relative phosphorylation was calculated from 4 independent experiments. Significance was calculated using a one-way ANOVA with a Dunnett’s multiple comparisons correction, *p < 0.05, **p < 0.01; error bars depict standard deviation. (B) The role of SFK activity in S1P-mediated barrier enhancement. HPAE cells were serum starved (1 hour) and analyzed using TER; 1 μM S1P and 100 nM Saracatinib or vehicle (DMSO) were added at time point 0. (C) Activation of SFKs following stimulation with thrombin. HPAE cells were serum starved (1 hour) and treated with 50 nM of Thrombin. Cell lysates were collected at the indicated time and analyzed using indicated antibody. (D) The role of SFK activity in barrier recovery following thrombin treatment. HPAE cells were serum starved (1 hour) and analyzed using TER. Thrombin (50 nM) was added at time point 0 and Saracatinib (100 nM, S) or vehicle (ethanol, V) was added 15 minutes later. (E and F) Effect of SRC and LYN activation on barrier recovery. HPAE cells co-expressing RapR-SRC-cerulean and mCherry-FRB (E) or RapR-LYN-cerulean and mCherry-FRB (F) were analyzed using TER. Thrombin (50 nM) was added at time 0 and rapamycin (500 nM) or vehicle (ethanol) (green line) was added 15 minutes later. All exogenous proteins were expressed using adenoviral transduction. (B, D-F) Graphs show an average resistance from 3 experiments and error bars depict the 90% confidence intervals. See also figures S6A–D.
The inability of Y731F VE cadherin expressing endothelial cells to exhibit SRC mediated barrier enhancement prompted us to evaluate the role of this phosphorylation site in the context of S1P mediated barrier enhancement. Immunoprecipitation of endogenous VE cadherin revealed a significant increase in VE cadherin phosphorylation on Y731 following S1P treatment (Figure 7A). S1P treatment of cells expressing the Y731F mutant of VE cadherin showed significantly reduced enhancement of endothelial barrier when compared to cells expressing either the WT VE cadherin or Y658F VE cadherin mutant (Figures 7B and S6E–F). Thus, our data show that phosphorylation of Y731 on VE cadherin is required for the S1P mediated endothelial barrier enhancement confirming its role downstream of physiological stimuli.
Figure 7: The role of VE cadherin phosphorylation on Y731 following S1P treatment.

(A) Phosphorylation of VE cadherin on Y731 following treatment with S1P. HPAE cells were serum starved for 1 hour and then treated with 1 μM S1P. Cell lysates were collected at the indicated time points, VE cadherin was immunoprecipitated, and samples were analyzed for phospho-Y731-VE cadherin and total VE cadherin. VE cadherin phosphorylation levels were normalized to time 0 to obtain relative phosphorylation values. The average relative phosphorylation was calculated from 3 independent experiments; error bars depict standard deviation. Significance was determined via a one-way ANOVA with a Dunnett’s multiple comparisons correction, *p < 0.05. (B) The role of VE cadherin phosphorylation in S1P-mediated endothelial barrier enhancement. HPAE cells expressing indicated VE cadherin-GFP construct wild-type (WT), Y658F, or Y731F mutant) were analyzed by TER. Cells were serum starved for 1 hour and then treated with S1P (1 μM) (0 min). Graph shows the average resistance from 3 independent experiments and the error bars show 90% confidence intervals. All exogenous proteins were expressed using adenoviral transduction. See also figures S6E–F.
DISCUSSION
The endothelial barrier disruptive role of SRC kinase is an accepted paradigm. However, recent studies have suggested that activation of SRC and its family members do not always lead to endothelial barrier disassembly (Birukova et al., 2013; Han et al., 2013; Knezevic et al., 2009). Our results obtained using an engineered system to directly activate SRC demonstrate that SRC kinase can play a dual role in regulating AJs and endothelial permeability. We propose an update to the existing model for SRC-mediated regulation of the endothelial cell-cell interactions and suggest a mechanism where activation of SRC regulates plasticity of AJs. SRC activation in a time variant manner regulates assembly or disassembly depending on duration of SRC activation and kinetics of distinct phosphorylation sites results in opposite effects on the endothelial cell barrier.
In agreement with previous studies, our data demonstrate that prolonged activation of SRC leads to disassembly of AJs at the level of VE cadherin (Dejana and Orsenigo, 2013; Komarova and Malik, 2010; Komarova et al., 2007; Lambeng et al., 2005; Lilien and Balsamo, 2005; Lum and Malik, 1994; Orsenigo et al., 2012). However, we also showed that the effect of acute SRC activation is transient accumulation of VE cadherin in AJs and reduction of barrier permeability (Figures 1A and D). This phenomenon demonstrates that the short-term effects of SRC activation are different from the changes induced by long-term exposure to SRC activity. Thus, SRC induces a transient phenotype in endothelial barrier regulation, in agreement with the time variant effects of SRC activation in other cell types (Karginov et al., 2014; Klomp et al., 2016). Prolonged activation of SRC mimics its role in pathological states where SRC activity is persistently elevated (Frame, 2002; Guo et al., 2013; Kim et al., 2009, 2002; Liu and Senger, 2004; Mukhopadhyay et al., 1995). Under physiological conditions relevant to activation of signaling pathways, however, SRC is activated for a defined period (Vouret-Craviari et al., 2002). The transient effects we observed upon activation of SRC may closer reflect physiological function of the kinase.
Activation of SRC not only induces accumulation of VE cadherin at endothelial cell-cell contacts, but also stimulates reorganization of AJs into broader reticular structures. These reticular AJs have been previously observed (Adam et al., 2010; Birukova et al., 2007a, 2016b, 2016a; Cain et al., 2010; Tian et al., 2016), but only recently they have been characterized (Fernández-Martín et al., 2012; Wilson and Ye, 2014). They are formed by a 3-dimensional mesh-like networks of VE cadherin at the interface of two overlapping endothelial cells. It was proposed that these junctions may exhibit reduced permeability when compared to linear AJs (Fernández-Martín et al., 2012). Our studies provide direct proof for this hypothesis and suggest a mechanism for formation of reticular AJs mediated by transient SRC activation. We observed that SRC-induced formation of membrane protrusions created overlapping regions between two interacting endothelial cells where VE cadherin re-arranged into reticular structures. Organization of VE cadherin could be guided by the actin cytoskeleton as suggested by their striking overlap in reticular AJs. Increased association between VE cadherin and the focal adhesion protein paxillin and their overlap within reticular AJs suggest that focal adhesions may also contribute to organization of reticular VE cadherin structures. Interestingly, previous studies showed that barrier-enhancing factors such as SIP induced association of paxillin with adherens junction proteins, and stimulated paxillin translocation to cell periphery (Birukova et al., 2007a; Dubrovskyi et al., 2012; Sun et al., 2009). Furthermore, some of these studies also showed that barrier enhancement was accompanied by formation of VE cadherin structures that closely resembled reticular AJs, and demonstrated that SFK activity was required for the endothelial barrier reduction (Birukova et al., 2007a, 2007b). Our study showed that SRC signaling pathways mediating membrane protrusion and focal adhesion formation were critical for SRC-mediated reduction in endothelial permeability. Overall, previous reports and our study indicate that reticular adherens junction represent an important physiological phenomenon mediated by transient SRC activation that contributes to regulation of endothelial permeability.
SRC-mediated phosphorylation of VE cadherin and its role in permeability has been extensively studied (Adam et al., 2010; Dejana and Orsenigo, 2013; Dejana et al., 2008; Komarova and Malik, 2010; Komarova et al., 2007; Lambeng et al., 2005; Lilien and Balsamo, 2005; Lum and Malik, 1994; Orsenigo et al., 2012; Wessel et al., 2014). However, several reports showed that VE cadherin phosphorylation may not be exclusively barrier disruptive and different phosphorylation sites may mediate distinct effects on cell-cell junctions. Recent reports showed that phosphorylation of Y685 did mediate an increase in endothelial permeability (Wallez et al., 2006; Wessel et al., 2014), whereas phosphorylation of Y731 was not required for VEGF and histamine induced vascular permeability (Wessel et al., 2014). Our results correlate with these data. Likewise, we found that phosphorylation of Y731 was required for SRC-mediated endothelial barrier enhancement. Interestingly, under basal conditions Y731 was found to be phosphorylated in vivo and phosphorylation of this site was required for leukocyte transmigration (Wessel et al., 2014). Leukocyte transmigration is mediated by platelet endothelial cell adhesion molecule 1 (PECAM-1) on endothelial cells (Muller, 2016; Muller et al., 1993) and PECAM-1 was found to be accumulated in reticular AJs (Fernández-Martín et al., 2012). It is tempting to speculate, based on these results, that SRC-mediated phosphorylation of Y731 contributed to formation of reticular junctions, which are used as gateways for leukocyte transmigration but remain impermeable to solutes. Thus, our study has uncovered a role for Y731 phosphorylation on VE cadherin.
Physiological processes such as barrier enhancement and recovery, leukocyte transmigration and angiogenesis require activation of SRC kinase, but at the same time do not compromise regulation of fluid exchange by the endothelial barrier. Our results may help to explain this phenomenon. SRC-mediated stimulation of reticular VE cadherin structures and signaling through phospho-Tyr731 on VE cadherin enables formation of dynamic AJs that retain their low permeability. This allows endothelial cells to rearrange AJs during leukocyte transmigration or tip cell migration in angiogenesis without disrupting barrier permeability. Further dissection of these signaling processes will allow identification of the pathways that play barrier protective function downstream of SRC and separate them from the barrier disruptive pathways. This will help in the development of selective therapeutic approaches targeting vascular leakage in inflammatory diseases.
METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andrei V. Karginov (karginov@uic.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture, transfection, and infection
HPAEC-Human Pulmonary Artery Endothelial Cells (Lonza, cat. no. CC-2530; gender = female) and HUVEC-Human Umbilical Vein Endothelial cells (Lonza, cat. no. C2517A; gender = female) were grown in EGM™−2 BulletKit™ (Lonza, cat. no. CC-2162) with 10-% FBS at 37°C and 5% CO2. Low passage cells were purchased from Lonza, amplified and frozen at passage 5, and subsequently used for experiments between passages 6 and 8. Cells were grown on surfaces coated with 0.2% gelatin (plastic dishes and electrodes) or 0.2% gelatin and fibronectin (5 mg/L) when plated on glass coverslips. Stargazin-mVenus transfections were done on cells at 70–80% confluency using Fugene 6 reagent (Promega Corporation, cat. no. E2691) according to manufacturer’s protocol; experiments were performed 24 hours after transfection. The transfection of mEOS-paxillin was done using electroporation. Trypsinized cells were spun at 300 x g for 5 minutes, rinsed with PBS, spun for another 5 minutes, and resuspended in Optimem. Sheared salmon DNA (ssDNA) (Thermo Fisher cat. no. 15632011) (10 μg) was combined with mEOS-paxillin (1 μg) and 2×106 cells in a total of 200 μl Optimem, and subsequently incubated with on ice for 5 minutes. The transfection/cell mixture was then transferred to a 4mm cuvette and electroporated using a BioRad Gene Pulser Xcell™ with the following settings: 190 V, 950 μF, infinity. For adenoviral infections, cells were exposed to the virus for 24 hours prior to experiments.
METHOD DETAILS
Plasmids and Adenoviruses
Previously described Stargazin-mVenus DNA plasmid construct (Klomp et al., 2016) was used for transient transfection. Adenoviral VE cadherin constructs were a gift from Dr. William Muller (Northwestern University). RapR-LYN-cerulean-myc DNA construct was produced from RapR-LYN-GFP-myc (Chu et al., 2014) by site-directed mutagenesis as described (Karginov and Hahn, 2011; Ray et al., 2017). To generate adenoviral constructs, RapR-SRC-cerulean-myc (Karginov et al., 2014), RapR-SRC-as2-mCherry-myc (Klomp et al., 2016), RapR-SRC-as2-cerulean-myc (Klomp et al., 2016), RapR-SRC-R175L-cerulean-myc, RapR-LYN-cerulean-myc, and mCherry-FRB genes (Ray et al., 2017) were cloned into a pShuttle plasmid obtained from Addgene (Plasmid no. 16402). The RapR-SRC-as2 constructs contain Threonine 338 to Alanine mutation making them sensitive to the allele specific inhibitor 1NA-PP1 (1-Naphthyl-PP1). To clone mCherry-FRB and Y66S-GFP-FRB (colorless) into pShuttle vector, mCherry-FRB and Y66S-GFP were amplified from mCherry-FRB (Karginov et al., 2014) and iPEP-Y66S-GFP-FRB (Klomp et al., 2016) constructs using NotI-XFP FWD and FRB-EcoRV REV primers and cloned into pShuttle vector using NotI and EcoRV restriction sites. RapR-kinase genes were excised from their parental vectors using XhoI and HindIII restriction enzymes and cloned into pShuttle vector using the same restriction sites for ligation. VE cadherin GFP constructs (WT, Y658F, and Y731F) were generated as described previously (Gonzalez et al., 2016). Adenovirus production and amplification was done in collaboration with Dr. Jody Martin (Vector Core Facility at University of Illinois – Chicago). mEos2-paxillin-22 DNA construct was obtained from Addgene (Plasmid no. 57409). Adenoviruses for RapR-SRC, FRB, and VE cadherin were used for all over-expression studies. Stargazin and mEos2-paxillin-22 were transiently transfected into cells.
Antibodies and chemical reagents
The following antibodies were used: anti-GAPDH (Ambion, cat. no. AM4300), anti-mCherry (Biovision, cat. no. 5993–100), anti-GFP (Clontech, cat. no. 632381), anti-paxillin (BD Biosciences, cat. no. 610568), anti-phospho-paxillin (Y118) (Invitrogen, cat. no. 44–722G), anti-phosphotyrosine (4G10) (Millipore, cat. no 05–321), anti-cortactin (p80/85) (Millipore, cat. no. 05–180), anti-phospho-cortactin (Y466) (Abcam, cat. no. ab51073), anti-p130Cas (BD Biosciences cat. no. 610272), anti-phospho-p130Cas (Y249) (BD Biosciences, cat. no. 558401), anti-FAK (Cell Signaling, cat. no. 3285), anti-phospho-FAK (Y576/577) (Cell Signaling cat. no. 3281), anti-VE cadherin (Santa Cruz, cat. no. sc-6458), anti-phospho-VE cadherin (Y658) (Thermo Fisher, cat. no. 44–1144G), anti-phospho-VE cadherin (Y731) (Thermo Fisher, cat. no. 44–1145G), anti-phospho-VE cadherin (Y685) was a gift from the laboratory of Dr. Elisabetta Dejana (IFOM), anti-β-catenin (Santa Cruz, cat. no. 1496), anti-SRC (Santa Cruz, cat. no. 8056), anti-SRC (Cell Signaling cat. no. 2108), anti-phospho-SRC family (Y416) (Cell Signaling cat. no. D49G4), anti-P120-catenin (Santa Cruz, cat. no. sc-1101), phalloidin Alexa Fluor™ 647 (Thermo Fisher, cat. no. A22287), Cy™5 anti-rabbit (Jackson ImmunoResearch Laboratories, cat. no. 711–175–152), Alexa Fluor® 488 anti-goat (Jackson ImmunoResearch Laboratories, cat. no. 705–545-003), Alexa Fluor® 647 anti-goat (Jackson ImmunoResearch Laboratories, cat. no 705–607-003), and Alexa Fluor® 647 anti-mouse (Jackson ImmunoResearch Laboratories, cat. no 715–605-151). The following reagents were used: Protein G-coupled agarose beads (Millipore, cat. no. IP04–1.5ML), Rapamycin (LC Laboratories, cat. no. R5000) Leupeptin hemisulfate (Gold Biotechnology, cat. no. L-010–5), and Aprotinin (Gold Biotechnology, cat. no. A-655–25), Pierce™ BCA Protein Assay Kit (Thermo Fisher, cat. no. 23225), Fluorescein isothiocyanate-dextra (Sigma-Aldrich cat. no. 46944), D-erythro-sphingosine-1-phosphate (Avanti Polar Lipids, cat. no. 860492), 1-NA-PP1 (1-Naphthyl-PP1) (Cayman, cat. no. 10954), Human Alpha Thrombin (Factor IIa) (Enzyme Research Laboratories, cat. no. HT1002a), Fluorescein isothiocyanate-avidin (Invitrogen, cat. no. 43–4411), gelatin (Sigma-Aldrich cat. no. G2500), Salmon Sperm DNA Solution (Thermo Fisher cat. no. 15632011), Saracatinib (Santa Cruz, cat. no. sc-364607), 2X Laemmli Buffer (Bio Rad 1610737), and donkey serum (Jackson ImmunoResearch, cat. no. 017–000-121).
Trans-endothelial Electrical Resistance Measurements
Trans Electrical Resistance (TER) assays (Figure 1A, 4E, 5F, 6B, 6D, 6E–F, 7B, SI 1A, SI 1E, SI 2D, SI 5C, SI 6A–E) were performed as follows. Cells were plated on 0.2% gelatin-coated 8W10E+ electrodes (Applied Biophysics) overnight at 37°C. Changes in electrical resistance were measured at an impedance of 4000 Hz and a constant voltage using an Electric Cell Substrate Impedance Sensing system (Applied Biophysics). Cell lysates were prepared in parallel for all cells to verify expression of exogenous constructs and activation of RapR-SRC (WT and R175L) and RapR-LYN constructs via rapamycin when appropriate.
Live cell imaging
HPAE cells were plated on glass coverslips, coated overnight at 37°C with 0.2% gelatin and fibronectin (5 mg/L), for 24 hours (Figures 2A, 2D, 3B, 3E, 5E, SI 5A–B, and Movies 1–2). For the VE cadherin wide-field imaging (Figures 2A, 5E, SI 5A–B, and Movie 1), HPAE cells were infected with adenoviruses expressing RapR-SRC-cerulean/RapR-SRC-as2-cerulean, mCherry-FRB, and the indicated VE cadherin-GFP construct. For the protrusive activity imaging (Figures 3E and Movie 2), cells were infected with adenoviruses expressing RapR-SRC-cerulean and mCherry-FRB, and 4 hours later cells were transfected with Stargazin-mVenus using Fugene6 (Promega Corporation, cat. no. E2691). HPAE cells evaluated via live cell 3D-SIM (Figure 2D and 3B) were infected with adenoviruses expressing RapR-SRC-as2-mCherry, GFP(Y66S)-FRB (colorless), and VE cadherin-GFP. For both wide-field and 3D-SIM imaging, coverslips were placed into an Attufluor Cell Chamber (Invitrogen, cat. no. A78–16) with Leibovitz (L-15) medium (Sigma Aldrich, cat. no. L1518–500mL) supplemented with 5% FBS. Imaging in all cases was performed 24 hours post adenovirus infections.
Samples were imaged either via wide-field or 3D-SIM. Wide-field imaging was done using an Olympus IX-83 microscope controlled by Metamorph software and equipped with a heated stage (Warner Instruments), PlanApo N 60x TIRFM objective (oil, NA 1.45), Xcite 120 LED (Lumen Dynamics) light source, and Image EMX2 CCD (Hamamatsu) camera. HPAE cells were imaged live every 2 min with the following filter sets: YFP (excitation 514/10, emission 540/30), CFP (excitation 455/10, emission 485/30), and mCherry (excitation 572/35, emission 632/60). VE cadherin-GFP constructs were imaged using the YFP filter set to separate from cerulean tagged RapR-SRC-cerulean/RapR-SRC-as2-cerulean. Live cell 3D-SIM imaging was collected every 5 minutes and performed as described (Shao et al., 2011) at Janelia Research Center Advanced Imaging Facility. Samples were maintained at 37 °C and 5% CO 2 using a stage-top incubator (H301, Okolabs, Naples, Italy). Excitation patterns were produced using a phase-only spatial light modulator (Bolder Vision Optik, BVO AHWP3). A mask system was employed to select the 0 and ±1 diffraction orders which were then focused onto the back focal plane of a Zeiss Plan-Apochromat × 100 1.46 NA objective. Optimal interference contrast was achieved in the sample, by rotation of the polarity of the light to match the angle of the pattern using a liquid crystal variable retarder (LC, Meadowlark, SWIFT) and wave plates. Interference filters were used to collect the emissions and a pair of sCMOS cameras (Hamamatsu, Orca Flash 4.0 v2 sCMOS) were used to collect images. Raw image sets were reconstructed as described (Gustafsson et al., 2008).
Immunofluorescence
HPAE cells (Figures 2B, 2C, 2E, 3A, 3F–H, 4A, and SI 2A–B) were plated on glass coverslips, coated overnight at 37°C with 0.2% gelatin and fibronectin (5 mg/L), for 24 hours. Cells were then infected with adenoviruses expressing RapR-SRC-cerulean/RapR-SRC-as2-cerulean and mCherry-FRB for 24 hours prior to treatment. Following treatment, cells were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100, blocked in 20% donkey serum and 2% Bovine Serum Albumin (BSA). Samples were incubated with the designated primary antibodies (1:100) in 10% donkey serum and 1% BSA for 1 hour at room temperature or overnight at 4°C and then in secondary antibodies (1:200) in 0.5% BSA for 45 minutes. For samples with F-actin staining, Phalloidin Alexa Fluor™ 647 (Thermo Fisher, cat. no. A22287) was added at a 1:200 dilution during the final 20 minutes of secondary staining. Prepared coverslips were mounted on glass slides using Fluoromount-G™ (Southern Biotech, cat. no. 0100–01). Fixed and stained samples were imaged using wide-field, confocal, or via Structured Illumination Microscopy (SIM). Figure legends describe what type of microscopy was used. Wide-field images were collected using an Olympus IX-83 microscope with a PlanApo N 60X TIRFM objective (oil, NA 1.45) or UPlanSAPO 40X (silicon oil, NA 1.25) objective. Confocal images were collected on a Zeiss 880 equipped with a PlanApo 63X (oil, NA 1.4) objective. SIM images were collected on a Nikon N-SIM microscope with a CFI Apochromat TIRF 60X objective (oil, NA 1.49). Images were processed using FIJI (Schindelin et al., 2012), Metamorph, and IMARIS software. The programs used are indicated in the appropriate figure legends or specific analysis description below.
For using interferometric photoactivation and localization microscope (iPALM) (Shtengel et al., 2009) imaging (Figures 4B–C and SI 2C), HPAE cells were transfected with mEOS-paxillin via electroporation, then 16 hours post transfection cells were plated on a 25 mm diameter coverslips containing gold nanorod fiducial markers coated with 0.2% gelatin and fibronectin (5 mg/L). Once cells were allowed to attach (8 hours later), they were infected with adenoviruses expressing RapR-Src-cerulean-as2 and Y66S-GFP-FRB for 24 hours. Cells were mounted in dSTORM imaging buffer (Jones et al., 2011), and illuminated with 647nm excitation (for Alexa Fluor 647 imaging) or 561nm excitation (mEOS imaging), with power intensities of 1–3 kW/cm2.
Low power 405nm laser (2–10 W/cm2) was used to photoconvert mEOS molecules. Interference images were collected using two Nikon 60x APO TIRF (oil NA 1.49) objectives and recorded using Andor iXon (DU-897) EMCCD cameras as described by Shtengel et al., 30,000–60,0000 frames were captured at 30–50ms exposure for each image channel, and subjected to localization analysis, channel registration, and image rendering using custom software (PeakSelector) developed at Janelia Research Campus (Sage et al., 2015).
Endothelial permeability assay using FITC-Avidin
Assessment of endothelial permeability using FITC-Avidin (Figures 3C–D) was performed as previously described (Dubrovskyi et al., 2013). Preparation of gelatin conjugated with biotin was carried out following previously described protocol (Dubrovskyi et al., 2013). HPAE cells were plated for 48 hours on sterilized glass coverslips coated with biotinylated gelatin. Cells were treated with FITC-avidin (1:200) for 2 minutes. Coverslips were quickly fixed with pre-warmed 4% formaldehyde and stained for endogenous VE cadherin using Alexa647-conjugated secondary antibody. Images of VE cadherin-Alexa647 and FITC-Avidin were collected using Olympus IX-83 microscope with a PlanApo N 60x TIRFM objective (oil, NA 1.45) and analyzed using Metamorph software.
Cell lysate preparation and immunoprecipitation
All protein samples (Figures 1B, 4D, 5A–D, 6A, 6C, 7A, SI 1B–D, SI 2E, SI 3A–D, SI 4B–E, SI 5D, SI 6F) were collected in lysis buffer (20 mM HEPES-KOH, pH 7.8, 50 mM KCl, 100 mM NaCl, 1 mM EGTA, 1% NP40, 1 mM NaF, 0.1 mM Na3VO4, 0.033% ethanol, aprotinin 16 g/ml, and Leupeptin hemisulfate 3.2 g/mL), and centrifuged at 4,000 rpm, 4°C, for 10 minutes. Cleared protein samples were quantified via Pierce™ BCA Protein Assay Kit (Thermo Fisher, cat. no. 23225). Equivalent protein amounts were then combined with in 2X Sample Laemmli Buffer + 2% 2-Mercaptoethanol (Figures 1B, 5D, 6A, 6C, SI 1B–D, SI 2E, SI 3C, SI 4C–D, SI 5D, and SI 6F) or used for immunoprecipitation (Figures 4D, 5A–D, 7A, SI 3A–B, SI 3D, and SI 4B–E). For immunoprecipitation, protein G-conjugated agarose beads (Millipore, cat. no. IP04–1.5ML) were incubated with the antibody targeting immunoprecipitated protein at 4°C for 1 hour in lysis buffer + 20 mg/ml BSA. Subsequently, beads were washed with lysis buffer prior to adding cleared cell lysates. Lysates were incubated with the antibody/bead complex for 1.5 hours at 4°C. Beads were washed with the wash Buffe r (20 mM Tris-HCl, 1 mM DTT, 40 mM NaCl, 30 mM MgCl2, 1 mM NaF, 0.1 mM Na3VO4, 0.033% ethanol, aprotinin 16 g/ml, and Leupeptin hemisulfate 3.2 g/mL) and then re-suspended in 2X Sample Laemmli Buffer + 2% 2-Mercaptoethanol.
FITC-Dextran Endothelial Permeability Assay
HPAE cells were plated on Transwells (Corning Inc, cat. no. 353495) for 48 h (Figure 1C). To test for construct(s) expression, an equal number of cells/cm2 was plated on a tissue culture plate, from which cell lysates were collected and analyzed by Western Blot. Cells were infected with RapR-SRC-as2-cerulean-myc and mCherry-FRB or mCherry-FRB alone for 24 hours. Cells in the Transwell plate were treated with rapamycin (500 nM) for the designated time. Dextran (400 μg/ml, 40 kDa) labeled with FITC (25 μl) was added to the top chamber (containing 500 μl of media) 15 min prior to sample collection. Aliquots from both the top and bottom chambers were collected and the amount of FITC fluorescence was measured in duplicates. Fluorescence signal from the lower chamber was divided by the signal from the upper chamber. Values for each condition were normalized to their respective time 0. The average and standard deviation from 3 independent experiments were calculated. Significance was evaluated using a two-way ANOVA with repeated measures and a post-hoc test with a Bonferroni’s multiple comparisons test.
QUANTIFICATION AND STATISTICAL ANALYSIS
Trans-endothelial Electrical Resistance Measurements
The TER graphs in Figures 1A, 4E, 5F, 6B, 6D–F, 7B, and SI 6D all show the average resistance and 90% confidence interval. SI Figures: 1A, SI 1E, 2D, 5C, 6A–C, and 6E all show the average relative resistance and standard deviations. The average relative resistance was calculated by dividing the values post treatment value via its respective 0 value. All TER experiments were done in triplicate with 3 independent biological replicates. Statistical significance between average relative resistance was evaluated via a two-way ANOVA with repeated measures and a post-hoc test with a Bonferroni’s multiple comparisons adjustment (SI Figures: 1A, 2D, 5C, 6A–C and 6E).
Image Analysis
VE cadherin average intensity and area of AJs (Figures 2C and 2E) were analyzed using Metamorph software. First, a mask of the nucleus was made and dilated by 6.7 μm. This was then subtracted from the VE cadherin image to remove the perinuclear accumulation of VE cadherin. Next, a binary mask of the AJs was generated using the VE cadherin signal. The average intensity of VE cadherin was multiplied by binary mask to calculate the average intensity in the AJs. This value was divided by the total average intensity of the original unmodified image. The average intensity of VE cadherin and standard deviation were then normalized by the median value for the 0 min time point to calculate the average relative intensity. The binary mask of the VE cadherin in the AJs was also used to calculate the average area of VE cadherin. The average area and standard deviation were then normalized to the median value for 0 min time point to calculate the average relative area. The following number of fields of view were analyzed: 62 fields of view from 3 independent experiments for each treatment timepoint were analyzed for FRB only expressing cells; 85 fields of view from 4 independent experiments for each treatment timepoint were analyzed for RapR-SRC and FRB expressing cells. A blocked-ANOVA test was used to compare 0 and 30 min samples for each condition.
iPALM images (Figures 4C and SI 2C) were analyzed using the PeakSelector software package developed at Janelia Research Campus (Sage et al., 2015). The axial distributions of VE cadherin and paxillin were each measured relative to the z-location of the gold nanorod fiducial markers embedded in the coverslip. The molecule counts for both proteins were normalized to a maximum of 1. The amount of total VE cadherin and paxillin was summed from 4–6 areas within each region of interest (ROI). The average of 3 ROIs and the SEM were calculated and graphed as the average amount of VE cadherin and paxillin versus axial position in the sample.
Changes in protrusive activity (Figure 3E) were calculated as previously described (Karginov et al., 2014). In brief, Stargazin-mVenus construct was used to label the cell plasma membrane. Cells expressing Stargazin-mVenus and the adenovirus constructs (RapR-SRC-cerulean-myc and mCherry-FRB) were selected using epifluorescence imaging. Cells were only imaged if they were in a confluent monolayer. Time-lapse movies (N= 30 cells) were generated by collecting images every 2 min. Analysis was performed using Metamorph and CellGeo software (Tsygankov et al., 2014). Stargazin-mVenus images were used to create a binary mask of a cell via MovThresh software (Tsygankov et al., 2014). Protrusive activity (the sum of areas which undergo local extensions between consecutive movie frames) was analyzed using ProActive software (Tsygankov et al., 2014). To determine the change in protrusive activity, the protrusive activity at a given time point was divided by the average protrusive activity prior to rapamycin treatment. The average and 90% confidence interval for each time point of all cells treated in the same conditions was calculated.
For the comparison of FITC-Avidin between reticular and linear AJs (Figure 3D) the VE cadherin channel was used to designate reticular AJs (>4.5 μm wide) and linear AJs (<2.5 μm wide). The total integrated intensity of the FITC-Avidin was measured directly underneath the two types of AJs. The average total integrated intensity and standard deviation for each type of junction was calculated. For each AJ type 30 regions of interest from 3 independent experiments were measured. An equal number of reticular and linear junctions were imaged from each sample preparation. A t-test was used to compare the two types of AJs across experiments.
The analysis of VE cadherin accumulation in AJs in live HPAE cells (Figures 5E and SI 5A–B) was analyzed as follows: Metamorph software was used to create a binary mask of the cell cytoplasm using mCherry-FRB image for each time point. mCherry-FRB localizes diffusely through the cell cytoplasm and is mostly absent from AJs. The cytoplasm binary mask was then multiplied by the VE cadherin YFP image for the corresponding time point. The resulting image was used to calculate the average intensity of VE cadherin in the cytoplasm. This value was divided by the average intensity of the total VE cadherin image, producing the relative average intensity of cytoplasmic versus total VE cadherin for each time point. The relative intensity for each time point was divided by the average value for the frames prior to rapamycin treatment, providing the normalized cytoplasmic VE cadherin signal. The following number of fields of view from 3 independent experiments were imaged for each VE cadherin construct: Wild-Type N=15, Y658F N=21, and Y731F N=28. For each VE cadherin construct, the average normalized cytoplasmic VE cadherin signal and the 90% confidence intervals were calculated for each time point. A two-way ANOVA with repeated measures and a post-hoc test with a Bonferroni’s multiple comparisons test was used for statistical analysis.
Protein expression and phosphorylation analysis
Western blot band intensities were analyzed using FIJI (Schindelin et al., 2012). All phosphorylation intensities were standardized to their appropriate total protein levels by dividing the signal intensity for the phospho-specific antibody in the sample by the signal intensity for antibody recognizing total analyzed protein in the sample. For comparing the ability of SRC and LYN to phosphorylate a panel of endogenous substrates (Figure 1B and SI 1B–C) all normalized phosphorylation values were standardized to the appropriate 4 h RapR-SRC value. Averages from 4 independent experiments were evaluated by a two-way ANOVA with repeated measures and a post-hoc test with a Bonferroni’s multiple comparisons test was used to compare the averages of all time points, except 4 h between SRC and LYN induced phosphorylation changes. A Wilcoxon-Mann-Whitney test was used to compare the 4 h time points between SRC and LYN. In the experiments assessing the effect of SRC and LYN activation on VE cadherin phosphorylation (Figures 5A–C and SI 4C–E), the basal level of VE cadherin phosphorylation was undetectable for some of the phosphorylation sites. Thus, all normalized phosphorylation values were standardized to the 4 h phosphorylation signal for each experiment, setting the 4 h value to 100%. An average of 3 experiments for LYN and 5 for SRC experiments were calculated and a two-way ANOVA with repeated measures and a post-hoc test with a Bonferroni’s multiple comparisons test were used to compare the averages of all time points, except 4 h, to time 0. For reversal of SRC activation with 1NA-PP1 (Figures 5D and SI 4B) the vehicle treated and rapamycin/1NA-PP1 treated values were standardized to the rapamycin/vehicle phosphorylation level for each phospho-site. A two-sample T-Test was used to compare the vehicle treated to the rapamycin/1NA-PP1 treated samples from an average of 3 independent experiments. Relative co-immunoprecipitation (percent) of p120-catenin and β-catenin with VE cadherin (Figures SI 3A–B) was determined by dividing the amount of each catenin in the immunoprecipitation sample by the amount of VE cadherin in the same sample for the corresponding time point. All time points for each experiment were then normalized to time 0 and time 0 was set to 100%. The average and standard deviation of 4 independent experiments was determined and a one-way ANOVA with repeated measures and a post-hoc test with a Dunnett’s multiple comparisons test. For S1P and α-thrombin treatments (Figures 6A, 6C, and 7A) all time points were standardized to time 0 and time 0 was set at 100%. The average and standard deviation for each time point was calculated for 3–4 independent experiments. Data was analyzed using a one-way ANOVA with a Dunnett’s multiple comparisons test to compare each time point to time 0.
Statistical Tests
All statistical analyses were performed using GraphPad (GraphPad Software Inc, www.graphpad.com). No methods were applied for determining whether the data met assumptions of the statistical approach.
Supplementary Material
Movie 1: Changes in VE cadherin localization following activation of SRC (related to Figure 2A). A monolayer of HPAE cells expressing RapR-SRC-cerulean-myc, mCherry-FRB, and VE cadherin-GFP was imaged live every 2 minutes for 6 hours. Activation of SRC is marked by the appearance of “Rapamycin Added” sign. All exogenous proteins were expressed using adenoviral transduction.
Movie 2: Changes in protrusive activity of endothelial cells induced by activation of SRC (related to Figure 3E). A monolayer of HPAE cells expressing RapR-SRC-cerulean-myc, mCherry-FRB and mVenus-Stargazin was imaged live every 2 minutes for 4 hours. Activation of SRC is marked by the appearance of “Rapamycin Added” sign. All exogenous proteins were expressed using adenoviral transduction.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-paxillin | BD Biosciences | cat. no. 610568; RRID: AB_397917 |
| anti-phospho-paxillin (Y118) | Thermo Fisher | cat. no. 44–722G; RRID: AB_2533733 |
| anti-phosphotyrosine (4G10) | Millipore | cat. no. 05–321; RRID: AB 309678 |
| anti-cortactin (p80/85) clone 4F11 | Millipore | cat. no. 05–180 RRID: AB_309647 |
| anti-phospho-cortactin (Y466) | Abeam | cat. no. ab51073 RRID: AB_869231 |
| anti-p130Cas | BD Biosciences | cat. no. 610272 RRID: AB_397667 |
| anti-phospho-p130Cas (Y249) | BD Biosciences | cat. no. 558401 RRID: AB 647286 |
| anti-SRC | Cell Signaling | cat. no. 2108; RRID: AB_10695298 |
| anti-SRC | Santa Cruz | cat. no. 8056; RRID: AB_627306 |
| anti-phospho-SRC family (Y416) | Cell Signaling | cat. no. D49G4; RRID: AB_10013641 |
| anti-VE cadherin | Santa Cruz | cat. no. sc-6458; RRID: AB_2077955 |
| anti-phospho-VE cadherin (Y658) | Thermo Fisher | cat. no. 44–1144G; RRID: AB_2533583 |
| anti-phospho-VE cadherin (Y731) | Thermo Fisher | cat. no. 44–1145G; RRID: AB_2533584 |
| anti-phospho-VE cadherin (Y685) | gift from the laboratory of Dr. Elisabetta Dejana (IFOM) | Orsenigo et al., 2012 |
| anti-β-catenin | Santa Cruz | cat. no. 1496; RRID: AB_1249372 |
| anti-P120-catenin | Santa Cruz | cat. no. sc-1101; RRID: AB_632091 |
| anti-GAPDH | Ambion | cat. no. AM4300; RRID: AB_437392 |
| anti-mCherry | Biovision | cat. no. 5993–100; RRID: AB_1975001 |
| anti-GFP | Clontech | cat. no. 63238; 1 RRID: AB_2313808 |
| Bacterial and Virus Strains | ||
| RapR-SRC-as2-cerulean-myc Adenovirus | Klomp et al., 2016 | |
| RapR-SRC-cerulean-myc Adenovirus | This Paper | N/A |
| RapR-SRC-R175L-cerulean-myc Adenovirus | This Paper | N/A |
| RapR-LYN-cerulean-myc Adenovirus | This Paper | N/A |
| RapR-SRC-as2-mCherry-myc Adenovirus | This Paper | N/A |
| mCherry-FRB Adenovirus | This Paper | N/A |
| GFP(Y66S)-FRB Adenovirus | This Paper | N/A |
| VE cadherin-GFP Adenovirus | Dr. William Muller (Northwestern University) | Gonzalez et al., 2016 |
| VE cadherin-GFP Y658F Adenovirus | Dr. William Muller (Northwestern University) | Gonzalez et al., 2016 |
| VE cadherin-GFP Y731F Adenovirus | Dr. William Muller (Northwestern University) | Gonzalez et al., 2016 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| D-erythro-sphingosine-1-phosphate (CAS 26993–30–6) | Avanti Polar Lipids | cat. no. 860492 |
| Human Alpha Thrombin (Factor lla) (CAS 9002–04–4) | Enzyme Research Laboratories | cat. no. HT1002a |
| Fluorescein isothiocyanate-avidin | Invitrogen | cat. no. 43–4411 |
| normal donkey serum | Jackson ImmunoResearch | cat. no. 017–000–121 |
| Cy™5 anti-rabbit | Jackson ImmunoResearch Laboratories | cat. no. 711–175–152 RRID: AB_2340607 |
| Alexa Fluor® 488 anti-goat | Jackson ImmunoResearch Laboratories | cat. no. 705–545–003 RRID: AB_2340428 |
| Alexa Fluor® 647 anti-goat | Jackson ImmunoResearch Laboratories | cat. no 705–607–003 RRID: AB_2340439 |
| Alexa Fluor® 647 anti-mouse | Jackson ImmunoResearch Laboratories | cat. no 715–605–151 RRID: AB_2340863 |
| Rapamycin (CAS 53123–88–9) | LC Laboratories | cat. no. R5000 |
| Saracatinib (CAS 379231–04–6) | Santa Cruz | cat. no. sc-364607 |
| 1-Naphthyl-PP1 (CAS 221243–82–9) | Cayman | cat. no. 10954 |
| Fluorescein isothiocyanate-dextra | Sigma-Aldrich | cat. no. 46944 |
| Gelatin | Sigma-Aldrich | cat. no. G2500 |
| Phalloidin Alexa Fluor™ 647 | Thermo Fisher | cat. no. A22287 RRID: AB_2620155 |
| Experimental Models: Cell Lines | ||
| HPAEC-Human Pulmonary Artery Endothelial cells - Passage 6–8 |
Lonza | cat. no. CC-2530 |
| HUVEC-Human Umbilical Vein Endothelial cells - Passage 6–8 |
Lonza | cat. no. C2517A |
| Oligonucleotides | ||
| Notl-XFP FWD 5’- atatatGCGGCCGCatggtgagcaagggcgagga −3’ | This Paper | N/A |
| FRB-EcoRV REV 5’- atatatGATATCttaactagtctttgagattc −3’ | This Paper | N/A |
| Recombinant DNA | ||
| RapR-LYN-cerulean-myc plasmid | This Paper | N/A |
| RapR-SRC-cerulean-myc plasmid | Karginov et al., 2014 | |
| RapR-SRC-as2-cerulean-myc plasmid | Klomp et al., 2016 | |
| RapR-SRC-as2-mCherry-myc plasmid | Klomp et al., 2016 | |
| RapR-SRC-R175L-cerulean-myc plasmid | Karginov et al., 2014 | |
| mCherry-FRB plasmid | Karginov et al., 2014 | |
| Y66S-GFP-FRB plasmid | This Paper | |
| pShuttle vector | Addgene | Plasmid no. 16402 |
| mEos2-Paxillin-22 | Addgene | Plasmid no. 57409 |
| Stargazin-mVenus | Klomp et al., 2016 | |
| Software and Algorithms | ||
| Metamorph Image Analysis Software | https://www.moleculardevices.com/products/cellular-imaging-systems/acquisition-and-analysis-software/metamorph-microscopy#gref | |
| Imaris Image Analysis Software | http://www.bitplane.com/ | |
| GraphPad Prism Software | www.graphpad.com | |
| FIJI - ImageJ | ||
| CellGeo software | Schindelin et al., 2012 | |
| MovThresh software | Tsygankov et al., 2014 | |
| ProActive software | Tsygankov et al., 2014 | |
| PeakSelector | Sage et al., 2015 | |
SIGNIFICANCE.
Despite an understanding of the function of SRC kinase in endothelial cells, significant gaps remain in defining its role in regulating the plasticity of cell-cell junctions. The present study shows the value of using a chemical biology method to identify a previously unrecognized function of SRC in regulating AJs and the endothelial barrier. We also established differences between SRC and its close homolog LYN, demonstrating that despite high similarity between SFKs, they play distinct roles in endothelial cells. Importantly, mechanisms uncovered by engineered tools were confirmed in physiologically stimulated endothelial cells showing the reliability of the approach in defining the biology of endothelial cells. Further development of approaches that enable activation of individual signaling cascades downstream of SRC will enable identification of molecular pathways specifically mediating barrier enhancement or disruption.
Highlights.
Highlights are 3–4 bullet points of no more than 85 characters in length, including spaces, and they summarize the core results of the paper in order to allow readers to quickly gain an understanding of the main take-home messages.
SRC activation causes temporally distinct effects on the endothelial cell barrier
Initially SRC causes endothelial barrier enhancement and VE cadherin rearrangement
VE cadherin phosphorylation on Y731 is required for SRC mediated barrier enhancement
Prolonged SRC activity cause barrier disruption
ACKNOWLEDGEMENTS
We extend our appreciation to Dr. John O’Bryan, Dr. Maulik Patel, and Dr. Anne-Marie Ray for helpful insights during the pursuit of this project; as well as to Dr. Jeffrey A. Klomp for assistance with statistical analysis. We would also like to thank Aaron Taylor and Satya Khuon at Janelia Research Campus for assistance with live cell SIM and technical support respectively. iPALM imaging was performed in collaboration with the Advanced Imaging Center at Janelia Research Campus, supported by the Gordon and Betty Moore Foundation and the Howard Hughes Medical Institute. We are grateful to NIH, the Chicago Biomedical Consortium, and University of Illinois at Chicago Center for Clinical and Translational Science for funding (R21CA159179 and CBC Pilot Grant, R01 grant R01GM118582 from NIGMS to A.V.K., NIH T32 HL007829–22 to A.B.M., UIC CCTS (grant # UL1TR000050) to J.E.K).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing financial interests
REFERENCES
- Adam AP, Sharenko AL, Pumiglia K, and Vincent PA (2010). Src-induced Tyrosine Phosphorylation of VE-cadherin Is Not Sufficient to Decrease Barrier Function of Endothelial Monolayers. J. Biol. Chem 285, 7045–7055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernheim-Groswasser A, Wiesner S, Golsteyn RM, Carlier M-F, and Sykes C (2002). The dynamics of actin-based motility depend on surface parameters. Nature 417, 308–311. [DOI] [PubMed] [Google Scholar]
- Birukova AA, Malyukova I, Poroyko V, and Birukov KG (2007a). Paxillin-beta-catenin interactions are involved in Rac/Cdc42-mediated endothelial barrier-protective response to oxidized phospholipids. Am. J. Physiol. Lung Cell. Mol. Physiol 293, L199–211. [DOI] [PubMed] [Google Scholar]
- Birukova AA, Chatchavalvanich S, Oskolkova O, Bochkov VN, and Birukov KG (2007b). Signaling pathways involved in OxPAPC-induced pulmonary endothelial barrier protection. Microvasc. Res 73, 173–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukova AA, Tian X, Tian Y, Higginbotham K, and Birukov KG (2013). Rap-afadin axis in control of Rho signaling and endothelial barrier recovery. Mol. Biol. Cell 24, 2678–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukova AA, Shah AS, Tian Y, Moldobaeva N, and Birukov KG (2016a). Dual role of vinculin in barrier-disruptive and barrier-enhancing endothelial cell responses. Cell. Signal 28, 541–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukova AA, Shah AS, Tian Y, Gawlak G, Sarich N, and Birukov KG (2016b). Selective Role of Vinculin in Contractile Mechanisms of Endothelial Permeability. Am. J. Respir. Cell Mol. Biol 55, 476–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain RJ, Vanhaesebroeck B, and Ridley AJ (2010). The PI3K p110α isoform regulates endothelial adherens junctions via Pyk2 and Rac1. J. Cell Biol. 188, 863–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu P-H, Tsygankov D, Berginski ME, Dagliyan O, Gomez SM, Elston TC, Karginov AV, and Hahn KM (2014). Engineered kinase activation reveals unique morphodynamic phenotypes and associated trafficking for Src family isoforms. Proc. Natl. Acad. Sci 111, 12420–12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deakin NO, and Turner CE (2008). Paxillin comes of age. J. Cell Sci. 121, 2435–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejana E (2004). Endothelial cell–cell junctions: happy together. Nat. Rev. Mol. Cell Biol. 5, 261–270. [DOI] [PubMed] [Google Scholar]
- Dejana E, and Orsenigo F (2013). Endothelial adherens junctions at a glance. J. Cell Sci. 126, 2545–2549. [DOI] [PubMed] [Google Scholar]
- Dejana E, Orsenigo F, and Lampugnani MG (2008). The role of adherens junctions and VE-cadherin in the control of vascular permeability. J. Cell Sci. 121, 2115–2122. [DOI] [PubMed] [Google Scholar]
- Dubrovskyi O, Tian X, Poroyko V, Yakubov B, Birukova AA, and Birukov KG (2012). Identification of paxillin domains interacting with β-catenin. FEBS Lett. 586, 2294–2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubrovskyi O, Birukova AA, and Birukov KG (2013). Measurement of local permeability at subcellular level in cell models of agonist-and ventilator-induced lung injury. Lab. Investig. J. Tech. Methods Pathol. 93, 254–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliceiri BP, Paul R, Schwartzberg PL, Hood JD, Leng J, and Cheresh DA (1999). Selective Requirement for Src Kinases during VEGF-Induced Angiogenesis and Vascular Permeability. Mol. Cell 4, 915–924. [DOI] [PubMed] [Google Scholar]
- Eliceiri BP, Puente XS, Hood JD, Stupack DG, Schlaepfer DD, Huang XZ, Sheppard D, and Cheresh DA (2002). Src-mediated coupling of focal adhesion kinase to integrin αvβ5 in vascular endothelial growth factor signaling. J. Cell Biol. 157, 149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Martín L, Marcos-Ramiro B, Bigarella CL, Graupera M, Cain RJ, Reglero-Real N, Jiménez A, Cernuda-Morollón E, Correas I, Cox S, et al. (2012). Crosstalk between reticular adherens junctions and platelet endothelial cell adhesion molecule-1 regulates endothelial barrier function. Arterioscler. Thromb. Vasc. Biol 32, e90–102. [DOI] [PubMed] [Google Scholar]
- Frame MC (2002). Src in cancer: deregulation and consequences for cell behaviour. Biochim. Biophys. Acta BBA -Rev. Cancer 1602, 114–130. [DOI] [PubMed] [Google Scholar]
- Gavard J, and Gutkind JS (2006). VEGF controls endothelial-cell permeability by promoting the β-arrestin-dependent endocytosis of VE-cadherin. Nat. Cell Biol. 8, 1223–1234. [DOI] [PubMed] [Google Scholar]
- Gavard J, Patel V, and Gutkind JS (2008). Angiopoietin-1 Prevents VEGF-Induced Endothelial Permeability by Sequestering Src through mDia. Dev. Cell 14, 25–36. [DOI] [PubMed] [Google Scholar]
- Gonzalez AM, Cyrus BF, and Muller WA (2016). Targeted Recycling of the Lateral Border Recycling Compartment Precedes Adherens Junction Dissociation during Transendothelial Migration. Am. J. Pathol 186, 1387–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Yuan SY, and Wu MH (2013). Involvement of FAK and Src in microvascular hyperpermeability caused by fibrinogen-gamma C-terminal fragments. FASEB J. 27, 896.9–896.9. [Google Scholar]
- Han J, Zhang G, Welch EJ, Liang Y, Fu J, Vogel SM, Lowell CA, Du X, Cheresh DA, Malik AB, et al. (2013). A critical role for Lyn kinase in strengthening endothelial integrity and barrier function. Blood 122, 4140–4149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y-T, Chen S-U, Chou C-H, and Lee H (2008). Sphingosine 1-phosphate induces platelet/endothelial cell adhesion molecule-1 phosphorylation in human endothelial cells through cSrc and Fyn. Cell. Signal 20, 1521–1527. [DOI] [PubMed] [Google Scholar]
- Karginov AV, Ding F, Kota P, Dokholyan NV, and Hahn KM (2010). Engineered allosteric activation of kinases in living cells. Nat. Biotechnol 28, 743–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karginov AV, Tsygankov D, Berginski M, Chu P-H, Trudeau ED, Yi JJ, Gomez S, Elston TC, and Hahn KM (2014). Dissecting motility signaling through activation of specific Src-effector complexes. Nat. Chem. Biol 10, 286–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim L, and Wong TW (1995). The cytoplasmic tyrosine kinase FER is associated with the catenin-like substrate pp120 and is activated by growth factors. Mol. Cell. Biol 15, 4553–4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim LC, Song L, and Haura EB (2009). Src kinases as therapeutic targets for cancer. Nat. Rev. Clin. Oncol 6, 587–595. [DOI] [PubMed] [Google Scholar]
- Kim Y-M, Kim Y-M, Lee YM, Kim H-S, Kim JD, Choi Y, Kim K-W, Lee S-Y, and Kwon Y-G (2002). TNF-related Activation-induced Cytokine (TRANCE) Induces Angiogenesis through the Activation of Src and Phospholipase C (PLC) in Human Endothelial Cells. J. Biol. Chem 277, 6799–6805. [DOI] [PubMed] [Google Scholar]
- Klomp JE, Huyot V, Ray A-M, Collins KB, Malik AB, and Karginov AV (2016). Mimicking transient activation of protein kinases in living cells. Proc. Natl. Acad. Sci 113, 14976–14981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knezevic N, Tauseef M, Thennes T, and Mehta D (2009). The G protein βγ subunit mediates reannealing of adherens junctions to reverse endothelial permeability increase by thrombin. J. Exp. Med 206, 2761–2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarova Y, and Malik AB (2010). Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu. Rev. Physiol 72, 463–493. [DOI] [PubMed] [Google Scholar]
- Komarova YA, Mehta D, and Malik AB (2007). Dual Regulation of Endothelial Junctional Permeability. Sci. Signal 2007, re8. [DOI] [PubMed] [Google Scholar]
- Lambeng N, Wallez Y, Rampon C, Cand F, Christé G, Gulino-Debrac D, Vilgrain I, and Huber P (2005). Vascular Endothelial–Cadherin Tyrosine Phosphorylation in Angiogenic and Quiescent Adult Tissues. Circ. Res 96, 384–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilien J, and Balsamo J (2005). The regulation of cadherin-mediated adhesion by tyrosine phosphorylation/dephosphorylation of β-catenin. Curr. Opin. Cell Biol. 17, 459–465. [DOI] [PubMed] [Google Scholar]
- Liu Y, and Senger DR (2004). Matrix-specific activation of Src and Rho initiates capillary morphogenesis of endothelial cells. FASEB J. 18, 457–468. [DOI] [PubMed] [Google Scholar]
- Lum H, and Malik AB (1994). Regulation of vascular endothelial barrier function. Am. J. Physiol. -Lung Cell. Mol. Physiol 267, L223–L241. [DOI] [PubMed] [Google Scholar]
- Mehta D, and Malik AB (2006). Signaling mechanisms regulating endothelial permeability. Physiol. Rev 86, 279–367. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay D, Tsiokas L, Zhou X-M, Foster D, Brugge JS, and Sukhatme VP (1995). Hypoxic induction of human vascular endothelial growth factor expression through c-Src activation. Nature 375, 577. [DOI] [PubMed] [Google Scholar]
- Muller WA (2016). Transendothelial migration: unifying principles from the endothelial perspective. Immunol. Rev 273, 61–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller WA, Weigl SA, Deng X, and Phillips DM (1993). PECAM-1 is required for transendothelial migration of leukocytes. J. Exp. Med 178, 449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins RD, Heuser JA, and Pollard TD (1998). The interaction of Arp2/3 complex with actin: Nucleation, high affinity pointed end capping, and formation of branching networks of filaments. Proc. Natl. Acad. Sci. U. S. A 95, 6181–6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Nieuw Amerongen GP, and van Hinsbergh VWM (2002). Targets for pharmacological intervention of endothelial hyperpermeability and barrier function. Vascul. Pharmacol 39, 257–272. [DOI] [PubMed] [Google Scholar]
- Okutani D, Lodyga M, Han B, and Liu M (2006). Src protein tyrosine kinase family and acute inflammatory responses. Am. J. Physiol. -Lung Cell. Mol. Physiol 291, L129–L141. [DOI] [PubMed] [Google Scholar]
- Orsenigo F, Giampietro C, Ferrari A, Corada M, Galaup A, Sigismund S, Ristagno G, Maddaluno L, Young Koh G, Franco D, et al. (2012). Phosphorylation of VE-cadherin is modulated by haemodynamic forces and contributes to the regulation of vascular permeability in vivo. Nat. Commun 3, 1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piedra J, Miravet S, Castaño J, Pálmer HG, Heisterkamp N, Herreros A.G. de, and Duñach M (2003). p120 Catenin-Associated Fer and Fyn Tyrosine Kinases Regulate β-Catenin Tyr-142 Phosphorylation and β-Catenin-α-Catenin Interaction. Mol. Cell. Biol 23, 2287–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter MD, Barbero S, and Cheresh DA (2005). Tyrosine phosphorylation of VE-cadherin prevents binding of p120-and beta-catenin and maintains the cellular mesenchymal state. J. Biol. Chem 280, 31906–31912. [DOI] [PubMed] [Google Scholar]
- Seebach J, Taha AA, Lenk J, Lindemann N, Jiang X, Brinkmann K, Bogdan S, and Schnittler H-J (2015). The CellBorderTracker, a novel tool to quantitatively analyze spatiotemporal endothelial junction dynamics at the subcellular level. Histochem. Cell Biol. [DOI] [PubMed] [Google Scholar]
- Sun X, Shikata Y, Wang L, Ohmori K, Watanabe N, Wada J, Shikata K, Birukov KG, Makino H, Jacobson JR, et al. (2009). Enhanced interaction between focal adhesion and adherens junction proteins: involvement in sphingosine 1-phosphate-induced endothelial barrier enhancement. Microvasc. Res 77, 304–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Y, Tian X, Gawlak G, Sarich N, Sacks DB, Birukova AA, and Birukov KG (2016). Role of IQGAP1 in endothelial barrier enhancement caused by OxPAPC. Am. J. Physiol. Lung Cell. Mol. Physiol ajplung.00095.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vouret-Craviari V, Bourcier C, Boulter E, and Obberghen-Schilling EV (2002). Distinct signals via Rho GTPases and Src drive shape changes by thrombin and sphingosine-1-phosphate in endothelial cells. J. Cell Sci. 115, 2475–2484. [DOI] [PubMed] [Google Scholar]
- Wallez Y, Cand F, Cruzalegui F, Wernstedt C, Souchelnytskyi S, Vilgrain I, and Huber P (2006). Src kinase phosphorylates vascular endothelial-cadherin in response to vascular endothelial growth factor: identification of tyrosine 685 as the unique target site. Oncogene 26, 1067–1077. [DOI] [PubMed] [Google Scholar]
- Wessel F, Winderlich M, Holm M, Frye M, Rivera-Galdos R, Vockel M, Linnepe R, Ipe U, Stadtmann A, Zarbock A, et al. (2014). Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-cadherin. Nat. Immunol 15, 223–230. [DOI] [PubMed] [Google Scholar]
- Wilson CW, and Ye W (2014). Regulation of vascular endothelial junction stability and remodeling through Rap1-Rasip1 signaling. Cell Adhes. Migr 8, 76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Craig AWB, Greer P, Miller M, Anastasiadis PZ, Lilien J, and Balsamo J (2004). Continuous association of cadherin with beta-catenin requires the non-receptor tyrosine-kinase Fer. J. Cell Sci. 117, 3207–3219. [DOI] [PubMed] [Google Scholar]
- Yuan SY (2002). Protein kinase signaling in the modulation of microvascular permeability. Vascul. Pharmacol 39, 213–223. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie 1: Changes in VE cadherin localization following activation of SRC (related to Figure 2A). A monolayer of HPAE cells expressing RapR-SRC-cerulean-myc, mCherry-FRB, and VE cadherin-GFP was imaged live every 2 minutes for 6 hours. Activation of SRC is marked by the appearance of “Rapamycin Added” sign. All exogenous proteins were expressed using adenoviral transduction.
Movie 2: Changes in protrusive activity of endothelial cells induced by activation of SRC (related to Figure 3E). A monolayer of HPAE cells expressing RapR-SRC-cerulean-myc, mCherry-FRB and mVenus-Stargazin was imaged live every 2 minutes for 4 hours. Activation of SRC is marked by the appearance of “Rapamycin Added” sign. All exogenous proteins were expressed using adenoviral transduction.